Embed Size (px)
Citation preview

Biochimica et Biophysica Acta 1852 (2015) 658–666
Contents lists available at ScienceDirect
Biochimica et Biophysica Acta
j ourna l homepage: www.e lsev ie r .com/ locate /bbad is
Review
Pathogenesis of immune-mediated neuropathies☆
Marinos C. Dalakas a,b,⁎a University of Athens Medical School, Athens, Greeceb Thomas Jefferson University, Philadelphia, PA, USA
☆ This article is part of a Special Issue entitled: NeuromMolecular Pathogenesis.⁎ Corresponding author at: Neuroimmunology Unit, De
University of AthensMedical School, 75Mikras Asias Str., A210 746 2616x2650; fax: +30 210 746 2664.
E-mail address: [email protected].
http://dx.doi.org/10.1016/j.bbadis.2014.06.0130925-4439/© 2014 Elsevier B.V. All rights reserved.
a b s t r a c t
a r t i c l e i n f oArticle history:Received 15 May 2014Accepted 9 June 2014Available online 17 June 2014
Keywords:AutoimmunityNeuropathyRanvierImmunotherapy
Autoimmune neuropathies occur when immunologic tolerance to myelin or axonal antigens is lost. Even thoughthe triggering factors and the underling immunopathology have not been fully elucidated in all neuropathy sub-sets, immunological studies on the patients' nerves, transfer experiments with the patients' serum or intraneuralinjections, andmolecularfingerprinting on circulating autoantibodies or autoreactive T cells, indicate that cellularand humoral factors, either independently or in concert with each other, play a fundamental role in their cause.The review is focused on the main subtypes of autoimmune neuropathies, mainly the Guillain–Barré syn-drome(s), the Chronic Inflammatory Demyelinating Polyneuropathy (CIDP), the Multifocal Motor Neuropathy(MMN), and the IgM anti-MAG-antibodymediated neuropathy. It addresses the factors associatedwith breakingtolerance, examines the T cell activation process including co-stimulatory molecules and key cytokines, and dis-cusses the role of antibodies against peripheral nerve glycolipids or glycoproteins. Special attention is given to thenewly identified proteins in the nodal, paranodal and juxtaparanodal regions as potential antigenic targets thatcould best explain conduction failure and rapid recovery. New biological agents against T cells, cytokines, Bcells, transmigration and transduction molecules involved in their immunopathologic network, are discussedas future therapeutic options in difficult cases. This article is part of a Special Issue entitled: NeuromuscularDiseases: Pathology and Molecular Pathogenesis.
© 2014 Elsevier B.V. All rights reserved.
1. Introduction
Autoimmune Peripheral Neuropathies (APN) develop when immu-nologic tolerance to key antigenic sites on the myelin, axon, nodes ofRanvier or ganglionic neurons is lost [1,2]. Current evidence supportsthe notion that in APN the autoimmunity is mediated by antibodies di-rected againstmyelin antigens, alongwith autoreactive T cells andmac-rophages that invade myelin sheath, axonal membranes or the nodes ofRanvier. In some APN the triggering factors have been identified andprogress has been made in understanding the key players involved inthe immunopathogenic network; in several others however, the exactimmunopathologic mechanisms remain still unclear, but autoimmunityis suspected based on the presence of antibodies, sensitized T cell infil-trates in the peripheral nerves, response to immunotherapies or coexis-tence with another autoimmune disease or viral infections. The APN areclinically important because they respond to immunotherapies basedon controlled studies, or have the potential to respond if more effective
uscular Diseases: Pathology and
pt. of Pathophysiology, Nationalthens, 11527 Greece. Tel.: +30
agents are used. The review addresses the autoimmune pathways in-volved in the pathogenesis of themost common autoimmune neuropa-thies and highlights the rationale for target-specific immunotherapies.
1.1. Main clinicopathologic features of common APN
The common autoimmune neuropathies include: 1) Acute Inflam-matory Polyneuropathy [the Guillain–Barré Syndrome(s)]; 2) Chronicinflammatory demyelinating polyneuropathy (CIDP) and its variants;3) Multifocal motor neuropathy with conduction block; and 4)Polyneuropathies associated with IgM monoclonal gammopathies andanti-MAG antibodies [1,2]. Other less common immune-mediated neu-ropathies that will not be addressed here because their pathogenesis isless clear include: the paraneoplastic neuropathies associatedwith anti-Hu antibodies, vasculitic neuropathies due to isolated peripheral nervevasculitis, certain diabetic demyelinating or inflammatory neuropathieswhere an immune component appears prominent, and the neuropa-thies associated with systemic autoimmune disorders.
As discussed below and elaborated previously [1,2], in some APN,like certain GBS variants and the anti-MAG neuropathy, themain targetantigens have been identified and the antibodies against them are wellcharacterized; in the most common neuropathies however, like the de-myelinating variant of GBS and the CIDP, the target antigens remain still

659M.C. Dalakas / Biochimica et Biophysica Acta 1852 (2015) 658–666
elusive in spite of the progress made in understanding their molecularimmunopathology.
The Guillain–Barré syndrome(s) (GBS) presents with an acute(within 1–3 weeks) ascending motor weakness, areflexia, and mild tomoderate sensory abnormalities [1–5]. The neuropathy is inflammatory,with perivascular and endoneurial lymphoid cell infiltrates throughoutthe nerves, roots or plexuses, and demyelinatingwith signs of segmentaldemyelination induced by complement-fixing antibodies and macro-phages as the final effector cells. GBS represents several syndromes –or variants – according to whether the main clinicopathologic involve-ment is centered onmotor or sensory nerve fibers andwhether it affectspredominantly the myelin or the axon [1–4]. Accordingly, the GBS syn-dromes comprise the following subtypes: i) the acute inflammatory de-myelinating polyneuropathy (AIDP), where the main target appears tobe the myelin and accounts for the majority of GBS patients; ii) theacute, motor axonal neuropathy (AMAN), where the primary pathologyis in the axon, either due tomassive acute demyelination and inflamma-tion, as occurs in experimental allergic neuritis when animals are im-munized with a high dose of myelin antigens [1,2,6], or due to aprimary attack on the axons and the nodes of Ranviermediated bymac-rophages and antibodies. A number of these cases have high GM1 anti-bodies and, as discussed later, report an antecedent infection withCampylobacter jejuni [2,3,7]; iii) the acute motor–sensory axonal neurop-athy (AMSAN), which is like AMAN but with concurrent involvement ofthe sensory axons; iv) the Miller-Fisher syndrome, characterized byophthalmoplegia, gait ataxia, and areflexia [1–5], a rather distinct vari-ant because of theunique clinical phenotype and the presence of IgG an-tibodies against GQ1b ganglioside [1–4,7,8]; vi) the sensory ataxic GBS,probably due to involvement of dorsal roots and ganglionic neurons;some of these patients have also IgG antibodies to GQ1b or GD1b gangli-oside and may be forming a continuumwith Miller-Fisher syndrome orshare autoantibodies with the same sialic groups [1,2,8,9]; and vii) theacute pandysautonomic neuropathywhere the target antigen is probablyin the sympathetic ganglionic neurons [1,2].
1.1.1. Chronic inflammatory demyelinating polyneuropathy (CIDP)This is the most common APN with prevalence as high as 9/100,000
[2,10,11], and the most gratifying neuropathy because it is treatable inthe majority of the cases [2]. CIDP is viewed as the chronic counterpartof GBS [1,2] because it shareswithGBS certain clinical, electrophysiolog-ic, histologic, laboratory and autoimmune features. It differs from GBSpredominantly by its tempo, mode of evolution, prognosis, and respon-siveness to steroids or immunosuppressants [1,2,10,12,16]. On electro-physiological grounds, CIDP demonstrates features of demyelination inmotor and sensory fibers with slow conduction velocity, dispersion ofthe compound muscle action potentials, conduction block in at leastone nerve, prolonged distal motor or sensory latencies and prolongedFwave latencies. On histological grounds, there is evidence of demyelin-ation associated with macrophages, complement and activated T cells[10,12–16]. The demyelination in CIDP is also multifocal, like the oneseen in GBS, affecting spinal roots, plexuses and proximal nerve trunks[13–15], accounting for the variable distribution of symptoms andsigns, which are clinically expressed as CIDP variants [10]. The mostnotable CIDP variables are the asymmetric, unifocal or multifocal motor–sensory form (the Lewis-Sumner syndrome); the pure motor form; thepure sensory form; the sensory ataxic form; and the pure distal [1,2,10,12,16].
1.1.2. Multifocal motor neuropathy (MMN) with conduction blockMMN has distinct clinical and electrophysiologic criteria, namely,
weakness in the distribution of individual motor nerves and multifocalconduction block limited to motor but sparing the sensory nerves [1,2,17]. In contrast to CIDP, where conduction in the sensory nerves isalso affected and sensory responses may not be elicited, in MMN thesensory conduction remains normal across the nerve segments thathave the motor block. The reasons for such a selective motor
involvement remain unclear. Differences in the myelin composition,leading to distinct antigenic specificities betweenmotor and sensory fi-bers, have been implicatedmainly because the ceramide content withinthe gangliosides differs between sensory and motor fibers [1,2,8]; thereasons however may be more complex and relate to the uniquenessof the targeted antigens not within the compact myelin but rather inthe nodal regions and axonal membrane at the nodes of Ranvier. Upto 50% of MMN patients have high IgM anti-GM1 antibody titers, butthe pathogenic role of these antibodies remains unclear [1,2,17], inspite of recent evidence that GM1 antibodies fix complement and mayinduce a complement-mediated nodal dysfunction that reverses afterIVIg therapy [18]. The suggestion that GM1 antibodies bind to GM1and interfere with saltatory conduction, has not been confirmed andGM1-binding sites have not been co-localized with voltage-gated sodi-um channels at the nodes of Ranvier. Even though the immunopatholo-gy remains uncertain, MMN patients respond remarkably well toimmunotherapy with IVIg.
1.1.3. IgM MGUS polyneuropathies with anti-MAG or ganglioside antibodiesThe majority of these patients present with a chronic, slowly pro-
gressive, large-fiber, sensory polyneuropathy of insidious onset, mani-fested as sensory ataxia [1,2,19–21]. Other times, it presents assensorimotor polyneuropathy with mixed features of demyelinationand axonal loss. Conduction velocity is slow with a rather characteristicprolonged distal motor and sensory latencies consistent with distal de-myelination. The serum protein electrophoresis shows a monoclonalIgM spike that recognizes antigenic components on the compact mye-lin, most of the times the Myelin Associated Glycoprotein (MAG) [21],as discussed later. Sural nerve biopsy demonstrates diminished numberof myelinated axons with a characteristic splitting of the outer myelinlamellae, linked to deposition of IgM that recognizes MAG in the samearea of the split myelin [1,2,21].
1.2. Immunopathogenesis
In general, complement-fixing antibodies, macrophages and T cellsare themain effectormechanisms in all APN. In vitro and in vivo studies,including immunization of animals withmyelin proteins, disease trans-fer experiments with the patients' serum or with intraneural injections,immunocytochemical studies on the patients' nerves, and T cell profil-ing studies in the peripheral blood, have provided evidence that bothcellular and humoral factors, either independently or in concert witheach other, play a central role in their pathogenesis [1,2].
1.3. GBS
1.3.1. Cellular factorsIn autopsy cases of GBS patients, perivascular and endoneurial in-
flammatory infiltrates are observed throughout the nerves, roots orplexuses along with segmental demyelination mediated by macro-phages, especially in areas with lymphoid infiltrates [1–4]. The macro-phages break through the basement membrane of healthy Schwanncells andmake direct contact with the outermostmyelin lamellae, lead-ing to destruction of the superficial myelin sheath [1–5]. Cytokines andchemokines released by the activated T cells or complement activation,may increase capillary permeability and facilitate transmigration of ad-ditional macrophages or T cells. Increased levels of IL-2 and soluble IL-2receptors are also noted in the serum during the acute phase of GBSsuggesting ongoing T-cell activation [3]. Further, lymphocytes fromGBS patients exert myelinotoxic activity when applied to cultures ofmyelinated axons [1,2]. The involvement of a T cell-mediated processin GBShas been strengthened byobservations in the experimental aller-gic neuritis (EAN)model induced in animals immunizedwith thewholehuman nerve or with various peripheral nerve myelin proteins, such asPo, P2, or galactocerebroside. These animals develop EANwith segmen-tal demyelination and mononuclear cell infiltrates consisting of

660 M.C. Dalakas / Biochimica et Biophysica Acta 1852 (2015) 658–666
macrophages and T cells; the T cells are sensitized against myelin andcan passively transfer the disease to healthy animals [1–4].
1.3.2. Humoral factors: the role of anti-ganglioside antibodies and molecularmimicry
There is a stronger supporting evidence that circulating serum fac-tors play a role in the pathogenesis of GBS. On clinical grounds, this isbased on the beneficial effect of plasmapheresis that removes putativepathogenic antibodies and other inflammatory mediators relevant todemyelination and conduction block [1–5]; on laboratory grounds it isbased on the variety of autoantibodies detected in the patients' serum.
It has been known for years that serum from the acute phase of GBSpatients can demyelinate rodent dorsal root ganglionic extracts in acomplement-dependent manner. Further, GBS serum injected into ratsciatic nerves can cause demyelination and conduction block [1–5]. Im-munocytochemical studies on peripheral nerves fromGBSpatients haveshown deposits of IgG, IgM and membranolytic attack complex, imply-ing complement-fixing antibodies against myelinated fibers [1–5]. Ad-ditionally, complement-fixing IgM antibodies against a peripheralnerve glycolipid that contains carbohydrate epitopes and various sulfat-ed or acidic glycosphingolipids, have been detected in the serum of GBSpatients [1–5,7–9].
The interest has been focused on gangliosides as the more likely an-tigenic targets because they are abundant in the peripheral nerve andcan elicit an immune response because their signature sugar residuesbearing one or more sialic acid molecules (such as ganglioside GM1that contains one sialic acid, GD1a with two, GT1a with three, or GQ1bwith four sialic acids), are exposed at the extracellular surface [1,2,7,19]. Different ganglioside antibodies associated with specific GBS sub-types appear pathogenic [1,2,8,9]; for example, immunization of rabbitswith GM1 or GD1b induces an acute neuropathy with histological fea-tures of AMAN, while GQ1b or GD1a antibodies may cause conductionblock at themotor nerve terminals in amouse phrenic nerve preparation[1–3,8,9]. An inadvertent experiment in humans has strengthened thepathogenic role of GM1 as a triggering factor because patients who re-ceived ganglioside injections for various maladies, developed AMANwith GM1 antibodies [20]. Overall, IgG antibodies that react with GM1,GD1a, GalNAc-GD1a and GM1b are found in 80% of patients with the ax-onal formsofGBS (AMANandAMSAN); in contrast, in themost commonGBS subtype, the AIDP, anti-ganglioside antibodies are infrequent andthe antigenic targets remain still elusive. One ganglioside that clearlycorrelates with a specific clinical syndrome is the GQ1b because IgG-anti-GQ1b antibodies are specifically associated with the Miller-Fishervariant and they are detected in more than 90% of these patients [1,2,8,9]. In contrast, anti-GQ1b antibodies of the IgM class are found in somepatients with chronic IgM paraproteinemic polyneuropathies [1,2,19],as discussed later. Anti-GQ1b IgG antibodies are also found in post-infectious ophthalmoplegias and in GBS patients with ophthalmoplegia,but not in GBS patients without ophthalmoplegia [1], probably becauseGQ1b is present in the paranodal regions of oculomotor nerves III, IV,and VI and the generated anti-GQ1b antibodies block impulse genera-tion at the nodes of Ranvier resulting in conduction block. Many patientswith antibodies to GQ1b also have antibodies to GD1a [1,2,8,9]. The
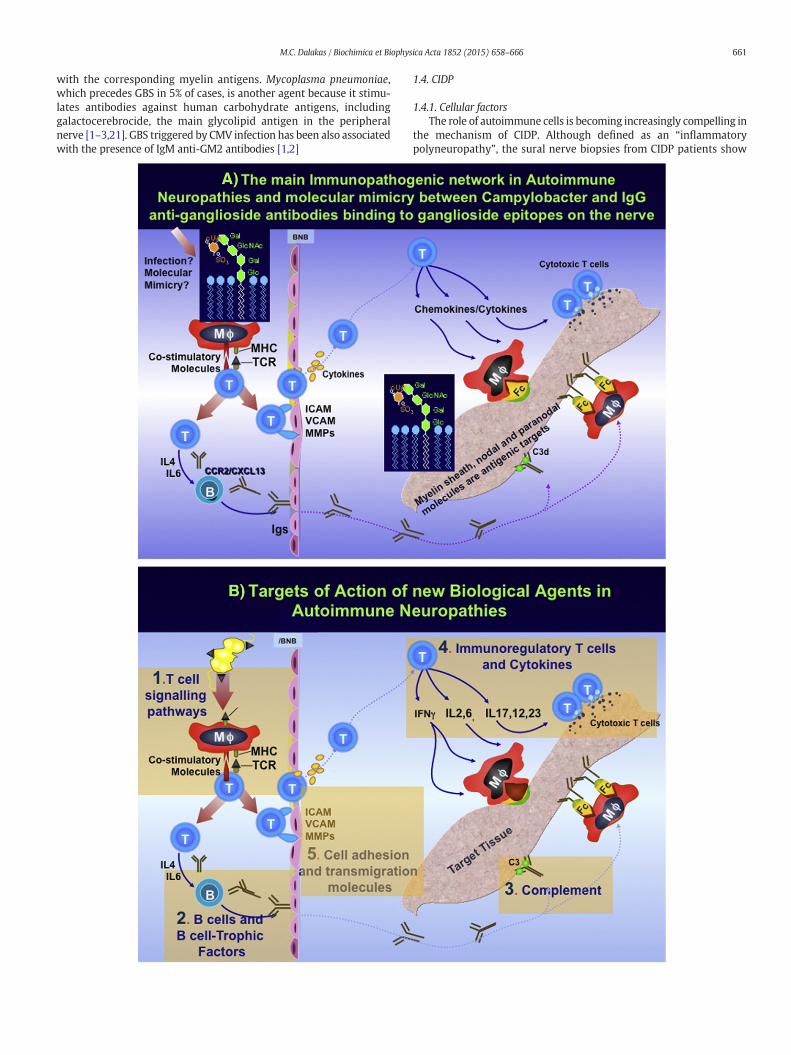
Fig. 1. A: Themain factors in the immunopathologyof autoimmuneperipheral neuropathies andinfections, such as Campylobacter jejuni, in a concept of molecular mimicry when glycoconjugazation of cross-reactive T cells and production of antibodies against myelin glycolipids. These aphages lead to demyelination and conduction block. Macrophages act as effector cells andupregulation of adhesion molecules (ICAM, VCAM, and MMP) on endothelial cells and transmibody production. B. Targets of action of new biological agents as future immunotherapies in Afollowing: 1) T cell activation and intracellular signaling pathways, such as Alemtuzumab direan oral Janus Kinase inhibitor, that inhibits interleukin-2-dependent differentiation of Th17 herituximab, ocrelizumab, ofatumumab (Arzera) and obinutuzumab (Gazyva), against CD20 moledrug Eculizumab, a monoclonal antibody against C5; 4) T cells and key Cytokines, such as Tocilizudirected against IL17; and Ustekinumab, a human monoclonal antibody against the p40 subuntransmigration and Fingolimod, that traps lymphocytes in the lymphoid organs.
reasons for different clinical syndromes in connection with specificgangliosides remain unclear, but distribution, accessibility, density andconformation of ganglioside epitopes at different sites may be criticalfactors [1,2,17]. For example, there is more GM1 in ventral than in dorsalroots, hence the predominantly motor neuropathy seen with GM1 anti-bodies, and more GQ1b in the ocular motor nerves which explains theophthalmoplegia in Miller-Fisher syndrome.
1.3.2.1. Triggering factors and molecular mimicry. Current evidence sug-gests that antecedent infectionswith certain viral or bacterial infectionscan break tolerance and trigger an autoimmune attack against myelin,in a phenomenon of molecular mimicry, when glycoconjugate epitopesare shared between bacterial and myelin proteins [1,2]. Two-thirds ofpatients with GBS give a history of a flu-like illness or acute dysentericepisodes that precede the development of GBS by 1–3 weeks [1–3]. Vi-ruses, such as Cytomegalovirus, Epstein–Barr virus (EBV), herpes, hepatitisA and E, or HIV, and bacteria, such as Hemophilus influenza,Mycoplasmapneumoniae and C. jejuni, are most commonly implicated. Among them,infectionwith C. jejuni, which is themain culprit for certain GBS subsets,has generated great interest because it provides the best example ofmolecular mimicry [1–3]. High titers of IgG or IgM C. jejuni-specific an-tibodies are seen in up to 30% of patientswith AMANand20%of patientswith Miller-Fisher syndrome and Campylobacter is isolated from thestools early in acute GBS from 44% to 88% of the patients [1–3,8,9].C. jejuni is a common cause of a diarrheal illness worldwide but it is acertain serotype, the Penner D:19 serogroup, that differs from theother enteritis-causing common serotypes because it contains thegenes for enzymes that synthesize sialic acid in the bacterial wallmimicking gangliosides GM1, GD1a or GQ1b [1–3,8,9,22]. Bacterialisolates from AMAN bear GM1-like or GD1a-like lipooligosaccharide,while those from patients with Miller Fisher syndrome havelipooligosaccharides mimicking GQ1b [2,3,8,9,22]. Further, injection oflipo-oligosaccharides extracted from C. jejuni into rabbits induces anacute neuropathy with GM1 antibodies, identical to AMAN, [1–3,22].Accordingly, a classic example of molecular mimicry takes place wheninfection by C. jejuni carrying GM1-like or GD1a-like lipooligosaccharideinduces antibodies to GM1 or anti-GD1a, which are expressed in motornerves, resulting clinically in AMAN [2,3,8,9]; in contrast, infection byC. jejuni bearing GQ1b-mimicking lipooligosaccharide, generates anti-GQ1b antibodies which, by binding to GQ1b expressed in oculomotornerves and muscle spindles, cause Miller Fisher syndrome [2,3,8,9].Cross-reactivity between epitopes in the lipo-oligosaccharide of thebacterial wall and the gangliosides on the peripheral nerve results insensitization of autoreactive T cells, production of complement-fixinganti-ganglioside antibodies and upregulation of cytokines and transmi-grationmolecules on the endothelial cell wall facilitating the entrance ofmore activated T cells to the endoneurial parenchyma [21]. The molec-ular mimicry phenomenon as a triggering factor and the main immuneplayers involved in the pathogenesis of GBS are diagrammaticallydepicted in Fig. 1A.
Molecular mimicry may be also involved with other bacteria.Hemophilus influenzae, which is a triggering factor in 5% of GBS patients,carries GM1 and GQ1b epitopes in its bacterial wall andmay cross-react
the concept ofmolecularmimicry. The immune attack beginswhen tolerance is brokenbyte epitopes are shared between Campylobacter and peripheral myelin resulting in sensiti-ntibodies by fixing complement on the nerve or by binding to the Fc receptors on macro-via co-stimulatory molecules, lead to clonal expansion of T-cells, release of cytokines,gration of T-cells to myelin sheath. Cytokines IL-4, IL-6 enhance B-cell-activation and anti-PN. Biological agents currently on the market for various autoimmune diseases target thected against the CD52 molecule; Daclizumab an IL-2 receptor antagonist; and Tofacitinib,lper T cells; 2) B cells, such as Belimumab (Benlysta) against the B cell trophic factor Blys;cules on B cells causing peripheral B cell depletion; 3) Complement, represented by themab, an IL6 receptor antagonist; Brodalumab and Ixekizumab, both monoclonal antibodiesit of IL12/1L-23; and 5) Cell adhesion and T cell migration, such as Natalizumab, that blocks
661M.C. Dalakas / Biochimica et Biophysica Acta 1852 (2015) 658–666
with the corresponding myelin antigens. Mycoplasma pneumoniae,which precedes GBS in 5% of cases, is another agent because it stimu-lates antibodies against human carbohydrate antigens, includinggalactocerebrocide, the main glycolipid antigen in the peripheralnerve [1–3,21]. GBS triggered by CMV infection has been also associatedwith the presence of IgM anti-GM2 antibodies [1,2]
1.4. CIDP
1.4.1. Cellular factorsThe role of autoimmune cells is becoming increasingly compelling in
the mechanism of CIDP. Although defined as an “inflammatorypolyneuropathy”, the sural nerve biopsies from CIDP patients show

662 M.C. Dalakas / Biochimica et Biophysica Acta 1852 (2015) 658–666
onlyminimal signs of T cell infiltrates, at least at the time nerve biopsiesare performed [1,2,10,20,22]. Thepredominant lymphoid cells consist ofmacrophages, found scattered or in clusters around endoneurial vessels[2,13–15,22,23]. Macrophages constitute the final effector cells associat-ed with demyelination because they express activationmarkers, proba-bly induced by cytokines released by autoreactive T cells in situ or in thecirculation [2,10–16,24,25], penetrate the basement membrane of theSchwann cell, displace the cytoplasm, split the myelin lamellae andresult in focal destruction of the myelin sheath (macrophage-mediateddemyelination). The macrophages, but also the Schwann cells, play arole in local antigen presentation because they express the co-stimulatory molecules B7-1, B7-2, while their counter-receptors CTLA-4 and CD28 are expressed on the rare endoneurial CD4+ T cells [26,27]. The role of B7-1/B7-2 is further supported by the development ofa spontaneous autoimmune polyneuropathy in a strain of non-obese di-abetic mice deficient in B7-2 co-stimulation that demonstrate clinical,electrophysiological and immunopathological features similar tohuman CIDP [28]. Preliminary data in a small series of patients, indicatethat the fewCD8+andCD4+T cells found in the nerve biopsies of CIDPpatients, have monoclonal or oligoclonal restrictions in their T-cell re-ceptor repertoire that overlap with those found in the same patients'peripheral blood lymphocytes, implying an antigen-driven T-cellresponse against peripheral nerve antigens [29]. After successful treat-ment with IVIg in a few of these patients, the oligoclonal expansionsof CD4+ and CD8+ populations appear reduced [30]; if these findingsare confirmed inmore prospectively studied patients and correlatewiththe clinical response, changes in Vβ elements of the T cell receptorsmight become interesting markers of CIDP responsiveness to IVIg [31].
Higher number of Th17-positive cells is also observed in the periph-eral blood and CSF of CIDP patients with increased levels of circulatinginterleukin-17 which augments the induction of co-stimulatory mole-cules and enhances further the immune process [32]. The noteddysfunction of immunoregulatory T cells may also play a role by affect-ing the local inflammatory microenvironment and sustaining the ongo-ing immune response [2,10,31–34]. Soluble adhesion molecules,chemokines, cytokines and metalloproteinases are increased in the pa-tients' sera, endothelial cells and CSF, facilitating T cell transmigrationacross the blood–nerve barrier [34–38]. Gene array studies have con-firmed the upregulation of genes for various inflammatory mediatorsnot only in the sural nerve biopsies [39] but also in the small nervefibersof the skin [40]. The skin biopsy is emerging as a powerful and accessibletool to further explore themolecular events associated with the inflam-matory process directly on the intradermal nerve fibers andmonitor thechanges over time or after therapies.
1.4.2. Humoral factorsHumoral factors play a more fundamental role in CIDP owing to the
beneficial effect of plasmapheresis that removes putative pathogenicantibodies or other inflammatory mediators [1,2,10]. It is the fast re-sponse of patients to plasmapheresis that implicates a circulating factor– probably an antibody – responsible for demyelination and conductionblock. In contrast to GBS however, where ganglioside antibodies play acausative role in the axonal and ataxic variants as discussed above, nospecific antibody has yet been identified as the causative factor inCIDP, in spite of the compelling indirect evidence. The first indicationthat antibodies are involved in CIDP was the presence of complement-fixing IgG and IgM deposits on the patient's myelin sheath [41]; thepresence of a band, probably IgG, in their CSF provided further credence[42]. Antibodies to glycolipids LM1, GM1, or GD1b were subsequentlydetected in some patients, but less frequently detected than in GBS, al-thoughmore frequently detected than in controls [43,44]. Passive trans-fer experiments have then demonstrated that serum IgG can induceconduction block in the rat nerves [45] implicating a 28 kDamyelin pro-tein Po as a putative antigen, but only in 20% of the tested patients [46].The recent observation that B cells from CIDP patients exhibit reducedexpression of FcγRIIB, an inhibitory receptor that prevents B cells
from entering the germinal centers to become IgG-positive plasmacells [47], if confirmed, may further support the role of B cells in thedisease.
In contrast to GBS where molecular mimicry with bacterial or viralantigens triggers the disease in some patient subsets, there is no con-vincing evidence that viral infections are antecedent events in CIDP. Ofinterest is the observation that the incidence of CIDP is higher in pa-tients with melanoma or after vaccination with melanoma lysates[48]. Because several carbohydrate epitopes, such as GM3, GM2, GD3,are shared between myelin and melanoma cells and the serum of onesuch patient recognized GM2 epitopes on her own melanomatoustumor, we have inferred that molecular mimicry may be a triggeringfactor in such clinical settings [49]. Overall, the immunopathogeneticscheme presented in Fig. 1A summarizing the proposed role of T cells,cytokines, B cells and autoantibodies for GBS, is also relevant in CIDP.
1.4.2.1. Emerging target antigens in the nodal regions: an explanation forconduction block and rapid recovery. In spite of the progress made inthe field, the target antigen(s) in CIDP and in the commonest form ofGBS, the acute inflammatory demyelinating polyneuropathy (AIDP), re-main still elusive. Recent studies however suggest that molecules not inthe compactmyelin aswehave been so far focused on, but ratherwithinthe nodal or paranodal regions and the Schwann cell/axonal interac-tionsmay be probable candidate targets of the immune attack [2,50] be-cause dysfunction in these regions can best explain the rapid changes inclinical symptomatology seen in CIDP patients after treatment withplasmapheresis or IVIg. Such a recovery is often noticeable withindays after treatment which cannot be explained on the basis ofremyelination but rather by the presence of a functional, “minute-to-minute”, blockade induced by humoral factors againstmolecules associ-ated with saltatory conduction at the nodes of Ranvier [2,10]. The sameis true for the worsening seen at the end of treatment effect, often pre-dictable at 3–5 weeks, which cannot be due to an another predictabledemyelinating episode but rather due to reappearance of the conduc-tion block across the nodes. Electron microscopy studies have now re-vealed multiple alterations in the nodal and paranodal regions in theSchwann cells of CIDP nerves compared to disease controls [51]. Thedistribution of KCNQ2, a potassium channel subunit present in nodal re-gions was found diminished in CIDP nerves, while paranodin, known asCASPR, an axonal membrane glycoprotein highly enriched at theparanodes, was more widespread in CIDP than in controls extendingalong the axon in the internodes [51]. Analogous alterations were ob-served in the demyelinated sciatic nerves of mice that exhibited in-creased levels of paranodin/CASPR and increased density of ankyrin Gclusters [51]. Additionally, inmicewith EAN disruption of sodium chan-nel clusters at the nodes of Ranvierwas associatedwith loss of adhesionmolecules gliomedin and neurofascin, that started before the loss ofsodiumchannels and the onset of paranodal demyelination, andwas ac-companied by antibodies to gliomedin and neurofascin [52]. According-ly, molecules in the nodal, paranodal or juxtaparanodal regions, asdepicted in Fig. 2, may be target antigens in human demyelinatingneuropathies that could best explain the conduction block. As depictedin Fig. 2, someof the putative antigenic proteinswithin these regions in-clude: at the node the neurofascin (NF186), gliomedin, sodium channels,ankyrin G or spectrin; at the paranode the neurofascin 155, contactin/Caspr 1 and connexins Cx31.3, Cx3232, Cx31.3, or 29; and at thejuxtaparanode the Transient Axonal Glycoprotein-1 (TAG-1)/CASPR 2and potassium channels [52,53]. Immune responses against such mole-culesmay change the fine structure at the nodes and induce conductionfailure that would best explain the rapid recovery seen after therapy [2,50].
A number of laboratories including ours, are actively searching forantibodies against such targets. In one study, 43% of patients with GBSand 30% of patients with CIDP showed IgG fixation at the nodes ofRanvier in an in vitro system; in eight of these patients IgG antibodiesrecognized the native extracellular domain of neurofascin NF186,

Fig. 2. Proteins in the nodal, paranodal and juxtaparanodal regions of myelinated fibers as target antigens in acquired demyelinating neuropathies with conduction block. Contactin 1, Na+ channel, Ankyrin G, and Neurofascin 186 are the most common putative antigens in the nodal region; Neurofascin 155, CASPR 1/contactin 1 in the paranodal region; and contactin 2/CASPR2 and K+ channel in the juxtaparanodal region. Additional ones (see text) include gliomedin, connexin, NCAM, cadherin and others. Part of thefigurewas adapted from Susuki [53]with permission.
663M.C. Dalakas / Biochimica et Biophysica Acta 1852 (2015) 658–666
gliomedin, or contactin [54]. In three other studies, high titer antibodiesto neurofascin have been observed in some patientswith CIDP and AIDPbut not in controls [55–57]. Although these antibodies were detected ina small number of patients, there is evidence that they may be patho-genic [57]. In another series, 4 of 46 (8.6%) CIDP patients reacted withhippocampal neurons and paranodal structures on teased nerves,while three of them (6.5%) had antibodies to contactin-1 and one tocontactin-associated protein 1 (CASPR 1) [58]. Of interest, the CASPR-positive patients had poor response to IVIg, more axonal involvementand aggressive course. The same group has also found in 4 other CIDPpatients who had predominant distal weakness, poor response to IVIgand a disabling tremor, IgG4 antibodies to neurofascin-155; these pa-tients' serum immunoreacted with paranodes in teased-nerve fibersand with the neuropil of rat cerebellum, brain, and brainstem [59]. Itis likely that different antibodies at the nodal regionmay identify differ-ent CIDP phenotypes. Polymorphisms in the TAG-1molecule have beenalso noted in CIDP patients and it was suggested that TAG-1 may be atarget antigen [60].We have tested 15 CIDP patients for TAG-1 antibod-ies using a cell-based assay in our laboratory, but we did not identifyany positive sera [61]. Further, testing for reactivity to CASPR 2, whichis localized in the juxtaparanodal region (Fig. 2), using a sensitive cell-based assay also failed to detect antibodies [62]. A more recent screenof 45 CIDP patients for contactin-2/TAG1, Connexins Cx31.3 and Cx32,as well as CASPR 2 was still disappointing except for one patient thatrecognized a heretofore-unidentified antigen at the node in a teasednerve fiber preparation [63]. In spite of the very small number of posi-tive sera, the plethora of proteins in the nodal regions, open the wayto explore other candidate target antigens, especially against proteinswith an extracellular domain, in an effort to identify distinct patientsubsets.
1.5. IgM-MGUS neuropathy with anti-mag or SGPG antibodies
Among patients with paraproteinemias, the best characterizedantibody-mediated neuropathy is the one associated with IgMmonoclonal gammopathy. The sera from approximately 50% of thesepatients react with myelin-associated glycoprotein (MAG), a 100-kDaglycoprotein of the central and peripheral nerve myelin, as well asother glycoproteins or glycolipids that share antigenic determinantswith MAG [1,2,64–68]. The antigenic determinant of MAG resides inthe carbohydrate component of the molecule because after deglycosyl-ation of MAG the IgM reactivity is lost [67]. Most importantly, the anti-MAG IgMparaproteins always reactwith an acidic glycolipid in the gan-glioside fraction of the human peripheral nerve that we had identifiedas a sulfoglucuronyl glycosphingolipid (SGPG) [68]. SGPG is more rele-vant in the pathogenesis of the neuropathy because, in contrast to MAGwhich is mostly present in the CNSmyelin, SGPG is found exclusively inthe peripheral nerve. In addition, animals we immunized with SGPGdevelop an ataxic neuropathy similar to the one seen in IgM-MGUSpatients. More than half of the IgM paraproteins in patients with neu-ropathy recognize MAG and SGPG, while 75% of the rest recognizeother ganglioside antigens, most commonly those that contain eithera disialosyl moiety, such as GD1b, GQ1b, GT1b, the GalNac-GM1b andGalNAc-GD1a, or two gangliosides that share epitopes with GM2, or acombination of GM2 and GM1, GM1 and GD1b [1,19,69,70]. Acidic gly-colipids are therefore, the most common antigenic epitopes in IgM-MGUS patients [1,19,69,70].
Because anti-MAG-reacting sera always recognize the SGPG glyco-lipid, the assay is often performed using SGPG as antigen instead of pu-rified human MAG [1]. It should be noted however that IgM binds toMAG 10–100 times more strongly than to SGPG, and checking for

664 M.C. Dalakas / Biochimica et Biophysica Acta 1852 (2015) 658–666
SGPG instead of MAGmay potentially miss some very low-affinity anti-MAG antibodies [1,2].
There is strong evidence that MAG/SGPG antibodies are causativelyrelated to the neuropathy because: a) IgM and complement are depos-ited on the myelinated fibers in the patient's sural nerve biopsies sug-gesting that activated complement is fixed in situ by the antibodies[71]; b) the patients' IgM recognizes the neural cell adhesion moleculeNCAM and co-localizes withMAG on the areas of the split myelin impli-cating a myelin disadhesion process induced by the circulating anti-MAG IgM [72]; c) in skin biopsies from the patients, there is depositionof IgM, complement and MAG on the intradermal myelinated fiberswith a concurrent loss of nerve fibers suggesting IgM-inducedfiber loss [72]; d) intraneural injections of the patients' serum supple-mented with fresh complement into peripheral nerve of cats results ina complement-dependent demyelination with conduction block, 2–9days after the injection; the injected serum also caused splitting of theouter layer of the myelin sheath resembling the human pathology[73]; d) systemic transfusion of anti-MAG IgM paraproteins causes seg-mental demyelination in chickens with deposition of IgM to the outerlayers of the myelin sheath and splitting of the myelin lamellae similarto the one seen in the patients' nerves [74]; and e) immunization ofcats with SGPG causes sensory ataxia, as seen in patients with anti-MAG neuropathy, and an inflammatory ganglionopathy with dorsalroot involvement consistent with the clinical signs of sensory ataxia[75].
1.5.1. Current therapies and emerging disease biomarkersThe autoimmune neuropathies are clinically important because they
respond to treatment with various immunomodulating or chemothera-peutic agents. Randomized clinical trials have shown that intravenousimmunoglobulin (IVIg) and plasma exchange are equally effective inGBS [76], although somepatients do not adequately respond for reasonsthat remain still unclear. In CIDP, corticosteroids, IVIg and plasma ex-change exert a short- or long-term meaningful clinical improvementin the majority of the patients, but maintenance therapy is required[2,10,77,78]. In MMN, the only effective therapy is IVIg but remainscurious why this neuropathy does not respond to any other immuno-therapy and worsens with corticosteroids. Anti-MAG neuropathy is re-fractory to available therapies [1,79], but a small number of patientsrespond verywell to Rituximab [80]. Immunosuppressants, such as aza-thioprine, cyclosporine, mycophenolate, or cyclophosphamide are vari-ably used as steroid- or IVIg-sparing agents in all APN [80], but theirefficacy is overall disappointing. Controlled trials with β-interferonand methotrexate were ineffective in CIDP [81–83]. Because someCIDP patients respond only to steroids, while others with pure motordisease worsen with steroids and still others respond only to plasma-pheresis or IVIg [10,84], biomarkers predicting response to therapiesfrom the outset are needed [84].
Pharmacokinetic studies in 174 GBS patients treated with IVIg haveshown that patients with a lower increase in serum IgG (DeltaIgG) re-covered more slowly and fewer of them reached the ability to walk un-aided at six months, concluding that a low DeltaIgG was independentlyassociatedwith poor outcome [85]. If confirmed, thiswould suggest thatpatients might benefit from a higher dosage or from a second IVIg infu-sion early in the disease course. Serum IgG levels after IVIg infusions donot seem to play a role in predicting responsiveness of CIDP to IVIg. Twopotential biomarkers, the TAG-1 (Transient Axonal Glycoprotein-1), anadhesion molecule involved in axonal maintenance, and the expressionof inhibitory FcγRIIB receptors on B cells [59,47], seem promising inconferring responsiveness to IVIg. Single nucleotide polymorphismsand haplotype studies in 100 Japanese patients, revealed an associationbetween IVIG responsiveness and TAG-1 polymorphism [59], suggest-ing that response to IVIg may be even genetically determined. The in-hibitory FcγRIIB receptors on B cells transduce inhibitory signals andprevent their transformation into IgG-producing plasma cells [47]. Pa-tients with CIDP were found to have lower FcγRIIB on naive B cells,
but after IVIg the FcγRIIB protein expression was upregulated; whethersuch an upregulatory effect of IVIg on FcγRIIB of B cells predicts respon-siveness to IVIg, remains to be proven [84].
1.5.2. Emerging biological agents as new targeted therapiesThere is a need for more effective and specific therapies for all APN
because: a) a number of patients with GBS or CIDP do not adequatelyrespond to the available therapies and left with significant disability;b) there is a need for steroid and IVIg-sparing effect in CIDP to diminishthe long-term steroid-related side effects and reduce the high cost ofmonthly IVIg maintenance; c) a number of patients with MMN do notadequately respond to IVIg or the efficacy wanes with time; and d)very few patients with anti-MAG neuropathy respond only to Rituxi-mab [80,86]. Emerging therapeutic agents in the form of monoclonalantibodies or fusion proteins offer target-specific therapy and are cur-rently used in other autoimmune disorders, as discussed [87,88].These agents may be of value as future therapies in all APN becausethey target key molecules involved in immunopathology of the diseasefrom the early T and B cell activation to the final induction of cytotoxic-ity including cytokines, complement, and adhesion or transmigrationmolecules, as depicted in Fig. 1B [2,10,87,88]. As emphasized [2], thesedrugs may be considered as future therapeutic options, provided thatthey are tested in control trials and the rare but potentially catastrophicside effects are being monitored. The new biological agents target thefollowing.
1.5.2.1. T cell intracellular signaling pathways. This group includes: a)Alemtuzumab, a monoclonal antibody against the CD52 transductionmolecule, that causes long-lasting lymphocyte depletion. Alemtuzumabhas been effective in multiple sclerosis and seems promising in CIDP [2,10,9,87,88] where a controlled trial is ready to begin; b) Daclizumab, amonoclonal antibody against CD25 (IL-2 receptor antagonist) that in-hibits T cell proliferation. The drug is well tolerated, has been approvedfor leukemia, and is very promising in patientswithMultiple Sclerosis intwo phase III clinical trials; c) Tofacitinib, an oral Janus Kinase inhibitor,that inhibits interleukin-2-dependent differentiation of type 2 and type17 helper T cells and attenuates signaling by proinflammatory cyto-kines, such as interleukin-6 and interferon-γ. Blockade of Janus kinasessuppresses both T and B cells and maintains Regulatory T-cell function.The drug has been effective in ulcerative colitis and rheumatoid arthritis[87,88].
1.5.2.2. B cells and B cell trophic factors. They include: i) Belimumab(Benlysta), a humanmonoclonal antibody against B-lymphocyte stimu-lator (BLyS) that has been approved for lupus; ii) rituximab, a chimericmonoclonal antibody directed against CD20, a membrane-associatedphosphoprotein present on B cells, causes peripheral B cell depletion[87–90]. Rituximab is given in two, 1 g each infusions, 15 days apart in-travenously or in 4 weekly infusions each at 375 mg/m2; up to 50% ofthe treated patients with CIDP seem to improve after 2–12 months[91] but controlled studies are needed. Rituximab also helps a numberof patientswith anti-MAGneuropathy, 6–8 months after one 2 gram in-fusion [80,86]; iii) ocrelizumab, the humanized version of rituximab; iv)ofatumumab (Arzera) that targets different CD20 epitopes and can begiven subcutaneously; and v) Obinutuzumab (Gazyva), another mono-clonal antibody against CD20 that causes more profound B cell deple-tion and has been approved for chronic lymphocytic leukemia.
1.5.2.3. Complement. Eculizumab, a monoclonal antibody against C5 in-tercepts the terminal formation of membranolytic attack complex(MAC) and blocks the generation of proinflammatory molecules [92,93]. The agent, approved for paroxysmal hemoglobinuria, is of interestin CIDP and GBS where complement is activated and deposited on thenerves. The drug was effective in an EANmodel, showed some promisein a small uncontrolled trial in MMN patients, and was effective in a

665M.C. Dalakas / Biochimica et Biophysica Acta 1852 (2015) 658–666
controlled study in human myasthenia gravis [92,93]. A study withEculizumab is ready to begin in GBS.
1.5.2.4. Cytokines and cytokine receptors. Themost relevant agents in thisgroup include: i) Tocilizumab, an IL6 receptor antagonist,which is prom-ising in SLE and is relevant to APN because IL6 affects the induction ofTregs to pathogenic Th1 cells; ii) Brodalumab and Ixekizumab, bothmonoclonal antibodies directed against IL17, which have been recentlyshown to be effective in psoriasis; and iii) Ustekinumab, a humanmono-clonal antibody against the p40 subunit of IL12/1L-23, which has showneffectiveness in psoriatic arthritis and has been approved for plaquepsoriasis [88].
1.5.2.5. Cell adhesion and T cell migration. The main drug in this categoryis Natalizumab, approved for multiple sclerosis and Crohn's disease,which prevents adhesion and transmigration of both T and B cells bybinding to α4β1 integrin (VLA4) on leucocytes. Although ineffective inone CIDP case [94], it is a reasonable drug to consider for experimentaltrials in APN. Fingolimod, an oral anti-T-cell-migration agent that trapslymphocytes in the lymphoid organs, has been approved for multiplesclerosis and is currently tested in an ongoing trial in CIDP.
References
[1] M.C. Dalakas, Autoimmune peripheral neuropathies, in: R.R. Rich, T.A. Fleisher, W.T.Shearer, et al., (Eds.), Clinical Immunology: Principles and Practice, edn. 3, MosbyElsevier, Philadelphia, 2011, pp. 977–994.
[2] M.C. Dalakas, Pathophysiology of Autoimmune Polyneuropathies Presse Medicale,2013.
[3] N. Yuki, H.P. Hartung, Guillain–Barré syndrome, N. Engl. J. Med. 366 (2012)2294–2304.
[4] R.A. Hughes, D.R. Cornblath, Guillain–Barré syndrome, Lancet 366 (2005)1653–1666.
[5] J.B. Winer, Guillain–Barré syndrome: clinical variants and their pathogenesis, J.Neuroimmunol. 231 (1–2) (2011) 70–72.
[6] T.E. Feasby, A.F. Hahn, W.F. Brown, C.F. Bolton, J.J. Gilbert, W.J. Koopman, Severe ax-onal degeneration in acute Guillain–Barré syndrome: evidence of two differentmechanisms? J. Neurol. Sci. 116 (1993) 185–192.
[7] N. Yuki, K. Susuki, M. Koga, Y. Nishimoto, M. Odaka, K. Hirata, et al., Carbohydratemimicry between human ganglioside GM1 and Campylobacter jejunilipooligosaccharide causes Guillain–Barré syndrome, Proc. Natl. Acad. Sci. U. S. A.101 (2004) 11404–11409.
[8] H.J. Willison, N. Yuki, Peripheral neuropathies and anti-glycolipid antibodies, Brain125 (2002) 2591–2625.
[9] S. Kusunoki, K. Kaida, Antibodies against ganglioside complexes in Guillain–Barrésyndrome and related disorders, J. Neurochem. 116 (2011) 828–832.
[10] M.C. Dalakas, Advances in the diagnosis, pathogenesis and treatment of CIDP, Nat.Rev. Neurol. 7 (2011) 507–517.
[11] M.P. Lunn, H. Manji, P.P. Choudhary, R.A. Hughes, P.K. Thomas, Chronic inflammato-ry demyelinating polyradiculoneuropathy: a prevalence study in south east En-gland, J. Neurol. Neurosurg. Psychiatry 66 (1999) 677–680.
[12] R.A.C. Hughes, D. Allen, A. Makowska, N.A. Gregson, Pathogenesis of chronic inflam-matory demyelinating polyradiculoneuropathy, J. Peripher. Nerv. Syst. 11 (2006)30–46.
[13] J.M. Vallat, C. Sommer, L. Magy, Chronic inflammatory demyelinatingpolyradiculoneuropathy: diagnostic and therapeutic challenges for a treatablecondition, Lancet Neurol. 9 (2010) 402–412.
[14] C. Sommer, S. Koch, M. Lammens, A. Gabreels-Festen, G. Stoll, K.V. Toyka, Macro-phage clustering as a diagnostic marker in sural nerve biopsies of patients withCIDP, Neurology 65 (2005) 1924–1929.
[15] C. Vital, A. Vital, A. Lagueny, X. Ferrer, D. Fontan, M. Barat, et al., Chronic inflamma-tory demyelinating polyneuropathy: immunopathological and ultrastructural studyof peripheral nerve biopsy in 42 cases, Ultrastruct. Pathol. 24 (2000) 363–369.
[16] H. Koller, B.C. Kieseier, S. Jander, H.P. Hartung, Chronic inflammatory demyelinatingpolyneuropathy, N. Engl. J. Med. 352 (2005) 1343–1356.
[17] J.T. Van Asseldonk, H. Franssen, R.M. Van den Berg-Vos, J.H. Wokke, L.H. Van denBerg, Multifocal motor neuropathy, Lancet Neurol. 4 (2005) 309–319.
[18] N. Yuki, H. Watanabe, T. Nakajima, P.J. Späth, IVIG blocks complement depositionmediated by anti-GM1 antibodies in multifocal motor neuropathy, J. Neurol.Neurosurg. Psychiatry 82 (1) (Jan 2011) 87–91.
[19] M.C. Dalakas, R.H. Quarles, Autoimmune ataxic neuropathies (sensoryganglionopathies): are glycolipids the responsible autoantigens?, Editorial Ann. Neurol.39 (1996) 419–422.
[20] I. Illa, N. Ortiz, E. Gallard, C. Juarez, J.M. Grau, M.C. Dalakas, Acute axonal Guillain–Barré syndrome with IgG antibodies against motor axons following parenteral gan-gliosides, Ann. Neurol. 38 (1995) 218–224.
[21] C.W. Ang, B.C. Jacobs, J.D. Laman, The Guillain–Barré syndrome: a true case ofmolec-ular mimicry, Trends Immunol. 25 (2004) 61–66.
[22] J.W. Prineas, J.G. McLeod, Chronic relapsing polyneuritis, J. Neurol. Sci. 27 (1976)427–458.
[23] M.C. Dalakas, Advances in chronic inflammatory demyelinating polyneuropathy:disease variants and inflammatory response mediators and modifiers, Curr. Opin.Neurol. 12 (1999) 403–409.
[24] K. Murata, M.C. Dalakas, Expression of the co-stimulatory molecule BB-1, the ligandsCTLA-4 and CD28 and their mRNAs in chronic inflammatory demyelinatingpolyneuropathy, Brain 123 (2000) 1660–1666.
[25] R. Kiefer, F. Dangond, M. Mueller, K.V. Toyka, D.A. Hafler, H.P. Hartung, Enhanced B7costimulatory molecule expression in infl ammatory human sural nerve biopsies, J.Neurol. Neurosurg. Psychiatry 69 (2000) 362–368.
[26] B.C. Kieseier, M. Tani, D. Mahad, N. Oka, T. Ho, N. Woodroofe, et al., Chemokines andchemokine receptors in inflammatory demyelinating neuropathies: a central rolefor IP-10, Brain 125 (2002) 823–834.
[27] W. Hu, A. Janke, S. Ortler, H.P. Hartung, C. Leder, B.C. Kieseier, et al., Expression ofCD28-related costimulatory molecule and its ligand in inflammatory neuropathies,Neurology 68 (2007) 277–282.
[28] B. Salomon, L. Rhee, H. Bour-Jordan, H. Hsin, A. Montag, B. Soliven, B. Soliven, et al.,Development of spontaneous autoimmune peripheral polyneuropathy in B7-2-deficient NOD mice, J. Exp. Med. 194 (2001) 677–684.
[29] T. Schneider-Hohendorf, N. Schwab, N. Uçeyler, K. Göbel, C. Sommer, H.Wiendl, CD8+T-cell immunity in chronic inflammatorydemyelinatingpolyradiculoneuropathy,Neu-rology 78 (2012) 402–408.
[30] A.K. Mausberg, M. Dorok, M. Stettner, M. Müller, H.P. Hartung, T. Dehmel, et al., Re-covery of the T-cell repertoire in CIDP by IV immunoglobulins, Neurology 80 (3)(2013) 296–303.
[31] M.C. Dalakas, Mechanistic effects of IVIg in neuroinflammatory diseases: conclusionsbased on clinicopathologic correlations, J. Clin. Immunol. 34 (Suppl 1) (jul 2014)120–126.
[32] L.J. Chi, W.H. Xu, Z.W. Zhang, H.T. Huang, L.M. Zhang, J. Zhou, Distribution of Th17cells and Th1 cells in peripheral blood and cerebrospinal fluid in chronic inflamma-tory demyelinating polyradiculoneuropathy, J. Peripher. Nerv. Syst. 15 (2010)345–356.
[33] L.J. Chi, H.B.Wang,W.Z.Wang, Impairment of circulating CD4+CD25+ regulatory Tcells in patients with chronic inflammatory demyelinating polyradiculoneuropathy,J. Peripher. Nerv. Syst. 13 (2008) 54–63.
[34] L. Sanvito, A. Makowska, N. Gregson, R. Nemni, R.A. Hughes, Circulating subsets andCD4(+)CD25(+) regulatory T cell function in chronic inflammatory demyelinatingpolyradiculoneuropathy, Autoimmunity 42 (2009) 667–677.
[35] H.P. Hartung, K. Reiners, B. Schmidt, G. Stoll, K.V. Toyka, Serum interleukin-2 con-centrations in Guillain–Barre syndrome and chronic idiopathic demyelinatingpolyradiculoneuropathy: comparison with other neurological diseases of presumedimmunopathogenesis, Ann. Neurol. 30 (1991) 48–53.
[36] D.J. Mahad, S.J. Howell, M.N. Woodroofe, Expression of chemokines in cerebrospinalfluid and serumof patientswith chronic inflammatory demyelinating polyneuropathy,J. Neurol. Neurosurg. Psychiatry 73 (2002) 320–323.
[37] D. Maimone, P. Annunziata, I.L. Simone, P. Livrea, G.C. Guazzi, Interleukin-6 levels inthe cerebrospinal fluid and serum of patients with Guillain–Barre syndrome andchronic inflammatory demyelinating polyradiculoneuropathy, J. Neuroimmunol.47 (1993) 55–61.
[38] N. Oka, I. Akiguchi, M. Nagao, T. Nishio, T. Kawasaki, J. Kimura, Expression of endo-thelial leukocyte adhesion molecule-1 (ELAM-1) in chronic inflammatory demye-linating polyneuropathy, Neurology 44 (1994) 946–950.
[39] S. Renaud, A.P. Hays, T.H. Brannagan III, H.W. Sander, M. Edgar, L.H. Weimer, et al.,Gene expression profiling in chronic inflammatory demyelinating polyneuropathy,J. Neuroimmunol. 159 (1–2) (2005) 203–214.
[40] G. Lee, Z. Xiang, T.H. Brannagan III, R.L. Chin, N. Latov, Differential gene expression inchronic inflammatory demyelinating polyneuropathy (CIDP) skin biopsies, J. Neurol.Sci. 290 (1–2) (2010) 115–122.
[41] M.C. Dalakas, W.K. Engel, Immunoglobulin and complement deposits in nerves ofpatients with chronic relapsing polyneuropathy, Arch. Neurol. 37 (1980) 637–640.
[42] M. Dalakas, S.A. Houff, W.K. Engel, D.L. Madden, J.L. Sever, CSF monoclonal bands inchronic relapsing polyneuropathy, Neurology 30 (1980) 864–867.
[43] A.A. Ilyas, F.A. Mitchen, M.C. Dalakas, Z.W. Chen, S.D. Cook, Antibodies to acidic gly-colipids in Guillain Barre' syndrome and chronic inflammatory demyelinatingpolyneuropathy, J. Neurol. Sci. 107 (1992) 1111–1211.
[44] Y. Tagawa, N. Yuki, K. Hirata, Anti-SGPG antibody in CIDP: a nosological position ofIgM anti-MAG/SGPG antibody-associated neuropathy, Muscle Nerve 23 (2000)895–899.
[45] W.X. Yan, J. Taylor, S. Andrias-Kauba, J.D. Pollard, Passive transfer of demyelinationby serum or IgG from chronic inflammatory demyelinating polyneuropathy pa-tients, Ann. Neurol. 47 (2000) 765–775.
[46] W.X. Yan, J.J. Archelos, H.P. Hartung, J.D. Pollard, P0 protein is a target antigen inchronic infl ammatory demyelinating polyradiculoneuropathy, Ann. Neurol. 50(2001) 286–292.
[47] B. Tackenberg, I. Jelcic, A. Baerenwaldt, W.H. Oertel, N. Sommer, F. Nimmerjahn,et al., Impaired inhibitory Fcgamma receptor IIB expression on B cells in chronicinfl ammatory demyelinating polyneuropathy, Proc. Natl. Acad. Sci. U. S. A. 106(2009) 4788–4792.
[48] S.J. Bird, M.J. Brown, M.E. Shy, S.S. Scherer, Chronic inflammatory demyelinatingpolyneuropathy associatedwithmalignantmelanoma, Neurology 46 (1996) 822–824.
[49] M.D. Weiss, C.A. Luciano, C. Semino-Mora, M.C. Dalakas, R.H. Quarles, Molecularmimicry in chronic inflammatory demyelinating polyneuropathy and melanoma,Neurology 51 (1998) 1738–1741.
[50] J.D. Pollard, P.J. Armati, CIDP-the relevance of recent advances in Schwanncell/axo-nal neurobiology, J. Peripher. Nerv. Syst. 16 (2011) 15–23.

666 M.C. Dalakas / Biochimica et Biophysica Acta 1852 (2015) 658–666
[51] Carmen Cifuentes-Diaz, O. Dubourg, T. Irinopoulou, M. Vigny, S. Lachkar, L. Decker,et al., Nodes of Ranvier and paranodes in chronic acquired neuropathies, PLoSONE 6 (2011) e14533.
[52] A. Lonigro, J.J. Devaux, Disruption of neurofascin and gliomedin at nodes of Ranvierprecedes demyelination in experimental allergic neuritis, Brain 132 (2009)260–273.
[53] K. Susuki, Node of Ranvier disruption as a cause of neurological diseases, ASN Neuro5 (3) (Aug. 7 2013) 209–219.
[54] J.J. Devaux, M. Odaka, N. Yuki, Nodal proteins are target antigens in Guillain–Barrésyndrome, J. Peripher. Nerv. Syst. 17 (2012) 62–71.
[55] W.X. Yan, E. Mathey, C. Yiannikas, J. Pollard, Antineurofascin antibodies are presentin patients with peripheral demyelinating neuropathies and mediate changes innerve conduction in animals, J. Peripher. Nerv. Syst. 15 (2010) 288–289.
[56] N. Kawamura, R. Yamasaki, T. Yonekawa, T. Matsushita, S. Kusunoki, S. Nagayama, Y.Fukuda, H. Ogata, D. Matsuse, H. Murai, J. Kira, Anti-neurofascin antibody in patientswith combined central and peripheral demyelination, Neurology 81 (2013)714–722.
[57] J.K. Ng, J. Malotka, N. Kawakami, T. Derfuss, M. Khademi, T. Olsson, et al., Neurofascinas a target for autoantibodies in peripheral neuropathies, Neurology 79 (2012)2241–2248.
[58] L. Querol, G. Nogales-Gadea, R. Rojas-Garcia, E. Martinez-Hernandez, J. Diaz-Manera,X. Suarez-Calvet, M. Navas, J. Araque, E. Gallardo, I. Illa, Antibody to contactin-1 inchronic inflammatory demyelinating polyneuropathy, Ann. Neurol. 73 (2013)370–380.
[59] L. Querol, R. Rojas-Garcia, J. Diaz-Manera, J. Pardo, M.J. Sedano, E. Gallardo, J.Berciano, R. Blesa, J. Dalmau, I. Illa, Neurofascin IgG4 antibodies in CIDP asso-ciate with disabling tremor and poor response to IVIg, Neurology 2014(2013).
[60] M. Iijima, M. Tomita, S. Morozumi, Y. Kawagashira, T. Nakamura, H. Koike, et al., Sin-gle nucleotide polymorphism of TAG-1 influences IVIg responsiveness of Japanesepatients with CIDP, Neurology 73 (2009) 1348–1352.
[61] H. Alexopoulos, P.P. Pavlakis, D. Karagogeos, C.E. Karageorgiou, M.C. Dalakas, Areanti-TAG-1 autoantibodies markers in autoimmune demyelinating disorders ofthe PNS and CNS? Ann. Neurol. 70 (Suppl. 15) (2011) S73.
[62] P.P. Pavlakis, H. Alexopoulos, D. Karagogeos, M.C. Dalakas, Search for nodaland paranodal autoantibodies in Chronic Inflammatory DemyelinatingPolyradiculoneuropathy (CIDP), Neurology 78 (2012) P05.159.
[63] P. Stathopoulos, H. Alexopoulos, A. Biba, D. Karagogeos, M.C. Dalakas, Search for au-toantibodies targeting the nodes of Ranvier in chronic inflammatory demyelinatingpolyradiculoneuropathy, Neurology 82 (10) (2014) (Suppl p.1.028).
[64] M.C. Dalakas, W.K. Engel, Polyneuropathy and monoclonal gammopathy: studies of11 patients, Ann. Neurol. 10 (1981) 45–52.
[65] N. Latov, P.E. Braun, R.B. Gross, W.H. Sherman, A.S. Penn, L. Chess, Plasma cell dys-crasia and peripheral neuropathy: identification of the myelin antigens that reactwith human paraproteins, Proc. Natl. Acad. Sci. U. S. A. 78 (1981) 7139.
[66] N. Latov, A. Hays, W.H. Sherman, Peripheral neuropathy and anti-MAG antibodies,Crit. Rev. Neurobiol. 3 (1988) 301–332.
[67] A.A. Ilyas, R.H. Quarles, T.D. McIntosh, M.J. Dobersen, B.D. Trapp, M.C. Dalakas, et al.,IgM in a human neuropathy related to paraproteinemia binds to a carbohydrate de-terminant in the myelin-associated glycoprotein and to a ganglioside, Proc. Natl.Acad. Sci. U. S. A. 81 (1984) 1225–1229.
[68] A.A. Ilyas, R.H. Quarles, M.C. Dalakas, R.O. Brady, Polyneuropathy with monoclonalgammopathy: glycolipids are frequently antigens for IgM paraproteins, Proc. Natl.Acad. Sci. U. S. A. 82 (1985) 6697–6700.
[69] G.C. Duane, R.G. Farrer, M.C. Dalakas, R.H. Quarles, Sensory neuropathy associatedwith monoclonal IgM to GD1b ganglioside, Ann. Neurol. 31 (1992) 683–685.
[70] R.H. Quarles, M.C. Dalakas, Do antiganglioside antibodies cause human peripheralneuropathies? J. Clin. Invest. 97 (1996) 1136–1137.
[71] S. Monaco, B. Bonetti, S. Ferrari, G. Moretto, E. Nardelli, F. Tedesco, et al., Comple-ment dependent demyelination in patients with IgM monoclonal gammopathyand polyneuropathy, N. Engl. J. Med. 322 (1990) 844–852.
[72] R. Lombardi, B. Erne, G. Lauria, G. Lauria, D. Pareyson, M. Borgna, M. Morbin, et al.,IgM deposits on skin nerves in anti-myelin-associated glycoprotein neuropathy,Ann. Neurol. 57 (2005) 180–187.
[73] H.J. Willison, B.D. Trapp, J.D. Bacher, M.C. Dalakas, J.W. Griffin, R.H. Quarles, Demye-lination induced by intraneural injection of human antimyelin associated glycopro-tein antibodies, Muscle Nerve 11 (1988) 1169–1176.
[74] A.H. Tatum, Experimental paraprotein neuropathy; demyelination by passivetransfer of human IgM anti-MAG, Ann. Neurol. 33 (1993) 502–506.
[75] A.A. Ilyas, Y. Gu, M.C. Dalakas, R.H. Quarles, S. Bhatt, Induction of experimental ataxicsensory neuronopathy in cats by immunization with purified SGPG, J.Neuroimmunol. 193 (2008) 87–93.
[76] R.A. Hughes, E.F. Wijdicks, R. Barohn, E. Benson, D.R. Cornblath, A.F. Hahn, et al.,Quality Standards Subcommittee of the American Academy of Neurology. Practiceparameter: immunotherapy for Guillain–Barré syndrome: report of the QualityStandards Subcommittee of the American Academy of Neurology, Neurology 61(2003) 736–740.
[77] R.A.C. Hughes, P. Donofrio, V. Bril, M.C. Dalakas, C. Deng, K. Hanna, et al., Intravenousimmune globulin (10% caprylate–chromatography purified) for the treatment ofchronic inflammatory demyelinating polyradiculoneuropathy (ICE study): arandomised placebo-controlled trial, Lancet Neurol. 7 (2008) 136–144.
[78] P.J. Dyck, W.J. Litchy, K.M. Kratz, G.A. Suarez, P.A. Low, A.A. Pineda, et al., A plasmaexchange versus immune globulin infusion trial in chronic infl amatory demyelinat-ing polyradiculoneuropathy, Ann. Neurol. 36 (1994) 838–845.
[79] M.C. Dalakas, R.H. Quarles, R.G. Farrer, J. Dambrosia, S. Soueidan, D.P. Stein, et al., Acontrolled study of intravenous immunoglobulin in demyelinating neuropathy withIgM gammopathy, Ann. Neurol. 40 (1996) 792–795.
[80] M.C. Dalakas, G. Rakocevic, M. Salajegheh, J.M. Dambrosia, A.F. Hahn, R. Raju, et al.,Placebo-controlled trial of rituximab in IgM anti-myelin associated glycoprotein an-tibody demyelinating neuropathy, Ann. Neurol. 65 (2009) 286–293.
[81] M. Mahdi-Rodgers, A.V. Swan, P.A. van Doorn, R.A.C. Hughes, Immunomodulatorytreatment other than corticosteroids, immunoglobulin and plasma exchange forCIDP, Cochrane Database Syst. Rev. (2010) (Issue 11).
[82] R.A. Hughes, K.C. Gorson, D. Cros, J. Griffin, J. Pollard, J.M. Vallat, et al., Intramuscularinterferon beta-1a in chronic inflammatory demyelinating polyradiculoneuropathy,Neurology 74 (2010) 651–657.
[83] RMC Trial Group, Randomised controlled trial of methotrexate for chronic inflammatory demyelinating polyradiculoneuropathy (RMC trial): a pilot, multicentrestudy, Lancet Neurol. 8 (2009) 158–164.
[84] M.C. Dalakas, Potential biomarkers for monitoring therapeutic responses to IVIg andother therapies in CIDP, J. Peripher. Nerv. Syst. (Suppl. 1) (2011) 63–67.
[85] K. Kuitwaard, J. de Gelder, A.P. Tio-Gillen, W.C. Hop, T. van Gelder, A.W. vanToorenenbergen, et al., Pharmacokinetics of intravenous immunoglobulin and out-come in Guillain–Barré syndrome, Ann. Neurol. 66 (2009) 597–603.
[86] J.M. Léger, K. Viala, G. Nicolas, A. Créange, J.M. Vallat, J. Pouget, P. Clavelou, C. Vial, A.Steck, L. Musset, B. Marin, RIMAG Study Group (France and Switzerland), Placebo-controlled trial of rituximab in IgM anti-myelin-associated glycoprotein neuropathy,Neurology 80 (24) (Jun 11 2013) 2217–2225, http://dx.doi.org/10.1212/WNL.0b013e318296e92b (Epub 2013).
[87] M.C. Dalakas, Biologics and other novel approaches as new therapeutic options inmyasthenia gravis: a view to the future, Ann. N. Y. Acad. Sci. 1274 (2012) 1–8.
[88] M.C. Dalakas, Novel future therapeutic options in myasthenia gravis, Autoimmun.Rev. 12 (9) (2013) 936–941.
[89] M.C. Dalakas, B cells as therapeutic targets in autoimmune neurological disorders,Nat. Clin. Pract. Neurol. 4 (2008) 557–567.
[90] L. Benedetti, C. Briani, D. Franciotta, R. Fazio, I. Paolasso, C. Comi, et al., Rituximab inpatients with chronic inflammatory demyelinating polyradiculoneuropathy: a re-port of 13 cases and review of the literature, J. Neurol. Neurosurg. Psychiatry 82(2011) 306–308.
[91] M. Kosmidis, M.C. Dalakas, Practical considerations on the use of rituximab in auto-immune neurological disorders, Ther. Adv. Neurol. Disord. 3 (2010) 93–105.
[92] A.M. Fitzpatrick, C.A. Mann, S. Barry, K. Brennan, J.R. Overell, H.J. Willison, An openlabel clinical trial of complement inhibition in multifocal motor neuropathy, J.Peripher. Nerv. Syst. 16 (2) (2011 Jun) 84–91.
[93] J.F. Howard Jr., R.J. Barohn, G.R. Cutter, M. Freimer, V.C. Juel, T. Mozaffar, M.L.Mellion,M.G. Benatar, M.E. Farrugia, J.J. Wang, S.S. Malhotra, J.T. Kissel, MG Study Group, Arandomized, double-blind, placebo-controlled phase II study of eculizumab in pa-tients with refractory generalized myasthenia gravis, Muscle Nerve 48 (1) (2013Jul) 76–84, http://dx.doi.org/10.1002/mus.23839. Epub 2013 Apr 30.
[94] C. Wolf, T. Menge, M.P. Stenner, G. Meyer zu Hörste, A. Saleh, H.P. Hartung, et al.,Natalizumab treatment in a patient with chronic inflammatory demyelinatingpolyneuropathy, Arch. Neurol. 67 (2010) 881–883.





























