Embed Size (px)
Citation preview

lable at ScienceDirect
Biomaterials 35 (2014) 9709e9718
Contents lists avai
Biomaterials
journal homepage: www.elsevier .com/locate/biomater ia ls
Bioinspired affinity DNA polymers on nanoparticles for drugsequestration and detoxification
Niancao Chen, Yike Huang, Yong Wang*
Department of Biomedical Engineering, College of Engineering, The Pennsylvania State University, University Park, PA 16802, USA
a r t i c l e i n f o
Article history:Received 24 June 2014Accepted 9 August 2014Available online 28 August 2014
Keywords:AffinityNanoparticleAptamerDNAPolymer
* Corresponding author.232 Hallowell Building, UnTel.: þ1 814 865 6867; fax: þ1 814 865 0490.
E-mail addresses: [email protected], yxw30@p
http://dx.doi.org/10.1016/j.biomaterials.2014.08.0170142-9612/© 2014 Elsevier Ltd. All rights reserved.
a b s t r a c t
Nanomaterials with the ability of sequestering target molecules hold great potential for a variety ofapplications. To ensure the stable sequestration, most of these nanomaterials have been traditionallydesigned with a clear boundary or compact structures and behave as closed systems. While this feature isbeneficial to applications such as drug delivery, it may pose a challenge to applications where fastmolecular transport from the environment to nanomaterials is critical. Thus, this study was aimed atexploring a nanomaterial with affinity DNA polymers and nanoparticles as an open systemwith functionsimilar to jellyfish tentacles in sequestering target molecules from surroundings. The results show thatthis nanomaterial can effectively and rapidly sequester both small molecule drugs and large moleculebiologics and resultantly mitigate their biological effects. Thus, this nanomaterial holds potential as auniversal nanoscale antidote for drug removal and detoxification. While this nanomaterial was evaluatedby using drug removal and detoxification as a model, the synthesis of periodically oriented affinitypolymers on a nanoparticle with the capability of sequestering target molecules may be tuned for broadapplications such as separation, sensing, imaging and drug delivery.
© 2014 Elsevier Ltd. All rights reserved.
1. Introduction
Nanomaterials with the ability of sequestering target moleculeshold great potential for a variety of applications such as drugodelivery [1,2], biological separation [3], chemical sensing [4,5],molecular imaging [6e8], and antidote development [9,10]. Diverseclasses of nanomaterials such as lipid-based nanoemulsions [11,12],coreeshell nanocapsules [13], magnetic nanoparticles [14,15] andmolecularly imprinted nanoparticles [16,17] have been widelystudied for these promising applications. Most of these nano-materials have a clear boundary or compact structures separatingtheir inside from the environment, behaving as closed systems toensure the stable sequestration of target molecules. This feature isbeneficial to certain applications such as drug delivery since theboundary and compact structures will ensure high efficiency ofmolecular loading. However, it may pose a challenge to applicationswhere fast molecular transport from the environment to nano-materials is critical. This challenge would be overcome by devel-oping nanomaterials as open systems without a boundary orcompact structures. However, open systems may lose sequestered
iversity Park, PA 16802, USA.
su.edu (Y. Wang).
molecules since there is no barrier to prevent their escape. Thisstudy was aimed at developing a nanomaterial that is open to theenvironment but has high affinity interactions with target mole-cules. This nanomaterial has long affinity DNA polymers (DPs)extended from the nanoparticle surface with a great capability ofsequestering target molecules from solution, which is functionallysimilar to jellyfish with long tentacles in catching prey fromsurroundings.
This nanomaterial was evaluated by the examination of its po-tential as a universal nanoscale antidote for drug sequestration anddetoxification. Drugs can cause detrimental or fatal side effects ifused inappropriately, despite their usefulness in treating virtuallyall human diseases. To mitigate or counteract toxic effects of drugs,molecular antidotes are used in the clinical practice to neutralizedrugs via the formation of molecular complexes [18]. However,molecular antidotes lack the functionalities of active entrapmentand fast removal of drugs from the circulation. As a result, drugmisuse, overuse and abuse remain a major public health problemworldwide [19]. In contrast, nanomaterials act as a sink to entraptoxic molecules [20]. They can also be optimized to acquire desiredproperties to allow active removal through detoxifying tissues (e.g.,liver) or extracorporeal assist devices [21,22]. Thus, nanomaterialsthat can rapidly sequester drugs with high binding strength wouldbe a promising platform for drug detoxification.

Table 1Oligonucleotides.
Name Sequence (50 to 30)
Sequences for Dox experimentsDI /biotin/-CACACCGAGCTCCCACCCGATCGTCACCTDM1 /Cy5/-CCCACCCGATCGTCACCTAGCTCGAGGTGACGATCGGGTGGGA
GCTCGDM2 CGAGCTAGGTGACGATCGGGTGGGCGAGCTCCCACCCGATCGTCACCTSequences for thrombin experimentsDI /biotin/-AAAAACAAAGTAGTCTAGGATTCGGCGTGDM1 TTTCCCTTATATTCTCTCTCTCTCC
AGTCTAGGATTCGGCGTGGGTTAACACGCCGAATCCTAGACTACTTTGDM2 TTAACCCACGCCGAATCCTAGACTCAAAGTAGTCTAGGATTCGGCGTGC-DM1 AGTCTAGGATTCGGCGTGGGTTAACACGCCGAATCCTAGACTACTTTGTA /TAMRA/-GAGAGAGAATATAAGGGAAAAAAAAGGTTGGTGTGGTTGG
Note: The letters between two slashes indicate the chemical modification of thesequence. DI: DNA initiator; DM: DNA monomer; C-DM1: control DM1 withoutoverhang; TA: anti-thrombin aptamer.
N. Chen et al. / Biomaterials 35 (2014) 9709e97189710
We synthesized the nanomaterial using affinity DPs and nano-particles. Affinity DPs were composed of synthetic DNA oligonu-cleotides with the molecular recognition capability; nanoparticleswere used as the molecular support of DPs (Fig. 1). Synthetic oli-gonucleotides have been recently used as structural elementsrather than genetic materials to build up a variety of materials[23,24]. They are attractive to the field of materials science as thecombination or permutation of basic nucleotides can generate anunlimited number of nucleic acid sequences and complementarybase-pairing interactions ensure high-fidelity hybridization.Numerous methods such as polymerase chain reaction [25] androlling circle amplification [26] can be used to synthesize linearDNA polymers or nanomaterials of high dimensions. In this study,DPs were synthesized through enzyme-free hybridization chainreaction [27] and intermolecular hybridization. Thus, the synthesisof both oligonucleotides and DPs does not need the involvement ofenzymes, which will make the synthesis of DPs and nanomaterialseasy and robust. DPs were evaluated by gel electrophoresis, dy-namic light scattering (DLS) and fluorescence imaging. Doxorubicin(Dox) and thrombin were used as the models of small moleculedrugs and large molecule biologics respectively to examine thefunctionality of the nanomaterial inmitigating the biological effectsof the drugs. Functionality assays included drug sequestration, celltoxicity and coagulation time.
2. Materials and methods
2.1. Chemical reagents
All oligonucleotides (Table 1) were ordered from IDT (Coralville, IA). 10�Phosphate buffered saline (PBS), agarose and fetal bovine serum (FBS) were pur-chased from Fisher Scientific (Suwanee, GA). Streptavidin-coated iron oxide nano-particles were ordered fromOcean Nanotech (San Diego, CA). The DNA ladder, SYBR-Safe, Hoechst 33342, human umbilical vein endothelial cells (HUVECs) and cellculture reagents were purchased from Invitrogen (Carlsbad, CA). The cell prolifer-ation assay kit was ordered from Promega (Madison, WI). Dox, human thrombin,sodium chloride, potassium chloride, sodium azide, magnesium chloride and Tween20 were obtained from SigmaeAldrich (Louis, MO). The pooled normal humanplasma (supplemented with sodium heparin) was ordered from Innovative research(Novi, MI). The Tris-acetate was ordered from Amresco (Solon, OH). The humanthrombin ELISA kit was purchased from Assaypro (St. Charles, MO).
2.2. Preparation of affinity DPs and DNA polymers with aptamers (PAs)
DIs and DMs were designed using NUPACK server (www.nupack.org) [28]. Thesecondary structures of the DNA sequences were also predicted using NUPACK.Before the synthesis of DPs or PAs, the oligonucleotides were heated to 95 �C andthen cooled to room temperature for 1 h. Themixture of the DI and the two DMswasincubated at room temperature overnight for the synthesis of DPs. The concentra-tion of the DI was 0.1 mM and the two DMswere 1 mM. To synthesize PAs, the aptamersolution of 1 mM was reacted with DPs for 8 h at room temperature.
Fig. 1. Schematic illustration of a nanomaterial with affinity D
To study the stability of DPs in serum, cy5-labeled DM1 was used to preparethe DPs. DP solution was mixed with the whole FBS with the final serum con-centration of 70% for different periods of time at 37 �C before running gelelectrophoresis.
2.3. Gel electrophoresis
DPs or PAs were prepared using the same procedure as described above. Theirsolutions were loaded into the wells of 1% agarose gel (Tris-borate-EDTA buffer,89 mM boric acid, 2 mM EDTA, pH 8.2), which was pre-stained with or without SYBR-Safe (0.1 mL of stock solution per mL of agarose gel). The gel electrophoresis was runat 100 V for 60 min and the gels were imaged using a CRI Maestro EX System(Woburn, MA).
2.4. Preparation of affinity DP-functionalized nanoparticles (DP-NPs) and PA-functionalized nanoparticles (PA-NPs)
Streptavidin-coated iron oxide nanoparticles (0.83 mg/mL) were mixed withbiotinylated DI (2 mM) at room temperature for 1 h. After the removal of free DI viacentrifugation, nanoparticles were incubated in the reaction buffer (PBS with 0.05%v/v Tween 20 and 0.02% w/v NaN3) containing the two DMs (2 mM) at room tem-perature overnight. The DP-NP solution was centrifuged to remove unreacted DMsand to harvest the DP-NPs. To synthesize PA-NPs, the aptamer solution (4.5 mM) wasincubated with the DP-NPs overnight at room temperature in the buffer containing20 mM Tris-acetate, 100 mM NaCl, 5 mM KCl, 0.1% v/v Tween 20, and 0.02% w/v NaN3.The PA-NPs were harvested via centrifugation.
DP-NPs were also prepared with Cy5-labeled DM1 to study their stability inserum. DP-NP suspensionwas carefully transferred into thewhole FBS with the finalserum concentration of 70% and the mixture was incubated at 37 �C on a shaker(300 rpm) for different periods of time. The DP-NPs were transferred into the re-action buffer for washing and centrifuged at 14,000 � g for 10 min for particleharvesting. DP-NPs were re-dispersed in 20 mL of reaction buffer before imagedwithMaestro. The fluorescence intensities of the DP-NP suspensions were measuredusing the software provided by the Maestro manufacturer.
NA polymers as an open system for target sequestration.

N. Chen et al. / Biomaterials 35 (2014) 9709e9718 9711
2.5. Fluorescence imaging of nanoparticles
DMs were labeled with either Cy5 or TAMRA at the 50 ends (Table 1) for theexamination of DP-NPs or PA-NPs. After the synthesis of the nanoparticles, theywere dispersed in PBS and transferred onto a Teflon-coated glass slide as droplets.The nanoparticle droplets were imaged to capture the fluorophore signals using theCRI Maestro EX System. The fluorescence intensity was determined by using thesoftware provided by the manufacturer.
2.6. Measurement of nanoparticle sizes via DLS
Nanoparticles (20 mg) were dispersed in 1 mL of PBS that was filtered through0.2 mm membrane (Millipore) prior to use. The nanoparticle dispersions weretransferred to a 12 mm Square Polystyrene Cuvette and were characterized usingZetasizer Nano (Malvern Instruments).
2.7. Study of the kinetics of Dox sequestration
To examine the kinetics of Dox sequestration by DPs, Dox was mixed with DPs atroom temperature. The molar ratio of DPs and Dox was 1:50. At the predeterminedtime points, the fluorescence intensity of the solution was examined using Nano-Drop 3300 Fluorospectrometer (Thermo Scientific). To examine the kinetics of Doxsequestration by DP-NPs, Dox and DP-NPs were mixed and immediately transferredto 96-well fluorescence plate (Perkin Elmer). The fluorescence of the mixture wasmeasured at 592 nm at different time points using fluorescence microplate reader(F200, Tecan). The capability of DP-NPs in sequestering Dox in serum was alsostudied. Briefly, the DP-NPs were mixed with FBS at 37 �C and the mixture wassampled at a predesigned time interval. The mixture was diluted in the reactionbuffer with 5 mM MgCl2 and immediately measured using the fluorescence micro-plate reader. The fluorescence intensity of the mixture was normalized to that of thebuffer that had Dox but had no nanoparticles.
2.8. Examination of the capability of Dox sequestration
To examine Dox sequestration by DPs, Dox was incubated with DPs for 30 minat room temperature with the molar ratio varied from 1:400 to 1:20. The fluo-rescence intensity of the solution was examined using NanoDrop 3300 Fluo-rospectrometer. To examine Dox sequestration by DP-NPs, 50 mL of nanoparticlesuspension (0.4 mg/mL) in PBS containing 0.05% v/v Tween 20, 0.02% w/v NaN3 and5 mM MgCl2 was mixed with 50 mL of Dox solution in the same PBS buffer at roomtemperature for 0.5 h. After the DP-NP suspension was centrifuged at 15,000 � gfor 10 min, the supernatant was harvested and transferred to the 96-well fluo-rescence plate. The fluorescence of Dox was measured at 592 nm using themicroplate reader. The Dox sequestration was calculated using this formula: Doxabsorbed% ¼ 100%�S%, where S% is the percentage of Dox left in supernatant. TheDox concentrations of the supernatants were normalized to the group that onlyDox was added (no nanoparticles). The fluorescence of the supernatants was alsoimaged using the CRI Maestro EX System.
2.9. Culture of human umbilical vein endothelial cells (HUVECs)
HUVECs were cultured at 37 �C in a humidified incubator containing 5% CO2 inMedium 200, in which low serum growth supplement (LSGS), gentamicin (10 mg/mL) and amphotericin B (0.25 mg/mL) were added. After trypsinization, HUVECswere seeded in an 8-well m-slide chamber (ibidi) at a density of 1.5e2.0 � 104 cells/well and cultured for 2 days to reach confluence prior to use. All the cells used werebetween passages 3 to 5.
2.10. Examination of Dox uptake by HUVECs
DP-NPs (0.2 mg/mL) were mixed with Dox (10 mM) in Medium 200 supple-mented with 5 mM MgCl2 for 0.5 h at room temperature and centrifuged at15,000 � g for 10 min. The supernatants were used to incubate with HUVECscultured in a m-slide chamber for 2 h at 37 �C in a humidified atmosphere. They werethen stained with Hoechst 33342 (2.5 mg/mL) and imaged using a fluorescencemicroscope (IX73, Olympus). The fluorescence signals of Hoechst 33342 wererecorded using the DAPI channel (350 ± 25 nm/460 ± 25 nm, Ex/Em). The fluores-cence signals of Dox were recorded using a Cy3 channel (535 ± 25 nm/610 ± 50 nm,Ex/Em). The acquired images were merged using the ImageJ software.
2.11. Examination of cell viability
HUVECs were seeded in 96-well plate at a density of 5000 cells/well andcultured overnight. The procedure for cell treatment was the same as describedabove in the Dox uptake experiment. After the 2 h treatment with the super-natant, the HUVECs were rinsed twice with the culture medium and cultured for48 h. Cell viability was assessed using a cell proliferation kit following themanufacturer's protocol. The value of cell viability was normalized to that of thegroup in which the HUVECs were only treated with the buffer (Medium 200,5 mM MgCl2).
2.12. Measurement of thrombin sequestration via enzyme-linked immunosorbentassay (ELISA)
Thrombin (322.5 ng) was incubated with nanoparticles (10 mg) in 150 mL ofbinding buffer (20 mM Tris-acetate, 100 mM NaCl, 5 mM KCl, 0.1% v/v Tween 20, 0.02%w/v NaN3, and 0.1%w/v BSA) for 0.5 h at 37 �C. The nanoparticles were centrifuged at10,000 � g for 0.5 h to collect the supernatant. The thrombin concentration in thesupernatant was measured using human thrombin ELISA kit according to the in-structions provided by the supplier.
2.13. Coagulation time analysis
The biological effect of the thrombin solution was examined by using thecoagulation time assay. The coagulation time was measured according to the pro-tocol provided by the supplier. In brief, the coagulometer (QuikCoag 1004, Bio-Medica, Nova Scotia, Canada) and the reagents were warmed up to 37 �C before themeasurement. The thrombin solution (4 unit/mL) was incubated with 10 mg nano-particles in 100 mL of binding buffer for 0.5 h at 37 �C to prepare the coagulation-triggering solution. Plasma of 50 mL was mixed with the equal volume of thebinding buffer in a cuvette. The cuvette was placed into the measuring channel ofthe coagulometer and incubated at 37 �C for 60 s. The coagulation-triggering solu-tion of 100 mL was added into the cuvette and the coagulation time was recorded bythe coagulometer.
3. Results and discussion
3.1. Synthesis of affinity DPs
The synthesis of the DPs involves three DNA sequences: DNAinitiator (DI), two DNA monomers (DMs). DI is a single-strandedoligonucleotide and DMs have similar hairpin structures with 6-nt (nucleotide) toeholds, 6-nt loops and 18-bp (base pair) stems(Fig. 2A). The procedure of the reaction is shown in Fig. 2B. In thepresence of DI, two DMs are transformed from the intramolecularhybridization state into the intermolecular hybridization state. DIfirst hybridizes with DM1 with the aid of the 6-nt toehold regionand opens half of the stem of DM1. The loop and unhybridized re-gions of the opened DM1 possess the same function as DI to furtherhybridize with DM2, which in turn leads to the opening of the stemof DM2 for the subsequent hybridization with DM1. This sequentialhybridization process is continued to form DPs. The gel electro-phoresis image shows that DPs were successfully synthesized(Fig. 2C). Meanwhile, it shows that the molecular weights of DPslargely fell into the range from 600 bp to 1500 bp, indicating thatthe molecular weight distribution of DPs was wide. This observa-tion may stem from the inherent probability whether an openedmonomer maintains the reactivity to hybridize with a closedmonomer or becomes a less active end. While the initiator andmonomers were purified via high performance liquid chromatog-raphy, their practical purity could not reach 100%. The impure oli-gonucleotides with base mismatches have lower reactivity [29,30]and therefore have negative effects on the growth of DPs. Studieshave also shown that DNA can form cyclic structures [31] thatwould terminate the polymerization. The ability to overcome thesepotential issues may lead to the formation of DPs with narrowermolecular weight distribution.
3.2. Examination of the ability of affinity DPs in Dox sequestration
Since the objective of this study was to demonstrate that affinityDPs on nanoparticles can sequester molecules of interest, we firstused Dox as a small model drug and 50TCG and 50AGC as affinitybinding sites of Dox to examine whether DPs could sequester drugsfrom solution (Fig. 2D). The 50TCG and 50AGC sequences were usedas the binding sites for rational design of the DMs and DPs (Table 1)since they were previously shown to strongly interact with Dox[32,33]. When Dox is intercalated into DNA stems, its fluorescencecan be effectively quenched [34]. The measurement of Dox fluo-rescence of the solution can indicate the amount of free Dox that isnot absorbed by DPs.
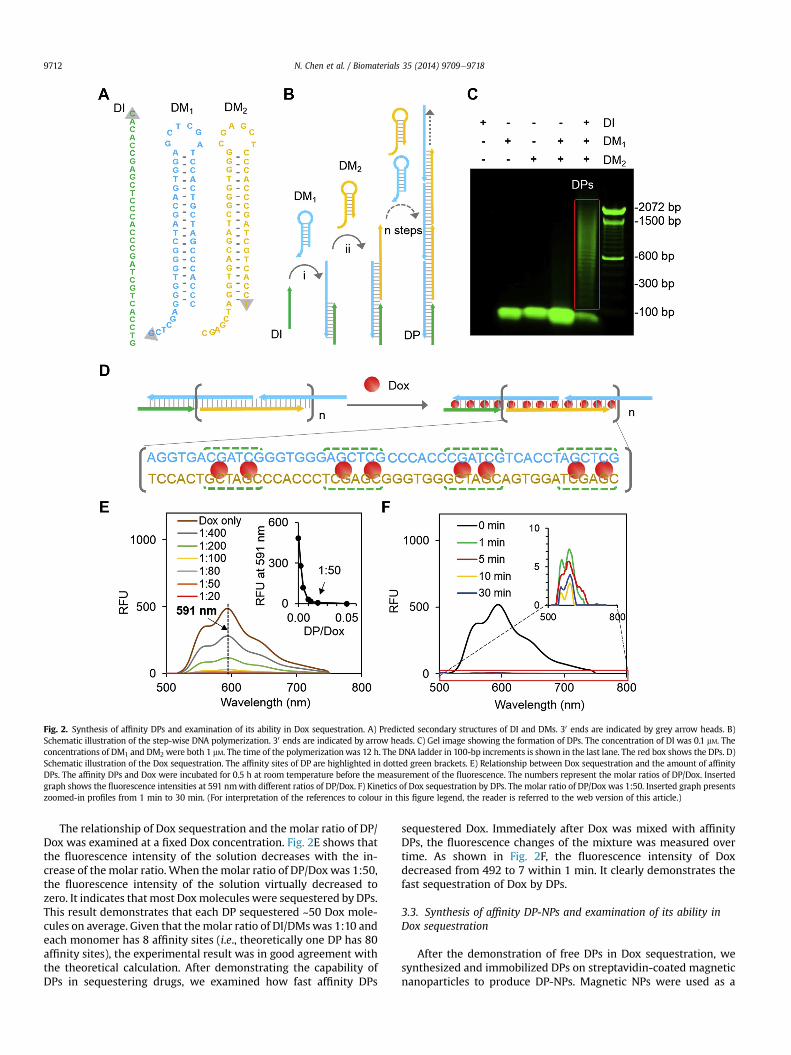
Fig. 2. Synthesis of affinity DPs and examination of its ability in Dox sequestration. A) Predicted secondary structures of DI and DMs. 30 ends are indicated by grey arrow heads. B)Schematic illustration of the step-wise DNA polymerization. 30 ends are indicated by arrow heads. C) Gel image showing the formation of DPs. The concentration of DI was 0.1 mM. Theconcentrations of DM1 and DM2 were both 1 mM. The time of the polymerizationwas 12 h. The DNA ladder in 100-bp increments is shown in the last lane. The red box shows the DPs. D)Schematic illustration of the Dox sequestration. The affinity sites of DP are highlighted in dotted green brackets. E) Relationship between Dox sequestration and the amount of affinityDPs. The affinity DPs and Dox were incubated for 0.5 h at room temperature before the measurement of the fluorescence. The numbers represent the molar ratios of DP/Dox. Insertedgraph shows the fluorescence intensities at 591 nmwith different ratios of DP/Dox. F) Kinetics of Dox sequestration by DPs. The molar ratio of DP/Dox was 1:50. Inserted graph presentszoomed-in profiles from 1 min to 30 min. (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
N. Chen et al. / Biomaterials 35 (2014) 9709e97189712
The relationship of Dox sequestration and the molar ratio of DP/Dox was examined at a fixed Dox concentration. Fig. 2E shows thatthe fluorescence intensity of the solution decreases with the in-crease of the molar ratio. When the molar ratio of DP/Dox was 1:50,the fluorescence intensity of the solution virtually decreased tozero. It indicates that most Doxmolecules were sequestered by DPs.This result demonstrates that each DP sequestered ~50 Dox mole-cules on average. Given that themolar ratio of DI/DMswas 1:10 andeach monomer has 8 affinity sites (i.e., theoretically one DP has 80affinity sites), the experimental result was in good agreement withthe theoretical calculation. After demonstrating the capability ofDPs in sequestering drugs, we examined how fast affinity DPs
sequestered Dox. Immediately after Dox was mixed with affinityDPs, the fluorescence changes of the mixture was measured overtime. As shown in Fig. 2F, the fluorescence intensity of Doxdecreased from 492 to 7 within 1 min. It clearly demonstrates thefast sequestration of Dox by DPs.
3.3. Synthesis of affinity DP-NPs and examination of its ability inDox sequestration
After the demonstration of free DPs in Dox sequestration, wesynthesized and immobilized DPs on streptavidin-coated magneticnanoparticles to produce DP-NPs. Magnetic NPs were used as a

Fig. 3. Synthesis of affinity DP-NPs and examination of its ability in Dox sequestration. A) Fluorescence images of nanoparticle suspensions including NP, DM1-NP and DP-NP. DM1
was labeled with Cy5 at the 50 end. The fluorescence intensity was examined using a CRI Maestro imaging system. B) Size characterization with DLS. The size of nanoparticles wasincreased from 69.0 to 190.0 nm after the synthesis of DPs on the nanoparticle surface. C) Examination of unreacted DMs with gel electrophoresis. After DNA polymerization,supernatants were collected via centrifugation and loaded into 1% agarose gel. The gel was pre-stained with SYBR-Safe prior to electrophoresis. The gel was imaged using the CRIMaestro imaging system. The amounts of unreacted DMs were estimated by measuring the fluorescence intensity of the bands. The control was the supernatant collected from themixture of DMs and nanoparticles without biotinylated DI. D) Schematic illustration of Dox sequestration by DP-NPs in solution. Dox in solution is captured by affinity segments ofDPs on the nanoparticle surface. E) Kinetics of Dox sequestration by DP-NPs. F) Fluorescence images of the supernatants of the Dox solution after different treatments. The Doxsolutions without NP treatment (No NP), treated with unmodified nanoparticles, and nanoparticles displaying only DM1 (DM1-NP) were used as controls. The supernatants werecollected via centrifugation and imaged using a CRI Maestro system. G) Quantification of Dox sequestration by DP-NPs in comparison to the control nanoparticles (n ¼ 3). The Doxconcentration was 10 mM. The amount of Dox sequestration was calculated by subtracting the amount of Dox in supernatant from that in the initial solution.
N. Chen et al. / Biomaterials 35 (2014) 9709e9718 9713
nanoparticle model to synthesize the nanomaterial for two reasons.First, magnetic NPs have been widely used in various biomedicalapplications such as bioimaging, drug delivery and diagnosis[14,35], which have shown the in vivo biocompatibility of magneticNPs [36]. Second, previous studies have demonstrated that externalmagnetic force can be applied to manipulate the behavior ofmagnetic NPs [22,37]. Thus, it is possible to integrate DP-NPs andmagnetic force for drug detoxification, which may provide moreflexibility during the drug detoxification procedure. Meanwhile, it
is important to note that there are a variety of conjugation methodsavailable to conjugate oligonucleotides to a supporting substrate.Thus, while we used biotin and streptavidin to immobilize DI onnanoparticles, any chemical conjugation methods can in principlebe applied to optimize the immobilization of DI onto nanoparticlesand therefore the synthesis of DPs on nanoparticles.
DM1 was labeled with Cy5 for the examination of DP synthesison nanoparticles. The images show that the fluorescence intensityof DP-NPs was significantly stronger than those of two controls
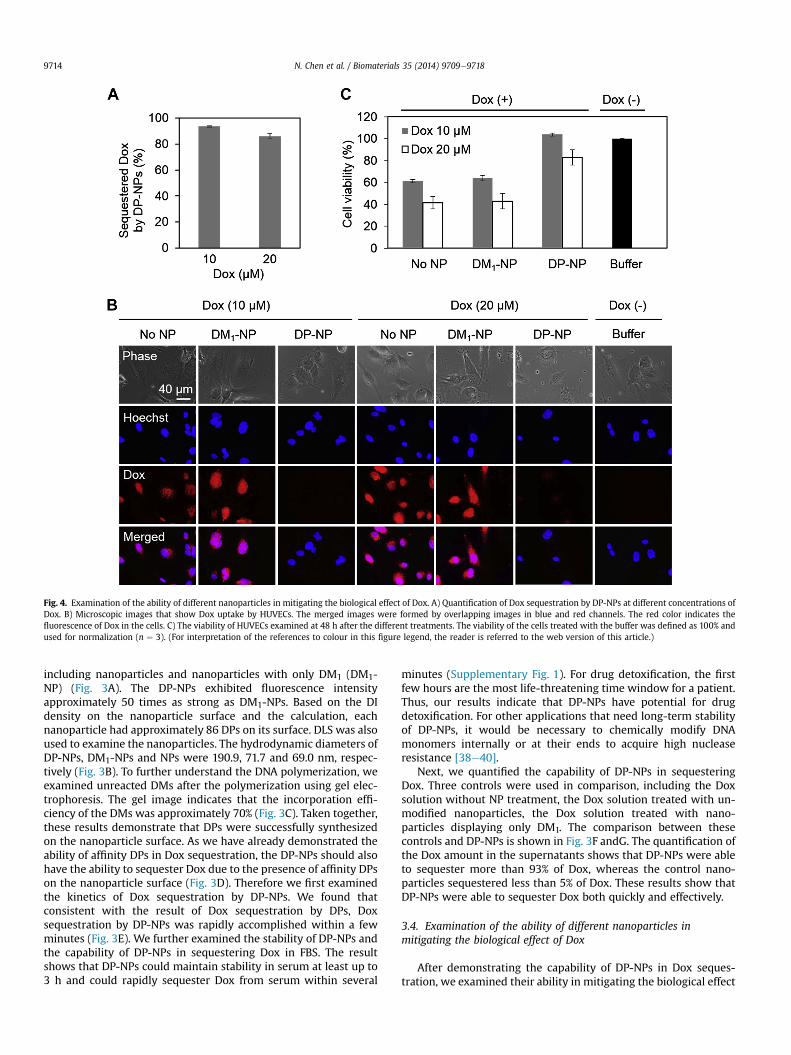
Fig. 4. Examination of the ability of different nanoparticles in mitigating the biological effect of Dox. A) Quantification of Dox sequestration by DP-NPs at different concentrations ofDox. B) Microscopic images that show Dox uptake by HUVECs. The merged images were formed by overlapping images in blue and red channels. The red color indicates thefluorescence of Dox in the cells. C) The viability of HUVECs examined at 48 h after the different treatments. The viability of the cells treated with the buffer was defined as 100% andused for normalization (n ¼ 3). (For interpretation of the references to colour in this figure legend, the reader is referred to the web version of this article.)
N. Chen et al. / Biomaterials 35 (2014) 9709e97189714
including nanoparticles and nanoparticles with only DM1 (DM1-NP) (Fig. 3A). The DP-NPs exhibited fluorescence intensityapproximately 50 times as strong as DM1-NPs. Based on the DIdensity on the nanoparticle surface and the calculation, eachnanoparticle had approximately 86 DPs on its surface. DLS was alsoused to examine the nanoparticles. The hydrodynamic diameters ofDP-NPs, DM1-NPs and NPs were 190.9, 71.7 and 69.0 nm, respec-tively (Fig. 3B). To further understand the DNA polymerization, weexamined unreacted DMs after the polymerization using gel elec-trophoresis. The gel image indicates that the incorporation effi-ciency of the DMs was approximately 70% (Fig. 3C). Taken together,these results demonstrate that DPs were successfully synthesizedon the nanoparticle surface. As we have already demonstrated theability of affinity DPs in Dox sequestration, the DP-NPs should alsohave the ability to sequester Dox due to the presence of affinity DPson the nanoparticle surface (Fig. 3D). Therefore we first examinedthe kinetics of Dox sequestration by DP-NPs. We found thatconsistent with the result of Dox sequestration by DPs, Doxsequestration by DP-NPs was rapidly accomplished within a fewminutes (Fig. 3E). We further examined the stability of DP-NPs andthe capability of DP-NPs in sequestering Dox in FBS. The resultshows that DP-NPs could maintain stability in serum at least up to3 h and could rapidly sequester Dox from serum within several
minutes (Supplementary Fig. 1). For drug detoxification, the firstfew hours are the most life-threatening time window for a patient.Thus, our results indicate that DP-NPs have potential for drugdetoxification. For other applications that need long-term stabilityof DP-NPs, it would be necessary to chemically modify DNAmonomers internally or at their ends to acquire high nucleaseresistance [38e40].
Next, we quantified the capability of DP-NPs in sequesteringDox. Three controls were used in comparison, including the Doxsolution without NP treatment, the Dox solution treated with un-modified nanoparticles, the Dox solution treated with nano-particles displaying only DM1. The comparison between thesecontrols and DP-NPs is shown in Fig. 3F andG. The quantification ofthe Dox amount in the supernatants shows that DP-NPs were ableto sequester more than 93% of Dox, whereas the control nano-particles sequestered less than 5% of Dox. These results show thatDP-NPs were able to sequester Dox both quickly and effectively.
3.4. Examination of the ability of different nanoparticles inmitigating the biological effect of Dox
After demonstrating the capability of DP-NPs in Dox seques-tration, we examined their ability in mitigating the biological effect
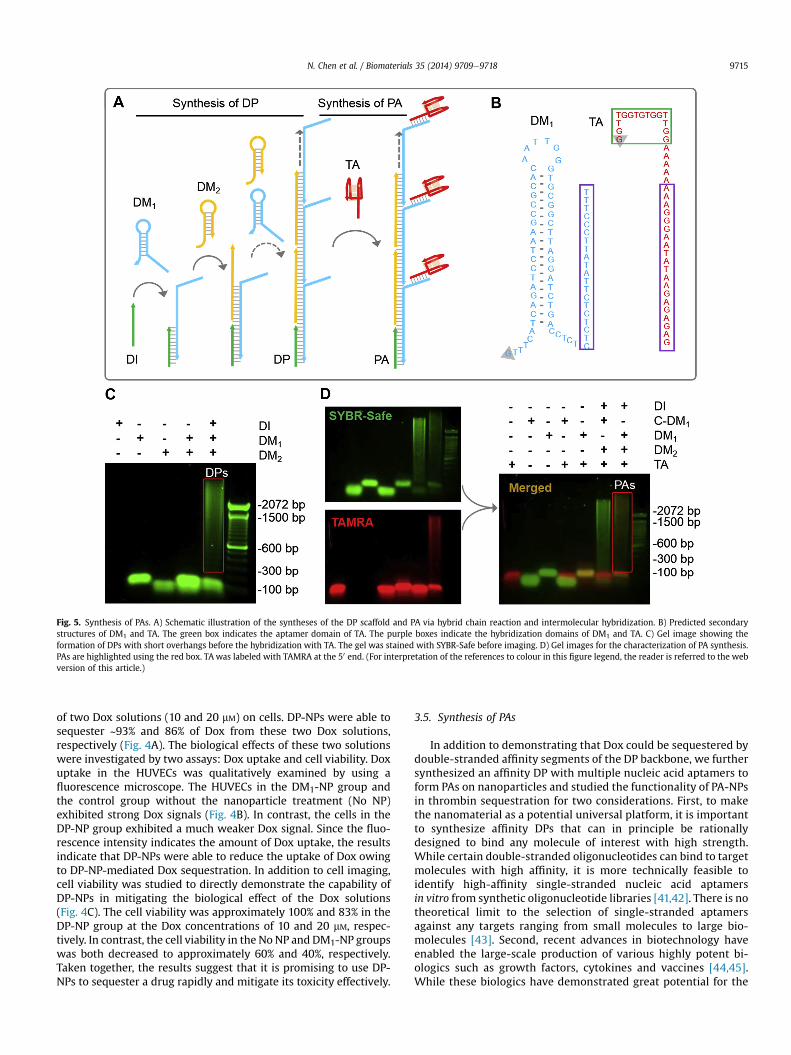
Fig. 5. Synthesis of PAs. A) Schematic illustration of the syntheses of the DP scaffold and PA via hybrid chain reaction and intermolecular hybridization. B) Predicted secondarystructures of DM1 and TA. The green box indicates the aptamer domain of TA. The purple boxes indicate the hybridization domains of DM1 and TA. C) Gel image showing theformation of DPs with short overhangs before the hybridization with TA. The gel was stained with SYBR-Safe before imaging. D) Gel images for the characterization of PA synthesis.PAs are highlighted using the red box. TA was labeled with TAMRA at the 50 end. (For interpretation of the references to colour in this figure legend, the reader is referred to the webversion of this article.)
N. Chen et al. / Biomaterials 35 (2014) 9709e9718 9715
of two Dox solutions (10 and 20 mM) on cells. DP-NPs were able tosequester ~93% and 86% of Dox from these two Dox solutions,respectively (Fig. 4A). The biological effects of these two solutionswere investigated by two assays: Dox uptake and cell viability. Doxuptake in the HUVECs was qualitatively examined by using afluorescence microscope. The HUVECs in the DM1-NP group andthe control group without the nanoparticle treatment (No NP)exhibited strong Dox signals (Fig. 4B). In contrast, the cells in theDP-NP group exhibited a much weaker Dox signal. Since the fluo-rescence intensity indicates the amount of Dox uptake, the resultsindicate that DP-NPs were able to reduce the uptake of Dox owingto DP-NP-mediated Dox sequestration. In addition to cell imaging,cell viability was studied to directly demonstrate the capability ofDP-NPs in mitigating the biological effect of the Dox solutions(Fig. 4C). The cell viability was approximately 100% and 83% in theDP-NP group at the Dox concentrations of 10 and 20 mM, respec-tively. In contrast, the cell viability in the No NP and DM1-NP groupswas both decreased to approximately 60% and 40%, respectively.Taken together, the results suggest that it is promising to use DP-NPs to sequester a drug rapidly and mitigate its toxicity effectively.
3.5. Synthesis of PAs
In addition to demonstrating that Dox could be sequestered bydouble-stranded affinity segments of the DP backbone, we furthersynthesized an affinity DP with multiple nucleic acid aptamers toform PAs on nanoparticles and studied the functionality of PA-NPsin thrombin sequestration for two considerations. First, to makethe nanomaterial as a potential universal platform, it is importantto synthesize affinity DPs that can in principle be rationallydesigned to bind any molecule of interest with high strength.While certain double-stranded oligonucleotides can bind to targetmolecules with high affinity, it is more technically feasible toidentify high-affinity single-stranded nucleic acid aptamersin vitro from synthetic oligonucleotide libraries [41,42]. There is notheoretical limit to the selection of single-stranded aptamersagainst any targets ranging from small molecules to large bio-molecules [43]. Second, recent advances in biotechnology haveenabled the large-scale production of various highly potent bi-ologics such as growth factors, cytokines and vaccines [44,45].While these biologics have demonstrated great potential for the

Fig. 6. Evaluation of PA-NPs in sequestering thrombin and mitigating its biological effect. A) Fluorescence images of nanoparticle suspensions. DP-NPs were hybridized with TA toform PA-NPs. Nanoparticles without modifications, with only DM1 hybridized with TA and with control DPs are denoted as NP, MA-NP and DP-NP respectively. The dotted circlesoutline the nanoparticle suspensions for clear legibility. B) Schematic illustration of thrombin sequestration by PA-NP. The image of thrombin was adopted from RCSB Portein DataBank. C) Measurement of thrombin sequestration from the thrombin solution (n ¼ 3). D) Measurement of coagulation time (n ¼ 3).
N. Chen et al. / Biomaterials 35 (2014) 9709e97189716
treatment of various human diseases, their high potency may alsoresult in more severe toxicity than small molecule drugs [46,47].However, in comparison to small molecule drugs, the develop-ment of antidotes to mitigate the toxicity of these highly potentbiologics has received very little attention. Since aptamers canbind biologics with high affinities and specificities comparable toantibodies, they are promising affinity ligands for the develop-ment of nanoscale antidotes for biologics. Thus, we synthesizedPA-NPs and further examined the functionality of PA-NPs inthrombin sequestration.
The synthesis of PAs involves two steps. In the first step, a DPscaffold was synthesized (Fig. 5A). Specifically, DM1 had a 25-nucleotide overhang that protrudes from the DP backbone andwas used to hybridize with the aptamer. The structural predictionshows that the 25-nucleotide overhang was single-stranded andhad no intramolecular hybridization with other regions of themonomer (Fig. 5B), indicating that the overhang would not inter-fere with the synthesis of DPs but had the capability to hybridizewith the anti-thrombin aptamer (TA). The gel image shows that DPscould be synthesized with the monomer with the overhang(Fig. 5C), which is consistent with the structural prediction. In thesecond step, DPs were hybridized with the aptamers (Fig. 5A). Thegel images show that the TAMRA-labeled TA hybridized with DPs toform PAs, as indicated by the overlap of green and red colors(Fig. 5D). Based on the measurement of the fluorescence intensityof free TA, each PA had approximately 7 TA.
3.6. Evaluation of PA-NPs in sequestering thrombin and mitigatingits biological effect
To illustrate whether PAs could be synthesized on nanoparticles,the nanoparticles were examined with fluorescence imaging. Theimages show that the fluorescence intensity of PA-NPs was signif-icantly stronger than those of three control nanoparticles (Fig. 6A).Specifically, the fluorescence intensity of PA-NPs was ~26-foldstronger than that of nanoparticles with only DM1 and TA (MA-NP).Clearly, our results show that PA-NPs were successfully synthesizedwith the DP, the aptamer and the nanoparticle.
Because of the presence of the PAs on nanoparticles, the PA-NPswere expected to have the capability of sequestering thrombin(Fig. 6B). The results show that PA-NPs sequestered 70% ofthrombin from the solution (Fig. 6C). In contrast, NPs and MA-NPssequestered 3% and 17%, respectively. These results demonstratethat the PA-NPs possessed the capability of sequestering the ma-jority of thrombin from the solution. Meanwhile, it is needed tonote that the molar ratio of aptamers to thrombin was approxi-mately 8:1. The anti-thrombin aptamer is the most commonly usedmodel aptamer and has a dissociation constant (Kd) of ~102.6 nM
[48,49]. In the situation of equilibrium, virtually all thrombinmolecules would be sequestered by the PA-NPs based on this Kdvalue. The difference between the experimental result and thetheoretical analysis may be due to the molecular dissociation dur-ing the high speed centrifugation that generates high shear stress

N. Chen et al. / Biomaterials 35 (2014) 9709e9718 9717
during the precipitation of nanomaterials, because previous studieshave indicated that high shear stress can induce biomoleculardissociation [50,51]. Such a problem may be solved by using high-affinity aptamers. Most aptamers identified against protein targetshave Kd values smaller than 10 nM. For instance, the anti-fibroblastgrowth factor 2 aptamer has a Kd of 0.35 nM [52]. Future work needsto be pursued to examine the effects of binding affinities on mo-lecular sequestration or to design hybrid aptamers for simu-latenous binding of two different regions of the same target. Wefurther performed a coagulation time assay to test the function ofPA-NPs. If more thrombin molecules are sequestered from solution,fewer free thrombin molecules will be present in the solution tocatalyze coagulation. As a result, the coagulation time will be pro-longed. Thus, this assay can be used to examine whether PA-NPscan effectively and stably sequester thrombin. The thrombin solu-tion without nanoparticles, the mixture of thrombin and nano-particles, and the mixture of thrombin and MA-NPs were used ascontrols. As shown in Fig. 6D, PA-NPs prolonged the coagulationtime from 24 to 103 s whereas the use of the control nanoparticlesprovided the coagulation time less than 35 s. Thus, the results showthat the PA-NPs can sequester target biologics and mitigate theirbiological effects.
4. Conclusions
In conclusion, a nanomaterial functionally similar to jellyfishtentacles in sequestering target molecules from surroundings hasbeen successfully synthesized with affinity DNA polymers andnanoparticles. This nanomaterial can effectively sequester bothsmall molecule drugs and large molecule biologics. As a result, thebiological effects of the sequestered drugs can be significantlymitigated. Thus, this nanomaterial holds potential as a universalnanoscale antidote for drug removal and detoxification. While thisnanomaterial was evaluated by using drug removal and detoxifi-cation as a model, the synthesis of periodically oriented affinitypolymers on a nanoparticle with the capability of sequesteringtarget molecules may find broad applications in the fields of sep-aration, sensing, imaging and drug delivery.
Acknowledgments
This workwas in part supported by the Penn State Start-up Fundand the U.S. National Science Foundation (DMR-1322332 andCMMI-1131587). N.C. would also like to acknowledge the supportfrom the China Scholarship Council. The authors would like toacknowledge Ms. Shihui Li and Mr. Xiaolong Zhang for technicalsupport.
Appendix A. Supplementary data
Supplementary data related to this article can be found at http://dx.doi.org/10.1016/j.biomaterials.2014.08.017.
References
[1] Peer D, Karp JM, Hong S, Farokhzad OC, Margalit R, Langer R. Nanocarriers asan emerging platform for cancer therapy. Nat Nanotechnol 2007;2:751e60.
[2] Kesharwani P, Gajbhiye V, Jain NK. A review of nanocarriers for the delivery ofsmall interfering RNA. Biomaterials 2012;33:7138e50.
[3] Wang H, Zhou W, Yin X, Zhuang Z, Yang H, Wang X. Template synthesizedmolecularly imprinted polymer nanotube membranes for chemical separa-tions. J Am Chem Soc 2006;128:15954e5.
[4] Cao YC, Jin R, Mirkin CA. Nanoparticles with Raman spectroscopic fingerprintsfor DNA and RNA detection. Science 2002;297:1536e40.
[5] Liu J, Lu Y. Fast colorimetric sensing of adenosine and cocaine based on ageneral sensor design involving aptamers and nanoparticles. In: Angew Chem,Intvol. 45; 2006. p. 90e4.
[6] Gao X, Cui Y, Levenson RM, Chung LWK, Nie S. In vivo cancer targeting andimaging with semiconductor quantum dots. Nat Biotechnol 2004;22:969e76.
[7] Chen N, Li S, Battig MR, Wang Y. Programmable imaging amplification viananoparticle-initiated DNA polymerization. Small 2013;9:3944e9.
[8] Haun JB, Devaraj NK, Hilderbrand SA, Lee H, Weissleder R. Bioorthogonalchemistry amplifies nanoparticle binding and enhances the sensitivity of celldetection. Nat Nanotechnol 2010;5:660e5.
[9] Dhanikula AB, Khalid NM, Lee SD, Yeung R, Risovic V, Wasan KM, et al. Longcirculating lipid nanocapsules for drug detoxification. Biomaterials 2007;28:1248e57.
[10] Hu C-MJ, Fang RH, Copp J, Luk BT, Zhang L. A biomimetic nanosponge thatabsorbs pore-forming toxins. Nat Nanotechnol 2013;8:336e40.
[11] Torchilin VP. Recent advances with liposomes as pharmaceutical carriers. NatRev Drug Discov 2005;4:145e60.
[12] Smith C, Shkumatov A, Withers S. A polymeric fastener can easily function-alize liposome surfaces with gadolinium for enhanced magnetic resonanceimaging. ACS Nano 2013;7:9599e610.
[13] Wang Y, Gao S, Ye W-H, Yoon HS, Yang Y-Y. Co-delivery of drugs and DNAfrom cationic core-shell nanoparticles self-assembled from a biodegradablecopolymer. Nat Mater 2006;5:791e6.
[14] Gupta AK, Gupta M. Synthesis and surface engineering of iron oxide nano-particles for biomedical applications. Biomaterials 2005;26:3995e4021.
[15] Pan Y, Du X, Zhao F, Xu B. Magnetic nanoparticles for the manipulation ofproteins and cells. Chem Soc Rev 2012;41:2912e42.
[16] Hoshino Y, Kodama T, Okahata Y, Shea KJ. Peptide imprinted polymer nano-particles: a plastic antibody. J Am Chem Soc 2008;130:15242e3.
[17] Hoshino Y, Koide H, Urakami T, Kanazawa H, Kodama T, Oku N, et al.Recognition, neutralization, and clearance of target peptides in the blood-stream of living mice by molecularly imprinted polymer nanoparticles: aplastic antibody. J Am Chem Soc 2010;132:6644e5.
[18] Bateman DN. Digoxin-specific antibody fragments: how much and when?Toxicol Rev 2004;23:135e43.
[19] Jones CM, Mack KA, Paulozzi LJ. Pharmaceutical overdose deaths, UnitedStates, 2010. JAMA J Am Med Assoc 2013;309:657e9.
[20] Leroux JC. Injectable nanocarriers for biodetoxification. Nat Nanotechnol2007;2:679e84.
[21] Cai K, Li J, Luo Z, Hu Y, Hou Y, Ding X. b-Cyclodextrin conjugated magneticnanoparticles for diazepam removal from blood. Chem Commun 2011;47:7719e21.
[22] Mertz CJ, Kaminski MD, Xie Y, Finck MR, Guy S, Rosengart AJ. In vitro studiesof functionalized magnetic nanospheres for selective removal of a simulantbiotoxin. J Magn Magn Mater 2005;293:572e7.
[23] Aldaye FA, Palmer AL, Sleiman HF. Assembling materials with DNA as theguide. Science 2008;321:1795e9.
[24] Seeman NC. DNA in a material world. Nature 2003;421:427e31.[25] Fire A, Xu SQ. Rolling replication of short DNA circles. Proc Natl Acad Sci U. S. A
1995;92:4641e5.[26] Saiki RK, Gelfand DH, Stoffel S, Scharf SJ, Higuchi R, Horn GT, et al. Primer-
directed enzymatic amplification of DNA with a thermostable DNA poly-merase. Science 1988;239:487e91.
[27] Dirks RM, Pierce NA. Triggered amplification by hybridization chain reaction.Proc Natl Acad Sci U S A 2004;101:15275e8.
[28] Zadeh JN, Steenberg CD, Bois JS, Wolfe BR, Pierce MB, Khan AR, et al. NUPACK:analysis and design of nucleic acid systems. J Comput Chem 2010;32:170e3.
[29] Letowski J, Brousseau R, Masson L. Designing better probes: effect of probesize, mismatch position and number on hybridization in DNA oligonucleotidemicroarrays. J Microbiol Meth 2004;57:269e78.
[30] Gotoh M, Hasegawa Y, Shinohara Y, Shimizu M, Tosu M. A new approach todetermine the effect of mismatches on kinetic parameters in DNA hybridi-zation using an optical biosensor. DNA Res 1995;2:285e93.
[31] Xu J, Fogleman EA, Craig SL. Structure and properties of DNA-based reversiblepolymers. Macromolecules 2004;37:1863e70.
[32] Roche CJ, Thomson JA, Crothers DM. Site selectivity of daunomycin.Biochemistry 1994;33:926e35.
[33] Chaires JB, Herrera JE. Preferential binding of daunomycin to 5'ATCG and5'ATGC sequences revealed by footprinting titration experiments. Biochem-istry 1990;29:6145e53.
[34] Tarasiuk J, Fr�ezard F, Garnier-Suillerot A, Gattegno L. Anthracycline incorpo-ration in human lymphocytes. Kinetics of uptake and nuclear concentration.Biochim Biophys Acta 1989;1013:109e17.
[35] Tassa C, Shaw SY, Weissleder R. Dextran-coated iron oxide nanoparticles: aversatile platform for targeted molecular imaging, molecular diagnostics, andtherapy. Acc Chem Res 2011;44:842e52.
[36] Reddy LH, Arias JL, Nicolas J, Couvreur P. Magnetic nanoparticles: design andcharacterization, toxicity and biocompatibility, pharmaceutical and biomed-ical applications. Chem Rev 2012;112:5818e78.
[37] Kaminski MD, Rosengart AJ. Detoxification of blood using injectable magneticnanospheres: a conceptual technology description. J Magn Magn Mater2005;293:398e403.
[38] Shaw JP, Kent K, Bird J, Fishback J, Froehler B. Modified deoxyoligonucleotidesstable to exonuclease degradation in serum. Nucleic Acids Res 1991;19:747e50.
[39] Verma S, Eckstein F. Modified oligonucleotides: synthesis and strategy forusers. Annu Rev Biochem 1998;67:99e134.

N. Chen et al. / Biomaterials 35 (2014) 9709e97189718
[40] Eder PS, DeVine RJ, Dagle JM, Walder JA. Substrate specificity and kinetics ofdegradation of antisense oligonucleotides by a 3’ exonuclease in plasma.Antisense Res Dev 1991;1:141e51.
[41] Tuerk C, Gold L. Systematic evolution of ligands by exponential enrichment:RNA ligands to bacteriophage T4 DNA polymerase. Science 1990;249:505e10.
[42] Andrew D, Ellington JWS. In vitro selection of RNA molecules that bind spe-cific ligands. Nature 1990;346:818e22.
[43] Wilson DS, Szostak JW. In vitro selection of functional nucleic acids. Annu RevBiochem 1999;68:611e47.
[44] Wurm FM. Production of recombinant protein therapeutics in cultivatedmammalian cells. Nat Biotechnol 2004;22:1393e8.
[45] Vicente T, Rold~ao A, Peixoto C, Carrondo MJT, Alves PM. Large-scale produc-tion and purification of VLP-based vaccines. J Invertebr Pathol 2011;107:S42e8.
[46] Sullivan DH, Carter WJ, Warr WR, Williams LH. Side effects resulting from theuse of growth hormone and insulin-like growth factor-I as combined therapyto frail elderly patients. J Gerontol Ser A 1998;53:M183e7.
[47] Vial T, Descotes J. Clinical toxicity of cytokines used as haemopoietic growthfactors. Drug Saf 1995;13:371e406.
[48] Bock LC, Griffin LC, Latham JA, Vermaas EH, Toole JJ. Selection of single-stranded DNA molecules that bind and inhibit human thrombin. Nature1992;355:564e6.
[49] Pasternak A, Hernandez FJ, Rasmussen LM, Vester B, Wengel J. Improvedthrombin binding aptamer by incorporation of a single unlocked nucleic acidmonomer. Nucleic Acids Res 2011;39:1155e64.
[50] Weisel J, Shuman H, Litvinov R. Proteineprotein unbinding induced by force:single-molecule studies. Curr Opin Struct Biol 2003;13:227e35.
[51] Farrance OE, Hann E, Kaminska R, Housden NG, Derrington SR, Kleanthous C,et al. A force-activated trip switch triggers rapid dissociation of a colicin fromits immunity protein. PLoS Biol 2013;11. e1001489.
[52] Jellinek D, Lynott CK, Rifkin DB, Janji�c N. High-affinity RNA ligands to basicfibroblast growth factor inhibit receptor binding. Proc Natl Acad Sci U. S. A1993;90:11227e31.





























