Embed Size (px)
Citation preview

Molecular and Cellular Pathobiology
BRCA1 Deficiency Exacerbates Estrogen-Induced DNADamage and Genomic Instability
Kienan I. Savage1, Kyle B. Matchett1, Eliana M. Barros1, Kevin M. Cooper2, Gareth W. Irwin1, Julia J. Gorski1,Katy S. Orr1, Jekaterina Vohhodina1, Joy N. Kavanagh1, Angelina F. Madden1, Alexander Powell1,2,LorenzoManti3, Simon S.McDade1, Ben Ho Park4, KevinM. Prise1, Stuart A.McIntosh1, Manuel Salto-Tellez1,Derek J. Richard5, Christopher T. Elliott2, and D. Paul Harkin1
AbstractGermline mutations in BRCA1 predispose carriers to a high incidence of breast and ovarian cancers. BRCA1
functions to maintain genomic stability through critical roles in DNA repair, cell-cycle arrest, and tran-scriptional control. A major question has been why BRCA1 loss or mutation leads to tumors mainly inestrogen-regulated tissues, given that BRCA1 has essential functions in all cell types. Here, we report thatestrogen and estrogen metabolites can cause DNA double-strand breaks (DSB) in estrogen receptor-a–negative breast cells and that BRCA1 is required to repair these DSBs to prevent metabolite-inducedgenomic instability. We found that BRCA1 also regulates estrogen metabolism and metabolite-mediated DNAdamage by repressing the transcription of estrogen-metabolizing enzymes, such as CYP1A1, in breast cells.Finally, we used a knock-in human cell model with a heterozygous BRCA1 pathogenic mutation to show howBRCA1 haploinsufficiency affects these processes. Our findings provide pivotal new insights into why BRCA1mutation drives the formation of tumors in estrogen-regulated tissues, despite the general role of BRCA1 inDNA repair in all cell types. Cancer Res; 74(10); 2773–84. �2014 AACR.
IntroductionBRCA1 is a tumor suppressor protein that functions to
preserve genomic stability by regulating key cellular processes,including homologous recombination (HR)-mediated DNArepair, cell-cycle checkpoint control, transcriptional regula-tion, chromatin remodeling, and postreplicative repair (1–3).Despite playing a role in processes essential to all cells, BRCA1mutation predisposes to tumors predominantly in estrogen-regulated tissues, such as the breasts and ovaries. Indeed,germline mutations in a single BRCA1 allele confer a lifetimerisk of up to 90% of developing breast cancer and 30% to 40% ofovarian cancer (4, 5).
Several observations suggest that estrogen has an importantrole in the development of BRCA1-dependent breast cancer.Pre- or postmenopausal oophorectomy in BRCA1 mutationcarriers significantly reduces the risk of breast cancer onsetand recurrence (6–8). Furthermore, pregnancy increases therisk of early-onset breast cancer in BRCA1mutation carriers, incontrast with noncarriers for whom pregnancy is protective(9). It has also been reported that BRCA1 represses the expres-sion of CYP19A1 (aromatase), which converts androgens tobioactive estrogens (10). Thus BRCA1 loss may increase CYP19expression and subsequent estrogen production, further driv-ing tumorigenesis (11).
Estrogen is postulated to promote tumorigenesis directlythrough stimulation of the estrogen receptor-a (ER-a) and thedownstream activation of promitogenic transcriptional pro-grams. However, this is confounded by observations thatapproximately 70% to 80% of BRCA1-mutated breast tumorsare ER-a negative (12, 13). Furthermore, BRCA1 drives ER-aexpression, suggesting the role of estrogen in BRCA1-depen-dent tumor, development may be independent of ER-a (14).Consistent with this, estradiol (E2; the predominant estrogen)induces tumor formation in ER-a knockout mice (15). In thesemice, reduction of endogenous E2, by either oophorectomy ortreatment with aromatase inhibitors, delayed tumorigenesis,whereas the ER-a antagonist fulvestrant had no effect (15).
The endogenous conversion of estrogen to genotoxic meta-bolites has been reported as an alternative, potentially ER-aindependentmechanism for estrogen-dependent breast tumor-igenesis. Estrogen is hydroxylated to form the catechol estro-gens 2-hydroxyestradiol (2-OHE1(E2)) and 4-hydroxyestradiol
Authors' Affiliations: 1Centre for Cancer Research and Cell Biology;2Institute for Global Food Security, Queen's University Belfast, Belfast,United Kingdom; 3Department of Physics, Radiation Biophysics Labora-tory, University of Naples Federico II, Naples, Italy; 4The Sidney KimmelComprehensive Cancer Center, The Johns Hopkins University School ofMedicine, Baltimore, Maryland; and 5Institute of Health and BiomedicalInnovation, Queensland University of Technology, Kelvin Grove, Brisbane,Australia
Note: Supplementary data for this article are available at Cancer ResearchOnline (http://cancerres.aacrjournals.org/).
K.I. Savage and K.B. Matchett contributed equally to this work.
Corresponding Authors: Kienan I. Savage and D. Paul Harkin, Queen'sUniversity Belfast, 97 Lisburn Road, Belfast, Northern Ireland, BT9 7BL,United Kingdom. Phone: 442890972934; Fax: 442890972776; E-mail:[email protected] and [email protected]
doi: 10.1158/0008-5472.CAN-13-2611
�2014 American Association for Cancer Research.
CancerResearch
www.aacrjournals.org 2773
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611

(4-OHE1(E2)), a process that is catalyzed by a number ofcytochrome (CYP) P450 enzymes, including CYP1A1, CYP1A2,CYP1B1, and CYP3A4. The catechol estrogens are furtheroxidized (by the same enzymes) into semiquinone and quinoneforms, the latter of which can react with DNA to form adducts.Interestingly, urinary levels of 2-OHE2 and 4-OHE2 are elevatedin patients with breast cancer compared with healthy controls(16), and 4-OHE2 concentrations have been reported to be up tothree times higher in breast cancer biopsies compared withnormal breast tissue (17). Moreover, in vivo studies have dem-onstrated that exogenous 2-OHE2 and 4-OHE2 can inducekidney and uterine cancers in mice (18, 19). The DNA adductsinduced by thesemetabolites produce apurinic sites in the DNAthat require repair, error-prone repair of which can lead to A-TtoG-Cmutations inDNA in the formofG.Theteroduplexes (20–22). Furthermore, high levels of depurinated estrogen adductshave been observed in serum and urine samples from patientswith breast cancer and women with a strong family history ofbreast cancer (23, 24). It has been suggested that these depur-inating adducts are repaired through the nucleotide excisionrepair (NER) and base excision repair pathways; however, astudy that examined chromosomal aberrations in DT40 cellsafter treatment with 4-OHE2 observed no difference betweenwild-type cells and cells depleted of XPA, a key protein in NER(25, 26). In contrast, there were enhanced chromosomal breaksfollowing 4-OHE2 treatment of rad54- and ku70-mutant DT40cells, both of which are required for repair of double-strandbreaks (DSB) by HR and non-homologous end joining, respec-tively. This suggests that estrogen metabolites may produceDNA DSBs.
The idea that estrogen metabolites may cause DNA DSBs,coupled with the role of BRCA1 in DSB repair, lead us tohypothesize that BRCA1-deficient cells may be more suscep-tible to estrogen metabolite-induced DNA damage and sub-sequent genomic instability. We therefore examined whetherestrogen and its metabolites 2-OHE2 and 4-OHE2 can causeDSBs in human breast cells and examined the role of BRCA1 inboth the induction and repair of estrogen metabolite-inducedDNA damage.
Materials and MethodsCell lines
MCF7 and MCF10A cells were obtained from the AmericanType Culture Collection and maintained according to therecommended instructions.MCF10ABRCA1þ/� 185delAG andmatched control BRCA1þ/þ cells were generated as previouslydescribed (27). All cell lines were verified by short tandemrepeat profiling.
siRNAssiRNAs were obtained from Qiagen and reverse transfected
into cells using RNAiMAX (Invitrogen) to a final concentrationof 10 nmol/L. See Supplementary Materials and Methods forsequences.
Immunofluorescence microscopyCells were transfected with siRNAs as above and incubated
for 48 hours. Cells were then plated onto coverslips and treated
with E2, 2-OHE2, or 4-OHE2 (Sigma), or mock treated withvehicle and incubated for indicated time points. Cells werethen fixed and stained with g-H2AX (Millipore), 53BP1 (Milli-pore), cyclin A (SCBT), or pATMSer1981 (Cell Signaling Tech-nology) primary antibodies and imaged using a Nikon EclipseTi microscope, using a �60 objective.
Comet assaysNeutral comet assays were carried out using the Cell Biolabs
SCGE Kit. Comets were scored using CometScore (TriTekCorp.).
Western blottingWestern blotting was carried out as previously described
(28).
Metaphase spreads, FISH staining, and chromosomalaberrations
Analysis was carried out as described previously (29).
Quantitative real-time PCR analysisQuantitative real-time PCR (qRT-PCR) was carried out
using Roche LightCycler 480 Real-Time ready catalogueassays for each gene (ACTB, CYP1A1, CYP1A2, CYP3A4,CYP1B1, COMT, and NQO1) as per the manufacturer'sinstructions. A matched qRT-PCR reaction was carried outusing the RT-ve control for each sample ensuring no geno-mic DNA contamination.
Chromatin immunoprecipitationsChromatin immunoprecipitations (ChIP) were performed
as described previously (28). See Supplementary Materials andMethods for complete protocol and primer sequences.
Immunohistochemical staining of CYP1A1Immunohistochemical staining was performed using a fully
automated BondMax immunostainer with a polymer-basedperoxidase detection system. [CYP1A1 (B4)] SCBT primaryantibody was used at a dilution of 1:50.
Ultra performance liquid chromatography/tandemmass spectrometry
Samples were extracted using liquid–liquid extraction withdiethyl ether, followed by dansyl chloride derivatization asdescribed by Xu and colleagues and analyzed using ultraperformance liquid chromatography/tandem mass spectrom-etry (UPLC/MS-MS). See Supplementary Materials and Meth-ods for complete protocol (30).
Isolation and culture or primary breast progenitor cellsSee Supplementary Materials and Methods for complete
protocol. Ethical approval to obtain primary breast tissue wasgranted through the Northern Ireland Biobank. Tissue wasdissociated and mammospheres cultured as previouslydescribed (31) in ultralow attachment 75 cm2
flasks for 7 days.Mammosphere cultured cells were then dissociated and platedinto Lab-Tek II CC2 treated chamber slides (Nunc) in themediaabove.
Savage et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2774
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611
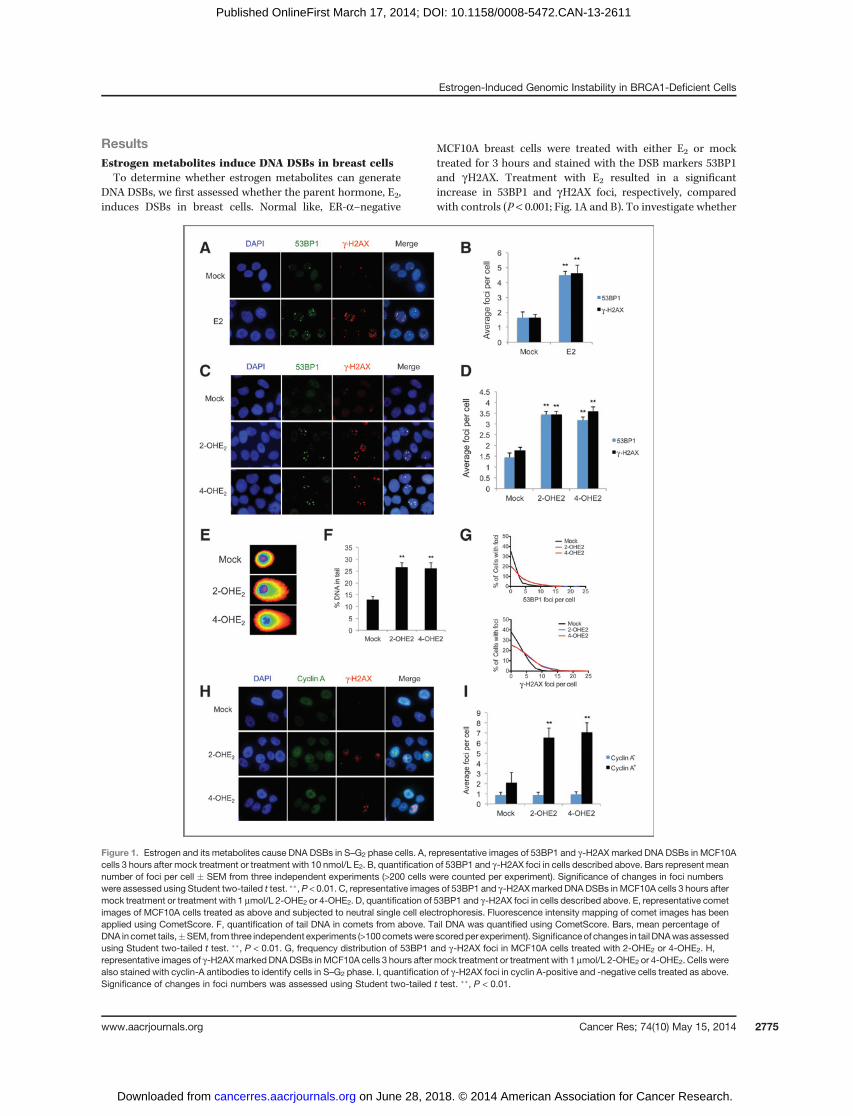
ResultsEstrogen metabolites induce DNA DSBs in breast cellsTo determine whether estrogen metabolites can generate
DNA DSBs, we first assessed whether the parent hormone, E2,induces DSBs in breast cells. Normal like, ER-a–negative
MCF10A breast cells were treated with either E2 or mocktreated for 3 hours and stained with the DSB markers 53BP1and gH2AX. Treatment with E2 resulted in a significantincrease in 53BP1 and gH2AX foci, respectively, comparedwith controls (P < 0.001; Fig. 1A and B). To investigate whether
Figure 1. Estrogen and its metabolites cause DNA DSBs in S–G2 phase cells. A, representative images of 53BP1 and g-H2AX marked DNA DSBs in MCF10Acells 3 hours after mock treatment or treatment with 10 nmol/L E2. B, quantification of 53BP1 and g-H2AX foci in cells described above. Bars represent meannumber of foci per cell � SEM from three independent experiments (>200 cells were counted per experiment). Significance of changes in foci numberswere assessed using Student two-tailed t test. ��, P < 0.01. C, representative images of 53BP1 and g-H2AXmarked DNA DSBs in MCF10A cells 3 hours aftermock treatment or treatment with 1 mmol/L 2-OHE2 or 4-OHE2. D, quantification of 53BP1 and g-H2AX foci in cells described above. E, representative cometimages of MCF10A cells treated as above and subjected to neutral single cell electrophoresis. Fluorescence intensity mapping of comet images has beenapplied using CometScore. F, quantification of tail DNA in comets from above. Tail DNA was quantified using CometScore. Bars, mean percentage ofDNA in comet tails,�SEM, from three independent experiments (>100cometswere scoredper experiment). Significanceof changes in tail DNAwasassessedusing Student two-tailed t test. ��, P < 0.01. G, frequency distribution of 53BP1 and g-H2AX foci in MCF10A cells treated with 2-OHE2 or 4-OHE2. H,representative images of g-H2AXmarked DNADSBs inMCF10A cells 3 hours after mock treatment or treatment with 1 mmol/L 2-OHE2 or 4-OHE2. Cells werealso stained with cyclin-A antibodies to identify cells in S–G2 phase. I, quantification of g-H2AX foci in cyclin A-positive and -negative cells treated as above.Significance of changes in foci numbers was assessed using Student two-tailed t test. ��, P < 0.01.
Estrogen-Induced Genomic Instability in BRCA1-Deficient Cells
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2775
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611

estrogen metabolites cause DSBs, we treated MCF10A cellswith the metabolic intermediates 2-OHE2 and 4-OHE2 for3 hours and stained with 53BP1 and gH2AX. Similar to E2treatment, we found that both metabolites induced a signifi-cant increase inDSB foci (P< 0.01; Fig. 1CandD).Thesefindingswere also confirmed in the ER-a–positive breast cancer cellline, MCF7, indicating that E2 metabolite-induced DSB induc-tion is independent of ER-a (Supplementary Fig. S1A and S1B).To further confirm that 2-OHE2 and 4-OHE2 induce DSBs, weperformed neutral comet assays. Indeed, MCF10A cells treatedwith 2-OHE2 and 4-OHE2 demonstrated a greater percentage oftail DNA, tail moment, and tail length, indicative of increasedDSBs (Fig. 1E and F and Supplementary Fig. S1C and S1D).
DNA DSBs are known to result in activation of the ATMkinase, which is activated through autophosphorylation ofATM at serine-1981. In keeping with this, 2-OHE2 and 4-OHE2treatment resulted in a 2.27-fold and 3.37-fold increase inATMpSer1981 foci (colocalized with 53BP1 foci) compared withmock controls (Supplementary Fig. S1E and S1F). Takentogether, these data suggest that 2-OHE2 and 4-OHE2 induceDNA DSBs in breast cells.
Interestingly, when carrying out these experiments, weconsistently observed uneven distribution of estrogen metab-olite-induced DSBs and rather than all cells containing slightlymore foci than mock-treated cells, we consistently observedthat a fraction of cells (� 25%–30%) incurred a greater numberof DNA DSBs following 2-OHE2 and 4-OHE2 treatment. Forexample, in cells treated with 2-OHE2, 23% had no 53BP1 foci,whereas 26% of cells had six or more foci (Fig. 1G). Thissuggested that estrogen metabolite-mediated DNA damagemay be affected by cell-cycle distribution. To test this, wecostained 2-OHE2 and 4-OHE2-treated MCF10A cells withgH2AX and cyclin A, which is specifically expressed during Sand G2 phases of the cell cycle. We found that the vast majorityof estrogen metabolite-mediated DNA damage occurred incyclin A-positive cells, suggesting that 2-OHE2 and 4-OHE2specifically induce DNA damage during S–G2 phase cells (Fig.1H and I). To confirm this, MCF10A cells treated with 2-OHE2and 4-OHE2 were pulse labeled with 5-ethynyl-20-deoxyuridine,which is incorporated into DNA during replication. Cells werethen costained for gH2AX and EdU. Indeed, significantly moregH2AX foci were observed in EdU-positive cells compared withEdU-negative cells following estrogen metabolite treatment(Supplementary Fig. S1G and S1H). This suggests that the DNADSBs produced by 2-OHE2 and 4-OHE2 treatment occur spe-cifically during S phase.
Estrogen metabolite-mediated DNA damage isexacerbated byBRCA1 loss andnot efficiently repaired inBRCA1-deficient cells
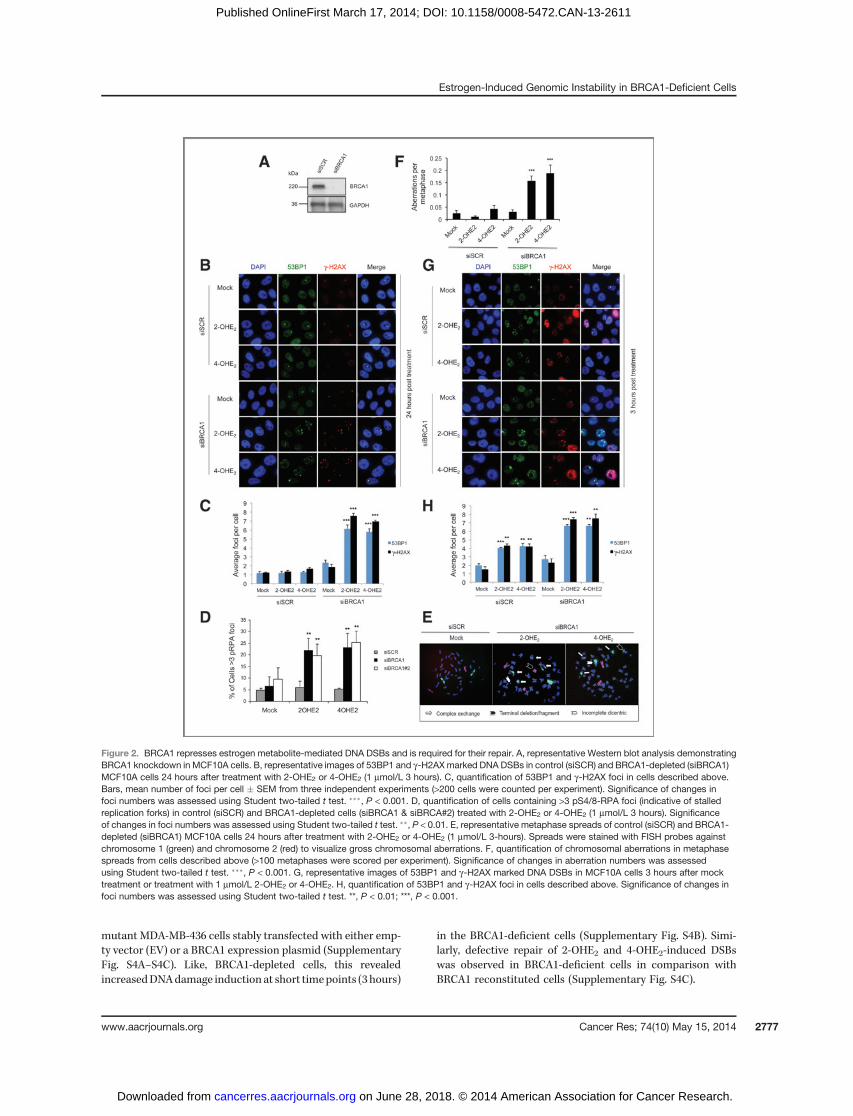
BRCA1 is known to play a pivotal role in DNA DSB repair. Inaddition, a role for BRCA1 in the postreplicative repair of bulkyDNA adducts induced by UV, which are structurally similar toE2 metabolite adducted bases, has been recently described (3).In light of this, we examined whether BRCA1 was required forthe repair of estrogen-induced DSBs. Endogenous BRCA1expression was depleted using two independent siRNAs(siBRCA1) compared with a nontargeting scrambled siRNA
(siSCR) and the cells treated with 2-OHE2 and 4-OHE2 for 3hours. Cells were then allowed to recover for 24 hours followingtreatment, before beingfixed and stained for 53BP1 and gH2AX(Fig. 2A–C and Supplementary Fig. S2A and S2B). No signif-icant difference in 53BP1 and gH2AX foci number wasobserved in siSCR cells treated with 2-OHE2 or 4-OHE2 relativeto untreated control cells, suggesting that DSBs generatedduring estrogen metabolite treatment had been efficientlyrepaired. In contrast, significantly more 53BP1 and gH2AXfoci remained in BRCA1-depleted cells treated with 2-OHE2and 4-OHE2 comparedwith controls (P< 0.001), indicating thatBRCA1 is required for efficient repair of E2 metabolite-inducedDSBs. This was visualized at multiple time points following3 hours of 2-OHE2 or 4-OHE2 treatment (SupplementaryFig. S2C and S2D). In addition, given that E2-metabolites induceDNA damage during S phase, taken together with the knownrole for BRCA1 in the postreplicative repair of bulky DNAadducts, we assessed whether depletion of BRCA1, using twoindependent siRNAs, caused replication fork stalling, markedby residual pS4/8 RPA32 foci (3). Consistent with a role forBRCA1 in repairing E2-metabolite-induced DNA damage in Sphase cells, we observed a dramatic increase in pS4/8 RPA32-positive cells upon BRCA1 depletion following 2OHE2 and4OHE2 treatment (Fig. 2D).
We next examined whether 2-OHE2 and 4OHE2 treatmentalso induced chromosomal instability in BRCA1-depleted cells.We assessed chromosomal aberrations in control and BRCA1-depleted MCF10A cells 24 hours following treatment with2-OHE2 and 4-OHE2. Structural rearrangements were visual-ized and quantified inmetaphase spreads using chromosome 1and 2 FISH staining (Fig. 2E and F). BRCA1 depletion resultedin a marked increase in chromosomal aberrations followingboth 2-OHE2 and 4-OHE2 treatment, demonstrating that E2metabolite treatment induces genomic instability in BRCA1-deficient cells. Intriguingly, when we examined DSB produc-tion in these cells immediately following 2-OHE2 and 4-OHE2treatment (3 hours), we found that 2-OHE2 and 4-OHE2 treat-ment resulted in a significant increase in 53BP1 and gH2AX fociin BRCA1-depleted cells compared with control cells (Fig. 2Gand H and Supplementary Fig. S2E). In addition, similar to thatdemonstrated earlier, E2 metabolite-mediated DNA damageoccurred specifically in S–G2 phase of the cell cycle in BRCA1-depleted cells (Supplementary Fig. S2F). Because of the rela-tively short treatment time (3 hours), it is unlikely that thisincrease inDSBs observed in BRCA1-depleted cells is solely dueto defective DNA repair. To confirm that this occurred at earlytime points and was not confounded by the dose and/or timepoints of E2-metabolite treatment used, we assessed gH2AXmarked DNA damage in control and BRCA1-depleted cellsfollowing treatment with 1 nm, 10 nm, 100 nm, and 1 mmol/LE2, 2-OHE2 and 4-OHE2 at various time points (SupplementaryFig. S3A–S3C). This revealed increasedDNAdamage in BRCA1-depleted cells at all doses and treatment time points assessed.
Taken together, these data indicate that loss of BRCA1expression results in delayed repair kinetics but also increasedlevels of DNA damage following treatment with E2metabolites.To further confirm this, we examined induction and repair ofDNA DSBs following 2-OHE2 and 4-OHE2 treatment in BRCA1-
Savage et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2776
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611
mutant MDA-MB-436 cells stably transfected with either emp-ty vector (EV) or a BRCA1 expression plasmid (SupplementaryFig. S4A–S4C). Like, BRCA1-depleted cells, this revealedincreasedDNAdamage induction at short timepoints (3 hours)
in the BRCA1-deficient cells (Supplementary Fig. S4B). Simi-larly, defective repair of 2-OHE2 and 4-OHE2-induced DSBswas observed in BRCA1-deficient cells in comparison withBRCA1 reconstituted cells (Supplementary Fig. S4C).
Figure 2. BRCA1 represses estrogen metabolite-mediated DNA DSBs and is required for their repair. A, representative Western blot analysis demonstratingBRCA1 knockdown in MCF10A cells. B, representative images of 53BP1 and g-H2AX marked DNA DSBs in control (siSCR) and BRCA1-depleted (siBRCA1)MCF10A cells 24 hours after treatment with 2-OHE2 or 4-OHE2 (1 mmol/L 3 hours). C, quantification of 53BP1 and g-H2AX foci in cells described above.Bars, mean number of foci per cell � SEM from three independent experiments (>200 cells were counted per experiment). Significance of changes infoci numbers was assessed using Student two-tailed t test. ���, P < 0.001. D, quantification of cells containing >3 pS4/8-RPA foci (indicative of stalledreplication forks) in control (siSCR) and BRCA1-depleted cells (siBRCA1 & siBRCA#2) treated with 2-OHE2 or 4-OHE2 (1 mmol/L 3 hours). Significanceof changes in foci numbers was assessed using Student two-tailed t test. ��, P < 0.01. E, representative metaphase spreads of control (siSCR) and BRCA1-depleted (siBRCA1) MCF10A cells 24 hours after treatment with 2-OHE2 or 4-OHE2 (1 mmol/L 3-hours). Spreads were stained with FISH probes againstchromosome 1 (green) and chromosome 2 (red) to visualize gross chromosomal aberrations. F, quantification of chromosomal aberrations in metaphasespreads from cells described above (>100 metaphases were scored per experiment). Significance of changes in aberration numbers was assessedusing Student two-tailed t test. ���, P < 0.001. G, representative images of 53BP1 and g-H2AX marked DNA DSBs in MCF10A cells 3 hours after mocktreatment or treatment with 1 mmol/L 2-OHE2 or 4-OHE2. H, quantification of 53BP1 and g-H2AX foci in cells described above. Significance of changes infoci numbers was assessed using Student two-tailed t test. **, P < 0.01; ***, P < 0.001.
Estrogen-Induced Genomic Instability in BRCA1-Deficient Cells
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2777
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611

Given that BRCA2 is also involved in HR-mediated DSBrepair and mutations in this gene also predispose to tumors inthe breast and ovaries, we examined the effect of BRCA2depletion on 2-OHE2 and 4-OHE2-induced DNA damage inboth MCF10A and MCF7 cells. Intriguingly, BRCA2 depletionresulted in slightly increased DSBs following treatment with 2-OHE2 and 4-OHE2 for 3 hours, which seemed to remainunrepaired at 24 hours following recovery from 2-OHE2 and4-OHE2 treatment (Supplementary Fig. S5). Nevertheless, theincreased level of DNA damage observed following treatmentwith 2-OHE2 and 4-OHE2 in BRCA2-depleted cells wasminimalin comparison with that observed in BRCA1-depleted cells. Inaddition, the defective repair of these DSBs in BRCA2-depletedcells is consistent with BRCA2's role in HR-mediated DSBrepair.
BRCA1 regulates the expression of estrogen-metabolizing enzymes
We have previously proposed that BRCA1 may transcrip-tionally repress the expression of estrogenmetabolizing genes.Specifically, we have demonstrated that CYP1A1, the enzymeresponsible for conversion of E2 to 2-OHE2 and semiquinone/quinone metabolites, is repressed by BRCA1 in a number ofbreast cancer cell lines (32). In addition, BRCA1 has been foundto transcriptionally activate a number of detoxification/anti-oxidant genes, including NAD(P)H quinone oxoreductase 1(NQO1), which reduces genotoxic quinones to nonreactivehydroquinones (33). This led us to hypothesize that the exac-erbated DNA damage observed in BRCA1-depleted cells atshort time points following 2-OHE2 and 4-OHE2 treatmentmaybe due to, at least in part, increased estrogen metabolism inthese cells, mediated by upregulated expression of estrogen-metabolizing enzymes, and/or downregulated expression ofdetoxification enzymes such as NQO1. To assess this, weexamined the expression of a panel of estrogen-metabolizingand detoxification genes, including CYP1A1, CYP1B1, CYP1A2,CYP3A4, NQO1, and COMT, using qRT-PCR, in MCF10A andMCF7 cells transfected with control and BRCA1-depletingsiRNAs (Fig. 3A and B). This revealed that CYP1A1 and CYP3A4are consistently upregulated upon BRCA1 loss, suggesting thatBRCA1 represses the expression of these genes. In addition,NQO1 was consistently downregulated upon BRCA1 depletionin both cell lines. We also examined the expression of thesegenes in BRCA1-mutant MDA-MB-436 cells, stably transfectedwith EV or BRCA1 Fig. 3C). Ectopic expression of BRCA1 inthese cells repressed expression of CYP1A1, but had a limitedeffect on CYP3A4 and NQO1 expression.
To assess whether BRCA1 regulates the transcription ofthese genes directly, we performed BRCA1 ChIP-qPCR assaysfrom MCF10A cells, using primers specific to the promoterregions of CYP1A1, CYP3A4, NQO1, and COMT as a negativecontrol (Fig. 3D). We observed enrichment of BRCA1 at all ofthese promoters with the exception of COMT, which is neithertranscriptionally regulated by BRCA1 nor promoter bound byBRCA1.
To confirm that this occurs in BRCA1-deficient tumors, weassessed the levels of CYP1A1, the most highly deregulatedgene upon BRCA1 loss, using immunohistochemistry (IHC) in
a panel of 21 BRCA1-mutant and 75 BRCA1 wild-type breasttumors. Intratumoral CYP1A1 expression was scored by apathologist and an independent scorer as very weak/absent¼ 1, moderate¼ 2, or strong¼ 3 (Fig. 3E and F). This revealedthat themean expression of CYP1A1 expression is significantlyupregulated in this cohort of BRCA1-mutant tumors comparedwith BRCA1 wild-type sporadic tumors (P ¼ 0.0007).
Given that we observed a slight increase in E2 metabolite-induced DNA damage in BRCA2-depleted cells treated with 2-OHE2 and 4-OHE2, we also assessed the role of BRCA2 inregulating the expression of CYP1A1 in both MCF10A andMCF7 cells (Supplementary Fig. S6A and S6B). This revealed nosignificant difference in CYP1A1 expression between controland BRCA2-depleted cells.
BRCA1 suppresses estrogen metabolite-mediated DNAdamage by suppressing estrogen metabolism
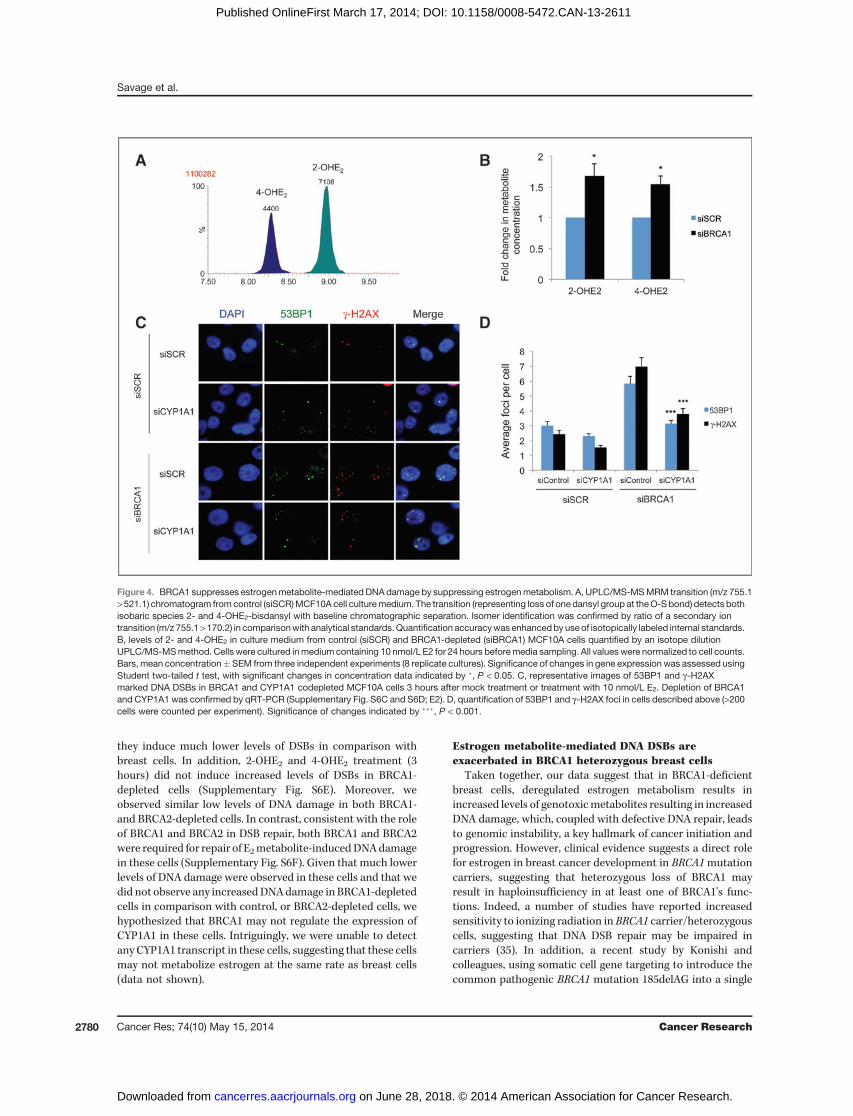
As BRCA1 depletion results in increased levels of CYP1A1and CYP3A4 enzymes and decreased NQO1 expression, wehypothesized that the levels of 2-OHE2 and 4-OHE2 and 2,3/3,4quinone products would be increased in BRCA1-depleted cellscompared with control cells following E2 treatment. Quinoneand semiquinonemetabolites are extremely unstable and havea very limited half-life making them difficult to quantify. Incontrast, 2-OHE2 and 4-OHE2 (which are generated by CYP1A1and CYP3A4 in breast cells) are relatively stable. We thereforedeveloped an UPLC/MS-MS method for the detection of 2-OHE2 and 4-OHE2. The method was optimized for baselinechromatographic separation and the accurate identificationand quantification of both isomeric forms of the hydroxyl-estradiolmetabolite (Fig. 4A). Using thismethod,we found thatthe relative concentration of 2-OHE2 and 4-OHE2 was signif-icantly higher in BRCA1-depleted cells compared with controlcells (P < 0.05; Fig. 4B).
As CYP1A1 was the most robustly upregulated estrogen-metabolizing gene upon BRCA1 depletion, we sought toexamine the role of this enzyme in estrogen-dependent DNAdamage. To investigate this, BRCA1 and CYP1A1 were code-pleted in MCF10A cells and DNA DSBs assessed 3 hoursfollowing treatment with E2 (Fig. 4C and D). BRCA1 andCYP1A1 depletion was assessed by qRT-PCR (SupplementaryFig. S6C and S6D). Strikingly, CYP1A1 depletion lead to amarked reduction in estrogen-mediated DSBs in BRCA1-depleted cells (P < 0.001), suggesting that estrogen-mediatedDNA damage in BRCA1-depleted cells is, at least in part, due toincreased CYP1A1 levels in these cells.
This suggests that BRCA1, apart frommediating repair of E2metabolite-mediated DNA damage, transcriptionally regulatesestrogen-metabolizing enzymes and subsequently repressesestrogen metabolism, thereby protecting cells against estro-gen-induced DNA damage. This is particularly important inbreast and ovarian cells, which are exposed to much higherlevels of estrogen than other tissues within the body (34).Nevertheless, to confirm whether this mechanism may drivegenomic instability in nonbreast cells, we assessed E2 metab-olite-induced DNA damage in HEK293, kidney cells (Supple-mentary Fig. S6E and S6F). This revealed that, although 2-OHE2and 4-OHE2 are capable of inducing DNA DSBs in these cells,
Savage et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2778
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611
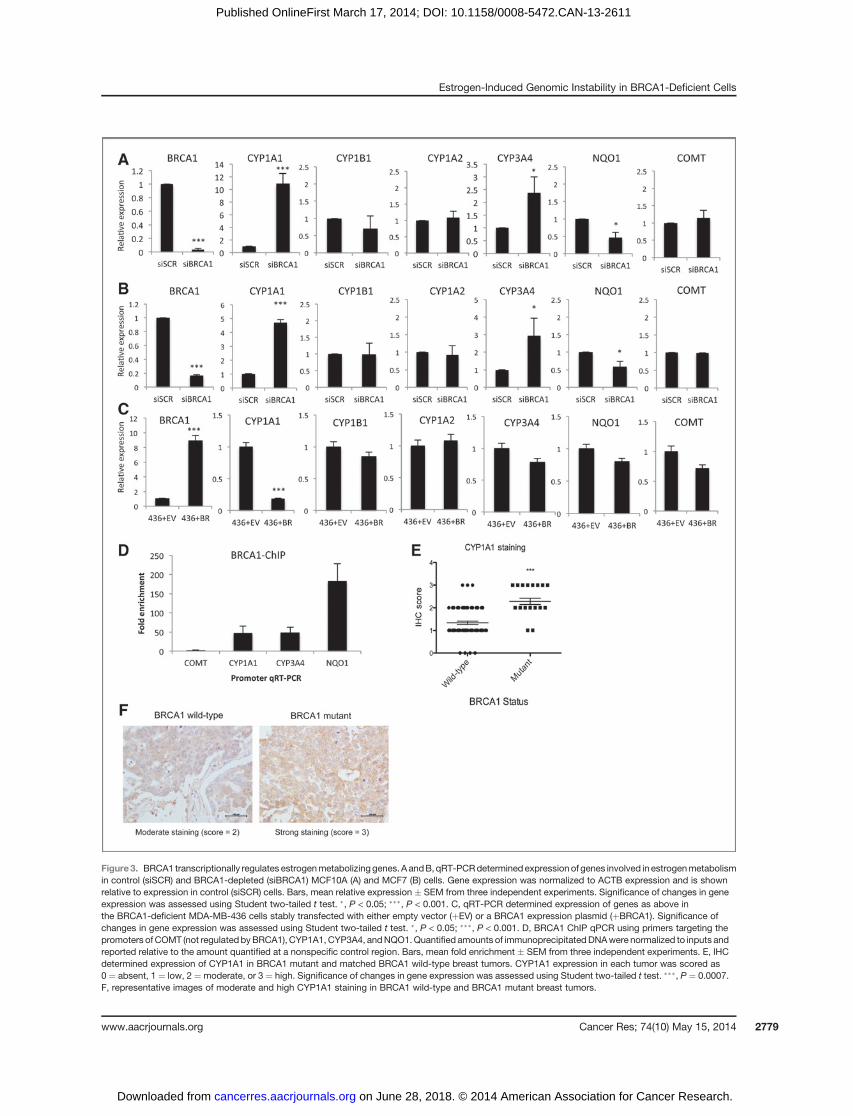
Figure3. BRCA1 transcriptionally regulates estrogenmetabolizinggenes. A andB, qRT-PCRdetermined expression of genes involved in estrogenmetabolismin control (siSCR) and BRCA1-depleted (siBRCA1) MCF10A (A) and MCF7 (B) cells. Gene expression was normalized to ACTB expression and is shownrelative to expression in control (siSCR) cells. Bars, mean relative expression � SEM from three independent experiments. Significance of changes in geneexpression was assessed using Student two-tailed t test. �, P < 0.05; ���, P < 0.001. C, qRT-PCR determined expression of genes as above inthe BRCA1-deficient MDA-MB-436 cells stably transfected with either empty vector (þEV) or a BRCA1 expression plasmid (þBRCA1). Significance ofchanges in gene expression was assessed using Student two-tailed t test. �, P < 0.05; ���, P < 0.001. D, BRCA1 ChIP qPCR using primers targeting thepromoters of COMT (not regulated byBRCA1), CYP1A1, CYP3A4, andNQO1.Quantified amounts of immunoprecipitatedDNAwere normalized to inputs andreported relative to the amount quantified at a nonspecific control region. Bars, mean fold enrichment � SEM from three independent experiments. E, IHCdetermined expression of CYP1A1 in BRCA1 mutant and matched BRCA1 wild-type breast tumors. CYP1A1 expression in each tumor was scored as0 ¼ absent, 1 ¼ low, 2 ¼moderate, or 3 ¼ high. Significance of changes in gene expression was assessed using Student two-tailed t test. ���, P ¼ 0.0007.F, representative images of moderate and high CYP1A1 staining in BRCA1 wild-type and BRCA1 mutant breast tumors.
Estrogen-Induced Genomic Instability in BRCA1-Deficient Cells
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2779
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611
they induce much lower levels of DSBs in comparison withbreast cells. In addition, 2-OHE2 and 4-OHE2 treatment (3hours) did not induce increased levels of DSBs in BRCA1-depleted cells (Supplementary Fig. S6E). Moreover, weobserved similar low levels of DNA damage in both BRCA1-and BRCA2-depleted cells. In contrast, consistent with the roleof BRCA1 and BRCA2 in DSB repair, both BRCA1 and BRCA2were required for repair of E2metabolite-inducedDNAdamagein these cells (Supplementary Fig. S6F). Given that much lowerlevels of DNA damage were observed in these cells and that wedid not observe any increasedDNAdamage in BRCA1-depletedcells in comparison with control, or BRCA2-depleted cells, wehypothesized that BRCA1 may not regulate the expression ofCYP1A1 in these cells. Intriguingly, we were unable to detectany CYP1A1 transcript in these cells, suggesting that these cellsmay not metabolize estrogen at the same rate as breast cells(data not shown).
Estrogen metabolite-mediated DNA DSBs areexacerbated in BRCA1 heterozygous breast cells
Taken together, our data suggest that in BRCA1-deficientbreast cells, deregulated estrogen metabolism results inincreased levels of genotoxicmetabolites resulting in increasedDNA damage, which, coupled with defective DNA repair, leadsto genomic instability, a key hallmark of cancer initiation andprogression. However, clinical evidence suggests a direct rolefor estrogen in breast cancer development in BRCA1mutationcarriers, suggesting that heterozygous loss of BRCA1 mayresult in haploinsufficiency in at least one of BRCA1's func-tions. Indeed, a number of studies have reported increasedsensitivity to ionizing radiation in BRCA1 carrier/heterozygouscells, suggesting that DNA DSB repair may be impaired incarriers (35). In addition, a recent study by Konishi andcolleagues, using somatic cell gene targeting to introduce thecommon pathogenic BRCA1 mutation 185delAG into a single
Figure 4. BRCA1 suppresses estrogenmetabolite-mediated DNAdamage by suppressing estrogenmetabolism. A, UPLC/MS-MSMRM transition (m/z 755.1> 521.1) chromatogram from control (siSCR)MCF10A cell culturemedium. The transition (representing loss of one dansyl group at theO-Sbond) detects bothisobaric species 2- and 4-OHE2-bisdansyl with baseline chromatographic separation. Isomer identification was confirmed by ratio of a secondary iontransition (m/z 755.1>170.2) in comparisonwith analytical standards.Quantification accuracywas enhancedbyuseof isotopically labeled internal standards.B, levels of 2- and 4-OHE2 in culture medium from control (siSCR) and BRCA1-depleted (siBRCA1) MCF10A cells quantified by an isotope dilutionUPLC/MS-MSmethod. Cells were cultured inmedium containing 10 nmol/L E2 for 24 hours beforemedia sampling. All valueswere normalized to cell counts.Bars, mean concentration� SEM from three independent experiments (8 replicate cultures). Significance of changes in gene expression was assessed usingStudent two-tailed t test, with significant changes in concentration data indicated by �, P < 0.05. C, representative images of 53BP1 and g-H2AXmarked DNA DSBs in BRCA1 and CYP1A1 codepleted MCF10A cells 3 hours after mock treatment or treatment with 10 nmol/L E2. Depletion of BRCA1and CYP1A1 was confirmed by qRT-PCR (Supplementary Fig. S6C and S6D; E2). D, quantification of 53BP1 and g-H2AX foci in cells described above (>200cells were counted per experiment). Significance of changes indicated by ���, P < 0.001.
Savage et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2780
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611

BRCA1 allele in MCF10A cells, demonstrated that this hetero-zygous BRCA1 mutation confers impaired HR-mediated DSBrepair, hypersensitivity to genotoxic stress, and increasedgenomic instability (27). We therefore set out to determinewhether heterozygous mutation of BRCA1 affects estrogenmetabolite-mediated DNA damage and estrogen metabolism.Using the same cell line model developed by Konishi and
colleagues, heterozygous BRCA1 185delAG (BRCA1þ/�) andcontrol (BRCA1þ/þ) MCF10A cells were treated with 2-OHE2and 4-OHE2 for 3 hours and the media replaced with normalmedia for 24 hours, before fixing and staining for 53BP1 andgH2AX. Consistent with a defect in BRCA1 function, a signif-icant number of unresolved DNA damage foci were visible in 2-OHE2- and 4-OHE2-treated BRCA1
þ/� cells in comparison withcontrolBRCA1þ/þcells (P< 0.001; Fig. 5A). Surprisingly, the levelof unrepairedDNADSBswas similar to that observed inBRCA1-depleted MCF10A cells, suggesting that BRCA1 haploinsuffi-ciency imparts a major defect in repair of E2 metabolite-mediated DNA damage. We next examined whether estrogenmetabolites generate more DSBs in BRCA1þ/� cells. Indeed,although short-term (3 hours) 2-OHE2 and 4-OHE2 treatmentinduced DNA damage in BRCA1þ/þ cells, significantly moreDNA DSB foci were observed in BRCA1þ/� cells (P < 0.01; Fig.5B). In support of this, we treated normal primary breastprogenitor cells, isolated from breast tissue obtained from awoman undergoing elective breast reduction, as well asBRCA1þ/� primary breast progenitor cells from a BRCA1muta-tion carrier undergoing a risk-reducing mastectomy, with both2-OHE2 and4-OHE2 andexaminedbothDNAdamage inductionat 3 hours posttreatment, as well as their ability to repair E2metabolite-inducedDNA damage 24 hours following treatmentwith 2-OHE2 and 4-OHE2. In keepingwith our previousfindings,2-OHE2 and 4-OHE2 treatment for 3 hours induced more DNAdamage in the BRCA1þ/�mammary progenitor cells comparedwith the BRCA1 wild-type cells and this DNA damage was notrepaired as efficiently in the BRCA1þ/� cells compared with theBRCA1 wild-type cells (Fig. 5C and D). Taken together thesedata suggest that like BRCA1-depleted cells, BRCA1þ/� cellsmay have increased rates of estrogen metabolism as well asdefective repair of E2 metabolite-induced DNA damage.To examine this, we assessed the expression levels of the same
panel of estrogen metabolizing and detoxification enzymes inBRCA1þ/þ and BRCA1þ/� cells. Indeed, as in BRCA1-depletedcells, CYP1A1 and CYP3A4 were upregulated in BRCA1þ/�
compared with BRCA1þ/þ cells (Fig. 5E). Intriguingly, NQO1expression was maintained at similar levels in both cells lines,suggesting that BRCA1 haploinsufficiency does not negativelyimpact all BRCA1-regulated transcriptional targets. Finally,consistent with the defective DSB repair observed in these cellsfollowing 2-OHE2 and 4-OHE2 treatment, we found that treat-ment with either of these metabolites induced genomic insta-bility in BRCA1þ/� cells but not BRCA1þ/þ cells (Fig. 5F).
DiscussionOne of the most perplexing features of BRCA1 biology is that
despite playing a central role in the DNA damage response andDSB repair pathways in all cells,mutation carriers predominantly
develop tumors in the breast and ovaries; both estrogen-driventissues exposed to high levels of estrogen. Here, we show thattreatment with both E2 and the E2 metabolites 2-OHE2 and 4-OHE2, induces DNADSBs in human breast cancer cells in an ER-a independent manner. We also found that E2 metabolite-mediated DSBs occur specifically in S phase cells, suggestingthat induction of these lesions is coupled toDNA replication.Wehypothesize that E2 metabolite adducted DNA bases representreplication barriers, which lead to replication fork stalling duringDNA synthesis. Indeed, a number of studies have shown that 4-hydroxyequilenin (4-OHEN), a metabolite of the equine estrogenequilenin (which is almost identical to 4OHE-2 in humans),causes identical DNA adducts to those caused by 4-OHE2 andthat these adducts cause replication fork stalling (36, 37).
In general, stalled replication forks do not collapse and formDSBs, but are instead stabilized by the ATR kinase through thesignaling and recruitment of a plethora of checkpoint signalingand repair proteins, resulting in resolution of the stalled forkthrough a HR-mediated repair process involving BRCA1. How-ever, recent studies have shown that E2 inhibits ATR signaling,suggesting that E2 and its metabolites may lead to replicationfork stalling and subsequent fork collapse and DSB formationthrough the combined effect of replication fork stalling andATR inhibition (38). Further to this, BRCA1 has been shown tobe required for both resolution of stalled replication forks aswell as HR-mediated repair of DSBs caused following stalledfork collapse (39). BRCA1 is also required for the removal andrepair of bulky base adducts, a mechanism through whichBRCA1 may suppress adduct-induced mutagenesis (3). Con-sistent with this, we observed a dramatic increase in pS4/8RPA32-positive cells, upon BRCA1 depletion in 2OHE2- and4OHE2-treated cells.
Interestingly, we found that BRCA1 depletion also resultedin increased levels of E2metabolite-inducedDNAdamage, evenat very early time points. This suggested that BRCA1 may alsoplay a more direct role in regulating the physical levels of DNAdamage induced by estrogen metabolites. Previous data fromour laboratory had indicated that BRCA1 loss leads to upre-gulation of CYP1A1, a major regulator of E2 metabolism inbreast tissues (32). We therefore tested whether BRCA1 mayalso regulate the expression of other estrogen metabolizingenzymes, thereby regulating the levels of estrogen-derivedmetabolites. This analysis revealed that BRCA1 directlyrepresses the transcription of CYP1A1 and CYP3A4 and pro-motes the expression of the NAD(P)H:quinone oxidoreductase,NQO1. We confirmed, using IHC in a cohort of BRCA1-mutantandmatched BRCA1wild-type tumors, that CYP1A1, themajorenzyme involved in conversion of E2 to 2-OHE2 in breasttissues, is significantly upregulated in BRCA1-mutant tumors.This is consistent with the increased levels of both 2-OHE2 and4-OHE2 observed in BRCA1-depleted cells. We also demon-strated that depletion of CYP1A1 significantly reduces theamount of DNA damage induced in BRCA1-depleted cellsexposed to short-term E2 treatment, confirming that E2-medi-ated DNA damage in BRCA1-depleted cells is, at least in part,due to increased estrogen metabolism. Intriguingly, whenexamining the impact of BRCA2 on estrogen metabolite-induced DNA damage, we found that although BRCA2 is
Estrogen-Induced Genomic Instability in BRCA1-Deficient Cells
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2781
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611
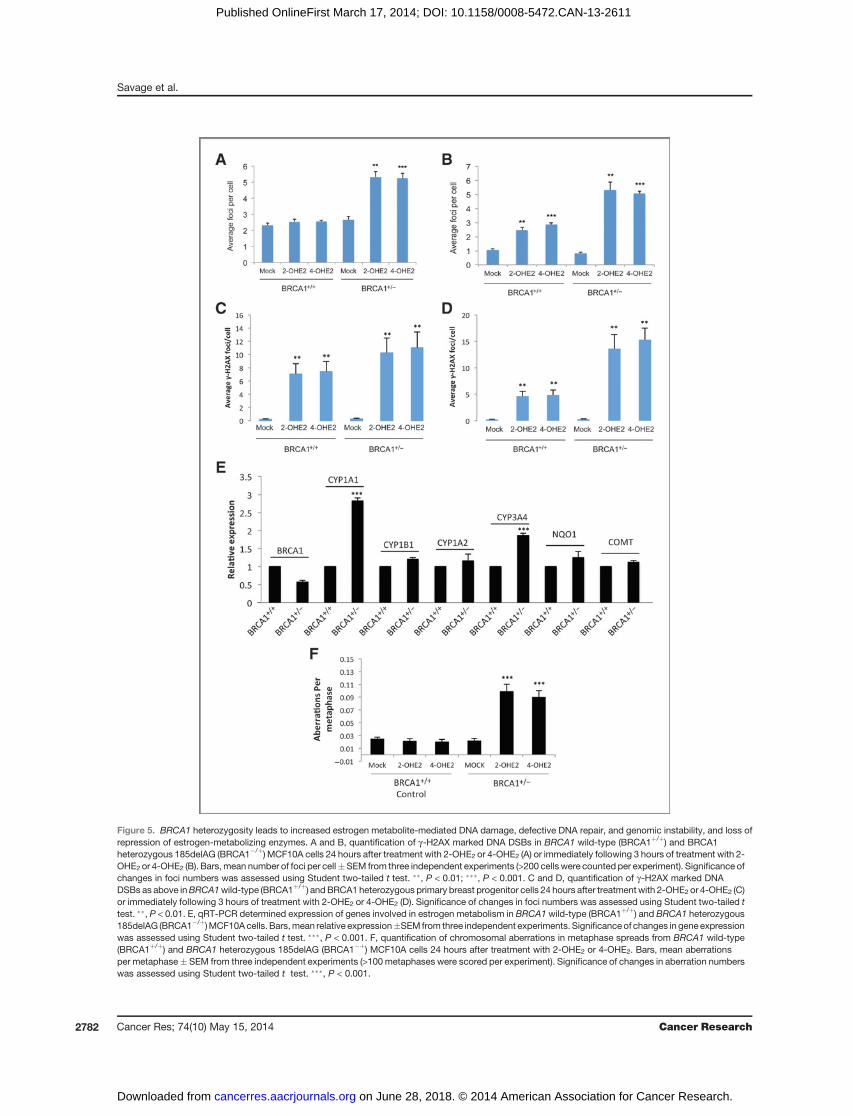
Figure 5. BRCA1 heterozygosity leads to increased estrogen metabolite-mediated DNA damage, defective DNA repair, and genomic instability, and loss ofrepression of estrogen-metabolizing enzymes. A and B, quantification of g-H2AX marked DNA DSBs in BRCA1 wild-type (BRCA1þ/þ) and BRCA1heterozygous 185delAG (BRCA1�/þ) MCF10A cells 24 hours after treatment with 2-OHE2 or 4-OHE2 (A) or immediately following 3 hours of treatment with 2-OHE2 or 4-OHE2 (B). Bars,mean number of foci per cell�SEM from three independent experiments (>200 cells were counted per experiment). Significance ofchanges in foci numbers was assessed using Student two-tailed t test. ��, P < 0.01; ���, P < 0.001. C and D, quantification of g-H2AX marked DNADSBs as above inBRCA1wild-type (BRCA1þ/þ) andBRCA1 heterozygous primary breast progenitor cells 24 hours after treatmentwith 2-OHE2 or 4-OHE2 (C)or immediately following 3 hours of treatment with 2-OHE2 or 4-OHE2 (D). Significance of changes in foci numbers was assessed using Student two-tailed ttest. ��, P < 0.01. E, qRT-PCR determined expression of genes involved in estrogen metabolism in BRCA1 wild-type (BRCA1þ/þ) and BRCA1 heterozygous185delAG (BRCA1�/þ)MCF10Acells. Bars,mean relative expression�SEM from three independent experiments. Significanceof changes in gene expressionwas assessed using Student two-tailed t test. ���, P < 0.001. F, quantification of chromosomal aberrations in metaphase spreads from BRCA1 wild-type(BRCA1þ/þ) and BRCA1 heterozygous 185delAG (BRCA1�þ) MCF10A cells 24 hours after treatment with 2-OHE2 or 4-OHE2. Bars, mean aberrationsper metaphase� SEM from three independent experiments (>100metaphases were scored per experiment). Significance of changes in aberration numberswas assessed using Student two-tailed t test. ���, P < 0.001.
Savage et al.
Cancer Res; 74(10) May 15, 2014 Cancer Research2782
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611

required for the repair of these breaks, loss of BRCA2 does notlead to deregulated estrogen metabolism and the associatedincreased DNA damage. Perhaps this explains why BRCA2mutations are less penetrant than BRCA1 mutations in pre-disposing carriers to breast and ovarian cancers.We have also demonstrated that 2-OHE2 and 4-OHE2 treat-
ment leads to increased DSB production in MCF10A cells andprimary breast cells harboring a pathogenic heterozygousBRCA1 mutation, and that like BRCA1-depleted cells, thesecells are unable to repair E2 metabolite-mediated DNA dam-age, leading to increased genomic instability. Importantly, wefound that BRCA1 heterozygous mutant cells also have upre-gulated levels of CYP1A1 and CYP3A4, suggesting that inc-reased estrogen metabolism may contribute to E2-mediatedDNA damage in these cells. Consistent with this, higher levelsof urinary excreted 2-OHE2 and 4-OHE2 have been observed inBRCA1 carriers compared with healthy control women with noBRCA1 mutation (40).Taken together, these findings suggest that exposure to
estrogen and its subsequent metabolism in BRCA1-deficientbreast cells is capable of driving genomic instability, a well-defined early event in breast cancer development. Given thatestrogen levels in normal/benign breast tissue are known to besix to seven times that of circulating estrogen levels, ourfindings suggest a mechanism through which BRCA1 carriers,through enhanced production of DNA-damaging estrogenmetabolites, may acquire the genetic alterations that initiateneoplastic transformation in breast tissue (34). Similarly, levelsof estrogen in ovarian tissues greatly exceed that of circulatingestrogen, suggesting that this model may also explain thesubstantially increased risk of ovarian cancer inBRCA1 carriers(41).A phase III trial termed, Prevention of Breast Cancer by
Letrozole in Postmenopausal Women carrying a BRCA1/2Mutation (LIBER), (ClinicalTrials.gov number, NCT00673335)is currently enrolling postmenopausal women for treatmentwith letrozole, an aromatase inhibitor, to evaluate its ability toprevent the development of breast cancer in patients with aBRCA1/2 mutation. Our results coupled with the finding thataromatase levels are substantially higher in prophylactic mas-tectomy and oophorectomy tissue from BRCA1 carriers (10),provides further mechanistic data to support this approach.However, aromatase inhibitors may have little preventative
effect in premenopausal women, in whom the majority ofBRCA1-linked tumors develop, and in whom estrogen produc-tion occurs predominantly in the ovaries through an aroma-
tase-independent biosynthesis pathway. In these women,oophorectomy has been shown to reduce the risk of breastcancer by up to 60% (42). Taking our findings into account, itmay also be worth considering the use of aromatase inhibitorsas an additional chemopreventative strategy in premenopausalwomen, who have undergone risk-reducing oophorectomywithout mastectomy.
Finally, in premenopausal women who have opted not toundergo risk-reducing oophorectomy ormastectomy, luteiniz-ing hormone releasing hormone agonists, may prove usefulas chemopreventative agents. These drugs cause reversibleovarian suppression/ablation and are currently used in com-bination with tamoxifen or aromatase inhibitors for the treat-ment of premenopausal women with ER-a–positive breastcancer (43, 44).
Disclosure of Potential Conflicts of InterestNo potential conflicts of interest were disclosed.
Authors' ContributionsConception and design: K.I. Savage, S. McIntosh, C.T. Elliot, D.P. HarkinDevelopment of methodology: K.I. Savage, K.B. Matchett, K.M. Cooper,G.W. Irwin, J.J. Gorski, M. Salto-Tellez, C.T. ElliotAcquisition of data (provided animals, acquired and managed patients,provided facilities, etc.): K.I. Savage, K.B. Matchett, E.M. Barros, K.M. Cooper,G.W. Irwin, K.S. Orr, J.N. Kavanagh, A. Powell, L. Manti, S. McIntosh, M. Salto-Tellez, D.J. RichardAnalysis and interpretation of data (e.g., statistical analysis, biostatistics,computational analysis): K.I. Savage, K.B. Matchett, E.M. Barros, K.M. Cooper,G.W. Irwin, A. Powell, L. Manti, S.S. McDade, D.P. HarkinWriting, review, and/or revision of the manuscript: K.I. Savage, K.B.Matchett, E.M. Barros, K.M. Cooper, G.W. Irwin, J.J. Gorski, A. Powell, S.S.McDade, B.H. Park, K.M. Prise, S. McIntosh, D.J. Richard, C.T. Elliot, D.P. HarkinAdministrative, technical, or material support (i.e., reporting or orga-nizing data, constructing databases): K.I. Savage, J. Vohhodina, A.F. Madden,B.H. ParkStudy supervision: K.I. Savage, D.P. Harkin
AcknowledgmentsThe authors thank the Northern Ireland Biobank for providing breast tumor
sections and fresh normal breast tissues.
Grant SupportThis work was supported by grants from Cancer Research UK (C538/A8132;
D.P. Harkin, K.B. Matchett, and E.M. Barros), the Research and DevelopmentOffice Northern Ireland (G.W. Irwin), and Cancer Focus Northern Ireland (K.I.Savage).
The costs of publication of this article were defrayed in part by the payment ofpage charges. This article must therefore be hereby marked advertisement inaccordance with 18 U.S.C. Section 1734 solely to indicate this fact.
Received September 10, 2013; revised January 31, 2014; accepted February 22,2014; published OnlineFirst March 17, 2014.
References1. Huen MS, Sy SM, Chen J. BRCA1 and its toolbox for the maintenance
of genome integrity. Nat Rev Mol Cell Biol 2010;11:138–48.2. Zhu Q, Pao GM, Huynh AM, Suh H, Tonnu N, Nederlof PM, et al.
BRCA1 tumour suppression occurs via heterochromatin-mediatedsilencing. Nature 2011;477:179–84.
3. Pathania S, Nguyen J, Hill SJ, Scully R, Adelmant GO, Marto JA, et al.BRCA1 is required for postreplication repair after UV-induced DNAdamage. Mol Cell 2011;44:235–51.
4. Chen S, Parmigiani G. Meta-analysis of BRCA1 and BRCA2 pene-trance. J Clin Oncol 2007;25:1329–33.
5. Risch HA, McLaughlin JR, Cole DE, Rosen B, Bradley L, Fan I, et al.Population BRCA1 and BRCA2 mutation frequencies and cancerpenetrances: a kin-cohort study in Ontario, Canada. J Natl CancerInst 2006;98:1694–706.
6. Kauff ND, Satagopan JM, Robson ME, Scheuer L, Hensley M, HudisCA, et al. Risk-reducing salpingo-oophorectomy in women with aBRCA1 or BRCA2 mutation. N Engl J Med 2002;346:1609–15.
7. RebbeckTR, LynchHT,NeuhausenSL,NarodSA,Van't Veer L,GarberJE, et al. Prophylactic oophorectomy in carriers of BRCA1 or BRCA2mutations. N Engl J Med 2002;346:1616–22.
Estrogen-Induced Genomic Instability in BRCA1-Deficient Cells
www.aacrjournals.org Cancer Res; 74(10) May 15, 2014 2783
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611

8. Narod SA, Kotsopoulos J, Lubinski J, Lynch H, Kim-Sing C, Neuhau-sen SL, et al. Oophorectomy after menopause and the risk of breastcancer in BRCA1 and BRCA2 mutation carriers? Cancer EpidemiolBiomarkers Prev 2012;21:1089–96.
9. Jernstrom H, Lerman C, Ghadirian P, Lynch HT, Weber B, Garber J,et al. Pregnancy and risk of early breast cancer in carriers of BRCA1and BRCA2. Lancet 1999;354:1846–50.
10. Chand AL, Simpson ER, ClyneCD. Aromatase expression is increasedin BRCA1 mutation carriers. BMC Cancer 2009;9:148.
11. Hu Y, Ghosh S, Amleh A, Yue W, Lu Y, Katz A, et al. Modulation ofaromatase expression by BRCA1: a possible link to tissue-specifictumor suppression. Oncogene 2005;24:8343–8.
12. Mavaddat N, Barrowdale D, Andrulis IL, Domchek SM, Eccles D,Nevanlinna H, et al. Pathology of breast and ovarian cancers amongBRCA1 and BRCA2 mutation carriers: results from the consortium ofinvestigators of modifiers of BRCA1/2 (CIMBA). Cancer EpidemiolBiomarkers Prev 2012;21:134–47.
13. Atchley DP, Albarracin CT, Lopez A, Valero V, Amos CI, Gonzalez-Angulo AM, et al. Clinical and pathologic characteristics of patientswith BRCA-positive and BRCA-negative breast cancer. J Clin Oncol2008;26:4282–8.
14. Hosey AM, Gorski JJ, Murray MM, Quinn JE, Chung WY, Stewart GE,et al.Molecular basis for estrogen receptor alpha deficiency inBRCA1-linked breast cancer. J Natl Cancer Inst 2007;99:1683–94.
15. YueW,Wang JP, Li Y, Fan P, Liu G, Zhang N, et al. Effects of estrogenon breast cancer development: role of estrogen receptor independentmechanisms. Int J Cancer 2010;127:1748–57.
16. Huang J, Sun J, Chen Y, Song Y, Dong L, Zhan Q, et al. Analysis ofmultiplex endogenous estrogen metabolites in human urine usingultra-fast liquid chromatography-tandem mass spectrometry: a casestudy for breast cancer. Anal Chim Acta 2012;711:60–8.
17. Rogan EG, Badawi AF, Devanesan PD, Meza JL, Edney JA,West WW,et al. Relative imbalances in estrogen metabolism and conjugation inbreast tissue of women with carcinoma: potential biomarkers ofsusceptibility to cancer. Carcinogenesis 2003;24:697–702.
18. Newbold RR, Liehr JG. Induction of uterine adenocarcinoma in CD-1mice by catechol estrogens. Cancer Res 2000;60:235–7.
19. Liehr JG, Fang WF, Sirbasku DA, Ari-Ulubelen A. Carcinogenicity ofcatechol estrogens in Syrian hamsters. J Steroid Biochem 1986;24:353–6.
20. Chakravarti D,MailanderPC, Li KM,HigginbothamS,ZhangHL,GrossML, et al. Evidence that a burst ofDNAdepurination inSENCARmouseskin induces error-prone repair and formsmutations in the H-ras gene.Oncogene 2001;20:7945–53.
21. Zhao Z, Kosinska W, Khmelnitsky M, Cavalieri EL, Rogan EG, Chak-ravarti D, et al. Mutagenic activity of 4-hydroxyestradiol, but not 2-hydroxyestradiol, in BB rat2 embryonic cells, and the mutationalspectrum of 4-hydroxyestradiol. Chem Res Toxicol 2006;19:475–9.
22. Mailander PC, Meza JL, Higginbotham S, Chakravarti D. Induction ofA.T toG.Cmutationsby erroneous repair of depurinatedDNA followingestrogen treatment of the mammary gland of ACI rats. J SteroidBiochem Mol Biol 2006;101:204–15.
23. Gaikwad NW, Yang L, Muti P, Meza JL, Pruthi S, Ingle JN, et al. Themolecular etiology of breast cancer: evidence from biomarkers of risk.Int J Cancer 2008;122:1949–57.
24. Gaikwad NW, Yang L, Pruthi S, Ingle JN, Sandhu N, Rogan EG, et al.Urine biomarkers of risk in the molecular etiology of breast cancer.Breast Cancer (Auckl) 2009;3:1–8.
25. Cavalieri E, Chakravarti D, Guttenplan J, Hart E, Ingle J, Jankowiak R,et al. Catechol estrogen quinones as initiators of breast and otherhuman cancers: implications for biomarkers of susceptibility andcancer prevention. Biochim Biophys Acta 2006;1766:63–78.
26. Mizutani A,OkadaT, Shibutani S, SonodaE,HocheggerH,Nishigori C,et al. Extensive chromosomal breaks are induced by tamoxifenand estrogen in DNA repair-deficient cells. Cancer Res 2004;64:3144–7.
27. Konishi H, Mohseni M, Tamaki A, Garay JP, Croessmann S, Karnan S,et al. Mutation of a single allele of the cancer susceptibility geneBRCA1 leads to genomic instability in human breast epithelial cells.Proc Natl Acad Sci U S A 2011;108:17773–8.
28. Gorski JJ, SavageKI,Mulligan JM,McDadeSS,Blayney JK,GeZ, et al.Profiling of the BRCA1 transcriptome through microarray and ChIP-chip analysis. Nucleic Acids Res 2011;39:9536–48.
29. Manti L, Durante M, Grossi G, Ortenzia O, Pugliese M, Scampoli P,et al. Measurements of metaphase and interphase chromosomeaberrations transmitted through early cell replication rounds in humanlymphocytes exposed to low-LET protons and high-LET 12C ions.Mutat Res 2006;596:151–65.
30. Xu X, Veenstra TD, Fox SD, Roman JM, Issaq HJ, Falk R, et al.Measuring fifteen endogenous estrogens simultaneously in humanurine by high-performance liquid chromatography-mass spectrome-try. Anal Chem 2005;77:6646–54.
31. Dontu G, AbdallahWM, Foley JM, Jackson KW, ClarkeMF, KawamuraMJ, et al. In vitro propagation and transcriptional profiling of humanmammary stem/progenitor cells. Genes Dev 2003;17:1253–70.
32. Harte MT, O'Brien GJ, Ryan NM, Gorski JJ, Savage KI, Crawford NT,et al. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1and regulates BRCA1-dependent transcription. Cancer Res 2010;70:2538–47.
33. Bae I, Fan S, Meng Q, Rih JK, Kim HJ, Kang HJ, et al. BRCA1 inducesantioxidant geneexpressionand resistance tooxidative stress.CancerRes 2004;64:7893–909.
34. Lonning PE, Helle H, Duong NK, Ekse D, Aas T, Geisler J. Tissueestradiol is selectively elevated in receptor positive breast cancerswhile tumour estrone is reduced independent of receptor status.J Steroid Biochem Mol Biol 2009;117:31–41.
35. ErnestosB,NikolaosP,KoulisG,EleniR,KonstantinosB,AlexandraG,et al. Increased chromosomal radiosensitivity in women carryingBRCA1/BRCA2 mutations assessed with the G2 assay. Int J RadiatOncol Biol Phys 2010;76:1199–205.
36. SuzukiN, YasuiM,Santosh LaxmiYR,OhmoriH,HanaokaF,ShibutaniS. Translesion synthesis past equine estrogen-derived 20-deoxycyti-dine DNA adducts by human DNA polymerases eta and kappa.Biochemistry 2004;43:11312–20.
37. Yasui M, Suzuki N, Liu X, Okamoto Y, Kim SY, Laxmi YR, et al.Mechanism of translesion synthesis past an equine estrogen-DNAadduct by Y-family DNA polymerases. J Mol Biol 2007;371:1151–62.
38. Pedram A, Razandi M, Evinger AJ, Lee E, Levin ER. Estrogen inhibitsATR signaling to cell cycle checkpoints and DNA repair. Mol Biol Cell2009;20:3374–89.
39. Feng Z, Zhang J. A dual role of BRCA1 in two distinct homologousrecombination mediated repair in response to replication arrest.Nucleic Acids Res 2012;40:726–38.
40. Berstein LM, Koskela A, Boyarkina MP, Adlercreutz H. Excretion ofestrogens,catecholestrogensandphytoestrogens incarriersofBRCA1gene mutations: effects of metformin. Neoplasma 2010;57:333–8.
41. Lindgren PR, Backstrom T, Cajander S, Damber MG, Mahlck CG, ZhuD, et al. The pattern of estradiol and progesterone differs in serum andtissue of benign and malignant ovarian tumors. Int J Oncol 2002;21:583–9.
42. Eisen A, Lubinski J, Klijn J, Moller P, Lynch HT, Offit K, et al. Breastcancer risk following bilateral oophorectomy in BRCA1 and BRCA2mutation carriers: an international case-control study. J Clin Oncol2005;23:7491–6.
43. Del Mastro L, Levaggi A, Giraudi S, Pronzato P. Luteinising hormonereleasing hormone agonists (LH-RHa) in premenopausal early breastcancer patients: current role and future perspectives.Cancer TreatRev2011;37:208–11.
44. Rossi E, Morabito A, DeMaio E, Di Rella F, Esposito G, Gravina A, et al.Endocrine effects of adjuvant letrozole þ triptorelin compared withtamoxifen þ triptorelin in premenopausal patients with early breastcancer. J Clin Oncol 2008;26:264–70.
Cancer Res; 74(10) May 15, 2014 Cancer Research2784
Savage et al.
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611

2014;74:2773-2784. Published OnlineFirst March 17, 2014.Cancer Res Kienan I. Savage, Kyle B. Matchett, Eliana M. Barros, et al. and Genomic InstabilityBRCA1 Deficiency Exacerbates Estrogen-Induced DNA Damage
Updated version
10.1158/0008-5472.CAN-13-2611doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2014/03/17/0008-5472.CAN-13-2611.DC1
Access the most recent supplemental material at:
Cited articles
http://cancerres.aacrjournals.org/content/74/10/2773.full#ref-list-1
This article cites 44 articles, 13 of which you can access for free at:
Citing articles
http://cancerres.aacrjournals.org/content/74/10/2773.full#related-urls
This article has been cited by 7 HighWire-hosted articles. Access the articles at:
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications Department at
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/74/10/2773To request permission to re-use all or part of this article, use this link
on June 28, 2018. © 2014 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Published OnlineFirst March 17, 2014; DOI: 10.1158/0008-5472.CAN-13-2611





























