Embed Size (px)
Citation preview


BLAST VIII MEETING
BOCA RATON/DEERFIELD BEACH, FLORIDA JANUARY 16-21, 2005
Meeting Chairperson:
Dr. Robert Kadner – University of Virginia, Charlottesville
Meeting Vice-Chairperson: Dr. Ikuro Kawagishi – Nagoya University, Nagoya, Japan
Program Committee: Dr. John S. Parkinson (Chairperson) – University of Utah
Robert Macnab Award Selection Committee: Dr. Judy Armitage (Chairperson) – Oxford University, UK Dr. Ikuro Kawagishi – Nagoya University, Nagoya, Japan
Dr. Michael Manson – Texas A&M University, College Station
Meeting Review Committee: Dr. Liz Sockett (Chairperson) – University of Nottingham, UK
Dr. Lotte Sogaard-Andersen – Max Planck Institute, Marburg, Germany Dr. Ruth Silversmith – University of North Carolina, Chapel Hill
Dr. Urs Jenal – University of Basel, Switzerland
Board of Directors – BLAST, Inc.: Dr. Joe Falke – University of Colorado, Boulder
Dr. Michael Manson – Texas A&M University, College Station Dr. Philip Matsumura (Chairperson) – University of Illinois at Chicago
Dr. John S. Parkinson – University of Utah, Salt Lake City
Administrative Assistants: Ms. Tarra Bollinger – Molecular Biology Consortium, Chicago Ms. Jasemin Brown – Molecular Biology Consortium, Chicago Ms. Peggy O’Neill – Molecular Biology Consortium, Chicago
ii

BLAST VIII MEETING SCHEDULE
TIME EVENT LOCATION Sunday, January 16, 20054:00 pm – 6:30 pm Meeting Registration Ballroom Foyer 6:30 pm – 8:00 pm Buffet Dinner Grand Ballroom 8:00 pm Poster room available after 8 pm for poster setup Hibiscus Room 9:00 pm – 11:00 pm Welcome Reception Pool Terrace/Ballroom Foyer Monday, January 17, 20058:00 am – 9:00 am Buffet Breakfast Hillsboro & Coral Rooms 9:00 am – 12:00 pm Meeting Session – “Flagella” Grand Ballroom 12:00 pm – 1:30 pm Lunch Hillsboro & Coral Rooms 2:00 pm – 4:00 pm Poster Session – even numbered posters Hibiscus Room 6:00 pm – 7:30 pm Buffet Dinner Hillsboro & Coral Rooms 7:30 pm – 10:00 pm Meeting Session – “Two-Component Signaling” Grand Ballroom Tuesday, January 18, 20058:00 am – 9:00 am Buffet Breakfast Hillsboro & Coral Rooms 9:00 am – 12:00 pm Meeting Session – “Surface Movements” Grand Ballroom 12:00 pm – 1:30 pm Lunch Hillsboro & Coral Rooms 2:00 pm – 4:00 pm Poster Session – odd numbered posters Hibiscus Room 6:00 pm – 7:30 pm Buffet Dinner Hillsboro & Coral Rooms 7:30 pm – 10:00 pm Meeting Session – “Chemotactic Signaling” Grand Ballroom Wednesday, January 19, 20058:00 am – 9:00 am Buffet Breakfast Hillsboro & Coral Rooms 9:00 am – 12:00 pm Meeting Session – “Chemoreceptors” Grand Ballroom 12:00 pm – 1:30 pm Lunch Hillsboro & Coral Rooms Thursday, January 20, 20058:00 am – 9:00 am Buffet Breakfast Hillsboro & Coral Rooms 9:00 am – 12:00 pm Meeting Session – “Regulation” Grand Ballroom 12:00 pm – 1:30 pm Lunch Hillsboro & Coral Rooms 6:00 pm – 7:30 pm Buffet Dinner Hillsboro & Coral Rooms 7:30 pm Robert Macnab Award Presentation Grand Ballroom 7:30 pm – 10:00 pm Meeting Session – “Microbial Biology” Grand Ballroom 10:00 pm – 12:00 am Reception Pool Terrace/Ballroom Foyer Friday, January 21, 20057:00 am – 8:30 am Buffet Breakfast Hillsboro & Coral Rooms
iii
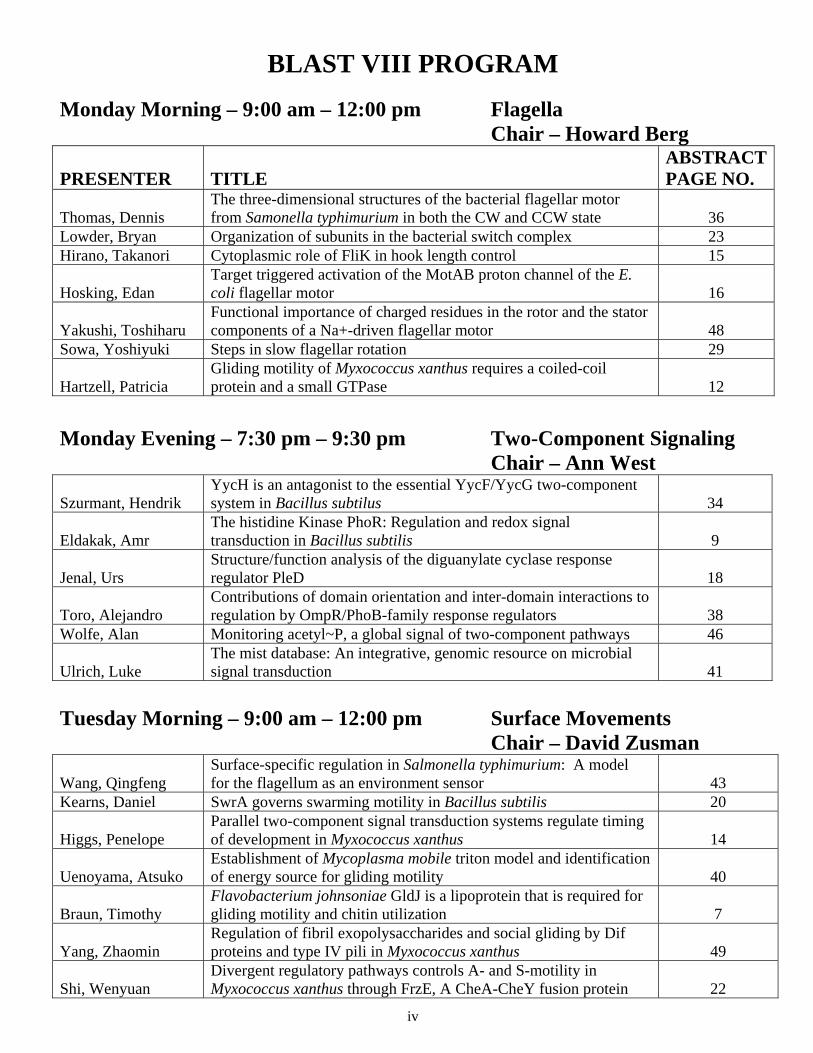
BLAST VIII PROGRAM Monday Morning – 9:00 am – 12:00 pm Flagella Chair – Howard Berg
PRESENTER TITLE ABSTRACT PAGE NO.
Thomas, Dennis The three-dimensional structures of the bacterial flagellar motor from Samonella typhimurium in both the CW and CCW state 36
Lowder, Bryan Organization of subunits in the bacterial switch complex 23 Hirano, Takanori Cytoplasmic role of FliK in hook length control 15
Hosking, Edan Target triggered activation of the MotAB proton channel of the E. coli flagellar motor 16
Yakushi, Toshiharu Functional importance of charged residues in the rotor and the stator components of a Na+-driven flagellar motor 48
Sowa, Yoshiyuki Steps in slow flagellar rotation 29
Hartzell, Patricia Gliding motility of Myxococcus xanthus requires a coiled-coil protein and a small GTPase 12
Monday Evening – 7:30 pm – 9:30 pm Two-Component Signaling Chair – Ann West
Szurmant, Hendrik YycH is an antagonist to the essential YycF/YycG two-component system in Bacillus subtilus 34
Eldakak, Amr The histidine Kinase PhoR: Regulation and redox signal transduction in Bacillus subtilis 9
Jenal, Urs Structure/function analysis of the diguanylate cyclase response regulator PleD 18
Toro, Alejandro Contributions of domain orientation and inter-domain interactions to regulation by OmpR/PhoB-family response regulators 38
Wolfe, Alan Monitoring acetyl~P, a global signal of two-component pathways 46
Ulrich, Luke The mist database: An integrative, genomic resource on microbial signal transduction 41
Tuesday Morning – 9:00 am – 12:00 pm Surface Movements Chair – David Zusman
Wang, Qingfeng Surface-specific regulation in Salmonella typhimurium: A model for the flagellum as an environment sensor 43
Kearns, Daniel SwrA governs swarming motility in Bacillus subtilis 20
Higgs, Penelope Parallel two-component signal transduction systems regulate timing of development in Myxococcus xanthus 14
Uenoyama, Atsuko Establishment of Mycoplasma mobile triton model and identification of energy source for gliding motility 40
Braun, Timothy Flavobacterium johnsoniae GldJ is a lipoprotein that is required for gliding motility and chitin utilization 7
Yang, Zhaomin Regulation of fibril exopolysaccharides and social gliding by Dif proteins and type IV pili in Myxococcus xanthus 49
Shi, Wenyuan Divergent regulatory pathways controls A- and S-motility in Myxococcus xanthus through FrzE, A CheA-CheY fusion protein 22
iv
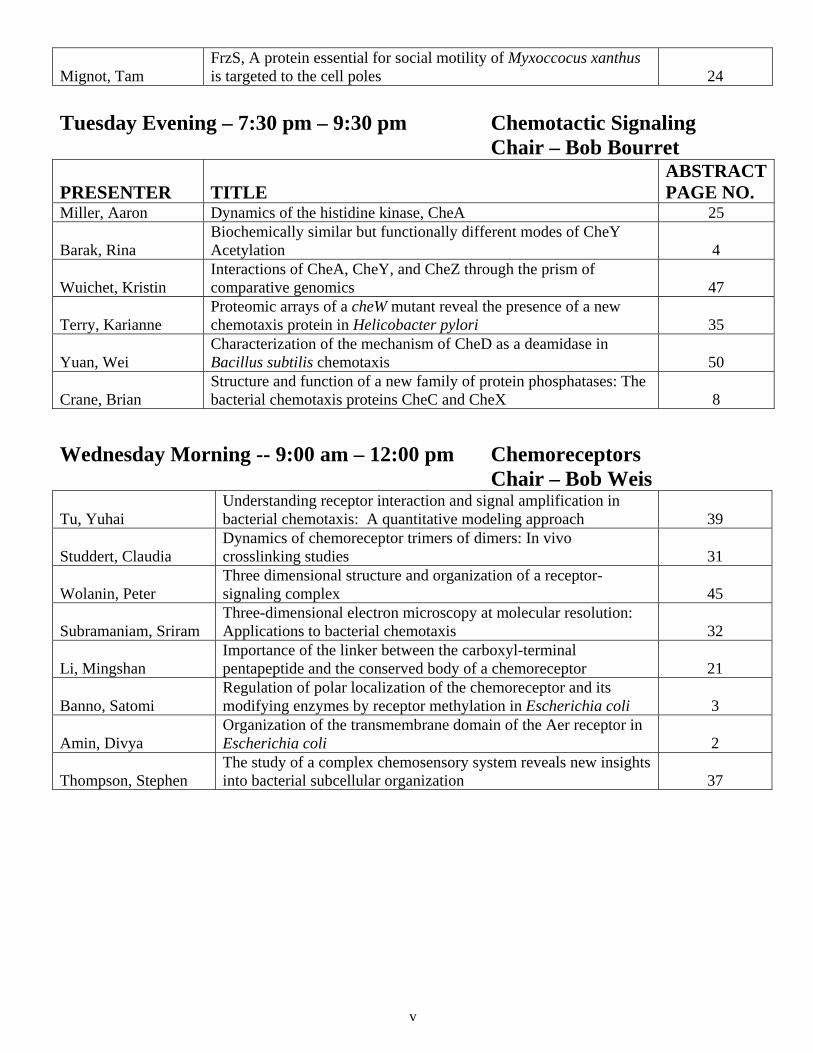
Mignot, Tam FrzS, A protein essential for social motility of Myxoccocus xanthus is targeted to the cell poles 24
Tuesday Evening – 7:30 pm – 9:30 pm Chemotactic Signaling Chair – Bob Bourret
PRESENTER TITLE ABSTRACT PAGE NO.
Miller, Aaron Dynamics of the histidine kinase, CheA 25
Barak, Rina Biochemically similar but functionally different modes of CheY Acetylation 4
Wuichet, Kristin Interactions of CheA, CheY, and CheZ through the prism of comparative genomics 47
Terry, Karianne Proteomic arrays of a cheW mutant reveal the presence of a new chemotaxis protein in Helicobacter pylori 35
Yuan, Wei Characterization of the mechanism of CheD as a deamidase in Bacillus subtilis chemotaxis 50
Crane, Brian Structure and function of a new family of protein phosphatases: The bacterial chemotaxis proteins CheC and CheX 8
Wednesday Morning -- 9:00 am – 12:00 pm Chemoreceptors Chair – Bob Weis
Tu, Yuhai Understanding receptor interaction and signal amplification in bacterial chemotaxis: A quantitative modeling approach 39
Studdert, Claudia Dynamics of chemoreceptor trimers of dimers: In vivo crosslinking studies 31
Wolanin, Peter Three dimensional structure and organization of a receptor-signaling complex 45
Subramaniam, Sriram Three-dimensional electron microscopy at molecular resolution: Applications to bacterial chemotaxis 32
Li, Mingshan Importance of the linker between the carboxyl-terminal pentapeptide and the conserved body of a chemoreceptor 21
Banno, Satomi Regulation of polar localization of the chemoreceptor and its modifying enzymes by receptor methylation in Escherichia coli 3
Amin, Divya Organization of the transmembrane domain of the Aer receptor in Escherichia coli 2
Thompson, Stephen The study of a complex chemosensory system reveals new insights into bacterial subcellular organization 37
v
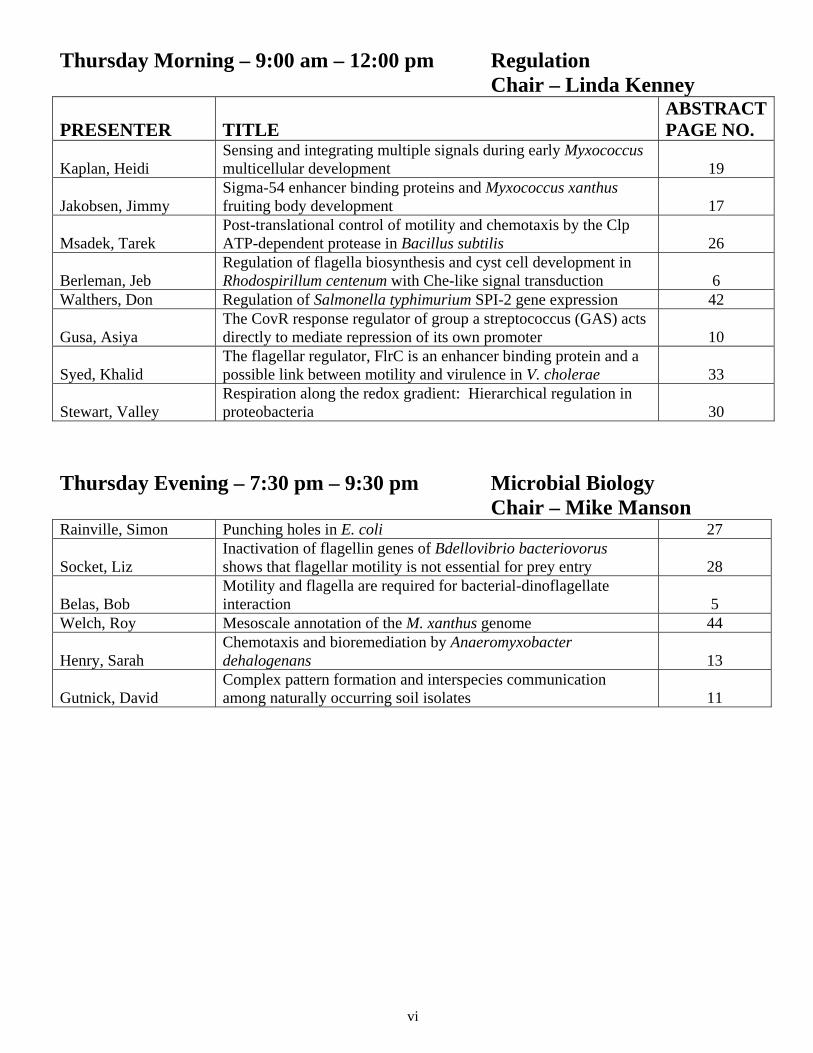
Thursday Morning – 9:00 am – 12:00 pm Regulation Chair – Linda Kenney
PRESENTER TITLE ABSTRACT PAGE NO.
Kaplan, Heidi Sensing and integrating multiple signals during early Myxococcus multicellular development 19
Jakobsen, Jimmy Sigma-54 enhancer binding proteins and Myxococcus xanthus fruiting body development 17
Msadek, Tarek Post-translational control of motility and chemotaxis by the Clp ATP-dependent protease in Bacillus subtilis 26
Berleman, Jeb Regulation of flagella biosynthesis and cyst cell development in Rhodospirillum centenum with Che-like signal transduction 6
Walthers, Don Regulation of Salmonella typhimurium SPI-2 gene expression 42
Gusa, Asiya The CovR response regulator of group a streptococcus (GAS) acts directly to mediate repression of its own promoter 10
Syed, Khalid The flagellar regulator, FlrC is an enhancer binding protein and a possible link between motility and virulence in V. cholerae 33
Stewart, Valley Respiration along the redox gradient: Hierarchical regulation in proteobacteria 30
Thursday Evening – 7:30 pm – 9:30 pm Microbial Biology Chair – Mike Manson Rainville, Simon Punching holes in E. coli 27
Socket, Liz Inactivation of flagellin genes of Bdellovibrio bacteriovorus shows that flagellar motility is not essential for prey entry 28
Belas, Bob Motility and flagella are required for bacterial-dinoflagellate interaction 5
Welch, Roy Mesoscale annotation of the M. xanthus genome 44
Henry, Sarah Chemotaxis and bioremediation by Anaeromyxobacter dehalogenans 13
Gutnick, David Complex pattern formation and interspecies communication among naturally occurring soil isolates 11
vi
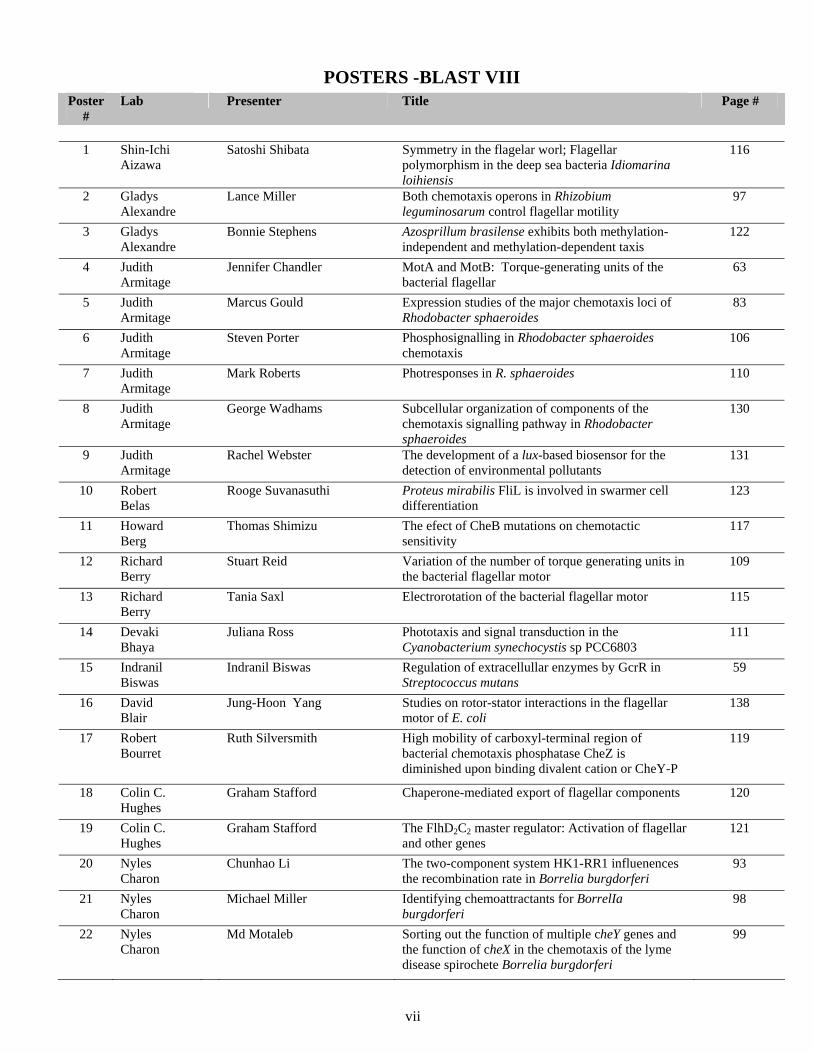
POSTERS -BLAST VIII
Poster #
Lab Presenter Title Page #
1 Shin-Ichi
Aizawa
Satoshi Shibata Symmetry in the flagelar worl; Flagellar polymorphism in the deep sea bacteria Idiomarina loihiensis
116
2 Gladys Alexandre
Lance Miller Both chemotaxis operons in Rhizobium leguminosarum control flagellar motility
97
3 Gladys Alexandre
Bonnie Stephens Azosprillum brasilense exhibits both methylation-independent and methylation-dependent taxis
122
4 Judith Armitage
Jennifer Chandler MotA and MotB: Torque-generating units of the bacterial flagellar
63
5 Judith Armitage
Marcus Gould Expression studies of the major chemotaxis loci of Rhodobacter sphaeroides
83
6 Judith Armitage
Steven Porter Phosphosignalling in Rhodobacter sphaeroides chemotaxis
106
7 Judith Armitage
Mark Roberts Photresponses in R. sphaeroides 110
8 Judith Armitage
George Wadhams Subcellular organization of components of the chemotaxis signalling pathway in Rhodobacter sphaeroides
130
9 Judith Armitage
Rachel Webster The development of a lux-based biosensor for the detection of environmental pollutants
131
10 Robert Belas
Rooge Suvanasuthi Proteus mirabilis FliL is involved in swarmer cell differentiation
123
11 Howard Berg
Thomas Shimizu The efect of CheB mutations on chemotactic sensitivity
117
12 Richard Berry
Stuart Reid Variation of the number of torque generating units in the bacterial flagellar motor
109
13 Richard Berry
Tania Saxl Electrorotation of the bacterial flagellar motor 115
14 Devaki Bhaya
Juliana Ross Phototaxis and signal transduction in the Cyanobacterium synechocystis sp PCC6803
111
15 Indranil Biswas
Indranil Biswas Regulation of extracellullar enzymes by GcrR in Streptococcus mutans
59
16 David Blair
Jung-Hoon Yang Studies on rotor-stator interactions in the flagellar motor of E. coli
138
17 Robert Bourret
Ruth Silversmith High mobility of carboxyl-terminal region of bacterial chemotaxis phosphatase CheZ is diminished upon binding divalent cation or CheY-P
119
18 Colin C. Hughes
Graham Stafford Chaperone-mediated export of flagellar components 120
19 Colin C. Hughes
Graham Stafford The FlhD2C2 master regulator: Activation of flagellar and other genes
121
20 Nyles Charon
Chunhao Li The two-component system HK1-RR1 influenences the recombination rate in Borrelia burgdorferi
93
21 Nyles Charon
Michael Miller Identifying chemoattractants for BorrelIa burgdorferi
98
22 Nyles Charon
Md Motaleb Sorting out the function of multiple cheY genes and the function of cheX in the chemotaxis of the lyme disease spirochete Borrelia burgdorferi
99
vii
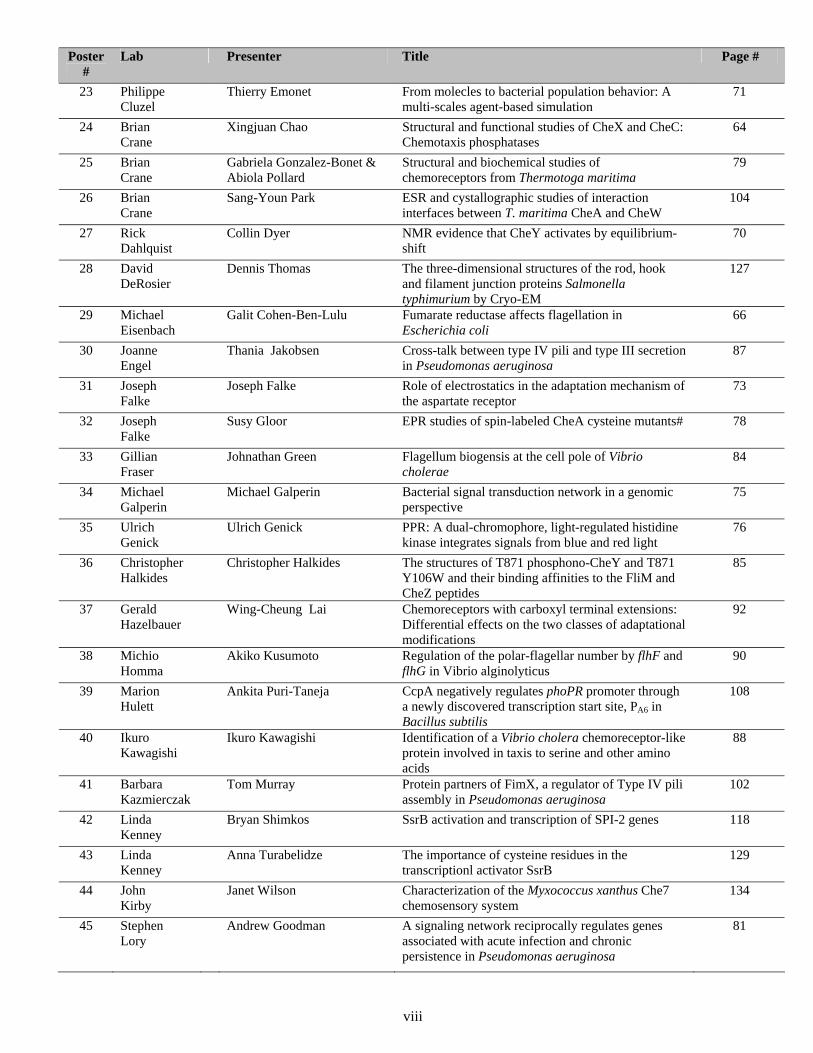
Poster #
Lab Presenter Title Page #
23 Philippe Cluzel
Thierry Emonet From molecles to bacterial population behavior: A multi-scales agent-based simulation
71
24 Brian Crane
Xingjuan Chao Structural and functional studies of CheX and CheC: Chemotaxis phosphatases
64
25 Brian Crane
Gabriela Gonzalez-Bonet & Abiola Pollard
Structural and biochemical studies of chemoreceptors from Thermotoga maritima
79
26 Brian Crane
Sang-Youn Park ESR and cystallographic studies of interaction interfaces between T. maritima CheA and CheW
104
27 Rick Dahlquist
Collin Dyer NMR evidence that CheY activates by equilibrium-shift
70
28 David DeRosier
Dennis Thomas The three-dimensional structures of the rod, hook and filament junction proteins Salmonella typhimurium by Cryo-EM
127
29 Michael Eisenbach
Galit Cohen-Ben-Lulu Fumarate reductase affects flagellation in Escherichia coli
66
30 Joanne Engel
Thania Jakobsen Cross-talk between type IV pili and type III secretion in Pseudomonas aeruginosa
87
31 Joseph Falke
Joseph Falke Role of electrostatics in the adaptation mechanism of the aspartate receptor
73
32 Joseph Falke
Susy Gloor EPR studies of spin-labeled CheA cysteine mutants# 78
33 Gillian Fraser
Johnathan Green Flagellum biogensis at the cell pole of Vibrio cholerae
84
34 Michael Galperin
Michael Galperin Bacterial signal transduction network in a genomic perspective
75
35 Ulrich Genick
Ulrich Genick PPR: A dual-chromophore, light-regulated histidine kinase integrates signals from blue and red light
76
36 Christopher Halkides
Christopher Halkides The structures of T871 phosphono-CheY and T871 Y106W and their binding affinities to the FliM and CheZ peptides
85
37 Gerald Hazelbauer
Wing-Cheung Lai Chemoreceptors with carboxyl terminal extensions: Differential effects on the two classes of adaptational modifications
92
38 Michio Homma
Akiko Kusumoto Regulation of the polar-flagellar number by flhF and flhG in Vibrio alginolyticus
90
39 Marion Hulett
Ankita Puri-Taneja CcpA negatively regulates phoPR promoter through a newly discovered transcription start site, PA6 in Bacillus subtilis
108
40 Ikuro Kawagishi
Ikuro Kawagishi Identification of a Vibrio cholera chemoreceptor-like protein involved in taxis to serine and other amino acids
88
41 Barbara Kazmierczak
Tom Murray Protein partners of FimX, a regulator of Type IV pili assembly in Pseudomonas aeruginosa
102
42 Linda Kenney
Bryan Shimkos SsrB activation and transcription of SPI-2 genes 118
43 Linda Kenney
Anna Turabelidze The importance of cysteine residues in the transcriptionl activator SsrB
129
44 John Kirby
Janet Wilson Characterization of the Myxococcus xanthus Che7 chemosensory system
134
45 Stephen Lory
Andrew Goodman A signaling network reciprocally regulates genes associated with acute infection and chronic persistence in Pseudomonas aeruginosa
81
viii
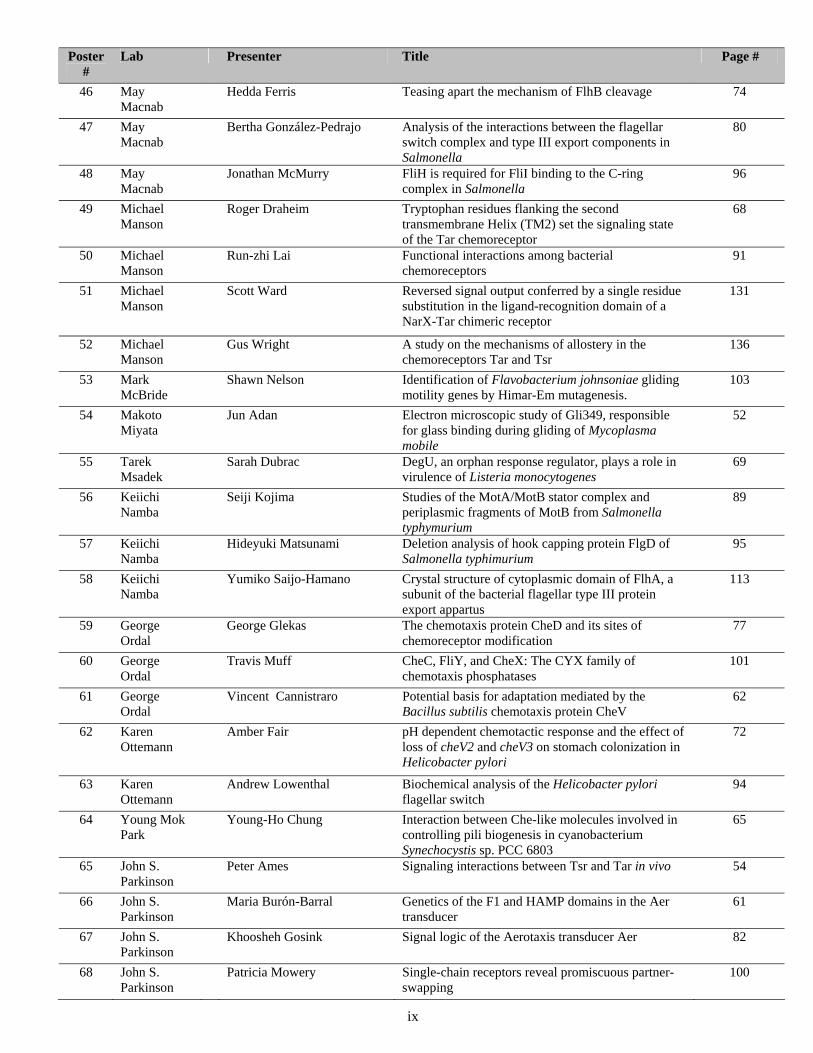
Poster #
Lab Presenter Title Page #
46 May Macnab
Hedda Ferris Teasing apart the mechanism of FlhB cleavage 74
47 May Macnab
Bertha González-Pedrajo Analysis of the interactions between the flagellar switch complex and type III export components in Salmonella
80
48 May Macnab
Jonathan McMurry FliH is required for FliI binding to the C-ring complex in Salmonella
96
49 Michael Manson
Roger Draheim Tryptophan residues flanking the second transmembrane Helix (TM2) set the signaling state of the Tar chemoreceptor
68
50 Michael Manson
Run-zhi Lai Functional interactions among bacterial chemoreceptors
91
51 Michael Manson
Scott Ward Reversed signal output conferred by a single residue substitution in the ligand-recognition domain of a NarX-Tar chimeric receptor
131
52 Michael Manson
Gus Wright A study on the mechanisms of allostery in the chemoreceptors Tar and Tsr
136
53 Mark McBride
Shawn Nelson Identification of Flavobacterium johnsoniae gliding motility genes by Himar-Em mutagenesis.
103
54 Makoto Miyata
Jun Adan Electron microscopic study of Gli349, responsible for glass binding during gliding of Mycoplasma mobile
52
55 Tarek Msadek
Sarah Dubrac DegU, an orphan response regulator, plays a role in virulence of Listeria monocytogenes
69
56 Keiichi Namba
Seiji Kojima Studies of the MotA/MotB stator complex and periplasmic fragments of MotB from Salmonella typhymurium
89
57 Keiichi Namba
Hideyuki Matsunami Deletion analysis of hook capping protein FlgD of Salmonella typhimurium
95
58 Keiichi Namba
Yumiko Saijo-Hamano Crystal structure of cytoplasmic domain of FlhA, a subunit of the bacterial flagellar type III protein export appartus
113
59 George Ordal
George Glekas The chemotaxis protein CheD and its sites of chemoreceptor modification
77
60 George Ordal
Travis Muff CheC, FliY, and CheX: The CYX family of chemotaxis phosphatases
101
61 George Ordal
Vincent Cannistraro Potential basis for adaptation mediated by the Bacillus subtilis chemotaxis protein CheV
62
62 Karen Ottemann
Amber Fair pH dependent chemotactic response and the effect of loss of cheV2 and cheV3 on stomach colonization in Helicobacter pylori
72
63 Karen Ottemann
Andrew Lowenthal Biochemical analysis of the Helicobacter pylori flagellar switch
94
64 Young Mok Park
Young-Ho Chung Interaction between Che-like molecules involved in controlling pili biogenesis in cyanobacterium Synechocystis sp. PCC 6803
65
65 John S. Parkinson
Peter Ames Signaling interactions between Tsr and Tar in vivo 54
66 John S. Parkinson
Maria Burón-Barral Genetics of the F1 and HAMP domains in the Aer transducer
61
67 John S. Parkinson
Khoosheh Gosink Signal logic of the Aerotaxis transducer Aer 82
68 John S. Parkinson
Patricia Mowery Single-chain receptors reveal promiscuous partner-swapping
100
ix
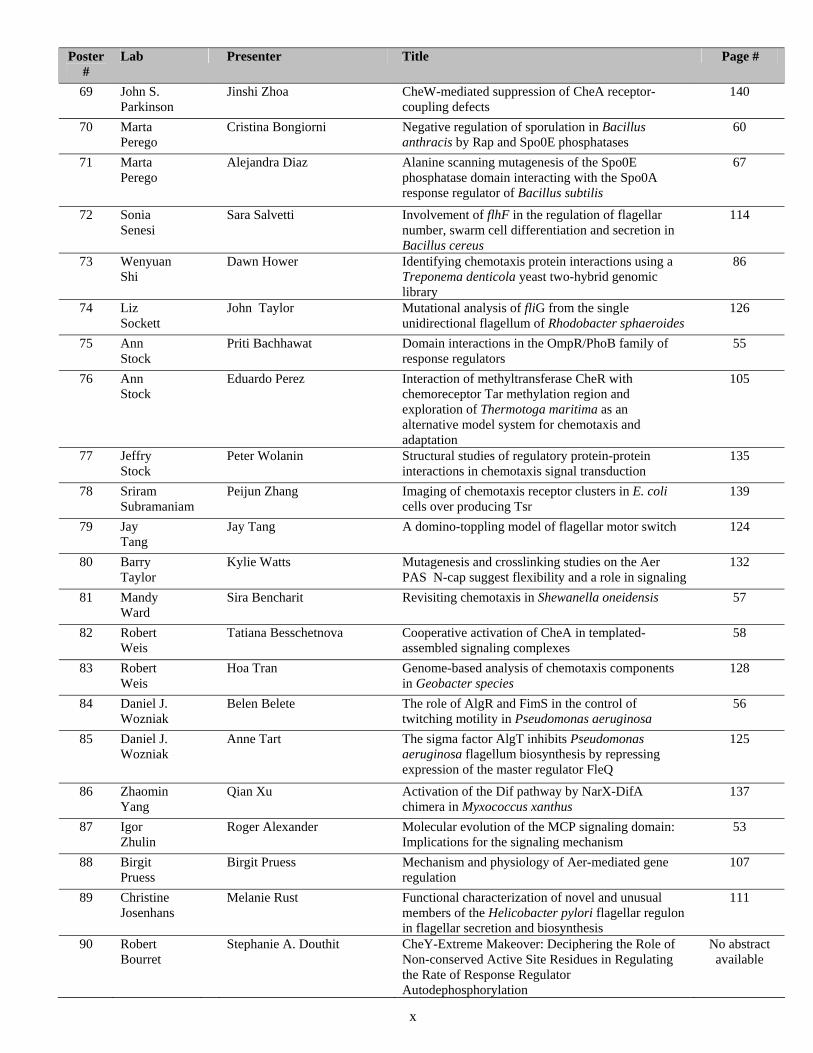
Poster #
Lab Presenter Title Page #
69 John S. Parkinson
Jinshi Zhoa CheW-mediated suppression of CheA receptor-coupling defects
140
70 Marta Perego
Cristina Bongiorni Negative regulation of sporulation in Bacillus anthracis by Rap and Spo0E phosphatases
60
71 Marta Perego
Alejandra Diaz Alanine scanning mutagenesis of the Spo0E phosphatase domain interacting with the Spo0A response regulator of Bacillus subtilis
67
72 Sonia Senesi
Sara Salvetti Involvement of flhF in the regulation of flagellar number, swarm cell differentiation and secretion in Bacillus cereus
114
73 Wenyuan Shi
Dawn Hower Identifying chemotaxis protein interactions using a Treponema denticola yeast two-hybrid genomic library
86
74 Liz Sockett
John Taylor Mutational analysis of fliG from the single unidirectional flagellum of Rhodobacter sphaeroides
126
75 Ann Stock
Priti Bachhawat Domain interactions in the OmpR/PhoB family of response regulators
55
76 Ann Stock
Eduardo Perez Interaction of methyltransferase CheR with chemoreceptor Tar methylation region and exploration of Thermotoga maritima as an alternative model system for chemotaxis and adaptation
105
77 Jeffry Stock
Peter Wolanin Structural studies of regulatory protein-protein interactions in chemotaxis signal transduction
135
78 Sriram Subramaniam
Peijun Zhang Imaging of chemotaxis receptor clusters in E. coli cells over producing Tsr
139
79 Jay Tang
Jay Tang A domino-toppling model of flagellar motor switch 124
80 Barry Taylor
Kylie Watts Mutagenesis and crosslinking studies on the Aer PAS N-cap suggest flexibility and a role in signaling
132
81 Mandy Ward
Sira Bencharit Revisiting chemotaxis in Shewanella oneidensis 57
82 Robert Weis
Tatiana Besschetnova Cooperative activation of CheA in templated-assembled signaling complexes
58
83 Robert Weis
Hoa Tran Genome-based analysis of chemotaxis components in Geobacter species
128
84 Daniel J. Wozniak
Belen Belete The role of AlgR and FimS in the control of twitching motility in Pseudomonas aeruginosa
56
85 Daniel J. Wozniak
Anne Tart The sigma factor AlgT inhibits Pseudomonas aeruginosa flagellum biosynthesis by repressing expression of the master regulator FleQ
125
86 Zhaomin Yang
Qian Xu Activation of the Dif pathway by NarX-DifA chimera in Myxococcus xanthus
137
87 Igor Zhulin
Roger Alexander Molecular evolution of the MCP signaling domain: Implications for the signaling mechanism
53
88 Birgit Pruess
Birgit Pruess Mechanism and physiology of Aer-mediated gene regulation
107
89 Christine Josenhans
Melanie Rust Functional characterization of novel and unusual members of the Helicobacter pylori flagellar regulon in flagellar secretion and biosynthesis
111
90 Robert Bourret
Stephanie A. Douthit CheY-Extreme Makeover: Deciphering the Role of Non-conserved Active Site Residues in Regulating the Rate of Response Regulator Autodephosphorylation
No abstract available
x

SPEAKER ABSTRACTS
1

ORGANIZATION OF THE TRANSMEMBRANE DOMAIN OF THE AER RECEPTOR IN ESCHERICHIA COLI D. Amin, BL Taylor and MS Johnson Loma Linda University, Loma Linda CA 92354
The Aer aerotaxis receptor in E. coli is a membrane-bound energy sensor that directs the cells to areas that generate maximum energy by the electron transport system. Each monomer of the dimeric Aer has an N-terminal PAS domain and a C-terminal signaling domain separated by a hydrophobic membrane-binding sequence of approximately 38 residues. Like MCPs, these dimers form trimers of dimers. To study the organization of the membrane domain we serially replaced 57 residues with cysteine using site-specific mutagenesis. In vivo crosslinking of these cysteine residues was performed using copper phenanthrolene to identify the topology and proximity of Aer monomers. Collisional interactions due to high flexibility were distinguished from stable interactions by comparing crosslinking rates at room temperature and at 4˚C, a temperature well below the membrane-lipid phase transition. Cytosolic and periplasmic boundaries of the membrane anchor were determined using the hydrophilic sulfhydryl reagent mPEG-MAL (PEG5000-maleimide), which binds covalently to accessible cysteine replacements. The Aer[PEG-maleimide] product migrated with an apparent molecular weight that was ~10kDa larger than Aer monomer. The complex was readily identifiable on Western blots, yielding high signal to noise ratios that obviated the necessity for high protein expression. Of unusual note, all residues from 182 to 188 crosslinked, indicating high flexibility and an undefined structure in this region. Furthermore, these same residues were accessible to the mPEG-MAL probe, suggesting that this region projects into the periplasm. These data are consistent with a membrane anchor that passes through the bilayer into the periplasm before returning to the cytosol.
2

REGULATION OF POLAR LOCALIZATION OF THE CHEMORECEPTOR AND ITS MODIFYING ENZYMES BY RECEPTOR METHYLATION IN ESCHERICHIA COLI Satomi Banno, Daisuke Shiomi*, Michio Homma and Ikuro Kawagishi Division of Biological Science, Graduate School of Science, Nagoya University, Chikusa-ku, Nagoya 464-8602, Japan; *Present address: Microbiology and Molecular Genetics, University of Texas Medical School, 6431 Fannin Street, Houston, TX 77030, U.S.A.
In the chemotaxis in Escherichia coli, it is essential for a cell to adapt to persisting stimulation. Adaptation involves methylation and demethylation of the chemoreceptors. The receptor forms a complex with the histidine kinase CheA and the adaptor protein CheW at a cell pole. Our previous studies using GFP fusions showed that the methyltransferase CheR and the methylesterase CheB co-localize with the polar receptor/kinase complex by binding to different targets: the C-terminal NWETF sequence of the receptor and the P2 domain of CheA, respectively (Shiomi et al., 2002; Banno et al., 2004). In this study, we examined the effects of receptor methylation on localization of the receptor itself, CheR and CheB. We found that methylation (amidation) of Tar-GFP slightly but significantly enhanced its polar localization. The polar localization of GFP-CheR was enhanced similarly by receptor methylation, presumably reflecting the change in receptor localization. On the other hand, GFP-CheB localized much more effectively when expressed with the methylated (amidated) receptor than with the demethylated one. Interestingly, however, the localization of the GFP fusion to another response regulator CheY, which also binds to the P2 domain of CheA and is phosphorylated by phospho-CheA, was not enhanced by receptor methylation. This regulation of CheB localization may provide a negative feedback loop, which might contribute to the termination of adaptation.
3

BIOCHEMICALLY SIMILAR BUT FUNCTIONALLY DIFFERENT MODES OF CheY ACETYLATION Rina Barak1 Jianshe Yan1, Alla Sheinskaya2 and Michael Eisenbach1
1Department of Biological Chemistry, The Weizmann Institute of Science, 76100 Rehovot, Israel. 2Biological Mass Spectrometry Facility, Department of Biological Services, The Weizmann Institute of Science, 76100 Rehovot, Israel
CheY, a response regulator of the chemotaxis system in Escherichia coli, can be activated by either phosphorylation or acetylation to generate clockwise flagellar rotation. Both covalent modifications are involved in chemotaxis, but the function of the latter remains obscure. We found in earlier studies that, in vitro, CheY can undergo acetylation by two modes: enzymatically by acetyl-CoA synthetase (Acs) with acetate or acetyl-CoA as the acetyl donor, or directly by autoacetylation with acetyl-CoA as the acetyl donor. In this study we investigated by biochemical, mass spectrometric and physiological approaches CheY autoacetylation and its involvement in chemotaxis. We found that the kinetics, stability and reversibility of CheY autoacetylation were similar to those obtained by enzymatic acetylation. Also, the phosphodonors CheA and AcP inhibited CheY autoacetylation whereas the phosphatase CheZ enhanced it; these effects were similar to the respective effects in the case of Acs-mediated acetylation, though smaller. Mass spectra confirmed the autoacetylation and indicated that like in the case of acetylation by Acs, the autoacetylation is at multiple sites. Unlike these biochemical similarities, these modes of acetylation were different with respect to their effect on chemotaxis. Deletion of Acs resulted in a defective response towards both attractants and repellents, whereas deletion of pyruvate dehydrogenase combined with a mutation in aspartate 1-decarboxylase, which causes a strong decrease in the acetyl-CoA level and, consequently, in CheY autoacetylation level, resulted in a defective response towards repellents only. Possible roles of both modes of acetylation in chemotaxis will be discussed.
4

MOTILITY AND FLAGELLA ARE REQUIRED FOR BACTERIAL-DINOFLAGELLATE INTERACTION
Robert Belas and Todd R. Miller
Center of Marine Biotechnology, 701 E. Pratt St., Baltimore, MD, USA
Since Pomeroy’s initial recognition of prokaryotes as essential components of the ocean food web, bacteria have been acknowledged as the force behind most major biogeochemical processes in the sea. Studying heterotrophic bacteria has been a challenge, however, as most of the major clades have never been cultured or only recently grown to low densities in unmodified seawater. An exception is the Roseobacter clade, members of the α-Proteobacteria, which comprises ~10-20% of coastal and oceanic mixed layer heterotrophic bacteria yet includes many members that can be readily cultured. Although roseobacters are cosmopolitan in the marine environment, their numbers and activity significantly rise with increases in the population density of phytoplankton and dinoflagellates. Little is known about the cellular factors and molecular mechanisms required for roseobacters to move towards and remain in the phycosphere surrounding dinoflagellates. The long-term goal of this research is to understand the signals and molecular mechanisms used to initiate and maintain the interaction between the roseobacter and its eukaryotic host. We have chosen a model system comprising the dinoflagellate Pfiesteria piscicida and the roseobacter Silicibacter sp. TM1040, originally isolated from a P. piscicida culture. TM1040 forms an ‘obligate’ interaction with this dinoflagellate such that axenic dinoflagellates fail to grow unless provided with the bacterium. P. piscicida produces the organosulfur compound, dimethylsulfoniopropionate (DMSP), while TM1040 catabolizes DMSP by demethylation producing 3-methylmercaptopropionate (MMPA). TM1040 is also chemotactic toward DMSP, MMPA and amino acids present in P. piscicida cells, suggesting that motility and chemotaxis play important roles in initiating the interaction between the bacteria and host cells. A bank of mutations constructed by random transposon mutagenesis was screened for defects in chemotaxis and motility. Two mutations affecting a homolog of cckA and a novel flagellar gene respectively, are defective in motility and do not produce flagella, while a third mutant, with an insertion in ctrA, produces flagella, but has an elongated cell phenotype and is poorly motile in broth and agar media. All mutants show reduced ability to form biofilms on abiotic surfaces and also have a reduced ability to attach to P. piscicida when compared to the wild type. An analysis of the ability of wild-type cells and motility mutants to complement growth defects of axenic P. piscicida suggests that a functional flagellum is essential for optimal interactions between TM1040 and dinoflagellate cells.
5

REGULATION OF FLAGELLA BIOSYNTHESIS AND CYST CELL DEVELOPMENT IN RHODOSPIRILLUM CENTENUM WITH CHE-LIKE SIGNAL TRANSDUCTION CASCADES Jeb Berleman1 and Carl Bauer2
1Georgia Institute of Technology, Atlanta, GA 30032, 2Indiana University, Bloomington, IN 47405
Rhodospirillum centenum is a purple photosynthetic bacterium that contains three che-like gene clusters. Previous studies demonstrated that chemotactic movement is dependent on the che1 operon. Here, we analyze the function of the R. centenum che2 and che3 gene clusters. In-frame deletion mutants in the che2 cluster exhibit either reduced/absent flagella (bald phenotype) or elevated amounts of flagella. Deletions of cheW2, cheB2, cheR2, cheY2, and of the entire cluster che2 exhibited a bald phenotype. In contrast, deletion of mcp2, ORF204, cheA2, and ORF74 were hyper-flagellated. The aflagellate (bald) phenotype is partially suppressed by growth at 42°C and under these conditions the cells were chemotactic. Additionally, the hyper-flagellated che2 mutants also remain chemotacticly and phototacticly competent. Deletion analysis of genes in the che3 cluster indicates that che3 mutants are defective in regulating resting cyst cell formation. Deletions of the che3 genes cheY3, cheB3, cheS3 and che3 resulted in cells that differentiate into cysts prematurely resulting in elevated levels of mature cyst production. In contrast, deletion of cheW3A, cheR3, cheW3B, mcp3, and cheA3 caused a deficiency in cyst cell formation. As is the case of che2 mutants, disruption of che3 genes do not affect chemotaxis or phototaxis. Thus, R. centenum contains three operons comprised of similar che-like components that are used to regulate three distinct cellular processes: chemotactic movement (che1 cluster), flagellar biosynthesis (che2 cluster) and encystment (che3 cluster).
6

FLAVOBACTERIUM JOHNSONIAE GLDJ IS A LIPOPROTEIN THAT IS REQUIRED FOR GLIDING MOTILITY AND CHITIN UTILIZATION Timothy F. Braun and Mark J. McBride Department of Biological Sciences, University of Wisconsin-Milwaukee Milwaukee, WI 53201
Flavobacterium johnsoniae exhibits rapid gliding motility over surfaces. Cells of F. johnsoniae lack flagella, pili, or other obvious appendages, and the mechanism responsible for gliding movement is not known. Eight genes required for gliding motility have been described. Complementation of the nonmotile mutant UW102-48 identified another gene, gldJ, that is required for gliding. gldJ mutants formed nonspreading colonies, and individual cells were completely nonmotile. They also failed to digest chitin and were resistant to bacteriophages that infect wild-type cells. Complementation with wild-type gldJ restored motility, bacteriophage sensitivity and ability to use chitin. gldJ encodes a 61 kDa protein which exhibits sequence similarity to Erwinia carotovora CarF and Bacillus stearothermophilus XaiF. CarF is involved in resistance to the β-lactam antibiotic carbapenem, while XaiF is involved in the production, export or activation of extracellular xylanase. Cell fractionation and labeling studies indicated that GldJ is an outer membrane lipoprotein. Mutations in gldA, gldB, gldD, gldF, gldG, gldH, or gldI resulted in normal levels of gldJ transcript but decreased levels of GldJ protein. GldJ may be part of a large complex of Gld proteins, and may be unstable in the absence of its partners. Pull-down experiments using His-tagged Gld proteins indicate that GldJ interacts directly or indirectly with GldB. Immunofluorescence microscopic analyses suggest that GldJ is part of a helical structure within the cell envelope. The mechanism of gliding motility and the role of GldJ remain uncertain. One model to explain gliding involves coordinated export and import of macromolecules that serve as 'conveyer belts' to move the cell.
7

STRUCTURE AND FUNCTION OF A NEW FAMILY OF PROTEIN PHOSPHATASES: THE BACTERIAL CHEMOTAXIS PROTEINS CHEC AND CHEX
Sang-Youn Park, Xingjuan Chao, Gabriela Gonzalez-Bonet, Bryan D. Beel, Alexandrine M. Bilwes and Brian R. Crane Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14850
In bacterial chemotaxis, phosphorylated CheY levels control the sense of flagella rotation and thereby determine swimming behavior. In E. coli, CheY dephosphorylation by CheZ extinguishes the switching signal. But, instead of CheZ, many chemotactic bacteria contain CheC, CheD, and/or CheX. The crystal structures of T. maritima CheC and CheX reveal a common fold unlike that of any other known protein. Unlike CheC, CheX dimerizes via a continuous β-sheet between subunits. T. maritima CheC, as well as CheX, dephosphorylate CheY, although CheC requires binding of CheD to achieve the activity of CheX. Structural analyses identified one conserved active site in CheX and two in CheC; mutations therein reduce CheY-phosphatase activity, but only mutants of two invariant asparagine residues are completely inactive even in the presence of CheD. Our structures indicate that the flagellar switch components FliY and FliM resemble CheC more closely than CheX, but attribute phosphatase activity only to FliY.
8

THE HISTIDINE KINASE PHOR: REGULATION AND THE REDOX SIGNAL TRANSDUCTION IN BACILLUS SUBTILIS Amr H. Eldakak and F. M. Hulett Laboratory for Molecular Biology, University of Illinois at Chicago, Chicago, IL 60607
In Bacillus subtilis, the Pho signal transduction network mediates the adaptive response to phosphate starvation conditions. This network encompasses three two-component systems PhoP/PhoR, ResD/ResE, and the sporulation phosphorelay. A positive feedback loop exists between the PhoP/PhoR and ResD/ResE two-component systems. The ResD/ResE system mediates aerobic and anaerobic respiration in B. subtilis. ResD controls ctaA expression that is required for the production of the a-type terminal oxidases, caa3 and aa3. The response regulator, ResD, is required for 80% of the Pho induction. Strains bearing a ∆resDE mutation spontaneously acquire secondary mutations that allow the expression of cytochrome bd to restore Pho induction to wild type levels. A ctaA mutant strain, deficient in the production of heme A, has the same Pho induction phenotype as a ∆resDE strain. Constitutive expression of cytochrome bd in a ∆ctaA background, restored Pho induction to wild type levels. This indicates that the impact of ResD on Pho induction, in a wild type strain, is through the a-type terminal oxidases. Our in vitro studies showed that the reduced state of quinones inhibits PhoR autophosphorylation, while the reducing agent alone or the oxidized quinone does not affect PhoR autophosphorylation. The addition of a reducing agent, dithiothreitol (DTT), to the growth medium reduced the level of Pho induction as indicated by PhoP activated promoter-lacZ fusions, in agreement with our in vitro data. Our current hypothesis is that the reduced form of quinones inhibits PhoR autophosphorylation under phosphate replete conditions and acts as an internal repression signal. This repression is relieved at the onset of phosphate starvation as terminal oxidases shift the quinone pool to an oxidized state. These data provide a novel link between aerobic respiration (electron transport) and Pho induction in B. subtilis.
9

THE COVR RESPONSE REGULATOR OF GROUP A STREPTOCOCCUS (GAS) ACTS DIRECTLY TO MEDIATE REPRESSION OF ITS OWN PROMOTER Asiya A. Gusa and June R. Scott Emory University School of Medicine, Atlanta, GA 30322
The group A streptococcus (GAS) or Streptococcus pyogenes is an important human pathogen that causes a wide range of diseases. These range from mild infections of the throat and skin, such as pharyngitis and impetigo, to more severe, life-threatening diseases such as necrotizing fasciitis and streptococcal toxic shock syndrome. The CovR/S (CsrR/S) two-component system is a global regulator of virulence gene expression in GAS. The response regulator, CovR, regulates about 15% of the genes of GAS, including the has operon (hyaluronic acid capsule synthesis), ska (streptokinase), sagA (streptolysin S), speMF/sda (streptodornase), and speB (cysteine protease). CovR also negatively regulates its own operon, although it is unclear whether this regulation is direct. Using in vitro DNA binding assays with purified CovR protein, we found that CovR binds a DNA fragment including the covR promoter (Pcov). DNase footprint analyses showed that phosphorylation of CovR enhanced and extended the protected regions. The proposed CovR consensus binding sequence (ATTARA) was present at least once in each protected region. The effect of replacing the two thymine residues in the consensus binding sequence with guanine residues was evaluated both in vitro and in vivo. Most, but not all consensus site mutations reduced binding of CovR in vitro. Using a transcriptional reporter introduced in single copy into the GAS chromosome, we found that each mutation completely or partially relieved CovR-mediated repression. This suggests that CovR regulation of Pcov is direct. Further support for this conclusion comes from use of an in vitro GAS transcription system. In this system, purified CovR was sufficient to mediate repression of Pcov and this repression was enhanced when CovR was phosphorylated.
10

COMPLEX PATTERN FORMATION AND INTERSPECIES COMMUNICATION AMONG NATURALLY OCCURING SOIL ISOLATES. Shira Omer+, Rina Avigad+, Leticia Marquez-Mangana*, Jimena Davila*, and David L. Gutnick+
+Department of Molecular Microbiology and Biotechnology, Tel-Aviv University, Tel-Aviv, 69978, Israel; * Department of Biology, San Francisco State University, San Francisco, 94132, CA. Paenibacillus dendritiformis forms complex and highly organized patterns during colonial development on hard agar surfaces. Physiological and biochemical characterization of mutants impaired in pattern formation coupled with time-lapse video cinematography and a generic modeling approach indicated that patterning is a cooperative process involving the collective movement of cells in response to both environmental (chemotaxis towards a food source) and cell-generated signals. Pattern formation is preceded by the formation of an organized “mother colony” . Cells accumulate at the periphery of the colony and subsequently emerge in multicellular branches encapsulated by a polysaccharide. We have isolated a number of patterning- defective mutants from a culture of parental cells grown through 1,000 generations on rich liquid media. Patterning in a “crippled” class of mutants was restored in the presence of a small soluble protein (MW 12,000) found in the broth of the wild-type cells. This protein (patterning factor –PF) was not produced by the mutant. Activity was also restored by any of 7 known proteases and reconstitution of patterning was inhibited by protease inhibitors. PF did not exhibit any protease activity in-vitro. A second patterning-defective mutant produces the active protein, but does not form patterns, perhaps owing to a defect in a putative receptor. Interestingly, the Bacillus subtilis strain 168 also appeared to produce a small amount of the PF, although the B. subtilis cells did not form patterns. These results suggested the possibility that PF and other extracellular factors associated with patterning might exhibit an extended host range. It was thus of interest to examine the possible diversity of pattern-forming microorganisms in soil. A series of 30 aerobic spore formers were isolated from four different soil samples. Analysis of these strains by 16S RNA sequencing revealed that the strains were closely related to known natural isolates of B. subtilis. No member of the Paenibacilli was isolated. Surprisingly, 50% of the natural isolates exhibited distinctive tip-splitting patterns. Moreover, 50% of the strains exhibited significant extracellular PF activity when tested with the crippled mutant. Several species of Paenibacilli also produced extracellular PF, while others, including those strains which produce the vortex pattern, were inactive. The results are in keeping with recent reports suggesting that “undomesticated” B. subtilis isolates may produce activities, such as swarming which are normally not associated with the classical laboratory strain. The results also suggest that pattern formation during colonial development may be more widespread than originally anticipated.
11

GLIDING MOTILITY OF MYXOCOCCUS XANTHUS REQUIRES A COILED-COIL
PROTEIN AND A SMALL GTPASE
Ruifeng Yang, Sarah Bartle, Rebecca Otto, and Patricia Hartzell
Department of Microbiology, Molecular Biology, and Biochemistry, University of Idaho,
Moscow, ID 83844-3052
Wild-type M. xanthus cells use two genetically independent motility systems - adventurous (A) and social (S) –to gliding over surfaces. Although the systems appear to be used simultaneously, little is known about how the two systems are coordinated. MglA, a small GTPase in M. xanthus has features that suggest it may be the coordinator. To test this hypothesis, protein partners of MglA were sought. One such partner, AglZ, was identified from a yeast two-hybrid assay in which MglA was used as bait. Disruption or deletion of aglZ abolished movement of isolated cells, showing that aglZ is part of the Adventurous gliding system. The aglZ gene encodes a 153kDa protein that has a N-terminal receiver domain characteristic of two-component response regulator proteins and a C-terminal coiled-coil similar to myosin. Like myosin, the coiled-coil domain of AglZ formed regular striated-lattice structures when expressed in E. coli and purified protein formed filament structures in vitro. Immunofluorescence suggests that AglZ forms a filament-like structure in vivo.
12

CHEMOTAXIS AND BIOREMEDIATION DURING REMEDIATION BY ANAEROMYXOBACTER DEHALOGENANS Sara Henry, Frank Löffler, and John Kirby
Georgia Institute of Technology Atlanta
Anaeromyxobacter dehalogenans was first isolated from various soil and sediment samples including stream and pond sediments, rainforest soil, compost, and flooded rice fields (Sanford et al., 2002; Treude et al., 2003). 16S rRNA sequencing identified this organism as a member of the myxobacteria. A. dehalogenans strain 2CP-C, isolated from Cameroon rainforest soil, is capable of both reductive dechlorination (chlororespiration) and growth coupled to dissimilatory metal reduction (e.g., Fe[III], uranium [IV]). It is the first member of the myxobacteria family found to thrive under anoxic conditions and degrade priority pollutants including chlorophenols and toxic metals.
Contaminant distribution in the natural groundwater environment is often heterogeneous due to variable solubility into the aqueous phase and sorption to the solid phase. Although the relevance of chemotactic behavior toward contaminants has been recognized, relatively little research has been done to explore chemotaxis in the context of bioremediation. We therefore wished to explore A. dehalogenans as a model organism for the relationship between chemotactic behavior, pilus-based motility, and bioremediation. Colonies of A. dehalogenans were observed to produce flares on solid R2A media characteristic of those observed for social motility by Myxococcus xanthus. Further analysis indicated that the A. dehalogenans genome likely contained homologs to both chemoreceptors and pilA genes. Recent acquisition of the genome sequence indicates that A. dehalogenans possesses seven Che clusters as well as genes for flagellar machinery and type IV pilus-based motility. Together, the data lead to the hypothesis that A. dehalogenans utilizes Type IV pilus-mediated motility and chemotaxis to terminal electron acceptors to expedite bioremediation of contaminated sites.
13

PARALLEL TWO-COMPONENT SIGNAL TRANSDUCTION SYSTEMS REGULATE TIMING OF DEVELOPMENT IN MYXOCOCCUS XANTHUS. Penelope I. Higgs*, Kyungyun Cho#, and David R. Zusman* *Dept. Molecular and Cell Biology, University of California at Berkeley, Berkeley, CA 94720-3204 #Department of Life Science, Hoseo University, Asan, Chungnam 336-795, Republic of Korea
Myxococcus xanthus is a social bacterium that undergoes a complex life cycle. Under starvation conditions, the bacteria initiate a developmental program in which groups of approximately 100,000 cells migrate into mounds (fruiting bodies) within which cells differentiate into environmentally-resistant myxospores. Previously, we demonstrated that deletion of espA, which encodes a sensor histidine kinase, results in a phenotype of accelerated development and smaller, more numerous, fruiting bodies relative to the wild-type strain. In our model, we envisioned that EspA functions to stay developmental progression until an unidentified signal is sensed. In a genetic screen designed to identify a cognate response regulator for EspA, we instead identified three distinct genetic loci encoding additional histidine kinases- the mutation of which also yielded an espA-like phenotype. Interestingly, one of these loci encoded two pairs of unusual two-component signal transduction (TCS) genes transcribed together in the red (regulation of early development) operon. While the developmental phenotype of the red TCS mutants was like that of espA mutants, the phenotype of red espA double mutants was additive- they developed extremely early. Together, these results suggest that the developmental pathway in M. xanthus is subject to control by parallel sets of TCS proteins that influence the timing of progression through the developmental pathway. We are currently using genetic and biochemical techniques to characterize each TCS system and to determine where it acts within the developmental pathway.
14

CYTOPLASMIC ROLE OF FliK IN THE HOOK LENGTH CONTROL Hirano, T. 1, Shibata, S. 1, Ohnishi, K. 2, Tani, T. 3, & Aizawa, S.-I. 1 1. CREST, Japan Science & Technology Agency (JST), 1064-18 Takahori, Hirata, Takanezawa, Shioya-gun, Tochigi 329-1206, Japan 2. Institute of Genetics, Kochi University, 200 Monobe, Nangoku 783-8502, Japan 3. Department of Molecular Physiology, The Tokyo Metropolitan Institute of Medical Science, Honkomagome 3-18-22, Bunkyo-ku, Tokyo 113-8613, Japan
Flagellar hook has a well-defined length of 55 nm in Salmonella. There are two key
players in the hook-length control: FliK, an export substrate of flagellar export system, and FlhB,
one of the inner membrane components of flagellar export system. Mutations in either fliK or
flhB genes give rise to abnormally elongated hooks, so-called polyhooks. Models that FliK
plays as a molecular ruler have been proposed. On the other hand, through extensive survey of
hook length control mutants, we found shorter hooks in switch (fliG, fliM or fliN.) mutants. We
have proposed that the flagellar hook length was determined by the amounts of the hook protein
measured at the flagellar base. However, the role of FliK has remained ambiguous in this event.
In this study we show that FliK can control the hook length from inside the cell, denying its
possibility as a physical ruler. FliK fused with fluorescent protein at the N terminus
complemented fliK mutations but was not secreted into the medium. Furthermore, fluorescence
microscopy showed that the fusion proteins homogeneously distribute in the cytoplasm. We will
discuss the cytoplasmic roles of FliK.
15

TARGET TRIGGERED ACTIVATION OF THE MotAB PROTON CHANNEL OF THE E. coli FLAGELLAR MOTOR Edan R. Hosking, Trey LaQuey, Catina Vogler, and Michael D. Manson Department of Biology, Texas A&M University, College Station, TX 77843
The energy for flagellar rotation in most bacteria is provided by proton flow through the transmembrane channel formed by the MotA and MotB proteins. MotB in E. coli consists of 308 residues and comprises a short N-terminal cytoplasmic extension, one transmembrane helix, and a large periplasmic domain. The latter domain contains a sequence motif, extending from residues 210 to 229, that is found in many proteins that bind peptidoglycan. Up to eight functional Mot-protein complexes surround the MS-ring, and it has long been postulated that the MotB component of these complexes anchors them to the cell wall so that MotB can act as stator components of the motor. Mutational analysis of MotB by introduction of UAG nonsense codons in place of select codons throughout the length of the gene demonstrated that all residues beyond position 267 can be removed without seriously impairing motility. MotAB can be overexpressed without the presence of a growth defect. A C-terminal fusion of the first 60 amino acids of MotB to TetA has been shown to cause a major growth defect, but cells containing fusions to longer framents of MotB do not show this defect. It has been proposed that there exists a “lid” that blocks proton flow through the MotAB proton channel in the absence of flagellar motors, but that once contact with motors is made, the “lid” is removed and allows protons to flow through the channels. For this to occur, there also needs to be a “trigger” within the motor that interacts with a “motor sensing” domain within the channel to allow for the removal of the “lid”. We have found that C-terminal PhoA fusions to fragments of MotB, namely fusions after residues 50 and 60 of MotB, cause such a large leak of protons that cell growth stops within one hour of induction of the fusion proteins. C-terminal fusions of PhoA after residues 65 and 70 of MotB do not cause as large a growth defect with the fusion after residue 70 showing the smallest defect. These defects are observed whether strains express motors or not. However, expression of a full-length MotB-PhoA fusion or a fusion to the first 190 residues of MotB, do not show a defect in the presence or absence of motors. Deletion analysis has also shown the same types of growth defects seen above. Deletion of residues 51-70, 51-80, 51-90, and 61-70 of MotB show the same major growth defect as the PhoA fusions at residues 50 and 60 of MotB, but a deletion of residues 71-90 does not show a growth defect and deletion of amino acids 51-60 shows a moderate growth defect. These defects are also observed in the presence or absence of flagellar motors. All growth defects can be fixed by a D32N mutation in MotB. Based on growth curves and the difference seen in the curves, we believe that the “trigger” within the motor is FliG and that the “motor sensing” domain is MotA, specifically two charged residues in MotA (R90 and E98). A previously characterized double mutant, MotAR90E/E98RB is non-motile, but has motility restored when a reversal of charged residues in FliG (D289/R281) are combined with the MotA double mutant. MotAR90E/E98RB has a growth curve identical to cells expressing the MotABD32N mutation or vector alone and cells expressing pmotAB grow slightly slower than those above when expressed in cells containing motors. However, all four growth curves are the same in a strain lacking flagellar motors. These results are analyzed here in relation to our model of the activation of the MotAB proton channel.
16

σ54 ENHANCER BINDING PROTEINS AND MYXOCOCCUS XANTHUS FRUITING BODY DEVELOPMENT. Jimmy S. Jakobsen,1,* Lars Jelsbak,1 Lotte Jelsbak,1 Roy D. Welch,2 Craig Cummings,3 Barry Goldman,4 Elizabeth Stark,4 Steve Slater,4 and Dale Kaiser1
Departments of Biochemistry and Developmental Biology, Stanford University,1 and Relman Laboratory, Stanford University School of Medicine, Department of Microbiology and Immunology, and VA Palo Alto Health Care System,3 Stanford, California 94305; Department of Biology, Syracuse University, Syracuse, New York;2 and Monsanto Company, St. Louis, Missouri 631674
* Present address: Max Planck Institute for Terrestrial Microbiology, D-35043 Marburg, Germany
Myxococcus xanthus is a soil dwelling Gram-negative bacterium that upon starvation
initiates a complex developmental program that results in spore containing fruiting bodies. This program depends on a series of intercellular signaling events that lead to changes in expression of specific genes.
Several σ54-promoters have been identified as crucial for the developmental program, therefore we searched the M. xanthus genome for sequence homologs of σ54-dependent enhancer binding proteins (EBPs). A DNA microarray was constructed from the genome that includes those homologs and 318 other M. xanthus genes. To screen the developmental program with this array, an RNA extract from growing cells was compared with one from developing cells at 12 h. Previous reporter studies had shown that M. xanthus had initiated development and begun to express many developmentally regulated genes by 12 h. The comparison revealed substantial increases in the expression levels of 11 transcription factors that may respond to environmental stimuli. Six of the 53 EBP homologs were expressed at significantly higher levels at 12 h of development than during growth. Three were previously unknown genes, and they were inactivated to look for effects on fruiting body development. One knockout mutant produced fruiting bodies of abnormal shape that depended on the composition of the medium. M. xanthus has two polar motility systems, called A and S, that allows its gliding motility. To assess the importance of movement during fruiting body development, several components of both engines were included on the array. CglB, a part of the A motility system, increased expression sevenfold during development. S-motility is produced by retraction of the polar type IV pili projecting from the leading end of the cell. Fibrils consisting of polysaccharide and protein provide distal anchor sites for pili when they retract and are among the S-motility genes. Four S-motility genes increased expression after 12 h of development: difB, difC, pilP, and pilR. These results confirm that motility plays an essential role in M. xanthus fruiting body development. We also confirm that the genes encoding the intercellular signaling molecules necessary for the developmental program increase expression during starvation. Specifically, we show that reporter genes or structural genes for four out of the five known signaling pathways increase gene expression during the developmental program.
This study confirmed a number of earlier observations, which show the validity of microarray experiments for this kind of study, and identified new regulators, which might lead to a more detailed understanding of the regulatory network.
17

STRUCTURE/FUNCTION ANALYSIS OF THE DIGUANYLATE CYCLASE RESPONSE REGULATOR PleD Sören Abel, Carmen Chan1, Beat Christen, Matthias Christen, Ralf Paul, Alexandra Schauerte, Tilman Schirmer1, Urs Jenal Division of Molecular Microbiology, Biozentrum, University of Basel, Switzerland; 1Division of Structural Biology, Biozentrum
The response regulator PleD controls pole development and motility in Caulobacter crescentus. Upon activation by phosphorylation of the N-terminal receiver domain, PleD is specifically sequestered to the differentiating cell pole. Targeting of PleD to the pole is coupled to the activation of a C-terminal di-guanylate cyclase (DGC) domain, which catalyzes the synthesis of the secondary messenger c-di-GMP. It has been postulated that the dynamic localization of activated PleD to the cell pole provides a mechanism to temporally and spatially control the signaling output of PleD during development. We have recently solved the 3-dimensional structure of fulllength PleD in complex with c-di-GMP by X-ray crystallography. In the crystal, PleD forms a dimer with two tandem N-terminal CheY-like receiver domains mediating weak monomer-monomer interactions. The DGC fold is similar to the adenylate cyclase with one product molecule bound in the active site. In addition, two mutually intercalated c-di-GMP molecules are tightly bound at the interface between the second receiver and the DGC domain. I-site binding of the product explains the observed allosteric inhibition, which might represent an overriding regulatory principle for the production of c-di-GMP by PleD and other DGC proteins. Mutational analysis of pleD was performed to test some of the predictions derived from the PleD structure. Mutant proteins were assayed with respect to activity, product inhibition, cdi-GMP binding properties, and subcellular localization. The results will be discussed in the context of the function of PleD during development.
18

SENSING AND INTEGRATING MULTIPLE SIGNALS DURING EARLY MYXOCOCCUS MULTICELLULAR DEVELOPMENT Jose Rivera, Mehdi Esmaeiliyan, Elena M. Barbu and Heidi B. Kaplan* Department of Microbiology and Molecular Genetics University of Texas Medical School, Houston, Texas 77030
Myxococcus xanthus is a Gram-negative soil bacterium that undergoes multicellular development when starved for nutrients at high density. The expression of many early developmental genes, including the class represented by the 4445 gene, requires starvation and high cell density. Interestingly, the high cell density requirement for 4445 expression can be bypassed by the presence of 1 mM isoleucine, 1 mM threonine, Mg++ (25 – 100 mM), or the absence of the lipopolysaccharide (LPS) O antigen. The addition of the specific amino acids appears to exacerbate the (p)ppGpp-dependent starvation response through negative feedback inhibition of methionine biosynthesis. The addition of high Mg++ and the absence of LPS O antigen may generate an envelope stress response.
A genetic approach, focusing on the transcriptional regulators of 4445 developmental gene expression, was used to identify the elements that integrate the information concerning the cell's nutrient status and cell density. A parent strain containing a 4445-lacZ transcriptional fusion was mutagenized using the eukaryotic transposable mariner element Himar1. Four mutants that bypassed the starvation and high cell density requirement for 4445 expression were isolated. These mutants were distinguished as dark blue colonies on nutrient plates when overlaid with X-gal. The insertions mapped to two adjacent genes encoding a putative anti-sigma factor and a negative regulator, designated reaA and reaB, respectively. These genes are immediately downstream of a gene encoding a putative extracytoplasmic function (ECF) sigma factor, designated ecfA. The insertion mutants expressed the 4445 fusion at levels almost 100 fold higher (>20,000 β-galactosidase specific activity units) than wild type during growth and development. RT-PCR analysis revealed that ecfA, reaA and reaB form one transcriptional unit. In an ecfA in-frame null mutant, 4445-lacZ was expressed at basal levels, indicating that EcfA is a positive regulator of 4445 expression. In a reaA in-frame null mutant, 4445-lacZ was expressed at very high levels during growth and development confirming that both ReaA and ReaB are negative regulators of 4445 expression and suggesting that the ecf operon is autoregulated. Starving LPS O-antigen mutants expressed 4445 at low density in an ecfA-dependent manner. These data indicate that the ecfA operon encodes the elements of a new regulatory pathway that integrates and transduces starvation and cell density cues during early M. xanthus development and may also sense cell surface alterations.
19

SWRA GOVERNS SWARMING MOTILITY IN BACILLUS SUBTILIS Daniel B. Kearns and Richard Losick Department of Molecular and Cellular Biology Harvard University 16 Divinity Ave Cambridge, MA 02138
Swarming is a social form of motility in which cells assemble in multicellular clusters to
rapidly disperse a population of bacteria across a solid surface. Like swimming motility, swarming is powered by the rotation of flagella, but swarming has a number of distinguishing characteristics. Swarming motility requires the production of the surfactant, surfactin, to reduce surface tension, and swarming cells are hyper-flagellate. Furthermore, swarming requires a period of adjustment following surface contact as a lag period precedes the initiation of surface migration, the duration of which depends on the cell-density of the inoculum. Finally, while laboratory strains swim in liquid media, they are unable to swarm, suggesting that other differences between swimming and swarming remained to be identified.
By employing an unbiased genetic approach, unexpected genes necessary for swarming
were identified and included two genes of unknown function (swrA and swrB), a gene involved in resistance to surfactin (swrC), and a gene encoding a ubiquitous translation factor (efp) that, unlike all other organisms in which it has been studied, appears to be dispensable for growth in B. subtilis. Using this genetic information, two mutations were identified that rendered laboratory strains non-swarming: a mutation in sfp, required for surfactin biosynthesis, and a mutation in the newly identified swrA gene. By correcting both of these mutations simultaneously, laboratory strains were restored to wild type swarming motility.
Swarming motility seems to be regulated at the level of SwrA activity. Recently, it has
been found that overexpression of swrA is sufficient to bypass both the surface requirement and cell-density requirement for swarming. Cells in which swrA is overexpressed become hyperflagellate in liquid medium and swarm without lag when presented with a solid surface. Paradoxically, in wild type cells, swrA expression is the same whether cells were grown in liquid medium or harvested from a mature swarming population. How does overexpression of swrA have such a dramatic effect if high level swrA expression is not observed in wild type strains? SwrA is not similar to any protein in the database and has no predicted motifs to suggest a possible function. What is the mechanism of SwrA action in the cell?
20

IMPORTANCE OF THE LINKER BETWEEN THE CARBOXYL-TERMINAL PENTAPEPTIDE AND THE CONSERVED BODY OF A CHEMORECEPTOR Mingshan Li and Gerald L. Hazelbauer Department of Biochemistry; University of Missouri-Columbia; Columbia, MO 65211
In E. coli and Salmomella some chemoreceptors carry a carboxyl-terminal pentapeptide sequence that interacts with both the methyltransferase CheR and the methylesterase CheB, substantially enhancing receptor adaptational modification catalyzed by those enzymes. Without the pentapeptide, a receptor itself cannot mediate effective adaptational modification or chemotaxis. The pentapeptide occurs at the end of a sequence of approximately 30 residues that are not significantly conserved among the receptors and that contains several prolines. The sequence is thought to serve as a flexible, perhaps unstructured tether between the pentapeptide and the conserved body of the chemoreceptor. Is this linker and apparent tether actually important for adaptational modification and receptor function? If so, does it have the same importance for modifications catalyzed by both the CheR and CheB? To address these questions we created a family of tar genes coding for receptors missing from 5 to 40 residues, in units of five amino acids from the pentapeptide, of the sequence between the carboxyl-terminal pentapeptide and the conserved body of Tar. We tested these altered receptors for binding to CheR, effectiveness of adaptational modification in vitro and the ability to mediate chemotactic migration in a spatial gradient in vivo. By isothermal titration calorimetry, even a deletion of 40 residues, which completely eliminated the presumed flexible linker and as well as a short sequence of the conserved body of Tar, had only a modest effect on binding of CheR to the pentapeptide. In contrast, functional assays revealed increasing and substantial defects with shorter linkers.
Receptors that carry the pentapeptide sequence assist adaptational modification of receptors lacking that sequence, and do so for their nearest neighbors in a receptor cluster. We tested the effects of shortened tethers on this adaptational assistance and found patterns that resembled but were not identical to effects on modification of the receptor carrying the pentapeptide. We will discuss the significance of all these observations for functional interaction among homologous and heterologous chemoreceptors.
21

DIVERGENT REGULATORY PATHWAYS CONTROL A- AND S-MOTILITY IN MYXOCOCCUS XANTHUS THROUGH FrzE, A CheA-CheY FUSION PROTEIN Yinuo Li1, Víctor H. Bustamante2,3, Renate Lux4, David Zusman2 and Wenyuan Shi1,4
1Molecular Biology Institute and 4School of Dentistry, University of California, Los Angeles, CA 90095, USA. 2Department of Molecular and Cell Biology, University of California, Berkeley, CA 94720, USA. 3Departamento de Microbiología Molecular, Instituto de Biotecnología, Universidad Nacional Autónoma de México, Cuernavaca, Morelos 62251, México.
Myxococcus xanthus moves on solid surfaces by gliding motility using two different motility systems: A-motility for individual cell movement and S-motility for coordinated group movements. The frz genes encode chemotaxis homologues that control directed motility of both motility systems. These genes control cellular reversal frequency, which allows cells to bias their movements. One of the components of the core Frz signal transduction pathway, FrzE, is homologous to both CheA and CheY from the enteric bacteria and is therefore a novel CheA-CheY fusion protein. In this study, we investigated the role of this fusion protein and, in particular, the CheY response regulator domain. The CheY domain of FrzE, FrzECheY, retains all the highly conserved residues of the CheY super family of response regulators, including Asp709, analogous to Asp57 of Escherichia coli CheY, the phospho-accepting residue. In-frame deletion of the entire frzE gene caused both motility systems to show a hypo-reversal or non-reversing phenotype. In contrast, a mutant containing an in-frame deletion of the FrzECheY domain of frzE showed divergent phenotypes for the two motility systems: hypo-reversals of the S-motility system and hyper-reversals of the A-motility system. Surprisingly, these cells were able to aggregate and form ‘messy’ fruiting bodies. To further investigate the role of FrzECheY in controlling A- and S-motility, point mutations were constructed such that the putative phospho-accepting residue, Asp709, was changed from D to A, and therefore never subject to phosphorylation, or D to E, possibly mimicking constitutive phosphorylation. The D709A mutant showed hyper-reversals for both A and S-motility, while the D709E mutant showed hyper-reversals for A-motility and hypo-reversal for S-motility. These results show that the FrzECheY domain plays a critical signaling role in coordinating A- and S- motility. Based on the reversal phenotypes of the frzE mutants generated in this study, a model is proposed for the divergent signal transduction through FrzE in controlling and coordinating A- and S-motility in M. xanthus.
22

ORGANIZATION OF SUBUNITS IN THE BACTERIAL SWITCH COMPLEX Bryan J. Lowder, Perry Brown, Mark Duyvesteyn, Moises Terrazas, David Blair Biology Department, University of Utah, 254 South 1400 East, Salt Lake City, Utah 84112-0842
Torque generation and switching in the flagellar rotor occur in a part of the rotor termed
the switch complex (SC), which is composed of multiple copies of each of the proteins FliG, FliM, and FliN. The shape of the SC is known at low resolution (~20Å) from EM, and x-ray crystal structures have been solved for portions of FliG and FliN. The organization of the FliG, FliM, and FliN subunits within the complex is not known. We are attempting to fill this gap by biochemical approaches.
The crystal structure of the middle and C-terminal domains of FliG (FliG-MC) shows two globular domains connected by a helix, and two well-conserved surface patches that were suggested to bind to FliM. To probe the configuration of FliG subunits within the flagellum, we have introduced cysteines in many positions on the surface of FliG-MC, and studied patterns of crosslinking by either bismaleimidohexane or oxidants that induce disulfide formation. Crosslinking was monitored on anti-FliG immunoblots. Certain pairs of cys residues in the middle domain allow crosslinking of FliG into dimers, trimers, and larger multimers, indicating that FliG subunits are adjacent to each other in the flagellum with their middle domains in close contact. Results for the C-terminal domains imply that it is more dynamic and less closely apposed than the middle domains.
To probe interactions with FliM, we made mutations sampling the surface of FliG, including the putative FliM-interaction sites, and measured the effects on function by swarming and swimming assays, flagellar staining, and cellular localization tests. Mutations in the hypothetical interaction sites had the largest effect, consistent with the model.
A recently solved crystal structure for FliN forms the basis of a model for FliN organization in the SC. A key feature of the model is the organization of FliN subunits into tetramers. We are testing this model using the crosslinking approach just described. Results of these studies will be presented.
23

FrzS, A PROTEIN ESSENTIAL FOR SOCIAL MOTILITY OF MYXOCCOCUS XANTHUS IS TARGETED TO THE CELL POLES Tâm Mignot, John Merlie and David Zusman 16, Barker Hall. Department of Molecular and Cell Biology. UC Berkeley. Berkeley CA94720-3204. USA.
In Myxoccocus xanthus, social motility (S-motility), like twitching motility, is due to the retractile activity of type IV pili. S-motility is achieved by extension of polarly localized type IV pili, attachment of the pili to an external substrate, followed by pilus retraction which pulls the cells forward in the direction of the attachment site. FrzS is an essential component of the S-motility system but how it functions is unknown.
Immunofluorescence microscopy showed that FrzS is localized at both cell poles. Deconvolution of the images indicated that FrzS could be part of large hollow tubular structures oriented perpendicular to the long axis of the cell. Electron microscopy confirmed that FrzS is localized in large polar clusters that can span the wide axis of the cell. A genetic screen to isolate loss of function mutations in FrzS led to the identification of stable mutants that are improperly localized in the cell. The structure of FrzS is such that a response regulator receiver-like domain is fused to an extended coiled-coil dimerization domain. The C-terminus of the protein contains 30 amino acids that are unlikely to be part of the coiled coil and thus define a C-terminal tail. We found that a region of 16 residues within the C-terminal tail is essential for proper localization. Interestingly, mutants missing this region were localized at only one pole of the cell, suggesting that bi-polar localization of the protein is essential for function. The C-terminal tail of FrzS may be required for shuttling between poles. Alternatively, the C-terminal tail may be a docking site with a target of FrzS that is only present at one pole. These possibilities and the perspectives opened by these results will be discussed.
24

DYNAMICS OF THE HISTIDINE KINASE, CHEA
Aaron S. Miller, Susy C. Kohout, Kaitlyn A. Gilman and Joseph J. Falke
Department of Chemistry and Biochemistry, University of Colorado, Boulder, Campus Box 215, Boulder, CO 80309 The goal of this work is to investigate the protein-protein interaction surfaces of the
bacterial histidine kinase, CheA. During E. coli and S. typhimurium chemotaxis,
chemoattractant binds to a cell surface receptor, inhibiting the activity of CheA. The sensory-
kinase complex consists of the receptor, CheA, the coupling protein CheW, and the response
regulator CheY. Although much structural information is known about the individual
components of this complex, little is known about the complex architecture, geometry, or
stoichiometry. A library of 71 surface cysteine mutations spread over all CheA domains has
been created for use in chemical and biochemical assays to elucidate which surfaces are
important for complex formation and function.
In the first surface-probing assay, the Protein-Interactions-by-Cysteine-Modification
(PICM) method is being used to map out regions on CheA essential for protein-protein
interactions required for a properly functioning kinase complex. PICM involves coupling of a
large bulky probe to engineered cysteine positions followed by activity assay of labeled and
unlabeled cysteine mutants. Surfaces that show perturbation of function upon modification by
the probe imply an important protein-protein interaction at that site.
A new surface-probing assay termed Protein-Accessibility-by-Cysteine-Modification
(PACM) is being used to determine which surfaces of CheA are accessible to collisions with
other proteins. In this assay, the cysteine library is probed with a single cysteine containing
protein, the C2 domain of PKCα. Under mild oxidizing conditions, the CheA-C2 domain
disulfide formation rate is quantified and used as a measure of accessibility to collisions with the
C2 domain. Surfaces which are inaccessible are generally protected by proximity to another
domain or subunit. Furthermore, changes in protein accessibility upon addition of other complex
components should yield insights into the structure of the receptor-kinase complex, especially
the locations of docking surfaces.
The results of these studies will be reported and are expected to expand the molecular
understanding of protein-protein interactions in the receptor-kinase complex as well as the
mechanism of receptor-mediated kinase regulation.
25

POST-TRANSLATIONAL CONTROL OF MOTILITY AND CHEMOTAXIS BY THE CLP ATP-DEPENDENT PROTEASE IN BACILLUS SUBTILIS Hiroki Yamamoto and Tarek Msadek Unité de Biologie des Bactéries Pathogènes à Gram Positif, Institut Pasteur, 25, rue du Dr. Roux, Paris 75015, France.
The B. subtilis Clp ATP-dependent protease plays a central role in the signal transduction
network controlling stationary phase responses such as competence, degradative enzyme
synthesis, stress response and sporulation (1). We previously reported that the B. subtilis clpP
mutant is non-motile as judged by swarm plate assays and displays a highly filamentous
morphology, growing as long chains of elongated cells during the exponential growth phase (2).
This suggested that expression of chemotaxis, motility and/or autolysin genes, most of which
require the sigma D alternative sigma factor for their expression, might be affected in the clpP
mutant. Fluorescence microscopy revealed very similar phenotypes for the clpP and sigD
mutants of B. subtilis: chromosome segregation and septation occur normally, but the cells fail to
separate, suggesting that expression of a specific sigmaD-dependent autolysin may be affected in
the clpP mutant. Among these, the major autolysin LytF plays an important role in cell
separation. We showed that lytF is not expressed in the clpP mutant. By placing the lytF gene
under the control of a sigmaD-independent inducible promoter, we were able to show that
increasing concentrations of LytF within the cell progressively reversed the filamentous
phenotype of the clpP mutant. SigmaD activity, and thus lytF expression, is known to be
controlled by the FlgM anti-sigma factor in B. subtilis. Accordingly, a flgM mutation completely
reverses the clpP filamentous phenotype, suggesting that the FlgM anti-sigma factor may be
specifically degraded by the Clp ATP-dependent protease, and that its accumulation in the clpP
mutant leads to inhibition of sigma D activity, and thus a loss of motility, chemotaxis and cell
separation.
References:
1. Msadek, T. (1999) Trends Microbiol. 7, 201-207. 2. Msadek, T., Dartois, V., Kunst, F., Herbaud, M.-L., Denizot, F. & Rapoport, G. (1998)
Mol. Microbiol. 27, 899-914.
26

PUNCHING HOLES IN E. COLI Simon Rainville, Aravinthan Samuel, Howard C. Berg
Department of Molecular and Cellular Biology and Physics, Harvard University, 16 Divinity Avenue, Cambridge, MA 02138
We are developing an in vitro system to study the bacterial flagellar motor. Tightly-focused
femtosecond laser pulses are used to vaporize a submicrometer-sized hole in the wall of
filamentous Escherichia coli. Since all three layers of the wall are damaged, the hole should be
stable and not reseal. If we then placed a punctured cell across a membrane dividing two
volumes, one side will correspond to the exterior of the cell and the other side (where the
bacterium is pierced) will correspond to the interior. Having a working flagellar motor whose
rotation speed can be monitored on the exterior side will then provide an ideal in vitro assay to
study the motor’s physical and chemical characteristics. Encouraging preliminary results will be
presented.
27

INSERTIONAL INACTIVATION OF THE SIX FLAGELLIN GENES OF BDELLOVIBRIO BACTERIOVORUS SHOWS THAT FLAGELLAR MOTILITY IS NOT ESSENTIAL FOR PREY ENTRY Liz Sockett1 Carey Lambert1, Katy J Evans1, Rob Till1, Laura Hobley1, Snjezana Rendulic2, Stephan C. Schuster2, Shin-Ichi Aizawa3 1Institute of Genetics, School of Biology, University of Nottingham, Queens Medical Centre, Nottingham NG7 2UH, UK. 2Max-Planck-Institut fur Entwicklungsbiologie, Spemannstrasse 37-39, D-72076 Tubingen Federal Republic of Germany 3CREST “Soft Nano-Machine Project”, c/o Kochi Sangyo, 1064-18 Takahori, Hirata, Takanezawa, Shioya-gun, Tochigi 329-1206, Japan
Bdellovibrio bacteriovorus is a Gram-negative motile bacterium that preys upon other
Gram-negative bacteria including pathogens, such as Salmonella, Serratia and Proteus. B.
bacteriovorus enters the periplasm of the host cell, where it grows, replicates and then lyses the
host cell to release the progeny. Bdellovibrio is highly motile with a single, polar, sheathed
flagellum and the high speeds of motility recorded suggest that motility may be important to its
predacious lifestyle. Individual gene deletions of the six flagellin genes found in the Bdellovibrio
HD100 genome show that while four of the six deletions have no discernable phenotype in both
the HD100 genome-sequenced strain and strain 109J; two fliC inactivations produced significant
effects. One mutant had a short flagellar filament and showed reduced predation efficiency
compared to wild type. The second mutant was non-flagellate and could only be efficiently
grown in the prey-free, Host-Independent (HI) state, and was weakly predatory only upon
immobilised hosts. Our finding that surface-applied, non-flagellate Bdellovibrio could enter prey
adds credence to a role for pilus-mediated prey entry by Bdellovibrio. Pili have been observed
using electron microscopy in predators both freshly incubated in the presence of prey and those
that have been incubated in prey absence, indicating that expression of pili may be constitutive.
The role of pili in predation is being tested experimentally.
28

STEPS IN SLOW FLAGELLAR ROTATION Yoshiyuki Sowa1, Alexander D. Rowe2, Toshiharu Yakushi3, Michio Homma3, Akihiko Ishijima1 and Richard M. Berry2
1Department of Applied Physics, Graduate School of Engineering, Nagoya University, Furocho, Chikusa-Ku, Nagoya, Aichi 464-8603, Japan 2Department of Physics, University of Oxford, Clarendon Laboratory, Oxford, OX1 3PU, UK 3Division of Biological Science, Graduate School of Science, Nagoya University, Furocho, Chikusa-Ku, Nagoya, Aichi 464-8602, Japan
The bacterial flagellar motor is a reversible rotary molecular machine that converts ion flux across the cytoplasmic membrane into torque. The motor consists of the rotor, which is a series of filament, hook and switch complex, and ~10 stators containing specific ion channels for H+ or Na+. Apparent flagellar rotation is very smooth, and processive steps in rotation have not been detected.
The proteins PomA and PotB have previously been reported to work in Escherichia coli as the stator unit of a Na+-driven chimeric motor1. Studying PomAPotB in E. coli has the advantages that we can regulate rotation rate with external Na+ concentration, or with specific inhibitors whilst investigating the rotational mechanism. We constructed strain YS34 that has neither flagella nor MotAMotB, and expressed PomAPotB and sticky-filaments in it. A 0.5 µm polystyrene bead was attached to a flagellar stub as a marker and its position was followed by optical trap nanometry2. By disrupting the sodium-motive force we found that the flagellar motor makes discrete steps when few stator units interact with the rotor during slow flagellar rotation. 1. Asai Y, Yakushi T, Kawagishi I and Homma M. (2003) J Mol. Biol. Ryu WS, Berry RM and Berg HC. (2000) Nature
29

RESPIRATION ALONG THE REDOX GRADIENT: HIERARCHICAL REGULATION IN PROTEOBACTERIA
Valley Stewart Section of Microbiology, University of California, Davis The facultative aerobe Escherichia coli uses a variety of electron acceptors for anaerobic respiration. Hierarchical regulation ensures that the acceptor with the highest standard redox potential is used preferentially. In the absence of oxygen, the Fnr protein activates transcription of operons encoding anaerobic respiratory enzymes. Transcriptional control in response to anaerobic acceptors is mediated by the orthologous NarX-NarL and NarQ-NarP two-component regulatory systems. Both systems respond equivalently to nitrate, the preferred anaerobic acceptor. Experiments with null mutants indicate that the two systems exhibit cross-regulation: during growth with nitrate, the NarX and NarQ sensors can both serve to phosphorylate the NarL and NarP response regulators. Why does E. coli use dual interacting systems to control response to nitrate? I will describe a variety of recent experimental observations that bear on this question. Experiments with E. coli narXL or narQ narP null mutants reveal distinct properties of the two systems. The NarX sensor is specialized, as transmitter autokinase activity is stimulated efficiently only by nitrate. By contrast, the NarQ sensor is generalized, responding to several signals including nitrate, nitrite and culture aeration. Likewise, the NarL and NarP response regulators exhibit discrete transcription activation functions at different promoters. Thus, the dissimilar properties of these paralogous regulatory systems represent independent determinants of anaerobic respiration physiology. Examination of complete genome sequences reveals that E. coli (and related enterobacteria) are unique in harboring both Nar regulatory systems. By contrast, the NarX-NarL system only is present in some species such as Pseudomonas aeruginosa and Ralstonia solanacearum, whereas the NarQ-NarP system only is present in other species such as Vibrio cholerae and Haemophilus influenzae. It seems likely that E. coli contains both Nar systems as a consequence of lateral gene transfer. Therefore, I hypothesize that these two regulatory systems serve discrete roles in regulating anaerobic respiration in other species: The NarX-NarL system regulates nitrate respiration in nitrate-abundant environments where the formate-nitrate respiratory chain (FdnGHI-NarGHI enzymes) is a direct generator of protonmotive force. In E. coli at least, the NarX-NarL system effects further hierarchical regulation of anaerobic respiratory enzyme synthesis by repressing transcription of genes encoding other anaerobic respiratory enzymes. This is the nitrate respiration specialist phenotype. Conversely, the homologous NarQ-NarP system regulates nitrate respiration in nitrate-limiting environments where periplasmic nitrate reductase (NapABC enzyme) is a less-efficient generator of protonmotive force. In E. coli at least, the NarQ-NarP system has little effect on further hierarchical regulation of anaerobic respiratory enzyme synthesis. This is the nitrate respiration generalist phenotype.
30

DYNAMICS OF CHEMORECEPTOR TRIMERS OF DIMERS: IN VIVO CROSSLINKING STUDIES Claudia A. Studdert and John S. Parkinson Biology Department, University of Utah, Salt Lake City, Utah 84112, USA
Transmembrane chemoreceptors of the methyl-accepting chemotaxis protein (MCP) family form signaling complexes through binding interactions with the cytoplasmic CheA and CheW proteins. In E. coli the receptor signaling complexes typically reside in tight clusters at the poles of the cell. In a previous study of cysteine-directed receptor crosslinking in vivo, we suggested that trimers of receptor dimers are an important building block of receptor clusters. We found that receptors of different detection specificities could join the same trimer, with probabilities proportional to their relative abundance in the receptor population. Moreover, trimer formation did not depend on the presence of CheA, CheW, or any other soluble component of the chemotaxis signaling pathway.
In the present work we extended our in vivo crosslinking methods to assess the contributions of CheA and CheW to the stability of receptor trimers of dimers. We used two different receptors (Tar and Tsr) with suitable cysteine reporters and a trifunctional maleimide reagent (TMEA) to capture two or three receptor subunits, ostensibly from the same trimer of dimers. The cells expressed the chromosomally-expressed Tar reporter constitutively, whereas expression of the the plasmid-borne Tsr reporter was induced at the start of the experiment. At various times thereafter, the cells were treated with TMEA to follow the kinetics of appearance of mixed dimer- and trimer-sized receptor crosslinking products.
We found that in the presence of both CheA and CheW, pre-formed trimers were very stable, as evidenced by slow incorporation of newly-synthesized receptors into mixed crosslinking products over the course of several cell generations. In the absence of ongoing protein synthesis, the proportion of mixed crosslinking products did not increase, even after long incubation times. In contrast, cells lacking CheA and/or CheW showed rapid appearance of mixed crosslinking products. Evidently, under these conditions, newly-synthesized receptors can freely mix with old ones to generate a population of trimers of dimers whose compositions reflect the overall proportions of the two receptor types. These findings suggest that receptors within polar clusters, which have formed ternary signaling complexes through CheA/CheW binding interactions, are somehow prevented from exchanging members with newly-made receptor molecules. Conceivably, in forming signaling teams, CheA/CheW bind to and stabilize receptor trimers of dimers, blocking further exchanges with the pool of receptor dimers. Alternatively, or in addition, the receptor cluster may physically shield many of its component signaling teams from encounters with newly-synthesized receptor molecules, which might only have access to receptors at the periphery of the cluster.
31

THREE-DIMENSIONAL ELECTRON MICROSCOPY AT MOLECUALR RESOLUTION: APPLICATIONS TO BACTERIAL CHEMOTAXIS
Sriram Subramaniam, Laboratory of Cell Biology, Center for Cancer Research, National Cancer Institute, NIH, Bethesda, MD 20892.
Emerging methods in cryo-electron microscopy allow determination of the three-dimensional architectures of objects ranging in size from small proteins to large eukaryotic cells, spanning a size range of more than 12 orders of magnitude. Advances in determining structures by “single particle” microscopy and by “electron tomography” provide exciting opportunities to describe the structures of subcellular assemblies that are either too large or too heterogeneous to be investigated by conventional crystallographic methods. Advances in electron crystallography allow determination of atomic resolution structures of membrane proteins while still embedded in a lipid bilayer and to analyze physiologically relevant conformational changes. We are very interested in applying these methods to investigate structural aspects of bacterial chemotaxis. A long-term goal of such studies is to arrive at plausible molecular models, at atomic resolution, of the various dynamic multi-protein complexes that collectively represent the bacterial chemotaxis machinery. Using high-resolution 3D electron microscopy, and advanced computational tools, we also hope to be able to obtain structural “snapshots” of chemotaxis-related multi-protein assemblies at various stages of the signaling process. I will present a review of the technological aspects of emerging tools in cryo-electron microscopy, and some recent applications to studying assemblies of the serine receptor Tsr. References: 1. Subramaniam, S. and Milne, J.L.S. (2004) Three-dimensional electron microscopy at molecular
resolution Ann. Rev. Biophys. Biomol. Struct. 33, 141-155.
2. Weis, R. M., Hirai, T., Chalah, A., Kessel, M., Peters, P. J. and Subramaniam, S. (2003) Electron microscopic analysis of membrane assemblies formed by the bacterial chemotaxis receptor Tsr J. Bact. 185, 3636-3643.
3. Lefman, J., Zhang, P., Hirai, T., Weis, R. M., Juliani, J. Bliss, D., Kessel, M., Bos, E., Peters, P. J. and Subramaniam, S. (2004) Three-dimensional imaging of chemotaxis receptor networks in a bacterial cell J. Bact. 186, 5052-5061.
4. Zhang, P., Bos, E., Heymann, J. A. W., Gnaegi, H., Kessel, M., Peters, P. J. and Subramaniam, S. (2004) Direct visualization of receptor arrays in frozen-hydrated sections and plunge-frozen specimens of E.coli engineered to overproduce the chemotaxis receptor Tsr J. Microsc. 216, 76-83.
32

THE FLAGELLAR REGULATORY PROTEIN, FLRC IS AN ENHANCER BINDING PROTEIN AND A POSSIBLE LINK BETWEEN MOTILITY AND VIRULENCE IN VIBRIO CHOLERAE. Nidia E. Correa, Khalid Ali Syed and Karl E. Klose
Dept. of Biology, University of Texas San Antonio, 6900 N. Loop 1604 W, San Antonio TX 78249
Vibrio cholerae is a highly motile organism by virtue of a polar flagellum, and motility has been inferred to be an important aspect of virulence for this human pathogen. We have previously demonstrated that the σ54-dependent activator FlrC is necessary for both flagellar synthesis and for enhanced intestinal colonization, suggesting that modulation of the transcriptional activity of FlrC is important for the colonization process. In order to identify the genes that are regulated by FlrC, we characterized the FlrC binding site. We analyzed two FlrC-dependent promoters, the highly transcribed flaA promoter and the weakly transcribed flgK promoter, utilizing transcriptional lacZ fusions, mobility shift assays and DNase I footprinting. Promoter fusion studies showed that the smallest fragment with wild type transcriptional activity for flaAp was from –54 to +137 with respect to the start site, and from –63 to +144 for flgKp. Gel mobility shift assays indicated that FlrC binds to a fragment containing the region +24 to +95 in the flaAp, and DNase I footprinting identified a protected region between +24 and +85. Mobility shift and Dnase I footprinting indicated weak binding of FlrC to a region downstream of the flgKp transcription start site. These results demonstrate a relatively novel �54-dependent promoter architecture, with the activator FlrC binding downstream of the σ54–dependent transcription start sites. When the FlrC binding site(s) in the flaA promoter was moved a large distance (285 bp) upstream of the transcription startsite of either flaAp or flgKp, high levels of FlrC-dependent transcription resulted, indicating that this binding region functions as an enhancer element. Our results suggest that the differences in FlrC binding to various flagellar promoters results in the differences in transcription levels that mirror the relative requirement for the flagellar components within the flagellum.
By using microarrays and comparing the transcriptome profile of the V.cholerae wild type and ∆flrC mutant we found that some known virulence genes (cholera toxin and toxin-corregulated pilus) and other putative virulence genes (hemolysin, tagD and pilO) were up-regulated in the absence of flrC. Thus, our results suggest that FlrC negatively regulates the expression of known and suspected virulence factors in V. cholerae.
33

YYCH IS AN ANTAGONIST TO THE ESSENTIAL YYCF/YYCG TWO-COMPONENT SYSTEM IN BACILLUS SUBTILIS Hendrik Szurmant, Kristin Nelson, and James A. Hoch Division of Cellular Biology, Department of Molecular and Experimental Medicine, The Scripps Research Institute, 10550 North Torrey Pines Road, La Jolla California
Of the numerous two-component regulatory systems among the bacteria and archaea only
very few have proven to be essential for cell viability. Among these is the YycF (response
regulator)-YycG (histidine kinase) system that is highly conserved in and specific to the low G +
C Gram-positive bacteria. Essentiality has been shown for Bacillus subtilis, Sthaphylococcus
aureus, Streptocoocus pneumoniae, Streptococcus pyogenes and Listeria monocytogenis. Given
the pathogenic nature of several members of this class of bacteria, the YycF-YycG system has
been suggested as a prime antimicrobial target. In an attempt to identify genes involved with this
two-component system a transposon mutagenesis study was designed to identify suppressors of a
temperature sensitive YycF mutant in B. subtilis. Suppressors could be identified and one prime
target was the yycH gene that is located adjacent to yycF and yycG within the same operon. A
lacZ reporter assay revealed that YycF regulated gene expression was elevated in a yycH strain
whereas deletion of any of the three downstream genes within the operon, yycI, yycJ, and yyxA,
showed no such effect. The yycH strain showed a characteristic cell wall defect consistent with
the previously suggested notion that the yycF-yycG system is involved in regulating cell wall
homeostasis. Copy numbers of both YycG and YycF -addressed immunologically- remained
unchanged between wild type and the yycH strain. YycH is predicted to be exported to the extra-
cellular space, which is currently investigated experimentally with a phoA-fusion approach.
These experiments strongly suggest that YycH has a YycG-antagonistic function.
34

PROTEOMIC ARRAYS OF A cheW MUTANT AND A cheW Che+ SUPPRESSOR REVEAL THE PRESENCE OF A NEW CHEMOTAXIS PROTEIN IN HELICOBACTER PYLORI Karianne Terry, Alvin Go, and Karen Ottemann University of California, Santa Cruz Depts. of Molecular, Cell, and Developmental Biology and Environmental Toxicology 1156 High Street, Santa Cruz, CA 95064
Helicobacter pylori is a Gram negative, spiral-shaped, Epsilon proteobacteria that uses motility and chemotaxis in the infection process. H. pylori infects approximately 50% of the world’s population and infection leads to diseases ranging from gastric and duodenal ulcers to gastric cancer. Genomic analysis of H. pylori predicted the presence of four chemoreceptors and the core chemotaxis proteins CheW, CheA, and CheY as well as three CheV orthologs. H. pylori is predicted to lack the adaptational proteins CheR, CheB, and CheZ. During characterization of ∆cheW::aphA3 H. pylori, which are nonchemotactic, a spontaneous, second-site suppressor arose that is able to form expanding colonies in soft agar that are indistinguishable from wild type. To locate this mutation, we catalogued all the proteins of the non-chemotactic mutant (cheW) with two-dimensional electrophoresis and compared this protein complement to that of the variant that had regained chemotaxis (cheW Che+). Using this approach, we found one protein that was abundant in the cheW mutant and absent in the cheW Che+ suppressor. This protein was identified by electrospray mass spectrometry as HP0170, a predicted protein with no known function. Sequencing verified that this ORF was mutated in several cheW Che+ suppressor isolates. Site-directed mutation of hp0170 in the cheW background recapitulated the Che+ suppressor phenotype. BLAST searches identified homologs of HP0170 in only the closely related species H. hepaticus, Wolinella succinogenes, and Campylobacter jejuni. SAM-T02, a program that predicts secondary structure and performs alignments based on these predictions, identified CheZ from E. coli as the closest match for HP0170, and PSI-BLAST also located this protein on the 4th iteration. As these programs imply an interaction between HP0170 and CheY, we analyzed a cheW Che+ cheY triple mutant and found that chemotaxis of the cheW Che+ suppressor requires CheY. These results suggest that HP0170 may either be a novel, CheY-interacting protein or a very remote homolog to CheZ. This work also shows that high-throughput analyses such as proteomics can be useful tools in identification of spontaneous mutations.
35

THE THREE-DIMENSIONAL STRUCTURES OF THE BACTERIAL FLAGELLAR MOTOR FROM SALMONELLA TYPHIMURIUM IN BOTH THE CW AND CCW STATE. Dennis R. Thomas1, Noreen R. Francis1, Xu Chen1, and David J. DeRosier1,2, 11 Rosenstiel Biomedical Sciences Research Center, Brandeis University, Waltham, MA 02454 2 Department of Biology, Brandeis University, Waltham, MA 02454
The bacterial flagellum used to swim by many motile bacteria, such as E. coli and S. typhimurium, is a remarkable nanomachine. It has a reversible rotary motor powered by the proton gradient across the cell’s plasma membrane. The motor is coupled to the flagellar filament, and transmits torque to the filament via the rod and the hook. More than 40 genes are known to be involved in motility in S. typhimurium and E. coli. There is evidence that as many as 24 proteins are components of the flagellum, including the export proteins FlhA, FlhB, FliO, FliP, FliQ and FliR. The motor is assembled from the inside out starting with the assembly of the MS ring (FliF). There is evidence that FlhA, FliP and FliR (export proteins) co-assemble with FliF forming the M ring. Next FliG assembles on the cytoplasmic face of the M ring followed by addition of the C ring (FliM and FliN). FliG, FliM, and FliN are called switch proteins since many mutations in fliG, fliM, and fliN lead to defects in control of direction of rotation but they are also required for torque generation and specific flagellar protein export. MotA and MotB are transmembrane proteins and form the proton channel. MotA and MotB are anchored to the peptidoglycan layer hence forming the stator of the motor and insert in the cytoplasmic membrane and assemble independently from the rest of the flagellum. It has been proposed that the motor domain of FliG resides in the C ring based on the crystal structure of the c terminal 2/3 of FliG. The motor domain (domain II) of FliG is connected to domain I by a long alpha helix with a flexible loop between the helix and domain II. Domain I is primarily required for assembly while domain II contains the sites of motor function and FliM. The C ring has been shown to vary in symmetry, but not subunit spacing with the numbers of subunits reported to range from 31-38. While much is known about the organization of the flagellar motor, detailed three-dimensional structures have not been available. There are a number of models for how the motor might work but what the models lack is a structure showing the spatial relationships between the key proteins. A number of assumptions are made about how the stator (Mot proteins) and rotor (FliG) interact. Most often it is viewed as a side to side interaction between the motor domain of FliG and the Mot proteins.
We present three dimensional reconstructions from electron micrographs of frozen hydrated bacterial flagellar motors of the C ring and M ring from both WT (counterclockwise default state) and a clockwise (CW) locked mutant of S. typhimurium (SJW2811). We have been able to sort edge on views C rings into symmetry classes and generate reconstructions from three of these symmetry classes (33-, 34- and 35-fold) that are nearly identical except for differing in the number of subunits and diameter. The same approach has also been applied to the M ring, and we find the M-ring symmetry also varies having 24-, 25- and 26-fold symmetry. We do not find that that the C ring symmetry correlates with the M ring symmetry. We have been able to sort images based on the combination of C ring and M ring symmetry and have reconstructed images of motors with specific symmetry combinations such as those having 34-fold C ring symmetry and 25-fold M ring symmetry. Several M ring/C ring symmetry combinations have been reconstructed. We have evidence that the inner domain of the C ring which lies nearest the M ring has the same symmetry as the M ring. This suggests the model of Brown et al., 2002 is correct and to confirm this we have used limited proteolysis to selectively cleave FliG which causes the C ring to dissociate leaving behind the M ring. These results suggest the motor domain of FliG is part of the C ring.
36

THE STUDY OF A COMPLEX CHEMOSONSORY SYSTEM REVEALS NEW INSIGHTS INTO BACTERIAL SUBCELLULAR ORGANISATION
Stephen R. Thompson, George H. Wadhams & Judith P Armitage
Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU, UK).
The purple, non-sulphur bacterium Rhodobacter sphaeroides contains multiple chemotaxis signalling proteins, some of which localise together to form a large discrete cluster within the cytoplasm. The use of chromosomal in-frame fluorescent protein fusions has enabled the study of these cytoplasmic protein clusters throughout the cell cycle. Together with expression analysis, our results reveal striking evidence for a system of regulating and positioning cluster formation in the cytoplasm. Our current understanding leads us to speculate that this system may be analogous to the type 1 DNA partitioning systems exhibited by a multitude of bacterial chromosomes and plasmids. Further study may reveal unified mechanisms for the partitioning of nucleic acids and proteins. The existence of this high degree of prokaryotic subcellular organisation has profound implications for the field of molecular microbiology.
37

CONTRIBUTIONS OF DOMAIN ORIENTATION AND INTER-DOMAIN INTERACTIONS TO THE MECHANISM OF REGULATION OF MEMBERS OF THE OMPR/PHOB SUB-FAMILY OF RESPONSE REGULATORS
Alejandro Toro1, 2, Priti Bachhawat2, 3, Timothy R. Mack2, 3, Victoria L. Robinson2, 4 and Ann M. Stock2, 3, 4
1Department of Chemistry and Chemical Biology, Rutgers University, 2Center for Advanced Biotechnology and Medicine, 3Department of Biochemistry, The University of Medicine and Dentistry of New Jersey - Robert Wood Johnson Medical School, and 4Howard Hughes Medical Institute, 679 Hoes Lane, Piscataway, New Jersey 08854, USA
Response regulators, together with histidine kinases, comprise two-component signal transduction systems in bacteria. Response regulators function as phosphorylation-dependent molecular switches, whose activities are modulated by those of the histidine kinases. In E. coli alone there are 34 response regulators, most of which are transcription factors composed of an N-terminal receiver domain and a C-terminal DNA-binding domain. These can be further divided into three sub-families based on their effector domain architecture. The largest of these is the OmpR/PhoB sub-family that has a C-terminal winged helix-turn-helix DNA-binding motif and whose most extensively characterized members are known to bind in tandem to DNA direct repeats. Phosphorylation triggers conformational changes in the receiver domain that ultimately results in regulation of transcription. Although the details of phosphoryl activation of the receiver domain are well understood, questions remain about how phosphorylation affects the DNA-binding domain, specifically with regard to domain orientations in dimers and oligomers, and the contributions of both intra- and inter-molecular interactions to transcriptional regulation. In order to address some of these questions we have used a combination of X-ray crystallography, NMR, analytical ultracentrifugation, mutational analysis and other biochemical techniques to probe domain orientations and interactions of several members of the OmpR/PhoB sub-family in their active and inactive states. Our results suggest a common mechanism in which the alpha4-beta5-alpha5 face of the receiver domain of these response regulators is used for dimerization upon activation. This interface has previously been shown to be involved in forming the interface between receiver and DNA-binding domains in the unphosphorylated state of one representative member of the OmpR/PhoB sub-family of response regulators.
38

UNDERSTANDING RECEPTOR INTERACTION AND SIGNAL AMPLIFICATION IN BACTERIAL CHEMOTAXIS: A QUATITATIVE MODELLING APPROACH Bernardo Mello and Yuhai Tu * Physical Sciences Department and Computational Biology Center, IBM T. J. Watson Research Center, Yorktown Heights, NY 10598
One of the most fascinating properties of E. Coli is its ability to amplify and detect minute change of environment over wide dynamic range. Motivated by the recent in vivo FRET measurements from Howard Berg’s lab, we propose a general framework for modeling bacterial chemotaxis signaling pathway, explicitly including the effects of covalent receptor modification and receptor interactions within the receptor cluster. We show that our model can quantitatively explain the in vivo measurements of the receptor sensitivity at different ligand concentrations for both the mutant and the wild type strains. Our model reveals the underlying mechanism for the high sensitivity of the E. Coli cell for over 4 orders of magnitude of dynamic range. Our study also show the existence of significant coupling between different types of chemoreceptor, in particular strong interactions between the aspartate (Tar) and serine (Tsr) receptors, which is crucial in explaining both the mutant and the wild type data. References:
• “Quantitative Modeling of Sensitivity in Bacterial Chemotaxis: The Role of Coupling Between Different Chemoreceptor Species”, B. Mello and Yuhai Tu, PNAS, 100(14), 8223-8228 (2003). • “Effects of receptor interaction in bacterial chemotaxis”, B. Mello, L. Shaw and Yuhai Tu, Biophysical Journal, 87(3), 1578-1595 (2004).
39

ESTABLISHMENT OF MYCOPLASMA MOBILE TRITON MODEL AND IDENTIFICATION OF ENERGY SOURCE FOR GLIDING MOTILITY
Atsuko Uenoyama1 and Makoto Miyata1, 2
Department of Biology, Graduate School of Science, Osaka City University, Sumiyoshi-ku Osaka 558-8585 JAPAN1 and PRESTO, JST2, JAPAN
Mycoplasmas are parasitic bacteria that lack a peptideglycan layer. A flask-shaped
species, M. mobile glides on glass in the direction of its tapered end at a rate of 3 to 7 times cell
length (2.0-4.5 micron) per second, with the maximum force of 27 pN. M. mobile has no
homologs of genes related to known bacterial motility or eukaryotic motor proteins, suggesting a
special motility mechanism. We previously identified three large gliding proteins, Gli349
(349kDa), Gli521 (521kDa) and Gli123 (123kDa), localizing at the head-like protrusion’s base,
“neck”. Gli349 and Gli521 are responsible for glass binding and force generation/transmission,
respectively. A simple model for gliding motility has been suggested, where spike-like structure
composed of Gli349 repeats the mechanical cycle, consisting of binding, force generation,
stroke, release, and displacement of Gli349 along glass, and then thrusts the cell. The energy is
likely to be supplied from ATP, because the decrease in ATP in a cell caused by arsenate
reduced the gliding speed.
To look inside of the mechanical cycle, in the present study, we constructed a triton-
permeabilized cell model. Treatment of gliding mycoplasmas by TritonX-100 stopped and
permeabilized the cells on glass. Addition of high concentration of ATP recovered the gliding
motility of the cell model at similar speeds with those of intact cells, while GTP and ATP-
gamma-S achieved slower speeds. The gliding speed depended on the ATP concentration, pH
and ionic strength, and was inhibited by ADP, AMP, AMP-PNP and ATP-gamma-S. These
observations demonstrated that the energy for gliding motility is provided from ATP hydrolysis
and have given a clue to elucidate this mysterious motility mechanism.
40

THE MIST DATABASE: AN INTEGRATIVE, GENOMIC RESOURCE ON MICROBIAL SIGNAL TRANSDUCTION Luke E. Ulrich and Igor B. Zhulin Center for Bioinformatics & Computational Biology, School of Biology, Georgia Institute of Technology, Atlanta, GA 30332-0230
We are developing the MIST database in order to provide a comprehensive genomic resource on microbial signal transduction to the scientific community. Currently, MIST contains data from more than 230 genomes of prokaryotes including over 60 draft genomes. In order to populate the database with data pertinent to signal transduction, each genome (along with relevant basic information) was downloaded from the National Center for Biotechnology Information and inserted into a local PostgreSQL (www.postgresql.org) relational database. Subsequently, each proteome (translated genome) was locally scanned against the Pfam (1) and SMART (2) domain libraries using the HMMER program to determine the domain architecture for every protein. Signal transduction proteins were then automatically extracted from each proteome based on the presence of a manually selected set of domains directly or indirectly implicated in signal transduction. MIST can be queried using the Standard Query Language (SQL) allowing precise and advanced searches, for example user-specified domain combinations and other filtering requests. The database also enables complex operations, such as analyzing genome neighborhood for any gene of interest.
Our preliminary analysis using MIST revealed approximately 25,000 signal transduction proteins in 230 genomes. There is a strong correlation between the number of signal transduction protein per genome and the genomic size, which in turn correlates with environmental complexity. Remarkably, the number of two-component regulatory systems found is nearly four times fewer than the number of single-component systems – signaling proteins which lack the signature histidine kinase or response regulator domains, but contain input and output domains implicated in signal transduction.
We are currently developing a biologist-friendly web interface to MIST. This will allow researchers to view and analyze the set of signal transduction proteins for a given organism(s). Users will be able to annotate any protein of interest and link it to appropriate literature. Common bioinformatic tools, such as BLAST and HMMER will also be integrated into the database. The MIST database will be an invaluable resource for genome-wide analysis of microbial signal transduction for the entire scientific community. 1. Bateman, A. et al. 2004. The Pfam protein families database. Nucleic Acids Res. 32:138-141. 2. Letunic, I. et al. 2004. SMART 4.0: towards genomic data integration. Nucleic Acids Res.
32:142-144.
41

REGULATION OF SALMONELLA TYPHIMURIUM SPI-2 GENE EXPRESSION Don Walthers1, Xiuhong Feng2 & Linda J. Kenney1
1Department of Microbiology & Immunology, University of Illinois at Chicago, Chicago, IL 2Department of Molecular Microbiology & Immunology, Oregon Health & Science University, Portland, OR
Salmonella typhimurium contains a pathogenicity island (SPI-2) required for systemic
infection in mice and survival within macrophages. SPI-2 genes encode components of a type-III secretion system, secreted effector proteins and a two-component regulatory system. SPI-2 genes are activated in the intracellular Salmonella-containing vacuole in response to signals such as low pH, osmolarity and possibly divalent cations. The SsrAB two-component system, which is comprised of the tripartite sensor kinase SsrA and the NarL/FixJ response regulator subfamily member SsrB, is required for regulation of genes both within and outside of SPI-2. Expression of ssrA and ssrB is controlled by at least four regulators, the nucleoid binding protein Fis, the MarA homologue SlyA, the response regulator OmpR and autogenously by SsrB. A further complexity of this system is that OmpR also directly participates in regulation of a subset of SsrB-dependent genes. Our goal is to understand how multiple transcription factors coordinately regulate expression of SPI-2 and SPI-2 co-regulated genes. We have examined the DNA binding properties of SsrB and SlyA using DNase I protection footprinting. Our results indicate that Sly binds to sites downstream of the ssrA transcription start site and both upstream and downstream of the ssrB transcription start site. SsrB binds to degenerate sites downstream of the ssrB transcription start site and both upstream and downstream of the ssrA transcription start site. Genetic evidence indicates that SsrB can also act as a repressor at ssrA. We are determining the sequence elements required for promoter function and clarifying the contribution of each binding site to regulation of ssrAB. A particularly interesting question is whether the requirement for autogenous control of ssrB expression is due to SsrB binding to the downstream site, expression of an ssrAB polycistronic transcript or indirect via expression of the ssrA kinase gene. Supported by NIH GM 58746 (LJK), NSF MCB 0243085 (LJK) and NIH F32-GM68364 (DW).
42

SURFACE-SPECIFIC REGULATION OF FLAGELLAR CLASS 3 GENES IN CHE MUTANTS OF SALMONELLA TYPHIMURIUM: A MODEL FOR THE FLAGELLUM AS AN ENVIRONMENT SENSOR Qingfeng (Tim) Wang, Asaka Suzuki, Susana Mariconda and Rasika M. Harshey. University of Texas at Austin, Austin, TX 78712
Swarming is a flagella-dependent form of bacterial surface motility. In Salmonella typhimurium, the chemotaxis system but not chemotaxis is essential for swarming. che mutants appear to have less flagella than wild-type bacteria on swarm media but not in broth. We report here that the che mutants adhere aberrantly to the agar and their swarm colonies appear less hydrated. Using microarray analysis, we show that the mutants down-regulate only class 3 flagellar genes on swarm media. Other down-regulated genes include putative class 3 motility genes, as well as some pathogenicity island SP-1 genes. These data can be explained by a model where the flagellar filament senses an external surface environment in che mutants that is unfavorable for subunit assembly, and signals the translation-coupled secretion apparatus TTSS located at the base of the flagellum to inhibit production of flagellin as well as stop secretion of FlgM, an inhibitor of class 3 transcription. External FlgM levels in swarm media vs broth were consistent with predictions of this model.
43

MESOSCALE ANNOTATION OF THE M. XANTHUS GENOME Balaji Srinivasen1, Nora Carboroy2, Farah Tengra2, Anthony Garza2, and Roy D. Welch2
Department of Developmental Biology, Stanford University School of Medicine, Stanford, CA Department of Biology, Syracuse University, Syracuse, NY
Myxococcus xanthus is a delta proteobacterium that can exist as both a free-living cell and a single-species biofilm called a swarm. Though each bacterium is autonomous with respect to metabolism and reproduction, M. xanthus has many of the characteristics of a multicellular organism; a swarm is a predatory collective that moves and feeds cooperatively, hunting together and pooling extracellular enzymes when digesting prey bacteria. The millions of cells within a swarm also exhibits a unified starvation stress-response, synchronizing a change in behavior and initiating a complex program of self-organization that culminates in the transition from an evenly distributed population of cells to densely packed aggregates called fruiting bodies. Each fruiting body is a spherical structure of approximately 1X10E5 cells that contain starvation-resistant spores. By aggregating before sporulating, the spores within a fruiting body can emerge as an “instant” swarm when nutritional conditions are conducive to growth
The formation of fruiting bodies is complicated from an movement and signaling standpoint, and even more complex when considering the requisite behavioral genetics. A swarm represents a distributed system, a geographically dispersed collection of (genetically) computing entities that must function together and coordinate their behavior through a communication network. The complexity of M. xanthus behavior is reflected in its 9.1 MB genome, which is the largest prokaryotic genome sequenced to date. Although pivotal genes involved in M. xanthus behavioral genetics have been identified, a global understanding of M. xanthus functional genomics is hindered by the fact that almost 50% of the predicted genes have no assigned function.
Our lab is assembling different genome-scale data sets into a single database that we are using to draw functional correlations between genes. This database is a powerful tool for the mesoscale annotation of the M. xanthus genome.
44

THREE DIMENSIONAL STRUCTURE AND ORGANIZATION OF A RECEPTOR-SIGNALING COMPLEX. Noreen R. Francis1, Peter M. Wolanin2, Jeffry B. Stock2,3, Dennis R. Thomas1 and David J. DeRosier1
1 Rosenstiel Biomedical Sciences Research Center, Brandeis University, Waltham, MA 02454 2 Department of Molecular Biology, Princeton University, Princeton NJ, 08544 3 Department of Chemistry, Princeton University, Princeton NJ, 08544
The core components of the Escherichia coli chemotaxis signal transduction system are
found in nearly all motile bacteria, and consist of transmembrane chemoreceptors, an associated histidine kinase, CheA, and an associated CheA-activator, CheW. These signaling complexes form within a large, densely packed patch of chemoreceptors, which is typically located at one (or both) poles of the cell. Recent studies and modeling of the chemotaxis system have emphasized the importance of interactions among large numbers of receptors in terms of the cell’s sensitivity and adaptive response. The chemoreceptor patches are mixed, containing receptors for different ligands, and receptors with different specificities communicate with one another. The lack of detailed information about how CheA and CheW interact with the chemoreceptors and each other in the receptor/signaling complexes is a critical gap in our understanding of how the signaling works at the molecular level. The X-ray crystal structure of the cytoplasmic domain of the serine receptor (TsrC) has been solved and shows the cytoplasmic domain to be a dimer which forms an extended 4 helix bundle. The dimers form trimers in the crystal and this “trimer of dimers” has become the basis for most current models of how large numbers of receptors might interact. In particular, this TsrC structure shows extensive inter-dimer contacts in the hairpin regions of the receptor signaling domains.
Soluble, active complexes can be formed from the cytoplasmic domain of the Tar receptor (TarC) fused to a leucine zipper (lz) with CheA and CheW. The stoichiometry of lzTarC: CheW: CheA monomers is about 24:6:4, which is similar to that found in vivo. The length of these complexes is 42 nm and the width varies from 12 nm at the ends to 18nm in the middle. The organization of this lzTarC/CheW/CheA complex has been determined by electron cryomicroscopy and single particle image analysis. The structure is elongated and bipolar with empty space between the pillars of density. The pillars are narrower at the ends and wider at the middle of the complex. The pillars meet roughly end to end near the middle. Each of the four pillars is composed of three dimers of the Tar cytoplasmic domain (i.e., a total of 24 lzTarC chains). The remaining density around the middle of the structure is sufficient to contain the CheA and CheW proteins. In these structures, we see clear indications that receptor four-helix bundles play an important role. However, these four-helix bundles are arranged with different dimer-dimer relationships than observed in the trimer of dimers of the X-ray crystal structure of TsrC. The hairpin regions of the receptor signaling domains appear to be widely separated, interacting with CheA and CheW, whereas the ends containing the N and C-termini are tightly packed.
45

MONITORING ACETYL ~ P, A GLOBAL SIGNAL OF TWO-COMPONENT PATHWAYS Alan J. Wolfe, David Keating, and Ana Shulla Department of Microbiology and Immunology, Stritch School of Medicine, Loyola University Chicago, Maywood, IL 60153
Acetyl phosphate (acetyl~P) is a high energy form of phosphate. An essential intermediate during the interconversion of acetyl-CoA and acetate, acetyl~P stores considerably more energy than does ATP. Consequently, acetyl~P donates phosphoryl groups to ADP to synthesize much of the ATP generated during growth on glucose and other acetate-producing (acetogenic) carbon sources. Acetyl~P also donates phosphoryl groups to the response regulators (RR) of certain two-component signal transduction (2CST) pathways. These include a subset that control flagellar biogenesis, capsule biosynthesis, type 1 pilus assembly and, accordingly, the development of biofilms (Wolfe et al., 2003).
These and other data suggest that acetyl~P coordinates subsets of 2CST pathways. If so, then mapping the extensive network of 2CST pathways, their interactions, their target genes and their gene products will require that we learn how acetyl~P impacts the entire network of signaling pathways. Such studies will guide future attempts to chart the influence of acetyl~P on the signaling networks of other eubacteria, and members of the other two kingdoms that possess both 2CST pathways and the capacity to make acetyl~P.
If we are to understand how acetyl~P impacts two-component signaling, we first must learn when cells accumulate acetyl~P and when they do not. Direct measurements of the intracellular acetyl~P pool have been reported only rarely. Most notably, Prüß and Wolfe (1994) and McCleary and Stock (1994) showed that acetyl~P levels in Escherichia coli depend upon acetyl-CoA availability, incubation temperature, and the acetogenic nature of the carbon source. Others have since accumulated indirect evidence implicating other nutritional and environmental factors in the control of acetyl~P. These factors include limiting oxygen, nitrogen, and carbon, each of which restricts the entry of acetyl-CoA into the TCA cycle. They also include aerobic growth on excess glucose and other sugars whose metabolism results in the excretion of large amounts of acetate. Finally, they include low pH and high osmolarity, environmental factors that influence the expression of certain genes in an acetyl~P-sensitive manner.
To assess the impact of acetyl~P upon two-component signaling networks, we have optimized a thin layer chromatographic assay, pioneered by Bochner and Ames (1982), and have begun to use it to complement indirect evidence with direct measurements.
46

INTERACTIONS OF CHEA, CHEY, AND CHEZ THROUGH THE PRISM OF COMPARATIVE GENOMICS Kristin Wuichet and Igor B. Zhulin Center for Bioinformatics & Computational Biology, School of Biology, Georgia Institute of Technology, Atlanta, GA 30332-0230
Amino acid residues that mediate protein-protein interactions are conserved due to the evolutionary pressure to maintain the interactions. The wealth of available microbial genomic data is conducive for the use of comparative analysis to predict and characterize protein-protein interactions.
We present a computational study of the interactions between CheA, CheY, and CheZ that takes advantage of comparative genomics and phylogenetics to predict the specific contact sites and use available structural information to validate the predictions. Our initial sequence analysis of the CheY-CheZ interaction identified the majority of contact sites previously revealed by X-ray crystallography (1) and pointed out additional residues that are likely to contribute to the interaction. Some of these residues have been implicated in the interaction by recent experimental data from the Bourret lab. Genomic analysis of the CheZ protein revealed new members of the CheZ phosphatase family present in α, δ, and ε-proteobacteria and expanded our understanding of the interaction between CheA and CheZ.
The binding of CheY to the P2 domain of CheA has been solved by X-ray crystallography in both E. coli (2) and T. maritima (3), but the nuances of this interaction are different. The low sequence conservation of the P2 domain makes this a challenging case for computational sequence analysis. Although the P2-CheY interaction cannot be simply extrapolated from one organism to another, our findings show patterns of sequence conservation specific to each of the known mode of interaction. Taken together our results indicate that a comparative analysis of protein sequences can complement and expand results obtained by structural studies, which appears to be especially important for refining protein-protein interactions.
. 1. Zhao et. al. (2002) Nat Struct Biol. 9:570-5. 2. McEvoy et. al. (1998) Proc Natl Acad Sci USA 95:7333-8. 3. Park et. al. (2004) Proc Natl Acad Sci USA 101:11646-51.
47

FUNCTIONAL IMPORTANCE OF CHARGED RESIDUES IN THE ROTOR AND THE STATOR COMPONENTS OF AN NA+-DRIVEN FLAGELLAR MOTOR Toshiharu Yakushi1, Jung-Hoon Yang2, Hajime Fukuoka1, Michio Homma1 and David F. Blair2
1Graduate school of Biological Science, Nagoya University, Chikusa-ku, Nagoya, 464-8602, Japan. 2Department of Biology, University of Utah, Salt Lake City, Utah 84112
Bacterial flagellar motors rotate using the energy from the transmembrane electrochemical potential of a specific ion, either H+ (e.g. E. coli) or Na+ (e.g. the polar flagellum of Vibrio alginolyticus). The stator complex formed from MotA and MotB (PomA/PomB for V. alginolyticus) and a rotor protein FliG are thought to be involved directly in torque generation. The outer membrane proteins MotX and MotY are also required for rotating the Na+-driven motor of V. alginolyticus. In the H+-driven flagellar motor, electrostatic interactions between the cytoplasmic region of MotA and the C-terminal domain of FliG have been proposed: Arg90 and Glu98 in MotA have been suggested to interact with Lys264, Arg281, Asp288, Asp289, and Arg297 in FliG. The residues Arg90 and Glu98 of MotA are conserved in PomA as Arg88 and Glu96, and PomA has additional charged residues, Lys89, Glu97, and Glu99, that are nearby in the sequence. Mutations that neutralize the five charged residues of PomA do not, however, affect torque generation by the Vibrio flagellar motor, nor do charge-neutralizing mutation in the conserved charged residues of FliG. It appears that a large change in charge is needed to affect the motor of Vibrio; certain mutants that reverse the charges do impair function of the Vibrio motor. Thus, the role of electrostatic interaction in the Na+-deriven motor, if any, remains unclear. The flagellar motor in E. coli can be made to work with Na+ by replacing MotA and MotB with PomA and PotB (a chimeric protein with N-terminal parts from PomB and C-terminal parts from MotB). Additionally, the C-terminal domain of E. coli FliG can be replaced with that from V. alginolyticus, allowing both the rotor and stator proteins of Vibrio to be studied in E. coli cells. In this system, several mutations of charged residues, both those that reverse charge and those that neutralize charge, were found to impair function. Moreover, certain mutations in the rotor suppressed defects caused by mutation in the stator. The suppression effects appear related to those seen with the E. coli proteins, but significant difference are also observed, pointing to the involvement of additional residues.
48

REGULATION OF FIBRIL EXOPOLYSACCHARIDES (EPS) AND SOCIAL GLIDING BY THE DIF CHEMOTAXIS-LIKE PROTEINS AND TYPE IV PILI (TFP) IN MYXOCOCCUS XANTHUS
Wesley Black1, Zhuo Li1, Qian Xu1, Michael Manson2 and Zhaomin Yang1
Department of Biology 1Virginia Polytechnic Institute and State University, Blacksburg, VA 24061 2Texas A&M University, College Station, TX 77843-3258
The developmental bacterium M. xanthus can move on solid surfaces using its TFP-mediated social gliding motility. Such movements also require the production of fibril EPS. Previous studies indicated that the Dif chemotaxis-like proteins are important for the regulation of EPS production. DifA (MCP-like), DifC (CheW-like) and DifE (CheA-like) regulate EPS production positively whereas DifD (CheY-like) and DifG (CheC-like) do so negatively. Both genetic epistasis and molecular biology techniques were recently used to analyze the functional and physical interactions among Dif proteins and to probe the nature of the input signal of the Dif pathway. Our studies led to the following conclusions. First, Dif proteins likely constitute a signal transduction pathway where the CheY homolog DifD may modulate the signaling strength through the central pathway instead of being directly downstream of the CheA homolog DifE. Second, the Dif pathway shares significant architectural similarities with the classical chemotaxis pathway in both inter-molecular interactions and signaling mechanisms. Lastly, TFP likely function upstream of the Dif pathway to positively regulate EPS production and it appears that TFP may constitute part of a sensor providing input signals to the Dif pathway. Results in support of the above conclusions will be presented and discussed at the meeting. Our current working model is depicted below.
Current working model for EPS regulation by Dif and TFP
CM
TFP
DifEDifEDifEDifEDifEDifE
DifA
DifA
DifA
DifACC
DifX’sDifX’s
EPSEPS
DifDDifD DifG
49

CHARACTERIZATION OF THE MECHANISM OF CHED AS A DEAMIDASE IN BACILLUS SUBTILIS CHEMOTAXIS
Wei Yuan, George W. Ordal
Department of Biochemistry, University of Illinois, Urbana, IL 61801
Bacteria chemotaxis occurs by controlling the direction of rotation of the flagella based
on a modified two-component system. B. subtilis contains several chemotaxis proteins that have
no counterparts in E. coli, including CheD, CheC, and CheV. It was recently reported that CheD
can specifically deamidate chemoreceptors with changing specific Gln into Glu prior to
methylation.
Elucidating the active sites of CheD can help us understand its function and biological
significance. In this work, the full-length CheD sequence was aligned with orthologues presented
in 17 other microorganisms. 100%-conserved residues were mutated into Ala, respectively. The
phenotypes of mutants were analyzed by the swarm plate assay. The changes S32A, D40A, or
H50A abolished the normal CheD function, which was verified by a chemoreceptor modification
assay. Interestingly, these results imply that CheD possesses the catalytic triad (H, D, S)
characteristic of serine proteases. To verify this, each mutated CheD protein was purified with
using GST-tag, then incubated with serine protease inhibitor dansyl fluoride (DNSF). MALDI
results showed that DNSF can specifically derivatize normal CheD, but not any of the three
mutated CheD’s. We think it likely that S32, D40, and H50 are in the active site of CheD, and its
deamidase function occurs through a serine protease-type mechanism.
50

POSTER ABSTRACTS
51

ELECTRON MICROSCOPIC STUDY OF GLI349, RESPONSIBLE FOR GLASS BINDING DURING GLIDING OF MYCOPLASMA MOBILE Jun O. Adan1*, Atsuko Uenoyama1, Makoto Miyata1, 2
(1Department of Biology, Graduate School of Science, Osaka City University, Sumiyosi-ku, Osaka, 558-8585 JAPAN 2PRESTO, JST, JAPAN) M. mobile lacks a cell wall but is featured with a protruded end, so-called head. It
attaches to solid surface and moves smoothly (gliding) at a speed of 2.5 micro m/sec. The
genome of M. mobile does not have any homologs of genes related to known bacterial motility or
motor proteins such as myosin, suggesting the existence of a special mechanism. We previously
identified a 349 kDa protein, named Gli349, which is located on the base of head, named neck,
and responsible for the glass binding during gliding. Rapid-freeze and fracture electron
microscopy of cells gliding on glass showed that spike-like structures sticking out from the neck
bind to the glass surface. In the present study, we focused on (i) identity between the spike and
Gli349, (ii) shape of the spike, and (iii) structure of Gli349 molecule.
To visualize the surface structure of the cell neck, the negative-staining method was
employed with using phosphotungstic acid. The cell-glass interface could not be seen in the
images of the normally growing cells. However, when the elongated cells frequently found in a
prolonged culture were observed, the spikes were found to stick out from the cell neck with
angles of 30 to 150 degrees relative to the cell axis. They were 52±10 nm (n=40) in length,
consistent with the spike previously observed by the rapid-freeze and fracture electron
microscopy. The identification of the spike structure is now undertaken.
To know the detailed structures of a Gli349 molecule, this protein was isolated from a M.
mobile culture by the successive procedures, as (i) solubilization of cells by 1% TritonX-100, (ii)
centrifugation at 200,000×g, (iii) salting out, (iv) anion-exchanger column chromatography, and
(v) hydrophobic column chromatography. The Gli349 protein was sprayed, rotary-shadowed,
and observed by electron microscopy. The images were classified into two large categories. The
first was 115nm-long rod-like structures featured with 3 hinges, and the other was smaller and
thicker particles. Detailed analyses will be discussed.
52

MOLECULAR EVOLUTION OF THE MCP SIGNALING DOMAIN: IMPLICATIONS FOR THE SIGNALING MECHANISM Roger P. Alexander and Igor B. Zhulin Center for Bioinformatics & Computational Biology, School of Biology, Georgia Institute of Technology, Atlanta, GA 30332-0230
We are performing comparative genomic analysis of the cytoplasmic signaling domain of methyl-accepting chemotaxis proteins (MCPs). Our goal is to leverage structural, biochemical, and functional information from well-studied MCPs and to develop an approach for accurate functional predictions for MCPs that have not been studied experimentally. The C-terminal signaling domain of MCPs is a conserved module that appears to have an unusual evolutionary history.
We collected 1250 protein sequences of known and predicted MCPs from the completely sequenced genomes of 75 bacterial and archaeal species. Using a series of bioinformatics tools, each MCP sequence was screened in order to delineate the position of the signaling domain with respect to upstream transmembrane regions and HAMP domains. A multiple alignment of several subfamilies of signaling domain sequences was constructed using the PCMA program (1) run on a supercomputer, followed by manual adjustments based on the structural information available for the Tsr transducer of E. coli (2). Conservation pattern of the signaling domain was determined at various thresholds at the level of individual amino acids and their groupings using sequence logos and consensus scripts. Using a much larger sequence sampling, we confirmed the earlier hypothesis by Le Moual and Koshland that a series of two unusual deletions of four turns of an alpha helix in the middle of the coiled coil structure drove the evolution of the C-terminal domain (3). However, we were able to better resolve the positions of these deletions, provide further evidence for the deletion versus insertion hypothesis, and identify more than three signaling domain subclasses.
Based on analysis of the conservation pattern within the signaling domain we present evidence that all MCPs (most of which will never be studied experimentally) form stable dimers as has been shown experimentally for the E .coli MCPs. This essentially generalizes the statement that ligand binding to the MCP sensory domain does not affect the equilibrium between monomer and dimer forms of the receptor as in many eukaryotic signaling systems. Mapping putative methylation sites with respect to deletions that occurred during the evolution of the C-terminal domain allowed us to further explain experimentally observed differences in methylation/demethylation patterns between MCPs from E. coli, B. subtilis and H. salinarum. A key feature of MCP signal transduction is that ligand binding and methylation lead to sampling of different conformational subspaces. Using a combination of genomics and molecular modeling we show that the ratio of flexible to rigid regions in the MCP signaling domain differs between subclasses and has implications for the signaling mechanism. 1. Pei, J. et al (2003) Bioinformatics 19:427 2. Kim, K.K. et al (1999) Nature 400:787 3. Le Moual, H. and Koshland, D.E., Jr. (1996) J Mol Biol 261:568.
53

SIGNALING INTERACTIONS BETWEEN TSR AND TAR IN VIVO Peter Ames and John S. Parkinson Biology Department, University of Utah, Salt Lake City, Utah 84112, USA
Tsr and Tar respectively mediate chemotaxis to serine and aspartate in E. coli. Recent
studies from several groups have shown that these chemoreceptors somehow collaborate with one another to amplify small stimuli into large motor-controlling signals. Our working model of receptor collaboration for high-gain signaling proposes that homodimeric receptors of different types form mixed trimer-of-dimer signaling teams, which may in turn build communication networks through shared connections to the CheA and CheW proteins. The epistatic behavior of some Tsr mutants (Tsr*) with mutations at trimer contact residues supports this view. Tsr* molecules cannot mediate serine chemotaxis, but also block the function of other receptors, such as Tar, in the cell. To explore the possibility that these epistatic effects occur in mixed trimers (or any higher-order receptor complex), we isolated compensatory mutations in Tar (Tar^) that restored signaling ability to Tsr* mutants. We characterized 19 different Tar^ mutations at 16 different residues. All had an amino acid replacement in the trimer contact region of Tar, three at contact sites themselves. In the absence of other receptors, Tar^ receptors exhibited a variety of signaling defects in their own right. None of them mediated a normal chemotaxis response to aspartate. Most were completely nonfunctional; five retained partial function. Some were epistatic to wild-type Tsr; some regained function in the presence of their Tsr* partner. The pattern of Tar^-Tsr* suppression was allele-specific, consistent with conformational suppression through direct protein-protein interaction. Moreover, we saw no correlation between the signaling biases of Tar^ mutants, as reflected by the flagellar rotation patterns they produced, and the Tsr* mutants they suppressed, which indicates that the suppression mechanism is not simply a balancing interaction between the Tar^ and Tsr* phenotypes. All Tar^ receptors, like their Tsr* counterparts, also formed polar clusters dependent on ternary complex interactions with CheA and CheW. We conclude that Tar^ and Tsr* mutations probably do not impair ternary complex formation, but rather block receptor function by altering the conformation of receptor signaling teams based on trimer-of-dimers geometry. In this view, mutually-suppressing Tar^/Tsr* pairs have compensatory trimer conformations that allow restoration of correct geometry and stimulus control in mixed trimer of dimer signaling teams.
54

DOMAIN INTERACTIONS IN THE OmpR/PhoB FAMILY OF RESPONSE REGULATORS
Priti Bachhawat1, 2, 3, GVTS Swapna3, 4, Gaetano T Montelione3, 4, Ann M Stock2, 3, 5 1Graduate School of Biomedical Sciences, 2Department of Biochemistry, University of Medicine and Dentistry New Jersey, 3Center for Advanced Biotechnology and Medicine, 4Rutgers University, 5Howard Hughes Medical Institute, 679 Hoes Lane, Piscataway, NJ 08854, USA
Bacteria use the two-component system of signaling, consisting of a sensory histidine kinase and a response regulator, to sense and adapt to environmental changes. The OmpR/PhoB family is the biggest family of response regulators that act as DNA binding transcription factors. These proteins are modular two-domain proteins consisting of a well conserved N-terminal regulatory domain and a C-terminal DNA-binding effector domain. In the inactive state these proteins have a low DNA binding ability. Upon phosphorylation at a conserved aspartate on the regulatory domain, these proteins undergo a conformational change often accompanied by a change in their oligomeric state. This allows them to bind with a greater affinity to DNA sites that are almost always arranged as tandem pairs. Although the changes that occur in the conserved receiver domain upon phosphorylation are well documented for various response regulators, it is not clear how this conformation change enhances the DNA binding ability upon activation in this family.
We have crystallized regulatory domains of PhoB and PhoP with and without an activating agent analog BeF3
-. PhoB is the master regulator of ~40 genes that comprise the pho regulon which is activated in phosphate deprivation conditions. These genes are involved in the metabolism, uptake and transport of phosphate from phosphorus sources. PhoP is a response regulator activated in low extra-cellular divalent ion conditions, and is involved in Mg++ homeostasis and virulence. In the active forms of both regulatory domains, the same α4−β5−α5 face is used for dimerization with a two-fold rotational symmetry. This is the same face that undergoes the greatest phosphorylation induced conformational change in all response regulators and is also involved in intra-molecular contacts with the C-terminal domain in the inactive state in another OmpR family protein DrrB. This seems to be a general mechanism as other regulatory domains of this family crystallized in our lab also use this same face in the activated state. These crystal structures have allowed us to propose a mechanism that is distinct from that of other response regulators, in which the N- and C-terminal domains adopt different symmetries in the activated DNA bound state. We have also performed NMR studies that confirm these symmetries in solution and provide a quick and simple way to assess domain interactions and symmetries.
55

THE ROLE OF ALGR AND FIMS IN THE CONTROL OF TWITCHING MOTILITY IN PSEUDOMONAS AERUGINOSA.
Belen Belete and Daniel J. Wozniak
Department of Microbiology and Immunology, Wake Forest University School of Medicine, Medical Center Boulevard, Winston-Salem, NC, 27157
Pseudomonas aeruginosa is an opportunistic pathogen that can infect immunocompromised individuals and cystic fibrosis patients. This bacterium produces a wide range of virulence factors including the exopolysaccharide alginate and type IV fimbriae. Type IV fimbriae retraction and extension mediate twitching motility, which is a flagella-independent mode of solid surface translocation. Both twitching motility and pilus production are essential for P. aeruginosa virulence. The FimS/AlgR sensor-regulator pair regulates the biosynthesis of type IV fimbriae (pili). At present, the mechanism by which AlgR controls twitching motility is unknown. Most response regulators require phosphorylation at an aspartate residue for their activity. AlgR is phosphorylated at Asp54 and mutating this residue results in the loss of twitching motility. Deletions of both algR and fimS also result in the loss of twitching motility. Western blot analyses on whole cell lysates of algR mutants show no defects in pili production. However, transmission electron microscopy and Western blot analyses on surface sheared samples reveal that algR mutants lack surface expressed pili. Cellular fractionation studies on the algR mutants show that pilin monomers are trapped in the cytoplasmic space and fail to reach the periplasmic space. Thus, while algR mutations do not affect pili production, it is clear that AlgR plays an important role in modulating proper pili localization. Data from adherence assays also show that both the algR deletion mutant and the algR phosphorylation mutant are significantly reduced in their ability to adhere to human bronchial epithelial cells. These findings indicate that algR is required for both twitching motility and adherence. Thus, the overall effects of mutating algR suggest a decrease in P. aeruginosa virulence.
56

REVISITING CHEMOTAXIS IN SHEWANELLA ONEIDENSIS Sira Bencharit and Mandy J. Ward Department of Geography and Environmental Engineering Johns Hopkins University, MD 21218 In 1995, Nealson et al. [1] published a study on the chemotactic responses of S. oneidensis (then known as S. putrefaciens) to a range of anaerobic electron acceptors using plug plate and capillary assays. These studies showed that S. oneidensis, a facultative anaerobe with considerable respiratory versatility, was capable of responding behaviorally to most of the electron acceptors tested. However, no behavioral responses to metals were observed, even though S. oneidensis is capable of reducing several metals during anaerobic respiration. The ability of bacteria to reduce metals is of considerable interest to geochemists, environmental engineers and microbiologists. Consequently, we decided to re-examine the ability of S. oneidensis to respond chemotactically to metals. Our preliminary results using swarm plate assays have shown that wild-type S. oneidensis does respond behaviorally to soluble Fe (III), along with several other anaerobic electron acceptors. The cells inoculated into the swarm plates first generate a gradient of Fe(III) through respiratory activity, then respond behaviorally to the self-generated gradient, suggesting that S. oneidensis may use energy-sensing behavior to respond to iron when it is being used as the terminal electron acceptor. The publication of the genome sequence of S. oneidensis in 2002 identified 28 genes encoding potential methyl-accepting chemotaxis proteins (MCPs) in this microbe [2]. Analysis of these proteins showed that five appear to have energy-, or redox-sensing PAS domains. We have constructed a set of mutants with insertions in the energy-sensing receptor genes. Mutants have also been constructed that have insertions in the cheA genes and in genes required for motility. Results of chemotaxis assays performed using these mutants will be discussed.
[1] Nealson et al. (1995). Anaerobic electron acceptor chemotaxis in Shewanella putrefaciens. Appl. Environ. Microbiol. 61: 1551-1554. [2] Heidelberg et al. (2002). Genome sequence of the dissimilatory metal iron-reducing bacterium Shewanella oneidensis. Nature Biotechnol. 20: 1118-1123.
57

COOPERATIVE ACTIVATION OF CHEA IN TEMPLATED-ASSEMBLED SIGNALING COMPLEXES Tatiana Y. Besschetnova, Frances M. Antommattei, Anthony L. Shrout, David J. Montefusco, and Robert M. Weis. Department of Chemistry, University of Massachusetts, 710 N. Pleasant St. LGRT 701, Amherst, MA 01003-9336.
Transmembrane signaling events in both eukaryotic and prokaryotic cells occur in the two-dimensional space defined by the bilayer membrane. Cellular responses are also regulated through processes that involve the specific recruitment of signaling proteins to the membrane surface. These phenomena suggest the importance of clustering and 2-D concentration in signaling. Previously, we used a template-directed self-assembly method to restore the kinase-activating and methyl-accepting properties of a histidine-tagged cytoplasmic fragment of Tar (CF) on phospholipid vesicles containing Ni-NTA lipids (Shrout, et. al. 2003. Biochemistry 42:13379-85). Here, this method is used to vary the 2-D concentration of CF, which was found to influence both CheA activation and methylation. Rates of templated CF methylation and the activity of CheA in CF/CheW/CheA complexes both increased substantially when the 2-D concentration of CF was increased. The activation of CheA was measured both as a function CF surface concentration and covalent modification (EEEE, QEQE, QQQQ), and was found to be consistent with a pre-clustering of CF at the higher levels of covalent modification. At both small and large surface densities of CFQQQQ, the activation of CheA occurred to a similar extent. For ternary complexes formed with CFEEEE, increasing the surface concentration of CFEEEE promoted a cooperative increase in CheA activity, which suggested immediately that cooperative interactions between cytoplasmic domains were involved in CheA activation. That the Hill coefficients were different for sonicated versus extruded unilamellar vesicles (4 versus 12) demonstrated that surface curvature influenced cooperative assembly in the small vesicle limit. Significantly, the large surface concentrations that resulted in the most effective activation of CheA activation did not correspond to the optimum condition for CF methylation, which occurred at a lower surface concentration. The difference in the surface density dependence for CheA activation and CF methylation suggests that under appropriate conditions the processes of clustering-mediated CheA activation and receptor methylation (and demethylation) counterbalance. The differing influences of CF density on CheA activation and CF methylation provide a means to regulate CheA activity through receptor clustering.
58

REGULATION OF EXTRACELLUAR ENZYMES BY GcrR IN STREPTOCOCCUS MUTANS Saswati Biswas, and Indranil Biswas. Division of Basic Biomedical Sciences, School of Medicine, University of South Dakota. Vermillion, SD 57069. Dental caries is one of the most common bacterial infections in humans and remains untreated in many underdeveloped countries. Based on biochemical, epidemiological and animal experiments, Streptococcus mutans is considered to be the principal etiological agent of dental caries. The ability to metabolize carbohydrates and to adhere to and form tenacious biofilms on the tooth surfaces is believed to be critically associated with the cariogenicity of this human pathogen. S. mutans synthesizes water-soluble and –insoluble glucans from sucrose by three glucosyltransferases (GTF), and adheres firmly to tooth surfaces with their cooperation. Moreover, S. mutans produces fructan from sucrose by fructosyltransferase (FTF) glucan-binding proteins (Gbps) and they appear to play major roles in smooth surface adherence. The specific mechanisms governing regulation of exopolysaccharide synthesis in S. mutans have yet to be discovered. However, it was shown that GcrR, a response regulator, modulates the expression of at least one Gtf and one Gbp gene. A recent report suggests that additional genes are subjected to GcrR regulation. This regulator is also required for biofilm formation and in cariogenesis as shown in rat models. Interestingly, GcrR shows extensive homology with the group A streptococcus (GAS) CovR, a response regulator that controls as much as 15% of the GAS genes including many important virulence factors. By sequence analysis of proximal region of gcrR locus and by Southern hybridization analysis of twelve different S. mutans strains, we found that GcrR is an orphan response regulator in all the strains tested. We have inactivated GcrR in three different strains (NG-8, UA159, and GS-5) of S. mutans. By analyzing the supernatant proteins in 1-D SDS-PAGE gels followed by mass spectrometry identification, we observed that GcrR affects expression/secretion of at least two GTFs, encoded by gftB and gtfC and FTF, encoded by sacB gene. Analysis of gftB transcription by RNA slot blot and transcriptional reporter fusion suggests that gtfB expression is directly controlled by GcrR. However analysis of sacB transcription by RNA slot blot and transcriptional fusion suggests that expression of FTF is indirectly regulated by GcrR. The expression of gtfC also appears to be indirectly regulated by GcrR. We have purified recombinant GcrR protein from E. coli. In EMSA analysis, our results showed that GcrR binds to the gtfB promoter region as well as to the gbpC promoter (a previously identified GcrR regulated gene). By comparing the DNaseI footprinting of GcrR binding to these promoters, we are trying to define the binding motif of GcrR. Once such a binding site is determined, in silico analysis will help us to identify putative genes that are regulated by GcrR. Our long term goal is to elucidate the molecular mechanisms of gene regulation by GcrR and how it controls progression of dental caries.
59

NEGATIVE REGULATION OF SPORULATION IN BACILLUS ANTHRACIS BY RAP AND Spo0E PHOSPHATASES. Cristina Bongiorni and Marta Perego The Scripps Research Institute; 10550 North Torrey Pines Road; MEM-116 La Jolla, CA 92037
Anthrax, a potentially fatal disease for animals and men, is caused by the Gram-positive endospore-forming bacterium Bacillus anthracis. Genetic and bioinformatic studies revealed that sporulation initiation is under control of a phosphorelay signal transduction pathway homologous to the one originally described as regulating the initiation of the developmental process in B. anthracis. In the phosphorelay, multiple histidine sensor kinases auto activate in response to a variety of environmental, metabolic or cell cycle signals. Following autophosphorylation, the sensor kinases transfer the phosphoryl group to an intermediate response regulator, Spo0F, which in turn transfers it to the Spo0A response regulator and transcription factor through the activity of the Spo0B phosphotransferase. Accumulation of a threshold level of Spo0A~P allows sporulation to initiate. The kinase activities are counteracted by protein phosphatases that respond to environmental and physiological signals antithetical to sporulation. In B. subtilis three proteins of the Rap family target the Spo0F~P intermediate of the phosphorelay while the three members of the Spo0E family, Spo0E, YisI and YnzD, dephosphorylate the Spo0A~P transcription factor. Bioinformatic analysis carried out on the genome of B. anthracis revealed the presence of 4 chromosomally encoded Rap proteins and 4 Spo0E proteins. An additional Rap protein is encoded by a gene located on the pX01 virulence plasmid. We carried out a genetic and biochemical characterization of each B. anthracis Rap and Spo0E protein and determined that one chromosomal-encoded and the plasmid-encoded Rap proteins are regulating the sporulation initiation pathway by targeting the Spo0F~P response regulator thus affecting its phosphorylation level. We also determined that all four Spo0E proteins of B. anthracis act on the Spo0A~P transcription factor thus regulating the output of the phosphorelay pathway.
Since the production of the toxin virulence factor, crucial for B. anthracis pathogenesis, is under control of the phosphorelay pathway, Rap and Spo0E proteins are likely to play a role in the pathogenesis of this organism. Analyses aimed at determining the extent of this role are underway and the results will be presented.
60

GENETICS OF THE F1 AND HAMP DOMAINS IN THE AER TRANSDUCER Maria Burón-Barral and John S. Parkinson Biology Department, University of Utah, Salt Lake City, Utah 84112, USA
The membrane-associated Aer protein of E. coli mediates aerotactic behavior. Aer has an N-terminal PAS domain, which is thought to sense aerotactic stimuli through the redox state of its FAD cofactor. The C-terminus of Aer resembles the signaling domain of methyl-accepting chemotaxis proteins (MCPs) and most likely forms ternary signaling complexes with the CheA and CheW proteins to communicate with the flagellar motors. A hydrophobic segment interposed between the input and output domains anchors the Aer molecule to the inner side of the cytoplasmic membrane. The membrane anchor is flanked by regions of poorly understood function, F1 (residues 121-167) and HAMP (residues 205-256). To elucidate the mechanism of input/output communication in Aer and what role(s) the F1 and HAMP segments might play, we isolated and characterized aerotaxis-defective F1 and HAMP mutations from a regulatable, plasmid-borne aer gene expressed in a transducer-less host strain.
Only seven amino acid replacements at six different sites were obtained in F1, consistent with its poorly conserved primary structure. All F1 mutants expressed low levels of Aer protein (less than 35% of wild-type) and were recessive to wild-type Aer. Upon maximal induction, the mutant F1 proteins, unlike wild-type Aer, did not cause a detectable increase in intracellular FAD content. Three of the seven F1 mutants were demonstrably leaky in the presence of other chemoreceptors, implying defects in protein maturation or stability. Turnover studies of one mutant protein confirmed a greatly increased degradation rate compared to wild-type Aer. Taken together, these properties suggest that the apparent FAD-binding defects of F1 mutants could be an indirect consequence of their reduced protein stability. We conclude that F1 plays a role in Aer maturation, but most likely has no direct role in input/output communication. Conceivably, the F1 segment interacts with other parts of the Aer molecule, but those interaction partners remain to be determined.
The HAMP domain, although not highly conserved in sequence, yielded mutations at 25 of 57 targeted codons, consistent with a more critical functional role than F1. The locations and phenotypes of the HAMP mutations roughly coincided with predicted HAMP secondary structure features (two amphipathic helices flanking a segment of unknown structure). Mutations in the putative helices reduced protein expression levels below 50% of wild-type with a concomitant reduction in FAD-binding and clockwise (CW) signaling ability. Mutations in the intervening region did not reduce protein or FAD-binding levels and caused excessive CW-signaling, as did one exceptional mutation at the C-terminus of the second predicted HAMP alpha-helix. All CW-biased HAMP mutants regained aerotactic ability in the presence of other chemoreceptors, demonstrating that the mutant proteins are still capable of aerosensing and output signal control. The putative HAMP alpha-helices might play more direct roles in Aer input-output signaling, but the apparent signaling defects caused by mutational lesions in these segments (no FAD-binding, no CW output signals) could be indirect consequences of a primary defect in protein maturation or stability.
61

POTENTIAL BASIS FOR ADAPTATION MEDIATED BY THE BACILLUS SUBTILIS CHEMOTAXIS PROTEIN CheV
Vincent Cannistraro and George Ordal University of Illinois at Champaign Urbana, 190 Medical Sciences Bldg. 506 S. Mathews Ave. Urbana, IL 61801 Previous studies have shown that CheV is a two-domain chemotaxis protein with an amino-terminal CheW-like coupling domain and a carboxyl-terminal response-regulator domain. However, the CheW-like coupling domain of CheV is not functionally identical to CheW. Expression of the truncated CheW-like coupling of CheV inhibited adaptation to the addition of attractant.
To test the effect of the coupling proteins CheV and CheW on the ability of CheA to phosphorylate CheY, CheA, CheY and ATP were mixed in the presence of CheW or CheV. The results show that the presence of CheW greatly inhibited CheY-P formation in the reaction. Conversely, the presence of CheV appears to slightly increase CheY-P formation. Therefore, the two coupling proteins in B. subtilis are not functionally identical.
Other previous work showed that CheV can be phosphorylated in vitro by CheA-P at aspartate 235. Mutation of residue 235 to an alanine (which cannot be phosphorylated) also inhibits adaptation. Phosphorylation of CheV must play a role in CheV-mediated adaptation. To characterize this, a competition assay was performed in the presence of CheY. This experiment showed that CheY is phosphorylated preferentially over CheV. However, when there is a ten-fold excess of CheV to CheY, then CheV can be phosphorylated at high levels in the presence of CheY. However, when CheV is phosphorylated in vitro before CheY, CheV-P appears to inhibit CheY-P formation. CheV-P may be inhibiting the formation of CheY-P by affecting CheA autophosphorylation or phospho-transfer from CheA-P to CheY. To help resolve this question, we are in the process of determining the effect of CheV and CheV-P on CheA autophosphorylation and the ability of CheY to bind CheA-P in the presence and absence of CheV/CheV-P.
62

MOTA AND MOTB: TORQUE-GENERATING UNITS OF THE BACTERIAL FLAGELLAR MOTOR Jennifer H. Chandler*, David F. Blair†, Richard M. Berry‡ and Judith P. Armitage* *Microbiology Unit, Department of Biochemistry, University of Oxford, South Parks Road, Oxford OX1 3QU †Department of Biology, University of Utah, Salt Lake City, Utah 84112 ‡Clarendon Laboratory, University of Oxford, Parks Road, Oxford OX1 3PU †Department of Biology, University of Utah, Salt Lake City, Utah 84112
The bacterial flagellar motor is composed of a rotor, which together with the rod and filament, rotates relative to the anchored torque-generating particles or ‘stator’. The stator is composed of two integral membrane proteins, MotA and MotB, which together form a proton-conducting channel.
Evidence for the location of Mot complexes is indirect, but electron microscopy suggests that they form a ring of 11-16 ‘studs’ around the rotor. ‘Studs’ are not observed in either motA or motB deletion mutants, but reappear when motA and motB are restored in motA and motB deletion mutants respectively and flagella begin to rotate, accelerating in up to eight equal speed increments (a ‘resurrection’ experiment) (Block and Berg, 1984; Blair and Berg, 1988). MotB-GFP fusion expressed at wild-type level confirms that MotB localizes to the motor. Using rotation measured by following a polystyrene bead attached to a filament, held in an optical trap, the composition and mechanism of the motor is being investigated. ‘Deresurrection’ experiments, with dominant-defective mutant MotB protein expressed in a wild-type background, suggest that newly-synthesized MotBs cannot displace wild-type MotBs from working motors and therefore that a synthesized motor is stable. Several plasmids have been constructed which allow co-expression of both wild-type and mutant Mot proteins from separate inducible plasmids in a mot deletion background. Experiments using this genetic system are investigating composition, assembly and the role of specific amino acids in the Mot complexes.
63

STRUCTURAL AND FUNCTIONAL STUDIES OF CHEX and CHEC: CHEMOTAXIS PHOSPHATASES Xingjuan Chao; Sang-Youn Park; Gabriela Gonzalez-Bonet; Bryan D Beel; Alexandrine Marie Bilwes, PhD; Brian R Crane, Ph.D. Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14850 Many non-enteric bacteria contain additional chemotaxis proteins than those found in E. coli. In particular, the thermophile, Thermotoga maritima contains CheC, CheD, and CheX. We have determined the crystal structures of CheX to 2.5 Å resolution and determined that CheX has CheY phosphatase activity. The CheX structure reveals a novel alpha-beta-alpha topology that dimerizes by forming two symmetric beta-sheets at the molecular interface. Structural analyses also indicate a putative active center for dephosphorylation. Mutation studies are currently underway to probe interactions with CheY.
64

INTERACTION BETWEEN CHE-LIKE MOLECULES INVOLVED IN CONTROLLING PILI BIOGENESIS IN CYANOBACTERIUM SYNECHOCYSTIS SP. PCC 6803 Soo Youn Kim, Mi-Sun Cho, Young Hye Kim, Jong-Soon Choi, Youn-Il Park,a Young Mok Park and Young-Ho Chung Proteome Analysis Team, Korea Basic Science Institute, Daejeon 305-333, Korea, aDepartment of Biology, Chungnam National University, Daejeon 305-764, Korea
The unicellular cyanobacterium Synechocystis sp. PCC 6803 displays gliding motility
that depends on the type IV-like thick pili. All disruptants of chemotaxis-like gene locus (slr1041-slr1044, called Tax3 by Bhaya et al.) did not show gliding motility. Predicted proteins of slr1041, slr1042, slr1043 and slr1044 are homologous to PatA, CheY, CheW and MCP, respectively. The missing cheA-like gene in this cluster was identified, as novel split genes, slr0073 and slr0322. The two disruptants of cheA-like genes did not show gliding motility on the agar surface. To elucidate functional relationship between two CheA-like molecules, we examined possible phosphorelay cascade between histidine kinase domain of Slr0322 and Hpt domain of Slr0073 using yeast two-hybrid and co-immunoprecipitation analyses. We detected the strong and specific interactions between Slr0322 and Slr0073. These results suggest that the phosphorelay signal of Slr0322-HK to Slr0073-Hpt exists in Synechocystis sp. PCC 6803. Also, we detected the interactions between each of two CheAs (Slr0322 & Slr0073) and a CheW (Slr1043) and a CheY (Slr1042). We will discuss the possible working model for a signal transduction pathway of the gliding motility.
65

FUMARATE REDUCTASE AFFECTS FLAGELLATION IN ESCHERICHIA COLI Galit Cohen-Ben-Lulu, Yael Sagi and Michael Eisenbach Department of Biological Chemistry, Weizmann institute of science, 76100 Rehovot, Israel
Fumarate is a cytoplasmic component that, by reducing the free energy difference
between the counterclockwise and clockwise states of the flagellar motor, enables counterclockwise to clockwise switching and promotes clockwise rotation in Escherichia coli and Salmonella typhimurium. To reveal the mechanism by which fumarate affects the switch complex of the flagella motor, we examined whether fumarate binds to a preparation of intact, isolated switch complexes. Being unable to detect such binding, we looked for a mediator that potentially links between fumarate and the switch complex. Succinate dehydrogenase (SDH), the only membrane-bound protein in the citric-acid cycle, is known to convert fumarate to succinate. While examining whether this enzyme mediates fumarate binding to the switch complex, we found that SDH itself binds to the isolated, intact switch complex (KD = 1 µM). Using BIAcore, we found that SDH binds to the switch protein FliG. In spite of this binding, we did not detect binding of fumarate to the switch complex in the presence of SDH, suggesting that SDH does not mediate fumarate binding to the switch complex.
In order to understand the physiological function of the interaction between FliG and SDH, we studied a ∆sdhABCD mutant for motility and chemotaxis. However, we did not detect any effect of this deletion on these functions. Knowing that fumarate reductase (FRD) is similar to SDH in function and sequence, we also deleted the frd gene so as to totally eliminate the SDH function. The double mutant had fewer flagella than its wild-type parent and it was chemotactically defective. Wishing to examine whether this phenotype was due to the absence of FRD or whether it was due to the absence of both FRD and SDH, we constructed an frdABCD-deleted strain. The ∆frd mutant strain had the phenotype of the double mutant: fewer flagella (2.3 ± 2.0 per cell compared to 6.0 ± 3.3 in the wild-type parent) and defective in chemotaxis. We are currently characterizing the interaction between FRD and FliG and the effect of fumarate on this interaction. This work may uncover additional parameters that affect flagellar formation and chemotactic response.
66

ALANINE SCANNING MUTAGENESIS OF THE SPO0E PHOSPHATASE DOMAIN INTERACTING WITH THE SPO0A RESPONSE REGULATOR OF BACILLUS SUBTILIS. Alejandra Diaz, Sophie Stephenson and Marta Perego. The Scripps Research Institute; 10550 North Torrey Pines Road; MEM-116; La Jolla, CA 92037
Spo0A~P is the essential response regulator and transcriptional factor for sporulation initiation in Bacillus subtilis. The phosphorylation level of Spo0A in the cell is determined by the opposing activities of sensor histidine kinases and aspartyl phosphate phosphatases acting on the phosphorelay, the multicomponent signal transduction system for sporulation initiation. The Spo0A~P response regulator and transcriptional factor is specifically dephosphorylated by the three members of the Spo0E family of phosphatases, Spo0E, YisI and YnzD. These are small proteins, ranging from 56 or 57 amino acids as for YisI and YnzD respectively, to 85 amino acids of Spo0E, with an overall low level of sequence identity (29-34% of identical residues, 12-15% of conserved substitution). Particularly striking, however, is the sequence conservation of a pentapeptide within the sequence of the three proteins. The conserved sequence is flanked by two conserved amino acid residues upstream and two hydrophobic residues downstream that seem to constitute the signature for Spo0E like phosphatases: TIxxSQELDxHyHy. This motif is indeed highly conserved among Spo0E orthologues identified in other spore-forming Gram-positive bacteria and this suggested a critical role for this sequence in Spo0E activity. We carried out an alanine scanning mutagenesis of the signature sequence of the Spo0E protein. Residues T35, I36, Q40, E41, L42, D43, C44, L45 and I46 of Spo0E were mutated to alanine and the effect of the substitiutions were first tested in an in vivo sporulation assay. We observed that the substitutions of E41 and C44 did not affect Spo0E activity while the T35A and Q40A mutants were partially inactive. Inactive protein resulted from the alanine substitutions of residues I36, S39, L42, D43, L45 and I46. Characterization of the biochemical properties of these mutants is ongoing and the results will be presented.
67

TRYPTOPHAN RESIDUES FLANKING THE SECOND TRANSMEMBRANE HELIX (TM2) SET THE SIGNALING STATE OF THE TAR CHEMORECEPTOR
Roger R. Draheim*, Arjan F. Bormans, Run-zhi Lai, and Michael D. Manson Department of Biology, Texas A&M University, College Station, TX. 77843
The chemoreceptors of Escherichia coli are homodimeric membrane proteins that cluster in patches near the cell poles. They convert environmental stimuli into intracellular signals that control flagellar rotation. The functional domains of a receptor are physically separated by the cell membrane. Chemoeffectors bind to the extracellular (periplasmic) domain, and the cytoplasmic domain mediates signaling and adaptation. These two domains communicate through the second transmembrane helix (TM2) that connects them. In the high-abundance receptors Tar and Tsr, TM2 is flanked by tryptophan residues, which should localize preferentially to the interfacial zone between the polar and hydrophobic layers of the phospholipid bilayer. To investigate the functional significance of the Trp residues that flank TM2 of Tar, we used site-directed mutagenesis to generate th� W192A and W209A substitutions. The W192A protein retains full activity in vivo and in vitro, but it increases the Ki for aspartate in the in vitro assay four fold. The W209A replacement eliminates receptor-mediated stimulation of CheA in vitro, and it leads to an increased level of adaptive methylation in vivo. This phenotype in some respects mimics the changes seen upon binding aspartate. Since the W209A substitution may cause the C-terminus of TM2 to protrude farther into the cytoplasm, these results reinforce the hypothesis that aspartate binding causes a similar displacement. Moving Trp to each position from residue 206 to residue 212 generated a wide variety of Tar signaling states that are generally consistent with the predictions of the piston model of transmembrane signaling. None of these receptors was completely locked in one signaling mode, although most showed pronounced signaling biases. Our findings suggest that the Trp residues flanking TM2, especially Trp-209, are important in setting the baseline activity and ligand sensitivity of the Tar receptor. We also conclude that Tyr-210 residue plays at least an auxiliary role in this control.
68

DEGU, AN ORPHAN RESPONSE REGULATOR, PLAYS A ROLE IN VIRULENCE OF
LISTERIA MONOCYTOGENES
Camille Cyncynatus1, Sarah Dubrac1, Alejandro Toledo Arana2, Olivier Dussurget3, Iñigo Lasa
Uzcudun2, Pascale Cossart3 and Tarek Msadek1
1 Unité de Biologie des Bactéries Pathogènes à Gram Positif, Institut Pasteur, 25, rue du Dr.
Roux, Paris 75015, France. 2 Laboratory of Bacterial Biofilms, Universidad Pública de Navarra, Pamplona, Spain 3 Unité des Interactions Bactéries-Cellules, Institut Pasteur, 25, rue du Dr. Roux, Paris 75015,
France.
Analysis of the recently completed genome of the Gram-positive intracellular pathogen
Listeria monocytogenes, reveals seventeen sets of genes encoding two-component systems
(TCS). All of these genes have now been inactivated and the analysis of the corresponding
mutants is in progress. L. monocytogenes is closely related to the Gram-positive model bacterium
Bacillus subtilis, in which the DegS/DegU TCS plays a central role, controlling degradative
enzyme synthesis, chemotaxis and motility, surfactin and polyketide biosynthesis as well as
competence gene expression (1). Interestingly, although an orthologue of the DegU response
regulator is present in L. monocytogenes, the gene encoding the cognate DegS kinase is absent.
DegU negatively regulates its own synthesis in L. monocytogenes, in contrast to the situation in
B. subtilis. Gel mobility shift and DNaseI footprinting assays were used to show direct binding in
vitro of purified L. monocytogenes DegU to its promoter region. A transcriptome analysis was
performed with cDNA synthesized from total RNA of Listeria monocytogenes EGDe and the
degU mutant, using high-density membranes corresponding to the entire L. monocytogenes
genome and approximately 80 DegU-regulated genes were identified, some of which are known
to play an important role in virulence. In a murine intravenous infection model, an 11-fold
increase in LD50 for the degU mutant was observed when compared to the EGDe parental strain.
References:
1. Msadek, T. (1999) Trends Microbiol. 7, 201-207.
69

NMR EVIDENCE THAT CHEY ACTIVATES BY EQUILIBRIUM-SHIFT
C. M. Dyer*, D. J. Hamel*, R. B. Bourret‡, and F. W. Dahlquist*
*Department of Chemistry and Biochemistry, University of California Santa Barbara, Santa Barbara, CA, 93106; ‡Department of Microbiology and Immunology, University of North Carolina, Chapel Hill, NC 27599-7290
CheY is able to modulate the direction of the flagellar motor by switching between two stable states (apoCheY and CheY~P) in a phosphorylation-dependent manner. Past work demonstrated that overexpressing either wild-type CheY or CheYD13K in E. coli caused an increase in tumbling bias that was phosphorylation-independent. This suggests that the phosphoryl group activates CheY by affecting a pre-existing conformational equilibrium (equilibrium-shift), rather than acting as a prerequisite for conformational change (induced fit). The work of Kern and collegues using NMR indicated that (phosphoryl-independent) activating mutations of the response regulator, NtrC, exist in a rapid equilbrium between inactive and active conformations, and that the relative population of active to inactive conformers qualitatively correlated with the degree of mutant activity.
In this study, we employ the notion that the population of CheYs can be driven towards the inactive conformation by binding the P2 domain of CheA, and towards the active conformation by binding to the FliM peptide. Using NMR techniques to probe the chemical environment of side-chain methyl groups, we show that the chemical shifts for most methyl groups in apoCheY lie roughly in between those of the inactive state and the active state, with a strong bias towards the inactive state. The same trend is evident for BeF3
--CheY alone (no FliM), though the bias in this case for most resonances is strongly towards the activated state. These results suggest that both apoCheY and BeF3
--activated CheY are composed of a combination of inactive and active conformers, supporting the equilibrium-shift view of activation. A notable exception to this trend is Ile 95, which has Cδ1 and Cγ2 methyl peaks movements that are not consistent with a simple shift in the active/inactive equilibrium when BeF3
- and subsequently FliM bind. We speculate that this additional process acting on Ile 95 is the “second-step” in FliM binding – the rotation of the Cδ1 methyl group about the Cβ-Cγ1 bond – which we hypothesized previously from several CheY crystal structures.
Biochemical assays conducted on the activated mutant CheYA113P suggest that this mutant may function by shifting the active/inactive equilibrium in the direction of the active population. Such an explanation is consistent with our NMR studies of CheYA113P, which show that most methyl peaks of CheYA113P lie in between the peaks corresponding to the active and inactive states, with a stronger bias toward the active state than apoCheY. In contrast, a second mutant activated to a similar degree, CheYD13KY106W (CheY**), did not show any evidence of equilibrium-shift activation, with most of the methyl peaks overlapping those of apoCheY. Instead, this mutant has large chemical shift differences for Ile 95 Cδ1 and Cγ2, consistent with our previous notion, motivated by the crystal structures of CheY** and the CheY**-FliM complex, that CheY** activates by affecting the “second-step” in FliM binding. These results, in combination with those of the Kern group, demonstrate that mutations which function by equilibrium-shift are likely a general, though not exclusive, route to activate response regulator proteins.
70

FROM MOLECULES TO BACTERIAL POPULATION BEHAVIOR: A MULTI-SCALES AGENT-BASED SIMULATION Thierry Emonet1, Michael J. North2, Charles M. Macal2, Charles Wickersham1, Philippe Cluzel1
1 The Institute for Biophysical Dynamics and the James Franck Institute, The University of Chicago, Cummings Life Science Center 403, 5640 S. Ellis Av., Chicago, IL 60637.
2 Center for Complex Adaptive Agent Systems Simulation, Decision and Information Sciences Division, Argonne National Laboratory, 9700 S. Cass Ave., Argonne IL 60439.
Most efforts in computational biology have isolated one particular scale of interest, concentrating on either intracellular, cellular or population dynamics. Our long-term goal is to develop a modular computational framework able to cross scales and relate stochastic events at the intracellular level to the behavior of a single cell and ultimately to the dynamics of a population of cells.
We present the preliminary version of AGENTCELL, an agent-based simulation software for the modeling of intracellular processes within cells and of cell behavior within a population. We used AGENTCELL to simulate the chemotactic behavior of more than 1000 independent E. Coli cells as they swim in a 3D environment. Each cell was an independent agent equipped with its own chemotaxis network, motors and flagella. Preliminary results compare favorably with published experimental data on the chemotaxis behavior of both single cells and bacterial populations.
71

pH DEPENDENT CHEMOTACTIC RESPONSE AND THE EFFECT OF LOSS OF cheV2 AND cheV3 ON STOMACH COLONIZATION IN HELICOBACTER PYLORI Amber S. Fair, Karianne Terry, and Karen Ottemann. Department of Environmental Toxicology, UC Santa Cruz, Santa Cruz, CA, USA.
Helicobacter pylori is a gram-negative bacterium that causes ulcers and uses chemotaxis, the biased movement towards or away from chemical stimuli, for colonization of the stomach. We have examined two aspects of H. pylori chemotaxis: taxis to pH and the roles of two putative signal transduction proteins. pH taxis, the movement from one pH to another, was assayed by measuring the number of bacteria attracted into capillary tubes containing medium of different pHs. We found H. pylori was somewhat attracted to the pH 7 capillary, and was moderately repelled from the pH 5 capillary, suggesting that H. pylori may move in response to pH, although these differences were not statistically significant. We also examined the roles of two putative chemotaxis proteins, CheV2 and CheV3, to better understand how the chemotactic signal transduction cascade of H. pylori operates. CheVs are hybrids of two well-characterized chemotaxis signaling proteins, CheW and CheY. H. pylori has three CheV paralogues, and none share more than 40% identity at the amino acid level, suggesting they each have a specific function. Previous work showed that H. pylori lacking cheV1 cannot chemotax, but no phenotype was found for cheV2 and cheV3 mutants (Pittman et al, 2001, Microbiology 147:2493). We have remade the cheV2 and cheV3 mutations in a distinct H. pylori strain and will report on the in vitro behavior of these mutants, and how they behave in a mouse infection model.
72

ROLE OF ELECTROSTATICS IN THE ADAPTATION MECHANISM OF THE ASPARTATE RECEPTOR Diane J. Starrett and Joseph J. Falke* Molecular Biophysics Program and Department of Chemistry and Biochemistry University of Colorado, Boulder CO 80309-0125 USA
The aspartate receptor of the Escherichia coli and Salmonella typhimurium chemotaxis pathway generates a transmembrane signal that regulates the activity of the cytoplasmic kinase CheA. Previous studies have identified a region of the cytoplasmic domain as critical to receptor adaptation and kinase regulation. This region, termed the adaptation subdomain, contains a high density of acidic residues including specific glutamate residues that serve as receptor adaptation sites. However, the mechanism of signal propagation through this region remains poorly understood. The present study uses site-directed mutagenesis to neutralize each acidic residue within the subdomain in order to probe the hypothesis that electrostatics in this region play a significant role in the mechanism of the kinase activation and modulation. Each point mutant was tested for its ability to regulate chemotaxis in vivo and kinase activity in vitro. Four point mutants (D273N, E281Q, D288N and E477Q) were found to super-activate the kinase relative to the wild type receptor, and all four of these kinase-activating substitutions are located along the same inter-subunit interface as the adaptation sites. These activating substitutions retained the wild type ability of the attractant-occupied receptor to inhibit kinase activity. When combined in a quadruple mutant (D273N/E281Q/D288N/E477Q), the four charge-neutralizing substitutions locked the receptor in a kinase-superactivating state that could not be fully inactivated by attractant. Similar lock-on character was observed for a charge reversal substitution, D273R. Together these results implicate the electrostatic interactions at the inter-subunit interface as a major player in signal transduction and kinase regulation. The negative charge in this region destabilizes the local structure in a way that enhances conformational dynamics, as detected by disulfide trapping, and this effect is reversed by charge neutralization of the adaptation sites. Finally, two substitutions (E308Q and E463Q) retained normal kinase activation in vitro but were unable to restore cellular chemotaxis in vivo, suggesting that these sites lie within the docking site of an adaptation enzyme, CheR or CheB. Overall, the present study highlights the importance of electrostatics in signal transduction and regulation of kinase activity by the cytoplasmic domain of the aspartate receptor.
73

TEASING APART THE MECHANISM OF FLHB CLEAVAGE. Hedda Ferris, Mary Kroetz, May Kihara and Robert Macnab Yale University, Department of Molecular Biochemistry and Biophysics New Haven, CT 06520-8114
The bacterial flagellum is a predominantly cell-external supermacromolecular structure whose components are exported by a flagellum-specific export apparatus. One of the export apparatus proteins, FlhB, regulates the substrate-specificity of the entire apparatus – i.e., it has a role in the ordered export of the two main groups of flagellar structural proteins such that the cell-proximal components (rod/hook proteins) are exported before the cell-distal components (filament proteins). The controlled switch between rod/hook protein export and filament protein export is postulated to be mediated by conformational changes in the structure of FlhB. The cytosolic portion of this protein (FlhBC) is consistently and specifically cleaved into two subdomains (FlhBCN and FlhBCC) that remain tightly associated with each other. Point-mutational analysis of the FlhB cleavage site has shown that this cleavage event is necessary for the switch in substrate-specificity to occur, though its mechanism is poorly understood. In an attempt to understand the mechanism and implications of FlhB cleavage, the following avenues are being investigated: 1) testing if, and under what experimental conditions, purified FlhBC cleavage occurs in vitro; 2) based on the assumption that it is highly unlikely that a heterologous host would have a protease that would consistently and specifically cleave a type III flagellar export component, assaying for cleavage of overexpressed Salmonella FlhBC in Saccharomyces cerevisiae.
74

BACTERIAL SIGNAL TRANSDUCTION NETWORK IN A GENOMIC PERSPECTIVE
Michael Y. Galperin
NCBI, NLM, National Institutes of Health, Bethesda, Maryland 20894, USA
The complexity of bacterial signaling systems makes comparative genome analysis a particularly valuable tool for their studies. Careful enumeration of bacterial and archaeal membrane receptors –- histidine kinases, methyl-accepting chemotaxis proteins (MCPs), diguanylate cyclases and c-di-GMP phosphodiesterases, adenylate cyclases, and Ser/Thr protein kinases – revealed some interesting trends in the organization of the signal transduction network in diverse microorganisms. General trends, common for all prokaryotes, include (i) modular structure of signaling proteins; (ii) common organization of signaling components with the flow of information from N-terminal sensory domains to the C-terminal transmitter or signal output domains (N-to-C flow); (iii) use of common conserved sensory domains by different membrane receptors; (iv) ability of some organisms to respond to one environmental signal by activating several regulatory circuits; (v) abundance of intracellular signaling proteins, typically consisting of a PAS or GAF sensor domains and various output domains; (vi) importance of secondary messengers, cAMP and cyclic diguanylate; (vii) crosstalk between components of different signaling pathways. Histidine kinases are by far the predominant type of sensory proteins. The relative abundance of other types of receptors varies in organisms of different phylogenetic lineages, from Ser/Thr protein kinases in actinobacteria and archaea to MCPs in firmicutes to diguanylate cyclases/phosphodiesterases and MCPs in proteobacteria. Escherichia coli turned out to be a surprisingly ‘dumb’ organism that encodes far fewer receptors than some of its close relatives and even its parasite Bdellovibrio bacteriovorus.
75

PPR: A DUAL-CHROMOPHORE, LIGHT-REGULATED HISTIDINE KINASE INTEGRATES SIGNALS FROM BLUE AND RED LIGHT Ulrich K. Genick, Eric C. Fontano, Michael DiPrima, Linda Hofmann, Stuart Endo-Streeter Brandeis University; Department of Biochemistry; Waltham, MA, 02453, USA
The bacterial photoreceptor PPR represents a fusion of two classes of bacterial photoreceptors. The C-terminal portion shows the classic domain structure of a bacterial phytochrome including a light-sensing domain carrying a tetra-pyrole chromophore as well as a his-kinase domain. Fused to the N-terminus of this pytochrome-related region is a PYP domain that carries a para-coumaric acid chromophore and confers sensitivity to blue light.
PYP proteins usually exist as stand-alone photoreceptors. How was PPR able to co-opt this PYP domain to regulate its kinase domain?
We use X-ray crystallography, time-resolved optical spectroscopy and protein engineering to study how information propagates from the light-sensing domains to the kinase domain and how signals from the two input channels (red light and blue light) are integrated into a single output channel (kinase activity) at the molecular level.
76

THE CHEMOTAXIS PROTEIN CHED AND ITS SITES OF CHEMORECEPTOR MODIFICATION
George D. Glekas and George W. Ordal Department of Biochemistry, University of Illinois, Urbana-Champaign, 190 Medical Sciences Bldg., 506 S. Mathews Ave., Urbana, IL 61801 The two-component chemotaxis system provides flagellated bacteria with the ability to move towards more favorable environments. At the heart of this system are transmembrane chemoreceptors that sense these environmental cues and transmit the signal to the histidine kinase CheA. Adaptation to the environment is achieved by reversible methylation of conserved glutamate residues on the chemoreceptors by the proteins CheR and CheB. Occasionally, these glutamate residues are encoded as glutamines, and, in Escherichia coli, are deamidated by CheB. Previously, work in our lab had shown that in Bacillus subtilis, CheD, and not CheB, is responsible for this deamidation reaction in the case of residues 593 and 594 in McpA. Using HPLC analysis of tryptic peptides, we have now identified a third site, at residue 586, which does not strictly conform to the consensus bacterial methylation sequence, the first such example identified to date, and we can also say that there are no other modifications.
Furthermore, we previously had tethered cell and capillary assay data that showed that B. subtilis cheD was completely unresponsive to proline, whose taxis is mediated by the chemoreceptor McpC, and only about half the cells responded to asparagine, whose taxis is mediated by McpB. Taxis to asparagine was also very impaired in the capillary assay. However, in these experiments all ten receptors were present. In a cheD mutant lacking all receptors except McpB or McpC, taxis in capillary assay to asparagine was nearly normal, although taxis toward proline was still very poor. Thus, the presence of other receptors significantly impaired taxis mediated by McpB. This result implies that receptor-receptor interactions, which normally are thought to enhance taxis sensitivity, can actually have very deleterious effects under certain circumstances.
We are currently working to identify CheD’s deamidation sites on both McpB and McpC to further elucidate the differences between these 2 chemoreceptors in their dependence on CheD. Mutational analysis of these CheD-mediated deamidation sites is also being used to analyze their importance.
77

EPR STUDIES OF SPIN-LABELED CHEA CYSTEINE MUTANTS#
Susy L. Gloor and Joseph J. Falke Department of Chemistry and Biochemistry, University of Colorado, Boulder, Campus Box 215, Boulder, CO, 80309
The goal of this work is to observe structural changes in the bacterial histidine kinase,
CheA, upon regulation by the chemoreceptor. CheA consists of five modular domains and each
serves a different purpose in regulation and activity of the kinase. It is thought that one or more
of these domains undergo conformational changes depending on the activation state of the
kinase. CheA forms a signaling complex with the coupling protein, CheW, and the
chemoreceptor. It is thought that CheA interacts with the chemoreceptor both directly and
through CheW. This study looks to observe structural changes in CheA and to locate regions of
the kinase that directly interact with other proteins of the signaling complex using electron
paramagnetic resonance (EPR) analysis: 1) upon binding to CheW in solution, 2) upon docking
of CheA and CheW to chemoreceptors in membranes, and 3) upon regulation by
chemoattractant. EPR analysis involves coupling a nitroxide radical containing spin-label to
engineered cysteine residues on the surface of the protein. This type of study can give
information about the motion of the spin label. EPR spectra are affected by both local tumbling
of the spin-label and by global tumbling of the whole protein. It is expected that conformational
and motional changes in CheA will give rise to changes in spin-label tumbling rates. By looking
at positions on each of CheA’s five domains, we seek to understand how the different domains
are affected when interacting with other chemotaxis components and during receptor-mediated
kinase regulation. The initial results of these EPR studies will be reported.
78

STRUCTURAL AND BIOCHEMICAL STUDIES OF CHEMORECEPTORS FROM THERMOTOGA MARITIMA Gabriela Gonzalez-Bonet, Abiola M. Pollard, Sang-Youn Park, Jawahar Sudhamsu, Alexandrine M. Bilwes and Brian R. Crane Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14850
Bacterial chemotaxis, the process by which bacteria travel towards an attractant or away
from a repellent, manifests remarkable sensitivity, gain and feedback control. Both the excitation and adaptation responses observed during chemotaxis depend on modulating the activity of the histidine kinase CheA. The rate of the CheA autophosphorylation depends on complexes formed with other chemotaxis proteins, which include cytoplasmic domains of chemoreceptors. Sensitivity of the receptors is regulated by their methylation state. We have cloned and expressed cytoplasmic fragments of transmembrane methyl-accepting chemoreceptor proteins (MCPs) from the thermophilic bacterium, Thermotoga maritima. Experiments were performed to evaluate the effects of these receptor fragments and other chemotaxis proteins (CheY, CheB, CheW) on T. maritima CheA activity. We have also generated site-directed mutants of receptor fragments designed to mimic different states of methylation. Two receptor fragments have been crystallized and we are currently determining their crystal structures by X-ray diffraction methods and their solution structures by small-angle X-ray scattering (SAXS).
79

ANALYSIS OF THE INTERACTIONS BETWEEN THE FLAGELLAR SWITCH COMPLEX AND TYPE III EXPORT COMPONENTS IN SALMONELLA Bertha González-Pedrajo1,2, Tohru Minamino3, John S. Van Arnam1, May Kihara1, Gillian Fraser1,4, Keiichi Namba3,5 and Robert M. Macnab1
1Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, CT, 06520-8114. 2Departamento de Genética Molecular, Instituto de Fisiología Celular, UNAM. Ap. Postal 70-243, México, D.F. 04510. 3Dynamic Nanomachine Project, ICORP, JST, 1-3 Yamadaoka, Suita, Osaka 565-0871, Japan. 4University of Cambridge, Department of Pathology, Tennis Court Road, Cambridge, CB2 1QP, UK. 5Graduate School of Frontier Biosciences, Osaka University 1-3 Yamadaoka, Suita, Osaka, 565-0871, Japan.
The cytoplasmic or C ring of the flagellum contains the FliM and FliN proteins, which together with FliG comprises the rotor/switch structure of the flagellar motor. All three proteins are essential for flagellar assembly and all are involved in controlling motor switching between clockwise and counterclockwise rotation. FliG also functions directly in torque generation, and FliM is the target for the output of the sensory transduction signal. The precise role of FliN remains mysterious; however, it has been implicated in flagellar protein export. The export apparatus component FliI is an ATPase, whose enzymatic activity is essential for flagellar assembly. FliH functions as a negative regulator of FliI.
With the aim of understanding the specific role of FliN, we have further investigated its interactions with export components in vitro using an affinity chromatography co-purification strategy and gel filtration chromatography. We show that FliN associates with FliH, and we have isolated multi-component complexes: FliN-FliM-FliH, FliN-FliM-FliH-FliI, and FliG-FliM-FliN-FliH-FliI. The FliN-FliM-FliH-FliI complex is stable during size exclusion chromatography.
We have also analyzed FliN using 10-amino-acid deletions throughout its C-terminal region, and looked at the physiological consequences in terms of flagellar assembly, binding to FliM and to FliH. A dissection of the FliN protein using the scanning deletion approach will be presented.
80

A SIGNALING NETWORK RECIPROCALLY REGULATES GENES ASSOCIATED WITH
ACUTE INFECTION AND CHRONIC PERSISTENCE IN PSEUDOMONAS AERUGINOSA
Andrew L. Goodman, Bridget Kulasekara, Arne Rietsch, Dana Boyd, Roger Smith, and Stephen
Lory
Department of Microbiology and Molecular Genetics, Harvard Medical School
200 Longwood Ave., Boston, MA 02115
The opportunistic pathogen Pseudomonas aeruginosa causes a variety of acute and
chronic infections. We identified a gene whose inactivation results in attenuation of virulence
due to premature activation of genes involved in biofilm formation and coordinate repression of
genes required for initial colonization. This gene, retS, encodes a hybrid sensor kinase/response
regulator with an unconventional arrangement of functional domains. Genomewide
transcriptional profiling indicates that the retS gene is required for expression of the Type III
secretion system and other virulence factors and for repression of genes responsible for
exopolysaccharide components of the P. aeruginosa biofilm matrix. These disparate phenotypes
are suppressed by transposon insertions in genes encoding the GacS/GacA/rsmZ signal
transduction pathway, a highly conserved system involved in the control of diverse adaptive
functions. This study defines RetS as a pleiotropic regulator of multiple virulence phenotypes
that orchestrates genes required for acute infection and genes associated with chronic
persistence.
81

SIGNAL LOGIC OF THE AEROTAXIS TRANSDUCER AER Khoosheh K. Gosink and John S. Parkinson Biology Department, University of Utah, Salt Lake City, Utah 84112
The Aer protein of E. coli promotes cell movement toward optimal concentrations of oxygen and other electron acceptors. The C-terminal portion of Aer resembles the signaling domain of conventional chemoreceptors and controls locomotor behavior by modulating the phosphorylation activity of CheA. In cells devoid of other receptors, wild-type Aer elicits episodes of CW flagellar rotation that produce random walk swimming movements. Oxygen increases suppress CW rotation; oxygen decreases enhance CW rotation. Aer is thought to sense aerotactic stimuli via redox changes in an FAD prosthetic group associated with its N-terminal PAS domain. If so, do the control signals from the PAS sensory domain activate or suppress generation of CW flagellar signals by the Aer output domain? To determine the sign of the PAS output control signal, we examined the signaling properties of Aer molecules with different PAS lesions. Aer∆[1-111], lacking nearly the entire PAS domain, was expressed at wild-type levels and seemed to be normally associated with the cytoplasmic membrane, but did not produce CW output signals, as determined by flagellar rotation and pseudotaxis assays. This suggests that the PAS domain in wild-type Aer actively elicits CW signal output. Consistent with this finding, most aerotaxis-defective PAS missense mutations, exemplified by Y93H, also reduced or abrogated CW signal output. In contrast, PAS mutations that enhance CW rotation, e.g., M112V, were relatively rare and most likely represent gain-of-function lesions.
The HAMP domain adjacent to the Aer signaling domain is the probable target of the CW-promoting PAS signals. Most aerotaxis-defective HAMP mutations cause reduced CW behavior, consistent with a defect in receiving or relaying PAS input signals. However, a small HAMP segment, residues 221-228, gives rise to CW-enhancing mutations, which might augment or bypass the PAS input signals. When combined with CCW-biased PAS mutations (∆[1-111] or Y93H), these HAMP mutations (K221E, N228S) no longer caused CW behavior, indicating that PAS input was required for their CW-enhancing properties. Consistent with this conclusion, we found that the input/output signaling path in CW-biased HAMP mutants remained intact: (i) The mutant proteins promoted normal aerotactic responses in cells containing methyl-accepting chemoreceptors (MCPs), which probably offset the excessive CW bias. Functional rescue of Aer mutants may require direct interaction with the helping MCP molecules and in vivo crosslinking studies showed that such interactions occur, most likely within mixed trimers of dimers. (ii) CW-biased HAMP mutations functioned normally in chimeric Aer molecules that had a methylation-tunable MCP signaling domain. (iii) CW-biased HAMP mutations were phenotypically suppressed by second-site mutations in the PAS domain or the downstream signaling domain.
We conclude that the Aer PAS domain is required for CW signal production by wild-type Aer molecules and that aerotactic stimuli that augment CCW flagellar rotation do so by turning off or suppressing CW-promoting interactions between the PAS domain and downstream portions of the Aer molecule.
82

EXPRESSION STUDIES OF THE MAJOR CHEMOTAXIS LOCI OF RHODOBACTER SPHAEROIDES Marcus Gould, Angela C. Martin, Elaine D. Byles and Judith P. Armitage Microbiology Unit, Department of Biochemistry, University of Oxford, South Parks Road, Oxford. OX1 3QU U.K.
Rhodobacter sphaeroides is a purple, non-sulphur photosynthetic bacterium that can grow under a wide range of conditions. It has a complex chemosensory system with multiple homologues of the Che proteins encoded by genes organised in three operons and at other unlinked loci. cheOp2 and cheOp3 are essential for chemosensing under laboratory conditions and are the main subjects of this study.
Differential expression of the Che proteins encoded in these operons was shown in cells grown in a variety of conditions that included aerobic heterotrophic and anaerobic photoheterotrophic using different light intensities. The copy numbers of these Che proteins were accurately determined by quantitative Western blot.
Transcriptional start sites for cheOp2 and cheOp3 were determined by primer extension studies. Promoter regions were identified and confirmed by the construction and analysis of appropriate deletion mutants and the presence or absence of putative internal promoters ascertained.
Translational fusions of putative promoter regions with a promoterless lacZ gene were constructed in the broad host range vector pUI523A and the expression of the operons compared under a variety of environmental conditions. In this study we show that expression of chemosensing components is tightly regulated in response to growth conditions and metabolic pathways. A number of transcriptional regulators and sigma factors are implicated in this regulation and these will be discussed.
83

FLAGELLUM BIOGENESIS AT THE CELL POLE OF VIBRIO CHOLERAE
Johnathan C. D. Green and Gillian M. Fraser.
University of Cambridge, Department of Pathology, Tennis Court, Cambridge, CB2 1QP, UK.
Vibrio cholerae swims by rotating a single flagellum located at its old cell pole.
Flagellum biogenesis requires accurate protein targeting to the appropriate pole, and must be
closely coupled to cell division to ensure that only one flagellum is built per cell. The
mechanisms underlying flagellum polar localization and coupling of flagellum biogenesis to cell
division are not known. Our current studies focus on flhF, flhG, and orf1, novel flagellar genes
implicated in flagellum placement and regulation of flagellar gene expression. FlhF is a putative
GTPase with 30% identity to the bacterial signal recognition particle (SRP) receptor, FtsY. FlhG
has 30% identity to MinD, an ATPase that oscillates between cell poles and determines the site
of cell division. Orf1 has 37% to the Bacillus subtilis ParA-like ATPase, Soj.
We have constructed chromosomal null mutations in V. cholerae flhF, flhG and orf1, and
determined their effect on motility and flagellum localization and number. In parallel, we have
overexpressed the genes in wild type V. cholerae to assess negative multicopy effects on
flagellum biogenesis and/or function. We have investigated flagellar protein targeting by
examining the localization of functional GFP fusions to flagellar structural components in wild
type V. cholerae and the null mutants. Using similar fluorescence microscopy techniques in live
cells we have determined the localization of functional GFP fusions to FlhF, FlhG and Orf1. To
establish whether NTPase activity is required for FlhF, FlhG, and Orf1 function we have
generated a range of point mutants encoding proteins with single amino acid substitutions in the
GTPase/ATPase active sites, and have screened these variants for function in the null mutants.
We have also initiated studies investigating the NTPase activities of these proteins in vitro. Our
results indicate that FlhF, FlhG and Orf1 are required for efficient biogenesis and/or function of
the V. cholerae polar flagellum.
84

THE STRUCTURES OF T87I PHOSPHONO-CHEY AND T87I Y106W AND THEIR BINDING AFFINITIES TO THE FLIM AND CHEZ PEPTIDES
The structures of T87I Phosphono-CheY and T87I/Y106W Phosphono-CheY and their binding affinities to the FliM and CheZ peptides.
Andrew Mesecar, Eric S. Casper, Ken McAdams, and Christopher J. Halkides Department of Medicinal Chemistry and Pharmacognosy, University of Illinois at Chicago and Department of Chemistry, University of North Carolina at Wilmington
The structure of T87I phosphono-CheY has been solved to 1.8 Å resolution using
molecular replacement. Phosphono-CheY proteins bear a D57C mutation, and have a
phosphonomethyl group on the sulfur atom, creating a structural mimic of the phosphoryl group
attached to Asp57 in the active (signaling) form of CheY. The purpose of this work is to
correlate the functional changes brought about by mutations with the structure of CheY in its
active form. The I87 side-chain forces the side chain of Y106, a residue known to be critical for
signaling, into a more solvent accessible conformation. The dissociation constants of the FliM
peptide (residues 1-16 of FliM, a protein of the flagellar motor) to phosphono-CheY, T87I
phosphono-CheY, and T87I Y106W phosphono-CheY were determined by fluorescence
quenching of W58. The FliM peptide binds to phosphono-CheY with a dissociation constant of
38 µM, almost as well as the peptide binds to CheY~P. However, this peptide binds to T87I
phosphono-CheY and T87I Y106W weakly or not at all. T87I CheY is phosphorylatable but
nonsignaling in vivo, and these results suggest that the nonsignaling phenotype is the result of a
failure of T87I CheY~P to bind to FliM tightly in vivo. These results are similar to the binding
of a peptide derived from the phosphatase CheZ, to the three variants of phosphono-CheY.
85

CONSTRUCTION OF A TREPONEMA DENTICOLA YEAST TWO-HYBRID GENOMIC LIBRARY FOR FUNCTIONAL ANALYSIS OF CHEMOTAXIS PROTEIN INTERACTIONS Dawn A. Hower*1, Rebeca Lopez, Renate Lux2, and Wenyuan Shi1,2
1 Molecular Biology Institute, University of California, Los Angeles, CA 90095 2 School of Dentistry, University of California, Los Angeles, CA 90095
Treponema denticola is an anaerobic spirochete implicated in periodontal disease. Signaling cascades, functioning via protein-protein interactions, are likely important in its pathogenicity. The chemotaxis pathway is one such signal transduction system implicated in the virulence of this motile pathogenic bacterium. The recent completion of the T. denticola genome sequencing project has revealed several unusual features of its chemotaxis system. These include a CheX protein, a CheR-like domain fused to the C-terminus of CheW, a second CheW homolog (CheW-2), and twenty putative methyl-accepting chemotaxis proteins (MCPs), some of which contain a protease-like domain at their N-termini. Experimental evidence must now be provided to further characterize these putative proteins and to investigate their roles in T. denticola chemotaxis. Thus, a T. denticola genomic library has been constructed for use in the yeast two-hybrid system, to provide a functional genomics tool for identifying new interacting proteins and to further examine protein-protein interactions in the chemotaxis system. The T. denticola genomic library was created by digesting genomic DNA and ligating genomic fragments into the yeast two-hybrid pGAD “prey” vector. The library appears to be complete and representative, covering the entire genome well over three times. Restriction digest analysis of isolated library pGAD plasmids reveals that approximately 90% of the vectors contain genomic library fragments. The genomic library was tested for functionality in the yeast two-hybrid system using established protein interactions in the chemotaxis pathway. Analysis of the completed genome of T. denticola has revealed new potential chemotaxis proteins, including twenty putative MCPs. The chemotaxis protein CheW is known to interact in a ternary complex with the chemosensory MCPs, as well as with the kinase CheA. Using CheW as bait in the yeast two-hybrid system, one previously known MCP and twelve new putative MCPs were pulled out from the T. denticola genomic library, providing the first experimental evidence for those twelve as functional MCPs. The known protein-protein interaction between CheW and CheA was also confirmed through this assay. The seven other putative T. denticola MCPs were not pulled out from the initial yeast two-hybrid screening experiments, although they were all shown to be present in the library. The highly conserved CheW-interaction domains of these seven MCPs were individually cloned and tested in one-on-one yeast two-hybrid experiments with CheW to determine if they are indeed likely to be functional MCPs.
As shown by these chemotaxis protein studies, the T. denticola yeast two-hybrid genomic library is proving to be a useful tool to discover unknown protein interactions in order to better understand the pathogenesis of this motile spirochete. Other potential spirochetal chemotaxis proteins are now being investigated in this system to determine their protein interactions and potential roles in chemotaxis signal transduction. These studies will supply essential functional support for genome sequence data in order to provide a more comprehensive view of the entire chemotaxis system of T. denticola and other spirochetes.
86

CROSS-TALK BETWEEN TYPE IV PILI AND TYPE III SECRETION IN PSEUDOMONAS AERUGINOSA
T. Jakobsen1, C. B. Whitchurch2, J. Bertrand3, N. Ghori4, J. N. Engel3
1Max Planck Institut für Terrestrische Mikrobiologie, 35043 Marburg, Germany 2Monash University-Clayton Campus, Microbiology Department, VIC 3800, Australia 3University of California, San Francisco, CA 94143-0654, USA 4Stanford University, Stanford, CA 94305, USA
Type IV pili (TFP) and the type III secretion system (TTSS) have been shown to be important virulence factors in vitro and in vivo in Pseudomonas aeruginosa (PA). TFP are the major adhesins of PA and are required for a form of solid surface translocation termed twitching motility. We have previously shown that several mutants of PA103 deficient in the assembly or function of TFP are defective for type III secretion mediated functions, suggesting that TFP are required for type III secretion. To test whether this is solely due to the adherence defect that the absence of pili is conferring, we now show that a pilin-deficient strain (PA103pilA) of PA103 is defective in twitching motility, adhesion, and translocation of type III secreted products. Adhesion to host cells could be fully restored by expression in trans of the pilA gene or of two heterologous adhesins, the pH6 antigen from Yersinia pseudotuberculosis or the F1845 fibril from diffuse adhering Escherichia coli. However, only PilA or the pH6 antigen could restore effector translocation, despite all three adhesins being surface expressed on P. aeruginosa. Unexpectedly, the pilA mutant was also defective for the production and secretion of type III effectors upon co-cultivation with host cells. Production and secretion could be restored by expression of pilA or the pH6 antigen, but not F1845, suggesting that TFP or other related surface adhesins can transmit signals that permit Type III secretion. We are currently focusing on the signal transduction cascade by which TFP transmits information to the type III secretion system. Together our results suggest the novel finding that molecular cross-talk occurs between these two surface structures that are critical in the virulence of this important human pathogen.
87

IDENTIFICATION OF A VIBRIO CHOLERA CHEMORECEPTOR-LIKE PROTEIN INVOLVED IN TAXIS TO SERINE AND OTHER AMINO ACIDS Yasuaki Ito1, Akihiro Hyakutake1, Noriko Nishioka1, Susan M. Butler2, Andrew Camilli2, Michio Homma1 and Ikuro Kawagishi1
1Division of Biological Science, Graduate School of Science, Nagoya University; 2Department of Molecular Biology and Microbiology, Tufts University School of Medicine, 150 Harrison Avenue, Jaharis 428, Boston, MA 02111, U.S.A.
Some chemotaxis genes of Vibrio cholerae have been implicated in pathogenicity. However, their precise roles have been eluded due to the lack of basic knowledge of the chemotaxis itself. Here we first screened for chemicals that attract a classical biotype strain O395N1. Ten amino acids, including serine, arginine and proline, were found to act as attractants. Next we wanted to identify chemoreceptor(s) responsible for amino acid taxis. V. cholerae has 45 proteins homologous to chemoreceptors (hereafter referred to as MCP-like proteins or MLPs). Based on the sequence comparison with MLPs from other bacterial species, we speculated that Mlp24 (also known as McpX), which is involved in the production of CTX upon infection in an El Tor biotype strain, may mediate attractant response(s) to amino acid(s). We therefore examined amino acid taxis of an mlp24 deletion derivative (AC-V1400) of an El Tor biotype strain (AC-V66). We found that (1) the mlp24-lacking mutant showed weaker responses to serine, arginine and proline than the parent strain; (2) the mlp24 gene form O395N1 partially complemented the defect of serine taxis in AC-V1400; and (3) the Mlp24 protein became more methylated upon addition of serine, arginine or proline. Therefore, we conclude that Mlp24 mediates taxis to serine, arginine and proline. However, taxis to serine may be mediated also by additional MLP(s), since it was not completely abolished by the deletion of the mlp24 gene.
88

STUDIES OF THE MOTA/MOTB STATOR COMPLEX AND PERIPLASMIC FRAGMENTS OF MOTB FROM SALMONELLA TYPHYMURIUM Seiji Kojima1, Tohru Minamino1 and Keiichi Namba1,2
1Dynamic Nanomachine Project, ICORP, JST, 2Graduate School of Frontier Biosciences, Osaka University, 1-3 Yamadaoka, Suita, Osaka, JAPAN 565-0871. MotA and MotB are integral membrane proteins that constitute the stator of the proton-
driven bacterial flagellar motor. They form a complex that conducts protons and couples proton
flow to motor rotation. A hypothesis of the rotation has been proposed, based on mutational and
biochemical studies, that protonation of critical Asp 32 of MotB triggers conformational changes
in the stator that act on the rotor to drive rotation. To confirm this hypothesis, we need to know
more about molecular and biochemical properties of the MotA/B complex. The aim of this study
is to establish an in vitro assay system, primarily to examine the proton-conducting activity
through the MotA/B complex. Then we ask questions, for example, whether there is a gate in
this proton channel complex and if so, how it affects the conformational changes in the stator. A
His tag was attached to the carboxyl terminus of MotA or MotB of Salmonella typhimurium to
facilitate purification of the complex, and detergents such as dodecylphosphocholine or sucrose
monolaurate were used for its efficient extraction. Currently we are trying to improve our
purification protocol to achieve better yield.
We are also interested in the mechanism of incorporation of the stator into the appropriate
position in the motor. However, little is known about the periplasmic domain of MotB, which is
believed to be responsible for association with peptidoglycan layer to anchor the stator around
the rotor. To investigate the role of this domain, we constructed several MotB fragments
encoding for different sizes of periplasmic domain. They were fused to a PelB leader sequence
at their amino termini to direct their periplasmic localization. Preliminary results showed that
one fragment, consisting of residues 55-309, was detected in the periplasmic fraction and
exhibited strong negative dominance in motility, suggesting that it was exported to the
periplasmic space and titrate MotB binding sites out from the motor. Further details of our
studies will be discussed.
89

REGULATION OF THE POLAR-FLAGELLAR NUMBER BY FLHF AND FLHG IN VIBRIO ALGINOLYTICUS Akiko Kusumoto1, Kenji Kamisaka2, Toshiharu Yakushi1, Hiroyuki Terashima1, Akari Shinohara3, S. Aizawa3 and Michio Homma1
1. Division of Biological Science, Graduate School of Science, Nagoya University, Chikusa,
Nagoya 464-8602, JAPAN 2. Department of Information and Biological Science, Graduate School of Natural Sciences,
Nagoya City University, Mizuho, Nagoya 467-8501, JAPAN 3. CREST, JST, JAPAN
The number and location of bacterial flagella vary among species. E. coli, S. typhimurium and B. subtilis have peritrichous flagella. V. cholerae, C. creescentus and P. aeruginosa have a single polar flagellum. V. fischeri, H. pylori and P. putida have multipolar flagella. Vibrio alginolyticus has two types of flagella: a single polar flagellum (Pof) driven by sodium ions is located at the cell pole and several lateral flagella (Laf) driven by protons are located around cell. The molecular mechanism for regulating the number and placement of the polar flagellum of V. alginolyticus is largely unknown. Cells possessing the polar flagellum but lacking lateral flagella (the Pof+ Laf- cells) were mutagenized with EMS and mutants were selected on the basis of reduced swarming ability on soft agar plates. Among these mutants, we found two mutants that possessed multiple Pofs, NMB155 and KK148. In Pseudomonas, FlhF and FleN are involved in regulating flagellar placement and flagellar number, respectively. We cloned the homologous genes of V. alginolyticus, flhF and flhG (the fleN homolog). As in Pseudomonas, the flhG gene is located immediately downstream of flhF. FlhF is predicted to be a soluble 57kDa protein with an ATP-binding motif at the N terminus. FlhG is predicted to be a soluble 32 kDa protein with a GTP-binding motif at the C terminus. NMB155 have no mutation in either flhF or flhG; cells expressing transgenic flhG did not recover swarming ability and remained multiflagellated. KK148 had a nonsense mutation in the flhG gene; cells expressing transgenic flhG recovered normal swarming ability and had a reduced number of the polar flagella. The KK148 cells expressing both flhF and flhG had the fewer polar flagella and showed a better swarming ability than the KK148 cells expressing flhG alone. In the parental Pof+ Laf- cells, overexpression of flhG decreased the number of polar flagella, and overexpression of flhF increased the number of polar flagella. These results suggest that the polar flagellar number is negatively regulated by FlhG and positively regulated by FlhF. FlhF and FlhG may function in a coordinated manner to regulate Pof number more precisely.
90

FUNCTIONAL INTERACTIONS AMONG BACTERIAL CHEMORECEPTORS Run-zhi Lai and Michael D. Manson Department of Biology, Texas A&M University, College Station, TX 77843
There are four chemoreceptors in Escherichia coli: Tar, whose primary attractant ligands are aspartate and maltose; Tsr, whose primary attractant ligand is serine; Trg, which senses ribose, galactose and glucose; and Tap, which mediates responses to a wide variety of of di- and tripeptides as attractants. The sugars and peptides are recognized by periplasmic substrate-binding proteins, which interact with the chemoreceptors in their closed, ligand-bound form. Amino acids, in contrast, bind directly at the subunit interface of the homodimeric receptors. Within the cytoplasm, chemoreceptors associate with the coupling factor CheW and the autophosphorylating CheA histidine-protein kinase. The activity of CheA is enhanced two orders of magnitude by association with Tar and Tsr. Attractants can inhibit the stimulation of CheA activity to a level below its unstimulated baseline level in a process known as squelching. The receptor dimers can form trimeric complexes (trimers of dimers), and these trimers can be joined by a bridge thought to consist of a CheW monomer, a CheA dimer, and a second CheW monomer. The primary contacts between the receptor trimers and the bridge are via CheW interacting at a dimer-dimer interface at the cytoplasmic tip of the trimer. It has been proposed that trimers of receptor dimers may be joined by CheW-CheA dimer-CheW links to form an extended hexagonal lattice that may be the structural basis of the chemoreceptor patches seen in E. coli. Finally, recent genetic experiments imply that trimers can contain both Tsr and Tar homodimers, and it seems likely that trimers composed of different receptors can be joined by CheW-CheA bridges. The collective biochemical, genetic and structural data raise the strong possibility that communication among receptors may be involved in signal amplification and crosstalk among receptors and provide one possible mechanism for signaling by the low-abundance receptors Trg and Tap. We have examined functional interactions among chemoreceptors using a receptor coupled in vitro phosphorylation assay. Here we show that there is significant receptor density effect present in the same kind receptors. And there is synergism present between Tar and Tsr. The synergism shows a pattern that fits a six order polynomial curve. The synergy pattern between Tar and Tsr indicates that receptor signaling complex forming is a not random event. We propose a model based on receptor affinity and activity that explains our results. Our results suggest a simple signaling principle that might be widely used in biological signaling systems.
91

CHEMORECEPTORS WITH CARBOXYL TERMINAL EXTENSIONS: DIFFERENTIAL EFFECTS ON THE TWO CLASSES OF ADAPTATIONAL MODIFICATIONS
Wing-Cheung Lai, and Gerald L. Hazelbauer
Department of Biochemistry, University of Missouri-Columbia, 117 Schweitzer Hall, Columbia, MO 65211
Sensory adaptation in bacterial chemotaxis is mediated by covalent modification of chemoreceptors. Specific glutamyl residues are methylated and demethylated in reactions catalyzed by methyltransferase CheR and methylesterase CheB. In the well-characterized chemosensory systems of Escherichia coli and Salmonella, efficient adaptational modification by either enzyme is dependent on a conserved pentapeptide sequence, NWETF or NWESF, present at the extreme carboxyl terminus of high-abundance chemoreceptors, a position distant from sites of modification. The pentapeptide enhances methyltransferase activity by serving as a docking site that increases enzyme concentration near the methyl-accepting sites. In contrast, interaction of pentapeptide and CheB enhances methylesterase activity allosterically. To what extent is location at the extreme carboxyl terminus important for the action of the pentapeptide in enhancing adaptational modification? Is carboxyl-terminal location equally important for enhancement of both enzyme activities? To address these questions, we created several forms of high-abundance receptors Tsr and Tar carrying one or more additional amino acids extending beyond their natural carboxyl termini and tested their propensities for methylation, demethylation and deamidation both in vivo and in vitro. In addition, we assessed the ability of altered receptors to mediate chemotaxis. In vitro carboxyl-terminal extensions significantly reduced the ability of the methylesterase/deamidase to modify high-abundance receptors but had a modest effect on the ability of CheR to methylate the same altered receptors. Interestingly, in intact cells receptors with carboxyl-terminal extensions appeared to mediate taxis as effectively as the wild-type forms and to be modified normally, illustrating the robustness of the chemosensory system.
92

THE TWO-COMPONENT SYSTEM HK1-RR1 INFLUENCES THE RECOMBINATION RATE IN BORRELIA BURGDORFERI.
CHUNHAO LI1, CHAD BROOKS 2, DARRIN AKINS 2, NYLES CHARON1
1. Dept. Microbiology, Immunology and Cell Biology, West Virginia University 2. Dept. Microbiology and Immunology, University of Oklahoma
Bacterial two-component systems typically consist of a histidine kinase sensor (HK) and a cognate response regulator (RR). These signaling pathways play a major role in the response of bacteria to both external and internal signals. Besides the multiple copies of chemotaxis genes cheA and cheY, there are two homologs of these regulatory two-component systems present in the genome of the Lyme disease spirochete Borrelia burgdorferi. These genes are HK1 (bb0420)-RR1 (bb419) and HK2 (bb0764)-RR2 (bb0763). Recently, RR2 was shown to act as an enhancer to control rpoS expression through the alternative sigma factor RpoN. HK2 lacks a trans-membrane domain and it possibly senses internal signals. In contrast, HK1 has an apparent trans-membrane domain that may sense environmental signals. However, the functions of HK1-RR1 are still unknown.
Sequence analysis indicates that HK1 is a typical histidine kinase: there are two trans-membrane regions that locate at the N-terminus (6~26 aa) and the central region (716~737aa). Between these two regions, there is an extracellular loop that contains a SBP-bac-3 domain (bacterial periplasmic substrate-binding proteins). HK1 also contains a well -conserved signal transduction histidine kinase-like ATPase domain (771~994aa), and a signal receiver domain (1190~1250aa). Consistently, RR1 contains a well -conserved CheY-like domain at the N-terminus, and a GGDEF domain at the C-terminus. The GGDEF domain is found in a variety of bacteria. The function of this domain is unknown. In addition, a DNA-binding domain (H-T-H) was found at the C-terminus (1303~1322aa) of HK1; typically this domain locates at RR in other bacteria. These results indicate that HK1-RR1 forms a signaling pathway that possibly mediates the bacterial responses to different environmental signals.
To further characterize the role of HK1-RR1 in B. burgdorferi, HK1was inactivated by allelic exchange mutagenesis. HK1 mutant had a higher recombinant rate (increase about 5-fold) than that of the wild-type. Microarray analysis indicated that inactivation of HK1 influenced the expression of a variety of genes. Among these genes, one encodes a DNA-binding protein HbbU, and another encodes a putative DNA mismatch repair protein. HbbU is a DNA-binding protein that is a homolog of IHF in Escherichia coli. Previous studies have shown that HbbU binds to the specific region on the chromosome, and changes the topology of DNA in B. burgdorferi.
Taken together, these results suggest that the two-component system HK1-RR1 may have an important role with respect to gene regulation and DNA recombination in B. burgdorferi. Our current hypothesis is that the HK1-RR1regulatory pathway increases the recombination rate through mediating the expression of HbbU and other proteins related to recombination.
93

BIOCHEMICAL ANALYSIS OF THE HELICOBACTER PYLORI FLAGELLAR SWITCH Andrew C. Lowenthal and Karen M. Ottemann Department of Molecular and Cell Biology, University of California at Santa Cruz 1156 High Street Santa Cruz, CA 95060 Helicobacter pylori is a gram-negative spiral shaped bacterium which is responsible for many gastric and duodenal ulcers, and is the only bacterium currently classified as a carcinogen by the WHO. Motility and chemotaxis are needed for full mammalian colonization by H. pylori. Thus, an understanding of the chemotactic pathway could lead to a therapeutic treatment. Flagellated bacteria chemotax using membrane bound chemosensors, which relay signals through several other proteins (designated Che proteins) to the flagellar switch. The flagellar switch is responsible for causing the flagella to spin in either one direction (causing “swimming”) or the other (causing “tumbling”). This molecular machinery is best understood in E. coli, where the phosphorylated-CheY protein interacts with the switch to change its direction. The situation in H. pylori appears to be more complex. There are five proteins that contain CheY-like domains, CheY, CheA-Y, CheV1, CheV2 and CheV3. Three of these, CheY, CheA and CheV1, cause defects in motility when eliminated. The function of the other CheY-domain-containing proteins is unknown. To determine whether the CheVs likely perform similar or different functions, we analyzed the sequence of these proteins and found they fall into separate families, in that they are more conserved between the cognate CheVs from other Helicobacter genus members than with each other. These cross-species similarities suggests that they each have an important conserved function. The Helicobacter switch appears to contain four proteins: FliM, FliY, FliN, and FliG. In other systems, FliM binds CheY. Using sequence analysis of FliM and CheY, we found that residues important for protein-protein interactions are conserved in the H. pylori proteins, suggesting these proteins do interact with each other. FliY is a chimeric protein containing FliN and CheC (responsible for dephosphorylating CheY) domains. FliN and FliG have structural and functional roles in the switch, and are not thought to mediate signaling. Our experimental goal is to determine which CheY domain-containing proteins and which other flagellar switch proteins interacts with FliM. To this end, we expressed and purified recombinant H. pylori FliM as a GST fusion. We then used this GST-FliM as bait to trap FliM-interacting proteins, and are determining the identity of these proteins using western blotting and mass spectrometry.
94

DELETION ANALYSIS OF HOOK CAPPING PROTEIN FLGD OF SALMONELLA TYPHIMURIUM Hideyuki Matsunami§ and Keiichi Namba§, †
§ Dynamic NanoMachine Project, ICORP, JST, and † Graduate School of Frontier Biosciences, Osaka University, 1-3 Yamadaoka, Suita, 565-0871 JAPAN The bacterial flagellum is composed of three parts: the filament; the hook; and the basal body. The flagellar hook is a universal joint for transmitting torque generated by the rotary motor to the filament. The hook capping protein FlgD is essential for hook assembly. It is transported from the cytoplasm to the tip of the rod by the flagellar-specific protein export pathway prior to the export of hook protein FlgE. With a help of FlgD, FlgE incorporates itself into the hook at growing end by self-assembly. FlgE cannot assemble into the hook in flgD mutant strains, in which FlgE is excreted out of the cell. In the filament assembly, HAP2 (hook associate protein 2) binds to the tip of the growing flagellum forming a pentameric structure called “star cap” and helping flagellin monomers to incorporate into the flagellum. We studied if FlgD forms pentamers as well by gel-filtration chromatography and chemical cross-linking and confirmed that it dose. Negatively stained samples of FlgD also showed plausible pentameric images by electron microscopy. Therefore, we infer that the pentameric form of FlgD is the hook capping structure for the hook assembly. FlgD of Salmonella typhimurium is composed of 232 amino acid residues. By secondary structure prediction, FlgD has an α-helix motif in its N terminal region, and about 90 residues fragment containing this motif can compliment flgD mutants of Salmonella. FlgD also has a distinct β-structure domain in its C terminal region where its folding feature resembles that of the core domains of FlgE lacking both terminal disordered regions. We systematically constructed 10-amino acids deletion variants through the N terminal region of FlgD and characterized them in terms of protein expression and secretion in the flgD mutant strain. These variants were expressed fairly well and secreted as well. We will discuss about pentamer formation and effect on the hook assembly of these FlgD variants.
95

FLIH IS REQUIRED FOR FLII BINDING TO THE C-RING COMPLEX IN SALMONELLA Jonathan L. McMurry1*, Bertha González-Pedrajo2, John S. Van Arnam1 and Robert M. Macnab1
1Department of Molecular Biophysics and Biochemistry, Yale University, New Haven, Connecticut, 06520-8144 and 2Departamento de Genética Molecular, Instituto de Fisiología Celular, Universidad Nacional Autónoma de México, Ap. Postal 70-243, México D.F. 04510, México
FliH regulates the flagellar export ATPase FliI, preventing nonproductive ATP hydrolysis. (FliH)2 and FliI form a heterotrimer. Recently, FliH has been shown to stably associate with C-ring protein FliN. A large FliG-FliM-FliN-FliH-FliI complex is stably and specifically formed upon overexpression of the proteins. Analysis of this complex reveals that FliH is required for FliI binding to the C-ring protein complex and is thus acting as both a negative regulator of ATPase activity and a chaperone. Purification of the GMNHI complex has allowed for analysis of binding affinities of FliH and FliI for each other and the complex. Within the C-ring the interactions among FliN, FliH and FliI are dynamic, which has implications for export function.
96

BOTH CHEMOTAXIS OPERONS IN RHIZOBIUM LEGUMINOSARUM CONTROL FLAGELLAR MOTILITY Lance D. Miller1,3, Christopher Yost2, Michael F. Hynes2 and Gladys Alexandre3
1School of Biology, Georgia Institute of Technology Atlanta, GA 30332-0230 2Department of Biological Sciences, The University of Calgary, Calgary, Alberta, Canada T2N 1N4 3Department of Biology, Georgia State University, Atlanta, Georgia 30302-4010
Chemotaxis in the nitrogen-fixing α-proteobacterium Rhizobium leguminosarum has been implicated in the symbiotic relationship between this organism and its plant host (1). In order to identify genes encoding the molecular machinery for chemotaxis we have analyzed the draft genome of Rhizobium leguminosarum biovar viciae strain 3841. Similarity searches using sequences of known chemotaxis proteins revealed the presence of two distinct chemotaxis operons, which we termed CheOp1 and CheOp2. Members of the α-proteobacteria are known to have multiple chemotaxis operons that are classified into three major groups based on genomic content and phylogenetic analysis of the CheA protein (2). We have found that CheOp1 is orthologous to the Group 1 chemotaxis operon from Sinorhizobium meliloti, whereas CheOp2 is orthologous to the Group 3 chemotaxis operon from Rhodobacter sphaeroides. In both organisms these are major operons controlling flagellar motility (3, 4). In order to reveal a role for each chemotaxis operon in R. leguminosarum, we have constructed mutant strains that carry in-frame deletions in each operon and analyzed their motile behavior under different conditions. The motility of the mutants was characterized using computerized motion analysis and other behavioral assays. R. leguminosarum wild type swimming behavior appears to comprise periods of smooth swimming punctuated by brief reversals that reorient the cell. Deletion of CheOp1 results in a smooth swimming phenotype whereas deletion of CheOp2 results in a tumbly phenotype. Interestingly, a double mutant lacking both Che operons has an intermediate swimming bias. This observation suggests that both operons contribute to the locomotive behavior of R. leguminosarum. Results from swarm plate and temporal gradient assays further support this hypothesis. We are currently measuring the relative expression of each chemotaxis operon under different growth conditions.
97

IDENTIFYING CHEMOATTRACTANTS FOR BORRELIA BURGDORFERI Michael R. Miller, Richard G. Bakker, Cynthia Cunningham and Nyles W. Charon Department of Microbiology, Immunology and Cell Biology, Health Sciences Center, PO Box 9177, West Virginia University, Morgantown, WV 26506
The etiologic agent of Lyme disease is the spirochete Borrelia burgdorferi. With 23,000 reported cases in 2002, Lyme disease is the most common vector-borne disease in the United States. Chemotaxis and motility are likely to be important in the pathogenesis and life cycle of B. burgdorferi; however, the unique morphology of spirochetes poses complex problems with respect to motility and chemotaxis. B. burgdorferi is 10 – 20 µm long, with bundles of flagella subterminally attached to both ends of the cell. These periplasmic flagella reside between the outer membrane and the cell cylinder, extending from each end toward the cell center where they overlap. While most bacteria move translationally by rotating the external flagella clockwise (CW) or counter clockwise (CCW), as viewed from the distal ends of the flagella to their insertion into the cell, spirochete motility is unique. Several lines of evidence indicate the B. burgdorferi periplasmic flagella at the opposite ends rotate asymmetrically during translation, i.e. the periplasmic flagella at one end rotate CCW while those at the other end rotate CW. Furthermore, most externally flagellated bacteria exhibit two modalities of movement (“runs” and “tumbles”), while spirochetes have three modalities. B. burgdorferi can translate in one direction, simultaneously reverse the rotation of motors at both ends and translate in the opposite direction, or “flex”, a non-translational mode when both motors rotate in the same direction (CCW or CW). Relatively little is known about the coordinate regulation of the motors at the opposite ends.
Identifying defined chemoattractants is fundamental to understanding B. burgdorferi
chemotaxis and motility – to investigate how individual cells respond to attractants, and to eventually determine if specific attractants function in their complex life cycle. Specifically, for feeding ticks to become infected, B. burgdorferi must concentrate at the site of the feeding tick. In addition, when an infected tick bites a new host, such as a human, B. burgdorferi must traverse the extracellular matrix and penetrate the vascular endothelial cell lining to become hematogeneous. Because B. burgdorferi are slow growing (~10 hr doubling time) and require a complex growth medium, most chemotaxis assays are time-consuming and/or labor-intensive. As a result, only a few complex attractants (rabbit serum and tick salivary gland extracts) have been reported. To begin to identify defined attractants, we developed a high-through-put capillary tube assay, in which B. burgdorferi entering capillary tubes filled with potential attractants are enumerated by flow cytometry. Among the attractants we have identified, some (chitosan dimers, N-acetylglucosamine, glucosamine) may be important for the B. burgdorferi life cycle and/or tissue invasion.
98

SORTING OUT THE FUNCTION OF MULTIPLE CHEY GENES AND THE FUNCTION OF CHEX IN THE CHEMOTAXIS OF THE LYME DISEASE SPIROCHETE BORRELIA BURGDORFERI Md. A. Motaleb, Richard Bakker, Chunhao Li, and Nyles W. Charon Department of Microbiology, Immunology, and Cell Biology, West Virginia University, Morgantown.
The spirochete Borrelia burgdorferi is the causative agent of Lyme disease. Infection is transmitted from one animal to another by the bite of a tick of the Ixodes class. B. burgdorferi invades a variety of host tissues. In humans, disease manifestations include skin rashes, neurological disorders, cardiac abnormalities, and acute and chronic arthritis. B. burgdorferi motility is provided by bundles of 7-11 periplasmic flagella that are subterminally attached to each end of the cell cylinder. A given periplasmic flagellum is attached only at one end. An outer-membrane sheath surrounds the entire cell. During translation, one bundle rotates counter-clockwise and the other clockwise. A given cell can run, reverse, or go into a non-translational mode referred to as flex. Flexing occurs when the periplasmic flagella bundles rotate in the same direction; the cell often bends in the center during a flex. We are beginning to understand B. burgdorferi chemotaxis using targeted mutagenesis. Genomic analysis indicates that B. burgdorferi has mutliple copies of chemotaxis genes: There are two cheA’s, three cheY’s, and three cheW’s. It lacks cheZ but has the cheC homologue cheX. Previous analysis indicated that cheA2, but not cheA1, was essential for chemotaxis. The cheA2 mutant constantly ran in one direction; it failed to flex and reverse. Similar results were found with the double cheA1cheA2 mutant. Here we report our results whereby we targeted other putative chemotaxis genes. cheY1, cheY2, and cheY3 each possess all of the essential functional residues present in E. coli CheY. We inactivated each of these genes by targeted mutagenesis. The only mutant that had an altered phenotype with respect to swimming and chemotactic behavior was a mutation in cheY3. This mutant was similar to the cheA2 mutant: it ran in only one direction, failed to reverse or flex, and was non-chemotactic. Complementation of the cheY3 mutant restored normal swimming and chemotactic behavior. A triple mutant of cheY1-cheY2-cheY3 exhibited the same phenotype as the cheY3 mutant. Our results indicate that CheY3, but neither CheY1 nor CheY2, is essential for chemotaxis in B. burgdorferi.
We also inactivated the cheC homologue cheX, which maps between cheA2 and cheY3. The cheX mutant had a markedly different phenotype than cheA2 and cheY3 mutants. Although it was non-chemotactic, it constantly flexed and did not translate. Complementation of cheX restored normal swimming and chemotactic behavior. In other bacteria, CheC has phosphatase activity that acts on CheY-P. Taken together, we hypothesize that under low CheY-P concentrations, cells constantly run and rotate their periplasmic flagellar bundles asymmetrically (cheA2 and cheY3 mutants), and under high CheY-P concentrations (cheX mutant), cells flex and thus rotate these bundles in the same direction.
99

SINGLE-CHAIN RECEPTORS REVEAL PROMISCUOUS PARTNER-SWAPPING Patricia Mowery and John S. Parkinson Biology Department, University of Utah, Salt Lake City, UT 84112
The signaling complex that mediates chemotactic behavior in bacteria contains homodimeric chemoreceptors (methyl-accepting chemotaxis proteins or MCPs), a histidine kinase (CheA), and a coupling protein (CheW). The architecture of the ternary complex is poorly understand, but recent evidence suggests that it may be based on MCPs forming trimers of dimers. To explore this possibility, we attempted to manipulate the geometry of Tsr (serine chemoreceptor) signaling teams by covalently joining two Tsr subunits with a flexible linker. Such “single-chain” receptors would allow us to introduce independent structural alterations in each subunit and thereby investigate the inside and outside structural environments predicted by the receptor trimer-of-dimers model. We designed single-chain genes to link the C-terminus of one Tsr subunit (Tsr-1) to the N-terminus of a second Tsr subunit (Tsr-2). In all constructs, Tsr-2 was encoded by a full-length gene (codons 1-551) to provide the C-terminal NWETF sequence needed for sensory adaptation. Three Tsr-1 join points were tested: after residue 546, eliminating only the NWETF segment; after residue 533, corresponding to the shortest of the E. coli MCP lengths; and after residue 517, which marks the end of the highly conserved portion of the receptor signaling domain. Polyglycine linkers of 5, 10, or 15 residues connected the Tsr-1 and Tsr-2 segments. Cells containing single-chain receptors exhibited serine swarm rings on soft agar plates and contained negligible amounts of monomer-sized proteolytic fragments, indicating that intact single-chain molecules can mediate receptor function. All but the shortest single-chain construct supported serine chemotaxis, demonstrating a minimal length requirement for the segment connecting the two subunits. Genetic tests indicated that both the Tsr-1 and Tsr-2 subunits could contribute to single-chain function. For example, a recessive mutation in either subunit did not eliminate function, but a dominant mutation in either subunit did. Surprisingly, monomeric Tsr subunits with dominant defects were found to block single-chain function in trans, suggesting that the dominant subunits might be able to invade and spoil single-chain molecules. To assess the extent of such subunit swapping, we constructed single-chain molecules with a cysteine residue in both subunits that was known to lie at the periplasmic dimer interface. More than half of the crosslinked products were strand-swapped, which may limit the utility of the single-chain approach. However, our results also raise some interesting questions: Do homodimeric receptors organized in trimers of dimers swap subunits? Do CheW/CheA binding, methylation state or ligand occupancy influence subunit swapping rates? Does subunit swapping cease when receptors become part of the polar cluster? Does subunit-swapping contribute to the high-gain signaling properties of bacterial chemoreceptors?
100

CHEC, FLIY, AND CHEX: THE CYX FAMILY OF CHEMOTAXIS PHOSPHATASES Travis Muff and George Ordal University of Illinois, Urbana-Champaign, 190 Medical Sciences Bldg; 506 S. Mathews Ave; Urbana, IL 61801
In bacterial two-component signal transduction systems many ways of removing the phosphorylated response regulator have been proposed. CheZ has been shown to remove the chemotaxis signal (CheY-P) in Escherichia coli, but most bacteria and the archaea do not contain a CheZ homolog. Recently we have shown that in Bacillus subtilis, which does not encode a homolog of CheZ, the proteins FliY and CheC are both able to increase the dephosphorylation rate of CheY-P. A third protein, CheX, is not found in B. subtilis but is found in other bacteria and shares the phosphatase activity. These proteins share areas of homology we have named the CYX-region. We suspect not only is the CYX-region responsible for the activity on CheY-P, but also defines a new family of proteins, found throughout the bacterial and archaeal kingdoms, involved in regulating CheY-P levels. Here we compare the activities of the three proteins and their effects on the chemotaxis system.
101

PROTEIN PARTNERS OF FIMX, A REGULATOR OF TYPE IV PILI ASSEMBLY IN PSEUDOMONAS AERUGINOSA Thomas Murray1 and Barbara Kazmierczak 2,3 Depts. of Pediatrics1, Internal Medicine2 and Microbial Pathogenesis3 Yale University School of Medicine, New Haven CT 06510
Type IV pili (TFP) are surface structures on Pseudomonas aeruginosa (PA) that play important roles in eukaryotic cell adhesion, biofilm formation, and twitching motility. Each of these functions contributes to the pathogenesis of clinical PA infections. Surface expression of TFP in response to environmental signals requires FimX (PA4959), a polarly localized protein whose role in pilus assembly is unknown. FimX has sequence homology to proteins with GGDEF and EAL motifs, suggesting that it may regulate levels of the second messenger di-cyclic GMP. FimX also possesses regions with homology to REC and PAS domains, arguing that FimX activity may be regulated by interactions with other proteins. The goal of this work was to identify potential protein partners of FimX as a first step in understanding the signaling pathways that govern TFP expression. To accomplish this goal, we used FimX as bait in a yeast two-hybrid screen of a PA genomic library. Three candidate interactors were identified by this method: PA3670, PA4461, and FlhF. PA3670 and PA4461 are predicted to encode components of two distinct ATPase Binding Cassette (ABC) transporters, while FlhF is a known regulator of flagellar assembly. We have made in-frame deletions of each of these genes in the PA103 and PAK backgrounds and have carried out assays for twitching and swimming motility. RFP fusions have been constructed for each protein, allowing us to demonstrate that both PA3670 and FlhF localize to the cell pole. We are currently confirming that these proteins physically interact with FimX. These preliminary data suggest a role for ABC transporters in the regulation of TFP expression in PA and argue that fimbrial and flagellar motility may be coordinately regulated in this organism.
102

IDENTIFICATION OF FLAVOBACTERIUM JOHNSONIAE GLIDING MOTILITY GENES BY HIMAR-EM1 MUTAGENESIS Timothy F. Braun, Shawn S. Nelson, Manjeet Uppal and Mark J. McBride Department of Biological Sciences, University of Wisconsin-Milwaukee Milwaukee, WI 53201
Flavobacterium johnsoniae moves rapidly over surfaces in a process known as gliding motility. The mechanism of gliding motility is not known but it does not involve flagella or pili. Nine genes that are required for F. johnsoniae gliding have been identified. A modified mariner transposon, HimarEm1, was constructed to allow the identification of additional motility genes. HimarEm1 transposed in F. johnsoniae and nonmotile mutants were identified and analyzed. HimarEm1 insertions were isolated in previously identified genes (gldI, gldJ) and in four novel genes (gldK, gldL, gldM, gldN), which are clustered together on the genome. GldK is similar in sequence to the outer membrane protein GldJ, which is also required for gliding. GldL, GldM, and GldN are predicted to reside in the outer membrane or periplasm but are not similar in sequence to proteins of known function. In addition to mutants that are completely nonmotile, mutants that form non-spreading colonies composed of cells that retain some ability to move were isolated. Many of these mutants had insertions in the 17 kb sprB gene, which encodes a putative outer membrane protein that is similar in sequence to RTX (repeats in toxin) proteins. SprB exhibits some similarity to Synechococcus SwmB, which is involved in non-flagellar cyanobacterial swimming motility, suggesting that there may be parallels between the motility systems of these bacteria. One model of gliding involves multiple transporters that move a macromolecular conveyor belt along the cell surface. The newly identified genes may encode components of this machinery.
103

ESR AND CRYSTALLOGRAPHIC STUDIES OF INTERACTION INTERFACES BETWEEN T. MARITIMA CHEA AND CHEW Sang-Youn Park, Peter P. Borbat, Alexandrine M. Bilwes, Jack H. Freed and Brian R. Crane Department of Chemistry and Chemical Biology, Cornell University, Ithaca, NY 14850
How chemoreceptors regulate the activity of the histidine kinase CheA is a central question in understanding the signaling pathway that controls bacterial chemotaxis. The interactions between receptors and CheA require the involvement of the coupling protein CheW. We have applied nitroxide spin-labeling and Double Quantum Coherence (DQC) - Electron Spin Resonance (ESR) to measure distances within T. maritima CheA and between CheA and T. maritima CheW. Nitroxide-labeled CheA yields high-quality dipolar spectra. By a variant of the DQC-ESR method developed by Freed and coworkers at Cornell, we limit the effects of nuclear-spin-diffusion on the electron spin phase-memory time, and thereby enable the accurate measurement of distances to up to 70 Å. Site-directed cysteine mutants of CheA and CheW allow the placement of spin-labels at specific positions on the individual domains, whose associations can then be mapped by multiple distance measurements. As a complementary tool, we are using X-ray crystallography to characterize in detail the molecular interface between CheA and CheW. We have cloned and expressed various constructs of CheA and determined their interaction properties with CheW. Crystallization trials and structure determinations are currently underway.
104

INTERACTION OF METHYLTRANSFERASE CHER WITH CHEMORECEPTOR TAR METHYLATION REGION AND EXPLORATION OF THERMOTOGA MARITIMA AS AN ALTERNATIVE MODEL SYSTEM FOR CHEMOTAXIS AND ADAPTATION. Eduardo Perez, Snezena Djordjevic, Ann H. West, Ann M. Stock, Center for Advanced Biotechnology and Medicine, Department of Biochemistry, University of Medicine and Dentistry of New Jersey-Robert Wood Johnson Medical School, and Howard Hughes Medical Institute, 679 Hoes Lane, Piscataway, NJ 08854, and Department of Biochemistry and Molecular Biology, University College London, Gower Street, London WC1E 6BT, United Kingdom and Department of Chemistry and Biochemistry, University of Oklahoma, 620 Parrington Oval, Norman, Oklahoma 73019
Bacterial chemoreceptors are posttranslationally modified by methylation of specific glutamate residues within their cytoplasmic domains. This highly regulated, reversible modification counterbalances the signaling effects of ligand binding and contributes to adaptation. Based on the crystal structure of methyltransferase CheR, we have postulated that positively charged residues in helix �2 in the N-terminal domain of the enzyme may be complementary to the negatively charged methylation region of the methyltransferase substrates, the bacterial chemotaxis receptors. To investigate this hypothesis, we have produced several single site Arg or Lys to Ala substitutions within S. enterica CheR and have characterized the methylation activities of these CheR proteins toward several receptor substrates containing different glutamates available for methylation. Our findings imply specific complementarity between the positively charged residues of CheR and the negatively charged methylatable glutamates and support the hypothesis that these residues are involved in the specific recognition of receptor methylation regions. Moreover, a more detailed view of the interaction between CheR and receptor methylation sites was pursued by utilizing computational docking programs. Results confirmed that the receptor could be docked within the active site cleft of CheR in an orientation that aligned the �2 helix of CheR roughly antiparallel to the methylation helix of the receptor. While, chemotaxis has been thoroughly characterized in E.coli and S.enterica, studies of chemosensory pathways from other organisms, have shown variations from the E. coli and S. enterica paradigm, suggesting that chemotaxis and adaptation may be more complex. The thermophilic eubacterium Thermotoga maritima, is an intriguing candidate to serve as an alternate model system for chemotaxis and adaptation. For example, T. maritima possess CheC and CheD which are not found in E. coli and S. enterica, but have been shown to play a role in methylation and adaptation. Secondly, the structure for three chemotaxis proteins have been solved from T. maritima, therefore the potential of solving the structures of other chemotaxis proteins from this organism, highlights the importance of understanding the biochemical functions of these proteins in T. maritima and how similar and or dissimilar they are to their E. coli and S. enterica counterparts. Lastly, efficient methylation in E. coli and S. enterica requires binding of CheR to an extreme C-terminal pentapeptide (NWETF) of the receptor. However, T. maritima, does not possess, nor does it require this sequence for efficient methylation. It is likely then, that CheR may bind tighter to the methylation region, opening up the possibility to further characterize CheR’s interactions with this region. However, before these steps can be taken, an in vitro methylation system for T. maritima needs to be established, with the first objective being the identification of the methylation sites in the T. maritima chemoreceptor.
105

PHOSPHOSIGNALLING IN RHODOBACTER SPHAEROIDES CHEMOTAXIS Steven L. Porter and Judith P. Armitage Microbiology Unit, Department of Biochemistry, University of Oxford, South Parks Road, Oxford. OX1 3QU. UK
Rhodobacter sphaeroides has a complex chemosensory system comprising two classical CheAs, two atypical CheAs and eight response regulators (6 CheYs and 2 CheBs). The classical CheAs, CheA1 and CheA2, have similar domain structures to Escherichia coli CheA, whilst the atypical CheAs, CheA3 and CheA4, lack some of the domains found in E. coli CheA. CheA3 possess only the Hpt (P1) and regulatory (P5) domains joined by a novel 751 amino acid sequence, while CheA4 has only the dimerisation (P3), kinase (P4) and regulatory (P5) domains. CheA2, CheA3 and CheA4 are all essential for chemotaxis.
In this investigation we demonstrate that CheA3 and CheA4 are both unable to undergo ATP dependent autophosphorylation, however CheA4 is able to phosphorylate CheA3. The in vitro kinetics of this phosphorylation reaction are consistent with a reaction mechanism in which CheA3 and CheA4 associate to form an enzyme substrate complex, CheA3A4.
Selective phosphotransfer was observed from CheA3A4-P to the response regulators CheY1, CheY6 and CheB2 . Therefore, CheA3A4 can perform all of the phosphorylation reactions that are common to all CheA proteins; to the best of our knowledge, CheA3A4 is the first characterized CheA where the Hpt and kinase domains are encoded by distinct genes.
Using phosphorylation site and kinase domain CheA mutants we show that phosphosignalling involving CheA2, CheA3 and CheA4 is essential for chemotaxis in R. sphaeroides. Interestingly, CheA3 was not phosphorylated in vitro by CheA1 or CheA2, although CheA1 and CheA2 mutants with defective kinase domains were phosphorylated by CheA4. Since in vivo CheA3 and CheA4 localize to the cytoplasmic chemotaxis cluster, whilst CheA2 localizes to the polar chemotaxis cluster, it is likely that the physical separation of CheA2 and CheA4 prevents unwanted crosstalk between these CheAs.
106

MECHANISM AND PHYSIOLOGY OF AER-MEDIATED GENE REGULATION
Birgit M. Prüß1
Veterinary & Microbiological Sciences Department, North Dakota State Univ., Fargo ND 58105 Escherichia coli FlhD/FlhC was initially discovered as an activator of flagellar gene expression in E. coli and Salmonella and subsequently as a global regulator of metabolism in several enteric bacteria (1-3). The regulation of FlhD/FlhC target genes that were involved in anaerobic respiration and the Entner-Doudoroff pathway was dependent on the aerotaxis signal transducer, Aer (2).
While the signaling pathway from the oxygen sensor Aer to the flagellar motor has been well investigated, little is known about the mechanism by which Aer regulates gene expression. Considering the large number of Aer homologs in some bacteria, this might constitute a new and important mechanism for gene regulation. Our hypothesis is that the mechanism by which Aer mediates gene regulation might have some overlap with components of the aerotaxis signaling pathway. Our approach to test this hypothesis is to examine the effect of individual components of the aerotaxis signaling pathway on the expression level of Aer target genes.
The first question is whether the gene regulatory effect of Aer was specific for Aer or whether another one of the four MCPs might also be involved in gene regulation. We determined the effect of Tsr (serine receptor) upon the expression of the Aer target genes using reporter gene fusions to the dmsA, nrfA and edd promoters. None of the Aer target genes was affected by the presence or absence of Tsr. Similarly, a plasmid expressing Tsr was unable to complement expression of the same Aer target genes in a ∆aer background. In addition, there was no effect of Tsr on genes, such as gltB, gcvT, serA and narK, that are regulated by FlhD/FlhC but not by Aer. An investigation of the Aer domain(s) required for gene regulation is currently in progress.
The second question deals with the physiological consequences of Aer-mediated gene regulation. The induction of anaerobic respiration genes indicates that Aer might be involved in a switch that takes place as bacteria grown in batch culture deplete the oxygen from the medium. We measured the concentration of oxygen with an electron electrode and related it to expression of cyoA (aerobic respiration) and dmsA (anaerobic respiration) throughout growth in wild-type cultures and aer mutants. After 3 h of growth, an optical density of 0.5 and 5% oxygen left, wild-type cells switched from the aerobic to the anaerobic enzyme, whereas aer mutants were unable to induce expression of the anaerobic enzyme. In a competition experiment, wild-type cells outnumbered the mutants by a factor of 100 within 4 to 5 days. The question whether wild-type cells might outnumber the mutants in their natural environment, the gut, remains to be answered.
1. B. M. Prüß, X. Liu, W. Hendrickson, P. Matsumura, FEMS Microbiol. Lett. 197, 91 (2001).
2. B. M. Prüß et al., J. Bacteriol. 185, 534 (2003).
3. V. Kapatral et al., Microbiol. 150, 2289 (2004).
107

CCPA NEGATIVELY REGULATES PHOPR PROMOTER THROUGH A NEWLY DISCOVERED TRANSCRIPTION START SITE, PA6 IN BACILLUS SUBTILIS
Ankita Puri-Taneja, Salbi Paul, Yinghua Chen and F. Marion Hulett Laboratory for Molecular Biology, Department of Biological Sciences, University of Illinois at Chicago, Chicago, IL, 60607.
The Bacillus subtilis PhoPR two-component system is directly responsible for the cellular
response to phosphate starvation, a condition frequently experienced by B. subtilis due to low
concentration of inorganic phosphate in the soil. We report data suggesting that catabolite
regulation plays a role in the expression of PhoP and PhoR, major signaling proteins required
during phosphate deprivation. The response regulator, PhoP, and the histidine kinase, PhoR, are
encoded in a single operon with a complex promoter region that contains 5 known transcription
start sites, which respond to at least 2 regulatory proteins. The catabolite repressor protein, CcpA
was also shown to repress phoPR transcription. Either coeffector of CcpA, HPr or Crh, was
sufficient to cause CcpA dependent repression of the phoPR promoter expression in vivo. A
mutation in the ccpA gene caused maximum relief of repression under excess phosphate
conditions, a growth condition where wild type phoPR transcription is normally low. In vivo
and in vitro studies showed that CcpA repressed the phoPR promoter by binding to the cre box
present in the promoter. Primer extension and in vitro transcription studies revealed that the
CcpA regulation was due to repression of PA6, a promoter positioned immediately upstream of
the cre box that is located upstream of the other 5 phoPR promoters. EσA was sufficient for
transcription of PA6 in vitro. Addition of CcpA to the in vitro transcription reaction repressed
transcription of PA6.
108

VARIATION OF THE NUMBER OF TORQUE GENERATING UNITS IN THE BACTERIAL FLAGELLAR MOTOR Stuart Reid, Mark Leake, Jennifer Chandler, Richard Berry Clarendon Laboratory, University of Oxford, OX1 3PU, UK
The bacterial flagellar motor is thought to contain up to eight independent torque-generating complexes of four MotA and two MotB proteins. The complex contains two proton channels and it is suggested that the complex is in fact a dimer consisting of two single proton channel units, each of two MotA and one MotB. Latex microspheres were attached to sheared flagellar filaments and rotation speed was measured in a weak optical trap. Inducible plasmids were used to vary the amount of torque-generating proteins. Resurrection traces were obtained where induction was increased during speed measurements with increases in speed corresponding to incorporation of units into the motor. Constant low induction measurements were made where the speeds of a population of cells group according to numbers of torque generators and the relationship between induction and mean speed of a population was explored. A resurrection dataset was analysed and evidence for more than eight speed levels is presented.
109

PHOTORESPONSES IN R. SPHAEROIDES Mark A J Roberts & Judith P Armitage. Microbiology Unit, Departmentt of Biochemistry, University of Oxford, South Parks Rd, Oxford, OX1 3QU
While a great deal is understood about bacterial responses to chemical agents very little is known about the responses to light and oxygen. Motile bacteria typically show a negative response to blue or UV light while photosynthetic bacteria also show a positive response to wavelengths of light that can be used for photosynthesis. Thus it is likely there are different receptors for the different responses.
R. sphaeroides is a purple non-sulphur bacterium that is tactic to a wide range of stimuli including light. Early experiments showed that positive phototaxis is linked to the photosynthetic pathway; a reduction in the rate of electron transport causes direction change (Grishanin et al., 1997). Proteins encoded by cheOp2 and cheOp3 (which are essential for chemotaxis) are also required for phototaxis (Porter et al., 2002). The primary signal leading from the photoreceptor to the chemotaxis pathway is unknown. More recently a negative photoresponse to blue / UV light has been observed in R. sphaeroides.
The R. sphaeroides genome contains a number of genes which may encode protein domains that have been implicated in light sensing in other species. There is a protein which includes a LOV domain, encoded just upstream from a gene that encodes the putative chemotaxis fusion protein cheBRA. LOV (Light, Oxygen, Voltage) domains are a subset of PAS domains and have been shown to be sensitive to light, particularly in the blue light region.
The deletion phenotype of this and other mutants will be described along with an assay for phototaxis and a transposon screen to identify novel genes involved in the photoresponse of R. sphaeroides. Grishanin, R.N., Gauden, D.E., and Armitage, J.P. (1997) Photoresponses in Rhodobacter
sphaeroides: role of photosynthetic electron transport. Journal of Bacteriology 179: 24-30.
Porter, S.L., Warren, A.V., Martin, A.C., and Armitage, J.P. (2002) The third chemotaxis locus of Rhodobacter sphaeroides is essential for chemotaxis. Molecular Microbiology 46: 1081-1094.
110

PHOTOTAXIS AND SIGNAL TRANSDUCTION IN THE CYANOBACTERIUM SYNECHOCYSTIS sp PCC6803.
Julianna Ross* and Devaki Bhaya Carnegie Institution of Washington, Department of Plant Biology, 260 Panama St., Stanford, CA 94305, USA.
Motility in the unicellular gliding cyanobacterium Synechocystis sp. PCC6803 requires Type IV pili. Phototaxis appears to be a complex and regulated light-directed phenomenon in which single cells and groups of cells participate. To study signal transduction during phototaxis we have isolated several mutants with an aberrant phototactic response (i.e they are non-motile or exhibit constitutive negative phototaxis). Several of these mutants mapped to che-like genes. Synechocystis sp has three loci containing che-like genes; all three of these (tax1, tax2 and tax3 loci) appear to play a role in phototaxis. Mutants in the tax1 and tax2 locus are negatively phototactic while mutants in tax3 are non-motile. Our analysis of some of these mutants using a novel slide-based phototaxis assay and time-lapse video microscopy suggests that there are at least two light inputs that regulate phototaxis. There is a red light (far red light reversible) positive phototaxis controlled by TaxD1 which fits with evidence that there are two phytochrome-like domains in TaxD1. These results suggest that while positive phototaxis is controlled by red light, negative phototaxis in Synechocystis sp. strain PCC6803 is mediated by one or more (as yet) unidentified photoreceptors. Analyses of specific site-directed mutants in the hybrid histidine kinase, TaxAY1, one of the key players in the signal transduction events, will be discussed. We will also describe some of the novel mutants that were isolated in the mutagenesis screen that suggest that group behavior and signaling may be involved in phototaxis. Our working model of the complex signal transduction networks that control phototactic movement will be presented.
111

FUNCTIONAL CHARACTER IZATION OF NOVEL AND UNUSUAL MEMBERS OF THE HELICOBACTER PYLORI FLAGELLAR REGULON IN FLAGELLAR SECRETION AND BIOSYNTHESIS Authors: Melanie Rust, Eike Niehus, Sarah Kühne, Sebastian Suerbaum, Christine Josenhans Institution: Department for Med. Microbiology and Hospital Epidemiology, Hannover Medical School ,Carl-Neuberg-Strasse 1, D-30625 Hannover, Germany Motility and chemotaxis are amongst the most important factors of persistence of H. pylori in its habitat, the human stomach mucus We have recently characterized in some detail the regulons, dependent on the two flagellar sigma factors FliA (sigma28) and RpoN (sigma54), as well as identified the H. pylori anti-sigma factor FlgM, which has an unusually short N-terminus, yet is functional in Salmonella. A number of novel protein-encoding genes were identified to belong to these flagellar regulons (Niehus et al., 2004, Mol Microbiol. 52:947-961). Since the requirements for flagellar biosynthesis, structural feedback inhibition, and type III secretion through the H. pylori flagellar apparatus seem to be different from the paradigm of Salmonella, the aim of the present study was to characterize in more detail novel and unusual flagellar proteins of H. pylori and their role in flagellar biosynthesis and structure. Our first aim was the investigation of flagellar secretion and coupled regulation mechanisms in the presence of a flagellar sheath. We determined that despite the unusual and very short N-terminus of H. pylori FlgM, this secretion substrate protein was secreted by Salmonella into the medium. With the aid of a H. pylori FlgM-specific antiserum, we showed that FlgM was also secreted through the flagellar type III secretion apparatus in wild type H. pylori, but was retained within the flagellar compartment and not released into the environment. Next, we determined functional requirements for FlgM secretion by detection and localization of FlgM in flagellar basal body mutants. FlgM secretion in H. pylori was found to occur in basal body mutants. Mechanisms of FlgM secretion and inactivation in H. pylori appeared to differ from Enterobacteriaceae and will be discussed. The hook structure formed by FlgE is not responsible for major transcriptional feedback regulation of flagellar biosynthesis in H. pylori. Therefore, we investigated the regulation of the unusual FlgE paralog FlgE2 (specific for ε-proteobacteria) and its localization and role in flagellar assembly, and addressed the question, whether FlgE2 might have a function in feedback regulation in this organism. A FlgM-independent role of FlgE2 in flagellar biosynthesis was demonstrated in this study, and will be discussed. We gratefully acknowledge the German Research Council, grant Jo344/2-1, for financial support.
112

CRYSTAL STRUCTURE OF CYTOPLASMIC DOMAIN OF FlhA, A SUBUNIT OF THE BACTERIAL FLAGELLAR TYPE III PROTEIN EXPORT APPARATUS Yumiko Saijo-Hamano,1 Katsumi Imada,1,2 Tohru Minamino1 and Keiichi Namba 1,2
1Dynamic NanoMachine Project, ICORP, JST & 2Graduate School of Frontier Biosciences Osaka University; 1-3, Yamadaoka, Suita, Osaka 565-0871 Japan The bacteria flagellum has the type III protein export apparatus as one part of the basal body. It selectively translocates flagellar axial proteins into the central channel of the flagellum. The export apparatus consists of six integral membrane components (FlhA, FlhB, FliO, FliP, FliQ and FliR) and three cytoplasmic components (FliH, FliI and FliJ). Genetic and biochemical information are available for some of these proteins. However, the export mechanism is not clear except that the energy released by ATP hydrolysis by FliI is used for the export process. FlhA (MW: 75 kDa), which is the largest subunit of the export apparatus, consists of two domains: a hydrophobic N-terminal transmembrane domain (FlhATMo, 34.5 kDa) and a hydrophilic C-terminal cytoplasmic domain (FlhAC, 40.5 kDa). We reported that FlhAC is directly involved in the export of flagellar proteins and contains predominately α-helical structure as well as β-structure. To reveal not only the function of FlhAC but also the export mechanism, we crystallized FlhAC for high-resolution structure analysis.
His-tagged FlhAC (43 kDa) and FlhAc38K (37.5 kDa), which is the smallest fragment of FlhAC produced by V8 proteolysis, were purified and crystallized. Both crystals were obtained by the sitting-drop vapour diffusion technique. X-ray diffraction data of a native crystal of His-tagged FlhAC were collected to 2.9 Å at a SPring-8 beamline, BL41XU, and reduced in space group I41, with unit-cell parameters a = b = 216.3 Å and c = 65.1 Å. We obtained the initial phase from multiwavelength anomalous diffraction data, and model building at 2.9 Å resolution is now under way.
113

INVOLVEMENT OF FLHF IN THE REGULATION OF FLAGELLAR NUMBER, SWARM CELL DIFFERENTIATION, AND SECRETION IN BACILLUS CEREUS
S. Salvetti, E. Ghelardi, F. Celandroni, S. Senesi
Dipartimento di Patologia Sperimentale, Biotecnologie Mediche, Infettivologia ed Epidemiologia, Università di Pisa, Italy.
The signal recognition particle (SRP) is an evolutionarily conserved complex that directs
integral membrane and secretory proteins to the cellular protein translocation machinery during
translation, in a GTP hydrolysis dependent manner. In some bacterial species, the SRP complex
comprises a small cytoplasmic RNA, the Ffh protein, and the SRP receptor-like protein FtsY;
both Ffh and FtsY belong to the widely conserved family of SRP-GTPases. Interestingly, it has
been shown that some species belonging to the Bacillaceae and Pseudomonadaceae families
contain a third gene, flhF, encoding a putative protein showing similarity with both Ffh and
FtsY. Although flhF mutants have been produced in Bacillus subtilis and Pseudomonas putida, it
has not yet been demonstrated whether the flhF gene product exerts any SRP-like function.
However, analysis of the flhF mutants revealed that this gene is required in B. subtilis for flagella
assembly, while in P. putida for polar flagellar placement and for the general stress response.
To investigate the role of Bacillus cereus flhF in motility and secretion, a flhF null mutant
was constructed. Our results show that the absence of flhF reduces the number of flagella and the
motility in liquid media. Interestingly, the mutant was unable to produce elongated and
hyperflagellated cells when propagated on solid media, thus resulting in impaired surface-
induced swarming differentiation exhibited by the parental strain. Secretion was increased in the
flhF mutant, the amount of total extracellular proteins being significantly higher in the mutant
compared to the parental strain. Nevertheless, some secreted proteins associated with the
virulence of B. cereus were found to be less abundant in the culture supernatants of the mutant
strain. The hypothetical role of FlhF in motility and secretion in B. cereus will be discussed.
114

ELECTROROTATION OF THE BACTERIAL FLAGELLAR MOTOR Tania Saxl, Mairi Sandison, Hywel Morgan, Richard Berry Clarendon Laboratory, University of Oxford, OX1 3PU, UK
A nanometre precision laser trapping and detection system has been combined with a microelectrode flow cell, for the purpose of electrorotating the bacterial flagellar motor. Asymmetric electrorotatable latex beads have been fabricated and electrorotation of these has been measured in the trap. The beads were then shown to attach reliably to sticky E. coli flagellar filaments. This allows us to apply external torque to the motor via the bead, as an alternative to electrorotating the body. This vastly reduces the viscous drag of the rotating system, enabling measurements to be made outside the narrow range of speeds available with cell body electrorotation. Microelectrode flow cells have been designed to facilitate continuous exchange of the medium during the experiment in order to prevent local ion depletion, and allow protein expression control factors to be added quantitatively. Extending knowledge of the torque speed relationship in the flagellar motor, which is currently only partially mapped, will test theories of motor operation.
115

SYMMETRY IN THE FLAGELLAR WORLD; FLAGELLAR POLYMORPHISM IN THE DEEP SEA BACTERIA Idiomarina loihiensis Shibata, S.,1 Alam, M.,2 and Aizawa, S.-I.1 1. Soft Nano-Machine Project, JST, 1064-18 Hirata, Takanezawa, Shioya-gun, Tochigi 329-1206, Japan 2. Department of Microbiology, University of Hawaii, Snyder Hall, 2538 The Mall, Honolulu, HI96822, USA
The helices of bacterial flagella so far studied seem to be universal in bacterial kingdom, either so-called Normal (left-handed) or Curly (right-handed) flagella dominates despite that the molecular size of the component protein, flagellin, distributes in a wide range from24 kDa to 62 kDa among species. However, these helices are asymmetrical; their mirror images are not identical. The helical parameters (the pitch and diameter) of Normal flagella are double those of Curly flagella. Universality of these two helices and their asymmetry in nature has been of the biological puzzles. The helical parameters are dependent on the packing patterns of flagellins that can be symmetrical. We found that the marine bacteria Idiomarina loihiensis had Curly-like flagella with left-handed helix. The helical parameters of the flagellar polymorphs appeared under different pH conditions agreed with theoretical calculation based on Calladine model (1978) and Hasegawa formula (1998). The results indicate that the left-handed Curly-like flagellum is actually Normal flagellum of I. loihiensis and there exists authentic right-handed Curly flagella. Symmetry in the flagellar world will be discussed.
116

THE EFFECT OF CHEB MUTATIONS ON CHEMOTACTIC SENSITIVITY Thomas Simon Shimizu1, Victor Sourjik2 and Howard C. Berg1 1Department of Molecular and Cellular Biology, Harvard University, 16 Divinity Avenue, Cambridge, MA 02138 USA 2ZMBH, University of Heidelberg, Im Neuenheimer Feld 282, 69120 Heidelberg, Germany
It has been found in multiple studies that Escherichia coli cells lacking the CheB protein have significantly higher thresholds of response to attractant (1-5). This could simply be due to the high methylation levels in these cells, since it has been demonstrated (5-8) that response sensitivity is negatively correlated with receptor modification levels. However, another possibility is that CheB acts directly in signal amplification, through a mechanism that does not involve deamidation/demethylation of receptors. A model for such a mechanism has previously been proposed (9), in which the phosphorylation state of CheB was postulated to play a crucial role.
To test whether such a mechanism is indeed at work, we have used fluorescence resonance energy transfer (FRET) to examine the sensitivity of a number of strains expressing different CheB mutants. Specifically, three CheB mutants were compared to the wild type: CheBC (non-phosphorylatable due to lack of entire regulatory domain), CheBD56E (non-phosphorylatable, due to a point mutation at the phosphorylation site) and CheBS164C (phosphorylatable, but with a point mutation in the catalytic site). We find that mutants that can not be phosphorylated (CheBC and CheBD56E) can still support high sensitivity if expressed at very high levels in a cheB- background. For cells expressing CheBS164C, subtle changes in the dose-response curves were observed with increasing levels of induction, but the maximal sensitivity was still >100-fold lower than cheB+ strains. Therefore, CheB’s involvement in the system’s sensitivity appears to occur predominantly through its catalytic activity as a methylesterase/deamidase, and not through an alternative mechanism that requires changes in its phosphorylation level.
REFERENCES
1. Parkinson JS. J Bacteriol 1978 135:45-53. 2. Block SM, Segall JE, Berg HC, Cell 1982 3:215-226. 3. Segall JE, Block SM, Berg HC. Temporal comparisons in bacterial chemotaxis. Proc
Natl Acad Sci U S A. 1986 83:8987-91. 4. Lux R, Munasinghe VR, Castellano F, Lengeler JW, Corrie JE, Khan S, Mol Biol
Cell 1999, 10:1133-1146. 5. Sourjik V, Berg, HC. Receptor Sensitivity in bacterial chemotaxis. Proc Natl Acad
Sci U S A 2002 99:123-127. 6. Borkovich, KA, Simon MI. Attenuation of sensory receptor signaling by covalent
modification. Proc Natl Acad Sci U S A 1992 89:6756–6760. 7. Li G, Weis RM. Covalent modification regulates ligand binding to receptor
complexes in the chemosensory system of Escherichia coli. Cell 2000 100:357-65. 8. Bornhorst JA, Falke JJ. Quantitative analysis of aspartate receptor signaling complex
reveals that the homogeneous two-state model is inadequate: development of a heterogeneous two-state model.
9. Barkai N, Leibler S. Robust amplification in adaptive signal transduction networks. C. R. Acad. Sci. Paris, Série IV 2001 2:871-877.
117

SSRB ACTIVATION AND TRANSCRIPTION OF SPI-2 GENES Bryan Shimkos, Don Walthers, and Linda J. Kenney University of Illinois Chicago, 835 S. Wolcott Av, Chicago, Illinois 60612
Salmonella enterica serovar typhimurium is capable of intracellular invasion and replication in intestinal mucosa due to large stretches of horizontally acquired genes organized into Salmonella pathogenicity islands, SPIs. Two of these, SPI-1 and SPI-2, are responsible for invasion and intracellular maintenance, respectively. OmpR activates the expression of the SsrA/B two-component regulatory system located in SPI-2. One of the identified signals that activates SPI-2 is low pH, presumably by mimicking the acidified vacuoles containing Salmonella, (SCV). We are interested in SsrB function and its mode of activation of SPI-2 genes. SsrB, a member of the NarL family of response regulators, has been shown to bind directly to secretory apparatus and secreted effector promoters stimulating transcription. SsrB appears to employ multiple mechanisms for activation. For example, binding sites for SsrB are not well conserved for SsrB binding and are located both upstream and downstream of the translational start sites with no predictable spacing. The questions that we are addressing include examination of the SsrB-dependent activation of SPI-2 genes sseA, ssaG, and ssaM, additional SsrB-dependent genes such as sifA, and the effects of transcriptional regulators SlyA and OmpR in combination with SsrB.
This work is supported by grants MCB 0243085 from the National Science Foundation and GM 58746 from the National Institutes of Health to L.J.K.
118

HIGH MOBILITY OF CARBOXYL-TERMINAL REGION OF BACTERIAL CHEMOTAXIS PHOSPHATASE CHEZ IS DIMINISHED UPON BINDING DIVALENT CATION OR CHEY-P Ruth E. Silversmith and Robert B. Bourret, Department of Microbiology and Immunology, University of North Carolina, Chapel Hill, NC 27599-7290
In E. coli chemotaxis, the CheZ phosphatase catalyzes the removal of the phosphoryl group from the signaling molecule, CheY. The co-crystal structure of CheZ with CheY·BeF3
-
·.Mg2+, (a stable analogue of CheY-P) revealed that CheZ is a homodimer with a multidomain, non-globular structure. To explore the effects of CheZ/CheY complex formation on CheZ structure, the rotational dynamics of the different structural domains of CheZ [the four-helix bundle, the N-terminal helix, the C-terminal helix, and the disordered linker between the C-terminal helix and the bundle] were evaluated. To monitor dynamics of the different regions, fluorescein probes were covalently attached at various locations on CheZ through reaction with engineered cysteine residues and the rotational behavior of the fluoresceinated derivatives were assessed using steady state fluorescence anisotropy. Anisotropy measurements at various solution viscosities (Perrin plot analysis) demonstrated large differences in global rotational motion for fluorophores located on different regions. Rotational correlation times for probes located on the four-helix bundle and the N-terminal helix agreed well with theoretical values predicted for a protein of the size and shape of the four-helix bundle. However, the rotational correlation times of probes located on the linker and C-terminal helix were 5-20x higher, indicating rapid motion independent of the bundle. The anisotropies of probes located on the linker and the C-terminal helix increased in the presence of divalent cation (Mg2+, Ca2+, or Mn2+) in a saturable fashion, consistent with a binding event (Kd ~ 1-2 mM) which results in decreased mobility of the probe. Furthermore, the anisotropies of probes located on the C-terminal helix and the C-terminal portion of the linker increased further as a result of binding CheY-P. In light of the recently available structural data and the high independent mobility of the C-terminus demonstrated here, we interpret the CheY-P-dependent increase in anisotropy to be a consequence of decreased mobility of the C-terminal region due to binding interactions with CheY-P, and not to the formation of higher order aggregates of the CheZ2CheY2 complex.
119

CHAPERONE-MEDIATED EXPORT OF FLAGELLAR COMPONENTS Graham P. Stafford, Lewis DB Evans; Rita Krumscheid; Gillian Fraser; Colin Hughes Cambridge University Department of Pathology, Tennis Court Road, Cambridge, UK
Our work focuses on flagellum assembly, in particular the key events involved in the transition of type III flagella export substrates from the cytosol to the membrane and beyond. We have characterised the interactions of the cytosolic chaperones with their substrates and shown that they are ‘bodyguards’ preventing premature interaction of newly synthesised flagellar subunits in the cytosol. By isolation of dominant-negative FlgN variants that are export-defective and arrest flagellar assembly in the wild type bacterium we were able to identify FlgN variants that strongly bound their specific export substrates (the HAPs, FlgK and FlgL) but severely reduced the export of all flagellar type III export substrates, including FlgK and FlgL. These variants were stalled at the hexameric membrane-associated flagellar type III export ATPase, FliI, and identifying a key docking event in the export of substrates and show the chaperones act as secretion ‘pilots’. We are trying to further understand the cycle of chaperoned substrate docking and release into the export machinery before incorporation into the nascent flagellum.
Thomas J, Stafford GP, Hughes C (2004) Docking of cytosolic chaperone-substrate complexes at the membrane
ATPase during flagellar type III protein export. Proc Natl Acad Sci U S A 101:3945-50. Claret L, Calder SR, Higgins M, Hughes C (2003) Oligomerization and activation of the FliI ATPase central to
bacterial flagellum assembly. Mol Microbiol 48:1349-55. Auvray F, Ozin AJ, Claret L, Hughes , C (2002) Intrinsic membrane targeting of the flagellar export ATPase FliI:
interaction with acidic phospholipids and FliH. J Mol Biol 318:941-50. Auvray F, Thomas J, Fraser GM, Hughes C (2001) Flagellin polymerisation control by a cytosolic export chaperone.
J Mol Biol 308:221-9.
120

THE FlhD2C2 MASTER REGULATOR: ACTIVATION OF FLAGELLAR AND OTHER GENES Graham Stafford, Tomoo Ogi, Laurent Claret, Colin Hughes Cambridge University Department of Pathology, Tennis Court Road, Cambridge CB2 1QP, UK
Flagellum assembly requires the tight control of over 50 genes organised in a three tier-regulon. At the apex of the transcriptional hierarchy is the flhDC operon, encoding the master transcriptional regulator, FlhD2C2, which binds and activates the class II flagellar promoters. We previously showed that FlhC2 homodimers bind to class II flagellar promoters, while the specificity, affinity and stability of its interaction with DNA was enhanced by FlhD2 in the complex. Reconstituted transcriptionally active FlhD2C2 heterotetramer was used in primer extension, band-shift assays, and footprinting experiments, a consensus FlhD2C2 binding site comprising 17-18bp inverted repeats, each a consensus FlhD2C2 box, separated by a 10-11bp spacer. To assess whether FlhD2C2 directly controls a wider regulon we searched the E.coli genome for putative FlhD2C2 binding sequences. Using DNA-band shift assays we found that FlhD2C2 binds non-flagellar target sequences with a range of KD values, some comparable to the affinity for class II promoters. These promoters are likewise dependent on FlhD2C2 for activation, but only in cases where the FlhD2C2 site overlaps the σ70 promoter -35 sequence. Stafford, GP, Ogi, T, Hughes C (2004) Binding and activation of non-flagellar gene promoters by the flagella master
regulator FlhD2C2 in preparation Claret L, Hughes C (2002) Interaction of the atypical prokaryotic transcription activator FlhD2C2 with early
promoters of the flagellar gene hierarchy. J Mol Biol 321:185-99. Claret L, Hughes C (2000) Functions of the subunits in the FlhD2C2 transcriptional master regulator of bacterial
flagellum biogenesis and swarming. J Mol Biol 303:467-78.
121

AZOSPRILLUM BRASILENSE EXHIBITS BOTH METHYLATION-INDEPENDENT AND METHYLATION-DEPENDENT TAXIS Bonnie Stephens and Gladys Alexandre Department of Biology, Georgia State University, Atlanta, GA 30303, USA
Azospirillum brasilense is a nitrogen-fixing bacterium that colonizes the rhizosphere of various grasses and cereals promoting plant growth. It has previously been reported that A. brasilense undergoes methylation-independent chemotaxis (1). However, the recently discovered chemotaxis operon in A. brasilense that controls both chemotaxis and aerotaxis contains genes encoding for a CheB methylesterase and a CheR methyltransferase (2). This suggests a role for methylation-dependent motile behavior in A. brasilense. The goal of this work is to determine the contribution of CheB and CheR to the behavioral response of A. brasilense. We have constructed ∆CheBR, ∆CheB, and ∆CheR mutants and characterized their locomotive behavior and demethylation activity. The ∆CheBR mutant had a motility bias similar to the wild type, while the ∆CheB mutant had a highly reversal motility bias. Neither cheB nor cheR mutants had a null phenotype in a swarm plate assay suggesting that CheB and CheR are not essential for chemotaxis. A null phenotype was observed for the aerotactic response of the ∆CheB (but not ∆CheBR) mutant indicating that both methylation-dependent and methylation-independent pathways contribute to the aerotactic response in A. brasilense. Measurements of the methanol release in response to the addition and removal of attractants were carried out for the wild type and the ∆CheBR mutant. The results suggest that CheB and CheR modulate responses to at least some chemoattractants in A. brasilense. The contribution of the methylation/demethylation system for chemotaxis to the plant colonization process will be discussed.
1. Zhulin, I.B., and J.P. Armitage. (1993). J Bacteriol. 175:952-8. 2. Hauwaerts, D., G. Alexandre, S.K. Das, J. Vanderleyden, and I.B. Zhulin. (2002).
FEMS Microbiol Lett. 208:61-7.
122

Proteus mirabilis FliL IS INVOLVED IN SWARMER CELL DIFFERENTIATION Rooge Suvanasuthi and Robert Belas, Center of Marine Biotechnology, 701 E. Pratt St., Baltimore, MD, USA
Proteus mirabilis is a gram-negative dimorphic bacterium that differentiates from a short swimmer cell to an elongated, highly flagellated swarmer cell in response to growth on surfaces or in viscous media. The inhibition of flagellar rotation appears to be critical for P. mirabilis swarmer cell differentiation, but the molecular mechanism whereby the signal is transduced into the cell to control gene expression remains a mystery. In this study, transposon mutagenesis was used to construct mutants defective in swarming and biofilm formation. Among these mutants is a class that produces nonswarming, constitutively elongated swarmer cells, suggestive of a defect in the surface sensing mechanism. The cause of this phenotype was identified as a mutation in fliL, a gene associated with the P. mirabilis flagellar biosynthesis and motility locus. FliL is a membrane-associated component of the flagellar basal body of Salmonella enterica serovar Typhimurium. Interestingly, fliL mutations in Escherichia coli and Salmonella are cryptic and do not affect swimming or swarming motility. However, in Caulobacter crescentus, fliL defects adversely affect flagellar motility and cell division, signifying that FliL may play a similar role in P. mirabilis swarmer cell differentiation. Complementation of the fliL mutation by plasmid-borne fliL restores wild-type cellular morphology, but not motility, indicating that the transposon insertion has negative effects on the flagellar biosynthetic and type three secretion genes (fliMNOPQR) downstream of fliL. Over-expression of fliL in a wild type background results in cells that are unable to differentiate under normally permissive conditions and are nonmotile. Over-expression of fliL also resulted in a marked reduction in flagellin (FlaA) synthesis, therefore FliL may also function in the regulation of flagellin synthesis. One interpretation of these data is that FliL is involved in repressing swarmer cell differentiation, specifically cell elongation and flagellin synthesis. The fliL mutation also affects the expression of zapA and hpmB, two virulence genes that are up-regulated during swarmer cell differentiation and swarming motility. These data suggest a possible mechanism whereby FliL may be involved in relaying the signal from the surface, through the flagellar filament, and into the cell.
123

A DOMINO-TOPPLING MODEL OF FLAGELLAR MOTOR SWITCH Guanglai Li, Qi Wen, and Jay X. Tang Physics Department, Brown University, Providence, RI 02912
Switch of flagellar motor plays a central role in the motility and chemotaxis of flagellated bacteria. For example, when all the flagella of E. coli rotate counterclockwise (CCW), the flagellar filaments form a bundle. The bundle rotates CCW and pushes the cell body forwards, leading to smooth swimming. When one or more flagella switch rotation to clockwise (CW), the bundle collapses and the cell starts tumbling. During the tumble, the cell reorients, and then swims along a new direction after all flagellar motors switch back to CCW rotation. The concentration gradient of attractant or repellent changes the frequency of tumbling and the cell moves up the gradient of the attractant or down the gradient of the repellent. To study the rotation of individual flagellar motor, a cell can be tethered to a surface by its flagellar filament and the rotation of the cell body is recorded. Alternatively, a micro-sized bead can be attached to a flagellar filament of a cell immobilized on a surface and the rotation of the bead is recorded. Two types of experimental data help understand the switch mechanism of the flagellar motor: (1) distribution of intervals of a flagellar motor rotating CW and CCW; (2) dependence of the switches in rotation on the concentration of CheY-p.
A well known switch model is the two-state model, assuming different free energies for CW and CCW rotations and a free energy barrier between them. This model predicts an exponential distribution for the intervals of a cell rotating CW and CCW, which has been observed for some bacterial species by measuring a population of cells. By assuming that the binding of CheY-p can change the free energy of each rotation, this model can also interpret the dependence of the switch probability on the concentration of CheY-p. However, a recent observation of individual E. coli cells shows that the interval distribution deviates from exponential distribution. The non-exponential distribution has also been observed in other species such as Halobacteria.
In our lab, we are studying the switch of the flagellar motor of Caulobacter crescentus. We find a completely different interval distribution from the exponential decay. Caulobacter crescentus is gram-negative bacterium and is believed to share similar structure and function of flagellar motor with E. coli. We propose a Domino Toppling model for the motor switch. In this model, the noise induces switches of FliM between CCW and CW conformations, at threshold fractions of binding of Che Y-p. The conformational transition of any FliM will drive the same transition of all the other FliMs in the switch complex, much like the collapse of a ring of Domino tiles. Consequently, the whole motor complex switches between CW and CCW states. This model can interpret the interval distribution of Caulobacter crescentus, as well as those reported in the literature for E. coli. This model can also interpret CheY-p dependence of flagellar motor of E. coli, and provide a molecular mechanism of how the binding of CheY-p affects the switch of a flagellar motor.
124

THE SIGMA FACTOR ALGT INHIBITS PSEUDOMONAS AERUGINOSA FLAGELLUM BIOSYNTHESIS BY REPRESSING EXPRESSION OF THE MASTER REGULATOR FLEQ Tart, A. H.1; Wolfgang, M. C.2; Blanks, M. J.1; and Wozniak, D. J.1 1Wake Forest University School of Medicine; Winston-Salem, NC 27157. 2University of North Carolina at Chapel Hill; Chapel Hill, NC27599.
Pseudomonas aeruginosa is the terminal pathogen in individuals suffering from cystic fibrosis (CF). Environmental strains colonizing the CF lung generally have a motile phenotype. However, as the disease progresses, these strains frequently convert to a non-motile phenotype. In many cases, this is coordinately regulated with the overproduction of the exopolysaccharide alginate. Both the expression of alginate (mucoidy) and loss of flagellum synthesis provide the bacterium with a selective advantage in the CF lung. We observed that in CF isolates, the mucoid and the non-motile phenotype occur predominantly, though not exclusively, together. Studies in our laboratory revealed that alginate and flagellum biosynthesis are inversely regulated by the alternative sigma factor AlgT (AlgU, σ22). The goal of the current study is to determine the AlgT-mediated regulatory mechanism involved in the inhibition of flagellum expression. Microarray analysis comparing mRNA levels of isogenic AlgT+ and AlgT- P. aeruginosa was performed to determine which genes within the flagellar pathway are controlled by AlgT. The results showed that the vast majority of flagellar genes were significantly downregulated in the presence of AlgT. A pronounced inhibitory AlgT-effect was observed in several genes essential for proper flagellum expression, including the gene coding for the flagellar master regulator, fleQ. This was confirmed using promoter fusion assays in isogenic AlgT+ and AlgT- strains. In addition, motility assays and transmission electron microscopy showed that overexpression of FleQ in mucoid, non-motile CF isolates restored both flagellum biosynthesis and motility. Electrophoretic mobility shift studies using purified AlgT and extracts derived from isogenic AlgT+ and AlgT- strains suggest that AlgT indirectly inhibits fleQ transcription. Together, these data show that AlgT mediates the negative control of flagellum biosynthesis by indirectly inhibiting the expression of the master regulator fleQ.
125

MUTATIONAL ANALYSIS OF FLIG FROM THE SINGLE UNIDIRECTIONAL FLAGELLUM OF RHODOBACTER SPHAEROIDES. John M. Taylor, Karen A. Morehouse, Richard D. Woods and Liz Sockett. Institute of Genetics, Queens Medical Centre, University of Nottingham, Nottingham, NG7 2UH, UK.
Bacteria are capable of movement through an aqueous environment by the rotation of protruding helical flagellar filaments. The direction of rotation has a profound effect on swimming behaviour and is influenced by chemosensory pathway, notably the interaction between CheY and FliM, consequently manipulating FliG by its close association with FliM. R. sphaeroides (Rs) has a medially located flagellum and modulates the swimming course by stopping and starting rotation. Conversely, E. coli (Ec) revise their swimming pattern by reversing the direction of their bi-directional motor, allowing for smooth motility, by virtue of counter clockwise (CCW) rotation, and tumbles by the clockwise (CW) rotation. Thus, contrary to enteric bacteria, Rhodobacter motility is the product of the stop-start action of a single flagellum.
Irrespective of the mode of motility, FliG proteins, with very few notable exceptions, contain a conserved diglycine motif located between the carboxyl terminal and linker middle domain. (Diglycine motifs considered between domains of other proteins are thought to act as flexible hinges, thus directed mutational studies were carried out on this region to elucidate whether the proposed flexibility is important for FliG-MotAB interactions during the torque-generating power stroke or the flagellin export. Subtle increments into the bulk of the glycine side chain revealed that steric hindrance has a profound effect on flagellin export, while the motility of the relatively conservative substitution G202A is not significantly perturbed, although percentage motility is slightly reduced. Thus the flexibility of the diglycine hinge may have a primary role in maintaining the elasticity of the C ring while flagellin subunits are exported.
126

THE THREE-DIMENSIONAL STRUCTURES OF THE ROD, HOOK and FILAMENT JUNCTION PROTEINS SALMONELLA TYPHIMURIUM BY CRYO-EM. Dennis R. Thomas1#, Noreen R. Francis1, Xu Chen1, and David J. DeRosier1,2,
11 Rosenstiel Biomedical Sciences Research Center, Brandeis University, Waltham, MA 02454 2 Department of Biology, Brandeis University, Waltham, MA 02454
The bacterial flagellum used to swim by many motile bacteria, such as E. coli and S. typhimurium, is a remarkable nanomachine. It has a reversible rotary motor powered by the proton gradient across the cell’s plasma membrane. The motor is coupled to the flagellar filament, the propeller, and transmits torque to the filament via the rod, drive shaft and the hook, the universal joint. More than 40 genes are known to be involved in motility in S. typhimurium and E. coli. There are 20 proteins that are known structural components of the flagellum. 9 (10?) of these form the axial components which make up the helical filament. The filament structure is assembled in order after the motor and export apparatus are in place. The axial proteins compose the rod or drive shaft (FlgB, FlgC, FlgF, FlgG, FliE?), the hook or universal joint (FlgE), the HAPs junction or the coupler (FlgK or HAP1, FlgL or HAP3), the filament (FliC or flagellin) and the cap (FliD or HAP2). The axial proteins all have both N and C terminal heptad repeats characteristic of coiled-coil regions and have all been found to form the same type of helical lattice. The exception is HAP2 which has only an N terminal heptad repeat consistent with its role in acting as a cap for the filament chaperoning the assembly of flagellin monomers underneath the cap. The filament has been solved to essentially atomic resolution. The hook has been recently solved to about 9Å resolution and recently docked with the atomic models of the D1 and the D2 domains (Shaikh et al 2004).
The HAPs junction structure is very important for the mechanical stability of the filament. The junction proteins share N and C terminal homology with the axial proteins of the filament, but the differences may be critical for the role of the junction in assembly of the filament. There is a cap for hook assembly and for filament assembly but not for the addition of junction subunits. Yet only one turn of each is added (Ikeda et al 1987). This suggests very specific interactions exist for HAP1 and HAP3. Cells with mutations in the middle of the HAP3 protein appear to form normal flagellar filaments, but cannot swim normally on soft agar plates (Fahrner, Block et al. 1994). The cause is a transformation of the filaments to a straight form that begins at the base and is propagated to the tip of the filament. HAP3 occupies the last helical turn just before flagellin in the helical filament, suggesting that the mechanical stability of the filament depends in part, on the structure that it is attached to.
We present three dimensional reconstructions of the rod, hook and filament junction proteins with the filament cap from electron micrographs of frozen hydrated bacterial flagellar motors. We can follow the transitions from one protein to the next through the three dimensional maps.
127

GENOME-BASED ANALYSIS OF CHEMOTAXIS COMPONENTS IN GEOBACTER SPECIES H. T. Tran1, F. M. Antommattei1, R. Glaven2, D. R. Lovley2 and R. W. Weis1
1Department of Chemistry, University of Massachusetts-Amherst, Amherst, MA 010032Department of Microbiology, University of Massachusetts-Amherst, Amherst, MA 01003
Geobacteraceae, which belong to the delta subdivision of proteobacteria, are the
predominant Fe(III)-reducing microorganisms in a variety of sedimentary environments. It has been subjected to extensive studies because of their potential application of these species in energy production and environmental bioremediation. In contrast to other iron-reducing microorganisms, Geobacter require direct contact to the surface of the Fe(III) oxide sources. The detailed mechanism for this process is still not well understood. Previous studies with Geobacter metallireducens demonstrated the successful effort to detect motility and chemotaxis under anaerobic conditions in iron(II) gradients, suggesting that this could be the key step in accessing Fe(III) oxide. These results provide compelling evidence that chemotaxis can play an essential role in Geobacter physiology, and provides the impetus for further investigation. To this first effort, the genome of Geobacter species was analyzed for chemotaxis components. The genomes of G. sulfurreducens and G. metallireducens contain multiple homologs of the chemotaxis genes found in E. coli, such as cheW, cheA, cheY, cheR, cheB, as well as some chemotaxis genes, such as cheC, cheD and cheV, that are not present in E. coli. G. sulfurreducens has 32 genes encoding methyl-accepting chemotaxis proteins (MCPs), and G. metallireducens has 24. These che genes are arranged in 5 putative che operons in G. sulfurreducens and 4 in G. metallireducens. Domain organization and topology of the MCPs demonstrated that Geobacter contain typical chemoreceptors (with 2 transmembrane regions, HAMP and MA domain), and some soluble MCPs. In addition, Geobacter sp MCPs contain domains that are not usually present in enteric bacteria, such as, the GAF, and globin. Altogether, our analysis and the available expression data provide useful information for the experimental design, in order to define and understand the role of chemotaxis in Geobacter ecology.
128

THE IMPORTANCE OF CYSTEINE RESIDUES IN THE TRANSCRIPTIONAL ACTIVATOR SsrB Anna Turabelidze, Alvin Go, and Linda J. Kenney Department of Microbiology and Immunology University of Illinois at Chicago, 835 S. Wolcott Avenue, M/C 790 Chicago, IL 60612.
Expression of genes located on Salmonella pathogenicity island 2 (SPI-2) is required for systemic infection in mice (1). A two-component regulatory system SsrA/B is located in this region. The sensor kinase is SsrA, and SsrB is the response regulator. SsrB is the predominant transcriptional activator of virulence genes in S. typhimurium within host cells. It is in the same subfamily as NarL, whose structure has been solved (3). Among response regulators SsrB is unique in that it contains three cysteine residues: C45, C46, and C203. Our laboratory has previously shown that C45 is essential for SsrB activity (2). The observation that C46 is not essential in SsrB activity eliminates the possibility of a vicinal disulfide bond formation in the SsrB regulation of transcription. The possibility that substitution of C45 with alanine prevents phosphorylation by phosphoramidate is under investigation. In order to further understand the importance of C45, we have isolated SsrB mutants in which all of the cysteines except C45 have been substituted with alanine. Based on the NarL structure, C45 is predicted to be at the interface between the N and C-terminal domains. This interface undergoes a substantial change upon phosphorylation before binding to DNA in the NarL structure. In contrast to NarL, using the double mutant, C46AC203A, we determined that C45 is surface exposed in denatured, unphosphorylated, and phosphorylated states. SsrB is distinct from NarL by this conformational analogy. Using the cross-linker, BMH, we have determined that C45 is not involved in homo-dimerization. Additional studies are aimed at further elucidating the role of this important cysteine in SsrB-dependent activation of transcription. This work is supported by GM 58746 from the National Institute of Health and MCB 0243085 from the National Science Foundation. References
1. Feng, X., Oropeza, R., and Kenney, L. (2003) Molecular Microbiology 48, 1131-1143.
129

SUBCELLULAR ORGANISATION OF COMPONENTS OF THE CHEMOTAXIS SIGNALLING PATHWAY IN RHODOBACTER SPHAEROIDES Wadhams, G.H., and Armitage, J.P. Microbiology Unit, Biochemistry Dept, University of Oxford, South Parks Road, Oxford. OX1 3QU. U.K.
Rhodobacter sphaeroides is a purple non-sulphur bacterium which demonstrates taxis to a wide range of stimuli. Genome sequencing of this bacterium has revealed the presence of three major chemotaxis operons containing multiple homologues of most of the chemotaxis genes found in E. coli. It has also been shown that genes encoded in both cheOp2 and cheOp3 are essential for chemotaxis. Recent data from our lab has revealed that the components of the signalling pathway encoded in cheOp2 mostly localise to the poles of the cell whereas those encoded in cheOp3 predominantly localise to a protein cluster within the cytoplasm.
The localisation of the individual components of the chemotaxis signalling pathways was studied by constructing in-frame fusions to either CFP or YFP and replacing the wild-type gene in the bacterial genome with the corresponding fusion gene. The localisation of the fusion proteins was then determined by fluorescence microscopy both in a wild-type background and in strains deleted for other components of the chemotaxis pathway.
Data will be presented on the requirements of specific components of the chemotaxis signalling pathway for the correct localisation of other chemotaxis proteins. The implications of this for our understanding of the assembly of these clusters will be discussed.
130

REVERSED SIGNAL OUTPUT CONFERRED BY A SINGLE RESIDUE SUBSTITUTION IN THE LIGAND-RECOGNITION DOMAIN OF A NARX-TAR CHIMERIC RECEPTOR Scott M. Ward and Michael D. Manson Department of Biology, Texas A&M University, College Station, TX 77843
Proteins that span the cytoplasmic membrane transmit information from the exterior of
the cell exterior to the interior. In E.coli, these signal transducers include sensor kinases, which
typically control gene expression via response regulators, and methyl-accepting chemoreceptors
(MCP’s), which control flagellar rotation via the CheA kinase and CheY response regulator. We
previously reported that a chimeric protein joining the ligand-binding, transmembrane, and linker
regions of the NarX sensor kinase to the signaling and adaptation domains of the Tar
chemoreceptor elicits a repellent response to nitrate and nitrite. As with NarX, nitrate evokes a
stronger response than nitrite. Here, we show that mutations in a highly conserved sequence (the
P-box) in the periplasmic domain of the hybrid receptor alter chemotactic signaling in a manner
consistent with their effects in the intact NarX protein. The most dramatic effect is associated
with the G51R substitution, which confers a reversed-signal phenotype that converts nitrate into
an attractant. Our results provide further evidence for the conservation of the mechanism of
transmembrane signaling in homodimeric sensor kinases and chemoreceptors and highlight the
plasticity of the coupling between ligand binding and signal output in these systems.
131

MUTAGENESIS AND CROSSLINKING STUDIES ON THE AER PAS N-CAP SUGGEST FLEXIBILITY AND A ROLE IN SIGNALING K. Sommer, K.J. Watts, M.S. Johnson, and B.L. Taylor Division of Microbiology and Biochemistry, Loma Linda University, Loma Linda, CA, 92350
Aer, the Escherichia coli aerotaxis-, energy- and redox-receptor, contains a cytosolic PAS domain that is separated from a HAMP domain and signaling region by a membrane anchor. The Aer-PAS domain contains an N-terminal segment, often called an “N-cap” which is poorly conserved in other PAS domains. The N-cap is disordered in both PYP and HERG, and can be deleted without incurring significant structural changes in PYP. To examine the importance of the N-cap in Aer, we studied the behavior of mutants generated by DNA mutagenesis. N-cap residues from 5-21 were mutated to all possible amino acid codons using degenerate primers, created by using an equimolar mix of all four nucleotides at a specific codon. Aerotaxis mutants were identified by their behavior on swarm plates and their response to oxygen in a temporal gradient. Amino acid substitutions in the N-cap caused four aerotaxis phenotypes: null, constantly tumbly, tumbly biased, and inverse. Non-aerotactic Aer mutants were unable to bind FAD, and could not be functionally resurrected by S28G, a suppressor that restores aerotaxis to both Aer-F1 and Aer-HAMP FAD- mutants. The proximity of two N-cap regions in an Aer dimer was then assessed by serially converting residues 4 to 22 into cysteine and performing in vivo crosslinking with copper phenanthroline. Cysteine replacements H4C and P5C crosslinked with their cognate cysteines at room temperature. However, this crosslinking was substantially reduced at 10ºC, suggesting that these interactions might be due to high flexibility in this region rather than due to stable structural proximity. The remaining N-cap residues crosslinked weakly or not at all at room temperature, suggesting that the two N-cap regions are not closely associated. The data show that mutations in the N-cap can critically alter signaling functions in Aer. Whether the N-cap stabilizes FAD binding and propagates signal transduction directly, or whether this region protects other residues critical for these functions, is not yet known.
132

THE DEVELOPMENT OF A LUX-BASED BIOSENSOR FOR THE DETECTION OF ENVIRONMENTAL POLLUTANTS
Rachel M. Webster,* Timothy Hart, † and Judith P. Armitage*
*Microbiology Unit, Department of Biochemistry, University of Oxford, South Parks Road, Oxford, OX4 3QU, U.K
†Cybersense Biosystems Limited, Milton Park, Unit 44C, Abingdon, OX14 4RU, U.K
The decontamination and assessment of soils is a priority in environmental legislation. Bioluminescent microbial biosensors are being developed to overcome the limitation of current technologies that require transport of the pollutant into the cell, and often subsequent metabolism.
To measure the extracellular presence of a pollutant, a functional chimeric protein combining the periplasmic ligand binding domain of a chemoreceptor with the conserved cytoplasmic domain of an osmosensor (EnvZ) has been engineered. Binding of the ligand induces activation of the EnvZ cytoplasmic domain and hence the cognate cytoplasmic transcriptional activator (OmpR). OmpR then drives transcription of the bioluminescent lux genes fused behind the ompC promoter. A plasmid containing the ompC-lux fusion was constructed and introduced into Escherichia coli. The osmoresponse of the luciferase circuit was assessed by measuring the light output response to osmotic strength and quantified using a luminometer. There is a dose-dependent response to osmotic strength that is absent in envZ and ompR mutant E. coli strains. In addition a receptor chimera between tar (chemoreceptor for aspartate) and the cytoplasmic domain of envZ, demonstrates the induction of lux genes in response to a specific extracellular chemical. The chimeric chemoreceptor thus senses the extracellular concentration of a specific pollutant and the level of bioluminescence from the ompC-lux fusion is a direct measure of the extracellular concentration.
133

CHARACTERIZATION OF THE MYXOCOCCUS XANTHUS CHE7 CHEMOSENSORY SYSTEM Janet Wilson and John Kirby Georgia Institute of Technology, School of Biology, 310 Ferst Dr, Atlanta, GA 30332
Analysis of the Myxococcus xanthus genome indicates the presence of eight cheA genes each defining a che cluster that also includes mcp, cheW, cheB, and cheR genes. In general, each of the eight che clusters have a unique organization of the chemotaxis-like genes as well as unique response regulators. Based on the previous characterization of frz, dif, and che3, it is clear that we are currently unable to predict the function of any given set of chemotaxis-like genes in M. xanthus based solely on sequence information. We therefore analyzed the M. xanthus CheA protein homologs in the context of the full set of known CheA homologs from the completed genomes in the publicly available database.
The M. xanthus che7 cluster encodes homologs to CheY, CheA, CheW, an MCP, CheR, and CheB in addition to a response regulator-histidine kinase fusion, a phycocyanobilin lyase (Cpc), and a fatty acid desaturase (Des). A divergently transcribed iron uptake regulator (Fur) was identified upstream of the putative promoter region for che7. Translational coupling is predicted for CheY7-CheA7-CheW7, Mcp7-Cpc7, and CheR7-CheB7 suggesting that these proteins may function together as part of a larger complex to regulate signal transduction.
To investigate a role for che7 during motility or development, mutations were made in the che7 cluster in the pilQ1 background. None of these mutants displayed any motility defects on either CF media or nutrient rich hard agar plates. All of the mutants were able to form aggregates when placed on starvation media. However, when starved, cells with either the cheA7 or the cpc7 insertion mutations were unable to form fruiting bodies. The fur7 and cheB7 mutants were delayed and only formed a few aggregates while the cheR7 mutant formed fruiting bodies earlier than the parent strain. Mutations in the regulator/kinase fusion, mcp7, and des7 all resulted in colonies that formed fruiting bodies around the perimeter, but not in the center of the spot. Because the mutants are able to aggregate and show no obvious motility defects we believe that the proteins encoded by the che7 cluster do not regulate motility. We conclude that the developmental defects are mediated through che7’s effect on either gene transcription or the generation/reception of a signal that influences the decision of the colony to stay on the developmental pathway.
134

STRUCTURAL STUDIES OF REGULATORY PROTEIN-PROTEIN INTERACTIONS IN CHEMOTAXIS SIGNAL TRANSDUCTION P.M. Wolanin1, N.R. Francis2, D.R. Thomas2, M.D. Baker3, D.J. DeRosier2, J.B. Stock1,3 1- Department of Molecular Biology, Princeton University, Princeton, NJ 08544 2- Rosenstiel Biomedical Sciences Research Center, Brandeis University, Waltham, MA 02454 3- Department of Chemistry, Princeton University, Princeton, NJ 08544
The bacterial chemotaxis system is an important model for understanding two-component signaling, as well as for the general problem of transmembrane signal transduction. We are currently using several approaches to understand the molecular details of the transmembrane signal transduction process in bacterial chemotaxis. In particular, we want to define the key protein-protein interactions that occur among the chemotaxis receptors, CheW, and CheA.
The approach that has been most productive is to use soluble chemotaxis receptor constructs consisting of a leucine zipper domain fused to the cytoplasmic part of the Tar receptor (lzTarC). These constructs have previously been used in both biochemical and structural studies of chemotaxis signaling complexes. Complexes of CheA and CheW with either lzTarC or lzTar516 (a C-terminal truncation) have been imaged using high-resolution transmission electron microscopy. Image analysis of the complexes has resulted in an initial three-dimensional reconstruction of the structure of the complexes that provides much more insight into the organization of the complex than the previous two-dimensional averages. In particular, the arrangement of receptor fragments within the complex is clearly very different than the “trimer of dimers” structure of soluble Tsr fragments determined through crystallization of the Tsr fragment in the absence of CheA and CheW.
135

A STUDY ON THE MECHANISMS OF ALLOSTERY IN THE CHEMORECEPTORS Tar and Tsr Gus Wright and Michael D. Manson, Department of Biology, Texas A&M University, College Station, TX 77843. This bacterial behavior toward chemicals is termed chemotaxis. Chemotaxis is mediated by transmembrane chemoreceptors, a SH3-like coupling protein (CheW), a histidine kinase (CheA) and the response regulator (CheY). There are five chemoreceptors known in E. coli. These are the serine receptor (Tsr), aspartate/maltose receptor (Tar), ribose/galactose receptor (Trg), dipeptide binding protein receptor (Tap), and the oxygen receptor (Aer). The aspartate/maltose receptor (Tar) and the serine receptor (Tsr) are the high abundance major transducers. Trg, Tap, and Aer are the low abundant minor transducers.
We are currently investigating the allosteric behavior of the two major transducers Tar and Tsr. Tar is known to bind aspartate with extremely high negative coorperativity, or half-of –sites binding. Tsr is known to bind serine with negative cooperativity. It is also known that when the receptors bind their respectively substr�tes they inhibit CheA kinase activity. The mechanism of how the binding of substrate to the receptor inhibits the CheA activity is not known. We hypothesize that there are conformational changes in the receptor upon substrate binding that influence the conformation of CheA to inhibit its activity. First, we are studying the affect of deleting the transmembrane regions from Tsr and Tar has on the CheA activity. Next we will study what conformational changes occur in the solublized Tar and Tsr upon substrate binding using fluorescence spectroscopy. We are also studying the conformational changes of reconstituted full length Tar and Tsr and CheA upon binding of substrate using fluorescence spectroscopy. Finally, we are investigating conformational changes of Tsr and Tar containing only the periplasmic, transmembrane and linker regions upon binding of substrate using fluorescence spectroscopy. We expect that the conformational changes of the transmembrane region and the linker region of Tar and Tsr are responsible for the inhibition of CheA activity. We also expect that the inhibition of CheA is caused by a conformational change in CheA imposed by the receptor when substrate is bound.
136

ACTIVATION OF THE DIF PATHWAY BY NARX-DIFA CHIMERA IN MYXOCOCCUS XANTHUS Qian Xu,1 Michael D. Manson2 and Zhaomin Yang1
Department of Biology 1Virginia Polytechnic Institute and State University, Blacksburg, Virginia 24061 2Texas A & M University, College Station, Texas 77843 The fibril expolysaccharide (EPS) is essential for the social gliding motility and development of Myxococcus xanthus. The chemotaxis-like Dif pathway is crucial for the regulation of M. xanthus EPS production. DifA, a homolog of methyl-accepting chemtaxis proteins (MCP), is proposed to mediate signal input into the Dif pathway. However, DifA lacks a prominent periplasmic domain and how DifA perceives and transmits signals remain enigmatic. In this study, steps were taken to investigate whether DifA shares similar signaling mechanism with other bacterial membrane sensors/receptors. We constructed a chimeric protein composed of the sensory module of Escherichia coli NarX and the methylation and signaling domains of DifA. The NarX-DifA (NafA) chimera was expressed in M. xanthus under the control of the E. coli tar promoter (Ptar) or the M. xanthus dif promoter (Pdif). Immunoblotting indicated that NafA under Pdif was expressed to a level similar to DifA expression in wild-type but undetectable under Ptar. Nevertheless, both the Ptar-nafA and the Pdif-nafA constructs were able to restore fruiting body formation to difA mutant in the presence of nitrate. Studies with difA difC, difA difD, difA difE and difA difG double mutants indicated that the cross-species NafA chimera still signals through the Dif pathway in M. xanthus. We also showed that NafA restored agglutination, S-motility and EPS production to difA mutants when nitrate was added. These results suggest that, despite the apparent structural differences, DifA shares similar transmembrane signaling mechanisms with the classical sensor kinases and chemoreceptors. Additionally, nafA was found to suppress the defects of pilA mutants in EPS production, implying that pili are upstream of the Dif proteins in the EPS regulation pathway.
137

STUDIES ON ROTOR-STATOR INTERACTIONS IN THE FLAGELLAR MOTOR OF E. COLI Jung-Hoon Yang1, Toshiharu Yakushi2, Hajime Fukuoka2, Michio Homma2 and David F. Blair1 1Department of Biology, University of Utah, Salt Lake City, UT84102 2Graduate school of Biological Science, Nagoya University, Chikusa-ku, Nagoya, 464-8602, Japan Bacterial flagellar rotation involves interaction between a rotor complex formed from FliG, FliM and FliN and stator complexes formed from MotA and MotB (or PomA and PomB in Vibrio). Previous studies in E. coli showed that electrostatic interactions between conserved charged residues in FliG and MotA are functionally important. Mutational experiments in Vibrio showed that electrostatic interactions might not be as important in the sodium-fueled motor, or that additional residues might participate in rotor-stator interactions. To probe the inter-species differences further, we are making use of chimeric proteins that contain key domains of Vibrio FliG or PomA, but which function in E. coli. Several charge-reversing and charge-neutralizing mutations in key residues were introduced in stator and in rotor proteins from Vibrio or E. coli. In a cell using the stator from E. coli and a rotor component from Vibrio, mutational defects and patterns of suppression were similar to what was observed previously with purely E. coli proteins. These results suggest that certain aspects of rotor-stator interactions are the same in proton-fueled and sodium-fueled motors. As an additional means of probing rotor-stator interactions, we are initiating crosslinking experiments that utilize Cys residues introduced in place of some of the interacting charge residues. Certain combinations of Cys residues on FliG and MotA appear to form disulfide crosslinks, consistent with close approach of these proteins within the flagellum. Additionally, certain mutations in MotA alter the patterns of crosslinking between FliG subunits, suggesting that rotor-stator interactions influence the arrangement or flexibility of FliG subunits on the rotor.
138

IMAGING OF CHEMOTAXIS RECEPTOR CLUSTERS IN E. COLI CELLS OVERPRODUCING TSR Peijun Zhang and Sriram Subramaniam Laboratory of Cell Biology, National Cancer Institute, NIH, Bethesda, MD 20817
Bacterial chemotaxis is a very well studied signal transduction system in biology. Many studies have suggested that the ability of bacterial cells to respond a wide range of chemical gradients can be achieved by amplifying the signal of ligand binding through receptor clustering. To elucidate the architecture and signaling role of chemoreceptor clusters, we are investigating receptor arrangements in E. coli cells over-producing Tsr using electron microscopy. We have shown previously that Tsr receptors are found in hexagonal micro crystalline arrays in detergent-treated membrane extracts with a repeating unit large enough to accommodate one trimer of receptor dimers (1). Tomograms constructed from fixed, cryo-sectioned cells reveal that the overproduction of Tsr leads to the formation of an extended internal membrane network composed of stacks and rounded structures similar to those found in membrane extracts (2). The Tsr receptor clusters were directly visualized in images recorded from plunge-frozen cells and from frozen-hydrated sections of high pressure-frozen E. coli cells (3). Further, tomograms reconstructed from frozen hydrated cells show detailed arrangements of Tsr receptors in these clusters. These studies suggest that the formation of the membrane stacks containing receptor clusters is stabilized by interactions between the cytoplasmic ends of Tsr as well as between the periplasmic ends. These interactions could be important in the mediation of signal amplification in bacterial chemotaxis. 1. Weis et al., (2003) J Bacteriol. 185:3636-3643 2. Lefman et al., (2004) J Bacteriol. 186:5052-5061 3. Zhang et al., (2004) J Microscopy 216: 76-83
139

CHEW-MEDIATED SUPPRESSION OF CHEA RECEPTOR-COUPLING DEFECTS Jinshi Zhao and John S. Parkinson Department of Biology, University of Utah, Salt Lake City, UT 84112, USA CheA, the histidine kinase for chemotactic signaling in bacteria, is regulated by transmembrane chemoreceptors in response to extracellular stimuli. CheA control takes place in a ternary complex with receptors and the cytoplasmic protein CheW. CheW binds to chemoreceptors and to CheA and is widely assumed to function as an adapter or linker in the ternary complex, possibly transmitting ligand-induced conformational changes between the receptors and CheA. To investigate the mechanism of CheA regulation in receptor signaling complexes, we first carried out mutational and cysteine-scanning analyses of the C-terminal receptor coupling domain (P5) of E. coli CheA. Those studies identified a partly exposed portion of the P5 surface, distinct from the CheW binding sites previously proposed by other groups, that was critical for CheW-CheA binding in vitro. Amino acid replacements and chemical modifications at P5 residues that pack against the CheA-P3 dimerization domain (in the T. maritima crystal structure) also impaired binding to CheW, suggesting that the in vitro binding interaction might require dynamic motions of the P5 domain. The cysteine-scanning studies also identified a P5 surface near the interface with the ATP-binding domain (P4), at which bulky modifications impaired receptor-mediated activation of CheA but not CheW binding. To further understand the role of the CheA-CheW binding interaction in vivo, we constructed chemotaxis-defective CheA mutants with missense mutations at the putative CheW-binding determinants in the P5 domain. We then asked whether CheW alterations could restore chemotactic ability to these CheA-P5* mutants. To our surprise, we found a variety of CheW missense mutations (designated CheW!) that phenotypically suppressed CheA-P5* defects. The CheW! mutations occurred at both buried and surface residues, including E13, P14, E18, F19, D32, I33, K35, Q37, Y42, T51, V66, V87, V105, and D107. Many of these residues lie at the interface between the two SH3-like subdomains of CheW. Many of them also coincide with sites previously implicated in the binding interaction between CheW and the chemoreceptors. All CheW! mutations retained wild-type CheW function, but alleviated the chemotaxis defects of many CheA-P5* mutations, including ones outside the proposed CheW-binding region. The lack of allele specificity in these interactions argues for a “global” suppression mechanism, perhaps based on co-folding interactions between CheA and CheW that only occur in vivo. We suggest that the current in vitro assays for CheA-CheW binding and receptor coupling do not fully reflect the in vivo situation. The existence of chemotaxis-defective CheA-P5* mutants with ostensibly normal in vitro activities lends support to this view.
140

BLAST VIII PARTICIPANT LIST
141

142
Jun Adan Osaka City University Sumiyosi-ku Osaka, Osaka 558-8585 JAPAN Phone: +81-06-6605-3157 Fax: +81-06-6605-3158 [email protected]
Shin-Ichi Aizawa CREST, JST 1064-18 Hirata Takanezawa, Tochigi 329-1206 JAPAN Phone: 028-676-8510 Fax: [email protected]
Roger Alexander Georgia Tech 310 Ferst Drive Atlanta, GA 30332-0230 Phone: (404) 217-5664 Fax: [email protected]
Gladys Alexandre Georgia State University 24 Peachtree Center Avenue, Kell Hall Atlanta, GA 30303 Phone: (404) 651-2786 Fax: (404) 651-2509 [email protected]
Peter Ames University of Utah 257 South 1400 East Salt Lake City, UT 84112-0840 Phone: (801) 581-3592 Fax: (801) 581-4668 [email protected]
Divyaben Amin Loma linda University AHBS 120, 24941 Stewart street Loma Linda, CA 92354 Phone: (909) 558-1000 ext 42748 Fax: (909) 558-4035 [email protected]
Judy Armitage University of Oxford South Parks Road Oxford OX1 3QU UK Phone: +44 1865 275299 Fax: +44 1865 275297 [email protected]
Ursula Attmannspacher University of Texas at Austin 100 W24th ESB 424A Austin, TX 78712 Phone: (512) 471-6799 Fax: (512) 471-7088 [email protected]
Priti Bachhawat UMDNJ 679 Hoes Lane Piscataway, NJ 08854 Phone: (732) 235-5164 Fax: (732) 235-5289 [email protected]
Melinda Baker Princeton University Lewis Thomas Laboratory, Princeton University Rm. 146 Princeton, NJ 08540 Phone: (609) 468-4136 Fax: (609) 258-7844

143
Satomi Banno Nagoya University Fro-cho, Chikusa-ku Nagoya, Aichi Prefecture 464-8602 JAPAN Phone: 81-52-789-2993 Fax: 81-52-789-3001 [email protected]
Rina Barak Weizmann Institute of Science Rehovot 76100 ISRAEL Phone: 972 8 9342710 Fax: 972 8 9344112 [email protected]
Sonia Bardy University of Michigan 830 N. University Ann Arbor, MI 48109 Phone: (734) 647-5677 Fax: (734) 647-0884 [email protected]
Bob Belas University of Maryland Biotechnology Institute 701 East Pratt Street Baltimore, MD 21202 Phone: (410) 234-8876 Fax: (410) 234-8896 [email protected]
Belen Belete Wake Forest University School of Medicine Medical Center Boulevard Winston-Salem, NC 27157 Phone: (336) 716-9333 Fax: (336) 716-9928 [email protected]
Sira Bencharit Johns Hopkins University 3501 St. Paul St. Apt. # 728 Baltimore, MD 21218 Phone: (443) 794-9295 Fax: [email protected]
Howard Berg Harvard University Bio Labs, 16 Divinity Ave Cambridge, MA 02138 Phone: (617) 495-0924 Fax: (617) 496-1114 [email protected]
James Berleman Georgia Institute of Technology 310 Ferst Dr. Atlanta, GA 30332-0230 Phone: (404) 385-4505 Fax: (404) 894-0519 [email protected]
Tatiana Besschetnova University of Massachusetts, Amherst 177 Rolling Green Dr. Apt# 177 Amherst, MA 01002 Phone: (413) 256-2348 Fax: (413) 545-4490 [email protected]
Alexandrine Bilwes-Crane Cornell University Baker Laboratory Ithaca, NY 14853-1301 Phone: (607) 255-1609 Fax: (607) 255-1248 [email protected]

144
Indranil Biswas University of South Dakota 414 E. Clark Street Vermillion, SD 57069 Phone: (605) 677-5163 Fax: (605) 677-6381 [email protected]
David Blair University of Utah 257 South 1400 East, Rm. 201 Salt Lake City, UT 84112-0840 Phone: (801) 585-3709 Fax: (801) 585-9735 [email protected]
Tarra Bollinger Molecular Biology Consortium 835 S. Wolcott Ave., M/C 790 Chicago, IL 60612 Phone: (312) 996-1216 Fax: (312) 413-2952 [email protected]
Cristina Bongiorni The Scripps Research Institute 10550 North Torrey Pines Road MEM-116 La Jolla, CA 92037 Phone: (858) 784-7909 Fax: (858) 784-7966 [email protected]
Pamela Bonner University of Georgia 527 Biological Sciences Bldg Athens, GA 30602 Phone: (706) 542-2682 Fax: (706) 542-2674 [email protected]
Bob Bourret University of North Carolina Chapel Hill, NC 27599-7290 Phone: (919) 966-2679 Fax: (919) 962-8103 [email protected]
Tim Braun University of Wisconsin-Milwaukee 3209 N. Maryland Ave Milwaukee, WI 53211 Phone: (414) 229-2910 Fax: (414) 229-3926 [email protected]
Jocelyn Brewster University of North Carolina-Chapel Hill Department of Microbiology and Immunology 804 MEJB, UNC Chapel Hill, NC 27599-7290 Phone: (919) 966-2679 Fax: (919) 962-8103 [email protected]
Jasemin Brown Molecular Biology Consortium 835 S. Wolcott Ave., M/C 790 Chicago, IL 60612 Phone: (312) 996-1216 Fax: (312) 413-2952 [email protected]
Maria del Carmen Buron Barral University of Utah 257 South 1400 East Salt Lake City, UT 84112-0840 Phone: (801) 581-3592 Fax: [email protected]

145
Vincent Cannistraro University of Illinois 506 S. Mathews St. Urbana, IL 61820 Phone: (217) 333-0268 Fax: (217) 333-8868 [email protected]
Brian Cantwell Texas A&M University Dept. of Biology 3258 TAMU College Station, TX 77843 Phone: (979) 845-1249 Fax: (979) 845-2891 [email protected]
Jennifer Chandler University of Oxford Merton College, Merton Street Oxford, Oxfordshire OX1 4JD UK Phone: 44 1865 275298 Fax: 44 1865 275297 [email protected]
Xingjuan Chao Cornell University 212 Cleveland Ave. Ithaca, NY 14850 Phone: (607) 351-2041 Fax: (607) 255-1248 [email protected]
Nyles Charon West Virginia University Health Sciences Center North Morgantown, WV 26506-9177 Phone: (304) 293-4170 Fax: (304) 293-7823 [email protected]
Gordon Churchward Emory University 1510 Clifton Road Atlanta, GA 30322 Phone: (404) 727-2538 Fax: (404) 727-3659 [email protected]
Galit Cohen-Ben-Lulu Weizmann Institute of Science Herzel Street Rehovot 76100 ISRAEL Phone: 972 8 934 3144 Fax: 972 8 934 4112 [email protected]
Julie Collins Emory University 1510 Clifton Road Atlanta, GA 30324 Phone: (404) 727-0522 Fax: [email protected]
Brian Crane Cornell University Baker Lab. Ithaca, NY 14853 Phone: (607) 254-8634 Fax: (607) 255-1248 [email protected]
Rick Dahlquist UC Santa Barbara UCSB Department of Chemistry - 9510 Santa Barbara, CA 93106 Phone: (805) 961-9641 Fax: [email protected]

146
David De Rosier Brandeis University MS029 - Rosenstiel, 415 South Street Waltham, MA 02454 Phone: (781) 736-2494 Fax: (781) 736-2419 [email protected]
Alejandra Diaz The Scripps Research Institute 10550 North Torrey Pines Road, MEM-116 La Jolla, CA 92037 Phone: (858) 784-7909 Fax: (858) 784-7966 [email protected]
Stephanie Douthit University of North Carolina 804 Mary Ellen Jones, UNC Chapel Hill, North Carolina 27599-7290 Phone: (919) 966-2679 Fax: [email protected]
Roger Draheim Texas A&M University 3258 TAMU College Station, Texas 77843 Phone: (979) 845-1249 Fax: (979) -845-2891 [email protected]
Collin Dyer UC Santa Barbara UCSB Department of Chemistry - 9510 Santa Barbara, CA 93106 Phone: (805) 961-9641 Fax: (805) 893-4120 [email protected]
Michael Eisenbach Weizmann Institute of Science P.O. Box 26 Rehovot 76100 ISRAEL Phone: 972-8-934-3923 Fax: 972-8-947-2722 [email protected]
Amr Eldakak University of Illinois-Chicago 900 S. Ashland Ave, MBRB Rm 4168, Chicago, IL 60607 Phone: (312) 996-2280 Fax: (312) 413 2691 [email protected]
Thierry Emonet University of Chicago 920 E. 58th St., CLSC 403 Chicago, IL 60608 Phone: (773) 834-4650 Fax: (773) 702-0439 [email protected]
Amber Fair University of California - Santa Cruz 124 Walti St. Santa Cruz, California 95060 Phone: (831) 459-4780 Fax: [email protected]
Joseph Falke University of Colorado 215 UCB Boulder, CO 80309-0215 Phone: (303) 492-3503 Fax: (303) 492-5894 [email protected]

147
Stephen Farrand University of Illinois at Urbana-Champaign B103 Chemistry and Life Sciences Laboratory 601 S. Goodwin Ave Urbana, IL 61801 Phone: (217) 333-1524 Fax: (217) 244-7830 [email protected]
Hedda Ferris Yale University PO Box 208114, 266 Whitney Ave. New Haven, CT 06520-8114 Phone: (203) 432-5589 Fax: (203) 432-9782 [email protected]
Noreen Francis Brandeis University MS029, 415 South Street Waltham, MA 02454 Phone: (781) 736-2439 Fax: (781) 736-2419 [email protected]
Michael Galperin National Institutes of Health, NLM 8600 Rockville Pike Bethesda, MD 20894 Phone: (301) 435-5910 Fax: (301) 435-7794 [email protected]
Ulrich Genick Brandeis University 415 South Street Waltham, MA 02453 Phone: (781) 736-2304 Fax: (781) 736-2349 [email protected]
George Glekas University of Illinois at Urbana-Champaign 409 Medical Sciences Bldg. 506 S. Mathews Ave. Urbana, IL 61801 Phone: (217) 333-0268 Fax: (217) 333-8868 [email protected]
Susy Gloor University of Colorado 215 UCB Boulder, CO 80309 Phone: (303) 492-3597 Fax: (303) 492-5894 [email protected]
Gabriela Gonzalez-Bonet Cornell University 37 Uptown Rd #3A Ithaca, NY 14850 Phone: (607) 351-4292 Fax: [email protected]
Bertha González-Pedrajo Universidad Nacional Autónoma de México Ap. Postal 70-243 Mexico, Distrito Federal 04510 MEXICO Phone: (5255) 56225965 Fax: (5255) 56225611 [email protected]
Andrew Goodman Harvard Medical School WAB 365/200 Longwood Ave Boston, MA 02115 Phone: (617) 432-5081 Fax: (617) 738-7664 [email protected]

148
Khoosheh Gosink University of Utah 247 South 1400 East Salt Lake City, UT 84112-0840 Phone: (801) 581-3592 Fax: [email protected]
Marcus Gould University of Oxford South Parks Road Oxford, Oxfordshire OX1 3QU England Phone: +44 (1865) 275298 Fax: +44 (1865) 275297 [email protected]
Johnathan Green University of Cambridge Tennis Court Road Cambridge CB1 2QP United Kingdom Phone: 0044 1223 766145 Fax: [email protected]
Carolin Groeger Justus-Liebig-University of Giessen Heinrich-Buff-Ring 26-32 Giessen, Hessen 35392 Germany Phone: 0641-99 35548 Fax: 0641-99 35549 [email protected]
Asiya Gusa Emory University 1510 Clifton Road Atlanta, GA 30322 Phone: (404) 727-0522 Fax: [email protected]
David Gutnick Tel-Aviv University Green Bldg 122 Tel-Aviv 69978 ISRAEL Phone: 972-3-6409834 Fax: 972-3-6409407 [email protected]
Christopher Halkides University of North Carolina at Wilmington 601 S. College Road Wilmington, NC 28403 Phone: (910) 962-7427 Fax: (901) 962-3013 [email protected]
Patricia Hartzell University of Idaho 142 Life Science Moscow, ID 83844-3052 Phone: (208) 885-0572 Fax: (208) 885-6518 [email protected]
Gerald Hazelbauer University of Missouri 117 Schweitzer Hall Columbia, MO 65211 Phone: (573) 882-9825 Fax: (573) 882-5635 [email protected]
Sara Henry Georgia Institute of Technology 310 Ferst Drive Atlanta, GA 30332-0230 Phone: (404) 385-4505 Fax: (404) 894-0519 [email protected]

149
Penelope Higgs UC Berkeley 16 Barker Hall #3204 Berkeley, CA 94720 Phone: (510) 643-5457 Fax: (510) 643-6334 [email protected]
Takanori Hirano JST 1064-18 Takahori, Hirata Takanezawa, Tochigi 329-1206 JAPAN Phone: +81 28 676 8520 Fax: +81 28 676 8520 [email protected]
James Hoch The Scripps Research Institute 10550 North Torrey Pines Road, MEM-116 La Jolla, CA 92037 Phone: (858) 784-7905 Fax: (858) -784-7966 [email protected]
Egbert Hoiczyk Johns Hopkins University 615 North Wolfe Street Baltimore, MD 21205 Phone: (443) 287-2898 Fax: (410) 955-0105 [email protected]
Edan Hosking Texas A&M University 3258 TAMU College Station, TX 77843 Phone: (979) 845-1249 Fax: (979) 845-2891 [email protected]
Dawn Hower UCLA 944 Tiverton Ave., #2 Los Angeles, CA 90024 Phone: (310) 825-9748 Fax: (310) 794-7109 [email protected]
Marion Hulett University of Illinois at Chicago 4170 MBRB, M/C 567 Chicago, IL 60612 Phone: (312) 996-5460 Fax: (312) 413-2691 [email protected]
Akihiko Ishijima NAGOYA University Furo-cho, Chikusa-ku Nagoya, Aichi 464-8603 JAPAN Phone: +81-52-789-4465 Fax: +81-52-789-4465 [email protected]
Thania Jakobsen Max Planck Institute Karl-von-Frisch Strasse Marburg 35043 GERMANY Phone: 0049/6421/178220 Fax: 0049/6421/178299 [email protected]
Jimmy Jakobsen Max Planck Institute for Terrestrial Microbiology Karl-von-Frisch Strasse Marburg D-35043 GERMANY Phone: ++49 06421 178220 Fax: ++49 06421 178299 [email protected]

150
Urs Jenal University of Basel Klingelbergstrasse 70 Basel CH-4056 SWITZERLAND Phone: +41 61 267 21 35 Fax: +41 61 267 21 18 [email protected], [email protected]
Mark Johnson Loma Linda University Alumni Hall Loma Linda, CA 92350 Phone: (909) 558-4480 Fax: (909) 558-4035 [email protected]
Christine Josenhans Hannover Medical School Carl-Neuberg-Strasse 1 Hannover D-30625 GERMANY Phone: +49 511 532 4348 Fax: +49 511 532 4355 [email protected]
Robert Kadner University of Virginia 1300 Jefferson Park Ave. Charlottesville, VA 22908-0734 Phone: (434) 924-2532 Fax: (434) 982-1071 [email protected]
Heidi Kaplan University of Texas Medical School at Houston 6431 Fannin, 1.765 JFB Houston, TX 77030 Phone: (713) 500-5448 Fax: (713) 500-5499 [email protected]
Ikuro Kawagishi Nagoya University Furo-cho, Chikusa-ku Nagoya, Aichi Prefecture 464-8602 JAPAN Phone: 81-52-789-2993 Fax: 81-52-789-3001 [email protected]
Tetsuya Kawamura University of California, Santa Barbara 345 Mathilda Dr. APT 4 Goleta, CA 93117 Phone: (805) 689-8543 Fax: [email protected]
Barbara Kazmierczak Yale University 333 Cedar Street, Box 208022 New Haven, CT 06520-8022 Phone: (203) 737-5062 Fax: (203) 785-3864 [email protected]
Daniel Kearns Harvard University 3029 Biological Laboratories, 16 Divinity Ave Cambridge, MA 02138 Phone: (617) 384-7622 Fax: (617) 496-4642 [email protected]
Linda Kenney University of Illinois at Chicago 835 South Wolcott Ave. M/C 790 Chicago, IL 60612 Phone: (312) 413-2014 Fax: [email protected]

151
John Kirby Georgia Institute of Technology 310 Ferst Drive Atlanta, GA 30332 Phone: (404) 894-8418 Fax: (404) 894-0519 [email protected]
Seiji Kojima Japan Science and Technology Agency (JST) 1-3 Yamadaoka Suita, Osaka 565-0871 JAPAN Phone: +81-6-6879-4625 Fax: +81-6-6879-4652 [email protected]
Akiko Kusumoto Nagoya University Furotyo, Chikusa-ku Nagoya, Aichi 464-8602 JAPAN Phone: +81(52)789-2992 Fax: +81(52)789-3001 [email protected]
Runzhi Lai Texas A&M University Biology Dept. BSBE 305, MS 3258, College Station, TX 77840 Phone: (979) 845-1249 Fax: [email protected]
Wing-Cheung Lai University of Missouri-Columbia 117 Schweitzer Hall Columbia, MO 65211 Phone: (573) 884-6334 Fax: (573) 882-5635 [email protected]
Matthew Levin University of Cambridge Downing Street Cambridge CB2 3DY UK Phone: +44 1223 333771 Fax: +44 1223 333786 [email protected]
Mingshan Li University of Missouri-Columbia 117 Schweitzer Hall Columbia, MO 65211 Phone: (573) 882-4845 Fax: (573) 882-5635 [email protected]
Chunhao Li West Virginia University 01 Medical Center Dr. Room 2079, HSCN Morgantown, WV 26506-9177 Phone: (304) 293-2585 Fax: (304) 293-7823 [email protected]
Angela Lilly University of Missouri 117 Schweitzer Hall Columbia, MO 65211 Phone: (573) 884-6334 Fax: [email protected]
Bryan Lowder University of Utah 257 S. 1400 E., Rm. 201 Salt Lake City, UT 84112-0842 Phone: (801) 585-3961 Fax: (801) 581-4668 [email protected]

152
Andrew Lowenthal University Of California at Santa Cruz 323 Monterey St Santa Cruz, CA 95060 Phone: (510) 541-9760 Fax: (831) 459-3524 [email protected]
May Macnab Yale University PO Box 208114, 266 Whitney Ave. New Haven, CT 06520-8114 Phone: (203) 432-5588 Fax: (203) 432-9782 [email protected]
Ashalla Magee University of North Carolina at Chapel Hill 804 Mary Ellen Jones Bldg., CB# 7290 Chapel Hill, NC 27599-7290 Phone: (919) 966-2679 Fax: (919) -962-8103 [email protected]
Michael Manson Texas A&M 3258 TAMU College Station, TX 77843 Phone: (979) 845-5158 Fax: (979) 845-2891 [email protected]
Phil Matsumura University of Illinois at Chicago Dept. of Microbiology/Immunology 835 S. Wolcott Ave., M/C 790 Chicago, IL 60612 Phone: (312) 996-2286 Fax: (312) 413-2952 [email protected]
Hideyuki Matsunami Japan Science and Technology Agency 1-3 Yamadaoka Suita, Osaka 565-0871 JAPAN Phone: +81-6-6879-4625 Fax: +81-6-6879-4652 [email protected]
Rosemary McAndrew Texas A&M 3258 TAMU College Station, TX 77843 Phone: (979) 845-1249 Fax: (979) 845-2891 [email protected]
Mark McBride University of Wisconsin-Milwaukee 3209 N. Maryland Ave. Milwaukee, WI 53211 Phone: (414) 229-5844 Fax: (414) 229-3926 [email protected]
Jonathan McMurry Yale University 266 Whitney Ave. New Haven, CT 06520-8144 Phone: (203) 432-5589 Fax: (203) 432-9782 [email protected]
Tam Mignot University of California 401, Barker Hall Berkeley, CA 94720 Phone: (510) 643-5457 Fax: (510) 643-6334 [email protected]

153
Lance Miller Georgia Institute of Technology 310 First Drive Atlanta, GA 30332 Phone: (404) 405-4134 Fax: (404) 894-0519 [email protected]
Aaron Miller University of Colorado at Boulder Department of Chemistry and Biochemistry 215 UCB Boulder, CO 80309-0215 Phone: (303) 492-3592 Fax: (303) 492-5894 [email protected]
Michael Miller West Virginia University PO Box 9142 Morgantown, WV 26506 Phone: (304) 293-7762 Fax: (304) 293-6846 [email protected]
Makoto Miyata Osaka City University Sugimoto, Sumiyoshi-ku Osaka, Osaka 558-8585 JAPAN Phone: +81-6-6605-3157 Fax: +81-6-6605-3158 [email protected]
Charles Moran Emory University School of Medicine 3015 Rollins Research Center 1510 Clifton Rd Atlanta, GA 30345 Phone: (404) 727-5969 Fax: (404) 727-3659 [email protected]
Karen Morehouse University of Nottingham Queens Medical Centre Nottingham NG7 2UH UK Phone: 4411 59194496 Fax: 4411 59709906 [email protected]
Md Motaleb West Virginia University Box 9177, Room 2078, Health sciences Center Morgantown, WV 26506-9177 Phone: (304) 293-2585 Fax: (304) 293-7823 [email protected]
Patricia Mowery University of Utah 257 S. 1400 East Salt Lake City, UT 84112 Phone: (801) 581-6307 Fax: [email protected]
Tarek Msadek Institut Pasteur 25, rue du Dr. Roux Paris 75015 FRANCE Phone: (33) 1-45-68-88-09 Fax: (33) 1-45-68-88-38 [email protected]
Travis Muff University of Illinois at Urbana-Champaign 506 S Matthews, 190 MSB, Rm 465 Urbana, IL 61801 Phone: (217) 333-0268 Fax: (217) 333-8868 [email protected]

154
Suman Mukhopadhyay University of Maryland at College Park 8075 Greenmead Dr., Room 1211 College Park, MD 20742 Phone: (301) 314-6812 Fax: (301) 314-6855 [email protected]
Tom Murray Yale University 333 Cedar St TAC S140 New Haven, CT 06520 Phone: (203) 737-1197 Fax: (203) 785-6961 [email protected]
Shawn Nelson University of Wisconsin Milwaukee 265 E Field Stone Circle Apt 6 Oak Creek, WI 53154 Phone: (414) 570-3418 Fax: [email protected]
Austin Newton Princeton University 334 Lewis Thomas Laboratory Princeton, NJ 08544 Phone: (609) 258-3854 Fax: (609) 258-6175 [email protected]
Noriko Ohta Princeton University 335 Lewis Thomas Laboratory Princeton, NJ 08544 Phone: (609) 258-5682 Fax: (609) 258-6175 [email protected]
Mayuko Okabe Yale University PO Box 208114, 266 Whitney Ave. New Haven, CT 06520-8114 Phone: (203) 432-5589 Fax: (203) 432-9782 [email protected]
Peggy O'Neill Molecular Biology Consortium 835 S. Wolcott Ave., M/C 790 Chicago, IL 60612 Phone: (312) 996-1216 Fax: (312) 413-2952 [email protected]
George Ordal University of Illinois 506 S. Mathews Urbana, IL 61801 Phone: (217) 333-9098 Fax: (217) 333-8868 [email protected]
Richard Park Cornell University S.T. Olin G63 Chemistry Research Bldg. Ithaca, NY 14853 Phone: (607) 255-4970 Fax: (607) 255-4810 [email protected]
John Parkinson University of Utah 257 South 1400 East Salt Lake City, UT 84112 Phone: (801) 581-7639 Fax: (801) 581-4668 [email protected]

155
Koushik Paul University of Utah 257 South 1400 East, Rm. 201 Salt Lake City, UT 84112-0840 Phone: (801) 585-3961 Fax: (801) 585-9735 [email protected]
Marta Perego The Scripps Research Institute 10550 North Torrey Pines Road, MEM-116 La Jolla, CA 92037 Phone: (858) 784-7912 Fax: (858) 784-7966 [email protected]
Eduardo Perez UMDNJ 679 Hoes Lane Piscataway, NJ 08854 Phone: (732) 235-5164 Fax: (732) 235-5289 [email protected]
Abiola Pollard Cornell University 190 Pleasant Grove Rd. #1-2 Ithaca, NY 14850 Phone: (607) 257-6741 Fax: [email protected]
Steven Porter University of Oxford South Parks Road Oxford OX1 3QU UK Phone: ++44 1865 275298 Fax: ++44 1865 275297 [email protected]
Birgit Pruess North Dakota State University 1523 Centennial Blvd. Fargo, ND 58105 Phone: (701) 231-7848 Fax: (701) 231-7514 [email protected]
Ankita Puri-Taneja University of Illinois at Chicago 900 S. Ashland ave, MBRB M/C 567, Chicago, IL 60607 Phone: (312) 996-2280 Fax: (312) 413-2691 [email protected]
Simon Rainville Harvard University 16 Divinity Ave Cambridge, MA 02138 Phone: (617) 495-4217 Fax: (617) 496-1114 [email protected]
Stuart Reid Oxford University Wadham College, Parks Road Oxford OX1 3PN UK Phone: 01865 272331 Fax: 01865 272400 [email protected]
Samantha Roberts Emory University 6003 Oak Park Circle Atlanta, GA 30324 Phone: (404) 727-0522 Fax: [email protected]

156
Mark Roberts University of Oxford South Parks Rd Oxford, Oxon OX1 3QU UK Phone: 44 1865 275298 Fax: 44 1865 275297 [email protected]
Melanie Rust Medical school of Hannover Carl-Neuberg-Str. 1 Hannover 30625 GERMANY Phone: 49-511-532-4346 Fax: 49-511-532-4355 [email protected]
Yumiko Saijo-Hamano Japan Science and Technology Agency (JST) 1-3 Yamadaoka Suita, Osaka 565-0871 JAPAN Phone: +81-6-6879-4625 Fax: +81-6-6879-4652 [email protected]
Daniel Salcedo Loma Linda University 11021 Campus St Loma Linda, CA 92350 Phone: (909) 558-1000 x42729 Fax: (909) 558-4035 [email protected]
Sara Salvetti University of Pisa Via San Zeno 35-39 Pisa 56127 ITALY Phone: 0039 050 2213698 Fax: 0039 050 2213711 [email protected]
Tania Saxl Oxford, England Merton College Oxford OX14JD UK Phone: 01865272331 Fax: [email protected]
Elmar Schilling University of North Carolina Dept. of Microbiology & Immunology Chapel Hill, NC 27599-7290 Phone: (919) 966-2679 Fax: (919) 962-8103 [email protected]
June Scott Emory Microbiology, Emory U Atlanta, GA 30322 Phone: (404) 722-0402 Fax: [email protected]
Wenyuan Shi UCLA 10833 le Conte Avenue Los Angeles, CA 90095 Phone: (310) 825-8356 Fax: (310) 794-7109 [email protected]
Satoshi Shibata Soft Nano-Machine Project, JST 1064-18 Takahori Hirata Takanezawa Shioya-gun, Tochigi 329-1206 JAPAN Phone: 81-28-676-8520 Fax: 81-28-676-8520 [email protected]

157
Thomas Shimizu Harvard University 16 Divinity Avenue (BL3063) Cambridge, MA 02138 Phone: (617) 495-4217 Fax: (617) 496-1114 [email protected]
Bryan Shimkos University of Illinois at Chicago Dept of Microbiology/Immunology 835 South Wolcott Ave., M/C 790 Chicago, IL 60612 Phone: (312) 413-2014 Fax: [email protected]
Eugenia Silva-Herzog University of Miami 1600 NW 10th Ave Miami, Fl 33149 Phone: 305-243-6592 Fax: 305-243-4623 [email protected]
Ruth Silversmith University of North Carolina Room 804 Mary Ellen Jones Chapel Hill, NC 27599-7290 Phone: (919) 966-2679 Fax: (919) 962-8103 [email protected]
Liz Sockett University of Nottingham Queens Medical Centre Nottingham NG7 2UH UK Phone: 4411 59194496 Fax: 4411 59709906 [email protected]
Lotte Sogaard-Andersen Max Planck Institute for Terrestrial Microbiology Karl-von-Frisch Str. Marburg 35043 GERMANY Phone: 49 6421 178 201 Fax: 49 6421 178 209 [email protected]
Yoshiyuki Sowa Nagoya University Furo-cho, Chikusa-ku Nagoya 464-8603 JAPAN Phone: 81 52 789 4467 Fax: 81 52 789 4465 [email protected]
Graham Stafford University of Cambridge Tennis Court Road Cambridge CB1 2QP UK Phone: 0044 1223 333733 Fax: [email protected]
Bonnie Stephens Georgia State University 4908 Clearstone Way Acworth, GA 30101 Phone: (770) 974-7706 Fax: [email protected]
Valley Stewart University of California, Davis One Shields Ave. Davis, CA 95616-8665 Phone: (530) 754-7994 Fax: (530) 752-9014 [email protected]

158
Ann Stock University of Medicine and Dentistry of New Jersey 679 Hoes Lane Piscataway, NJ 08854-5627 Phone: (732) 235-4844 Fax: (732) 235-5289 [email protected]
Claudia Studdert University of Utah 257 S 1400 E Salt Lake City, UT 84112 Phone: (801) 581-6307 Fax: [email protected]
Sriram Subramaniam NIH 50 Memorial Drive Room 4306 Bethesda, MD 20892 Phone: (301) 594-2062 Fax: (301) 480-3834 [email protected]
Rooge Suvanasuthi University Maryland 701 E. Pratt st. Baltimore, MD 21202 Phone: (410) 234-8877 Fax: [email protected]
Khalid Ali Syed University of Texas at San Antonio 11600 Huebner Rd. #3205 San Antonio, TX 78230 Phone: (210) 690-7915 Fax: (210) -458-4468 [email protected]
Hendrik Szurmant The Scripps Research Institute 10550 North Torrey Pines Road, MEM-116 La Jolla, CA 92037 Phone: (858) 784-7909 Fax: (858) 784-7966 [email protected]
Jay Tang Brown 182 Hope Street Providence, RI 02912 Phone: (401) 863-2292 Fax: (401) 863-2024 [email protected]
Anne Tart Wake Forest University School of Medicine Medical Center Boulevard Winston-Salem, NC 27157 Phone: (336) 716-9333 Fax: (336) -716-9928 [email protected]
Barry Taylor Loma Linda University 24888 Prospect Street Loma Linda, CA 92350 Phone: (909) 558-8544 Fax: (909) 558-0244 [email protected]
John Taylor University of Nottingham Queens Medical Center Nottingham NG7 2UH UK Phone: 44-(0)115-9194496 Fax: 44(0)115-9709906 [email protected]

159
Karianne Terry University of California, Santa Cruz 1156 High Street Santa Cruz, CA 95064 Phone: (831) 459-4780 Fax: (831) 459-3524 [email protected]
Dennis Thomas Brandeis University 415 South st. Waltham, MA 02454 Phone: (781) 736-2608 Fax: (781) -736-2419 [email protected]
Stephen Thompson University of Oxford South Parks Rd Oxford, Oxon OX1 3QU UK Phone: +441865275298 Fax: [email protected]
Alejandro Toro Rutgers University 679 Hoes Lane Piscataway, New Jersey 08854 Phone: (732) 235-5164 Fax: (732) 235-5289 [email protected]
Hoa Tran University of Massachusetts, Amherst 710 North Pleasant St. LGRT 701 Amherst, MA 01003 Phone: (413) 210-4481 Fax: (413) 545-4490 [email protected]
Billyana Tsvetanova The Scripps Research Institute 10550 North Torrey Pines Road, MEM-116 La Jolla, CA 92037 Phone: (858) 784-7909 Fax: (858) 784-7966 [email protected]
Yuhai Tu IBM Research 1101 KITCHAWAN ROAD Yorktown Heights, NY 10598 Phone: (914) 945-2762 Fax: (914) 945-4506 [email protected]
Anna Turabelidze University of Illinois at Chicago 835 South Wolcott Ave., M/C 790 Chicago, IL 60612 Phone: (312) 413-2014 Fax: [email protected]
Atsuko Uenoyama Osaka City University Sumiyoshi-ku Osaka City, Osaka 558-8585 JAPAN Phone: 81(6)6605 3157 Fax: 81(6)6605 3158 [email protected]
Luke Ulrich Georgia Institute of Technology 310 First Drive Atlanta, GA 30332 Phone: (404) 385-6169 Fax: (404) 894-0519 [email protected]

160
Kottayil Varughese The Scripps Research Institute 10550 North Torrey Pines Road, MEM-116 La Jolla, CA 92037 Phone: (858) 784-7945 Fax: (858) 784-7966 [email protected]
Jovanka Vuksanovic West Virginia University Health Sciences Center Morgantown, WV 26506-9177 Phone: (304) 293-2585 Fax: (304) 293-7823 [email protected]
George Wadhams University of Oxford South Parks Road Oxford OX1 3QU UK Phone: 01865 275298 Fax: 01865 275297 [email protected]
Don Walthers University of Illinois at Chicago 835 S Wolcott Chicago, IL 60612 Phone: (312) 413-2014 Fax: [email protected]
Qingfeng Wang The University of Texas at Austin 100 W 24th St Austin, TX 78712 Phone: (512) 471-6799 Fax: (512) 471-7088 [email protected]
Mandy Ward Johns Hopkins 3404 N. Charles St Baltimore, MD 21210 Phone: (410) 516-4201 Fax: (410) 516 8996- [email protected]
Kylie Watts Loma Linda University AHBS Building, Rm 120 Loma Linda, CA 92350 Phone: (909) 558-1000 x42758 Fax: (909) 558-4035 [email protected]
Rachel Webster University of Oxford Linacre College, St Cross Road Oxford, Oxfordshire OXL 3JA UK Phone: 44 1865 275298 Fax: 44 1865 275297 [email protected]
Robert Weis University of Massachusetts Amherst 710 North Pleasant St Amherst, MA 01003-9336 Phone: (413) 545-0464 Fax: (413) 545-4490 [email protected]
Roy Welch Syracuse University 130 College Place, BRL Room 702A Syracuse, NY 13244 Phone: (315) 443-2159 Fax: [email protected]

161
Ann West University of Oklahoma 620 Parrington Oval Norman, OK 73019 Phone: (405) 325-1529 Fax: (405)-325-6111 [email protected]
Andrea White The Scripps Research Institute 10550 North Torrey Pines Road, MEM-116 La Jolla, CA 92037 Phone: (858) 784-7909 Fax: (858) 784-7966 [email protected]
Janet Wilson Georgia Institute of Technology 310 Ferst Drive Atlanta, GA 30332 Phone: (404) 385-4505 Fax: [email protected]
Peter Wolanin Princeton University 146 LTL Princeton, NJ 08544 Phone: (609) 258-6112 Fax: (609) 258-7844 [email protected]
Alan Wolfe Loyola University of Chicago Maguire Blgd 105, Rm 3823 2160 S. First Ave. Maywood, IL 60153 Phone: (708) 216-5814 Fax: (708) 216-9574 [email protected]
Gus Wright Texas A&M University 3258 TAMU College Station, TX 77843 Phone: (979) 845-1249 Fax: (979) -845-2891 [email protected]
Kristin Wuichet Georgia Institute of Technology 310 Ferst Drive Atlanta, GA 30332 Phone: (404) 405-4134 Fax: (404) 894-0519 [email protected]
Qian Xu Virginia Polytechnic Institue and State University 2119 Derring Hall Blacksburg, VA 24061 Phone: (540) 231-9381 Fax: (540) 231-9370 [email protected]
Toshiharu Yakushi Nagoya University Furo-cho Chikusa-ku, Nagoya 464-8602 JAPAN Phone: 81-52-789-2992 Fax: 81-52-789-3001 [email protected]
Jung Yang University of Utah 257 S 1400 E Salt Lake City, UT 84112 Phone: (801) 585-3961 Fax: (801) -585-9735 [email protected]

162
Zhaomin Yang Virginia Tech 2119 Derring Hall/Virginia Tech Blacksburg, VA 24061-0406 Phone: (540) 231-1350 Fax: [email protected]
Wei Yuan University of Illinois, Champaign-Urbana 190 Med Sci Bldg. 506 South Mathew's Ave. Urbana, IL 61801 Phone: (217) 333-0268 Fax: [email protected]
Peijun Zhang National Cancer Institute, NIH Bldg 50, Rm 4312, 50 South Drive, MSC 8008 Bethesda, MD 20892 Phone: (301) 594-2206 Fax: (301) 480-3834 [email protected]
Jinshi Zhao University of Utah 257 South 1400 East Salt Lake City, UT 84112 Phone: (801) 581-7639 Fax: (801) 581-4668 [email protected]
Igor Zhulin Georgia Institute of Technology 310 Ferst Drive Atlanta, GA 30332-0230 Phone: (404) 385-2224 Fax: (404) 894-0519 [email protected]
David Zusman University of California 16 Barker Hall Berkeley, CA 94720-3204 Phone: (510) 642-2293 Fax: (510) 643-6334 [email protected]
: