Embed Size (px)
Citation preview

Biochemical analysis of a multifunctional cytochromeP450 (CYP51) enzyme required for synthesis ofantimicrobial triterpenes in plantsKatrin Geislera,b, Richard K. Hughesc, Frank Sainsburyc,1, George P. Lomonossoff c, Martin Rejzekc, Shirley Fairhurstc,Carl-Erik Olsenb, Mohammed Saddik Motawiab, Rachel E. Meltona, Andrew M. Hemmingsd,e, Søren Bakb,and Anne Osbourna,2
Departments of aMetabolic Biology and cBiological Chemistry, John Innes Centre, Norwich NR4 7UH, United Kingdom; bDepartment of Plant andEnvironmental Sciences, VKR Research Centre Pro-Active Plants, Faculty of Sciences, University of Copenhagen, Thorvaldsensvej 40, DK-1871 Frederiksberg C,Copenhagen, Denmark; and Schools of dChemistry and eBiological Sciences, University of East Anglia, Norwich NR4 7TJ, United Kingdom
Edited by Joseph R. Ecker, The Salk Institute, La Jolla, CA, and approved July 18, 2013 (received for review May 15, 2013)
Members of the cytochromes P450 superfamily (P450s) catalyzea huge variety of oxidation reactions in microbes and higherorganisms. Most P450 families are highly divergent, but in contrastthe cytochrome P450 14α-sterol demethylase (CYP51) family is oneof the most ancient and conserved, catalyzing sterol 14α-demethy-lase reactions required for essential sterol synthesis across thefungal, animal, and plant kingdoms. Oats (Avena spp.) produceantimicrobial compounds, avenacins, that provide protectionagainst disease. Avenacins are synthesized from the simple triter-pene, β-amyrin. Previously we identified a gene encoding a mem-ber of the CYP51 family of cytochromes P450, AsCyp51H10 (alsoknown as Saponin-deficient 2, Sad2), that is required for avenacinsynthesis in a forward screen for avenacin-deficient oat mutants.sad2 mutants accumulate β-amyrin, suggesting that they areblocked early in the pathway. Here, using a transient plant expres-sion system, we show that AsCYP51H10 is a multifunctional P450capable of modifying both the C and D rings of the pentacyclictriterpene scaffold to give 12,13β-epoxy-3β,16β-dihydroxy-olea-nane (12,13β-epoxy-16β-hydroxy-β-amyrin). Molecular modelingand docking experiments indicate that C16 hydroxylation is likelyto precede C12,13 epoxidation. Our computational modeling, incombination with analysis of a suite of sad2 mutants, providesinsights into the unusual catalytic behavior of AsCYP51H10 andits active site mutants. Fungal bioassays show that the C12,13epoxy group is an important determinant of antifungal activity.Accordingly, the oat AsCYP51H10 enzyme has been recruited fromprimary metabolism and has acquired a different function comparedto other characterized members of the plant CYP51 family—asa multifunctional stereo- and regio-specific hydroxylase in plantspecialized metabolism.
CPMV-HT transient expression | cytochrome P450 monooxygenase CYP51family | disease resistance | neofunctionalization | terpenes
Higher plants produce a huge array of low molecular weightspecialized compounds (natural products) that have im-
portant functions in biotic and abiotic stress tolerance (1, 2) andthat also provide a matchless starting point for drug and agro-chemical discovery (3). The cytochrome P450 (P450) superfamilyis the largest family of plant metabolic enzymes. The majority ofplant P450 families are highly divergent, reflecting diversificationand neofunctionalization as new metabolic pathways evolve (4–6). In contrast, the cytochrome P450 14α-sterol demethylase(CYP51) family is one of the most ancient of the P450 families,and the function of CYP51 enzymes is highly conserved acrossfungi, plants, and animals (7, 8). These enzymes are steroldemethylases required for the synthesis of essential sterols (9–14). Although different sterol substrates are used (e.g., lanosterolin mammals and yeast and obtusifoliol in plants), the reactionmechanism—14α-demethylation and subsequent formation ofa Δ14–15 double bond—is preserved. In plants, CYP51 enzymes
with sterol 14α-demethylase activity are known as obtusifoliol14α-demethylases and constitute the CYP51G subfamily (7).The triterpenes are one of the largest classes of plant-derived
natural products. Previously we reported the discovery of a geneencoding a divergent plant CYP51 (AsCyp51H10) that is dis-pensable for the synthesis of essential sterols but is required forproduction of specialized antimicrobial triterpene glycosidesknown as avenacins that confer disease resistance in oats (15).AsCyp51H10 (also known as Saponin-deficient 2 or Sad2) wasfirst identified in a screen for mutants of diploid oat that wereunable to make avenacins (16). Subsequent analysis revealedthat this gene forms part of a metabolic gene cluster for avenacinsynthesis (15). AsCYP51H10 (SAD2) belongs to a newly definedand as yet functionally uncharacterized subfamily of CYP51enzymes, the CYP51H subfamily, which also includes ninemembers of unknown function from rice (7, 15, 17). The firstcommitted step in the synthesis of avenacins is the cyclization of2,3-oxidosqualene to β-amyrin, catalyzed by the oat β-amyrinsynthase AsbAS1 (also known as SAD1) (18, 19). Biochemicalanalysis has shown that sad2 mutants accumulate β-amyrin,suggesting that β-amyrin may be the substrate for AsCYP51H10(15, 20). Partial characterization of AsCYP51H10 in yeast isconsistent with this observation (21). However, full character-
Significance
We carried out functional analysis of the oat enzyme AsCYP51H10,which is a divergent member of the CYP51 cytochrome P450family and showed that this enzyme is able to catalyze bothhydroxylation and epoxidation of the simple triterpene β-amyrin to give 12,13β-epoxy-3β,16β-dihydroxy-oleanane (12,13β-epoxy-16β-hydroxy-β-amyrin). In contrast, the canonical CYP51enzymes are highly conserved and catalyze only sterol deme-thylation. We further show that the C12,13 epoxy group iscritical for antifungal activity, a discovery that has importantimplications for triterpene metabolic engineering for food,health, and industrial biotechnology applications.
Author contributions: K.G., A.M.H., S.B., and A.E.O. designed research; K.G., R.K.H., S.F.,C.-E.O., M.S.M., R.E.M., and A.M.H. performed research; K.G., F.S., G.P.L., and R.E.M.contributed new reagents/analytic tools; K.G., M.R., S.F., C.-E.O., M.S.M., R.E.M., A.M.H.,S.B., and A.E.O. analyzed data; and K.G., G.P.L., M.R., A.M.H., S.B., and A.E.O. wrotethe paper.
Conflict of interest statement: A.E.O. is a co-inventor on a patent filing on AsCYP51H10.
This article is a PNAS Direct Submission.
Freely available online through the PNAS open access option.1Present address: Australian Institute for Bioengineering and Nanotechnology, Universityof Queensland, St Lucia QLD 4072, Australia.
2To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1309157110/-/DCSupplemental.
E3360–E3367 | PNAS | Published online August 12, 2013 www.pnas.org/cgi/doi/10.1073/pnas.1309157110

ization of the biochemical function of AsCYP51H10 has not yetbeen carried out.Triterpenes have a wide range of commercial applications as
agrochemicals, food additives, and pharmaceuticals and asfoaming agents in the beverage, food, and cosmetics industries(22). Commercial exploitation of triterpenes has been limitedthus far by their recalcitrance to synthetic chemistry and theiroccurrence in low abundance in complex mixtures in plants (23).The availability of enzymes that can stereo- and regio-specificallyfunctionalize triterpene scaffolds will open opportunities for theproduction of novel triterpenes using synthetic biology ap-proaches. Several triterpene-modifying P450s from eudicots havebeen characterized recently by heterologous expression in yeast.These include CYP93E1 from soybean, which hydroxylatesβ-amyrin and sophoradiol at position C24 (24); two P450s fromliquorice, one (CYP88D6) that converts β-amyrin to 11-oxo-β-amyrin and a second (CYP72A154) that converts 11-oxo-β-amyrin to glycyrrhizin acid (25, 26); and CYP716A12 fromMedicago truncatula, a β-amyrin 28-oxidase that converts β-amyrinto oleanolic acid (27, 28). These enzymes belong to differentP450 families, indicating that the ability to oxygenate β-amyrinhas arisen multiple times during evolution.Here we show that AsCYP51H10 is a multifunctional CYP51
that is able to convert β-amyrin to a product that we determinedby NMR spectrometry (NMR) to be 12,13β-epoxy-3β,16β-dihy-droxy-oleanane (12,13β-epoxy-16β-hydroxy-β-amyrin). Molecularmodeling and docking experiments indicate that C16 hydroxyl-ation is likely to occur first, followed by C12,13 epoxidation. Ourcomputational modeling in combination with mutant analysis hasyielded insights into the structural features that are important forAsCYP51H10 function. We further show that the C12,13 epoxygroup is critical for antifungal activity, a discovery that has im-portant implications for triterpene metabolic engineering forfood, health, and industrial biotechnology applications.
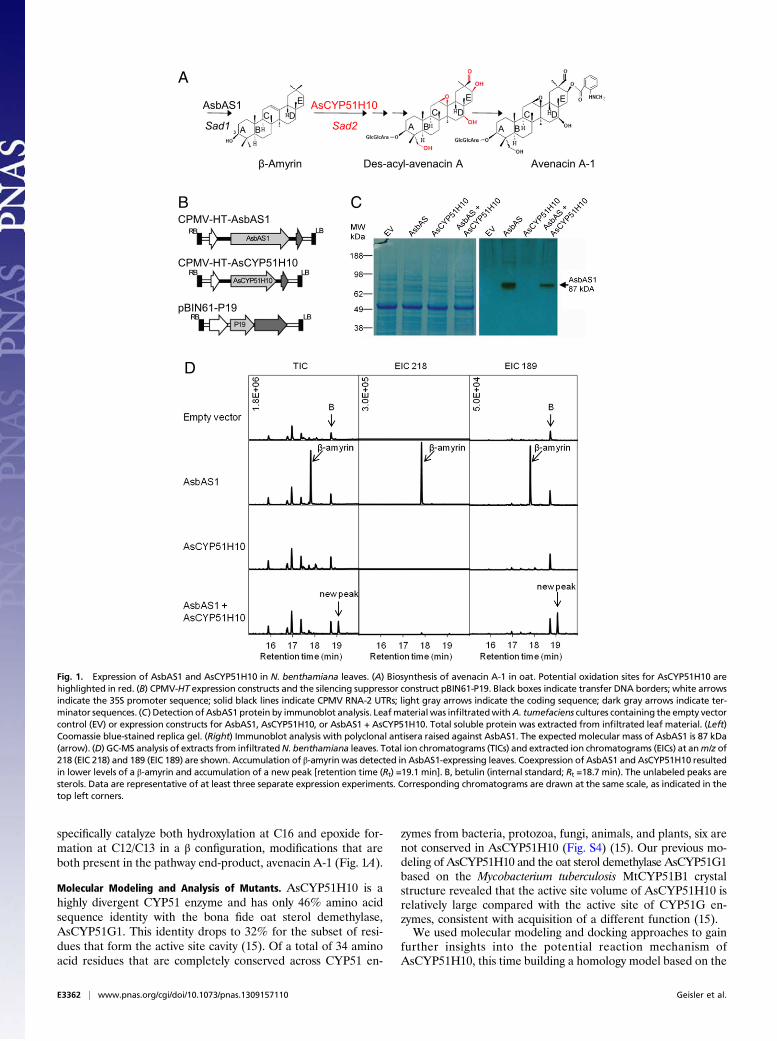
ResultsCoexpression of β-Amyrin Synthase and AsCYP51H10 in Nicotianabenthamiana Leaves. We investigated the biochemical functionof AsCYP51H10 using the cowpea mosaic virus hyper-transcassette (CPMV-HT), a plant expression system based on cow-pea mosaic virus. For CPMV-HT, constructs are generated sothat the sequence to be expressed is flanked by modified 5′ and 3′UTR sequences from CPMV RNA-2 (the CPMV-HT cassette)under the control of the 35S promoter. The presence of theCPMV-derived UTR sequences enhances mRNA translatability,thereby substantially increasing the amount of protein expressed.Constructs then are transformed into Agrobacterium tumefaciensand infiltrated into leaves of N. benthamiana for transient ex-pression in the presence of the gene-silencing suppressor, P19.CPMV-based systems previously have been used very success-fully for production of structural proteins such as vaccines andantibodies (29, 30). We also have shown this approach to be ef-fective for expression of three triterpene biosynthetic enzymes(an acyltransferase, a methyl transferase, and a sugar transferase)required for avenacin acylation (31, 32).Our previous analysis of oat mutants defective in avenacin
synthesis indicated that mutations in the AsCyp51H10 (Sad2)gene lead to accumulation of the first committed intermediate inthe avenacin pathway, β-amyrin, suggesting that β-amyrin may bethe substrate for AsCYP51H10 (Fig. 1A) (15, 20). We thereforecoexpressed AsCYP51H10 with AsbAS1, the oat β-amyrin syn-thase that catalyses the first step in avenacin synthesis (18), to in-vestigate the biochemical function of AsCYP51H10. Constructsfor transient expression were made containing the AsbAS1 andAsCyp51H10 coding sequences, each under the control of the35S promoter (Fig. 1B). N. benthamiana leaves were infiltratedwith A. tumefaciens containing the empty vector control, AsbAS1,and AsCYP51H10 constructs separately or were coinfiltrated with
the AsbAS1 and AsCYP51H10 constructs. Leaf tissue was har-vested after 6 d, and protein extracts were analyzed by immu-noblot analysis using polyclonal antisera raised against AsbAS1(attempts to raise antisera specific for AsCYP51H10 were un-successful). AsbAS1 protein was readily detectable in proteinextracts from leaves infiltrated with the AsbAS1 construct aloneor the AsbAS1 and AsCYP51H10 constructs together, as expec-ted (Fig. 1C). The triterpene content of infiltrated leaf materialthen was analyzed using GC-MS. The total ion chromatogram(TIC) from AsbAS1-infiltrated leaf extracts revealed a peak witha retention time of 17.8 min that was not observed in the emptyvector control (Fig. 1D). The fragmentation pattern of this peakwas identical to that of β-amyrin (Fig. S1A). β-Amyrin was notdetected in the empty vector-treated control leaves or leaves thathad been infiltrated with the AsCYP51H10 construct alone (Fig.1D). Coexpression of AsbAS1 and AsCYP51H10 resulted ina substantial reduction in β-amyrin and the appearance of a newpeak with a retention time of 19.1 min (Fig. 1D), indicating thatAsCYP51H10 is able to modify β-amyrin.
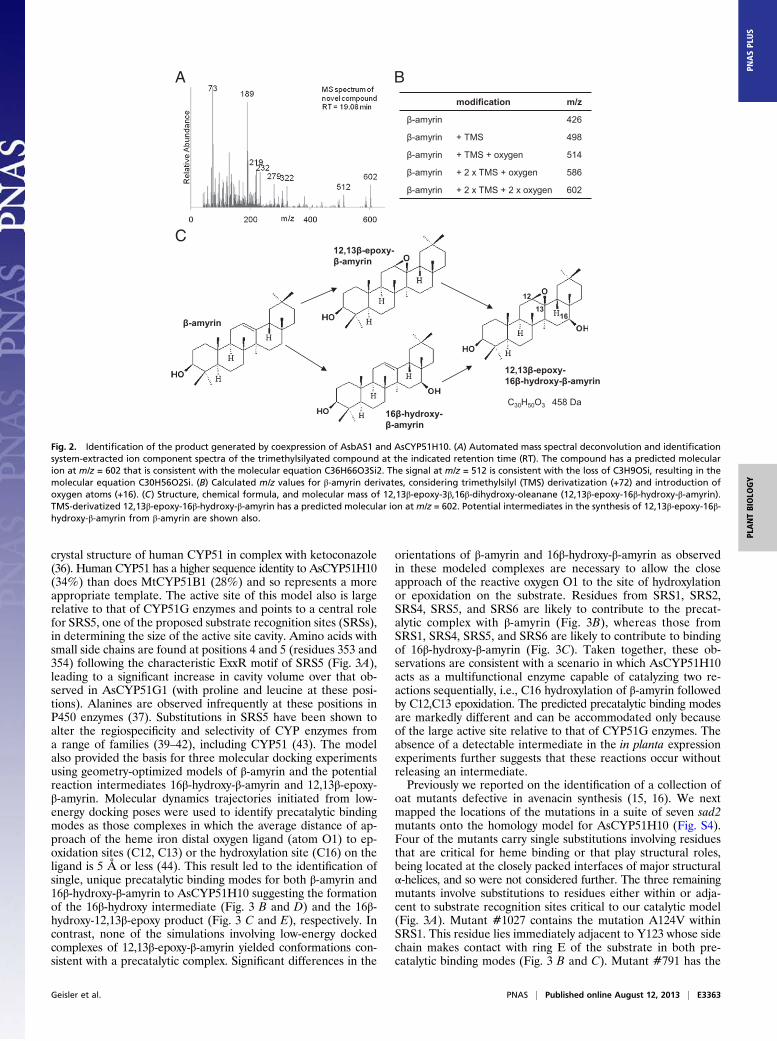
Identification of the AsCYP51H10 Product. The extracted ionizationspectrum for the new peak revealed a molecular ion at m/z 602(retention time = 19.08 min; Fig. 2A). Given that we analyzedtrimethylsilyl (TMS)-derivatized extracts and the AsCYP51H10product is derived from β-amyrin, a mass of 602 is consistent withthe molecular formula C36H66O3Si2. This result suggests that twoadditional oxygen atoms have been added to the β-amyrinbackbone, of which only one can be TMS-derivatized (Fig. 2B).Under GC-MS conditions, the β-amyrin mass spectrum showsa characteristic base peak at m/z 218 because of retro-Diels–Alder fragmentation typical for triterpenes that contain a C12/C13 double bond (Fig. S1A) (33). Because this typical β-amyrinbase peak at m/z 218 was not prominent in the mass spectrumof the AsCYP51H10 product (Figs. 1D and 2A), loss of theβ-amyrin double bond at C12/C13 is likely.Approximately 20 mg of the AsCYP51H10 product was puri-
fied from AsbAS1/AsCyp51H10 coinfiltrated plant material (∼17g dry weight) (Materials and Methods) and was analyzed by MSand NMR to determine the structure. A molecular mass of 458Da was determined using high-resolution MS. This result sug-gests a chemical formula of C30H50O3 and is consistent with thepreviously obtained GC-MS data. The 1H and 13C NMR spectraof the purified compound were compared with available NMRdata for β-amyrin (34, 35). Characteristic signals for the olefiniccarbons in β-amyrin (C12 δ 123; C13 δ 145) (34) were missing inthe 13C NMR spectrum of the analyzed compound, indicatingthe loss of the double bond. Further distortionless enhancementby polarization transfer, heteronuclear multiple bond correla-tion, and selective NOE experiments revealed an epoxide be-tween C12 and C13 and a hydroxyl group attached to C16, leadingto the identification of the compound as 12,13β-epoxy-3β,16β-dihydroxy-oleanane (12,13β-epoxy-16β-hydroxy-β-amyrin) (Fig.2C and Table S1).Potential reaction intermediates in the conversion of β-amyrin
to 12,13β-epoxy-16β-hydroxy-β-amyrin are 12,13β-epoxy-β-amyrinand 16β-hydroxy-β-amyrin (Fig. 2C). Neither of these was de-tectable in leaf extracts expressing AsbAS1 alone (Fig. S2), in-dicating that N. benthamiana is not able to modify β-amyrin. Wecannot exclude the possibility that AsCYP51H10 may carry outone modification to generate either 12,13β-epoxy-β-amyrin and16β-hydroxy-β-amyrin and that an endogenous N. benthamianaenzyme then may mediate the second modification. However,this possibility is unlikely, because previous preliminary analysisin yeast suggested that AsCYP51H10 is able to oxygenate β-amyrinat two positions (although the full structure was not determined)(21). Comparisons of the MS spectrum of our product with thatof the partially characterized product from yeast were in agree-ment (Fig. S3). Thus, AsCYP51H10 is able to stereo- and regio-
Geisler et al. PNAS | Published online August 12, 2013 | E3361
PLANTBIOLO
GY
PNASPL
US
specifically catalyze both hydroxylation at C16 and epoxide for-mation at C12/C13 in a β configuration, modifications that areboth present in the pathway end-product, avenacin A-1 (Fig. 1A).
Molecular Modeling and Analysis of Mutants. AsCYP51H10 is ahighly divergent CYP51 enzyme and has only 46% amino acidsequence identity with the bona fide oat sterol demethylase,AsCYP51G1. This identity drops to 32% for the subset of resi-dues that form the active site cavity (15). Of a total of 34 aminoacid residues that are completely conserved across CYP51 en-
zymes from bacteria, protozoa, fungi, animals, and plants, six arenot conserved in AsCYP51H10 (Fig. S4) (15). Our previous mo-deling of AsCYP51H10 and the oat sterol demethylase AsCYP51G1based on the Mycobacterium tuberculosis MtCYP51B1 crystalstructure revealed that the active site volume of AsCYP51H10 isrelatively large compared with the active site of CYP51G en-zymes, consistent with acquisition of a different function (15).We used molecular modeling and docking approaches to gain
further insights into the potential reaction mechanism ofAsCYP51H10, this time building a homology model based on the
AsbAS1
Sad1 Sad2
AsCYP51H10
β-Amyrin Avenacin A-1Des-acyl-avenacin A
B
A
D
A BC D
E
pBIN61-P19
CPMV-HT-AsbAS1
CPMV-HT-AsCYP51H10
A BC D
E
A BC D
E
C
Fig. 1. Expression of AsbAS1 and AsCYP51H10 in N. benthamiana leaves. (A) Biosynthesis of avenacin A-1 in oat. Potential oxidation sites for AsCYP51H10 arehighlighted in red. (B) CPMV-HT expression constructs and the silencing suppressor construct pBIN61-P19. Black boxes indicate transfer DNA borders; white arrowsindicate the 35S promoter sequence; solid black lines indicate CPMV RNA-2 UTRs; light gray arrows indicate the coding sequence; dark gray arrows indicate ter-minator sequences. (C) Detection ofAsbAS1 protein by immunoblot analysis. Leafmaterial was infiltratedwithA. tumefaciens cultures containing the empty vectorcontrol (EV) or expression constructs for AsbAS1, AsCYP51H10, or AsbAS1 + AsCYP51H10. Total soluble protein was extracted from infiltrated leaf material. (Left)Coomassie blue-stained replica gel. (Right) Immunoblot analysis with polyclonal antisera raised against AsbAS1. The expected molecular mass of AsbAS1 is 87 kDa(arrow). (D) GC-MS analysis of extracts from infiltratedN. benthamiana leaves. Total ion chromatograms (TICs) and extracted ion chromatograms (EICs) at anm/z of218 (EIC 218) and 189 (EIC 189) are shown. Accumulation of β-amyrin was detected in AsbAS1-expressing leaves. Coexpression of AsbAS1 and AsCYP51H10 resultedin lower levels of a β-amyrin and accumulation of a new peak [retention time (Rt) =19.1 min]. B, betulin (internal standard; Rt =18.7 min). The unlabeled peaks aresterols. Data are representative of at least three separate expression experiments. Corresponding chromatograms are drawn at the same scale, as indicated in thetop left corners.
E3362 | www.pnas.org/cgi/doi/10.1073/pnas.1309157110 Geisler et al.
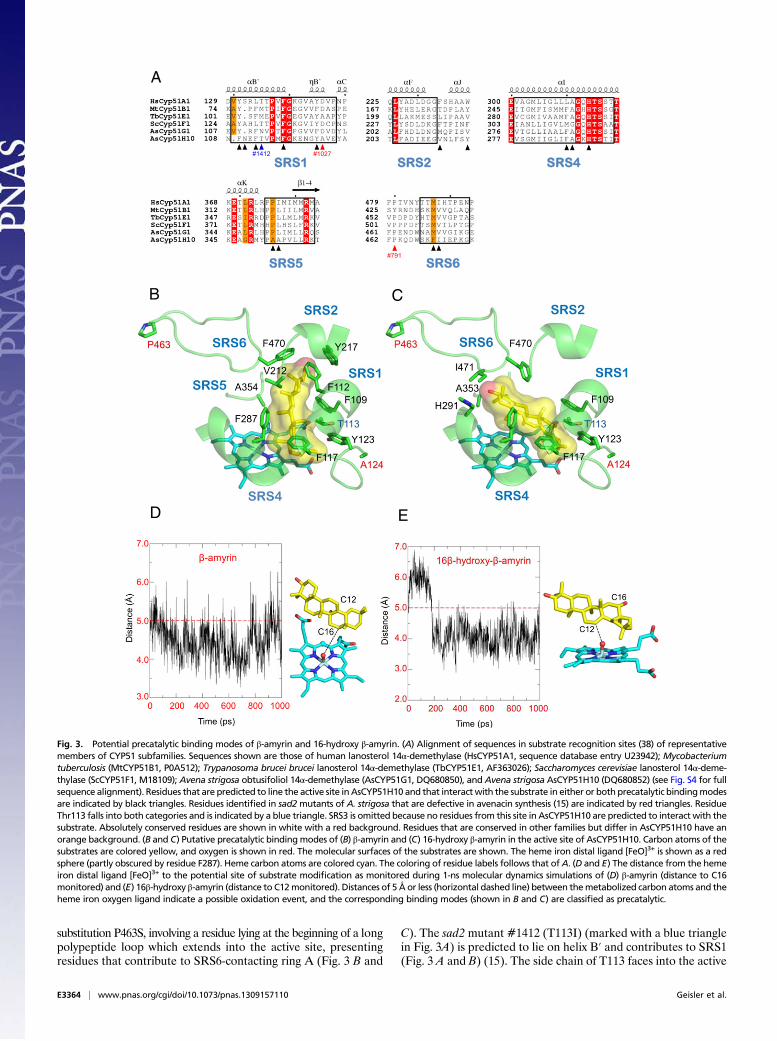
crystal structure of human CYP51 in complex with ketoconazole(36). Human CYP51 has a higher sequence identity to AsCYP51H10(34%) than does MtCYP51B1 (28%) and so represents a moreappropriate template. The active site of this model also is largerelative to that of CYP51G enzymes and points to a central rolefor SRS5, one of the proposed substrate recognition sites (SRSs),in determining the size of the active site cavity. Amino acids withsmall side chains are found at positions 4 and 5 (residues 353 and354) following the characteristic ExxR motif of SRS5 (Fig. 3A),leading to a significant increase in cavity volume over that ob-served in AsCYP51G1 (with proline and leucine at these posi-tions). Alanines are observed infrequently at these positions inP450 enzymes (37). Substitutions in SRS5 have been shown toalter the regiospecificity and selectivity of CYP enzymes froma range of families (39–42), including CYP51 (43). The modelalso provided the basis for three molecular docking experimentsusing geometry-optimized models of β-amyrin and the potentialreaction intermediates 16β-hydroxy-β-amyrin and 12,13β-epoxy-β-amyrin. Molecular dynamics trajectories initiated from low-energy docking poses were used to identify precatalytic bindingmodes as those complexes in which the average distance of ap-proach of the heme iron distal oxygen ligand (atom O1) to ep-oxidation sites (C12, C13) or the hydroxylation site (C16) on theligand is 5 Å or less (44). This result led to the identification ofsingle, unique precatalytic binding modes for both β-amyrin and16β-hydroxy-β-amyrin to AsCYP51H10 suggesting the formationof the 16β-hydroxy intermediate (Fig. 3 B and D) and the 16β-hydroxy-12,13β-epoxy product (Fig. 3 C and E), respectively. Incontrast, none of the simulations involving low-energy dockedcomplexes of 12,13β-epoxy-β-amyrin yielded conformations con-sistent with a precatalytic complex. Significant differences in the
orientations of β-amyrin and 16β-hydroxy-β-amyrin as observedin these modeled complexes are necessary to allow the closeapproach of the reactive oxygen O1 to the site of hydroxylationor epoxidation on the substrate. Residues from SRS1, SRS2,SRS4, SRS5, and SRS6 are likely to contribute to the precat-alytic complex with β-amyrin (Fig. 3B), whereas those fromSRS1, SRS4, SRS5, and SRS6 are likely to contribute to bindingof 16β-hydroxy-β-amyrin (Fig. 3C). Taken together, these ob-servations are consistent with a scenario in which AsCYP51H10acts as a multifunctional enzyme capable of catalyzing two re-actions sequentially, i.e., C16 hydroxylation of β-amyrin followedby C12,C13 epoxidation. The predicted precatalytic binding modesare markedly different and can be accommodated only becauseof the large active site relative to that of CYP51G enzymes. Theabsence of a detectable intermediate in the in planta expressionexperiments further suggests that these reactions occur withoutreleasing an intermediate.Previously we reported on the identification of a collection of
oat mutants defective in avenacin synthesis (15, 16). We nextmapped the locations of the mutations in a suite of seven sad2mutants onto the homology model for AsCYP51H10 (Fig. S4).Four of the mutants carry single substitutions involving residuesthat are critical for heme binding or that play structural roles,being located at the closely packed interfaces of major structuralα-helices, and so were not considered further. The three remainingmutants involve substitutions to residues either within or adja-cent to substrate recognition sites critical to our catalytic model(Fig. 3A). Mutant #1027 contains the mutation A124V withinSRS1. This residue lies immediately adjacent to Y123 whose sidechain makes contact with ring E of the substrate in both pre-catalytic binding modes (Fig. 3 B and C). Mutant #791 has the
A
C
modification m/z
β-amyrin 426
β-amyrin + TMS 498
β-amyrin + TMS + oxygen 514
β-amyrin + 2 x TMS + oxygen 586
β-amyrin + 2 x TMS + 2 x oxygen 602
B
C30H50O3 458 Da
12,13β-epoxy-16β-hydroxy-β-amyrin
16β-hydroxy-β-amyrin
12,13β-epoxy-β-amyrin
β-amyrin
12
1316
Fig. 2. Identification of the product generated by coexpression of AsbAS1 and AsCYP51H10. (A) Automated mass spectral deconvolution and identificationsystem-extracted ion component spectra of the trimethylsilyated compound at the indicated retention time (RT). The compound has a predicted molecularion at m/z = 602 that is consistent with the molecular equation C36H66O3Si2. The signal at m/z = 512 is consistent with the loss of C3H9OSi, resulting in themolecular equation C30H56O2Si. (B) Calculated m/z values for β-amyrin derivates, considering trimethylsilyl (TMS) derivatization (+72) and introduction ofoxygen atoms (+16). (C) Structure, chemical formula, and molecular mass of 12,13β-epoxy-3β,16β-dihydroxy-oleanane (12,13β-epoxy-16β-hydroxy-β-amyrin).TMS-derivatized 12,13β-epoxy-16β-hydroxy-β-amyrin has a predicted molecular ion at m/z = 602. Potential intermediates in the synthesis of 12,13β-epoxy-16β-hydroxy-β-amyrin from β-amyrin are shown also.
Geisler et al. PNAS | Published online August 12, 2013 | E3363
PLANTBIOLO
GY
PNASPL
US
substitution P463S, involving a residue lying at the beginning of a longpolypeptide loop which extends into the active site, presentingresidues that contribute to SRS6-contacting ring A (Fig. 3 B and
C). The sad2 mutant #1412 (T113I) (marked with a blue trianglein Fig. 3A) is predicted to lie on helix B′ and contributes to SRS1(Fig. 3 A and B) (15). The side chain of T113 faces into the active
A
B
D E
C
#1412
#791
#1027
Fig. 3. Potential precatalytic binding modes of β-amyrin and 16-hydroxy β-amyrin. (A) Alignment of sequences in substrate recognition sites (38) of representativemembers of CYP51 subfamilies. Sequences shown are those of human lanosterol 14α-demethylase (HsCYP51A1, sequence database entry U23942); Mycobacteriumtuberculosis (MtCYP51B1, P0A512); Trypanosoma brucei brucei lanosterol 14α-demethylase (TbCYP51E1, AF363026); Saccharomyces cerevisiae lanosterol 14α-deme-thylase (ScCYP51F1, M18109); Avena strigosa obtusifoliol 14α-demethylase (AsCYP51G1, DQ680850), and Avena strigosa AsCYP51H10 (DQ680852) (see Fig. S4 for fullsequence alignment). Residues that are predicted to line the active site inAsCYP51H10 and that interactwith the substrate in either or both precatalytic bindingmodesare indicated by black triangles. Residues identified in sad2mutants of A. strigosa that are defective in avenacin synthesis (15) are indicated by red triangles. ResidueThr113 falls into both categories and is indicated by a blue triangle. SRS3 is omitted because no residues from this site in AsCYP51H10 are predicted to interact with thesubstrate. Absolutely conserved residues are shown in white with a red background. Residues that are conserved in other families but differ in AsCYP51H10 have anorange background. (B and C) Putative precatalytic bindingmodes of (B) β-amyrin and (C) 16-hydroxy β-amyrin in the active site of AsCYP51H10. Carbon atoms of thesubstrates are colored yellow, and oxygen is shown in red. The molecular surfaces of the substrates are shown. The heme iron distal ligand [FeO]3+ is shown as a redsphere (partly obscured by residue F287). Heme carbon atoms are colored cyan. The coloring of residue labels follows that ofA. (D and E) The distance from the hemeiron distal ligand [FeO]3+ to the potential site of substrate modification as monitored during 1-ns molecular dynamics simulations of (D) β-amyrin (distance to C16monitored) and (E) 16β-hydroxy β-amyrin (distance to C12monitored). Distances of 5 Å or less (horizontal dashed line) between themetabolized carbon atoms and theheme iron oxygen ligand indicate a possible oxidation event, and the corresponding binding modes (shown in B and C) are classified as precatalytic.
E3364 | www.pnas.org/cgi/doi/10.1073/pnas.1309157110 Geisler et al.

site cavity and is predicted to be in van der Waals contact withbound β-amyrin and 16β-hydroxy-β-amyrin at rings D and E. Thesemutants were tested individually by computational modeling, andnone was predicted to be able to form precatalytic complexes(Fig. S5). Consistent with the computational analysis, although theseven sad2 mutants all accumulate β-amyrin (15, 20), none wasfound to accumulate either 16β-hydroxy-β-amyrin or 12,13β-epoxy-β-amyrin. Our computational structural model in combi-nation with mutant analysis thus provides insights into the unusualcatalytic behavior of AsCYP51H10 and its active site mutants.
The Epoxide Group Is Required for Antifungal Activity of Avenacin A-1. β-Amyrin itself is not antifungal. We therefore were interestedin establishing whether the action of AsCYP51H10 was likely toinfluence the antimicrobial properties of the triterpene scaffold.Structural variants of avenacin A-1 lacking one or more mod-ifications are not commercially available, and so the significanceof the C12/13 epoxide and the C16 hydroxide for the antimi-crobial activity of avenacin A-1 was not known. However, we wereable to generate and purify 12-oxo-avenacin A-1, in which theC12/13 epoxide is replaced by a C12 carbonyl group (Fig. 4A).Assays of antifungal activity were carried out using Gaeumanno-myces graminis var. tritici, a fungus that is sensitive to avenacin A-1(45). Avenacin A-1 was clearly inhibitory to fungal growth, but12-oxo-avenacin A-1 had no effect (Fig. 4B), indicating thatthe presence of the C12/C13 epoxide group is critical for an-tifungal activity. Thus, AsCYP51H10 is important in determiningthe potency of avenacin A-1 through modification of thetriterpene scaffold.
DiscussionCYP51 enzymes are regarded as one of the most ancient and highlyconserved cytochrome P450 families. These enzymes are wellknown to have functions in the synthesis of essential sterols inanimals, fungi, and plants and are important targets for antifungalagents, herbicides, and cholesterol-lowering drugs. Here we showthat AsCYP51H10 has acquired a different function comparedto other characterized members of the CYP51 family and carriesout two modifications to the β-amyrin scaffold: addition of a hy-
droxyl group at C16 of the D ring and introduction of an epoxideat the C12/C13 positions of the C ring. Our molecular modelingand docking approaches demonstrate that both possible inter-mediates, 12,13β-epoxy-β-amyrin and 16β-hydroxy-β-amyrin, fitinto the active site cavity. However, when a distance of hemeiron distal ligand to reaction sites of 5 Å or less is used as a cri-terion (44), the proposed first reaction step is likely to be thehydroxylation of β-amyrin at C16, followed by an epoxidation of16β-hydroxy-β-amyrin at C12/C13. The active site of AsCYP51H10is large compared with that of conserved CYP51G proteins (15),consistent with our findings that AsCYP51H10 introduces twosuccessive modifications to the β-amyrin backbone. We furthershow that none of the seven sad2 mutants examined was predictedto be able to form precatalytic complexes. Consistent with thisobservation, these mutants all accumulated β-amyrin but not 16β-hydroxy-β-amyrin or 12,13β-epoxy-β-amyrin. That a relatively largeactive site allows sequential reactions at different positions hasbeen discussed previously for CYP71D20, a 5-epiaristolochene1,3-dihydroxylase that catalyzes two successive hydroxylations ona sesquiterpene without releasing an intermediate (46). Wefurther show that the epoxide group is critical for the antifungalactivity of avenacin A-1. This result has important implicationsfor metabolic engineering of crop plants for enhanced diseaseresistance and also for the generation of antimicrobial triterpenesfor other applications.Multifunctional P450 enzymes from other P450 families that
catalyze both hydroxylation and epoxidation reactions havebeen described recently from bacteria (47–51) and fungi (52, 53).Examples of both possible reaction orders (hydroxylation fol-lowed by epoxidation and epoxidation followed by hydroxyl-ation) have been reported (Fig. 5). Here, we have shown thata plant P450 AsCYP51H10 is also able to catalyze both of thesetypes of modification. An exciting future challenge will be to es-tablish which amino acid residues are important in convertinga canonical CYP51 into an enzyme with the properties ofAsCYP51H that is able to catalyze both hydroxylation and ep-oxide formation of β-amyrin stereo- and regio-specifically. Giventhe many differences between these two types of enzymes, thisconversion in activity is unlikely to be trivial and may require
Avenacin A-1 (A1) 12-Oxo-avenacin A-1 (OA)A
B 2μg 5μg 10μgA1
OA
c c
A1
OA
c c
A1
OA
c c
2μg 5μg 10μgA1
OA
c c
A1
OA
c c
A1
OA
c c
Fig. 4. The C12/C13 epoxide is important for antifungal activity. (A) Structures of avenacin A-1 and the 12-oxo-avenacin A-1 derivative (OA). (B) Disk assaysfor antifungal activity. The avenacin-sensitive fungus G. graminis var. tritici was grown in presence of avenacin A-1 (A-1) and 12-oxo-avenacin A-1 (OA). Theamounts of compound applied to the discs are indicated. The control disk (c) was treated with 75% methanol only. A concentration-dependent zone ofinhibition can be observed for avenacin A-1 but not for the modified compound.
Geisler et al. PNAS | Published online August 12, 2013 | E3365
PLANTBIOLO
GY
PNASPL
US

systematic combinatorial mutagenesis of multiple amino acidresidues. The development of methods for the synthesis of thetwo possible intermediates 12,13β-epoxy-β-amyrin and 16β-hy-droxy-β-amyrin, neither of which is commercially available,also will be important for further mechanistic insights intoAsCYP51H10 function.Our experiments demonstrate that the CPMV-HT expression
system provides a rapid and effective means of expressing en-zymes for the synthesis of low molecular weight specialized com-pounds alone and in combination. Previous work on triterpenebiosynthetic enzymes by other groups has focused on yeast asa heterologous expression system (24–28). Here, we have usedthe CPMV-HT system for the synthesis and modification of thesimple triterpene β-amyrin, thereby reconstructing the first twocommitted steps in the avenacin biosynthetic pathway. Unlikeyeast, there was no requirement for introduction of a heterologouscytochrome P450 reductase because endogenous N. benthamianacytochrome P450 reductase activity was able to provide sufficientelectron transfer to enable catalysis. Furthermore, even withoutsubstantial scale-up and optimization, we were able to purify ∼20mg of the triterpene 12,13β-epoxy-16β-hydroxy-β-amyrin and deter-mine its structure by NMR. The CPMV-HT expression systemrecently has been refined to give the pEAQ series of transientexpression vectors (30), some of which are Gateway-based. It alsocan be made to be modular, permitting the expression of severalgenes from multiple CPMV-HT cassettes within the same transferDNA. Our proof-of-concept experiments now open the possi-bility of using the CPMV-HT system for synthetic biology-basedmetabolic engineering approaches to enable the production ofknown and novel triterpenes and other high-value compounds in“green factories.”
Materials and MethodsExpression of AsbAS1 and AsCYP51H10 in N. benthamiana. CPMV expressionconstructs carrying the AsbAS1 or AsCyp51H10 coding sequence weretransformed into A. tumefaciens strain LBA4404 and leaves of N. benthamianaplants were infiltrated as previously described (29–32). Full details of cloningprocedures, including primer sequences (Table S2) and expression in N.benthamiana are provided in SI Materials and Methods.
Protein Extraction and Immunoblot Analysis. Details of methods for proteinextraction and immunoblot analysis are provided in SI Materials and Methods.
Triterpene Analysis and Purification of 12,13β-Epoxy-16β-Hydroxy-β-Amyrin.The triterpene content of N. benthamiana leaves expressing AsbAS1 and/or AsCYP51H10 cDNA was analyzed by GC-MS as previously described (54).For further purification of 12,13β-epoxy-16-hydroxy-β-amyrin from N. ben-thamiana leaf material, large-scale triterpene extraction was performed.Further purification was achieved using medium pressure liquid chroma-tography. Full details of triterpene extraction methods, purification of12,13β-epoxy-16β-hydroxy-β-amyrin, and analysis of oat mutants are pro-vided in SI Materials and Methods.
Molecular Modeling. Development of an AsCYP51H10 model was carriedout as previously described (15), except the crystal structure of humanCYP51 in complex with ketoconazole (Protein Data Bank ID code 3LD6)(37) was used as a template. Docking of substrate molecules was per-formed using Autodock Vina (55). Molecular dynamics simulations werecarried out using GROMACS (56). Full details are provided in SI Materialsand Methods.
Fungal Growth-Inhibition Assays. Assays were carried out as described previously(57). Plugs of mycelium from an actively growing colony of the ascomycetefungus G. graminis var. tritici isolate T5 (45) were placed onto potato dextroseagar. Avenacin A-1 and carbonyl-avenacin A-1 were dissolved in 70%methanol(1 mg/mL stock solution) and applied to filter paper discs. The discs were placed
A MycG MycG
M-IV M-V M-II
BGfsF
25-O-Methyl FD-892
GfsF
FD-891
TirC
CTamI
TirA TirBTirDTirE
TamL TamI TamI
D Tri4
Trichodiene
Fig. 5. Examples of microbial multifunctional P450s catalyzing epoxidations. (A) MycG, a P450 involved in mycinamicin biosynthesis in the actinomycete,Micromonspora griseorubida. MycG catalyzes hydroxylation of mycinamycin IV (M-IV) followed by epoxidation of mycinamycin V (M-V) to mycinamycin II (M-II). (B) GfsF is required for biosynthesis of the antibiotic FD-891 in Streptomyces. GfsF first catalyzes the epoxidation of 25-O-methyl FD-892 and then thehydroxylation, yielding FD-891. (C) TamI is required for tirandamycin (Tir) biosynthesis in Streptomyces and catalyzes the hydroxylation of TirC to TirE. TirE isfurther modified to TirD by the flavoprotein TamL. Subsequently TamI catalyzed epoxidation and hydroxylation of TirD to form TirA and TirB. (D) The P450Tri4 is involved in four successive oxygenation reactions during trichothecene synthesis in Fusarium. After the trichodiene formation, Tri4 catalyzes hy-droxylation at C2, epoxidation at C12/13, hydroxylation at C11, and hydroxylation at C3.
E3366 | www.pnas.org/cgi/doi/10.1073/pnas.1309157110 Geisler et al.

equidistant from the fungal plug of inoculum. Plates were incubated at 22 °Cin the dark, and growth was monitored after 1 wk. Details of avenacin A-1 andcarbonyl-avenacin A-1 preparation are provided in SI Materials and Methods.
ACKNOWLEDGMENTS. We thank Alan Jones and Lionel Hill of John InnesMetabolite Services for metabolite analysis. The computer modeling and sim-ulation presented in this paper were carried out on the High Performance Com-
puting Cluster supported by the Research and Specialist Computing SupportService at the University of East Anglia. This work was supported by the UnitedKingdom Biotechnological and Biological Sciences Research Council InstituteStrategic Programme Grant “Understanding and Exploiting Plant and MicrobialSecondary Metabolism” BB/J004561/1 and by the John Innes Foundation (R.K.H.,G.P.L., M.R., S.F., R.E.M., and A.E.O.). K.G. was supported by a Danish ResearchSchool for Biotechnology International PhD studentship.
1. Dixon RA (2001) Natural products and plant disease resistance. Nature 411(6839):843–847.
2. Kliebenstein DJ, Osbourn A (2012) Making new molecules - evolution of pathways fornovel metabolites in plants. Curr Opin Plant Biol 15(4):415–423.
3. Newman DJ (2008) Natural products as leads to potential drugs: An old process or thenew hope for drug discovery? J Med Chem 51(9):2589–2599.
4. Mizutani M, Ohta D (2010) Diversification of P450 genes during land plant evolution.Annu Rev Plant Biol 61:291–315.
5. Nelson D, Werck-Reichhart D (2011) A P450-centric view of plant evolution. Plant J66:1):194–211.
6. Hamberger B, Bak S (2013) Plant P450s as versatile drivers for evolution of species-specific diversity. Phil Trans R Soc B, 19 February 368(1612):20120426.
7. Nelson DR, Schuler MA, Paquette SM, Werck-Reichhart D, Bak S (2004) Comparativegenomics of rice and Arabidopsis. Analysis of 727 cytochrome P450 genes andpseudogenes from a monocot and a dicot. Plant Physiol 135(2):756–772.
8. LepeshevaGI, Virus C,WatermanMR (2003) Conservation in the CYP51 family. Role of theB’ helix/BC loopandhelices F andG in enzymatic function. Biochemistry 42(30):9091–9101.
9. Yoshida Y, Aoyama Y (1984) Yeast cytochrome P-450 catalyzing lanosterol 14 alpha-demethylation. I. Purification and spectral properties. J Biol Chem 259(3):1655–1660.
10. Aoyama Y, Yoshida Y (1991) Different substrate specificities of lanosterol 14a-demethylase (P-45014DM) of Saccharomyces cerevisiae and rat liver for 24-methylene-24,25-dihydrolanosterol and 24,25-dihydrolanosterol. Biochem Biophys Res Commun178(3):1064–1071.
11. Trzaskos JM, Fischer RT, Favata MF (1986) Mechanistic studies of lanosterol C-32demethylation. Conditions which promote oxysterol intermediate accumulationduring the demethylation process. J Biol Chem 261(36):16937–16942.
12. Strömstedt M, Rozman D, Waterman MR (1996) The ubiquitously expressed humanCYP51 encodes lanosterol 14 alpha-demethylase, a cytochrome P450 whose expressionis regulated by oxysterols. Arch Biochem Biophys 329(1):73–81.
13. Bak S, Kahn RA, Olsen CE, Halkier BA (1997) Cloning and expression in Escherichia coliof the obtusifoliol 14 alpha-demethylase of Sorghum bicolor (L.) Moench, a cytochromeP450 orthologous to the sterol 14 alpha-demethylases (CYP51) from fungi and mammals.Plant J 11(2):191–201.
14. Cabello-Hurtado F, et al. (1997) Cloning and functional expression in yeast of a cDNAcoding for an obtusifoliol 14alpha-demethylase (CYP51) in wheat. Biochem BiophysRes Commun 230(2):381–385.
15. Qi X, et al. (2006) A different function for a member of an ancient and highlyconserved cytochrome P450 family: From essential sterols to plant defense. Proc NatlAcad Sci USA 103(49):18848–18853.
16. Papadopoulou K, Melton RE, Leggett M, Daniels MJ, Osbourn AE (1999) Compromiseddisease resistance in saponin-deficient plants. Proc Natl Acad Sci USA 96(22):12923–12928.
17. Inagaki Y-S, et al. (2011) Investigation of the potential for triterpene synthesis in ricethrough genome mining and metabolic engineering. New Phytol 191(2):432–448.
18. Haralampidis K, et al. (2001) A new class of oxidosqualene cyclases directs synthesisof antimicrobial phytoprotectants inmonocots. ProcNatl Acad Sci USA98(23):13431–13436.
19. Qi X, et al. (2004) A gene cluster for secondary metabolism in oat: Implications for theevolution of metabolic diversity in plants. Proc Natl Acad Sci USA 101(21):8233–8238.
20. Qin B, et al. (2010) High throughput screening of mutants of oat that are defective intriterpene synthesis. Phytochemistry 71(11-12):1245–1252.
21. Kunii M, et al. (2012) β-Amyrin oxidation by oat CYP51H10 expressed heterologouslyin yeast cells: the first example of CYP51-dependent metabolism other than the 14-demethylation of sterol precursors. Biol Pharm Bull 35(5):801–804.
22. Sawai S, Saito K (2011) Triterpenoid biosynthesis and engineering in plants. FrontPlant Sci 2:25, 10.3389/fpls.2011.00025.
23. Osbourn A, Goss RJ, Field RA (2011) The saponins: Polar isoprenoids with importantand diverse biological activities. Nat Prod Rep 28(7):1261–1268.
24. ShibuyaM, et al. (2006) Identification of beta-amyrin and sophoradiol 24-hydroxylase byexpressed sequence tag mining and functional expression assay. FEBS J 273(5):948–959.
25. Seki H, et al. (2008) Licorice beta-amyrin 11-oxidase, a cytochrome P450 with a keyrole in the biosynthesis of the triterpene sweetener glycyrrhizin. Proc Natl Acad SciUSA 105(37):14204–14209.
26. Seki H, et al. (2011) Triterpene functional genomics in licorice for identification ofCYP72A154 involved in the biosynthesis of glycyrrhizin. Plant Cell 23(11):4112–4123.
27. Carelli M, et al. (2011) Medicago truncatula CYP716A12 is a multifunctional oxidaseinvolved in the biosynthesis of hemolytic saponins. Plant Cell 23(8):3070–3081.
28. Fukushima EO, et al. (2011) CYP716A subfamily members are multifunctional oxidasesin triterpenoid biosynthesis. Plant Cell Physiol 52(12):2050–2061.
29. Sainsbury F, Lomonossoff GP (2008) Extremely high-level and rapid transient proteinproduction in plants without the use of viral replication. Plant Physiol 148(3):1212–1218.
30. Sainsbury F, Thuenemann EC, Lomonossoff GP (2009) pEAQ: Versatile expressionvectors for easy and quick transient expression of heterologous proteins in plants.Plant Biotechnol J 7(7):682–693.
31. Mugford ST, et al. (2009) A serine carboxypeptidase-like acyltransferase is requiredfor synthesis of antimicrobial compounds and disease resistance in oats. Plant Cell21(8):2473–2484.
32. Mugford ST, et al. (2013) Modularity of plant metabolic gene clusters: A trio of linkedgenes that are collectively required for acylation of triterpenes in oat. Plant Cell 25:1078–1092.
33. Burnouf-Radosevich M, Delfel NE, England R (1985) Gas chromatography-mass spec-trometry of oleanane- and ursane-type triterpenes – application to Chenopodium quinoatriterpenes. Phytochemistry 24(9):2063–2066.
34. Jhoo JW, et al. (2001) Characterization of the triterpene saponins of the roots andrhizomes of blue cohosh (Caulophyllum thalictroides). J Agric Food Chem 49(12):5969–5974.
35. Kajikawa M, et al. (2005) Cloning and characterization of a cDNA encoding beta-amyrin synthase from petroleum plant Euphorbia tirucalli L. Phytochemistry 66(15):1759–1766.
36. Strushkevich N, Usanov SA, Park HW (2010) Structural basis of human CYP51inhibition by antifungal azoles. J Mol Biol 397(4):1067–1078.
37. Seifert A, Pleiss J (2009) Identification of selectivity-determining residues in cytochromeP450 monooxygenases: A systematic analysis of the substrate recognition site 5.Proteins 74(4):1028–1035.
38. Podust LM, Stojan J, Poulos TL, Waterman MR (2001) Substrate recognition sites in14α-sterol demethylase from comparative analysis of amino acid sequences and X-raystructure of Mycobacterium tuberculosis CYP51. J Inorg Biochem 87(4):227–235.
39. Born SL, John GH, Harlow GR, Halpert JR (1995) Characterization of the progesterone21-hydroxylase activity of canine cytochrome P450 PBD-2/P450 2B11 through recon-stitution, heterologous expression, and site-directed mutagenesis. Drug Metab Dispos23(7):702–707.
40. Schalk M, Croteau R (2000) A single amino acid substitution (F363I) converts theregiochemistry of the spearmint (-)-limonene hydroxylase from a C6- to a C3-hydroxylase.Proc Natl Acad Sci USA 97(22):11948–11953.
41. Liu J, et al. (2004) The effect of reciprocal active site mutations in human cytochromesP450 1A1 and 1A2 on alkoxyresorufin metabolism. Arch Biochem Biophys 424(1):33–43.
42. Lentz O, et al. (2006) Altering the regioselectivity of cytochrome P450 CYP102A3 ofBacillus subtilis by using a new versatile assay system. ChemBioChem 7(2):345–350.
43. Cools HJ, et al. (2010) Heterologous expression of mutated eburicol 14α-demethylase(CYP51) proteins of Mycosphaerella graminicola to assess effects on azole fungicidesensitivity and intrinsic protein function. Appl Environ Microbiol 76(9):2866–2872.
44. Yuki H, Honma T, Hata M, Hoshino T (2012) Prediction of sites of metabolism ina substrate molecule, instanced by carbamazepine oxidation by CYP3A4. Bioorg MedChem 20(2):775–783.
45. Bryan GT, Daniels MJ, Osbourn AE (1995) Comparison of fungi within theGaeumannomyces-Phialophora complex by analysis of ribosomal DNA sequences.Appl Environ Microbiol 61(2):681–689.
46. Takahashi S, et al. (2005) Kinetic and molecular analysis of 5-epiaristolochene 1,3-dihydroxylase, a cytochrome P450 enzyme catalyzing successive hydroxylations ofsesquiterpenes. J Biol Chem 280(5):3686–3696.
47. Anzai Y, et al. (2008) Functional analysis of MycCI and MycG, cytochrome P450enzymes involved in biosynthesis of mycinamicin macrolide antibiotics. Chem Biol15(9):950–959.
48. Li S, et al. (2012) Substrate recognition by the multifunctional cytochrome P450 MycGin mycinamicin hydroxylation and epoxidation reactions. J Biol Chem 287(45):37880–37890.
49. Kudo F, Motegi A, Mizoue K, Eguchi T (2010) Cloning and characterization of thebiosynthetic gene cluster of 16-membered macrolide antibiotic FD-891: Involvementof a dual functional cytochrome P450 monooxygenase catalyzing epoxidation andhydroxylation. ChemBioChem 11(11):1574–1582.
50. Carlson JC, et al. (2010) Identification of the tirandamycin biosynthetic gene clusterfrom Streptomyces sp. 307-9. ChemBioChem 11(4):564–572.
51. Carlson JC, et al. (2011) Tirandamycin biosynthesis is mediated by co-dependentoxidative enzymes. Nat Chem 3(8):628–633.
52. McCormick SP, Alexander NJ, Proctor RH (2006) Fusarium Tri4 encodes a multifunctionaloxygenase required for trichothecene biosynthesis. Can J Microbiol 52(7):636–642.
53. Tokai T, et al. (2007) Fusarium Tri4 encodes a key multifunctional cytochrome P450monooxygenase for four consecutive oxygenation steps in trichothecene biosynthesis.Biochem Biophys Res Commun 353(2):412–417.
54. Field B, Osbourn AE (2008) Metabolic diversification—independent assembly ofoperon-like gene clusters in different plants. Science 320(5875):543–547.
55. Trott O, Olson AJ (2010) AutoDock Vina: Improving the speed and accuracy ofdocking with a new scoring function, efficient optimization, and multithreading.J Comput Chem 31(2):455–461.
56. Van Der Spoel D, et al. (2005) GROMACS: Fast, flexible, and free. J Comput Chem26(16):1701–1718.
57. Carter JP, Spink J, Cannon PF, Daniels MJ, Osbourn AE (1999) Isolation, characterization,and avenacin sensitivity of a diverse collection of cereal-root-colonizing fungi. ApplEnviron Microbiol 65(8):3364–3372.
Geisler et al. PNAS | Published online August 12, 2013 | E3367
PLANTBIOLO
GY
PNASPL
US





























