Embed Size (px)
Citation preview

1
Better Co-ordination Better Care
A Review of Services for people living with Neuromuscular Conditions in the South East Coast

2
INFORMATION READER BOX
Document Purpose Service Review
Title Better Co-ordination; Better Care - A review of services for people with Neuromuscular Conditions in the South East Coast
Author Elizabeth Sear, Dr Brijender Rana, Andrew Bibby
Publication Date June 2010
Target Audience PCT CEs, PCT Directors of Commissioning, Acute & Tertiary Neurology Providers
Description This review examines arrangements for the provision of specialised services for patients with Neuromuscular Disorders in the South East Coast and makes recommendations for future local commissioning policy.
Superseded Docs N/A
Contact Details Andrew Bibby, Associate Director (Commissioning), South East Coast SCG, 1 The Causeway, Goring-by-sea, West Sussex, BN12 6BT Tel: 01903 707453

3
Contents Page
1. Executive Summary 5
1.1 Introduction 5 1.2 Key Findings 5 1.3 Recommendations 5
2. Introduction 8
3. Neuromuscular Disorders 9
3.1 Classification of Neuromuscular Disorders and Disease Descriptions 9
4. Burden of Disease 17
4.1 Incidence of Neuromuscular Disorders 17 4.2 Prevalence of Neuromuscular Disorders 17 4.3 Life Expectancy and Trends in Mortality 18 4.4 Morbidity 20
5. Current Services, Models of Care, Health Economic Assessment and Gaps in Services 25
5.1 Current Services 25
5.1.1 Specialised Centres 25 5.1.2 Acute Providers 26 5.1.3 Community and Primary Care Services 26
5.2 Models of Care 27
5.2.1 National Policy and Standards of Care 27 5.2.2 Best Practice Treatment and Care 28 5.2.3 Best Practice Models of Care 28 5.2.4 Muscular Dystrophy Campaign Reports 29 5.2.5 Walton Report 29
5.3 Economics of Care 29 5.4 Service Gap Analysis 31
6. Issues Identified with Current Arrangements 32
6.1 National Work by the Muscular Dystrophy Campaign 32 6.2 Specific Local Recommendations for the South East Coast 32 6.3 Findings of hearings by the All Party Parliamentary Group on Muscular Dystrophy 33 6.4 The Patient Perspective – view of patients and their carers 33 6.5 Views of Clinician 36
7. Discussion 38 8. Recommendations 39 9. Glossary 41 10. References 42 11. Appendices 48

4

5
1. Executive Summary
1.1 Introduction
This report by the South East Coast Specialised Commissioning Group examines the arrangements for services for people living with Neuromuscular Disorders in Kent, Surrey and Sussex. The study that informed this report included engagement with local patients living with a Neuromuscular Disorder; their families and carers; the Muscular Dystrophy Campaign (the leading charity for people with these conditions); and clinicians at specialised centres. It also considered current arrangements in the context of reports published by the Muscular Dystrophy Campaign, and more recently by the All Party Parliamentary Group on Muscular Dystrophy. In drawing together evidence from all of these areas of study, this report sets out a vision and service model for the treatment and care of patients with neuromuscular disorders in the future. This document is the formal response to the requirement of the All Party Parliamentary Group for Muscular Dystrophy and the requirement set out in Lord Walton’s report that called for prompt action to address failings in services across the country.
1.2 Key Findings
Neuromuscular Disorders include a wide variety of diseases and conditions. They are complex in aetiology, presentation and progression. Many of these disorders do not have definitive treatments. They present with a range of medical and other issues for patients and their families. The management of these conditions is challenging throughout the patient pathway; from diagnosis to ongoing care across a range of healthcare settings. An assessment of the epidemiological evidence suggests that there are approximately 4,300 patients living with a neuromuscular condition in the South East Coast. A key recurrent theme identified from a number of sources (and echoing the findings of the All Party Parliamentary Group on Muscular Dystrophy inquiry) was the lack of co-ordination of services, which if resolved would both improve the patient experience but also have a beneficial effect on outcomes and increase efficiency. The current configuration of specialised hospital care for neuromuscular disorders for South East Coast patients is fragmenting care pathways and compounding difficulties in providing a consistent and co-ordinated package of care for affected patients. Access to appropriate physiotherapy and rehabilitation services is problematic for many patients and current service delivery models for community physiotherapy services pose problems with ongoing need for physiotherapy to maintain current health status and help prevent deterioration. Access to appropriate wheelchairs is also problematic. Particular issues relating to neuromuscular conditions (e.g. curved spines resulting from scoliosis) need to be factored into specifications for bespoke chairs to meet the individual needs of patients. As part of this review we identified circumstances where patients waited a considerable length of time for a specialised wheelchair to be provided only for this to be unsuitable for their needs both causing distress to the patient and being wasteful of NHS resources.
1.3 Recommendations 1.3.1 Management
• This report highlights the complex and specialised nature of services for patients with neuromuscular conditions and the relatively small population affected by these conditions across the Health Authority area. As such, we recommend that the SCG assumes responsibility for the commissioning of

6
specialised neuromuscular services in line with the national decision to include these in the SSNDS and recommendations from the All Party Parliamentary Group that these services are commissioned at SCG level.
• The SCG Team will build on the strategic direction articulated in this document by developing a detailed service specification for these services including agreement on referral pathways, outcome measures, clinical standards and performance monitoring.
• The detail of service development should fit strategically with the NSF for Long Term Neurological Conditions and with the NSF for Childrens services
1.3.2 Health Intelligence
• There is a clear need to improve the data available for planning purposes to better inform Commissioners in the future. Detailed plans should be developed to enhance data collection. This should include the development of a regional patient registry for neuromuscular disorders; work with the South East Public Health Observatory; and a robust mechanism for collection of local mortality data informed by analysis of death certificates.
1.3.3 Diagnosis
• That the SCG engage the Audit, Information and Analysis Unit to undertake work to examine to what degree appropriate and timely diagnosis of neuromuscular conditions occurs amongst South East Coast patients. The findings of this study will then inform an assessment of whether an education programme is required for primary care and community health staff who are likely to identify these patients and if so, how to target this in terms of professional groups and content.
1.3.4 Specialised Care
• The fragmentation and lack of consistency of specialised care is problematic in terms of forming a consistent base upon which to form the hub of an effective clinical network to better provide appropriate care for patients from the South East Coast. We recommend that the services at Great Ormond Street Hospital and Guy’s and St Thomas’s for Children and the adult service at Guy’s and St Thomas’s working in collaboration with the service at King’s College Hospital under the auspices of the King’s Health Partners Academic Health Sciences Centre are afforded preferred provider status and form the basis of a hub for a clinical network to better provide co-ordinated care for patients from the South East Coast with a neuromuscular condition.
• That all patients with a neuromuscular condition should be jointly managed by a preferred specialised centre working within a shared care framework with a local management centre.
• That patients currently receiving their care at a centre outside of these arrangements are given the opportunity to transfer into the South East Coast preferred care model.
1.3.5 Local Acute Care
• There is a need for local neurologists to develop closer working relationships with teams from specialised centres in order to provide more joined up ongoing care for patients as close to their homes as possible. Formal shared care arrangements through a clinical network model should be developed to optimise care for patients.
• Local services to better support ventilated patients need to be established both to minimise delayed discharges from specialised centres, but also to better support vulnerable patients closer to their homes.
• Consistent region-wide protocols need to be developed for the management of Emergency Care for patients with Neuromuscular Disorders. Evidence heard in the production of this report indicated that there are safety concerns with the particular needs of neuromuscular patients being overlooked when they are admitted via A&E departments, resulting in avoidable morbidity.
1.3.6 Co-ordinated & Consistent Care
• That defined and documented pathways are developed for patients, including for counselling and support.

7
• That co-ordination is enhanced to reduce fragmentation and mitigate risks that patients will “fall through the gaps” through the employment of a Care Co-ordinator role similar to that serving other SHA areas utilising monies identified in the SCG operating framework in 2010/11 for the purposes of improving co-ordination of neuromuscular care.
1.3.7 Rehabilitation & Therapies
• That, where they are provided or commissioned by the NHS, PCTs make hydrotherapy facilities available to patients with neuromuscular conditions on an equal basis to patients with other disabling conditions.
• That interested parties are engaged in a feasibility study in relation to the possible development of a specialist neuromuscular rehabilitation centre along the same lines as the national neuromuscular centre in Nantwich, Cheshire established as a social enterprise bringing together a partnership of the NHS, voluntary and business sectors. In addition to physiotherapy, the centre could generate income from commercial activities by people with neuromuscular conditions such as graphic design. Such a model could offer more innovative day and respite care support.
• That the SCG work with the PCT commissioners of physiotherapy services to ensure that community physiotherapy services better meet the ongoing needs of this patient group rather than through inappropriate short-term input which is the standard care package in a number of PCT areas.
1.3.8 Specialised Aids and Adaptations
• That an in-depth review of the provision arrangements for specialist wheelchairs is undertaken with a view to a specification being developed that accurately reflects the complex needs and considerations required when assessing for and providing appropriate chairs for neuromuscular patients.

8
2. Introduction
This service review into provision of care for patients in the South East Coast living with a neuromuscular disorder has been undertaken by the South East Coast Specialised Commissioning Group in response to a number of studies highlighting that the NHS in England is failing to adequately meet the healthcare needs of patients living with these conditions. Chiefly, the reports that have presented findings suggesting a deficit in service provision are the following documents:
• Building on Foundations: The Need for a Specialist Neuromuscular Service Across England, produced by the Muscular Dystrophy Campaign in December 2007.1
• Building on the Foundations: The Need for a Neuromuscular Service Serving Patients in the NHS South East Coast Region produced by the Muscular Dystrophy Campaign in May 20092,
• the Neuromuscular Service Development Strategy’, which was produced by the South West Specialised Commissioning Group (SWSCG) in February 2008.3
• Access to Specialist Neuromuscular Care: The Walton Report, published by the All Party Parliamentary Group for Muscular Dystrophy in July 2009.4
This document is the formal response to the requirement of the All Party Parliamentary Group for Muscular Dystrophy and the requirement set out in Lord Walton’s report that called for prompt action to address failings in services across the country. South East Coast Specialised Commissioning Group (SECSCG) is one of 10 SCGs in England, established following Sir David Carter’s review of Specialised Commissioning in the NHS. SECSCG is a formal joint sub-committee of the eight Primary Care Trusts in Kent & Medway; Surrey; and Sussex. The SCG is supported by a team of specialist staff from commissioning, finance and public health disciplines. Specialised services for adults with Neuromuscular Conditions are contained within Specialised Service National Definition Set (3rd Edition) No.8 – Specialised Neurosciences (Adult) and services for Children are contained within Specialised Service National Definition Set (3rd Edition) No.23 – Specialised Services for Children. The Department of Health has outlined in statute that specialised services are defined as being services that require a planning population of greater than 1 million people. These are often high cost, low volume interventions and treatments provided by relatively few specialist centres. Not all healthcare services required by patients living with neuromuscular disorders will be specialised or within the definition set (No. 8 for adults and 23 for children). One of the roles of Specialised Commissioners however is to consider the interface between specialised and non-specialised elements of care and make recommendations to the responsible commissioner of the non-specialised elements on how best care pathways can be co-ordinated. The aim of this review is to support the future commissioning of healthcare services for people living with a neuromuscular condition and as such, it’s recommendations, which set out a vision and a service model for the future, should be considered in the planning of these services.

9
3. Neuromuscular Disorders
3.1 Classification of Neuromuscular Disorders and Disease Descriptions Neuromuscular disorders cover a wide range of conditions including neuropathies (either acquired or inherited), muscular dystrophies, spinal muscular atrophy (SMA), as well as a range of very rare muscle disorders. There are many causes of progressive muscle weakness, which can strike any time from infancy through adulthood. Neurodegenerative diseases can affect different parts of the body in different ways. There are more than 60 different types of muscular dystrophy and related neuromuscular disorders (see Appendix 1). Neuromuscular disorders (NMDs) can be genetic or acquired, and may present in childhood (e.g. Duchenne muscular dystrophy), or young adult life. Others can be late onset conditions (e.g. inclusion body myositis).
There are few curative treatment options for most of these diseases. A number, such as Duchenne muscular dystrophy, are aggressive and cause progressive muscle wasting and weakness, orthopaedic deformity, cardiac and respiratory compromise, dependency on others for day-to-day care and usually result in premature death. Others cause life-long disability.1 Classification of neuromuscular disorders is challenging in that there is no firm basis on which to work. The causes of most of the disorders are unknown and for this reason, optimal classification is not possible. We have, in this document, classified neuromuscular disorders on the basis of primary anatomical site affected by the condition.
Table 1:Classification of Neuromuscular Disorders
Inherited neuromuscular disorders Acquired neuromuscular disorders
• Muscular dystrophies
• Spinal muscular atrophies
• Congenital and syndromal neuropathies
• Congenital myopathies
• Metabolic myopathies
• Inherited myasthenic syndromes
• Channelopathies
• Mitochondrial disorders
• Myotonias
• Inherited neuropathies
• Myasthenia gravis
• Autoimmune neuropathies
• Inflammatory myopathies
3.1.1 Disorders of the Anterior Horn Cells
3.1.1.1 Spinal Muscular Atrophy Spinal Muscular Atrophy (SMA) is the general name for a number of different disorders, all having in common a genetic cause and the manifestation of weakness due to loss of the motor neurons of the spinal cord and brainstem.1
The most common form of SMA is caused by mutation of the SMN gene and manifests over a wide range of severity affecting infants through to adults. This spectrum has been divided into three groups, depending on the age of onset: infantile SMA, intermediate SMA and adult SMA, for details of each of these see Appendix 2.1
In general, the earlier the symptoms appear, the shorter the lifespan. The onset is sudden and dramatic because the motor neuron cells deteriorate quickly following the appearance of symptoms. SMA can be

10
fatal and there is no known cure. The major management issue in Type I SMA is the prevention and early treatment of respiratory infections since pneumonia is the most common cause of mortality.1
Other gene mutations can cause different forms of spinal muscular atrophy. All forms of SMA have in common weakness caused by denervation, i.e. the muscle atrophies because it has lost the nerve which signals it to contract. Only motor nerves are affected. Heritable disorders that affect both motor and sensory denervation, and thus cause both muscle weakness and sensory impairment, are known by the inclusive label Charcot-Marie-Tooth or hereditary motor sensory neuropathy. The term spinal muscular atrophy thus refers to atrophy of muscles due to loss of motor neurons within the spinal cord.1
3.1.2 Disorders of the Peripheral Nerves
3.1.2.1 Metabolic Neuropathies3
Metabolic neuropathies are caused by the disruption of chemical processes in the body (such as an inability to use energy effectively in the body, or the build-up of toxins), which leads to nerve damage. Some are inherited, whilst others are acquired as secondary complications of various diseases (e.g. diabetes mellitus, alcoholism, kidney failure, sepsis, thyroid disease, or vitamin deficiencies). Symptoms of metabolic neuropathies include clumsy walk (gait), numbness, loss of coordination, pain (burning, pins and needles, or shooting pains), and weakness. These occur because the damaged nerves cannot send and receive signals to and from the brain. Symptoms tend to be first apparent in the toes and feet, with gradual progression upwards.
The most appropriate treatment for a metabolic neuropathy is the correction of the metabolic problem. For instance, vitamin deficiencies are treated with a dietary plan; and thyroid and blood sugar problems are resolved using medication. However, in some cases it is the symptoms that are treated, for example, pain can be reduced using medications to dull abnormal pain signals from the nerves. Also, weakness may be treated with physical therapy, whilst balance can be improved through the use of a cane or walker.
3.1.2.2 Guillain-Barre Syndrome
Guillain-Barre syndrome (GBS) is an autoimmune disorder affecting the peripheral nervous system, which is usually triggered by an acute infectious process. GBS is rare, with an incidence of around 1-2 per 100,000 people. It is frequently severe and usually exhibits as an ascending paralysis noted by weakness in the legs that spreads to the upper limbs and the face along with complete loss of deep tendon reflexes. With prompt treatment by plasmapheresis or intravenous immunoglobulins, and supportive care, the majority of patients regain full functional capacity. However, death may occur if severe pulmonary complications and autonomic nervous system problems are present. Guillain-Barre is one of the leading causes of non-trauma-induced paralysis in the world.
3.1.3 Neuromuscular Transmission Disorders
3.1.3.1 Myasthenia Gravis
Myasthenia gravis (MG) is an auto-immune disorder characterised by weakness. This is caused by circulating antibodies blocking acetylcholine receptors at the post-synaptic neuromuscular junction; thereby inhibiting the stimulative effect of acetylcholine. With a prevalence of between 200 to 400 cases per million population, it is one of the less common autoimmune disorders.1 MG gives rise to varying muscle weakness and tiredness, and can include respiratory failure and swallowing difficulties. It should be noted that MG is not usually a progressive disease (although the symptoms may come and go). For some, the symptoms decrease after a span of 3–5 years.3 Up to 75% of cases may be linked the thymus abnormalities, although there is also a slight genetic predisposition. Quality of life can vary depending on the severity and the cause of the condition.1
3.1.3.2 Lambert-Eaton Syndrome4
Lambert-Eaton syndrome (LES) is a rare, autoimmune disorder of the neuromuscular junction, in which faulty communication between muscles and their associated nerves leads to muscle weakness.LES can occur either as a paraneoplastic disorder in association with an underlying cancer (CA-LES), or without

11
cancer and as part of a more general autoimmune state (NCA-LES). Both types are caused by the presence of circulating antibodies against voltage-gated calcium channels, which impair neuromuscular transmission by inhibiting inward calcium current and subsequently the release of acetylcholine into the synaptic cleft. Symptoms of LES include insidious and gradual onset of fatigue, muscle weakness, vision changes, and a dry mouth. Proximal muscle weakness in hip girdle and thigh muscles, absent or reduced tendon reflexes that may ease after brief exercise, and dilated, poorly reactive pupils are all clinical symptoms of LES. Treatment of the underlying cancer, or plasmapheresis (to remove the antibodies from the blood), may both lead to the improvement of symptoms. However, long-term treatment usually involves drugs which facilitate neuromuscular transmission (e.g. neostigmine or 3,4-diaminopyridine), as well as immunomodulators (such as prednisolone). Many patients experience long-term disability due to weakness.
3.1.4 Disorders of the Muscles 3.1.4.1 Muscular Dystrophy Muscular dystrophy (MD) refers to a group of inherited (genetic) muscle diseases that cause progressive muscle weakness. In some instances the inheritance is recessive (both parents must have the causative gene); whereas in others there is dominant inheritance (only one parent need have the causative gene).1 Muscular dystrophies (MDs) are not only characterised by progressive skeletal muscle weakness, but also by defects in muscle proteins, and the death of muscle cells and tissue. Duchenne Muscular Dystrophy is the most common form of muscular dystrophy in children. 1 MDs are multi-system disorders with manifestations in body systems including the heart, gastrointestinal and nervous systems, endocrine glands, skin, eyes and other organs. The principle symptoms include muscle wasting and weakness, resulting in poor balance, falls, difficulty in walking, muscle contractures, pain, drooping eyelids and curvature of the spine (scoliosis).1
3.1.4.2 Duchenne Muscular Dystrophy
Duchenne Muscular Dystrophy (DMD) is the most common childhood form of muscular dystrophy. About a third of cases are caused by sporadic mutations in the DMD gene, whilst the remaining two-thirds of cases are inherited in X linked patterns. Because of this pattern of inheritance it is far more common in boys than in girls, and female carriers tend to have milder symptoms. 1
DMD usually becomes clinically evident when a child begins walking, with age of onset being between two and six years. The symptoms include general muscle weakness and wasting, which initially affects the pelvis, upper arms, and upper legs; and eventually involves all voluntary muscles. Patients typically require a wheelchair by age 7 to 12, and historically, most patients died in their late teens or early 20s. However, with appropriate, proactive medical care, these patients now have the potential to live until their late 20s and 30s, making it even more critical that any future service can deal with the additional demand on provision and the widening demographic that are accessing the service.1
3.1.4.3 Myotonic Disorders (Myotonic Muscular Dystrophy) Myotonic muscular dystrophy (MMD) is the most common adult form of MD although age of onset is very variable, with symptoms first appearing any time from birth to old age. MMD follows an autosomal dominant pattern of inheritance, meaning that on average half of the children of an affected person are themselves affected. Men and women have an equal chance of inheriting and of passing on the disorder, but affected women are more likely to have a severely affected child. In general (though not always), the disorder tends to be more severe in successive generations.1
Muscle stiffness or ‘myotonia’ is characteristic, especially affecting the hands. Involvement in other body systems (the heart, endocrine organs, eyes, and gastrointestinal tract) is frequent. Associated problems include cataracts, disturbance of heart rhythm, hormonal problems and, in children, learning difficulties.1
Since many doctors are unfamiliar with the condition, it is essential that people who have myotonic dystrophy are themselves aware of the problems and risks they may face. For example, operations and

12
anaesthetics can pose risks, even for mildly affected people. Problems usually occur when doctors are unaware of the disorder.1
3.1.4.4 Glycogen Storage Diseases
Glycogen storage diseases (GSD) are inherited metabolic disorders; this typically means that affected individuals are lacking in or have a dysfunction of one of the enzymes responsible for making or breaking down glycogen. There are many different types of GSD, which are either named according to the enzyme which is deficient in each case, according to the doctor that first identified them, or numerically. Generally it is either the liver, the muscles, or both, that are affected by the deficiency, although there are several other areas of the body that can be affected (e.g. red blood cells, heart, kidneys).5
Because glycogen is the storage molecule for glucose, an inability to either make or break down glycogen means that either glucose cannot be stored in the first place, or it cannot be released. This means that there is not a steady supply of glucose available in the area that is affected.5
For a brief comparison of the different types of glycogen storage disorder, refer to table 2 below:
Table 2: Types of Glycogen Storage Disorder
Type of GSD Dysfunction Areas
Affected Pattern of
Inheritance Symptoms Treatment
Other Information
1: von Gierke: Ia and Ib
Ia: deficiency of the glucose-6-phosphatase (G6Pase) enzyme Ib: deficiency in glucose-6-phosphate translocase, or transporter (G6PT) enzyme These both break
down glycogen6
Liver, kidney, other areas
6
Autosomal recessive
7
Poor growth, short stature,
rachitic changes, gingivitis,
xanthoma on elbows and
knees, hypoglycaemia7
Small, frequent feedings during the day, restriction of fructose and galactose, and feeding of uncooked cornstarch, which is digested slowly and so provides a steady release of glucose in between feedings. In some cases, an overnight tube feeding is required to provide a continuous delivery of glucose.
6
Most common – accounts for 25% of all cases in Europe and USA
6
2: Pompe: Infantile onset
and Late onset
Dysfunction of the enzyme acid alpha-1,4-glucosidase (also called acid maltase) prevents break-down of glycogen
8
Lysosomes of heart and muscles
8
Autosomal recessive
8
In the infantile-onset form symptoms start before 12 months of age; these can include: feeding and breathing difficulties, weight gain low muscle tone and weak muscles (meaning that the infants cannot perform motor skills and have developmental delay. In the late onset form symptoms can start anytime between 1 yr of age and adulthood, and can include progressive muscle weakness, starting in the muscles of the trunk and hip, and then affecting the legs (which can cause walking difficulties). Also common is scoliosis, shortness of breath and tiredness.
9
Treatment focuses on relieving symptoms instead of the underlying dysfunction. This may include therapy or mechanical ventilation to assist breathing, physical therapy to alleviate muscle weakness, occupational or speech and language therapy to improve development and dietetic input.
9
3: Cori
Dysfunctional Glycogen debrancher enzyme = build up of glycogen in tissue = damage to this
10
Liver, muscles, heart
10
Autosomal recessive
10
Hypoglycaemia, mild muscle weakness during childhood, becomes more evident in adults aged 30-40, slowly progressing weakness and distal muscle deterioration; may eventually require the use of a wheelchair.
10
As yet no effective treatment, although hypoglycaemia can be controlled with an appropriate diet
10
Types 3a and 3b - Only the former is associated with muscle disorders, as it is the only type with an enzyme deficiency in the muscle as well

13
as the liver. 10
4: Andersen
deficient branching enzyme - causes the accumulation of glycogen with very long outer branches, which in turn triggers the immune and surrounding tissues
11
Liver and muscle
11
Autosomal recessive
12
enlargement of the liver, below average weight gain, and poor muscle tone. Ultimately results in death, frequently by the age of five.
11
The only effective treatment is liver transplant, although muscular problems will still be evident following such surgery
11
5: McArdle
phosphorylase deficiency in the muscles - Phosphorylase mediates the breakdown of glycogen to produce glucose. Hence, a deficiency corresponds with a lack of glucose, leading to particular problems during anaerobic exercise. Consequently, energy is obtained by the breakdown of muscle.
13
Muscle13
Autosomal recessive
14
muscle pain, cramping, fatigue and tenderness, as well as myoglobinuria (myoglobin in the urine).
13
Advising patients with the condition against excessive exercise (in order to prevent muscle breakdown).
13
6: Hers
Liver phosphorylase enzyme deficiency, leading to problems with glycogen breakdown
15
Liver16 Autosomal
recessive17
Similar to GSD type I in terms of clinical presentation, although tends to be milder. Muscles are not affected in this form.
16
Nothing specific. Frequent meals containing carbohydrates to prevent hypoglycaemia and mediate normal growth. In severe cases, liver transplantation may be performed.
17
7: Tarui
Lack of sufficient phosphofructokinase enzyme in their muscle, which is responsible for mediating the breakdown of glycogen. Hence, glycogen breakdown during periods of muscle stress (such as anaerobic respiration), cannot occur
18
Muscle18
Autosomal recessive
19
pain, weakness and cramping in affected muscle. The muscle itself is broken down in order to produce energy, causing further muscular pain and myoglobinuria
18
Glucagon can be administered in order to increase blood sugar level and sustain muscle tissue. To prevent painful and dangerous rhabdomyolytic episodes patients are instructed to avoid high-carbohydrate meals and most forms of physical activity.
18
9
phosphorylase kinase deficiency. PHK metabolises glycogen, which means that a deficiency in this leads to the accumulation of glycogen
20
Liver, muscle, blood cells, heart (rarely)
20
either in an autosomal recessive or X-linked pattern, the former is most common accounting for 75% of all cases
20
Similar to those of type VI GSD. GSD IX can cause hypoglycaemia, especially after long periods of fasting. Other common symptoms are growth retardation, mild delay in motor development, and elevated blood lipids. The symptoms usually improve as a child ages, and children usually reach their full potential height and weight by adulthood.
20
Maintenance of a high carbohydrate diet, adequate amounts of protein in the diet, and avoiding long periods of not eating.
20
0
deficient glycogen synthase enzyme. This prevents the formation of glycogen stores in the liver and muscles, for instance. This means that hypoglycaemia can occur between meals, when there is a lack of glucose available from food.
21
Liver. muscles
21
Autosomal recessive disorder
21
muscle cramps caused by the body’s attempts to make energy from lactic acid
21
Preventing hypoglycaemic episodes by consuming food frequently, and ingesting raw cornstarch to act as a slow release of glucose (especially overnight). A high protein diet may alleviate cramping, tiredness and

14
fatigue. 21
3.1.4.5 Periodic Paralysis22
Periodic paralysis is a rare inherited disorder that causes temporary muscle weakness or paralysis and can onset at any age. The paralysis can be mild (e.g. involving only one limb), or so severe that patients are unable to breath unaided. It can last for a few moments, or can go on for several days. Some forms of paralysis may involve some muscle stiffness or rigidity during attacks, whilst others may be characterised by permanent muscle weakness. It is caused by channelopathies, which are disruptions of the ion channels involved in neuromuscular conduction (due to mutations of the genes for these ion channels). The type of periodic paralysis inherited is determined by the nature of the mutation; to date there have been 30 mutations identified, affecting either the genes for sodium or calcium channels.
There are several types of periodic paralysis; hypokalaemic periodic paralysis, which occurs following the consumption of sweet or starchy foods and eases following potassium treatment; thyrotoxic hypoKPP, which is associative with an overactive thyroid gland and is most common in asian males; normokalaemic periodic paralysis, in which people with the condition experience weakness when potassium levels rise; and paramyotonia congenital, which is characterised by muscle stiffness or rigidity following exposure to cold or activity. The final form is Andersen’s syndrome, in which the potassium shifts are inconsistent, and people affected may experience generalised weakness between attacks, as well as irregular heart rhythms. People with Andersen’s syndrome may also have unusual facial and hand characteristics.
3.1.4.6 Inflammatory Myopathies23
The inflammatory myopathies are characterised by chronic muscle inflammation (or myositis), accompanied by weakness. There are three main types of chronic inflammatory myopathy: polymyositis, dermatomyositis and inclusion body myositis. These are thought to be autoimmune disorders, although the cause is not known for certain. Inflammatory myopathies can affect both adults and children, but dermatomyositis is the most common form in children.
Symptoms of the chronic inflammatory myopathies include slow, progressive muscle weakness (starting in the proximal muscles), fatigue following walking or standing, falling, and difficulty swallowing or breathing. The former is caused by damage to the muscle fibres as a result of the inflammation. Polymyositis affects skeletal muscles bilaterally, and is most common in people between the ages of 31 and 60. Other symptoms of polymyositis include arthritis, shortness of breath, difficulty climbing stairs and lifting objects, and heart arrhythmias. Inclusion body myositis (IBM) is similar to polymyositis, with its own distinctive features. For instance, the onset of muscle weakness is gradual, and it affects both proximal and distal muscles. Muscle weakness in IBM may affect only one side of the body. The first symptoms of IBM tend to be falling and tripping, although some first experience weakness of the wrists and fingers, leading to problems with gripping, pinching and buttoning actions. Other common symptoms include swallowing difficulties and atrophy of the forearm and quadriceps muscles. Symptoms do not usually present until after the age of 50.
Dermatomyositis, on the other hand, is characterised by a skin rash which accompanies the muscle weakness and commonly develops on the eyelids, over the muscles used to extend and flex joints, and also on the face, neck, upper chest and back. Other symptoms can include weight loss, a low-grade fever, inflamed lungs and sensitivity to light. Dermatomyositis may be accompanied by calcium deposits under the skin or in the muscle (calcinosis), which most often occurs with a few years after onset of the disease. As polymyositis and dermatomyositis progress, the distal muscles may also be affected (as well as the more proximal ones). Polymyositis and dermatomyositis may be associated with collagen-vascular or autoimmune diseases.
Juvenile myositis, another inflammatory myopathy, usually affects children aged between 2 and 15 years. Common symptoms include proximal muscle weakness and inflammation, oedema, muscle pain, fatigue, skin rashes, abdominal pain, fever and contractures. Around 20 to 30 percent of children with juvenile dermatomyositis develop calcinosis.

15
Treatment options for the symptoms of chronic inflammatory myopathies can include medication, physical therapy (with the aim of preventing muscular atrophy and regaining muscle strength and motion), heat therapy, exercise, orthotics and rest. In addition, polymyositis and dermatomyositis can be treated with high doses of corticosteroids. In patients with a poor response to corticosteroids, immunosuppressant drugs may reduce inflammation. Recovery in patients with polymyositis and dermatomyositis can be improved with the administration of intravenous immunoglobulin. Dermatomyositis may be relieved with a topical ointment (e.g. corticosteroids or tacrolimus). Surgery may be beneficial in order to remove the calcium deposits which cause nerve pain and infections. IBM is mostly unresponsive to corticosteroids, immunosuppressive drugs, and intravenous immunoglobulin. Physical therapy may help to maintain mobility, whilst other therapies may improve certain symptoms.
3.1.4.7 Muscle Disorders Associated with Endocrine Diseases24
Various endocrine diseases, including those affecting the thyroid, parathyroid, suprarenal and pituitary glands, as well as some affecting the ovaries, testes and the islets of Langerhans (pancreas), can cause myopathies. Examples are those associated with dysfunction of the adrenal (e.g. Cushing’s disease), thyroid, parathyroid, pituitary, and Islets of Langerhans glands, and (cortico) steroid myopathy (which is the most common endocrine myopathy). Myopathies associated with endocrine disorders are being recognised more and more frequently, although precise prevalence is difficult to ascertain as many patients with symptoms of fatigue or weakness are being diagnosed as having a myopathy without relevant histological or electrophysiological testing to confirm this.
Symptoms of myopathy can include weakness and pain, which tends to affect the proximal more than the distal muscles in a symmetrical pattern. These can have a negative impact both on quality of life and the ability to cope with day-to-day tasks. Muscle atrophy is also a common feature, although not universally observed. The most effective treatment of myopathies associated with endocrine disease is to treat the underlying endocrine disorder by administering beta-adrenergic blockers to improve muscle strength.
3.1.4.8 Other Muscles Disorders
There are a number of other muscle disorders, such as myotonia congenita.
Myotonia congenita affects muscle relaxation (i.e. causes muscle stiffness). It is caused by a channelopathy of the chloride channels within skeletal muscle cells as a result of a mutation in the gene for these channels. A person with myotonia congenita is born with the condition, but may not develop symptoms until the age of 2 or 3. Early symptoms can include swallowing difficulties, gagging, and shortness of breath during exercise, although the most prominent symptom is stiff and slow movements (which are improved upon repetition). This stiffness is only evident during the start of a movement.25
The two most common types of myotonia congenita are Becker and Thomsen diseases.25 These differ both in the severity of the symptoms experienced by different individuals, and also by the way in which they are inherited.26 For instance, Becker disease tends to have later presentation than Thomsen disease and causes more severe stiffness which is often accompanied by periods of muscle weakness (particularly in the arms and hands) during movement following rest.26 Thomsen is an autosomal dominant disorder, whereas Becker is autosomal recessive.26 Treatment for myotonia congenita is not always required, but symptoms can be relieved using drug therapy.25
Myositis ossificans, otherwise known as fibrodysplasia ossificans progressiva, is a rare genetic disorder characterised by malformations of the big toes, and the progressive ossification of soft connective tissue. Although flare-ups of this disease are sporadic, it worsens with age, with ossification beginning by 10 years of age (this can be spontaneous or induced by injury). Myositis ossificans usually begins as swellings of soft connective tissue (including tendons, ligaments, fascia and skeletal muscle, not including cardiac and smooth muscle, the tongue, and the diaphragm), and appears to be caused by an immune/inflammatory response. The death of skeletal muscle seems to be due to macrophages and lymphocytes since there are macrophages, lymphocytes and mast cells present in early lesions, and there are flare-ups of the disease following viral infections. Some of the lesions/swellings regress, but most go on to form bone (by ossification). The pattern of disease progression generally follows that of embryonic skeletal formation, that is, it is first observed in the dorsal (towards the back), axial (within the CNS), cranial (towards the head) and proximal (towards the midline) regions of the body.27

16
Affected individuals are commonly wheelchair-bound by the third decade of life. There is currently no treatment available to prevent ossification, however, it has been reported that a 4-day course of high-dose corticosteroids, started within 24 hours of a flare-up, may reduce the inflammation and oedema (only to be used for flare-ups involving the joints and jaw region). Non-steroidal anti-inflammatories can be used as an alternative to corticosteroids, if the latter are not suitable. Muscle relaxants may be given to decrease muscle spasms and maintain function in skeletal muscle surrounding the swelling. Surgery is not a viable treatment option as it may provoke new bone growth. Finally, analgesics can be given for pain caused by the inflammation. 27

17
4. Burden of Disease
4.1 Incidence of Neuromuscular Disorders
Incidence refers to the number of new cases of a disease occurring per unit of population per unit of time.31 The incidence of neuromuscular diseases in general is around 1 in every 2,400 births, meaning that there were around 21 babies born with a neuromuscular disorder in the South East Coast SHA area in 2008 (based on 51,565 births in SEC that year). The incidence of muscular dystrophy (MD) in general is close to 1 in 3700 births, meaning that there were roughly 14 babies born in the South East Coast in 2008 with muscular dystrophy. In individual diseases the greatest incidence is found in Duchenne Muscular Dystrophy (the most common and severe), which is 1 in every 3,500 males born.32 This means that there would have been about 7 babies born in the SEC in 2008 with DMD. More data on the incidence of specific diseases is outlined in Appendix 3. Appendix 4 of this document provides estimates of the prevalence of neuromuscular diseases, muscular dystrophies, and Duchenne muscular dystrophy within the South East Coast. Table 3: Incidence Rates for Neuromuscular Disorders within the South East Coast by PCT (for 2008 – birth rates based on live births by Local Authority from document by Office of National Statistics)
PCT Birth Rate
Incidence of NMDs
Incidence of MDs
Incidence of DMD
Brighton and Hove 3303 1.38 0.89 0.47
East Sussex Downs and Weald 3306 1.38 0.89 0.47
Eastern and Coastal Kent 8720 3.63 2.36 1.25
Hastings and Rother 1851 0.77 0.50 0.26
Medway 3419 1.42 0.92 0.49
Surrey 13710 5.71 3.71 1.96
West Kent 9785 4.08 2.64 1.40
West Sussex 8881 3.70 2.40 1.27
4.2 Prevalence of Neuromuscular Disorders
Prevalence refers to the total number of cases of a disease in a given population at a specific time. The prevalence of NMDs in the UK in general is estimated at around 70,000 people, or 1 in 1000 people (i.e. there are 70,000 people in the UK with a type of neuromuscular disorder).
The South East Coast has a population of 4.37 million (ONS mid-year estimate, 2008), suggesting that a total of around 4,370 individuals are affected with neuromuscular disorders in the South East Coast region. For data on the prevalence of specific diseases see Appendix 3.
The number of people with neuromuscular disorders within the South East Coast is estimated to be 4370.
The number of new cases of neuromuscular disorders within the South East Coast is estimated to be 21 per year.
The number of new cases of Duchenne muscular dystrophy within the South East Coast is estimated to be 7 per year.

18
4.3 Life Expectancy and Trends in Mortality
4.3.1 Disorders of the Anterior Horn Cells
4.3.1.1 Spinal Muscular Atrophy SMA type I (the most common form), has the earliest age of onset (before 6 months) and of death, with most affected not living past their second birthday.1,33,34,35,36 SMA type II has an age of onset of between 6 and 18 months36, and people with SMA type II can live well into adolescence, with some living longer.37,38 The onset of SMA type III is after 18 months of age11, and the life expectancy is near normal.1,38,39 4.3.2 Disorders of the Peripheral Nerves
4.3.2.1 Metabolic Neuropathies
Metabolic neuropathies are by definition caused by underlying metabolic disorders, which tend to lead to a number of other complications/symptoms (such as macrovascular disease and kidney failure, for instance). It is usually one or more of these other symptoms which result in death. Hence, life expectancy for metabolic neuropathies ultimately depends upon the life expectancy for the underlying metabolic disorders. 4.3.2.2 Guillain-Barre Syndrome
The overall prognosis of GBS is good with approximately 85% of patients making a good functional recovery.40 However, prognosis worsens with older age,40,41,42 and is poorer in those with more severe weakness, rapid onset, muscle wasting, electrically inexcitable nerves and/or preceding diarrhoeal illness.40,43 In addition, Miller-Fisher syndrome has a poorer prognosis compared to other GBS subtypes.40
Most severely disabled patients with acute motor axonal neuropathy have been found to walk independently within a few years.40,44 The majority of patients with a poor outcome have been mechanically ventilated, but a mortality rate of 20% has unfortunately been demonstrated in these patients.40,45 The recovery period from severe disease may be prolonged, but most patients are eventually able to walk independently.40,45 4.3.3 Neuromuscular Transmission Disorders
4.3.3.1 Myasthenia Gravis
The majority of people with myasthenia gravis enjoy a good a quality of life and a normal lifespan as a result of advances in diagnosis and immunosuppressive treatment. However, approximately 15% to 20% of patients may experience a myasthenic crisis necessitating mechanical ventilation. Such disease exacerbations can occur due to infections, surgery, medicine exposures, malignancy, pregnancy or other stressors. Myasthenic crisis can sometimes be averted with aggressive early intervention during an exacerbation.46 In addition; those patients with a malignant thymoma will have a lesser life expectancy due to the thymoma itself, and not due to the myasthenia. Quality of life can vary depending on the severity and the cause of the condition.
4.3.3.2 Lambert-Eaton Syndrome
Prognosis of Lambert-Eaton syndrome (LES) is determined by the type of underlying cancer or the severity of the associated autoimmune disease, and the severity and distribution of weakness.47 That is, prognosis for LES associated with cancer depends upon the prognosis and likely recurrence of the cancer and is improved by effective cancer treatment,47,48 whilst prognosis for LES associated with an autoimmune disease often improves with correct administration of immunosuppressive medications,47,49 although the underlying autoimmune disorders do not generally decrease life expectancy.47
4.3.4 Disorders of the Muscles

19
4.3.4.1 Muscular Dystrophy The most common cause of morbidity and mortality for patients with muscular dystrophies is the progressive weakness of respiratory muscles.50 However, a study demonstrated that the use of the oximetry/respiratory muscle aid protocol almost entirely eliminates respiratory mortality and that those patients that were trained and equipped to non-invasively support inspiratory and expiratory muscle function instead tend to die from cardiac failure.51 Patients with muscular dystrophy can live into their mid to late 40s.52
4.3.4.2 Duchenne Muscular Dystrophy
DMD usually becomes clinically evident when a child begins walking, with age of onset being between two and six years. The symptoms include general muscle weakness and wasting, which initially affects the pelvis, upper arms, and upper legs; and eventually involves all voluntary muscles. Patients typically require a wheelchair by age 7 to 12, and used to die in their late teens or early 20s. However, with appropriate, proactive medical care, these patients now have the potential to live until their late 20s and 30s. The life expectancy for people with untreated DMD is 19 years. However, with the right treatment, life expectancy can rise above 25 years making it even more critical that any future service can deal with the additional demand on provision and the widening demographic that are accessing the service.1.32,53 A review of notes from patients with DMD over a period from 1967 to 2002 showed that the life expectancy of the cohort increased from 14.4 in the 1960s to 25.3 in the 1990s.53,54 This rise in life expectancy is mainly due to improved ventilation, such as NIPPV.54,55,56,57 Life expectancy may also be improved by long-term daily corticosteroid use.57,58
4.3.4.3 Myotonic Disorders (Myotonic Muscular Dystrophy)59
The infant mortality rate amongst babies with congenital myotonic dystrophy is 141 per 1000 births. Average life expectancy for those with the disorder is otherwise between 45 and 55 years of age.
4.3.4.4 Glycogen Storage Diseases60
GSD type I can be fatal due to the hypoglycaemia it causes although mortality is rare, particularly early on in life, as treatment is, for the most part, effective. The infantile form of GSD type II has a severely reduced life expectancy (due to a progressive cardiorespiratory defect), whereas people affected by the juvenile form can live into their thirties and those with the adult form have almost normal lifespans. People with GSD type III have a relatively normal lifespan, as do those with type VI, whilst those with type IV tend not to live past two years of age as a result of extensive cirrhosis of the liver.
4.3.4.5 Periodic Paralysis61
The hyperkalaemic form of periodic paralyses does not reduce life expectancy, however hypokalaemic periodic paralyses can reduce life expectancy as a result of complications with pneumonia or an inability to clear secretions.
4.3.4.7 Inflammatory Myopathies62
Patients with dermatomyositis generally respond to treatment, whilst some patients with polymyositis respond well and others do not respond so well, resulting in a significant disability. Inclusion body myositis tends not to respond to treatment.
4.3.4.8 Muscle Disorders Associated with Endocrine Diseases
These muscle disorders are caused by an underlying endocrine disease and so life expectancy is dependant upon the life expectancy for the particular underlying condition. The muscular problems themselves rarely cause mortality.
4.3.4.9 Other Muscles Disorders

20
Myositis Ossificans is characterised by poor prognosis, with almost total immobility being the inevitable result of disease progression. In addition, the development of thoracic insufficiency syndrome is typical and can lead to pneumonia and failure of the right side of the heart - complications which can be fatal. The average life expectancy is around 41 years of age.63
People with myotonia congenita typically lead fairly normal lives, since movement tends to return to normal following a few repetitions of the muscle. In addition, the symptoms of myotonia congenita may improve with age.64
4.4 Morbidity Morbidity refers to the degree to which a disease or condition impacts on the health of the patient. To comprehensively determine morbidity due to NMD would require longitudinal studies to detail the complications that are experienced by the patients and their impact on the quality of life. There is a paucity of such studies and instead, the usage of health services by patients with a condition is often used as a proxy for morbidity in that disorder. In NMD there is lack of information on the usage of primary and community health services. Even in the case of hospital activity, the information avaialble is limited to in-patient episodes since outpatient activity is rarely coded with diagnosis. As a result, we have a limited picture of the morbidity caused by NMD.
In the following section the in-patient activity, both elective and emergency, is analysed for NMDs.
Figure 1 shows the number of finished consultant episodes (FCE) due to NMDs in all age groups in the South East Coast. There is a gradual increase over the three years in both the elective and emergency FCEs . The numbers include FCEs where NMD was a secondary diagnosis.
Figure 2 shows the number of FCEs for patients of all ages with selected NMD diagnoses (SMA and related syndromes, systemic atrophies, hereditary and idiopathic neuropathy, myositis, myasthenis gravis and other myoneural disorders and primary disorders of muscle). In this group of patients the number of emergency FCEs is slightly higher than elective FCEs. There is gradual increase in both types of FCEs over the three years.
Figure 1: Number of Finished Consultant Episodes (both Emergency and Elective) for Patients of all ages, where the primary or secondary diagnosis is a NMD
0
500
1000
1500
2000
2500
3000
Elective Emergency Elective Emergency Elective Emergency
2006/07 2007/08 2008/09
Financial Year
Financial Year and Type of Admissions
Nu
mb
er
of
Fin
ish
ed
Co
nsu
ltan
t E
pis
od
es Surrey PCT
West Sussex PCT
Hastings and RotherPCT
East Sussex Downsand Weald PCT
Brighton and HoveCity PCT
Eastern & CoastalKent PCT
West Kent PCT
Medway PCT

21
Figure 2: Number of Finished Consultant Episodes (both Emergency and Elective) for Patients of all ages, where the primary or secondary diagnosis is a specific NMD (SMA and related syndromes, systemic atrophies, hereditary and idiopathic neuropathy, myositis, myasthenia gravis and other myoneural disorders, and primary disorders of muscles)
0
100
200
300
400
500
600
700
800
Elective Emergency Elective Emergency Elective Emergency
2006/07 2007/08 2008/09
Financial Year
Financial Year and Type of Admissions
Nu
mb
er
of
Fin
ish
ed
Co
nsu
ltan
t E
pis
od
es
Surrey PCT
West Sussex PCT
Hastings and RotherPCT
East Sussex Downs andWeald PCT
Brighton and Hove CityPCT
Eastern & Coastal KentPCT
West Kent PCT
Medway PCT
Figure 3: Number of Finished Consultant Episodes (both Emergency and Elective) for patients aged 35 and under where the primary diagnosis is a specific NMD (SMA and related syndromes, systemic atrophies, hereditary and idiopathic neuropathy, myositis, myasthenia gravis and other myoneural disorders, and primary disorders of muscles)
0
20
40
60
80
100
120
140
Elective Emergency Elective Emergency Elective Emergency
2006/07 2007/08 2008/09
Financial Year
Financial Year and Type of Admissions
Nu
mb
er
of
Fin
ish
ed
Co
ns
ult
an
t E
pis
od
es
Surrey PCT
West Sussex PCT
Hastings and RotherPCT
East Sussex Downsand Weald PCT
Brighton and HoveCity PCT
Eastern & CoastalKent PCT
West Kent PCT
Medway PCT
22
The number of FCEs from 2006/07 to 2008/09, for patients aged 35years and under with specific NMDs are shown in Figure 3. There is a higher ratio of elective to non-elective FCEs in this group.
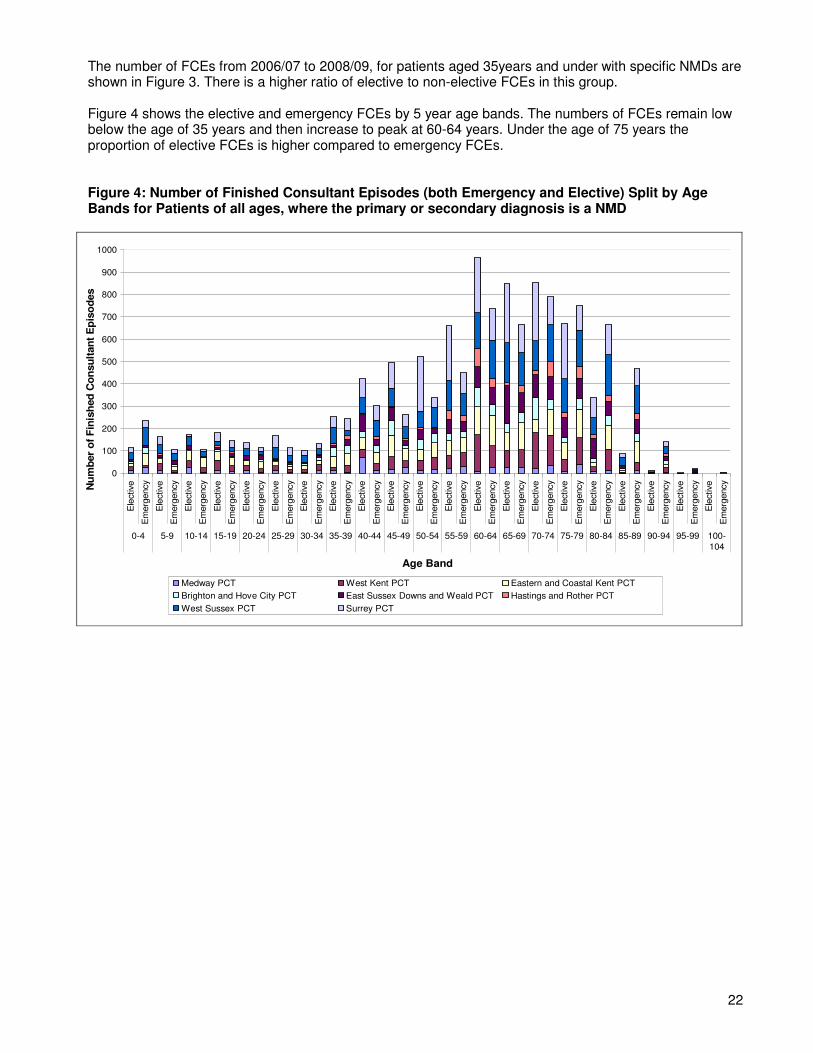
Figure 4 shows the elective and emergency FCEs by 5 year age bands. The numbers of FCEs remain low below the age of 35 years and then increase to peak at 60-64 years. Under the age of 75 years the proportion of elective FCEs is higher compared to emergency FCEs.
Figure 4: Number of Finished Consultant Episodes (both Emergency and Elective) Split by Age Bands for Patients of all ages, where the primary or secondary diagnosis is a NMD
0
100
200
300
400
500
600
700
800
900
1000
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
Ele
ctive
Em
erg
ency
0-4 5-9 10-14 15-19 20-24 25-29 30-34 35-39 40-44 45-49 50-54 55-59 60-64 65-69 70-74 75-79 80-84 85-89 90-94 95-99 100-
104
Age Band
Nu
mb
er
of
Fin
ish
ed
Co
nsu
ltan
t E
pis
od
es
Medway PCT West Kent PCT Eastern and Coastal Kent PCT
Brighton and Hove City PCT East Sussex Downs and Weald PCT Hastings and Rother PCT
West Sussex PCT Surrey PCT

23
Figure 5: Number of Finished Consultant Episodes (both Emergency and Elective) at Tertiary/Specialist and Local/Acute Providers from 2006-2009 for Patients of all ages, where the primary or secondary diagnosis is a NMD
0
1000
2000
3000
4000
5000
6000
7000
Elective Emergency Elective Emergency
Tertiary/Specialist Local/Acute
Location of Finished Consultant Episodes
Nu
mb
er
of
Fin
ish
ed
Co
nsu
ltan
t E
pis
od
es
Surrey PCT
West Sussex PCT
Hastings and RotherPCT
East Sussex Downs andWeald PCT
Brighton and Hove CityPCT
Eastern & Coastal KentPCT
West Kent PCT
Medway PCT
Figure 5 shows the number of elective and emergency FCEs for South East Coast over 3 years (2006/07 to 2008/09) for local acute NHS and tertiary/specialist hospitals. The number of both elective and emergency FCEs is significantly higher for the local acute Trusts in comparison to the tertiary providers which are all based in London. Elective FCEs are a significantly a higher proportion of the total FCEs at the tertiary /specialist providers and may indicate that these FCEs are predominantly at the early stage for establishing diagnosis. It is worth noting that the majority of elective and emergency care is provided at the local acute Trusts.
Table 4 shows the five NMDs in Kent & Medway, Surrey and Sussex that resulted in the most hospital admissions from 2006/07 to 2008/09. The most commonly presenting neuromuscular condition of all is muscular dystrophy, with other commonly diagnosed NMDs including juvenile dermatomyositis, Guillain-Barre syndrome and polyneuropathies.

24
Table 4: The Top 5 Neuromuscular Disorders (by Number of FCEs) in Kent & Medway, Surrey and Sussex for the Financial Years 06/07, 07/08 and 08/09¥
Kent & Medway Surrey Sussex
Primary Diagnosis ICD Code
Primary Diagnosis ICD Description
Primary Diagnosis ICD Code
Primary Diagnosis ICD Description
Primary Diagnosis ICD Code
Primary Diagnosis ICD Description
1 G710 Muscular dystrophy G710 Muscular dystrophy G710 Muscular dystrophy
2 M330 Juvenile
dermatomyositis R068
Other and unspecified
abnormalities of breathing
M330 Juvenile
dermatomyositis
3 G610 Guillain-Barre
syndrome M331
Other dermatomyositis
E740 Glycogen storage
disease
4 G628 Other specified
polyneuropathies G610
Guillain-Barre syndrome
G610 Guillain-Barre
syndrome
5 G629 Polyneuropathy,
unspecified M330
Juvenile dermatomyositis
G618 Other inflammatory polyneuropathies

25
5. Current services, models of care, health economic assessment, and gaps in services.
5.1 Current Services
5.1.1 Specialised Centres
There are no specialised centres for neuromuscular disorders within the South East Coast SHA area. Patients are either referred to neuromuscular clinics at the specialist centres in London (especially in more complex cases and during diagnosis); with follow-up predominantly at local neurology centres, or they attend local neurology clinics; which may or may not involve specialists from the tertiary centres (outreach clinics are available at some centres on a quarterly or half-yearly basis). An assessment of best practice standards at the tertiary/specialised centres in detailed in Appendix 6. The location of the specialist centres are shown in figure 6 (with the key for this map detailed in Appendix 7).
Specialist clinics for NMDs are provided at the tertiary centres. Details of the various clinics available for patients with subgroups of NMDs are outlined in Appendix 8. There are a number of specialist neuromuscular centres in London, which provide tertiary services to patients throughout London and the South East, as well as to some patients from further afield.
Specialised paediatric centres treating patients from the South East Coast are located at:
• Great Ormond Street Hospital (Dubowitz Neuromuscular Centre)
• Evelina Children’s Hospital
Figure 6

26
Referrals for specialised neuromuscular care for adults are made from South East Coast providers to:
• Guys and St Thomas’s Hospitals
• Kings College Hospital
• St Georges Hospital
• Hammersmith Hospital
• Charing Cross Hospital
• The National Hospital for Neurology and Neurosurgery, UCLH
• Southampton Hospital*
* - Referrals from Western Sussex Hospitals NHS Trust only. Southampton do not view themselves as a specialised provider of neuromuscular services.
5.1.2 Acute Providers Following diagnosis, patients with NMDs tend to be seen locally at general neurology clinics (with the exception of patients from the Gravesend area, who attend the specialist clinics at Kings for all their care). Details of these clinics are provided in Appendix 10, for more specific information about each acute trust see Appendix 9, and for location of each hospital please refer to figure 7 (keys in Appendix 7).
Figure 7
5.1.3 Community and Primary Care Services
In addition to hospital based services, patients with neuromuscular disorders require ongoing input from a range of community based services provided by PCT provider arms or equivalents. These include services such as physiotherapy, orthopaedics, counselling/psychological assessment, occupational therapy, speech and language therapy, dietetics, respiratory care, hydrotherapy, wheelchair services, equipment services and orthotic services.

27
Arrangements for provision of these services differ in each of the eight primary care trusts within the South East Coast. Services may be provided via the community rehabilitation/intermediate care team (e.g. at South Downs Health, Eastern and Coastal Kent PCT, West Sussex Health and Hastings and Rother PCT), or as a stand alone service (such as in East Sussex Downs and Weald PCT, Hastings and Rother PCT, Medway PCT, Surrey PCT and West Kent PCT). However, the services provided for patients with neuromuscular disorders by some PCTs (e.g. West Kent PCT) are undetermined, meaning that patients are referred straight from their GP to the acute trusts and will only receive additional care from the community services team if this is specifically requested by the patient’s hospital doctor. For a clearer picture of the services provided and gaps in provision see Appendices 11 and 12. Figure 8
5.2 Models of Care
5.2.1 National Policy and Standards of Care
Progress is being made by the clinical community in developing an agreement on standards of care for neuromuscular conditions through the TREAT-NMD initiative, which is co-ordinated from the MRC Centre for Neuromuscular Diseases in Newcastle.
Consensus has been agreed for a standard of care in Spinal Muscular Atrophy (SMA). This has been published in a document entitled Consensus Statement for Standard of Care in Spinal Muscular Atrophy65. A copy of the précis outlining the objectives, methods, and the results of the work by the International Standard of Care Committee (SCC) for SMA, which identified guidelines for standards of care is included in Appendix 13. This details the suggested diagnostic testing and care of new SMA patients, the pulmonary care of people with SMA, the appropriate GI and nutritional care, the appropriate orthopaedic and rehabilitatory care, and the palliative care of such patients.

28
The Centre for Disease Control in the United States recently co-ordinated a project – ‘Care considerations for Duchenne Muscular Dystrophy’. The project brought together 84 international experts to draw up a comprehensive set of recommendations for standards of care in Duchenne Muscular Dystrophy. The findings of this working group were published in The Lancet in November 2009. Both TREAT-NMD and the CDC led Care considerations for Duchenne Muscular Dystrophy both describe input required from a number of disciplines to meet the diverse health needs of patients with DMD. These include input from Neurologists; Gastroenterologists; Dieticians; Respiratory Physicians and Sleep Study specialists; Cardiologists; Orthopaedic Surgeons; Psychosocial Support; Physiotherapists; Occupational Therapists; Orthotists; Dentists; and equipment specialists. These are detailed in Appendix 14 which outlines a short version of their interim findings66. The National Service Framework (NSF) for Children, Young People and Maternity Services67 sets standards aimed at improving the quality of services and care available to children, young people and their families, and to reducing variations in health and social care. Standard 8 states “children and young people who are disabled or who have complex health needs receive co-ordinated, high quality child and family centred services which are based on assessed needs, promote social inclusion and, where possible, enable them and their families to live ordinary lives”. In general, the NSF aims to ensure access to seamless care pathways to the range of health and social care services based on assessed need.
The National Service Framework for Long Term Conditions68 aims to improve the lives of people living with neurological conditions by providing health and social care services. Severe inherited life limiting neuromuscular conditions are multi-system disorders that require complex long term surveillance and care to optimise quality of life and life expectancy.
Aiming High for Young People - a ten year strategy for positive activities69 highlights the importance of considering social and educational opportunities for all young people.
Better Care: Better lives - Improving outcomes and experiences for children, young people and their families living with life-limiting and life-threatening conditions70 sets out a framework for end of life care support for children. 5.2.2 Best Practice Treatment and Care A summary of best practice information provided by BMJ Evidence can be found in Appendix 15. Further best practice information for muscular dystrophies, SMA, DMD and myasthenia gravis can be found in Appendices 14, 16,17 and 18, respectively. 5.2.3 Best Practice Models of Care
Any model of care needs to reflect the importance of both timely access to specialised services, local provision through secondary care clinical teams supported by a well developed local infrastructure of health and social care together with voluntary agencies and other organisations in the Third Sector.1
Figure 9 illustrates the commissioning arrangements required to support a co-ordinated approach to Neuromuscular care. Figure 9: Diagram of tiered commissioning arrangement required to support co-ordinated care.

29
National NCG
Supra-RegionalSpecialised
Commissioning Groups
RegionalSpecialised Commissioning
Group
Local(PCT)
PracticePractice-based
commissioning
•Rare Neuromuscular Disorders
•Pompe Disease•McArdles Disease
NHS Levels of Commissioning
• Specialised care for Neuromuscular disorders
• Specialised Neuro-rehab
• Physiotherapy• Occupational Therapy
• Speech & Language Therapy
• Primary Care services
5.2.4 Muscular Dystrophy Campaign reports
The Muscular Dystrophy Campaign Report, ‘Building on the Foundations: The Need for a Specialist Neuromuscular Service across England December 2007’28 and the Muscular Dystrophy Campaign report ‘Building on the Foundations: The Need for a Neuromuscular Service Serving Patients in the NHS South East Coast Region’ (May 2009)2 detailed a set of key elements of care, which are summarised in Appendix 19. 5.2.5 The Walton Report The recommendations set out by the Walton Report on Access to Specialist Neuromuscular Care 30 produced by the All Party Parliamentary Group on Muscular Dystrophy following their inquiry in 2009 are detailed in Appendix 20.
5.3 Economics of Care There Is an overwhelming evidence base for investing in, and improving the care pathway for these conditions. This is demonstrated in various sections of this report that show that investment can achieve significantly improved outcomes, particularly in improving life expectancy, and quality of life for patients with neuromuscular conditions. The care pathway for neuromuscular conditions (in general), can be split into the following elements:
• Diagnostics, accompanied by Genetic Counselling.
• Orthopaedics for the management of scoliosis (primarily)
• Dietetic care, to assist with nutrition and swallowing difficulties
• Promoting independence and improving quality of life (i.e. rehabilitation), via orthotics, wheelchairs, physio and hydro therapies, fall prevention and occupational therapy.
• Management of osteoporosis
• Monitoring and treating respiratory and cardiac complications
• Psychological support, to enable patients and their families to develop management strategies during loss of ambulation, bereavement, diagnosis, and transition. Psychological support may also be required to manage both the behavioural side effects of corticosteroids, and disorders such as autism, OCD and ADHD, which occur more frequently in male DMD patients.

30
More effective management of this care pathway should lead to improved outcomes and value for money – i.e. effective care, provided in the most appropriate location, at the right time, by the correct professional.
It is economical for PCTs to provide specialised services within a managed clinical network, where local services receive support from specialist services in order to provide local care for people with neuromuscular conditions.
Appropriate and timely provision of multi-disciplinary care should prevent unnecessary emergency admissions. For instance, a lack of specialist physiotherapy input can lead to respiratory complications resulting in a prolonged, unplanned hospital stay. Since this is far more costly than physiotherapy support, there is a clear health economic case for providing appropriate physiotherapy in the first instance in order to prevent more resource intensive non-elective hospital admissions. Also important to note is that delayed diagnosis means that there will be a period of time where appropriate treatment is not being received, which may result in an accelerated deterioration of the patient, and a need for an increase in the intensity of interventions required in order to deal with the resulting complications. Therefore early diagnosis is key to reducing unplanned spend on neuromuscular disorders.
Table 5 shows the cost to South East Coast PCTs of the numerous emergency admissions accounted for by people with neuromuscular conditions. Table 5: Cost of Emergency (and Non-Elective) Admissions for 2008-09 - All Ages, All Diagnoses in any Diagnosis field
PCT Name Resident
Population
Prevalence of neuromuscular
conditions (1:1000)
Number of emergency admissions
Total cost of emergency admissions
Number of emergency admissions per patient
Cost of emergency admissions per patient
Medway 252,200 252 90 £259,886 0.36 £1,030
West Kent 668,100 668 252 £737,126 0.38 £1,103
Eastern & Coastal Kent 726,700 727 352 £915,143 0.48 £1,259
Brighton & Hove City 253,500 254 123 £278,608 0.49 £1,099
East Sussex Downs & Weald
331,500 332 173 £499,846 0.52 £1,508
Hastings & Rother 176,800 177 87 £263,518 0.49 £1,490
West Sussex 776,300 776 404 £1,096,030 0.52 £1,412
Surrey 1,086,200 1,086 430 £1,333,277 0.40 £1,227
Grand Total (columns 1-4), Average (columns
5&6) 4,271,300 4,271 1,911 £5,383,432 0.46 £1,260
N.B. Admissions data was extracted from SUS (Secondary Uses Service): Admitted Patient Care Finished Consultant Episodes using a query that
pulled out any record from the system in which an ICD 10 Code relating to a neuromuscular condition appeared, for patients aged 35 and under.
Data was then sorted and used to produce tables. Prevalence and population data was from the South West report1 and ONS (mid 2007 pop
estimates), respectively. The number of emergency admissions per patient was then calculated by dividing the no. of admissions by the prevalence
of NMDs, whilst the cost of emergency admissions was calculated by dividing the total cost of emergency admissions by the prevalence.
Some of the highest cost emergency admissions are those relating to cardiac problems, respiratory crisis and falls (such as those commonly experienced by patients with neuromuscular disorders). In this paper we propose that a significant proportion of these could be prevented in patients with neuromuscular disorders, meaning that spend on emergency admissions for this patient group could be greatly reduced. There is general consensus that those patients who receive regular Specialised Neuromuscular care have fewer emergency hospital admissions due to complications of their conditions.

31
Furthermore, evidence from elsewhere in the UK and internationally suggests that when best practice models of care are in place, outcomes for patients with neuromuscular disorders (i.e. life expectancy and quality of life) are greatly improved.
Hence, the cost-benefit of providing the model of care outlined in this document is two-fold, the first being an improvement in patient outcome, the second being a decrease in the amount spent by PCTs on non-elective hospital admissions over time.
5.4 Service Gap Analysis
Initial evaluation of current service provision against the best practice guidelines indicates the following gaps in neuromuscular services:
• Specialist multidisciplinary teams providing regular local clinics – There is only one local/acute provider with specialists in attendance at paediatric clinics, and only two with specialists in attendance at adult clinics, meaning that there is a distinct overall deficit in outreach clinics. These outreach clinics are not provided by multidisciplinary teams, but by only one or two clinicians.
• Although tertiary centres do support secondary and community care for patients with NMDs, the level of support provided does not seem to be uniform. This is particularly the case for paediatric services, especially when patients are referred to GOS for specialist provision.
• Not all local centres have adequate respiratory services (using techniques such as NIPPV), although most do provide at least some service for neuromuscular patients. Unfortunately, those local centres which do not provide adequate respiratory services (MTW, MFT and BSUH) seem to be located in the PCT areas which do not provide adequate community services (i.e. WKPCT, Medway PCT and B&HPCT), leading to a clear deficit in provision of respiratory services.
• There is variation in the provision of neurorehabilitation clinics based in the acute and community settings. Neurorehabilitation clinics are available at some specialist centres but these are not ideally located for most patients. In some areas there is a lack of community-based rehabilitation team to refer adolescents onto when they reach adulthood; and they may therefore be referred to acute service teams, or not receive any specific rehab services at all.
• Orthopaedic care and scoliotic monitoring are not provided throughout the SEC, and where they are available are not always provided on a regular basis. Specialist orthopaedic care is not available at all tertiary centres.
• There is variation in the cardiac screening for patients with NMDs within the SEC (where it is available, it is mostly provided in acute trusts), and where it is provided it is not necessarily carried out regularly. However, cardiac screening is mostly a matter of routine at specialist centres. Furthermore, cardiac screening for carriers of the mutated dystrophin gene seems to be severely lacking – it has been identified that carriers may instead be referred to their GP for this.
• There is access to physiotherapy in most areas of the SEC, but it has been identified that therapies in general seem to be contracting, so it is not always possible to get the necessary amount of therapy required for patients with neuromuscular diseases. Access to hydrotherapy is not so common, with many local centres and community services lacking such care. Access at specialist centres is variable, with hydrotherapy being extremely limited, but physiotherapy being far more widely available.
• There are no specialist muscle nurses or neuromuscular care coordinators in the region, either at acute trusts or within the community services. Not all specialist centres have access to specialist muscle nurses or NM care coordinators).
• There are no key workers to support transition either at acute trusts or within the community. It appears that only one tertiary centre has a key worker for transition.
• There is a clear lack of psychological support from trained psychologists at tertiary centres. The provision at acute trusts and by community services is variable across the SEC, with many areas lacking psychological support. Psychological help is instead often organised by support groups, with palliative advice alternatively being provided by CCNs (community care nurses).

32
6. Issues identified with current arrangements 6.1 National work by the Muscular Dystrophy Campaign A number of reports have been produced in recent years highlighting the issues associated with services for those affected by NMD, including three by the Muscular Dystrophy Campaign:
• ‘Building on the Foundations ‘The Need for a Specialist Neuromuscular Service across England’28
• ‘Building on the Foundations: The Need for a Neuromuscular Service Serving Patients in the NHS South East Coast Region’2
• ‘State of the Nation - the 2008 National Survey’29
• ‘Access to Specialist Neuromuscular Care: The Walton Report’30 produced by the All Party Parliamentary Group for Muscular Dystrophy.
A number of the generic issues identified are outlined below:
• Current commissioning arrangements through Primary Care Trusts are not resulting in the effective collaboration of services in every region, meaning that patients are having to rely on charitable or self funding in some areas in order to receive certain core services.28
• Neuromuscular services in many parts of the country currently fall well below a minimum acceptable level, and this compromises patient life expectancy and well being.2,30 For instance, the clinical audit data from the South West region shows that the mean age of death is 19 years of age for patients with Duchenne Muscular Dystrophy, which compares starkly with published life expectancy data showing the average age of death for similar patients in the North East region has reached 30 years of age and is extending beyond this age.30
• The UK also lags behind some European countries in the provision of specialist services, supporting independent living, and life expectancy for men with Duchenne muscular dystrophy.30
• Research infrastructure must be developed in the UK to enable trials to be conducted at additional centres and thereby to allow more patients to participate in the trials. The goal should be to develop a national network of research and trial centres alongside specialist neuromuscular clinical services in each region.30
• A named transitional coordinator should be in place for each young person with a neuromuscular condition that is moving from paediatric to adult services.30
• A lack of systematically collected data regarding the number of children and adults with a particular neuromuscular condition for the different regions in England and for England as a whole. Also, there is a lack of life expectancy for people with neuromuscular conditions.30
6.2 Specific local recommendations for the South East Coast
The Muscular Dystrophy Campaign Report which focused on the South East Coast2 set out a number of recommendations (for the South East Coast specifically), which are detailed below:
• There should be a major shift in the way services are commissioned in the South East Coast region, in line with the Department of Health’s guidance that services for patients with this group of rare conditions should be regarded as specialised and therefore subject to collaborative commissioning arrangements.
• A short life working group should be established to carry out an in-depth review of current service provision and its vulnerability in South East Coast. This review would involve families, clinicians, PCTs, and the SCG; and would bring forward proposals in autumn 2009 to secure and develop the comprehensive, multidisciplinary service for children and adults, including transition services for young people.
• Since patients with neuromuscular conditions require specialist diagnosis, treatment and on-going care, it is recommended that services should be delivered through specialised centres, which can give support and leadership to a network of local clinics. Such a

33
neuromuscular network should be established with a managed clinical network as the proposed model. Commissioners in the South West have agreed to take forward this model which has already been successful in Scotland with the Scottish Muscle Network and it also reflects the model set out in the National Definition Set for specialist neuromuscular services.
• At least four full-time Care Advisors/neuromuscular care coordinators with expertise in muscular dystrophy and related neuromuscular conditions should be established and embedded in the NHS to serve the 4,300 people in the area living with these conditions.
• The existing specialist physiotherapist (based at the tertiary centre at Kings College Hospital) and local physiotherapists across the region should be supported as part of a managed clinical network to ensure that ongoing physiotherapy is provided to all adults and children with a neuromuscular condition in each PCT, supported and developed by enhanced specialist physiotherapy support from the tertiary centres.
• Resources to be allocated to ensure that a structure is in place to ensure that vital respiratory and cardiac support can be accessed by all neuromuscular patients who require it. These are key factors in extending life.
• Psychological support is a vital element of a comprehensive service that should be provided for patients and families living with a neuromuscular condition in South East Coast. These are typically genetic conditions, meaning that families require psychological support throughout the lifetime of the condition; and particularly during key times such as diagnosis, genetic counselling, when becoming wheelchair dependent, at transition stage, prior to spinal surgery and when end of life issues need to be addressed.
6.3 Findings of hearings by the All Party Parliamentary Group on Muscular Dystrophy
In August 2009 the All Party Parliamentary Group for Muscular Dystrophy published a report entitled ‘Access to Specialist Neuromuscular Care: The Walton Report’.4 This report has been produced using information obtained from an inquiry which was launched by the All Party Parliamentary Group (APPG) in December 2008 and looked at all aspects of specialist neuromuscular care. The inquiry began with leading neuromuscular consultants giving evidence setting out the staffing and relevant specialisms required to provide patients with a comprehensive, multi-disciplinary service. In subsequent sessions, evidence was presented which highlighted the fact that neuromuscular services in many parts of the country currently fall well below a minimum acceptable level, and this compromises patient life expectancy and well being. Indeed, the clinical audit data from the South West region shows that the mean age of death at 19 years of age for patients with Duchenne Muscular Dystrophy compares starkly with published life expectancy data showing the average of death for similar patients in the North East region has reached 30 years of age and is extending beyond this age. These variances were deemed unacceptable in any decent, civilised society, and were used as evidence of service failures, to be addressed with urgency. Evidence received by the APPG for Muscular Dystrophy also shows that the UK lags behind some European countries in the provision of specialist services, support for independent living and life expectancy for men with Duchenne muscular dystrophy. The report suggested that such second-rate care results in shorter lives, and this was identified as being not acceptable. It therefore proposed a number of recommendations which call for prompt action to address the failings in services in this country, which are detailed in Appendix 20 of this report.
6.4 The Patient Perspective – views of patients and their carers.
As part of the work to inform this review, the SCG undertook a focus group with patients, their carers and families organised through the South East Coast Muscle Group. At this focus group, we sought feedback on their experiences of living with a neuromuscular disorder and of the care that they receive. A key emerging theme was that many people continue to encounter poor care, inadequate support and lack of provision or access to specialised NHS services. This is mirrored by a similar lack of co-ordination with regard to social care services provided by local authorities. Whilst this is not directly within the gift of the NHS to resolve, it is important to note that many patients and their carers see that the lack of a seamless interface between health and social care services is, at the very least, frustrating.

34
6.4.1 Diagnosis
There is no uniform or consistent availability of specialist expertise for early diagnosis. Patients have had errors or delays in diagnosis which have in some cases resulted in inappropriate treatment. The experiences reported by members of the SEC Muscle Group shows that Ambulance, Primary and Secondary Care clinicians do not always recognise the conditions, or underlying conditions, when presenting and are then unable to treat patients correctly. Particularly evident for Non-Elective episodes, the situation has resulted in some patients carrying information booklets with them. Case study: Patient 1 from Surrey is affected by limb girdle muscular dystrophy type 2b. She travels to Kings College Hospital once a year to see her consultant and physio assessment, but receives no other care. She was wrongly diagnosed and given aggressive treatments at her local hospital. She herself argued for a referral into a London hospital. Patient 2 is affected by Hereditary Body Inclusion Myopathy Type 1, a very rare neuromuscular condition. Other immediate members of her family also have the condition, and she was only diagnosed when she recently accompanied one member to their appointment with a neuromuscular specialist at Kings College Hospital.
6.4.2 Physiotherapy, Hydrotherapy and Respiratory services
Reported as previously being available across most areas, access to these services is becoming more difficult as services are withdrawn. Patients also feel that there is a de-skilling of local therapists as providers work to make services more efficient, thus they become homogenous and are less able to deliver a specialist service. Where they have physiotherapy, patients are usually limited to the number of session they receive and how often. In 2008, 40% of the PCTs and over half of the Trusts within SEC did not provide ongoing physiotherapy where required2. Less than one-third of PCTs and only half of the Trusts have specifically trained physiotherapists.
Hydrotherapy is generally not available. Patients report that they have in some cases been told to use local authority pools, however this is not appropriate. There are only two respiratory clinics within SEC.
Case Study: Patients 3 and 4 from West Sussex have Duchenne Muscular Dystrophy. Their main problem they face is lack of physiotherapy. “The lady who sees the boys is very good but she just doesn’t have time to visit us very regularly.” The family travels to Great Ormond Street for their specialist care, but finds the journey difficult: “I have previously travelled by train and bus which takes about 4 hours each way. It is impossible to travel by tube as most stations don’t have lifts. It would be much easier to go to a closer hospital.” Patient 5 from West Sussex is affected by facioscapulohumeral muscular dystrophy. She travels to the National Hospital at Queens Square, but does not receive local support. She says: “It would be really useful to have regular physio appointments rather than once or twice a year.” Patient 6 has a form of muscular dystrophy. He broke his arm when he had an accident with his wheelchair while on holiday, was flown back to the UK and taken to his local hospital in Surrey. Without specialist knowledge of his condition the local physiotherapist conducted inappropriate exercises which led to the patient losing mobility in his arm.( This could have been avoided if the local physiotherapist had contacted the specialist neuromuscular physiotherapist at Kings College Hospital. A managed clinical network would have ensured strong links between the tertiary centres and primary and secondary care so that specialist knowledge about these rare conditions is shared between clinicians. (Reported by Clinician - April 2009)

35
6.4.3 Paediatric/Adult transition Rather than being a smooth change as the patients get older, the change in services between paediatric and adult is seen as a barrier. This is in spite of the evidence that young people with muscle disease are living longer as therapies and medical care improve. Particular barriers mentioned locally are the different age criteria to access services and the lack of communication between services. Nationally, 60% of people with muscle disease rate the transition as very poor. Essential services, such as physiotherapy, are often withdrawn immediately once young people reach ages of 16 to18.
Case Study: Patient 7 from Kent was transferred to another clinic, but that clinic is hard to access and not as personal. Patient 8 had fortnightly physiotherapy sessions to monitor changes in his condition. These ended at 18 and the transition was confusing and uncoordinated. He remains reliant on children’s services 3 months after turning 18.
6.4.4 Community equipment and wheelchairs People with NMD and their carers report that access to wheelchair services and community equipment is significantly less than needed. They describe the service delivered as poor and that attempting to access appropriate services can be a battle; the effort involved resulting in significant distress. A number of people with NMD have conditions that require those giving the assessment and providing wheelchairs to have specific knowledge. This is mostly unavailable in local wheelchair services and combined with the general nature of the services, people with NMD compete with patients requiring short term use of standard equipment. This leads to delays in receiving specialist equipment, an example of which is an FOI request in 2009 that showed the average wait for children in Eastern and Coastal Kent to receive a wheelchair was 9 months. It is also reported that the equipment received is often unsuitable and that even should a specialist Occupational Therapist be involved in the chair design, the final product will meet basic needs but is not sympathetic to the persons needs. Furthermore NHS wheelchair services will only provide one chair, whereas most patients require two for use in different circumstances. As a result of these issues almost half of people with NMD fund wheelchairs themselves, or seek the assistance of a charity, which some feel degrading. The costs of individual chairs vary but can be up to £20,000 for powered wheelchairs.
Case Study: Patient 9, from Surrey, is in his twenties and is affected by Duchenne Muscular Dystrophy, a severe and life-limiting neuromuscular condition. He is a full-time wheelchair user, and has had spinal surgery. His family purchased his own chair, and he receives no physiotherapy. He visits the Oxford Muscle and Nerve Centre for specialist care. Patient 10, from Surrey, is affected by Charcot Marie Tooth disease type 1a. She is powered wheelchair user, but paid for her own chair, and receives no medical care at all.
6.4.5 Care Coordination Underlying many of these themes is a lack of co-ordination of their care. People with NMD and their carers feel that they struggle with the “system” and often do not receive the help they need from the services who are in contact with them. Sometimes these services are not aware of each other and it will be the person themselves, the parents or the carers who act as care coordinator. This lack is not solely relating to Health needs and the SEC Muscle Group expressed a significant frustration with Local Authority services including housing, OT and education.

36
Case Study: Patient 11, West Sussex, is a ten-year old with Duchenne Muscular Dystrophy. The family travel to Great Ormond Street for care but say: “it has been very difficult to get counselling for my son when he gets depressed about his condition.” Patient 12 is from Kent and was recently diagnosed with Duchenne Muscular Dystrophy. The family travel to the Evelina Children’s Hospital for specialist care. “What we have found lacking is someone who coordinates and helps you through all the various services and professionals and makes sure you have access to everyone it is appropriate to meet for health, education and grants etc.”
6.5 Views of clinician We also sought the views of clinicians about current service arrangements and asked what could be done to improve quality and productivity of services in the future. 6.5.1 Paediatric Services The consensus from clinicians is that the specialised centres are well equipped to meet the needs of the children that are referred to them and between Guy’s & St Thomas’s and Great Ormond Street are attracting referrals from across the totality of the South East Coast area. The specialised services run outreach clinics in a number of settings locally mainly for initial outpatient and confirmatory diagnoses as it is felt that follow-up clinics (at least in the early stages when investigations are required) need to be held at the London centres. There is a debate currently as to what proportion of the specialised centre MDT are needed to attend outreach clinics. There is an inherent conflict in that there is a need for these skillsets to be available at clinics however local providers are reluctant to free up staff to attend which would enable them to be trained up in order to meet the ongoing needs of these patients. As a result, the immediate need for input is drawn from the specialised centre although no skill transfer occurs. There are particular issues with regard to meeting the needs of ventilated patients locally and delayed discharges are common as a result of difficulties in repatriating these patients from the specialised centre once they are stable. The team at Guy’s and St Thomas’ highlighted that currently respiratory complications of paediatric neuromuscular disorders have to be managed elsewhere, complicating the patient pathway and providing a less joined-up service for patients and their families. They propose that this could be resolved by moving this aspect of the service from the Royal Brompton & Harefield to be co-located with the other aspects of the service which would have the effect of improving the overall quality of care. 6.5.2 Adult Services Referral patterns for Adult care are less straightforward than for paediatrics. This complicates the ability of the various clinicians involved in providing services that meet the breadth of needs for the patient in working in a co-ordinated way and communicating effectively to jointly plan care for a patient. Discussions are at an early stage with a view to establishing outreach clinics at some providers in the South East Coast health economy, however there is a reluctance by some local clinicians to engage in these proposals. There is a perception amongst clinicians that there is considerable scope to improve arrangements for patient care. In addition, there are significant gaps in arrangements for end-of-life and respite care.

37
6.5.3 Transition from Paediatric to Adult Services Where a patient transitions from GOSH or Evelina Children’s Hospital to the Adult service at GSTT, transition arrangements are in place and seem to be working. There are, however, no transition arrangements where these patients do not move to GSTT and it is feared that many of these patients fall between gaps in the system and are lost to follow-up. Transition for Duchenne Muscular Dystrophy has additional complications as there is a distinct change in the presentation of the condition at around the age that patients will be moving from paediatric to adult services with a marked deterioriation in respiratory capability between 14 and 18 and in endocrine function between 20 and 25 years old. The challenges of transition and deterioration of condition are compounded by happening concurrently with the cessation of formal education, potentially resulting in increasing social isolation and lack of peer support. There is some evidence that patients who get lost to follow-up during transition reappear as a result of emergency admission into local ITU beds, It is likely that if we improved transition arrangements that there would be a reduction in these patients, who currently get lost to follow-up, getting admitted as emergency cases.

38
7. Discussion Neuromuscular Disorders include a wide variety of diseases and conditions. They are complex in aetiology, presentation and progression. Most of the disorders do not have definitive treatments. They present with a wide range of issues for the patients and their families. The management of these conditions is challenging throughout the patient pathway; from diagnosis to on-going care, in the different health care settings.
There is limited knowledge on the incidence, prevalence and morbidity of the NMD; the information on the different types of NMD varies and in most cases the local incidence and prevalence is derived from studies undertaken elsewhere. The best estimates indicate that incidence of NMD is 21 cases per year in South East Coast and prevalence is estimated to be 4300 people with neuromuscular disorders. For the future planning and delivery of services for the NMD patients it is important that accurate local information be collected.
The information on morbidity due to NMD is limited to hospital in-patient activity. This is largely provided in the local acute hospitals in South East Coast, although the specialist expertise is confined to the tertiary/specialist centres in London. People with NMD access a wide range of services; primary care, community services, out patients and outreach. However, there is hardly any activity information on the usage of these services for NMD patients. Commissioners and the Providers are not fully aware of the range of services that are accessed by the NMD patients. Increased knowledge of morbidity due to NMD and the available services would be helpful in improving the quality of care provided for this group of patients.
Patients with NMD require specialist diagnosis and on-going management. They require services that are provided by different health care providers in different settings at different stages of the disease progression. This potentially leads to variation in the services available for NMD in different parts of south East Coast. Commissioners are also not fully aware of the services available or accessed by NMD patientsand, services should be better co-coordinated and sign-posted. This problem is compounded by the disparate nature of current services and the lack of consistency of what is offered in different settings. There is also a need for health care providers to work much more collaboratively to provide comprehensive and joined up services for NMD patients. A more consistent and structured network model of services which streamlined arrangements and improved consistency of care would be a beneficial development. There is also a strong case for the establishment of neuromuscular care coordinators to assist patients and families to appropriately access services to effectively meet their needs.
Various reports have highlighted gaps in services for NMD patients, particularly community services, i.e. specialist physiotherapy, dietetics, and speech and language therapy. Improved access to respiratory and cardiac support for NMD patients has also been indicated as a priority.
There is evidence which indicates that a significant number of emergency admissions for NMD patients could be prevented with better coordinated care in the community setting. This could also be a cost effective investment, with significant potential health gain.
In conclusion, there is much variation and gaps in services provided for patients with neuromuscular disorders. There is opportunity to improve the quality of care provided for neuromuscular disorder patients by collaborative working between the Commissioners, providers and the different health care professionals.

39
8. Recommendations
8.1 Management
• This report highlights the complex and specialised nature of services for patients with neuromuscular conditions and the relatively small population affected by these conditions across the Strategic Health Authority area. As such, we recommend that the SCG assumes responsibility for the commissioning of specialised neuromuscular services in line with the national decision to include these in the SSNDS and recommendations from the All Party Parliamentary Group that these services are commissioned at SCG level.
• The SCG Team will build on the strategic direction articulated in this document by developing a detailed service specification for these services including agreement on referral pathways, outcome measures, clinical standards and performance monitoring, together with linkages to and impacts on other neurorehabilitation services.
• The detail of service development should fit strategically with the NSF for Long Term Neurological Conditions and with the NSF for Childrens services.
8.2 Health Intelligence
• There is a clear need to improve the data available for planning purposes to better inform Commissioners in the future. Detailed plans should be developed to enhance data collection. This should include the development of a regional patient registry for neuromuscular disorders; work with the South East Public Health Observatory; and a robust mechanism for collection of local mortality data informed by analysis of death certificates.
8.3 Diagnosis
• That the SCG engage the Audit, Information and Analysis Unit to undertake work to examine to what degree appropriate and timely diagnosis of neuromuscular conditions occurs amongst South East Coast patients. The findings of this study will then inform an assesment of whether an education programme is required for primary care and community health staff who are likely to identify these patients and if so, how to target this in terms of professional groups and content.
8.4 Specialised Care
• The fragmentation and lack of consistency of specialised care is problematic in terms of forming a consistent base upon which to form the hub of an effective clincal network to better provide appropriate care for patients from the South East Coast. We recommend that the services at Great Ormond Street Hospital and Guy’s and St Thomas’s for Children and the adult service at Guy’s and St Thomas’s working in collaboration with the service at King’s College Hospital under the auspices of the King’s Health Partners Academic Health Sciences Centre are afforded preferred provider status and form the basis of a hub for a clinical network to better provide co-ordinated care for patients from the South East Coast with a neuromuscular condition.
• That all patients with a neuromuscular condition should be jointly managed by a preferred specialised centre working within a shared care framework with a local management centre.
• That patients currently receiving their care at a centre outside of these arrangements are given the opportunity to transfer into the South East Coast preferred care model.
8.5 Local Acute Care
• There is a need for local neurologists to develop closer working relationships with teams from specialised centres in order to provide more joined up ongoing care for patients as close to their homes as possible. Formal shared care arrangements through a clinical network model should be developed to optimise care for patients.
• Local services to better support ventilated patients need to be established both to minimise delayed discharges from specialised centres, but also to better support vulnerable patients closer to their homes.

40
• Consistent region-wde protocols need to be developed for the management of Emergency Care for patients with Neuromuscular Disorders. Evidence heard in the production of this report indicated that there are safety concerns with the particular needs of neuromuscular patients being overlooked when they are admitted via A&E departments, resulting in avoidable morbidity.
8.6 Co-ordinated & Consistent Care
• That defined and documented pathways are developed for patients, including for counselling and support.
• That co-ordination is enhanced to reduce fragmentation and mitigate risks that patients will “fall through the gaps” through the employment of a Care Co-ordinator role similar to that serving other SHA areas utilising monies identified in the SCG operating framework in 2010/11 for the purposes of improving co-ordination of neuromuscular care.
8.7 Rehabilitation & Therapies
• That, where they are provided or commissioned by the NHS, PCTs make hydrotherapy facilities available to patients with neuromuscular conditions on an equal basis to patients with other disabling conditions.
• That interested parties are engaged in a feasability study in relation to the possible development of a specialist neuromuscular rehabilitation centre along the same lines as the national neuromuscular centre in Nantwich, Cheshire established as a social enterprise bringing together a partnership of the NHS, voluntary and business sectors. In addition to physiotherapy, the centre could generate income from commercial activites by people with neuromuscular conditions such as graphic design. Such a model could offer more innovative day and respite care support.
• That the SCG work with the PCT commissioners of physiotherapy services to ensure that community physiotherapy services better meet the ongoing needs of this patient group rather than through inappropriate short-term input which is the standard care package in a number of PCT areas.
8.8 Specialised Aids and Adaptations
• That an in-depth review of the provision arrangements for specialist wheelchairs is undertaken with a view to a specification being developed that accurately reflects the complex needs and considerations required when asssessing for and providing appropriate chairs for neuromuscular patients.

41
9. Glossary NM neuromuscular NMD(s) neuromuscular disorder(s) MD(s) muscular dystrophy(ies) SMA(s) spinal muscular atrophy(ies) DMD Duchenne muscular dystrophy MMD myotonic muscular dystrophy PROMM proximal myotonic myopathy DM2 type 2 myotonic dystrophy MG myasthenia gravis SMN survival motor neurone (gene) SEC South East Coast SCG Specialised Commissioning Group SECSCG South East Coast Specialised Commissioning Group PCT(s) Primary Care Trust(s) Incidence the number of new cases of a disease in the population Prevalence the total number of cases of a disease in the population at a specific time Morbidity the degree to which a disease impacts the patient NIPPV non-invasive positive pressure ventilation UCLH University College London Hospitals NHS Foundation Trust NHNN National Hospital for Neurology and Neurosurgery (UCLH) ECH Evelina Children’s Hospital (GSTT) CC Charing Cross Hospital (Imperial) GSTT Guy’s and St Thomas’s Hospitals NHS Foundation Trust KCH King’s College Hospital NHS Foundation Trust BSUH Brighton and Sussex University Hospitals NHS Trust DGH District General Hospital GOSH Great Ormond Street Hospital NHS Trust MTW Maidstone and Tunbridge Wells NHS Trust MFT Medway NHS Foundation Trust WKPCT West Kent PCT B&HCPCT Brighton and Hove City PCT GP General Practitioner CCN(s) community care nurse(s) TREAT-NMD a network for people with neuromuscular diseases and professionals working in the
field GI gastro-intestinal NSF national service framework BMJ British Medical Journal NSCG National Specialised Commissioning Group NCG National Commissioning Group for Highly Specialised Services

42
10. References
1. South West Specialised Commissioning Group. Neuromuscular Service Development Strategy: A development plan for services in the South West. 2008.
2. Muscular Dystrophy Campaign. Building on the Foundations: The Need for a Neuromuscular
Service Serving Patients in the NHS South East Coast Region. 2009. 3. Medline Plus [homepage on the Internet]. Metabolic Neuropathies: Causes, Symptoms and
Treatment sections [cited 2009 Dec 30]. Available from: http://www.nlm.nih.gov/medlineplus/ency/article/001161.htm
4. BMJ Evidence Centre [homepage on the Internet]. BMJ Best Practice for Lambert-Eaton
syndrome, Summary and Definition sections [cited 2009 Dec 24]. Available from: http://bestpractice.bmj.com/best-practice/monograph/1052.html http://bestpractice.bmj.com/best-practice/monograph/1052/basics/definition.html
5. Association for Glycogen Storage Disease [homepage on the Internet]. What is GSD? [cited 2009
Dec 31]. Available from: http://www.agsdus.org/html/whatisglycogenstoragedisease.html 6. Association for Glycogen Storage Disease [homepage on the Internet]. Type 1 GSD [cited 2009
Dec 31]. Available from: http://www.agsdus.org/html/typeivongierke.htm 7. emedicine [homepage on the Internet]. Glycogen-Storage Disease Type I [cited 2010 Jan 05].
Available from: http://emedicine.medscape.com/article/949937-overview 8. Association for Glycogen Storage Disease [homepage on the Internet]. Type 2 GSD [cited 2009
Dec 31]. Available from: http://www.agsdus.org/html/typeiipompe.htm 9. Madisons Foundation [homepage on the Internet]. Pompe Disease [cited 2010 Jan 05]. Available
from: http://www.madisonsfoundation.org/index.php/component/option,com_mpower/Itemid,70/diseaseI
D,199/ 10. Association for Glycogen Storage Disease [homepage on the Internet]. Type 3 GSD [cited 2009
Dec 31]. Available from: http://www.agsdus.org/html/typeiiicori.htm 11. Association for Glycogen Storage Disease [homepage on the Internet]. Type 4 GSD [cited 2010
Jan 05]. Available from: http://www.agsdus.org/html/typeivandersen.htm 12. National Organisation for Rare Disorders [homepage on the Internet]. Andersen Disease (GSD IV)
[cited 2010 Jan 05]. Available from: http://www.rarediseases.org/search/rdbdetail_abstract.html?disname=Andersen%20Disease%20
%28GSD%20IV%29 13. Association for Glycogen Storage Disease [homepage on the Internet]. Type 5 GSD [cited 2010
Jan 05]. Available from: http://www.agsdus.org/html/typevmcardle.htm 14. NCBI [homepage on the Internet]. Glycogen Storage Disease V [cited 2010 Jan 05]. Available
from: http://www.ncbi.nlm.nih.gov/entrez/dispomim.cgi?id=232600

43
15. National Organisation for Rare Disorders [homepage on the Internet]. Hers Disease [cited 2010
Jan 05]. Available from: http://www.rarediseases.org/search/rdbdetail_abstract.html?disname=Hers%20Disease 16. Association for Glycogen Storage Disease [homepage on the Internet]. Type 6 GSD [cited 2010
Jan 05]. Available from: http://www.agsdus.org/html/typevihers.htm 17. Madisons Foundation [homepage on the Internet]. Glycogen-Storage Disease Type VI [cited 2010
Jan 05]. Available from: http://www.madisonsfoundation.org/index.php/component/option,com_mpower/Itemid,70/diseaseI
D,582/ 18. Association for Glycogen Storage Disease [homepage on the Internet]. Type 7 GSD [cited 2010
Jan 05]. Available from: http://www.agsdus.org/html/typeviitarui.htm 19. Madisons Foundation [homepage on the Internet]. Tarui Disease [cited 2010 Jan 05]. Available
from: http://www.madisonsfoundation.org/index.php/component/option,com_mpower/Itemid,70/diseaseI
D,593/ 20. Association for Glycogen Storage Disease [homepage on the Internet]. Type 9 GSD [cited 2010
Jan 05]. Available from: http://www.agsdus.org/html/typeix.htm 21. Association for Glycogen Storage Disease [homepage on the Internet]. Type 0 GSD [cited 2010
Jan 05]. Available from: http://www.agsdus.org/html/type0.html 22. Periodic Paralysis News Desk [homepage on the Internet]. General Periodic Paralysis FAQ [cited
2009 Dec 30]. Available from: http://www.hkpp.org/faq/periodic_paralysis.html 23. National Institute of Neurological Disorders and Stroke [homepage on the Internet]. Inflammatory
Myopathies Fact Sheet [cited 2009 Dec 30]. Available from: http://www.ninds.nih.gov/disorders/inflammatory_myopathies/detail_inflammatory_myopathies.htm 24. emedicine [homepage on the Internet]. Endocrine Myopathies [cited 2009 Dec 30]. Available from: http://emedicine.medscape.com/article/1170469-overview 25. Medline Plus [homepage on the Internet]. Myotonia Congenita [cited 2010 Jan 05]. Available from: http://www.nlm.nih.gov/medlineplus/ency/article/001424.htm 26. Genetics Home Reference [homepage on the Internet]. Myotonia Congenita [cited 2010 Jan 05].
Available from: http://ghr.nlm.nih.gov/condition=myotoniacongenita
27. emedicine [homepage on the Internet]. Fibrodysplasia Ossificans Progressiva (Myositis
Ossificans) [cited 2010 Jan 05]. Available from: http://emedicine.medscape.com/article/1007104-overview
28. Muscular Dystrophy Campaign. Building on Foundations: The Need for a Specialist
Neuromuscular Service Across England. 2007. 29. Muscular Dystrophy Campaign. State of the Nation - the 2008 National Survey. 2008. 30. All Party Parliamentary Group for Muscular Dystrophy. Access to Specialist Neuromuscular Care:
The Walton Report. 2009.

44
31. Scherer K, Bedlack RS, Simel DL. Does this patient have myasthenia gravis? JAMA.
2005;293(15):1906–14. 32. Iannaccone ST, Browne RH, Samaha FJ, Buncher CR. Prospective study of spinal muscular
atrophy before age 6 years. DCN/SMA Group. Pediatric Neurology. 1993;9(3):187–93. 33. Thomas NH, Dubowitz V. The natural history of type I (severe) spinal muscular atrophy.
NeuromuscularDisorders 1994;4(5-6):497–502. 34. Cobben JM, Lemmink HH, Snoeck I, Barth PA, van der Lee JH, de Visser M. Survival in SMA type
I: a prospective analysis of 34 consecutive cases. Neuromuscular Disorders 2008;18(7):541–44. 35. Bosboom WMJ, Vrancken AFJE, van den Berg LH, Wokke JHJ, Iannaccone ST. Drug treatment
for spinal muscular atrophy types II and III (Review). Cochrane Database of Systematic Reviews, Issue 3, 2009.
36. Russman BS, Buncher CR, White M, Samaha FJ, Lannaccone ST. Function changes in spinal
muscular atrophy II and III. The DCN/SMA Group. Neurology 1996;47(4):973–6. 37. Zerres K, Rudnik-Schoneborn S, Forrest E, Lusakowska A, Borkowska J, Hausmanowa-
Petrusewicz I. A collaborative study on the natural history of childhood and juvenile onset proximal spinal muscular atrophy (type II and III SMA): 569 patients. Journal of the Neurological Sciences 1997;146(1):67–72.
38. Russman BS, Iannacone ST, Buncher CR, Samaha FJ, White M, Perkins B, et al. Spinalmuscular
atrophy: new thoughts on the pathogenesis and classification schema. Journal of Child Neurology 1992;7(4):347–53.
39. BMJ Evidence Centre [homepage on the Internet]. BMJ Best Practice for Muscular Dystrophies,
Prognosis section [cited 2009 Sept 29]. Available from: http://bestpractice.bmj.com/best-practice/monograph/969/follow-up/prognosis.html
40. BMJ Evidence Centre [homepage on the Internet]. BMJ Best Practice for Guillain-Barre syndrome,
Prognosis Details [cited 2009 Dec 24]. Available from: http://bestpractice.bmj.com/best-practice/monograph/176/follow-up/prognosis.html 41. Hadden RD, Karch H, Hartung HP, et al. Preceding infections, immune factors, and outcome in
Guillain-Barre syndrome. Neurology. 2001;56:758-765. 42. Guillain-Barre Syndrome. Study Group. Guillain-Barre syndrome: an Italian multicentre case-
control study. Guillain-Barre Syndrome Study Group. Neurol Sci. 2000;21:229-234. 43. Hadden RD, Hughes RA. Management of inflammatory neuropathies. J Neurol Neurosurg
Psychiatry. 2003;74(suppl 2):ii9-ii14. 44. Hiraga A, Mori M, Ogawara K, et al. Recovery patterns and long term prognosis for axonal
Guillain--Barre syndrome. J Neurol Neurosurg Psychiatry. 2005;76:719-722. 45. Fletcher DD, Lawn ND, Wolter TD, et al. Long-term outcome in patients with GBS requiring
mechanical ventilation. Neurology. 2000;54:2311-2315. 46. BMJ Evidence Centre [homepage on the Internet]. BMJ Best Practice for Myasthenia Gravis,
Prognosis section [cited 2009 Sept 29]. Available from: http://bestpractice.bmj.com/best-practice/monograph/238/follow-up/prognosis.html
47. BMJ Evidence Centre [homepage on the Internet]. BMJ Best Practice for Lambert-Eaton
syndrome, Prognosis Details [cited 2009 Dec 24]. Available from: http://bestpractice.bmj.com/best-practice/monograph/1052/follow-up/prognosis.html

45
48. Chalk CH, Murray NM, Newsom-Davis J, et al. Response of the Lambert-Eaton myasthenic syndrome to treatment of associated small-cell lung carcinoma. Neurology. 1990;40:1552-1556.
49. Maddison P, Lang B, Mills K, et al. Long term outcome in Lambert-Eaton myasthenic syndrome
without lung cancer. J Neurol Neurosurg Psychiatry. 2001;70:212-217. 50. Gomez-Merino E, Bach JR. Duchenne muscular dystrophy: prolongation of life by noninvasive
ventilation and mechanically assisted coughing. Am J Phys Med Rehabil. 2002;81:411-415. 51. Bach JR. A comparison of long-term ventilatory support alternatives from the perspective of the
patient and care giver. Chest. 1993;104:1702-1706. 52. Bushby, K, Bourkeb J, Bullockc R, Eaglea M, Gibsond M, Quinbyd J. The multidisciplinary
management of Duchenne muscular dystrophy. Current Paediatrics 2005;15:292-300. 53. Health and Care Partnerships Analysis, Department of Health. Palliative Care Statistics for
Children and Young Adults. 2007. pp.11. 54. Eagle M, Baudouin SV, Chandler C, Giddings DR, Bullock R, Bushby K. Survival in Duchenne
muscular dystrophy: improvements in life expectancy since 1967 and the impact of home nocturnal ventilation. Neuromuscular Disorders. 2002;2(10):926-929.
55. Eagle M, Bourke J, Bullock R, Gibson M, Mehta J, Giddings D, et al. Managing Duchenne
muscular dystrophy--the additive effect of spinal surgery and home nocturnal ventilation in improving survival. Neuromuscul Disord 2007;17:470-5.
56. Konagaya M, Sakai M, Wakayama T, Kimura S, Kuru S, Yasuma F. Effect of intermittent positive
pressure ventilation on life-span and causes of death in Duchenne muscular dystrophy. Rinsho Shinkeigaku 2005;45:643-6.
57. Yiu E and Kornberg A. Duchenne muscular dystrophy. Neurology India. 2008;56 (3):236-46. 58. Biggar WD, Harris VA, Eliasoph L, Alman B. Long-term benefits of deflazacort treatment for boys
with Duchenne muscular dystrophy in their second decade. Neuromuscul Disord. 2006;16:249-55. 59. Medical News Today [homepage on the Internet]. Myotonic Muscular Dystrophy : Another Step
Toward a First Treatment [cited 2010 Jan 13]. Available from: http://www.medicalnewstoday.com/articles/32531.php 60. emedicine [homepage on the Internet]. Glycogen Storage Diseases Types I-VII [cited 2010 Jan
13]. Available from: http://emedicine.medscape.com/article/1116574-overview 61. emedicine [homepage on the Internet]. Periodic Paralyses: Follow-up, prognosis section [cited
2010 Jan 13]. Available from: http://emedicine.medscape.com/article/1171678-followup 62. National Institute of Neurological Disorders and Stroke [homepage on the Internet]. Inflammatory
Myopathies Factsheet, What is the prognosis for these diseases? [cited 2010 Jan 13]. Available from:
http://www.ninds.nih.gov/disorders/inflammatory_myopathies/detail_inflammatory_myopathies.htm#1331527280
63. emedicine [homepage on the Internet]. Fibrodysplasia Ossificans Progressiva (Myositis
Ossificans): Follow-up, Prognosis section [cited 2010 Jan 15]. Available from: http://emedicine.medscape.com/article/1007104-followup 64. Medline Plus [homepage on the Internet]. Myotonia Congenita, Outlook (prognosis) section [cited
2010 Jan 15]. Available from http://www.nlm.nih.gov/medlineplus/ency/article/001424.htm

46
65. Wang CH, Finkel RS, Bertini ES, Schroth M, Simonds A, Wong B, et al. Consensus Statement for
Standard of Care in Spinal Muscular Atrophy. J Child Neurology 2007; 22: 1027. 66. TREAT-NMD. Standards of care for Duchenne muscular dystrophy: Brief TREAT-NMD
recommendations. 2007. 67. Department of Health. National Service Framework for Children, Young People and Maternity
Services. 2004; (particularly) Standard 8. 68. Department of Health. The National Service Framework for Long Term Conditions. 2005. 69. HM Treasury. Aiming High for Young People - a ten year strategy for positive activities. 2007. 70. Department of Health. Better Care: Better lives - Improving outcomes and experiences for
children, young people and their families living with life-limiting and life-threatening conditions. 2008.
71. BMJ Evidence Centre [homepage on the Internet]. BMJ Best Practice for Muscular Dystrophies,
Step by Step Treatment Details [cited 2009 Sept 28]. Available from: http://bestpractice.bmj.com/best-practice/monograph/969/treatment/step-by-step.html
72. Biggar WD, Politano L, Harris VA, et al. Deflazacort in Duchenne muscular dystrophy: a
comparison of two different protocols. Neuromuscul Disord. 2004;14:476-482. 73. King WM, Ruttencutter R, Nagaraja HN, et al. Orthopedic outcomes of long-term daily
corticosteroid treatment in Duchenne muscular dystrophy. Neurology. 2007;68:1607-1613. 74. Bach JR, McKeon J. Orthopedic surgery and rehabilitation for the prolongation of brace-free
ambulation of patients with Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1991;70:323-331.
75. Bach JR, Valenza J, Ogin S. Stage 2: stage of loss of ambulation. In: Bach JR. Management of patients with neuromuscular disease. Philadelphia, PA: Hanley & Belfus; 2004:125-154.
76. Bach JR, Zeelenberg AP, Winter C. Wheelchair-mounted robot manipulators: long term use by
patients with Duchenne muscular dystrophy. Am J Phys Med Rehabil. 1990;69:55-59. 77. Bach JR. Stage 1: the ambulatory stage. In: Bach JR. Management of patients with
neuromuscular disease. Philadelphia, PA: Hanley & Belfus; 2004:107-124. 78. Brown JC, Zeller JL, Swank SM, et al. Surgical and functional results of spine fusion in spinal
muscular atrophy. Spine. 1989;14:763-770. 79. Ramirez N, Richards BS, Warren PD, et al. Complications after posterior spinal fusion in
Duchenne's muscular dystrophy. J Pediatr Orthop. 1997;17:109-114. 80. McDonald CM, Abresch RT, Carter GT, et al. Profiles of neuromuscular diseases: Duchenne
muscular dystrophy. Am J Phys Med Rehabil. 1995;74:70-92. 81. Rideau Y, Glorion B, Delaubier A, et al. Treatment of scoliosis in Duchenne muscular dystrophy.
Muscle Nerve. 1984;7:281-286.
82. Bach JR. Update and perspectives on noninvasive respiratory muscle aids. Part 1: the inspiratory aids. Chest. 1994;105:1230-1240.
83. Bach JR, Alba AS. Management of chronic alveolar hypoventilation by nasal ventilation. Chest.
1990;97:52-57.

47
84. Bach JR, Alba AS, Saporito LR. Intermittent positive pressure ventilation via the mouth as an alternative to tracheostomy for 257 ventilator users. Chest. 1993;103:174-182.
85. Gomez-Merino E, Bach JR. Duchenne muscular dystrophy: prolongation of life by noninvasive
ventilation and mechanically assisted coughing. Am J Phys Med Rehabil. 2002; 81:411-415. 86. Bach JR. Update and perspectives on noninvasive respiratory muscle aids. Part 2: the expiratory
aids. Chest. 1994;105:1538-1544. 87. BMJ Evidence Centre [homepage on the Internet]. BMJ Best Practice for Myasthenia Gravis, Step
by Step Treatment Details [cited 2009 Sept 28]. Available from: http://bestpractice.bmj.com/best-practice/monograph/238/treatment/step-by-step.html
88. Benatar M, Kaminski H. Medical and surgical treatment for ocular myasthenia. Cochrane
Database Syst Rev, Issue 2. 2006. 89. Schneider-Gold C, Gajdos P, Toyka KV, et al. Corticosteroids for myasthenia gravis. Cochrane
Database Syst Rev, Issue 2. 2005. 90. Hart IK, Sathasivam S, Sharshar T. Immunosuppressive agents for myasthenia gravis. In: The
Cochrane Library, Issue 4. 2007. Chichester, UK: John Wiley and Sons Ltd. 91. Gronseth GS, Barohn RJ. Practice parameter: thymectomy for autoimmune myasthenia gravis (an
evidence-based review): report of the Quality Standards Subcommittee of the American Academy of Neurology. Neurology. 2000;55:7-15. http://www.neurology.org/ (last accessed 12 December 2008).
92. Bershad EM, Feen ES, Suarez JI. Myasthenia gravis crisis. South Med J. 2008;101:63-69. 93. Gajdos P, Chevret S, Toyka K. Intravenous immunoglobulin for myasthenia gravis. Cochrane
Database Syst Rev, Issue 1. 2008. 94. Ponseti JM, Gamez J, Azem J, et al. Tacrolimus for myasthenia gravis: a clinical study of 212
patients. Ann N Y Acad Sci. 2008;1132:254-263. 95. Zinman L, Ng E, Bril V. IV immunoglobulin in patients with myasthenia gravis: a randomized
controlled trial. Neurology. 2007; 68:837-841. 96. Manzur AY, et al. Glucocorticoid corticosteroids for Duchenne muscular dystrophy. Cochrane
Systematic Review. 2008. 97. Cheuk DKL, et al. Surgery for scoliosis in Duchenne muscular dystrophy. Cochrane Systematic
Review. 2007.

48
10. Appendices Appendix 1: ICD 10 Codes Relating to Neuromuscular Conditions
G12 Spinal muscular atrophy and related syndromes
G12.0 Infantile spinal muscular atrophy, type I [Werdnig-Hoffman] G12.1 Other inherited spinal muscular atrophy
Progressive bulbar palsy of childhood [Fazio-Londe] Spinal muscular atrophy:
· adult form · childhood form, type II · distal · juvenile form, type III [Kugelberg-Welander] · scapuloperoneal form
G12.8 Other spinal muscular atrophies and related syndromes G12.9 Spinal muscular atrophy, unspecified
G13* Systemic atrophies primarily affecting central nervous system in diseases classified elsewhere
G13.0* Paraneoplastic neuromyopathy and neuropathy Carcinomatous neuromyopathy (C00-C97+) Sensorial paraneoplastic neuropathy [Denny Brown] (C00-D48+)
G60 Hereditary and idiopathic neuropathy
G60.0 Hereditary motor and sensory neuropathy Disease:
· Charcot-Marie-Tooth · Déjerine-Sottas
Hereditary motor and sensory neuropathy, types I-IV Hypertrophic neuropathy of infancy Peroneal muscular atrophy (axonal type) (hypertrophic type) Roussy-Lévy syndrome
G60.1 Refsum's disease G60.2 Neuropathy in association with hereditary ataxia G60.3 Idiopathic progressive neuropathy
G61 Inflammatory polyneuropathy
G61.0 Guillain-Barré syndrome Acute (post-) infective polyneuritis
G61.1 Serum neuropathy Use additional external cause code (Chapter XX), if desired, to identify cause.
G61.8 Other inflammatory polyneuropathies G61.9 Inflammatory polyneuropathy, unspecified
G62 Other polyneuropathies
G62.0 Drug-induced polyneuropathy Use additional external cause code (Chapter XX), if desired, to identify drug.
G62.1 Alcoholic polyneuropathy G62.2 Polyneuropathy due to other toxic agents
Use additional external cause code (Chapter XX), if desired, to identify toxic agent.
G62.8 Other specified polyneuropathies Radiation-induced polyneuropathy Use additional external cause code (Chapter XX), if desired, to identify cause.
G62.9 Polyneuropathy, unspecified Neuropathy NOS
G63* Polyneuropathy in diseases classified elsewhere

49
G63.0* Polyneuropathy in infectious and parasitic diseases classified elsewhere Polyneuropathy (in):
· diphtheria ( A36.8+ ) · infectious mononucleosis ( B27.-+ ) · leprosy ( A30.-+ ) · Lyme disease (A69.2+) · mumps ( B26.8+ ) · postherpetic ( B02.2+ ) · syphilis, late ( A52.1+ ) · congenital syphilis, late ( A50.4+ ) · tuberculous ( A17.8+ )
G63.1* Polyneuropathy in neoplastic disease (C00-D48+) G63.2* Diabetic polyneuropathy (E10-E14+ with common fourth character .4)
G63.3* Polyneuropathy in other endocrine and metabolic diseases ( E00-E07+ , E15-E16+ , E20-E34+ , E70-E89+ )
G63.4* Polyneuropathy in nutritional deficiency ( E40-E64+ ) G63.5* Polyneuropathy in systemic connective tissue disorders (M30-M35+) G63.6* Polyneuropathy in other musculoskeletal disorders (M00-M25+, M40-M96+) G63.8* Polyneuropathy in other diseases classified elsewhere
Uraemic neuropathy (N18.8+) G70 Myasthenia gravis and other myoneural disorders
Excludes: botulism (A05.1) and transient neonatal myasthenia gravis (P94.0) G70.0 Myasthenia gravis
Use additional external cause code (Chapter XX), if desired, to identify drug, if drug-induced.
G70.1 Toxic myoneural disorders Use additional external cause code (Chapter XX), if desired, to identify toxic agent.
G70.2 Congenital and developmental myasthenia G70.8 Other specified myoneural disorders G70.9 Myoneural disorder, unspecified
G71 Primary disorders of muscles
Excludes: arthrogryposis multiplex congenita (Q74.3), metabolic disorders (E70-E90), myositis (M60.-) G71.0 Muscular dystrophy
Muscular dystrophy: · autosomal recessive, resembling Duchenne or Becker · benign [Becker] · benign scapuloperoneal with early contractures [Emery-Dreifuss] · distal · facioscapulohumeral · limb-girdle · ocular · oculopharyngeal · scapuloperoneal · severe [Duchenne]
Excludes: congenital muscular dystrophy: · NOS (G71.2) · with specific morphological abnormalities of the muscle fibre (G71.2)
G71.1 Myotonic disorders Dystrophia myotonica [Steinert] Myotonia:
· chondrodystrophic · drug-induced · symptomatic
Myotonia congenita: · NOS · dominant [Thomsen]

50
· recessive [Becker] Neuromyotonia [Isaacs] Paramyotonia congenita Pseudomyotonia Use additional external cause code (Chapter XX), if desired, to identify drug, if drug-induced.
G71.2 Congenital myopathies Congenital muscular dystrophy:
· NOS · with specific morphological abnormalities of the muscle fibre
Disease: · central core · minicore · multicore
Fibre-type disproportion Myopathy:
· myotubular (centronuclear) · nemaline
G71.3 Mitochondrial myopathy, not elsewhere classified G71.8 Other primary disorders of muscles G71.9 Primary disorder of muscle, unspecified Hereditary myopathy NOS
G72 Other myopathies
Excludes: arthrogryposis multiplex congenita (Q74.3) dermatopolymyositis ( M33.- ) ischaemic infarction of muscle ( M62.2 ) myositis ( M60.- ) polymyositis ( M33.2 )
G72.0 Drug-induced myopathy Use additional external cause code (Chapter XX), if desired, to identify drug.
G72.1 Alcoholic myopathy G72.2 Myopathy due to other toxic agents
Use additional external cause code (Chapter XX), if desired, to identify toxic agent.
G72.3 Periodic paralysis Periodic paralysis (familial):
· hyperkalaemic · hypokalaemic · myotonic · normokalaemic
G72.4 Inflammatory myopathy, not elsewhere classified G72.8 Other specified myopathies G72.9 Myopathy, unspecified
M33 Dermatopolymyositis
M33.0 Juvenile dermatomyositis M33.1 Other dermatomyositis M33.2 Polymyositis M33.9 Dermatopolymyositis, unspecified
M60 Myositis
M60.0 Infective myositis Tropical pyomyositis Use additional code (B95-B97), if desired, to identify infectious agent.
M60.1 Interstitial myositis M60.2 Foreign body granuloma of soft tissue, not elsewhere classified
Excludes: foreign body granuloma of skin and subcutaneous tissue (L92.3) M60.8 Other myositis M60.9 Myositis, unspecified

51
M61 Calcification and ossification of muscle M61.0 Myositis ossificans traumatica M61.1 Myositis ossificans progressiva
Fibrodysplasia ossificans progressiva M61.2 Paralytic calcification and ossification of muscle
Myositis ossificans associated with quadriplegia or paraplegia M61.3 Calcification and ossification of muscles associated with burns
Myositis ossificans associated with burns M61.4 Other calcification of muscle
Excludes: calcific tendinitis (M65.2) of shoulder ( M75.3 )
M61.5 Other ossification of muscle M61.9 Calcification and ossification of muscle, unspecified
G73* Disorders of myoneural junction and muscle in diseases classified elsewhere
G73.0* Myasthenic syndromes in endocrine diseases Myasthenic syndromes in:
· diabetic amyotrophy ( E10-E14+ with common fourth character .4) · thyrotoxicosis [hyperthyroidism] ( E05.-+ )
G73.1* Eaton-Lambert syndrome (C80+) G73.2* Other myasthenic syndromes in neoplastic disease (C00-D48+) G73.3* Myasthenic syndromes in other diseases classified elsewhere G73.4* Myopathy in infectious and parasitic diseases classified elsewhere G73.5* Myopathy in endocrine diseases
Myopathy in: · hyperparathyroidism ( E21.0-E21.3+ ) · hypoparathyroidism ( E20.-+ )
Thyrotoxic myopathy (E05.-+) G73.6* Myopathy in metabolic diseases
Myopathy in: · glycogen storage disease ( E74.0+ ) · lipid storage disorders ( E75.-+ )
G73.7* Myopathy in other diseases classified elsewhere Myopathy in:
· rheumatoid arthritis ( M05-M06+ ) · scleroderma ( M34.8+ ) · sicca syndrome [Sjögren] ( M35.0+ ) · systemic lupus erythematosus ( M32.1+ )
E74.0 Glycogen storage disease
Cardiac glycogenosis Disease:
· Andersen · Cori · Forbes · Hers · McArdle · Pompe · Tauri · von Gierke
Liver phosphorylase deficiency
Appendix 2: Sub-classification of Spinal Muscular Atrophy

52
Group Name General age of onset
Description
Infantile SMA
Type I or Werdnig-Hoffmann disease
0-6 months
SMA Type I, also known as severe infantile SMA or Werdnig Hoffmann disease, is the most severe, and manifests in the first year of life. This type generally onsets quickly and unexpectedly after birth; babies diagnosed with Type I SMA do not generally live past one year of age. Pneumonia is considered the ultimate cause of death due to deterioration of survival motor neurons; motor neuron death causes insufficient functioning of the major bodily organ systems, particularly respiratory (e.g. breathing, ridding of pooled secretions inside lungs).
Intermediate SMA
Type II 7-18 months
Type II SMA, or intermediate SMA, describes those children who are never able to stand and walk, but who are able to maintain a sitting position at least some time in their life. The onset of weakness is usually recognized some time between 6 and 18 months. Weakness slowly and gradually increases over the life of the individual.
Adult SMA
Type III or Kugelberg-Welander disease
>18 months
SMA Type III describes those who are able to walk at some time.

53
Appendix 3: The Incidence and Prevalence of Neuromuscular Diseases
Disease Incidence Prevalence Reference
Neuromuscular diseases (in general)
1 in every 2,400 births
1 1 in 1,000 people2
1Calculated from the individual
incidence figures from this table; 2Taken from Table 2 in the
Muscular Dystrophy Campaign document on the Incidence and Prevalence of Neuromuscular Conditions, which combines the patient numbers for all the separate muscular dystrophies and related neuromuscular conditions, resulting in a total prevalence of 70,000. This equates to about 0.1% of the population of the United Kingdom (which is 60,512,000 according to table 1 of this document), leading to an expected prevalence of 1 in 1000 people.
Muscular dystrophies (in general)
1 in every 3,700 births
1
Estimated 8,000-10,000 people (in UK)
2
1Calculated from the individual
incidence figures from this table; 2Norwood et al., 2009; Mostacciuolo
et al., 1996
Duchenne muscular dystrophy
1 in every 3,500 male births
Bushby et al., 2005
Myotonic disorders Estimated 7,500 people (in UK)
Hilton-Jones, 2009
Myotonic dystrophy (DM1)
1 in every 8,000 births
1 2-14 per 100,000 people
2
1Larkin and Fardaei, 2001;
2Rüegg
et al., 2005
Becker muscular dystrophy
1 in every 30,000 male births
Map of Medicine, US data
Oculopharyngeal muscular dystrophy
1 per 100,000 people Rüegg et al., 2005
Spinal muscular atrophies
1 in 6,000 to 10,000
1
Estimated 3,000 people (in UK)
2
1Cobben et al., 2001; Nicole et al.,
2002; Bosboom et al., 2009; 2Reilly,
2009; Dalakas, 2006
Adult onset spinal muscular atrophy
0.32 per 100,000 people
Rüegg et al., 2005
Spinal bulbar muscular atrophy
2.5 per 100,000 people
Rüegg et al., 2005
Inflammatory and autoimmune neuropathies
Estimated 6,400 people (in UK)
Connor, 2008
Guillain-Barre syndrome
1.3 new cases per 100,000 people per annum
Kuwabara, 2004
Disorders of the neuromuscular junction
Estimated 10,500 people (in UK)
Buckley, 2008; Hilton Jones, 2009

54
Myasthenia gravis 0.25-20 new cases per 1,000,000 people per annum
1
1 in every 5,000 people
2
1Pekmezovic et al., 2006;
2Juel and
Massey, 2007; Phillips, 2003
X-linked myotubular myopathy
2 in every 100,000 male births
Jungbluth et al., 2008
Eaton-Lambert myasthenic syndrome
0.5 per 100,000 people
Rüegg et al., 2005
Congenital myopathies Estimated 1,000 people (in UK)
Jungblut(H?), 2009?; Darin and Tulinius, 2000; Hughes et al., 1996; Turnbull, 2009
Proximal myotonic myopathy
2-14 per 100,000 people
Rüegg et al., 2005
Distal myopathies Estimated 300 people (in UK)
Norwood et al., 2009
Mitochondrial myopathies
Estimated 3,500 people (in UK)
Quinlivan, 2009
Metabolic myopathies Estimated 700 people (in UK)
Dalakas, 2006
Periodic paralysis (channelopathies)
Estimated 900 people (in UK)
Cusin, 2003; Muntoni, 2009
Myositis (in general) Estimated 5,000 to 6,000 people (in UK)
Martyn et al., 1997; Muntoni, 2009
Inclusion body myositis 0.5-1 per 100,000 people
Rüegg et al., 2005
Juvenile dermatomyositis
4 new cases per 1,000,000 people per annum
Sell-Salazar, 2002
Charcot-Marie Tooth 1 in every 2,500 people
Kochanski, 2005
Myositis ossificans progressiva (MOP)
Estimated 60 people (in UK)
Connor, 2008; Muntoni, 2009
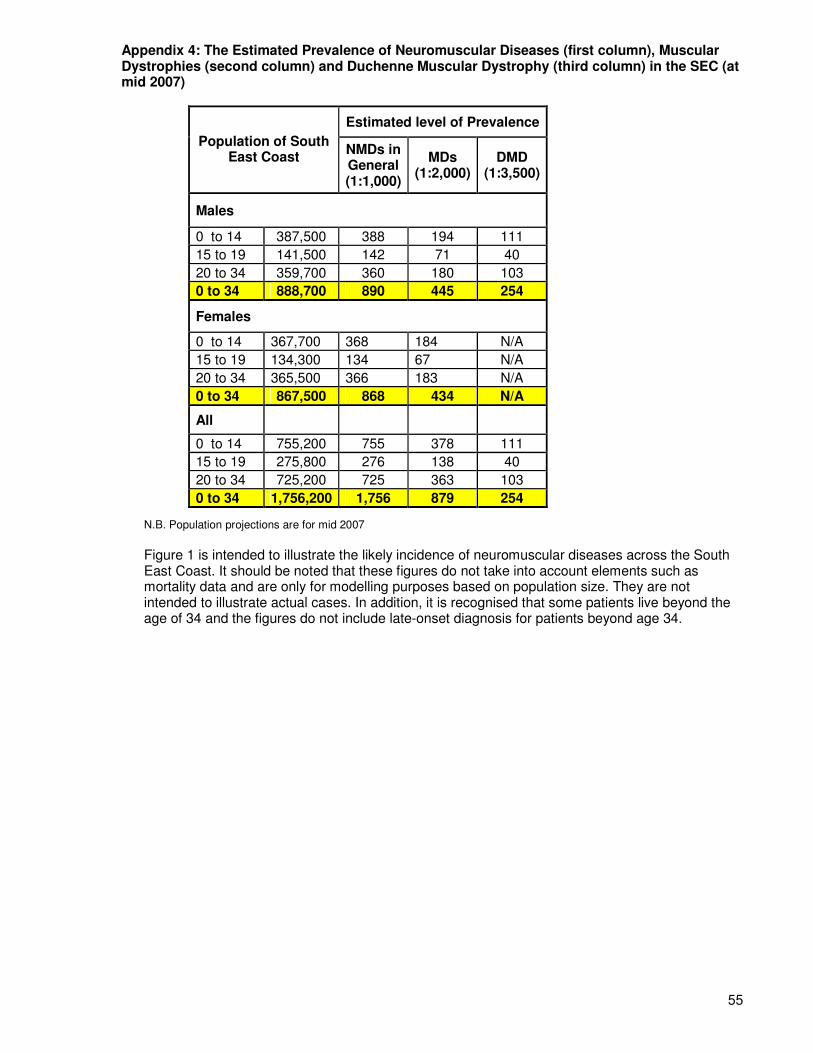
55
Appendix 4: The Estimated Prevalence of Neuromuscular Diseases (first column), Muscular Dystrophies (second column) and Duchenne Muscular Dystrophy (third column) in the SEC (at mid 2007)
Estimated level of Prevalence
Population of South East Coast
NMDs in General (1:1,000)
MDs (1:2,000)
DMD (1:3,500)
Males
0 to 14 387,500 388 194 111
15 to 19 141,500 142 71 40
20 to 34 359,700 360 180 103
0 to 34 888,700 890 445 254
Females
0 to 14 367,700 368 184 N/A
15 to 19 134,300 134 67 N/A
20 to 34 365,500 366 183 N/A
0 to 34 867,500 868 434 N/A
All
0 to 14 755,200 755 378 111
15 to 19 275,800 276 138 40
20 to 34 725,200 725 363 103
0 to 34 1,756,200 1,756 879 254
N.B. Population projections are for mid 2007
Figure 1 is intended to illustrate the likely incidence of neuromuscular diseases across the South East Coast. It should be noted that these figures do not take into account elements such as mortality data and are only for modelling purposes based on population size. They are not intended to illustrate actual cases. In addition, it is recognised that some patients live beyond the age of 34 and the figures do not include late-onset diagnosis for patients beyond age 34.

56
Appendix 5: Population Estimates for Primary Care Trusts within the South East Coast
PCT Population
Brighton and Hove 251,000
East Sussex Downs and Weald 326,000
Eastern and Coastal Kent 710,000
Hastings and Rother 170,000
Medway 270,000
Surrey 1.2 million
West Kent 655,700
West Sussex 776,300
Total 4.359 million

57
Appendix 6: A Table Showing the Services Provided by the Specialist/Tertiary Centres for Patients with NMDs from the South East Coast
Paediatric Adult Best Practice
Recommendation GOS ECH GST KCH StG Hamm C Cross NHNN, UCLH Sthmptn
The care of all patients with a neuromuscular condition should be led from a regional Specialist Neuromuscular Centre with specialist multi-disciplinary teams providing regular local clinics.
X Y Y ? ? X ? O O
Specialist multi-disciplinary care may be led by neurologists, clinical geneticists, paediatricians, paediatric neurologists or rehabilitation physicians. This specialist supervision supports and oversees local provision.
X Y Y Y ? ? X O O
Judicious use of expert respiratory services (including non invasive positive pressure ventilation) to improve quality of life, to reduce impromptu admissions to hospitals, and to delay the onset of respiratory failure to prolong life.
O X Y Y ? ? X O ?
Access to neurorehabilitation clinics; which can include physiotherapy, occupational therapy, speech and language therapy, wheelchair services and orthotics; to maintain independence, adapt to changes in disease severity, improve quality of life and delay progression of the condition.
? ? Y X ? ? X O ?
Provision of specialist orthopaedic care for the treatment of spinal deformities which are common in patients with neuromuscular disorders, to correct spinal deformity, improve posture, increase comfort, and prolong life.
Y ? Y X ? ? X ? ?
Monitoring of scoliotic development at a specialist muscle clinic every 3 to 6 months; including a spinal inspection, chest x-rays, radiographs, swallow studies, pulse oximetry and polysomnography; to ensure that surgery is performed when the spine is mobile and at a Cobb angle of twenty to forty degrees, when optimal success rates will be achieved.
Y ? Y ? ? ? X ? ?

58
Screening for heart problems every two years up to age 10, every year after this, and prior to any surgery, since many neuromuscular conditions affect the heart and cardiomyopathy can progress almost entirely without symptoms until signs of heart failure emerge.
Y Y Y Y ? ? X ? ?
Cardiac screening available for women who are carriers of mutations in the dystrophin gene, as they are at increased risk of cardiomyopathy.
O O Y ? ? ? X ? ?
Access to specialist physio and hydrotherapies to maintain mobility, independence, improve quality of life, reduce pain, reduce emergency admissions, and perhaps even delay disease progression. {Where a specialist physiotherapist has training in both neurological and musculoskeletal physiotherapies, experience of treating muscle conditions, and who can support outreach clinics and provide training and professional development for community physiotherapists (who are then able to treat neuromuscular patients themselves)}.
O O Y O ? ? ? O ?
Coordination of care by a specialist muscle nurse, to support self-management of the disease; reducing admissions, length of stay, and improving wellbeing.
Y O Y X ? ? X Y ?
Input from a neuromuscular care advisor to coordinate health and social care needs, transition service; to provide information and support to families of patients, and during diagnosis. This may decrease hospital admissions, length of stays, re-admissions and GP intervention.
Y ? X X ? ? ? ? ?

59
Support from psychologists to enable patients and their families to develop coping mechanisms and management strategies during loss of ambulation, bereavement, diagnosis, and transition. Additionally, psychological support may be required to manage the behavioural side effects of corticosteroids, as well as disorders such as autism, OCD and ADHD which occur more frequently in male DMD patients. Such psychological support will reduce the need for consultants to take on the role, as is happening currently, and therefore save time for other aspects of care.
X ? Y X ? ? O O ?
Access to a dedicated key worker to support transition to adulthood: the Government and commissioners should take urgent action to make sure that a named transition co-ordinator is in place for each young person with a neuromuscular condition who is moving from paediatric to adult services.
O Y Y O ? ? ? ? ?
It is essential for patients’ participation in future trials to ensure that our standards of care are harmonised with the best available elsewhere in Europe.
Y Y Y Y ? ? O Y ?
Key Recommendation Covered Y Recommendation Somewhat Covered O Recommendation Not Covered X Could not be ascertained from information supplied ?

60
Appendix 7: A Key for Figures 7, 8 and 9 Detailing the Centres Present at each Reference Number Category Hospital Postcode Index
Acute Provider Brighton General Hospital BN2 3EW 1
Acute Provider Royal Alexandra Children's Hospital BN2 5BE 2
Acute Provider Royal Sussex County Hospital BN2 5BE 3
Acute Provider Eastbourne District General Hospital BN21 2UD 4
Acute Provider Southlands Hospital BN43 6TQ 5
Acute Provider Kent and Canterbury Hospital CT1 3NG 10
Acute Provider Buckland Hospital CT17 0HD 11
Acute Provider Royal Victoria Hospital CT19 5BN 12
Acute Provider Queen Elizabeth the Queen Mother Hospital CT9 4AN 13
Acute Provider Darent Valley Hospital DA2 8DA 15
Acute Provider Frimley Park Hospital GU16 7UJ 16
Acute Provider Royal Surrey County Hospital GU2 7XX 17
Acute Provider St Peter's Hospital KT16 0PZ 19
Acute Provider Epsom Hospital KT18 7EG 20
Acute Provider Maidstone Hospital ME16 9QQ 21
Acute Provider Medway Maritime Hospital ME7 5NY 22
Acute Provider St Richard's Hospital PO19 6SE 23
Acute Provider Worthing Hospital PO19 6SE 24
Acute Provider East Surrey Hospital RH1 5RH 25
Acute Provider Princess Royal Hospital RH16 4EX 26
Acute Provider Hurstwood Park Neurosciences Centre RH16 4EX 27
Acute Provider Queen Victoria Hospital RH19 3DZ 28
Acute Provider Sutton Hospital SM2 5NF 34
Acute Provider St Helier Hospital SM5 1AA 35
Acute Provider Queen Mary's Hospital for Children (on St Helier site) SM5 1AA 36
Acute Provider Pembury Hospital TN2 4QJ 40
Acute Provider William Harvey Hospital TN24 0LZ 42
Acute Provider Conquest Hospital TN37 7RD 43
Acute Provider Kent and Sussex Hospital TN4 8AT 44
Acute Provider Bexhill Hospital TN40 2DZ 45
Acute Provider Ashford Hospital TW15 3AA 47
Community / Primary Care Centre Chailey Heritage Clinical Services BN8 4JN 6
Community / Primary Care Centre Haslemere and District Hospital GU27 2BJ 18
Community / Primary Care Centre Uckfield Hosptial TN22 5AW 41
Community / Primary Care Centre Crowborough War Memorial Hospital TN6 1HF 46
Specialist Centre Beckenham Beacon BR3 3QL 7
Specialist Centre Princess Royal University Hospital BR6 8ND 8
Specialist Centre Orpington Hospital BR6 9JU 9
Specialist Centre Queen Mary's, Sidcup DA14 6LT 14
Specialist Centre Evelina Children’s Hospital SE1 7EH 29
Specialist Centre St Thomas’s Hospital SE1 7EH 30
Specialist Centre Guys Hospital SE1 9RT 31
Specialist Centre Queen Elizabeth, Woolwich SE18 4QH 32
Specialist Centre Kings College Hospital SE5 9RS 33
Specialist Centre Southampton General Hospital SO16 6YD 37
Specialist Centre St George's Healthcare NHS Trust SW17 0QT 38
Specialist Centre Royal Brompton Hospital SW3 6NP 39
Specialist Centre Hammersmith Hospital W12 0HS 48
Specialist Centre St Mary's Hospital W2 1NY 49
Specialist Centre Charing Cross Hospital W6 8RF 50
Specialist Centre Centre for Neuromuscular Disease, at the NHNN, UCLH WC1N 3BG 51
Specialist Centre Dubowitz NM Centre, GOSH WC1N 3JH 52

61
Appendix 8: A Detailed Narrative of the Services Provided by Specialist/Tertiary Centres for Patients with NMDs from the South East Coast Paediatric:
The Dubowitz Neuromuscular Centre at Great Ormond Street (GOS) is a paediatric specialised centre which accepts referrals from the SEC for the majority of neuromuscular disorders, as well as accepting patients with congenital myopathy and congenital muscular dystrophy from all over the country as a ministerially designated and nationally commissioning centre for these conditions. FPH and West Sussex PCT currently refer children with neuromuscular disorders here.
Specialist staff available here:
• Two clinical consultants and a professor.
• A neuromuscular care advisor.
• A clinical nurse specialist.
• A speech and language therapist and a dietitian (who each work for 2 days a week and provide support to regional services).
• Physiotherapists (who are experts in NMDs and provide assessment and advice to regional services).
Other services provided here:
• Orthopaedic clinics: a spinal clinic and one for feet and ankles.
• Cardiac screening as per best practice, with female carriers being referred to GPs for screening on a five-yearly basis.
• Orthotic services.
• A transition clinic – run jointly with St Thomas’s (N.B. if the patient chooses to switch to regional neurologists for adult treatment then there are no transition clinics available and no-one to follow through the process to ensure its effectiveness).
The Evelina Children’s Hospital (ECH) is a paediatric specialised centre which takes neuromuscular referrals from the SEC region. MTW and Eastern and Coastal Kent PCT currently refer children with neuromuscular disorders here. Following initial assessment and diagnosis, children are referred back to regional centres, where they will receive the majority of their treatment with support from the specialists, including regional outreach clinics.
Specialist staff available here:
• Two neuromuscular consultants (Dr Elizabeth Wraige and Dr Heinz Jungbluth) – carry out assessments for diagnosis.
• Two specialist physiotherapists – are involved in the assessment process and support local physiotherapists.
• Outreach nurse – may be involved in the assessment process, is not a neuromuscular specialist but is involved in care for all neurology patients.
Other services provided here:
• Occupational therapy (although this is run by non-specialists)
• Cardiac screening (which is carried out on a regular basis for patients by the paediatric cardiologist, although parents are referred to their GP for screening).
• A transitional care worker (who is a trained physio) - splits her time between the adult service at St Thomas’s and the paediatric service here, with transition clinics taking place on a regular basis.
Adult: Guy’s and St Thomas’s (GSTT) provide a specialised service for adults with neuromuscular disorders and takes referrals from South East Coast for patients with myotonic dystrophy. Medway and FPH currently refer patients with neuromuscular disorders here.
Clinics provided here:
• A number of different clinics serving children and adults with complex neuromuscular diseases, including those below.
• Monthly myotonic dystrophy clinics which involve surveillance for signs and symptoms of the condition, genetic counselling, and family follow-up, as well as advice about benefits/support available for people with myotonic dystrophy.

62
• Monthly hereditary motor and sensory neuropathy clinics including a diagnostic service, genetic counselling, and family studies for patients.
• DMD clinics. Specialist staff available here:
• Consultant geneticist – leads the myotonic dystrophy service.
• Consultant neuromyologist (Fiona Norwood) leads the hereditary motor and sensory neuropathy clinics.
• A physiotherapist who coordinates the care of muscle patients from St Thomas's hospital.
• A clinical nurse specialist (muscle nurse) – supervises/coordinates ongoing local neuromuscular services (may act in place of a NM care coordinator).
• A consultant who deals specifically with the care of transitional patients. Other services provided here:
• Outreach clinics within the SEC for more common NMDs.
• Respiratory services provided by the Lane Fox Respiratory Unit who have an unparalled international experience in the provision of respiratory support for neuromuscular diseases.
• Neurorehab, either within the neurology service at Guy’s & St Thomas’ or within the Lane Fox Rehabilitation Unit.
• Orthopaedic care - GSTT is a world centre in the provision of spinal orthopaedic services and scoliotic development in children is monitored regularly.
• Cardiac screening as per guidelines, including screening of carriers.
• Hydrotherapy (on top of the physio) support provided by staff who are very experienced in management NMDs.
• Considerable psychological support for children, with support available for adults if needed.
• Limited participation in clinical trials provided they do not impinge upon the care of patients.
Kings College Hospital (KCH) is a specialised centre for adults with neuromuscular disorders and takes referrals from SEC. MTW currently refer patients with peripheral nerve diseases here. EKHT currently refer all adult neuromuscular patients here.
Clinics provided here:
• Three supraregional muscle clinics per week run by the consultant neuromyologists.
• Weekly multidisciplinary/investigatory clinic for patients with long-term muscle disease, run by the specialist muscle physiotherapist and attended by either MR or FN.
• Weekly needle muscle biopsy clinic performed by either Mr, FN or a registrar.
• Fortnightly muscle/myasthenia and MND clinic which is held at the South London NHS Trust (SLH) and is run by Dr Norwood.
Specialist staff available here:
• Three consultant neuromyologists - Dr M Rose, Dr P Barnes (who will be leaving mid-September), and Dr F Norwood.
• Specialist muscle physiotherapist – attends weekly muscle clinics. And runs the weekly multidisciplinary/investigatory clinic for patients with long-term muscle disease.
Other services provided here:
• Cardiac care is provided as an integral part of the King's multidisciplinary follow-up clinic.
• Respiratory care – also provided as an integral part of the King's multidisciplinary follow-up clinic.
• A peripheral nerve service – is one of the largest in the UK and is supported by the largest clinical neurophysiology department in the UK.
• Skin biopsy - for the diagnosis of small fibre peripheral neuropathy. This is the only department in the UK offering this as an NHS clinical service. This is a specialist supraregional referral service for patients who have already seen a consultant elsewhere and are known to have a peripheral neuropathy. Specialist tests can be performed if necessary to clarify the diagnosis, as well as a full range of treatments, including plasma exchange. The department takes part in many clinical trials. Consultants involved in this service include Dr Robert Hadden, who specialises in inflammatory neuropathy, and Professor Kerry Mills, who is head of the Clinical Neurophysiology Unit.
The Centre for Neuromuscular Disease at the National Hospital for Neurology and Neurosurgery (NHNN) aims to provide a regional, supraregional and national centre of clinical excellence for the diagnosis and management of NMDs, with a particular focus on diseases of

63
peripheral nerves and muscles. BSUH and QVH currently refer patients with neuromuscular disorders here. The Centre runs a number of specialist clinics, including peripheral nerve clinics, peripheral nerve genetic clinics, muscle disease clinics and myotonic dystrophy clinics.
Clinics provided here:
• Four outpatient peripheral nerve clinics per week; these clinics are multidisciplinary and run by one of the three consultants. The clinical nurse specialist for peripheral nerve disease also attends, providing support and advice.
• A clinic for patients with peripheral nerve diseases that are on immunosuppressive drugs and need frequent monitoring, which is run by the peripheral nerve nurse specialist.
• A weekly peripheral nerve genetic clinic which is the only one of its kind in the country. Is run by Dr Reilly and is also attended by the neuromuscular nurse specialist and a physiotherapist. There is a genetic counselling service which runs alongside the clinic.
• There are four muscle disease clinics per week which are run by Professors Hanna, Shapiro, and by Dr Parton. Patients are referred to these clinics from all over the UK with a wide range of muscle diseases, including conditions such as muscular dystrophy, congenital myopathy, metabolic myopathy and autoimmune myositis.
• National clinic for muscle channelopathies and mitochondrial disease, which takes place weekly and is run by Professor Hanna. The muscle nurse specialist runs a weekly clinic alongside this clinic.
• A once-monthly myotonic dystrophy clinic, which involves a consultant neurologist (either Dr Turner, Dr Parton or Prof Hanna), a muscle clinical nurse specialist with a special interest in respiratory support (Ms Parry) and the specialised muscle physiotherapist. There is a plan to extend the service to include genetic counselling, psychiatric services, OT, fertility services and social worker input.
• A myasthenia gravis clinic run by Dr Howard. Specialist staff available here:
• Consultants include: Dr Reilly, Dr Manji, Dr Lunn, Professor Koltzenberg and Dr Houlden (who all specialise in peripheral nerve diseases); Professor Hanna, Professor Shapiro, Dr Rahman and Professor Schapira (who all specialise in muscle diseases); Dr Howard, Professor Kullman and Dr Hirsch (who specialise in myasthenia gravis); and Dr Turner and Dr Parton.
• Dr Blake provides neurophysiology for the outpatient peripheral nerve clinics.
• There are three clinical nurse specialists; one for muscle disease, one for peripheral nerve disease, and one for respiratory support in neuromuscular disease.
• Three specialist muscle physiotherapists. Other services provided here:
• OT and SALT – provided in the peripheral nerve clinics as required.
• Clinical trials are carried out as standard.
Charing Cross Hospital (CCH) takes referrals from regional centres all over the country for neuromuscular patients. BSUH and QVH refer patients with neuromuscular disorders here.
Clinics provided here:
• Neuromuscular clinics take place four times a week, each run by a different consultant with different specialisms.
Hammersmith Hospital (HaH) runs one specialised neuromuscular clinic a week, as does St Mary’s Hospital (SMH). BSUH and QVH refer patients with neuromuscular disorders to Hammersmith. S&SH and FPH also refer children to Hammersmith.
Southampton leads on care of neuromuscular patients referred from WSHT, although there is some controversy as to whether this is a specialised centre for NMDs, as the providers perception of themselves is that they don’t seem to think so. S&SH also refer children to Southampton for more specialised investigations.
• Services are led from here with regular regional outreach clinics, supporting and overseeing local provision.

64
Appendix 9: A Table Showing the Services Provided by the Acute Trusts for Patients with NMDs within the South East Coast
Best Practice Recommendation
ASPH D&G ESH EStH FPH RSCH S&SH QVH WSH EKUH MTW MFT BSUH SDH
The care of all patients with a neuromuscular condition should be led from a regional Specialist Neuromuscular Centre with specialist multi-disciplinary teams providing regular local clinics.
? X ? ? X ? O Y Y O X X Y O
Specialist multi-disciplinary care may be lead by neurologists, clinical geneticists, paediatricians, paediatric neurologists or rehabilitation physicians. This specialist supervision supports and oversees local provision.
? X ? ? ? ? ? ? Y O X Y ? O
Judicious use of expert respiratory services (including non invasive positive pressure ventilation) to improve quality of life, to reduce impromptu admissions to hospitals, and to delay the onset of respiratory failure to prolong life.
? Y Y ? O ? ? O O Y X O O Y
Access to neurorehabilitation clinics; which can include physiotherapy, occupational therapy, speech and language therapy, wheelchair services and orthotics; to maintain independence, adapt to changes in disease severity, improve quality of life and delay progression of the condition.
? X O ? O ? X O Y Y X O ? Y

65
Provision of specialist orthopaedic care for the treatment of spinal deformities which are common in patients with neuromuscular disorders, to correct spinal deformity, improve posture, increase comfort, and prolong life.
? X Y ? Y ? Y O Y Y X X Y Y
Monitoring of scoliotic development at a specialist muscle clinic every 3 to 6 months; including a spinal inspection, chest x-rays, radiographs, swallow studies, pulse oximetry and polysomnography; to ensure that surgery is performed when the spine is mobile and at a Cobb angle of twenty to forty degrees, when optimal success rates will be achieved.
? X ? ? O ? ? ? ? O X O ? Y
Screening for heart problems every two years up to age 10, every year after this, and prior to any surgery, since many neuromuscular conditions affect the heart and cardiomyopathy can progress almost entirely without symptoms until signs of heart failure emerge.
? X O ? Y ? Y X O Y O O ? X
Cardiac screening available for women who are carriers of mutations in the dystrophin gene, as they are at increased risk of cardiomyopathy.
? X ? ? ? ? ? X O ? O X ? X

66
Access to specialist physio and hydrotherapies to maintain mobility, independence, improve quality of life, reduce pain, reduce emergency admissions, and perhaps even delay disease progression. {Where a specialist physiotherapist has training in both neurological and musculoskeletal physiotherapies, experience of treating muscle conditions, and who can support outreach clinics and provide training and professional development for community physiotherapists (who are then able to treat neuromuscular patients themselves)}.
? X O ? ? ? X O O Y X X O Y
Coordination of care by a specialist muscle nurse, to support self-management of the disease; reducing admissions, length of stay, and improving wellbeing.
? X X ? X ? X X X X X X ? X
Input from a neuromuscular care advisor to coordinate health and social care needs, transition service; to provide information and support to families of patients, and during diagnosis. This may decrease hospital admissions, length of stays, re-admissions and GP intervention.
? X ? ? X ? X X X X X X ? X

67
Support from psychologists to enable patients and their families to develop coping mechanisms and management strategies during loss of ambulation, bereavement, diagnosis, and transition. Additionally, psychological support may be required to manage the behavioural side effects of corticosteroids, as well as disorders such as autism, OCD and ADHD which occur more frequently in male DMD patients. Such psychological support will reduce the need for consultants to take on the role, as is happening currently, and therefore save time for other aspects of care.
? O Y ? X ? ? X O Y X O ? O
Access to a dedicated key worker to support transition to adulthood: the Government and commissioners should take urgent action to make sure that a named transition co-ordinator is in place for each young person with a neuromuscular condition who is moving from paediatric to adult services.
? X X ? X ? ? X X X X X ? X
It is essential for patients’ participation in future trials to ensure that our standards of care are harmonised with the best available elsewhere in Europe.
? Y ? ? Y ? ? ? ? ? X ? ? Y
Key
Recommendation Covered Y Recommendation Not Covered X
Recommendation Somewhat Covered O Could not be ascertained from information supplied ?

68
Appendix 10: A Detailed Narrative of the Services Provided by the Acute Trusts for Patients with NMDs from the South East Coast
Ashford and St Peter's Hospitals NHS Trust: Run neurorehab clinics. No further information made available. Dartford and Gravesham NHS Trust:
Services provided here:
• Respiratory services - are provided as necessary.
• Support from psychologists - is not provided as standard but requests for it are met.
• The trust has an active research programme which mainly involves projects supported by the Comprehensive Local Research Network.
East Sussex Hospitals NHS Trust:
Services provided here:
• Rehabilitation is available from the Community Rehabilitation Service provided by the PCT (see East Sussex Downs and Weald info), although this is not specialist neurorehabilitation.
• Orthopaedic care is provided if patients are referred by their GP to the orthopaedic consultant.
• A regional cardiac service is available to patients with cardiac complications of neuromuscular diseases, although this may not include regular monitoring as specified by best practice guidelines.
• A very good regional respiratory service which is available for patients with respiratory complications of neuromuscular diseases.
• Non-specialist physiotherapy and hydrotherapy is provided regionally (at the Conquest Hospital, Eastbourne).
• Local psychological services.
Epsom and St Helier University Hospitals NHS Trust: No information provided.
Frimley Park NHS Foundation Trust:
Services provided here:
• Patients are generally seen at regional non-specialised neurology clinics, although they may be sent to the Atkinson Morley's Wing at St George's Hospital, for more complex investigations such as EMGs, muscle biopsies, with follow up at FPH. There is a visiting neuro-geneticist from St George's providing a more specialised diagnostic process.
• Cardiac monitoring and treatment is carried out at FPH, but more complex treatment is provided at St George's. There is no mention of regularity of screening. Cardiac screening is also available for children locally. Cardiac screening for carriers was not specified.
• Orthopaedic care is provided by the internal orthopaedic department, when required, although paediatric orthopaedic care is provided by supraregional centres. Scoliotic monitoring is provided at a regional level by the orthopaedic department but this does not seem to happen regularly.
• Neurorehab clinics are provided at Ashford and St Peter's Hospital and at the Haslemere Unit, which provide a full multi-disciplinary team. However, such provision is only made available to children if there is a specific problem, and care is not coordinated in a multidisciplinary manner.
• Some patients are currently participating in clinical trials and all are encouraged to do so if possible.
• Non-invasive ventilation is being developed locally (at a regional level).
Royal Surrey County Hospital NHS Trust: No information provided.

69
Surrey and Sussex Healthcare NHS Trust: Services provided here:
• Children are usually referred here from general paediatric outpatients and may have initial investigations organised by the Trust, with many of the more specialised investigations being carried out in supraregional units such as Southampton General Hospital and Hammersmith Hospital. Longer-term follow up and support is usually provided locally by community children’s teams in Surrey and West Sussex, depending on where the child lives.
• Paediatric cardiac clinics which children can be referred to if necessary, although there is no mention as to whether screening occurs as standard, nor as to whether carriers are screened within these clinics.
• There are some paediatric orthopaedic clinics provided here within which scoliotic monitoring may take place, although this is not stated.
Queen Victoria Hospital NHS Foundation Trust:
Services provided here:
• There is regional access to neurorehabilitation services such as physiotherapy, speech and language therapy, and OT (although there is only limited SALT cover in the community, meaning that patients who are too frail/poorly to attend as out-patients only receive a domiciliary swallowing assessment & one follow up).
• Respiratory services are run by a respiratory physician, who works alongside a consultant anaesthetist and neurologist. The anaesthetist and neurologist are particularly involved in the sleep studies which are required as the major part of a patient’s respiratory assessment. The neurologist is keen to develop home ventilation services for all SEC patients via QVH (as a specialist centre).
Western Sussex Hospitals NHS Trust:
Services provided here:
• Children with neuromuscular conditions are seen at the consultant neurologist's general clinics, with a regional dedicated neuromuscular clinic being provided at St Richard's by Southampton General Hospital clinicians three times a year. Daily neurology clinics are provided at St Richard's Hospital within which adult neuromuscular patients are seen. Adults are seen by general consultant neurologists at Worthing and Southlands neurology clinics and at clinics led by neurology nurses. Children are referred to specialist neuromuscular services (at GOS) upon diagnosis, with regional 3-6 monthly consultant-led outreach clinics (and physio attendance). There are also regional paediatric 6-12 monthly multi-disciplinary clinics held at Worthing and Southlands Hospitals.
• Regional neurorehabilitation services are provided at Donald Wilson House, St Richard’s Hospital. This is a multi-disciplinary unit with inpatient facilities for 9 inpatients of working age who have neurological disorders that require specialist multidisciplinary rehabilitation in order to achieve specific goals. The unit is for patients from the St Richard’s and Worthing hospitals’ catchment areas. Any inpatient neurorehabilitation services for patients from the north of the county would be provided by Princess Royal Hospital, Haywards Heath. There are also three 5-day beds which can be used as a “step-down” for patients preparing for discharge or for brief admissions for patients who require a short spell of inpatient rehabilitation. Patients who have been through the service may then attend as “day-patients” which involves them seeing various therapy groups on a couple of days a week for a set period of time. Again, this is limited to achieving specific goals. The unit has a consultant in rehabilitation medicine, specialist nurses, physiotherapists, occupational therapists, speech and language therapists, dieticians and minimal psychology input. Specialist Neurorehabilitation outpatient clinics are conducted twice weekly by the consultant in rehabilitation medicine, usually seeing 8-12 patients a clinic. These may have been former inpatients on the

70
unit or may be referrals from the community. Many of these patients will be receiving therapy in a community setting from one of the community teams that cover the Worthing and Chichester areas. Patients from the North of the County and the Hampshire border (Emsworth) are also seen, although there is no co-ordinated service for patients from the North of the County.
• A limited psychology service available through the neurorehabilitation service at St Richard's Hospital.
• There are respiratory services within St Richard's for patients with chronic neurological diseases, and specifically a monthly joint clinic run with the cardiologists and urologists for patients with complex disabilities.
• Consultant led paediatric orthopaedic clinics at Worthing and Southlands Hospitals, within which scoliotic monitoring presumably takes place.
• There are also consultant led paediatric cardiac clinics at Worthing and Southlands Hospitals.
• Cardiology services within St Richard's for patients with chronic neurological diseases, with a monthly joint clinic run by cardiologists, urologists and respiratory consultants, for patients with complex disabilities. Within these cardiology services there is cardiac screening for adults, but it is unclear where this takes place.
• Hydrotherapy is available at Bognor Regis War Memorial Hospital, but demand outstretches supply.
• Neuro therapists (physiotherapists, OTs, SALTs) assess and treat patients with neurological disorders on critical care and the acute wards, as well as in DWH neurological rehabilitation centre.
• There is a weekly neuro-physiotherapy out-patient service seeing 2 new patients and 4 follow-ups each Wednesday, which is currently heavily oversubscribed with a waiting list of 8–12 weeks. There is limited scope for frequent hands-on treatment; instead the focus is on assessment, advice, problem solving, onward referrals and self management advice. This service accepts referrals from hospital consultants and supraregional centres. All GP referrals are redirected to the physiotherapy service within the PCT. There are also occasional, ad-hoc handovers of particular patients by paediatric physiotherapy to neuro-physiotherapy out-patient service, but no formal pathways for transfer.
• An orthotics service on the St Richard's site with joint therapist/orthotist assessments being possible, dependent on patient need. Additionally, neuro orthotic clinics are held at Southlands Hospital for adults and involve an orthotist and a neuro-physiotherapist.
• There are also paediatric orthotic clinics held twice-monthly at Roehampton or Chailey heritage, along with wheelchair and non-standard equipment services.
East Kent Hospitals University NHS Foundation Trust:
Services provided here:
• Rehabilitative care of patients with NMDs is led from the regional specialist centre at Kent and Canterbury.
• Multi-disciplinary care is lead by the neurorehabilitation consultant, supporting and overseeing community? provision. However, there are no neurologists involved in this care (as far as we know).
• Neurologists here have agreed to organise a fast track clinic for diagnosis.
• Staff at the multidisciplinary inpatient neurorehabilitation unit at Kent and Canterbury Hospital in East Kent include a consultant rehabilitationist, a neurorehabilitation coordinator (secretary), a social service care manager, a neuro-physiotherapist, an occupational therapist, a nurse, palliative nurse specialists, a speech therapist, a dietitian, and a consultant neuropsychologist.
• The neurorehab unit at K&C also offers respite, continuing, and palliative care for neuromuscular patients if necessary.

71
• Non-Invasive ventilation and gastrostomy is provided to patients with NMDs where necessary, carried out at the neurorehabilitation unit at Kent & Canterbury Hospital.
• Specialist orthopaedic care, including monitoring of scoliotic development.
• Cardiac services are available both from a regional paediatrician trained in paediatric echocardiography (Dr Vinit Shah), and through supraregional outreach clinics provided by cardiologists from Guy's and the Brompton Hospitals, although it is not mentioned whether this includes screening for carriers of the dystrophin gene.
• Access to hydrotherapy services.
• There is also good access to orthotics and wheelchair services.
• The neurorehabilitation unit in East Kent arranges fast-track equipment delivery by liaising with necessary services, such as Assistive Technology for Speech and Environmental Control Systems. In addition, liaison between East Kent Hospitals and the Kings supraregional service has resulted in repatriation over the last 3-4 years, whilst also providing a source for help with diagnostic dilemmas.
• On top of the inpatient services available, outpatient neurorehabilitation clinics are held at Kent and Canterbury and Queen Elizabeth Queen Mother Hospitals in East Kent.
• Any communications from patients or professionals at East Kent Hospitals go through the coordinator to be brought to the attention of the team.
• Any proposed improvements to the service must be coordinated by the community respiratory nurse. In order to obtain feedback regarding the services provided, there are monthly focus groups held at various locations, with input from both carers and patients. Furthermore, The East Kent Development Group; which is composed of relatives of both current and previous patients, the care manager, the consultant and the speech therapist; proposes ways in which to develop/improve the service.
Maidstone and Tunbridge Wells NHS Trust:
Services provided here:
• Specialist multidisciplinary care will be led by experts from Kings if the patients are in regular attendance here.
• Children have access to physiotherapists and other therapists as needed. Medway NHS Foundation Trust:
Services provided here:
• General therapeutic services in the community for adults with neurological conditions; this includes physiotherapy, occupational therapy, dietetics, and speech and language therapy.
• Regional cardiac screening for myotonic dystrophy patients. Patients with other neuromuscular conditions may or may not have access to cardiology services on an individual basis.
• Neuro physiotherapy services are provided locally by the community team, although access to specialist physiotherapy is only at supraregional centres.
• There is also a small physical disabilities psychology service available for people in Medway, but no dedicated psychologists available at present, although there may be provision in future from the therapy unit for long-term conditions.
Brighton and Sussex University Hospitals NHS Trust:
Services provided here:
• General adult neurology clinics run by one of 5 permanent consultant neurologists. Inflammatory muscle disorders such as polymyositis are normally managed alongside the rheumatologists (e.g. Prof Kevin Davies at RSCH). Patients are sent to supraregional centres in London for particular aspects of care, such as confirmation of diagnosis, genetic counselling and clinical trials, but much of the routine follow-up and care is provided locally.

72
• General physiotherapy and neurorehabiliatation services.
• Rehabilitation services – although these are non-specialist.
• Orthopaedic care – although this is not specialist.
• General cardiac care.
• Physiotherapy services are available but these are not specialist. South Downs Health NHS Trust:
Services provided here:
• The Community Respiratory and Heart Failure Service see a small number of neuromuscular patients.
• The Integrated Community Equipment Service (ICES) see a small number of neuromuscular patients.
• The Community Physiotherapy Service see a small number of neuromuscular patients from SDH.
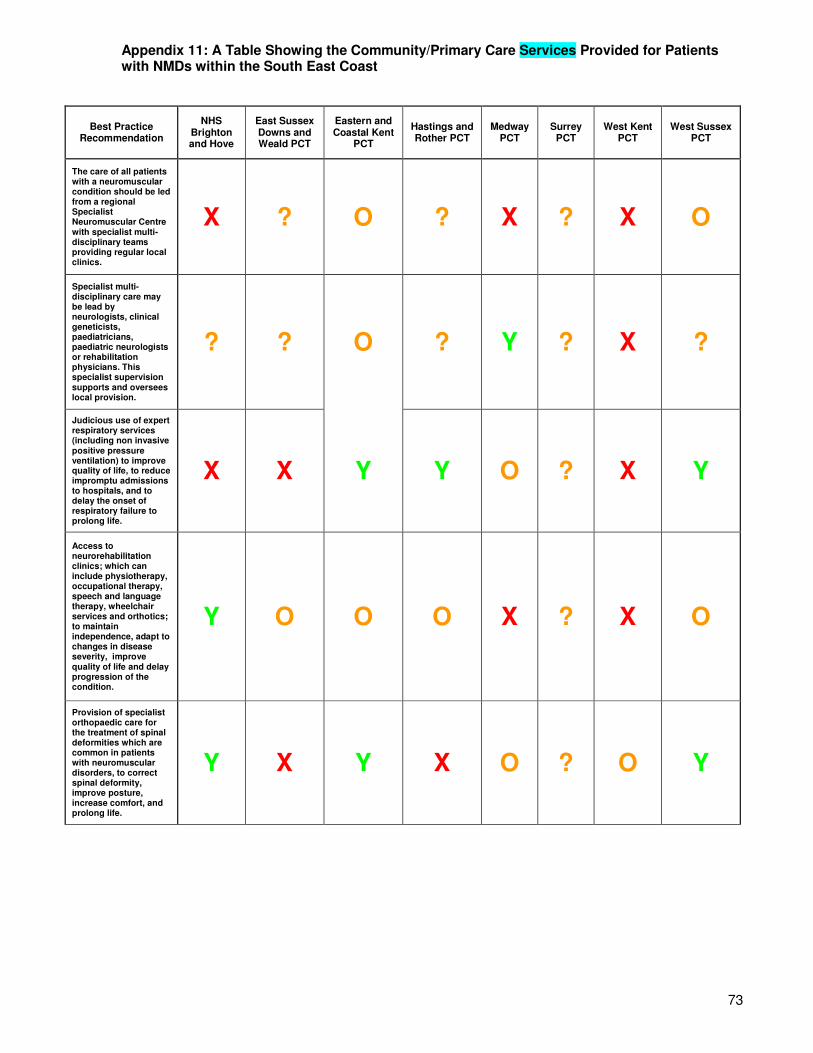
73
Appendix 11: A Table Showing the Community/Primary Care Services Provided for Patients with NMDs within the South East Coast
Best Practice Recommendation
NHS Brighton and Hove
East Sussex Downs and Weald PCT
Eastern and Coastal Kent
PCT
Hastings and Rother PCT
Medway PCT
Surrey PCT
West Kent PCT
West Sussex PCT
The care of all patients with a neuromuscular condition should be led from a regional Specialist Neuromuscular Centre with specialist multi-disciplinary teams providing regular local clinics.
X ? O ? X ? X O
Specialist multi-disciplinary care may be lead by neurologists, clinical geneticists, paediatricians, paediatric neurologists or rehabilitation physicians. This specialist supervision supports and oversees local provision.
? ? O ? Y ? X ?
Judicious use of expert respiratory services (including non invasive positive pressure ventilation) to improve quality of life, to reduce impromptu admissions to hospitals, and to delay the onset of respiratory failure to prolong life.
X X Y Y O ? X Y
Access to neurorehabilitation clinics; which can include physiotherapy, occupational therapy, speech and language therapy, wheelchair services and orthotics; to maintain independence, adapt to changes in disease severity, improve quality of life and delay progression of the condition.
Y O O O X ? X O
Provision of specialist orthopaedic care for the treatment of spinal deformities which are common in patients with neuromuscular disorders, to correct spinal deformity, improve posture, increase comfort, and prolong life.
Y X Y X O ? O Y

74
Monitoring of scoliotic development at a specialist muscle clinic every 3 to 6 months; including a spinal inspection, chest x-rays, radiographs, swallow studies, pulse oximetry and polysomnography; to ensure that surgery is performed when the spine is mobile and at a Cobb angle of twenty to forty degrees, when optimal success rates will be achieved.
? ? X ? X ? ? X
Screening for heart problems every two years up to age 10, every year after this, and prior to any surgery, since many neuromuscular conditions affect the heart and cardiomyopathy can progress almost entirely without symptoms until signs of heart failure emerge.
? ? X ? X ? X Y
Cardiac screening available for women who are carriers of mutations in the dystrophin gene, as they are at increased risk of cardiomyopathy.
? ? Y ? X ? ? ?
Access to specialist physio and hydrotherapies to maintain mobility, independence, improve quality of life, reduce pain, reduce emergency admissions, and perhaps even delay disease progression. {Where a specialist physiotherapist has training in both neurological and musculoskeletal physiotherapies, experience of treating muscle conditions, and who can support outreach clinics and provide training and professional development for community physiotherapists (who are then able to treat neuromuscular patients themselves)}.
O O Y O O ? O O
Coordination of care by a specialist muscle nurse, to support self-management of the disease; reducing admissions, length of stay, and improving wellbeing.
? X X X X ? X ?

75
Input from a neuromuscular care advisor to coordinate health and social care needs, transition service; to provide information and support to families of patients, and during diagnosis. This may decrease hospital admissions, length of stays, re-admissions and GP intervention.
X X X X X ? X X
Support from psychologists to enable patients and their families to develop coping mechanisms and management strategies during loss of ambulation, bereavement, diagnosis, and transition. Additionally, psychological support may be required to manage the behavioural side effects of corticosteroids, as well as disorders such as autism, OCD and ADHD which occur more frequently in male DMD patients. Such psychological support will reduce the need for consultants to take on the role, as is happening currently, and therefore save time for other aspects of care.
? X Y X Y ? O X
Access to a dedicated key worker to support transition to adulthood: the Government and commissioners should take urgent action to make sure that a named transition co-ordinator is in place for each young person with a neuromuscular condition who is moving from paediatric to adult services.
? X X X X ? X X
Key Recommendation Covered Y Recommendation Somewhat Covered O Recommendation Not Covered X Could not be ascertained from information supplied ?

76
Appendix 12: A Detailed Narrative of the Services Provided by the Community/Primary Care Services for Patients with NMDs from the South East Coast
NHS Brighton and Hove:
Services provided here:
• Patients may receive orthopaedic care and access physiotherapy either in the acute hospital (Brighton & Sussex University Hospital) or at the community rehabilitation services at South Downs Health.
For more information see section above for Brighton and Sussex University Hospitals NHS Trust and South Downs Health NHS Trust. East Sussex Downs and Weald PCT:
Services provided here:
• Orthopaedic care – via GP referral to the orthopaedic consultant.
• A physiotherapist that specialises in the treatment of neuromuscular conditions.
• Counselling and psychological assessment can be provided by referral from a GP.
For more detail about certain aspects of care see information above for East Sussex Hospitals NHS Trust. Eastern and Coastal Kent PCT:
Services provided here:
• Agreements are currently being finalised for a local (regional?) paediatric outreach neuromuscular clinic, involving consultants from the supraregional centre (and possibly the physiotherapists that they work with), which should start in the New Year.
• A multidisciplinary neuro-disability clinic is held locally?? every month and children with neuromuscular conditions are included in this, receiving 6 monthly or annual reviews, depending on their needs. Included in this clinic are the child's therapists and a paediatrician.
• Adults with NMDs are offered multidisciplinary assessment, rehabilitation and equipment provision via the intermediate care team. This service is provided on a needs led basis to patients within their home/work environment. The Intermediate Care comprises teams of nurses, physiotherapists, occupational therapists, speech and language therapists, dieticians, a neuropsychologist, and mental health clinicians.
• In order to provide respiratory, cardiac and orthopaedic services for adults with NMDs, intermediate care professionals will liaise with these specialist services as required.
• Respiratory care is provided by the specialist physiotherapist (who has a specific interest in respiratory care and neurology and can assist with the use of home ventilators and other respiratory equipment) and the community children’s nursing team.
• Necessary equipment is provided by the equipment store (new and recycled) following identification by the child’s physiotherapist/occupational therapist.
• Orthopaedic needs are monitored by local physiotherapists and the regional team at the Evelina Children’s Hospital, with their spinal surgeon offering regional outreach clinics.
• Paediatric physiotherapists work in conjunction with OTs, offering clinic appointments as well as home and school visits to advise and educate parents/carers and school staff in addition to providing and adjusting equipment that is needed. Children are reviewed as needed throughout the year. They are often offered treatment blocks throughout the year, when this is thought to be of benefit, to work towards goals that are set jointly by therapists, children and their parents/carers.
• Blocks of hydrotherapy are also provided for children who need it.
• A clinical psychologist based in Eastern and Coastal Kent. The paediatricians can also refer patients to the local CAHMS team.

77
• There are patients participating in a clinical trial at present, but they both moved from Worcester two years ago and were put onto the trial there.
Hastings and Rother PCT:
Services provided here:
• There is a community respiratory team who can provide respiratory services to neuromuscular patients.
• Patients are visited by physiotherapists in their own home for assessment and treatment on a weekly or fortnightly basis, depending on the needs of the patient and the time needed to develop a movement/mobility programme or advice on respiratory care. The aim is to promote self-management (where possible), and/or provide advice to carers. Liaison with the local Community Rehabilitation Team occurs if a multidisciplinary approach is required.
For more information see section above on East Sussex Hospitals NHS Trust. Medway PCT:
Services provided here:
• Specialist multidisciplinary care is coordinated at supraregional centres and oversees local and regional provision.
• Neuro physiotherapy services are provided locally by the community team.
• There is a small physical disabilities psychology service available for people in Medway.
Surrey PCT:
Services provided here:
• Dietetic help is provided via general outpatient clinics at Crawley Hospital, Horsham Hospital and some GP practices, upon referral by the patient's GP or consultant. A home enteral nutrition service is also provided for any patients requiring tube feeding at home or in a residential placement setting.
For more information see information for Ashford and St Peter’s Hospitals NHS Trust, Epsom and St Helier University Hospitals NHS Trust, Frimley Park NHS Foundation Trust, Royal Surrey County Hospital NHS Trust, Surrey and Sussex Healthcare NHS Trust, Queen Victoria Hospital NHS Foundation Trust. West Kent PCT:
Services provided here:
• Mark Shepperd, Medical Director of West Kent Community Services, advised that they are (in headline terms) a “Nil return”. That is, the little provision that there is in West Kent is provided as outreach from the regional acute providers, i.e. there is no dedicated service provided as standard by West Kent PCT itself.
• Support from community services (such as physios, SALTs etc.) may be provided if requested by the patient. Where patients with underlying neuromuscular problems have need of a district nurse or physiotherapist (post surgery for instance) they would get this, but as part of the general service and not as part of their particular underlying condition.
West Sussex PCT:
Services provided here:
• Paediatric supraregional centres advise regional/local centres.
• Although there are multidisciplinary teams that provide local clinics, these are not specially trained in NMDs.
• Children are referred to GOS upon diagnosis, with regional 3-6 monthly consultant led clinics (and physio attendance) at St Richard’s Hospital, and regional multi-disciplinary 6-12 monthly clinics at Worthing and Southlands Hospital.
• Paediatric OT, speech and language therapy and physio and hydrotherapies, which tend to be provided via weekly or fortnightly clinics, some of which are multi-disciplinary.

78
• Respiratory care is available at the Brompton, with regular regional respiratory clinics being held in Crawley, which a paediatric respiratory consultant from the Brompton visits every 4 months.
• Regional paediatric orthopaedic clinics held at Crawley, although these are not specialist.
• Children are offered cardiac screening as routine, in consultant led cardiac clinics.
• There is paediatric access to physio and hydro therapies, although these are not staffed by neuromuscular specialists.
• Specialist muscle nurses who liase with paediatric staff from Southampton, although it is unclear whether care is coordinated by them.
• Wheelchair, equipment and orthotic services, although these are not specialised to neuromuscular diseases.

79
Appendix 13: Best Practice Guide for SMA65
DIAGNOSTIC TESTING AND CARE OF NEW SMA PATIENTS
Diagnostic procedures The stepwise algorithm of the diagnostic procedure is summarized in the diagram below. Briefly, the first diagnostic test for a patient suspected to have SMA should be the SMN gene deletion test. A homozygous deletion of the SMN1 gene exon 7 (with or without deletion of exon 8) confirms the diagnosis of SMN-associated SMA (5q-SMA). The other diagnostic tests should be ordered only after a negative SMN gene test has been obtained.
Clinical management of newly diagnosed SMA patients
Many care issues arise when a patient is newly diagnosed with SMA. Clinicians need to address the various aspects of care issues as soon as possible.
Family education and counselling:
Because of the complexity of medical problems associated with a diagnosis of SMA, medical providers should designate a person to meet with the family.

80
During the first meeting with parents it is important to explain:
• The disease process
• Pathogenesis
• Phenotype classification
• Prognosis of the patient’s SMA
• Online information on SMA and SMA patient advocacy groups
• Referral to clinical trial studies
The physician should also formulate a plan of multi-disciplinary intervention with the family. This usually includes referral to the following services:
• Pediatric neuromuscular clinic
• Genetics
• Pulmonary
• GI/nutrition
• Orthopedic/rehabilitation
Genetic Topics:
Several genetic topics should be addressed with the diagnosis of SMA.
• Genetics of SMA such as autosomal recessive inheritance and genomic structure of the SMN genes - SMN1 and SMN2 copies.
• While higher SMN2 gene copy number is correlated with milder phenotype, predicting clinical severity using SMN2 copy number is not currently recommended because there can be substantial variation of clinical phenotype for any given SMN2 gene copy number.
• Recurrence risk.
• Carrier testing.
• Information for reproductive planning (prenatal or pre-implantational diagnosis).
PULMONARY CARE
Overview of pulmonary problems in SMA
The key respiratory problems in SMA are:
1. Impaired cough resulting in poor clearance of lower airway secretions
2. Hypoventilation during sleep 3. Chest wall and lung underdevelopment 4. Recurrent infections that exacerbate muscle weakness Pulmonary disease is the major cause of morbidity and mortality in SMA types I and II, and may occur in a small proportion of patients with SMA type III. Swallowing dysfunction and reflux are important contributors to pulmonary morbidity. Individuals tend to progress to daytime respiratory failure via a sequence of recurrent chest infections, nocturnal oxygen desaturation, followed by nocturnal hypoventilation and then daytime hypercarbia. Ventilatory support should be added at night if sleep disordered breathing is present, and cough assistance provided if cough efficiency is reduced. Airway clearance is very important in both acute and chronic management of all patients with SMA.

81
II: Assessment and monitoring
Suggested frequency of evaluation is every 3-6 months, less frequently in stable walkers, more frequently in clinically unstable non-sitters.
A. Non-Sitters:
• Physical examination: monitor cough effectiveness, chest wall deformity, work of breathing, respiratory rate, paradoxical breathing, and skin color.
• Polysomnography: to document signs of hypoventilation.
• Pulse oximetry: to monitor oxygen saturation through transcutaneous sensor
• Pneumonias: monitor frequency of infection and antibiotic treatments over past 6 months.
• Chest x-ray: baseline and during respiratory deterioration.
• Swallow studies: in unexplained acute respiratory deterioration and recurrent pneumonia.
B. Sitters:
• Physical examination: monitor cough effectiveness, chest wall deformity, work of breathing, respiratory rate, paradoxical breathing, and skin color.
• Polysomnography: to document signs of hypoventilation.
• Pulse oximetry: to monitor oxygen saturation through transcutaneous sensor
• Pneumonias: frequency of infections and antibiotic treatments over the past 6 months.
• Scoliosis: inspection of spine and radiographic evaluations of scoliosis.
C. Walkers:
In general, SMA walkers have relatively preserved pulmonary function until late into their disease course.
• Physical examination: monitor cough effectiveness, chest wall deformity, work of breathing, respiratory rate, and skin color.

82
• Pulmonary function testing: spirometry, lung volumes, and respiratory muscle function.
• Pneumonias: frequency of infections and antibiotic treatments over the past 12 months.
III: Anticipatory Respiratory Care
Critical to respiratory management of SMA is to provide families with information about options for chronic care, acute illness management, and perioperative care.
• Non-sitters are the most fragile group and early discussions should include the option of noninvasive ventilation (NIV) and secretion management due to the rapid progression of the disease.
• Ongoing discussion of the family’s desires for support should occur, and the result should be a negotiated care plan with maximums and minimums outlined.
Day to day management should include:
• Understanding the child’s baseline and deviations from his/her baseline
• Understanding hypoventilation and intervention
• Acute illness management including rapid access to specialty medical care providers
• Airway clearance and secretion management techniques
• Respiratory support including NIV
• Nutrition and hydration
• A low threshold to start antibiotics
• Routine immunizations including influenza vaccine, pneumococcus vaccine, and RSV prophylaxis (palivizumab).
IV: Chronic management
Discussion of the family’s goals is essential. This may include balancing caring for the child at home for as long as possible, life expectancy, quality of life and comfort, and the availability of resources. Goals of chronic management are to: normalize gas exchange, improve sleep quality, facilitate home care, reduce hospitalizations and ICU care, and reduce the burden of illness. Early aggressive and proactive intervention may prolong life without compromising quality of life.
Airway Clearance:
• Assisted cough, manually or assisted with mechanical insufflation-exsufflation, is recommended daily in more severely affected patients. Caregivers of patients with SMA should learn to assist coughing.
• Secretion mobilization techniques are helpful and include chest physiotherapy and postural drainage.
• Oximetry should be used to guide therapy. Oral suctioning can assist in secretion management after assisted coughing.
Respiratory Support: This is clearly indicated in daytime hypercapnia. Nocturnal NIV reduces symptoms of disordered breathing during sleep and increases life quality.
• NIV should be combined with airway clearance techniques.
• In non-sitters, care without ventilation support is an option if the burden of treatment outweighs benefit.
• CPAP may be an option, with the goal of transition to bi-level positive airway pressure (BiPAP). • Use of NIV with high span BiPAP, even for short daytime periods, may improve chest wall and
lung development, and reduce ribcage and sternal deformity in non-sitters and sitters.
• Tracheotomy: In non-sitters, this is controversial and an ethical dilemma. There is a large spectrum of options that can be provided, ranging from no respiratory support, to NIV, to tracheotomy, and mechanical ventilation.

83
• Palliative care is an option for non-sitters. NIV can be used as a routine therapy or as a palliative tool. A key goal is to prevent PICU stays and avoid tracheotomy if possible. If supportive ventilation is chosen by the family, NIV is recommended.
V: Perioperative care
Patients with SMA are at high risk for post-anesthesia complications, which may lead to prolonged intubation, nosocomial infections, tracheotomy, and death. It is critical that the patient’s respiratory status be optimized before surgery.
Pre-operative evaluation:
Physical examination
• Measurements of respiratory function and cough effectiveness
• Chest x-ray
• Evaluation for sleep disordered breathing
• Consider complicating factors including jaw ankylosis, oropharyngeal aspiration, gastroesophageal reflux, nutritional status, and asthma.
If measurements of respiratory function and/or sleep study are abnormal, nocturnal NIV and assisted coughing techniques may be indicated before surgery. The patient should become familiar with these techniques prior to surgery. If jaw ankylosis is present, intubation should be performed by fiberoptic bronchoscopy.
Post-operative management:
• If cough clearance is normal and muscle function is relatively preserved, there is not an increased risk for post-operative complications.
• If decreased respiratory muscle strength is present pre-operatively, close monitoring and aggressive respiratory management is required.
• If respiratory support is required pre-operatively during sleep, similar respiratory support in the immediate postoperative course is required.
• Extubation in the recovery room to NIV should be planned as a bridge to weaning to the patient’s baseline respiratory support. This requires careful planning and coordination. If the patient requires continuous ventilator support pre-operatively (via non-invasive interface or via tracheotomy tube) or use of muscular blocking agent during surgery then it is best to transferred patient directly from OR to ICU.
• Patients are encouraged to bring their • personal devices such as NIV and MI-E machines to use in the post-operative period because hospitals may be limited in the availability of these devices.
• Oxygen must be applied with caution in the patient with SMA. Hypoxemia secondary to hypoventilation may be mistaken with hypoxemia due to other causes such as mucus plugging and atelectasis. ETCO2 or TcCO2 monitoring or arterial blood gas analysis will facilitate appropriate oxygen use.
• Adequate pain control will aid in preventing hypoventilation secondary to splinting. Pain management should be titrated to promote airway clearance and minimize respiratory suppression. Transient increased respiratory support may be needed while controlling post-operative pain.
VI: Acute care management
The goal of management during acute illness is to normalize gas exchange by reducing atelectasis and enhancing airway clearance where possible by non-invasive respiratory support. Blood gas monitoring may be of benefit.

84
Airway Clearance:
• Airway clearance with manual cough assist or MI-E, oral or airway suctioning. Assisted cough techniques are preferred over deep suctioning and bronchoscopy.
• Oximetry feedback to guide airway clearance.
• Chest physiotherapy.
• Postural drainage.
Respiratory Support:
(i) For non-sitters and sitters:
• Acute use of NIV reverses ventilatory decompensation caused by vicious cycle of added ventilatory load, increased respiratory muscle weakness, and ineffective secretion clearance.
• If already using nocturnal NIV, daytime NIV may be required, and airway clearance techniques carried out during NIV.
• Oxygen therapy entrained into the NIV circuit should be used to correct oxygen desaturation, after inspiratory and expiratory positive pressure settings are optimized and airway clearance techniques are optimally utilized.
• If a non-invasive approach fails, intubation and mechanical ventilation is a short-term measure. After recovery from the acute illness and arterial oxygen saturation on room air has normalized, they should be extubated back to NIV.
• Decision-making about escalation to intubation should be carried out in advance as part of anticipatory care planning.
• Tracheotomy and ventilation can be considered in case of frequent acute pulmonary infections in non-sitters, but may not improve quality of life or reduce hospitalizations. A tracheotomy is not an acute intervention. Tracheotomy is not appropriate in sitters.
• With deteriorating function it may be appropriate to redirect care to a palliative approach, particularly for non-sitters.
(ii) For walkers:
• NIV may be needed during an acute illness, in combination with airway clearance techniques.
• Oxygen therapy and need for transient intubation should be carried out as outlined above for non-sitters/sitters.
• NIV for home use should be considered if NIV was needed during an acute illness.
Additional Management: For non sitters, sitters, and walkers, recommended additional therapies are antibiotics, adequate nutritional support, hydration, and gastroesophageal reflux management.
GASTROINTESTINAL (GI) AND NUTRITIONAL CARE
Overview of gastrointestinal and nutritional care
The key clinical problems associated with GI and nutritional complications in SMA are:
1. Feeding and swallowing problems. Bulbar dysfunction is universal in SMA patients with severe weakness and can result in aspiration pneumonia, which is a common cause of death.
2. Gastrointestinal dysfunction. GI dysmotility problems include constipation, delayed gastric emptying and potentially life-threatening gastro-esophageal reflux (GER).
3. Growth and under/over nutrition problems. Without optimal management, growth failure is universal in non-sitters while excessive weight gain is more common in sitters and walkers.
4. Respiratory problems. The presence of respiratory complications (weak cough, increased work of breathing, dyspnea, pneumonias and cyanosis or desaturation with feeds) raises concerns for feeding difficulty and increased risk of aspiration which can be life threatening. Increased work of breathing may also result in increased energy expenditure.

85
a. Feeding and swallowing problems
Feeding and swallowing difficulties are common in non-sitters and sitters but are rarely a concern in walkers.
1. Key symptoms of feeding and swallowing problems:
• Prolonged mealtime.
• Fatigue with oral feeding.
• Choking or coughing during or after swallowing.
• Recurrent pneumonias potential indicator of aspiration, which may be silent, i.e. without evident choking or coughing.
• Vocal cord paralysis may be diagnostic sign of silent laryngeal aspiration.
2. Causes of feeding difficulty:
Pre-oral phase
• Limited mouth opening due to reduced range of mandibular motion.
• Difficulties in getting food to the mouth for self-feeding.
Oral phase
• Weak bite force.
• Increased fatigue of masticatory muscles.
Swallowing phase
• Poor head control.
• Inefficient pharyngeal phase of swallowing.
• Poor co-ordination of the swallow with airway closure.
3. Evaluation of feeding and swallowing problems:
• Feeding assessment by feeding specialist.
• A feeding case history with mealtime observation is desirable.
• Examination of oral structures which impact on feeding efficiency and consideration of the effect of positioning and head control on feeding and swallowing is essential.
• Videofluoroscopic swallow studies (VFSS) if concerns on concerns about swallow function and safety, and opportunity to evaluate therapeutic strategies.
4. Management of feeding and swallowing difficulties: Treatment should aim at reducing the risk of aspiration and optimizing efficiency of feeding and promote enjoyable mealtimes.
Changing food consistency and optimizing oral. A semi-solid diet can compensate for poor chewing and reduce length of mealtimes. Thickened liquids may protect against aspiration of thin fluids. Preferably, this intervention would be evaluated objectively on VFSS.
Positioning and seating alterations and orthotic devices (e.g. Neater Eater®, elbow support, valved straw) to enhance self-feeding ability may improve swallow safety and efficiency. Plan in liaison with an occupational therapist and/or physiotherapist as required.
Proactive nutritional supplementation as soon as inadequate oral intake is recognized. Whether or not a g-tube is placed in a particular child often requires extensive discussion with multiple caregivers. Nutritional supplementation via nasogastric (NG) or nasojejunal (NJ) feeding is desirable in the interim before g-tube placement. NJ feeding may be preferable in circumstances when GER with aspiration is a concern, especially when the patient is on ventilatory support. However, technical difficulty may prevent its feasibility.
G-tube feeding is the optimal method of feeding for insufficient caloric intake or unsafe oral feeding. It prevents the potential morbidity and poor ventilatory mask fit associated with prolonged use of either NG or NJ tubes. A laparoscopic surgical technique for g-tube placement provides the best possible setting for immediate or early post-operative extubation. Care should be taken to minimize

86
the amount of fasting preoperatively, and to quickly resume full nutritional support following the procedure.
II. Gastrointestinal dysfunction
Children with SMA will have the following GI problems: gastroesophageal reflux (GER), constipation, and abdominal distension and bloating. GER is an important determinant of mortality and morbidity in SMA patients. High fat foods delay gastric emptying and increase the risk of GER.
1. Key symptoms of GER:
• Frequent “spitting up” or vomiting post meals
• Emesis
• Complaints of chest or abdominal discomfort
• bad breath
• Obvious regurgitation of feeds
• Refusal of feeds when developing discomfort with swallowing
2. Evaluation of Gastrointestinal Dysfunction:
• Search early for symptoms of GER (emesis, regurgitation, gurgling after feeds).
• A routine upper gastrointestinal (UGI) series for presurgical evaluation for gastrostomy tube (g-tube) placement to primarily rule out anatomical anomalies and secondarily to document reflux.
• Motility studies including scintigraphy can be helpful in documenting delayed gastric emptying which may contribute to GER and early satiety.
3. Management of gastroesophageal reflux (GER):
• Short-term use of acid neutralizers (e.g. magnesium or calcium carbonate) and/or inhibitors of acid secretion (e.g. histamine blockers and proton pump inhibitors (e.g. famotidine, ranitidine, omeprazole) for symptomatic management. However, prolonged use may be associated with a greater risk for gastroenteritis and pneumonia.
• When delayed gastric emptying or diminished motility is present, prokinetic agents (e.g.metaclopramide, erythromycin) may be useful.
• Use of probiotics such as acidophilus or lactobacillus to help maintain a healthy gastrointestinal flora, particularly following antibiotic treatment or in the setting of prolonged use of acid inhibitors, is an area deserving further study.
• Laparoscopic anti-reflux Nissen fundoplication during g-tube placement may be of value in the SMA patient with medically refractory GER, and in whom the benefit is deemed to outweigh the associated surgical and anesthetic risks.
III. Growth and under or over nutrition problems
Children with SMA are at risk for growth failure or excessive weight gain. Growth failure is commonly seen in non-sitters and some sitters, while obesity is a problem in the stronger sitters and walkers. Decreased activity and lean body mass will lead to reduced resting energy expenditure and increased risk of obesity.
Management of growth and under or over nutrition problems:
• The goal is to maintain each child on his/her own growth velocity.
• Follow growth velocity curves (weight, height/length, weight/height) followed over a period of time. Recumbent length, segmental measurements or arm span may be useful if contractures complicate length measurement.
• Assessment of nutritional intake by a dietician or other health care provider proficient in nutrition is recommended at each visit. A 3 day dietary record is a simple and accurate tool to assess nutritional intake. A 24 hour food recall is a practical method to highlight major nutritional concerns and inquire regarding use of any special supplements.

87
• With a reduction in lean body mass, calculated body mass index (BMI) will significantly underestimate body fat. This may result in inappropriate dietary recommendations which could lead to relative obesity.
• SMA patients at risk of obesity should have growth parameters in the lower percentiles for weight/height and BMI.
• It is important to document appropriate intake of calcium and vitamin D.
• Checking pre-albumin levels may help assess adequate protein status.
IV. Management of nutrition in acutely sick SMA patients
• SMA patients, particularly non-sitters and sitters, are particularly vulnerable to catabolic and fasting states, and are more likely to develop hypoglycemia in the setting of fasting. It is therefore necessary to avoid prolonged fasting, particularly during acute illness, in all SMA patients.
• Nutritional intake should be optimized to meet full caloric needs within 4-6 hours following admission for acute illness, via enteral feeding, parenteral feeding or a combined approach as necessary.
• Prompt post-operative caloric supplementation is recommended to avoid muscle catabolism, particularly in a child with reduced fat store. If enteral intake is not imminent then IV caloric feeding should be considered.
ORTHOPEDICS CARE AND REHABILITATION
Overview of orthopedic care and rehabilitation strategies in SMA
A. Key problems: Muscle weakness resulting in contractures, spinal deformity, and increased risk of pain, osteopenia and fractures.
B. Key evaluation procedures:
• Range of motion (ROM)
• strength, function
• seating and mobility
• orthotics
• radiographs (spine and other joints)
• DEXA scan
• Orthopedic surgery
I. Recommendations on evaluation and treatment by functional levels
A. Non-sitters:
Assessments:
• Physical and occupational therapy evaluation of function (CHOP-INTEND) •
• Speech therapy evaluation if swallowing is impaired or speech affected by jaw contracture • or inadequate voice.
Key interventions:
• Nutritional support
• Posture management: Patient’s primary posture should direct choice of equipment that supports function. Ensure comfortable seating.
• Contracture management: Splinting to preserve ROM and prevent pain may be indicated.
• Pain management

88
• Therapy for ADL and assistive equipment: Play and occupational support should include lightweight toys and assistive technology with variable controls and a myriad of activation systems.
• Wheelchair: Ensure optimal independence and seating comfort.
• Limb orthotics: Upper extremity (UE) orthotics to aid in function includes the use of mobile arm supports or elastic slings that augment active range of motion and functional abilities.
• Environmental controls and home modifications to allow for safe accessibility and optimal independence.
B. Sitters:
Assessments:
• Functional assessment (Hammersmith Functional Motor Scale for SMA, the Modified-Hammersmith functional motor scale for SMA, Gross Motor Function Measure (GMFM), and the Motor Function Measurement (MFM) scale for neuromuscular disease).
• Contracture measurement by goniometry.
• Strength measurement by manual muscle testing or myometry.
• Spine and hip radiographs.
• Equipment evaluation of seating, mobility, positioning, and self care equipment. Evaluations for manual and power mobility may be conducted as early as 18 to 24 months of age.
Key interventions (PT, OT, and orthopaedics):
• Wheelchair mobility. Ensure optimal independence and seating comfort.
• Environmental controls and home modifications to allow for safe accessibility and optimal independence.
• Contracture management constitutes major focus of treatment with regular stretching and bracing program to preserve flexibility. Serial casting for contractures may improve standing and improve tolerance of bracing. AFO orthotics may delay development of Achilles tendon contractures. Upper extremity orthotics with mobile arm supports or slings augment active range of motion and functional abilities.
• Regular exercise should be encouraged to maintain fitness and endurance and might include swimming and adaptive sports.
• Standing is encouraged. Light weight ischial weight bearing KAFOs or reciprocal gait orthoses (RGO’s) for standing or assisted ambulation for those with sufficient strength. Where this is not possible a standing frame or mobile stander with AFOs should be considered.
• Spine orthotics and surgery (see below).
C. Walkers:
Assessments:
• Balance and ambulation evaluations include a specific survey of environmental adaptability and access.
• Evaluation of joint ROM and spinal alignment.
• PTand OTassessments to determine appropriate mobility aides, adaptive equipment, assistive technology and environmental access.
• ADLassessment for equipment and adaptation.
• Non-spine x-rays and DEXA• are considered in the event of acute musculoskeletal injures, as a result of overuse, an accident or a fall.
Key interventions:
• Wheelchair for longer distance transportation adds mobility and independence.
• Contracture management and education to maximize joint protection.
• PTand OT to maximizes safety, endurance and independence or to prolong ambulation.
• Walking should be encouraged• with appropriate assistive devices and orthotics.

89
• Regular exercise to maintain fitness and stamina. May include swimming, aquatic therapy, horseback riding and adaptive sports.
• Driver’s education alternatives and consideration of customized driving controls.
• Environmental controls and home modifications to allow for safe accessibility and optimal independence.
• Spine and limb orthotics if scoliosis and contractures start to develop
• Spine surgery (see below).
II. Orthotics
• It is important that the orthotist, therapist and family work together to ensure that the appropriate orthosis is fabricated and allows wearers to meet their functional goal.
• The orthotist should have a good background and experience in working with patients with neuromuscular disorders to choose proper materials and to make adaptations that allow for “best” fit and function.
• Spinal orthoses may be used for postural support but there is insufficient evidence to support delayed curve progression. When used, spinal orthoses should be fabricated with an abdominal cut out to allow appropriate diaphragmatic excursion and access to gastrostomy tubes where present.
III. Orthopedic surgery
1. Hip subluxation and contractures:
• Hip subluxation in SMA is rarely painful. Surgical reduction and osteotomy is frequently followed by redislocation. In most circumstances, this surgery is avoidable.
• Ankle and foot deformities make conventional shoes difficult to wear, and may be an indication for soft tissue releases. In walkers, if soft tissues releases are performed, rapid and aggressive physical therapy may improve outcome.
2. Scoliosis surgery:
• Scoliosis surgery provides benefits in sitting balance, endurance, and cosmesis. Earlier surgery results in better outcome.
• Scoliosis surgery appears to be beneficial in patients that live beyond two years of age when curves are severe and progressive and should be performed while pulmonary function is adequate.
• Beneficial effects of scoliosis surgery on pulmonary function remain controversial, but the rate of pulmonary decline may be slowed.
• Complications of intraoperative excessive bleeding may occur. Postoperative complications include loss of correction, pseudarthrosis, a requirement for prolonged ventilatory support, and chest and wound infections.
• Careful consideration is warranted for the ambulatory SMA patient, since altered function, balance and respiration may result in loss of independent walking.
IV. Perioperative management in SMA
1. Pre-operative management:
• A plan for orthotic intervention including timing and modification of orthoses.
• New wheelchair or wheelchair modification (seat, back, arm, leg or head rests) likely to be required.
• Instruction in transfer including arrangements for a mechanical lift, if necessary.
• Arrangements for bathing, toileting and dressing equipment and potential modifications to clothes.
• Pre-operative spirometry, noninvasive (NIV) pulmonary supports such as BiPAP and, if necessary, cough-assist devices.

90
2. Post-operative management:
• Confirm timing of appropriate casting and fitting of orthoses, allowed ROM, and activity, and that appropriate adaptive equipment is available.
• Appropriate use of incentive spirometry and NIV pulmonary support.
• Instruction of nursing staff and family on bed mobility, transfers, dressing, bathing and toileting.
• Mobilization as soon as possible as allowed by the procedure and surgeon.
PALLIATIVE CARE
• Optimal clinical care for SMA patients should be mindful of potential conflict of therapeutic goals. This conflict is made more difficult by the natural involvement of surrogate decision makers for a dependent infant (parents, siblings, other relatives, caregivers, payers and the wider community).
• There is a deep responsibility to present care options in an open, fair and balanced manner, to be started soon after diagnosis.
• A choice for or against interventional supportive care is not a single binary choice, nor must it be unchanging with circumstance. Sufficient time, honest appraisal of the choices, openness to revisiting decisions made, and personal rapport are essential.
• Placement of gastrostomy tube is better done relatively early when associated risks are lower in order to provide more stable and comfortable nutritional support later when feeding is more tenuous.
• Discuss and determine early the appropriate response to potential life-threatening respiratory insufficiency, as emergency resuscitation and endotracheal intubation during times of crisis without prior respiratory support is associated with many more problems than when decisions are made in advance. If appropriate, other forms of non-invasive respiratory aid should be introduced in time and according to increasing need.
• End of life care decisions need to be defined, neither delayed nor aggressively foisted upon unsuspecting, grieving, stunned parents.
• Care is often best accomplished with a multi-specialty team approach, including appropriate medical, social, and spiritual assistance as appropriate. In addition, hospice referral or other provision for the specific issues regarding terminal care, grief and bereavement support are important.
• In the circumstance of a choice against mechanical ventilatory support, appropriate provision for management of terminal dyspnea can be of comfort to patient and family alike. Use of nebulized narcotics can avoid much of the concern that overdosing contributes to death and provide comfort to the patient.

91
Appendix 14: A Short Version of the TREAT-NMD interim findings
Diagnosis of DMD:
Clinical examination must include seeing the child try to run, jump, climb stairs and get up from the floor. Common presenting symptoms include abnormal gait with frequent falls, difficulties in rising from the floor, tip-toe walking, and pseudohypertrophy of calves. Examination may reveal decreased or lost muscle reflexes, and commonly a positive Gower sign, i.e. the need to make use of arms to push to erect position from lying by moving hands up the thighs. Many signs of proximal muscle weakness will be detected much more easily in the corridor than the consulting room.
Massive elevation of the serum CK (at least 10–20 x normal and often much more) is nonspecific but always present. The finding of a high CK level should prompt urgent specialist referral for confirmation of the diagnosis. The clinician should be made aware of the association of non-hepatic elevation of AST and ALT in DMD. Unexpected elevation of these enzymes should raise the suspicion of high CK.
Genetic testing should be performed. A deletion of the dystrophin gene will be found in around 70% of cases, a duplication in around 6% and the remaining cases will have a point mutation. Readily available genetic tests for DMD are not always exhaustive and a negative result on initial testing does not exclude the disease. It is very important to understand the tests offered by a particular laboratory and their limitations—further specialist input may be necessary. A laboratory diagnosis should be possible in > 95% of cases.
General signs of muscular dystrophy will be seen in a muscle biopsy, including muscle fibre degeneration, muscle regeneration, and increased content of connective tissue and fat. Dystrophin analysis on a muscle biopsy specimen will always be abnormal and offers a route to confirm the diagnosis, complementary to genetic testing. Dystrophin analysis needs to be followed by molecular genetic testing in order to be able to offer genetic counselling to other family members.
An integral part of the diagnostic process is offering, through professional genetic counselling, determination of the carrier status of the mother by molecular genetic testing. Even if the condition has arisen as a result of a new mutation, there is an average 10% risk of recurrence due to germline mosaicism. Genetic counselling should also be offered to sisters and aunts (mother’s side) in reproductive age if the mother carries the mutation. Around the time of diagnosis it is useful to provide contact with a named member of support staff and imperative to offer details of parent/patient support groups such as the national muscular dystrophy charities and the Parent Project.
Neurology:
Corticosteroids should be given for DMD at or before the point at which the physical typically seen around the age of 4-6. Less functional gain may be seen if initiation of steroids is delayed until close to the loss of ambulation. The most common daily dosage regimes are 0.75 mg/kg/day prednisone/prednisolone and 0.9 mg/kg/day deflazacort. They are likely to be equally effective, but have slightly different side-effect profiles. Deflazacort may produce less weight gain but has a higher risk of asymptomatic cataracts. Other regimes suggested to reduce the incidence of steroid-associated side-effects include alternate day dosing, lower dose daily regimes and intermittent regimes (e.g. 10 days on/10 days off; high dose on weekends). It is important to note that none of these regimes have been tested against the daily dosing schedules so that their relative efficacy in the long term is not known.
Immunity to chicken pox (and in high risk populations, tuberculosis) should be ensured ahead of starting steroids. Boosters of vaccinations should be up to date, and consider giving the 6 year booster early if needed. Monitoring for efficacy should include tests of muscle function and strength (e.g. timed function tests, Hammersmith motor ability score, MRC muscle strength score), FVC and parent and child perception of the value of the treatment.
Monitoring and prophylaxis of the predictable side-effects of steroid use should go hand in hand (http://enmc.org/workshop/?id=21&mid=88). Major side effects to consider are behavioural changes, failure to gain height, excessive weight gain, osteoporosis, impaired glucose tolerance, immune/adrenal suppression, dyspepsia/peptic ulceration, cataract, and skin changes. It is for this

92
reason important to monitor weight, height, blood pressure, urinary dipstix (glucose), cushingoid features, mood/behaviour/personality/GI skin changes, red reflex of eyes, bone fractures, and recurrent infections. Many side effects can be dealt with without dose reduction or withdrawal of the corticosteroid. Monitoring for weight gain should be accompanied by dietary advice and support prior to starting steroids, behavioural changes should be supported by psychological input and advice on behaviour management, advice on bone health should be provided alongside monitoring of fracture frequency. Concomitant treatment with non-steroidal anti-inflammatory agents should be avoided. Abdominal pain/peptic ulcerations can be treated with antacids.
Despite prophylactic measures above; certain events may necessitate dose reduction. These include behavioural changes disrupting family/school life, weight gain 25% or 3 centile increase from baseline, failure to gain height or skin changes (e.g. acne, striae, hirsuitism) unacceptable to child/family, fasting blood glucose >110 mg/dl (>6.1 mmol/l) or blood glucose 2 hours after meal >140 mg/dl (7.8 mmol/l), unusually high frequency of infections/unusual organism, persistent GI symptoms (abdominal pain, heartburn, GI bleeding) despite treatment with antacids.
Corticosteroids should be stopped if severe and/or unacceptable side effects occur. Events that may necessitate this include severe behaviour changes disrupting family/school life, weight gain/failure to gain height or skin changes unacceptable to the child/family despite dose reduction, diabetes mellitus defined as fasting blood glucose >126 mg/dl (7.0 mmol/l) or blood glucose 2 hours after meal >200 mg/dl (>11.1 mmol/l), or confirmed hypertension (systolic blood presure increased 15-30 mm Hg over 97th centile or diastolic blood pressure increased 10-30 mm Hg over 97th centile for height), unusually high frequency of infections/unusual organism on lowered dose corticosteroids, or GI symptoms not satisfactorily controlled by antacids and lowered dosage of corticosteroids. If corticosteroids need to be stopped, they should be tapered/stopped slowly over weeks and not suddenly. Suggested tapering of drug dosage is to take ½ the regular corticosteroid dose the first week, ¼ the dose during the second week, the dose during the third week and thereafter stop corticosteroid medication.
Continuation of steroids beyond the loss of ambulation is common practice in some centres for the possible protective effect on spinal alignment, respiratory and cardiac function. There is, as yet, no evidence for any benefit of starting steroids after a boy has stopped walking. However, some patients may notice an improvement in function and forced vital capacity.
GI-Nutrition:
Adequate dietary advice should be offered from a young age, with focus on healthy eating habits that the whole family could benefit from, with specific focus on weight control, adequate calcium and vitamin D intake and controlled sodium intake. Particular emphasis should be placed on appetite control around time the corticosteroids are started. The weight in boys without nutritional problems should be measured 1-2 times a year. If there is active concern about overweight or underweight the weight should be measured more frequently. Situations where changes in weight are to be expected should also initiate weight monitoring (e.g. loss of walking ability, before major surgery).
A child’s ideal weight is determined by his height and is influenced by loss of lean body mass (such as in Duchenne Muscular Dystrophy). Tracking a child’s weight and height on a centile chart gives an indication of excessive weight gain. Body Mass Index (BMI) body weight divided by the square of height (kg/m, centile adjusted for age and sex) is a more reliable measure of adiposity and can also be tracked on a chart. Clinical judgment, taking into account all issues such as emotional, psychosocial and familial aspects, will influence the dietary advice. To prevent excessive weight gain a dietician should be involved at diagnosis, at initiation of steroids and at loss of ambulation. A dietician should also be involved if there is a tendency to underweight. In case of overweight it is preferable to aim for a weight loss of 0.5 kg per month, or stabilization of weight in cases when a long-term normalization is preferred.
Problems with undernutrition are most likely after the boy starts using a wheelchair (approximately 12-13 years of age) and may be multifactorial. The first step is to evaluate intake and to optimize if necessary the existing diet with energy and protein. The next step with more severe malnutrition is enteral nutrition during night time. Nutritional status should always be examined before major surgery and in particular undernutrtion should be addressed before surgery. Particularly overweight patients

93
may have respiratory dysfunction during sleep so may require additional sleep evaluation of oxygen saturation prior to surgery.
Especially in steroid treated boys, if the diet is not adequate in supply of calcium and vitamin D, these should be added separately to reach a recommended intake of calcium (4-8 years: 800 mg/d; 9-18 years: 1300 mg/d) and vitamin D (400 IU). In later stages of the disease there can be difficulty swallowing and where this leads to aspiration and/or undernutrition, discussion of feeding by tube or percutaneous endoscopic gastrostomy (PEG) is indicated.
Respiratory care:
Serial measurement of forced vital capacity (FVC: absolute values and as predicted for height, arm span, or ulna length) provides an easy way to document the progression of respiratory muscle weakness. Once clinical signs of nocturnal hypoventilation develop or FVC drops to 1.25 l or <40% predicted value, then serial measurement of overnight oximetry allows the recognition of the development of nocturnal respiratory failure. This can be done easily at home through the use of small portable machines. Symptoms should also be sought for at every clinic attendance. Serial measurement of peak cough flow will enable monitoring of cough effectiveness. Methods of augmenting cough such as assisted coughing, volume recruitment techniques, cough assist machine should be considered when the PCF is below 270 l/min in non-ambulant boys and introduced before PCF isless than 160 l/min. Once a patient’s FVC begins to drop, boys are susceptible to chest infections and should be offered flu, pertussis, and pneumonococcal vaccination. When coughing is ineffective, antibiotics should be provided promptly. Chest physiotherapy such as postural drainage and assisted coughing should be taught when coughing is ineffective and may need to be supplemented with cough assist machine or other volume recruitment techniques such as glossopharyngeal breathing.
Ask for symptoms of nocturnal hypoventilation at every visit. Symptomatic nocturnal hypoventilation is an indication for elective non-invasive nocturnal ventilation (NIV). NIV should also be considered if nocturnal oxycapnography /polisomnography shows low SaO2 or elevated pCO2. Extending ventilation to the daytime should be considered if the patient presents elevated pCO2 lowered or SaO2 while awake. Greater patient comfort is achieved with intermittent ventilation with positive pressure, using a mouthpiece. Structured education of the ventilator user and the carers and regular follow-up should be an integral part of the treatment, and there should be surveillance of NIV complications, e.g. air leaks, gastric distension, mucosal dryness, facial bone deformation. Anaesthetic techniques must be tailored to minimize intra and post-operative respiratory and cardiovascular depression and may require invasive monitoring and access to intensive care. Depolarizing muscle relaxants should be avoided because of the risk of hyperkalaemia.
Cardiac care:
Cardiac investigation (echocardiogram and ECG) is indicated at diagnosis, every 2 years thereafter to age 10 and then annually, or more often, if abnormalities are detected. Cardiac investigation should be done prior to general anaesthesia at any age. Cardiac MRI may be useful in patients with limited echocardiographic acoustic window. Abnormalities of cardiac rhythm should be promptly investigated and treated. Periodic Holter monitoring should be considered for patients with demonstrated cardiac dysfunction.
ACE-inhibitors should be started at a subclinical deterioration of cardiac function as detected by echocardiography. One long term study suggests that even earlier start of ACE-inhibitor medication prevents later deterioration. Early prophylactic treatment with ACE-inhibitors at a pre-clinical stage is therefore recommended by several centers from 5-10 years of age, although there is still no general consensus on this. Dependent on type and stage of cardiomyopathy. Dilated cardiomyopathy is the most common form. ACE-inhibition and beta-blocker both at once or ACE inhibitors first followed, when indicated, by beta-blockers should be initiated in the presence of progressive abnormalities, with the addition of diuretics and other with onset of heart failure.
Anticoagulation therapy should be considered in patients with severe cardiac dysfunction to prevent systemic thromboembolic events. Ventricular arrhythmias may occur at any time, but occur more frequently in the late stage of dystrophinopathic cardiomyopathy. Periodic Holter monitoring should

94
therefore be considered for patients with demonstrated cardiac dysfunction. Isolated premature ventricular beats require no treatment, but it is important to monitor the cardiac status carefully. If major ventricular arrhythmias occur, antiarrhytmic treatment should be introduced, bearing in mind the possible negative inotropic effect of agent chosen. Carriers should have cardiac investigation (echocardiogram and ECG) every five years, or more frequently if abnormalities are found.
Orthopaedics:
In ambulant children night splints should be provided when there is loss of dorsiflexion at the ankle when there is loss of normal grade of dorsiflexion and before the foot can only achieve plantagrade. Daytime AFOs are not recommended before loss of ambulation. In non-ambulant children sitting AFOs are recommended as painful contractures will develop that also impact negatively on posture. Some children will require tenotomies but AFOs are still needed after surgery. KAFOs can be considered to delay contracture development and prolong ambulation. Standing frames or swivel walkers can delay contracture development in non-ambulant children. DMD spine deformity presents an onset in the early teens if not long term treated with corticosteroids. A fusion surgery can be recommended as soon as there is clear progression and the Cobb angle passes 25 – 30 degrees.
Psychosocial:
Every family should be offered a home visit at diagnosis to help come to terms with the emotional and practical problems that the knowledge of the diagnosis bring up. For example, feelings of loss, guilt, anger, talking to affected child and siblings about the disease. Issues about access for home, school, leisure and barriers to independence. Social (information, advocacy and advice) and psychological support should be offered at times of changing needs and crises. For example, considering more children, moving/adapting house, loss of ambulation, surgery, cardiac & respiratory problems, starting university/employment, end of life. Psychological support should be offered to affected children and their families during times of emotional/behavioral problems. Learning difficulties/ autism spectrum disorders should be identified early and recommendations given to parents and educators about managing these difficulties.
Rehabilitation:
Annual neurological, respiratory and cardiological assessments should ideally be co-ordinated via a centralized DMD rehabilitation unit. From the time of diagnosis the boys must be assessed once to twice a year by therapists (physiotherapists and occupational therapists) with special experience in neuromuscular disorders. The interval between the assessments depends on the boy’s age, the progression of the disease and his functional ability. The aim of the assessments is to set up a plan for interventions for the child to optimize his physical, social and intellectual abilities. The plan should ensure that professionals and parents are ahead of the events and prepared in advance on the next stage of disease.
Assessments of physical abilities, that are repeated with fixed intervals, are needed to determine the rate of progression of the disease. Major aims of the physiotherapist and occupational therapist are to encourage activity and promote function. This includes interventions to delay or reduce complications due to the deterioration of muscle strength, and to give guidelines regarding activities, possibilities, adaptations and adjustments enabling the boys/men to live a socially active life together with family and friends. Annual home visit / assessments performed by an interdisciplinary team from a special DMD rehabilitation unit is recommended to support the family and the local team of physiotherapists, occupational therapists, social worker and teacher. Resisted exercises should not be prescribed as there is no evidence that they are useful but there are concerns that they may accelerate muscle damage. Moderate levels of active exercise particularly in the hydrotherapy pool is recommended. Children taking steroids may acquire additional motor skills such as riding a bike and this encourages independent play and interaction with peers.
Wheelchairs should be supplied to improve mobility and independence. Tilt-in-space / recline electric wheelchairs with supportive seating should be supplied early to avoid postural contractures and poor sitting posture. Annual, centralized courses for young and adults with DMD and their families are recommended, arranged by the NMD-association in co-operation with the centralized rehabilitation unit.

95
Oral care:
Boys with DMD should see a dentist with extended experience and detailed knowledge of the disease, preferably at a centralized or specialist clinic. The dentist’s mission should be to strive for high-quality treatment, oral health and wellbeing and to function as a resource for the families and the boy’s own dentist in his home community. This dentist should be aware of the specific differences in dental and skeletal development in boys with DMD and collaborate with a well-informed and experienced orthodontist. Oral and dental care is to be based on prophylactic measures with a view to maintaining good oral and dental hygiene. Individually adapted assistive devices and technical aids for oral hygiene are of particular importance when the muscular strength of the patient’s hands, arms and neck begins to decrease.

96
Appendix 15: Best Practice Information by BMJ Evidence
Muscular Dystrophies71
• In the first (ambulatory) stage of treatment for the muscular dystrophies, the focus should be on preserving muscle strength, namely using glucocorticoids. ‘Glucocorticoid therapy can delay wheelchair dependence by more than 3 years and the need for scoliosis surgery and mechanical ventilation’.72,73 The patients and their families should also be given psychological support to discourage overprotection, prevent sibling resentment, encourage maturation, and to aid adherence to treatment options. Finally, physiotherapists should be provided in order to administer muscle strengthening exercises (although there is no evidence that such exercises cause patients to remain functional for longer), to reverse musculotendinous contractures (alongside lower limb surgery; together these may increase brace-free ambulation by more than 1 year).74
• In the second (wheelchair-bound) stage of treatment the focus should be on maintaining daily activities. This can be achieved through use of either standard or motorised wheelcairs, robotic arms75,76, lower limb resting splints, computers for environmental control, and/or tendon-release surgery. The majority of studies indicate that moulded ankle-foot orthotics are not worthwhile.77 Nutrition should also be maintained using non-invasive ventilation to increase swallowing time as tachypneoa increases. Finally, scoliosis should be monitored and treated if necessary using Luque rod instrumentation before the curve exceeds 45°78,79, but after the patient has become wheelchair dependent80. Around 90% of patients that have been treated with glucocorticoids do not need surgery to correct scoliosis; hence, this should be the first line approach taken.72,81
• The third (ventilatory supported) stage of treatment should focus on muscle rest and support. This can be achieved through use of IPPV, with a mouthpiece during the day and a nasal or oro-nasal attachment at night.82 Patients should be introduced to IPPV if they show symptoms of nocturnal low ventilation.83 As the disease progresses, patients may require more ventilatory support; depending upon nocturnal IPPV 24 hours a day.84 The use of non-invasive IPPV, along with MAC to reverse airway secretion congestion, prevents the need to undertake a tracheotomy.85 Expiratory muscles can be supported by use of mechanical insufflation-exsufflation.86
Myasthenia Gravis87
• There are a broad range of therapeutic strategies available for myasthenia gravis, with strategy generally varying according to symptom severity.
• Patients with mild disease severity may not require any treatment, but if symptoms are frequent they should be given a cholinesterase inhibitor, such as pyridostigmine, with corticosteroids given to those who fail pyridostigmine monotherapy.88
• Patients with moderate disease severity should be given immunosuppressants on top of the pyridostigmine that mild patients are given. Examples are low-dose corticosteroids, azathioprine, mycophenolate mofetil, cyclosporin and tacrolimus.88,89,90 Such immunosuppressants should begin to be administered at a low dose, and the dose should be gradually increased as necessary up to a maximum dose, which is set to prevent the worsening of symptoms associated with a high dose.
• In addition to drug therapies, patients with moderate and severe forms of the disease may benefit from a thymectomy; this can even be performed in the absence of a thymoma.91 Any benefits are not usually seen until 2-5 years after the surgery, and remission rates tend to be positively correlated with the extent of thymus removal. Patients may also benefit from plasma exchange, in order to avoid peri-operative corticosteroids or other immunosuppressive drugs. However, effects of this are temporary, lasting no more than a few weeks.
• Patients are classed as having a severe disease if they are unable to swallow or if they have breathing difficulties, particularly if their breathing becomes so poor that they require mechanical

97
ventilation (myasthenic crisis). This normally accounts for around 15% to 20% of patients92, and can be provoked by infections, aspiration, medicines (including high-dose corticosteroids), malignancy, surgery, or trauma.
• Treatment for such patients should initially consist of removal of the provoker, optimising ventilatory settings, and acute therapy with immunoglobulins or plasma exchange.93 Acute therapy is expensive, requiring hospitalisation. However, IV immunoglobulin is easy to administer94, and response is rapid, reaching a maximal within a matter of weeks.
• Following recovery from a myasthenic crisis, immunosuppressants should be administered on a long-term basis89,90,91,93, with an aim to establish a minimally effective dose. Tacrolimus tends to be better tolerated than ciclosporin.99 As an alternative to immunosuppressive therapy (if it is not tolerated or if symptoms worsen despite it), chronic IV immunoglobulin therapy should be administered every 4 to 6 weeks.93,95

98
Appendix 16: Best Practice Information for Muscular Dystrophies
Unless there is a known risk in the baby boy, a diagnosis of Duchenne muscular dystrophy is unlikely to be made until between two and three years of age. Diagnosis is usually through observation of posture and “waddling” gait, and may be confirmed by testing for blood levels of creatine phosphokinase (usually five times greater than the upper limit found in non-affected boys).1
If the blood levels of creatine phosphokinase are elevated a muscle biopsy may be performed to test for dystrophin. In addition electromyography should be available to measure the electrical activity that is generated in muscles when the muscle contracts.1
Ultrasonography and electrocardiograms will show whether there is any abnormal heart activity.1
Care management options for muscular dystrophies should include:
• Monitoring of sleep and early intervention with non-invasive home ventilation as this is the ONLY intervention that has been shown to improve life expectancy.
• Exercise to keep the muscles working.
• Physiotherapy to maintain muscle strength and help prevent muscle contractures and joint deformities
• Physical aids such as wheelchairs and splints/calipers; major and minor house adaptations e.g. en-suite ground floor, wheelchair accessible accommodation, through floor lifts to showers, taps, lowered steps etc. Also school adaptations from major to minor and equipment e.g. tables, chairs, IT equipment to class room and personal assistants.
• Pacemaker for irregular heart rhythms.
• Glucocorticoid corticosteroids may be used cautiously in the short-term (up to two years) to stabilise or improve muscle strength and walking in boys with DMD, but the overall benefit must be weighed against the long-term side effects of these drugs.96
• Surgery to correct postural deformities, although currently there is no quality evidence for scoliosis surgery for people with Duchenne muscular dystrophy97, but has helped with care and management of the patient and relieved back pain symptoms in those having surgery.
• Emotional support for the individual patients, their families and carers.1

99
Appendix 17: Best Practice Information for SMA In most cases a diagnosis of SMA can be made by the SMN gene test, which determines whether there is at least one copy of the SMN1 gene by looking for its unique sequences (that distinguish it from the almost identical SMN2) in exons 7 and 8. In some cases, when the SMN gene test is not possible or does not show any abnormality, other tests such as an electromyography (EMG) or muscle biopsy may be indicated.1 The major management issue in Type I SMA is the prevention and early treatment of respiratory infections as pneumonia is the cause of death in the majority of the cases.1

100
Appendix 18: Best Practice Information for Myasthenia Gravis
Diagnosis of MG can be difficult because the symptoms can be subtle and hard to distinguish from both normal variants and other neurological disorders. A thorough physical examination can reveal easy fatigability, with the weakness improving after rest and worsening again on repeat of the exertion testing. A good response to medication can also be considered a sign of auto-immune pathology. Alternatively, if the diagnosis is suspected, a serological blood test can be performed to identify antibodies against the acetylcholine receptor. The test has a reasonable sensitivity of 80–96%.1
As this is an auto-immune condition that affects voluntary muscles (i.e. ones that the person is able to control) treatment options are different to those for muscular dystrophies. Initially symptoms are mild, worsening over months and reaching most severe within one year.1
There is no cure for this condition. The care management programme should reflect the need to control the symptoms, which may result in patients becoming symptom free and able to lead a normal or a near normal life with few symptoms. For some people this will simply be rest and a good night’s sleep. For others, it should include:
Medication through the use of:
• cholinesterase inhibitors, drugs that can (where symptoms are mild) improve muscle contractions and strengthen affected muscles
• steroids or immunosuppressants, drugs that alter the body’s immune system and reduce the production of antibodies. Over time, for some people, there may be a significant reduction in symptoms and in some cases complete relief from symptoms. This may be a treatment option for people unable to have thymectomy or for whom the symptoms have not improved following thymectomy.1
Thymectomy – surgical removal of the thymus gland. This treatment is only recommended for people under 60 years of age unless they have a Thymoma. It improves symptoms in some 70% of people affected by MG, and in some 30% the symptoms disappear altogether. Improvement is usually noted in the first year after surgery, but there may be continued improvement for up to three years post-operatively. There is an international randomised control trial of Thymectomy on-going (as at November 2008).1
For some people whose muscle weakness is so severe as to cause life-threatening breathing or swallowing symptoms the following treatment should be available. It is noted that both produce rapid improvement, but the benefits only last for a few weeks. These treatments are not suitable for long-term treatment, only for when the patient is seriously ill:
a) plasmophoresis – where the plasma containing the harmful antibodies is removed and
replaced with antibody free plasma b) intravenous immunoglobulin therapy – the patient is injected with normal antibodies so as to
change the way the immune system works.1
There are additional self-help activities that patients may use to effectively manage the impact of their symptoms, which may include:
• Eating softer food or taking more frequent smaller meals so as to reduce chewing if the muscles affected are those used for speaking, chewing and swallowing
• Conserve energy by using electrical devices for day-to-day activities (e.g. electric toothbrush)
• Plan activities for when energy levels are highest.1
The use of neuro-physiology is necessary in the management of MG. In addition, treatment of MG can consist of medication and/or surgery. Medication consists mainly of cholinesterase inhibitors to directly improve muscle function or immunosuppressant drugs to reduce the auto-immune process. It is common for patients to be treated with a combination of these two groups of drugs. The drugs used to control MG either diminish in effectiveness over time (cholinesterase inhibitors) or cause severe side effects of their own (immunosuppressants). Most patients will require drug treatment for the remainder

101
of their lives. Thymectomy is a surgical intervention to remove tumours of the thymus gland and can also be performed for those with benign thymic enlargement, to improve the myasthenia.1

102
Appendix 19: Muscular Dystrophy Campaign Reports The Muscular Dystrophy Campaign report ‘Building on the Foundations: The Need for a Specialist Neuromuscular Service across England December 2007’28 set out the following key elements of care:
• The care of all patients with a neuromuscular condition should be led from a regional Specialist Neuromuscular Centre with specialist multi-disciplinary teams providing regular local clinics.
• Specialist multi-disciplinary care may be lead by neurologists, clinical geneticists, paediatricians, paediatric neurologists or rehabilitation physicians. This specialist supervision supports and oversees local provision.
• Agreed standards of diagnosis and care should be developed and agreed and disseminated across all regions including to patients and patient groups to ensure equity of care.
• Provision of expert physiotherapy, early cardiac monitoring and intervention and corticosteroids to improve muscle function and maintain independent mobility.
• Judicious use of spinal surgery and expert respiratory services (including non invasive positive pressure ventilation to improve quality of life and delay the onset of respiratory failure to prolong life.
The Muscular Dystrophy Campaign report ‘Building on the Foundations: The Need for a Neuromuscular Service Serving Patients in the NHS South East Coast Region’ (May 2009)2 set out the following additional elements of care:
• Access to neurorehabilitation clinics; which can include physiotherapy, occupational therapy, speech and language therapy, wheelchair services and orthotics; to maintain independence, adapt to changes in disease severity, improve quality of life and delay progression of the condition.
• Provision of specialist orthopaedic care for the treatment of spinal deformities which are common in patients with neuromuscular disorders, to correct spinal deformity, improving posture, increasing comfort, and prolonging life.
• Monitoring of scoliotic development at a specialist muscle clinic every 3 to 6 months; including a spinal inspection, chest x-rays, radiographs, swallow studies, pulse oximetry and polysomnography; to ensure that surgery is performed when the spine is mobile and at a Cobb angle of twenty to forty degrees, when optimal success rates will be achieved.
• Access to respiratory services (i.e. ventilation) to improve quality and length of life, and to reduce impromptu admissions to hospitals.
• Screening for heart problems every two years up to age 10, every year after this, and prior to any surgery, since many neuromuscular conditions affect the heart and cardiomyopathy can progress almost entirely without symptoms until signs of heart failure emerge.
• Cardiac screening available for women who are carriers of mutations in the dystrophin gene, as they are at increased risk of cardiomyopathy.
• Access to specialist physio and hydrotherapies to maintain mobility, independence, improve quality of life, reduce pain, reduce emergency admissions, and perhaps even delay disease progression.
• Inappropriate physiotherapy by a non-specialist may hinder the mobility of the patient, so the treatment of neuromuscular patients should be managed by a specialist physiotherapist who has training in both neurological and musculoskeletal physiotherapies, and experience of treating muscle conditions and who can support outreach clinics and provide training and

103
professional development for community physiotherapists (who are then able to treat neuromuscular patients themselves).
• Coordination of care by a specialist muscle nurse, to support self-management of the disease; reducing admissions, length of stay, and improving wellbeing.
• Input from a neuromuscular care advisor to coordinate health and social care needs,
transition services; and to provide information and support to families of patients. This may decrease hospital admissions, length of stays, re-admissions and GP intervention.
• Support from psychologists to enable patients and their families to develop coping mechanisms and management strategies during loss of ambulation, bereavement, diagnosis, and transition. Additionally, psychological support may be required to manage the behavioural side effects of corticosteroids, as well as disorders such as autism, OCD and ADHD which occur more frequently in male DMD patients. Such psychological support will reduce the need for consultants to take on the role, as is happening currently, and therefore save time for other aspects of care.
• Access to a dedicated key worker to support transition to adulthood.
• Support from a neuromuscular care advisor during diagnosis.

104
Appendix 20: The recommendations set out by the Walton Report on Access to Specialist Neuromuscular Care30
• The Department of Health and the NHS (together with each of the National Health organisations in the devolved countries) should ensure that specialised neuromuscular services are given their own place in the National Definition Set to reflect the urgent need to treat these services as a priority following years of under-investment and weak coordination.
• SCGs should follow the lead of the South West SCG and West Midlands SCG and conduct an urgent in-depth review of neuromuscular services in the regions.
• The responsibility for ensuring that access to specialised, multi-disciplinary neuromuscular services is available for all patients should lie with a named individual postholder in the National Specialised Commissioning Group and with a named individual in each of the English regions and devolved Health administrations.
• The lack of Workforce Planning must be addressed as a priority as highlighted in the NHS Next Stage Review. While there are some excellent services provided at the four national muscle centres, they are vulnerable through their reliance largely on the research interests and drive of the individual leading clinician. These services must be protected and strategically developed with succession planning.
• A summit meeting should be organised by NHS Workforce Planning with the leading neuromuscular clinicians, the Royal Colleges, the other professional bodies and the Muscular Dystrophy Campaign to develop and implement a Neuromuscular Workforce Plan by June 2010.
• The DH and the NHS should introduce a funded development plan (working with the Muscular Dystrophy Campaign) by the end of March 2010 to retain the existing posts and to ensure that a national network of Care Coordinators is established within 5 years. There are currently only 13 Neuromuscular Care Coordinators, but to provide adequate support to the 60,000 people living with these conditions a network of some 60 Care Coordinator posts is required. Investment in these posts is cost-effective as they have been shown to save consultants’ time, reduce emergency admissions and re-admissions, reduce hospital stays and coordinate care locally.
• It should be ensured that vital support is accessible from professionals such as physiotherapists, speech and language therapists, occupational therapists and dieticians, due to fact that muscular dystrophy and related neuromuscular conditions are complex, multi-system disorders requiring specialist multi-disciplinary care. In order to achieve this health commissioners and managers should assess the needs and gaps in professional support in their local area and produce a development plan to address service weaknesses by March 2010.
• The DoH should commission an urgent comparative study to identify international best practice and ensure this is disseminated in this country to clinicians, commissioners and patients, since it has become clear that the UK falls well behind some other countries internationally in the provision of care for patients with a neuromuscular condition. Life expectancy in the UK for individuals with Duchenne muscular dystrophy, for example, lag behind those achieved elsewhere in Europe in countries such as Denmark and the Netherlands. In those countries, specialist multi-disciplinary care with excellent links to supported, independent living is the norm.
• NICE should put in place a Clinical Guideline for Duchenne muscular dystrophy, building on the existing Standards of Care developed by the TREAT-NMD European consortium; this will help to drive up standards and also give patients and families a clear understanding of the standards of care to which they are entitled.
• It is essential for patients’ participation in future trials to ensure that our standards of care are harmonised with the best available elsewhere in Europe.

105
• The Government and commissioners should take urgent action to make sure that a named transition co-ordinator is in place for each young person with a neuromuscular condition who is moving from paediatric to adult services.
• Health service commissioners, local authorities, and local government associations should ensure that they develop effective plans to guarantee meeting the comprehensive health and social care needs of the growing number of adults with neuromuscular disorders. Good multi-disciplinary services should be well co-ordinated, involving specialities such as genetics, a muscle pathology service, cardiology, respiratory and cardiac support and specialist physiotherapy.
• The Department of Health should set up a Short-Life Review Group involving the Muscular Dystrophy Campaign and representatives from other patients’ organisations in order form proposals by June 2010 which will address the failings in the provision of vital services such as wheelchair, equipment and orthotic support.
• The Department for Communities and Local Government should undertake an urgent review of access to hydrotherapy pools across the country, involving the Local Government Association, the Muscular Dystrophy Campaign and other organisations. The review should be completed and recommendations brought forward within 12 months to ensure that all people living with a neuromuscular condition have the opportunity to use a fully accessible, local hydrotherapy pool, if they wish.
• The NHS should develop and implement a plan by December 2010 to gather data systematically regarding the numbers of children and adults with a particular neuromuscular condition and also to collect data regarding life expectancy, in order to plan services effectively and also to enable comparisons to be made both between UK regions and countries and also on a comparative, international basis.
• The Department of Health, the Social Care Institute for Excellence and the local government associations should undertake a systematic review of social care support for people living with a neuromuscular condition by September 2010, as stark failings in social care have been highlighted. Failings identified include significant problems in: accessing support for taking up and retaining employment, securing home adaptations in a reasonable timeframe, independent use of public transport, and accessing higher and further education.

106

107

108





























