Embed Size (px)
Citation preview

doi.org/10.26434/chemrxiv.12552998.v1
Basis Set Extrapolations for Density Functional TheoryPeter Kraus
Submitted date: 24/06/2020 • Posted date: 06/07/2020Licence: CC BY-NC-ND 4.0Citation information: Kraus, Peter (2020): Basis Set Extrapolations for Density Functional Theory. ChemRxiv.Preprint. https://doi.org/10.26434/chemrxiv.12552998.v1
Improving results of correlated wavefunction theory calculations by extrapolating from successive basis sets isnowadays a common practice. However, such approaches are uncommon in density functional theory,especially due its faster convergence towards the basis set limit. In this work I present approaches for basisset extrapolation in density functional theory that enable users to obtain results of 4-zeta quality from 3- and2-zeta calculations, i.e. at a fraction of the computational cost. The extrapolation techniques work well withmodern density functionals and common basis sets.
File list (2)
download fileview on ChemRxivmain_v1.0.pdf (408.61 KiB)
download fileview on ChemRxivsi_v1.0.pdf (201.81 KiB)

Basis set extrapolations for density functional theory
Peter Kraus∗
School of Molecular and Life Sciences,Curtin University,
GPO Box U1987, Perth 6845, WA, Australia
Abstract1
Basis set extrapolation is a common technique in wavefunction theory, used to squeeze bet-2
ter performance out of the highest affordable level of theory by combining it with lower quality3
calculations. In this work, I present analogous techniques for basis set extrapolation in density4
functional theory, focusing on [2,3]–ζ calculations, and including double hybrid and dispersion5
corrected functionals. My recommendations are based on basis set limit data from finite element6
calculations, estimates of basis set limits for double hybrid corrections, and they are validated using7
the GMTKN55 and NCDT datasets. A short review of extrapolation methods for Hartree-Fock8
calculations based on modern finite element data is carried out to inform this work. Extrapolation9
of [2,3]–ζ calculations in cc-pvXz-pp and def2-Xvpd basis sets with the proposed recipes routinely10
matches and sometimes outperforms 4–ζ calculations at a fraction of the cost. The methods are11
implemented in Psi4, allowing for an automated and intuitive application.12
1 Introduction13
The concept of estimating complete basis set (CBS) energies using basis set extrapolation methods14
is nothing new for practitioners of wavefunction theory (WFT). Usually, such extrapolation methods15
involve calculating absolute energies using correlation-consistent basis sets of consecutive ζ-qualities,16
with Helgaker’s power formula and cubic scaling (Eq. (1)) being one common example. [1]17
∗E-Mail: [email protected]
1

Ecorr∞ = Ecorr
X − AX−α, α = 3 (1)
Here, Ecorr∞ is the correlation (i.e. post-Hartree-Fock) energy at an infinite basis set size, while X denotes18
the cardinal number of the finite basis set, with this number usually equivalent to the ζ-ness of the basis19
set. The parameter A is system-dependent and is always fitted to EcorrX data. Many different variants of20
Eq. (1) have been proposed since, sometimes replacing the power scaling AX−α term with exponential21
(Ae−αX), [2] exponential–square-root (Ae−α√X , expsqrt in the following) or other functions. Results of22
such [X − 1, X]–ζ extrapolations are usually comparable to the results from a X + 1–ζ calculation,23
provided sufficiently large basis sets are used (X >= 4). [3] Smaller basis sets might perform better with24
a specifically tailored α. [4]25
The Hartree-Fock (H-F) component of WFT calculations converges to the basis set limit rather26
quickly when compared to the correlation energy. In Truhlar’s H-F extrapolation scheme, derived from27
estimates of Ne, HF, and H2O basis set limits, Eq. (1) is applied with α = 3.4 which is optimal for a28
[2,3]–ζ extrapolation. [4] Halkier et al. have proposed the use of Eq. (2) for extrapolating Hartree-Fock29
energies:30
EH−F∞ = EH−F
X − Ae−αX , α = 1.63 (2)
This “global” α parameter has been determined from fits of [2-6]–, [3-6]–, and [2-5]–ζ energies of diatomic31
molecules using correlation-consistent basis sets, with EH−F∞ values sourced from numerical H-F results32
of 9 species available at the time. [5] This extrapolation method performs well, and it is the default33
H-F extrapolation method in Psi4. [6] A three-point variant of Eq. (2), where both α and A are fitted34
to results from [X − 2, X − 1, X]–ζ calculations, is used in cfour, [7] despite the lower generality of35
this variant. [5] The exponential–square-root (expsqrt) function, shown in Eq. (3), is used by default in36
ORCA,37
2

EH−F∞ = EH−F
X − Ae−α√X (3)
with values of α depending on basis set family as well as size. [8] Many other extrapolation schemes are38
available, for a more detailed overview see the recent review of Varandas. [9]39
In density functional theory (DFT), extrapolation methods have gained little traction. Raymond and40
Wheeler reported that while a [3,4]–ζ extrapolation applied to a hybrid density functional approximation41
(DFA) produces reasonable values, the improvements over 4–ζ results are negligible. [10] Jensen showed42
that three-point extrapolation schemes (i.e. where both A and α are fitted) applied to a semi-local43
DFA do not perform well when contracted polarisation-consistent basis sets are used, and two-point44
schemes perform similarly to the non-extrapolated results. [11] This is attributed to the fast convergence45
of DFT with respect to basis set size, which is comparable to H-F, and is therefore much quicker than for46
correlated WFT methods. [11,12] Furthermore, the fitting of functional parameters during the development47
of DFAs may correct for basis set incompleteness, [11] which is why it’s recommended to use basis sets48
comparable to the ones used during the development of such methods. [13]49
With the advent of double hybrid DFAs, which include a correlation component based on second-50
order Møller-Plesset perturbation theory (MP2), the outlook for basis set extrapolation might have51
changed. Similarly, various treatments of dispersion forces have different dependencies on basis set52
sizes. To the best of my knowledge, basis set extrapolation results have been published to-date for only53
one double hybrid DFA, with a focus on chemical kinetics. [14,15] The changes in the density functional zoo54
therefore warrant another visit to the extrapolation pavillion. The goals of this work are the following:55
i) revisit extrapolation schemes for H-F, obtaining values of α derived from modern numerical H-F data;56
ii) investigate basis set convergence for double-hybrid and dispersion-corrected density functionals as57
such, i.e. would an extrapolation work and does it need to be different for single and double hybrids;58
iii) propose, implement, and validate a reasonable and transferrable extrapolation scheme for DFAs;59
and iv) gauge whether such methods are worth it, i.e. whether a [2,3]–ζ extrapolation can approach60
4–ζ results. I have decided to focus on [2,3]–ζ extrapolations, as performance of DFAs does not seem to61
improve significantly past 4–ζ, and there are many large systems for which DFT applied with 3–ζ basis62
3

sets is the currently highest affordable level of theory.63
2 Computational methods64
In this study I investigated the convergence behaviour of three families of basis sets: i) the correlation-65
consistent “Dunning” series supplemented by effective core potentials (ECP), cc-pvXz-pp (X ∈ [d, t, q, 5]);66
ii) the polarisation-consistent contracted and segmented variant of the “Jensen” sets, pcseg-N (N ∈67
[0, 1, 2, 3, 4], with X ≈ N + 1); and iii) the “Karlsruhe” family of basis sets, including their ECPs,68
in (singly-)polarised and augmented variants, def2-Xzvp and def2-Xzvpd, respectively (X ∈ [s, t, q]).69
I have included the Dunning basis sets for comparison with previous works, as well as due to their70
widespread use in DFT calculations. However, some recent works showed that more modern basis sets71
constructed with DFT in mind can be superior to the original Dunning sets for DFT applications. [11,16,17]72
The Jensen basis sets were included as they are modern, and 5 levels of quality are available. Unfortu-73
nately, Jensen basis sets for heavy atoms are not available. Finally, the two variants of the Karlsruhe74
basis sets were included as several functionals have been parametrised using these basis sets, and aug-75
mented variants are available across the whole periodic table. Their main downside compared to the76
Dunning and Jensen basis sets is the lack of a 5-ζ variant.77
To obtain estimates of basis set limits, the H-F energies of two databases of diatomic molecules, and78
MP2 energies of the 15 reference species were calculated using Psi4 1.4a2.dev532. [6] The direct algorithm79
for the evaluation of the electron repulsion integrals was used throughout. For all DFT calculations a80
(150, 974) quadrature grid was used. The MP2 and DFT basis set limits were estimated from calculations81
in [3,4,5]ZaPa-NR-CV basis sets. [18] All MP2 and double-hybrid DFA calculations were carried out with82
core electrons frozen, including the 4f shell for post-lanthanides. Tightened convergence criteria for the83
density and energy (< 10−10 Eh) were applied in all of the above calculations.84
Sampling the large collection of available DFAs and basis sets is always going to introduce biases: an85
exhaustive study is not feasible. For a list of the DFAs that were selected, see Table 1. I have included86
the functionals BLYP-D3(BJ), PBE-D3(BJ), B3LYP-D3(BJ), and B2PLYP-D3(BJ) for their popularity87
in their respective classes. The remaining two generalised gradient approximation (GGA) functionals,88
revPBE-D3(BJ) and B97-D3(BJ), are good performers in the GMTKN55 benchmark, [19] which war-89
4

ranted inclusion. The three meta-GGAs were selected to investigate different dispersion correction90
forms, as well as the differences in approaches to their construction: a “non-empirical” representative91
in SCAN-D3(BJ), a combinatorially optimized B97M-V, as well as an “empirical” representative in92
M06L-D3. My selection of the four single hybrids followed similar patterns, with dlDF+D10 included93
as a counterpoint to M052X-D3: both DFAs are empirically fitted to data, but while in M052X short-94
and medium-range dispersion contributions are included, in dlDF they are excluded by design. Fi-95
nally, the four double hybrids that I selected also range from DFAs with few optimized parameters96
(PBE0DH-D3(BJ)) to methods obtained from extensive fitting to data (DSD-BLYP-D3(BJ)).97
Table 1: Overview of studied DFAs. References correspond to functionals and parametrizations ofdispersion corrections.
Method name DFA type Disp. type Reference
BLYP-D3(BJ) GGA D3(BJ) [20–22]
PBE-D3(BJ) GGA D3(BJ) [22,23]
revPBE-D3(BJ) GGA D3(BJ) [22–24]
B97-D3(BJ) GGA D3(BJ) [22,25]
SCAN-D3(BJ) mGGA D3(BJ) [26,27]
M06L-D3 mGGA D3(0) [28,29]
B97M-V mGGA VV10 [30]
B3LYP-D3(BJ) single hyb. D3(BJ) [22,31,32]
ωB97X-V single hyb. VV10 [33]
M052X-D3 single hyb. D3(0) [29,34]
dlDF+D10 single hyb. DAS2010 [35,36]
DSD-BLYP-D3(BJ) double hyb. D3(BJ) [37]
B2PLYP-D3(BJ) double hyb. D3(BJ) [29,38]
PWPB95-NL double hyb. VV10 [39–41]
PBE0DH-D3(BJ) double hyb. D3(BJ) [42,43]
To obtain HF and DFT energies of species at the basis set limit, I have carried out numerical98
finite element calculations in HelFEM. [44] As neither range-separated hybrids nor double hybrids are99
implemented in HelFEM, the selection of DFAs is limited to BLYP, PBE, revPBE, B97-D, SCAN,100
M06L, B97M-V, B3LYP, M052X, and dlDF. Note that the non-local “VV10” component of B97M-101
V is also not yet implemented in HelFEM. The parameters that I used to obtain total energies with102
the diatomic code are determined for each molecule by the diatomic cbasis tool, requesting 10−10103
accuracy. A 150-point quadrature rule is used with each DFA.104
To investigate the performance of the proposed extrapolation schemes, I chose the GMTKN55 bench-105
5

mark containing 1505 relative energies or 2462 single point calculations. [19] Due to the large size of106
GMTKN55, I used its “diet” subset containing 100 relative energies or 240 single point calculations107
(denoted diet100). [45] The latter has been shown to reproduce the results of the former both qualita-108
tively and quantitatively, but its use is nonetheless re-validated below. Unlike in the original GMTKN55109
study, [19] here I used the same basis sets for all calculations, without augmentation in selected database110
subsets. All calculations are performed with Psi4 using development versions of 1.4, [6] with the default111
convergence criteria for energy and a (75, 302) point integration grid. Double hybrid functionals are112
applied without the frozen core approximation. The figure of merit for the GMTKN55 and diet100113
benchmarks is the weighted mean absolute deviation (WTMAD), which corresponds to the average of114
the weighted absolute devations of computed relative energies from the reference energies in the dataset,115
with weights taken from the original references. [19,45]116
To check performance of the extrapolation methods for energies of van der Waals complexes, I have117
turned to the NCDT dataset [46] in it’s revised form. [47] A modified version of Psi4’s cbs() driver function118
is used to obtain the interaction energies in an automated fashion.119
All raw input and output files, collated results in json format, and Jupyter notebooks used to analyse120
the results and prepare the figures and tables in this manuscript are included in the Supplemental121
information archive.122
3 Results123
As modern DFAs contain various components with presumably different scalings, this is a good point124
to outline the nomenclature I use below. The total energy of a density functional (EDFA), such as125
B2PLYP-D3(BJ), contains a single-reference energy component obtained from the self-consistent cycle126
including the exchange-correlation functional (Efctl), double-hybrid correlation contribution (∆Edh),127
and dispersion correction (Edisp):128
EDFA = Efctl + ∆Edh + Edisp (4)
6

For the B2PLYP-D3(BJ) example above, Efctl is the self-consistent density functional energy, which129
contains the nuclear repulsion, one- and two-electron energies, and the exchange-correlation component130
of the DFA. This single-reference energy contribution contains all components that affect the Kohn–131
Sham orbitals, including the non-local correlation (Enl) term, if present. The ∆Edh is non-zero only132
for double-hybrid functionals and contains the scaled MP2-like correlation. The dispersion term for the133
functionals in this study, including B2PLYP-D3(BJ), depends only parametrically on the functional,134
i.e. it is not affected by basis set size, and therefore shouldn’t be further extrapolated. For MP2, the135
components in the above equation are simply equivalent to the H-F energy, the MP2 correlation energy,136
and zero, respectively.137
3.1 Hartree-Fock extrapolation revisited138
First I focus on the global α = 1.63 value determined for the exponential extrapolation (Eq. (2)) by139
Halkier et al., which was fitted using a set of 9 closed-shell diatomics. [5] While the H-F energy converges140
exponentially with basis set size, [48,49] this is a good point to check whether a power (Eq. (1)) or an141
expsqrt function (Eq. (3)) performs better than the exponential function when used with smaller basis142
sets. For this purpose I used an updated database of H-F energies of 70 diatomic species, obtained143
from numerical basis set calculations, which has been recently published. [44] This dataset includes many144
charged species, open shell diatomics, as well as species with elements from the third row of the periodic145
table. Due to the difficulty in convergence of 3VO− and 1CrMn+, these two molecules are excluded in146
the current study, forming a dataset of 68 species.147
Fig. 1 presents the αs obtained from fits of the logarithms of Eqs. (1–3) to cc-pvXz-pp data (X ∈148
[d, t, q, 5]). Species, for which the total energy doesn’t converge smoothly, or for which the basis set149
contains an ECP, are excluded from this analysis. The average values are α = 1.35±0.24 for exponential,150
α = 4.36± 0.75 for power, and α = 4.91± 0.86 for expsqrt functions, with n = 57. The median values151
(α) are close to the means. As the scatter of the individual αs for the three extrapolation functions is152
very similar, with relative σα of around 18%, the following discussion is focused only on the exponential153
extrapolation function.154
While the α (as well as α) obtained here is significantly lower than the α = 1.63 proposed by Halkier155
7

0.8 1.0 1.2 1.4 1.6
AeX
3 4 5
AX
3 4 5 6Ae
X
Figure 1: Values of α for various extrapolation functions obtained for the set of 68 diatomics, for thecc-pvXz-pp basis set family. The means (α, N) and medians (α, |) also shown. Colours correspond tocationic (red), neutral (green), or anionic (blue) species.
et al., [5] the distribution of charged species indicated by the colour-coding in Fig. 1 shows that anionic156
species converge to the basis set limit slower than neutral or cationic species. This has also been shown157
by Varandas, who highlighted that diffusely augmented basis sets increase the speed of convergence of158
anions, but that it is hard to predict which basis set will yield the best relative energies. [48] The effect159
of different combinations of charges and multiplicities in this dataset is shown as a matrix in Fig. 2.160
There is no correlation of α with multiplicity, but there is a significant correlation with charge. For161
all three charge classes, the relative σα is significantly smaller than the 18% for the overall dataset.162
Therefore, at least for the cc-pvXz-pp basis sets, it may be appropriate to use different α values for163
cationic (1.59), neutral (1.45), or anionic (1.08) species. As this charge-dependent effect is likely to164
be most pronounced in electron affinities and ionisation potentials, the G21EA and G21IP subsets of165
GMTKN55 can be used to validate this charge-dependent approach. In Table 2 I compare the [2,3]–ζ166
and [3,4]–ζ exponential extrapolations using a charge-independent α = 1.35 to an extrapolation using167
the charge-dependent values above. For the electron affinities, the WTMADs of the charge dependent168
extrapolation are lower than for the global approach; for the ionisation potentials the charge dependent169
approach is only beneficial at the [3,4]–ζ level.170
The α values for exponential extrapolation with the four basis set families investigated in this study171
are presented in Table 3. The pcseg-N basis set family (where X ≈ N+1 with N ∈ [0−4]) shows trends172
similar to the cc-pvXz-pp basis sets, but the magnitude of the variations in α results between the charge173
8

Table 2: Comparison of WTMAD values for databases of elecron affinities (G21EA) and ionisationpotentials (G21IP) with exponential global and charge-dependent extrapolation of Hartee-Fock resultsusing cc-pvXz-pp basis sets, with cc-pv5z-pp as reference.
GMTKN55 subsetWTMAD [kJ/mol]
Global α Charge dep. α[2,3]–ζ [3,4]–ζ [2,3]–ζ [3,4]–ζ
G21EA 25.331 9.188 23.192 8.919G21IP 53.815 0.755 53.861 0.616
¹AB
¹AB
²AB
²AB
³AB
³AB
all
all
Multiplicity
AB AB
AB AB
AB + AB +
all all
Char
ge
= 1.13 = 0.13
n = 9
= 1.25 = 0.05
n = 2
= 1.02 = 0.12
n = 8
= 1.08 = 0.15
n = 19
= 1.44 = 0.14
n = 19
= 1.44 = 0.12
n = 6
= 1.46 = 0.11
n = 7
= 1.45 = 0.13
n = 32
= 1.60 = 0.06
n = 5
= 1.57
n = 1 n = 0
= 1.59 = 0.06
n = 6
= 1.37 = 0.22
n = 33
= 1.40 = 0.14
n = 9
= 1.17 = 0.26
n = 15
= 1.35 = 0.24
n = 57
Figure 2: Matrix of values of α, σα, and n for all combinations of charges and multiplicities in thedataset of diatomic molecules, for the cc-pvXz-pp basis set family.
classes is significantly smaller. The overall α = 1.26 is comparable to the results from cc-pvXz-pp basis174
sets. However, the distribution of the α values is almost bimodal as shown in Fig. 3, so the use of a single175
α may still be problematic. The Karlsruhe families converge much faster, with α being almost double176
that of pcseg-N and cc-pvXz-pp. This could also be a consequence of fitting to [2,3,4]–ζ data. The177
inclusion of diffuse functions (def2-Xzvp→ def2-Xzvpd) does not eliminate the scatter in the results for178
anions, but merely shifts the distribution to a higher mean. The extrapolations with power and expsqrt179
functions behave similarly.180
Another approach to H-F extrapolation was proposed recently by Varandas. [48] In summary, the181
method amounts to anchoring an exponential function (Eq. (2)), applicable for all basis set families and182
chemical systems with a universal α = 2.284, by adjusting A to ensure that the fitting function passes183
9

Table 3: Comparison of α± σα values for charge classes and basis set families using exponential extrap-olation.
Basis set familyCharge group
AB+ AB AB− Allcc-pvXz-pp 1.59+/-0.06 1.45+/-0.13 1.08+/-0.15 1.35+/-0.24pcseg-N 1.57+/-0.08 1.26+/-0.19 1.21+/-0.19 1.26+/-0.19def2-Xvp 2.58+/-0.11 2.65+/-0.32 2.11+/-0.38 2.58+/-0.41def2-Xvpd 2.58+/-0.11 2.71+/-0.12 2.59+/-0.61 2.70+/-0.37
0.5 1.0 1.5 2.0 2.5 3.0 3.5cc-pvXz-pp n = 57
pcseg-N n = 43
def2-Xzvp n = 66
0.5 1.0 1.5 2.0 2.5 3.0 3.5def2-Xzvpd n = 65
Figure 3: Values of α for the set of 68 diatomics shown for all studied basis set families. Colourscorrespond to cationic (red), neutral (green), or anionic (blue) species.
through the Hartree–Fock complete basis limit as well as the energy obtained with a STO-2G basis. The184
basis set cardinalities (X) are then renormalised to hierarchical numbers (x), with x = 1 for a STO-2G185
basis set. For the cc-pvXz basis sets, the 〈x〉 values obtained from averaging over a set of 18 neutral,186
closed shell species are 3.02, 3.64, 4.28, and 5.14 for X ∈ [d, t, q, 5], respectively. [48] Unfortunately,187
neither Jensen nor Karlsruhe basis sets were investigated, and the regression was performed with [6,7]–ζ188
extrapolated data as reference values instead of numerical basis set limits. Here, I have re-fitted the189
hierarchical numbers x using the dataset of the 68 diatomic species, by first calculating A for each190
diatomic using the STO-2G result, and then calculating x for each larger basis set, and finally averaging191
and obtaining 〈x〉 over the whole dataset. The 〈x〉s are 3.59±0.54, 4.08±0.53, 4.58±0.58, and 5.26±0.64192
for the cc-pvXz-pp basis sets, which is significantly higher and with a significantly wider σx than the193
results of Varandas. This is likely due to the inclusion of anionic species in the fitting process.194
The performance of the discussed H-F extrapolation methods is presented in Table 4, using cc-195
pv[d,t]z-pp data. In the diatomics dataset, the extrapolated values are compared to and normalised by196
the reference EH−F∞ values obtained from Lehtola, [44] with each species in the dataset weighted equally,197
10

Table 4: Performance of various H-F extrapolation methods in the diatomics dataset and the diet100subset of GMTKN55, using cc-pv[d,t]z-pp data.
Extrapolation method Diatomics MRE diet100 WTMADFunction α [Xd, Xt] Ref. % kJ/mol
exponential 1.630 [2.00, 3.00] [5] 0.0061 12.21exponential 1.450 [2.00, 3.00] [5] 0.0060 12.07power 3.400 [2.00, 3.00] [4] 0.0016 5.76exponential 2.284 [3.02, 3.64] [48] 0.0062 12.41exponential 1.349 [2.00, 3.00] c.w. 0.0060 11.95power 4.361 [2.00, 3.00] c.w. 0.0034 7.29expsquare 4.912 [2.00, 3.00] c.w. 0.0026 6.18exponential 2.284 [3.59, 4.08] c.w. 0.0059 12.07
obtaining a mean relative error (MRE). For the diet100 subset of the GMTKN55 database, the respective198
cc-pv5z-pp values are taken as the reference for each of the 100 reactions, as the goal of this benchmark is199
to estimate the performance of the extrapolation method, not H-F theory as such. The best performing200
scheme overall is Truhlar’s power extrapolation with α = 3.4 (see Eq. (1)), which was developed to match201
estimates of the H-F limit for Ne, HF, and H2O from [2,3]–ζ basis data. [4] The exponential function202
performs significantly worse, whether a global α = 1.63 or [2,3]–ζ specific α = 1.45 is applied. [5] Fitting203
extrapolation constants using a larger dataset, or using the approach of Varandas, [48] fails to improve204
the performance in [2,3]–ζ extrapolation. From the four global fits to the 68 diatomics performed in this205
study, the best function for extrapolating cc-pv[d,t]z-pp data is the expsqrt function (see Eq. (3)).206
3.2 Extrapolation of functional energy in DFAs207
The H-F results show it is not necessary to use the large dataset of 68 diatomics, and that a well-208
performing extrapolation formula can be obtained from fitting to basis set limit estimates of as little as209
3 species, provided it is tailored for a [2,3]–ζ extrapolation. To determine the universality of α values in210
DFT, I have calculated the numerical basis set limit energies for the smaller set of 9 diatomics used by211
Halkier et al. [5] The basis set limits are then used to obtain α for each functional using the cc-pvXz-pp212
basis sets, analogously to the above work for H-F. In addition to a “global” extrapolation using all213
available basis sets within the family, I have also included a “[d,t]” extrapolation using only results from214
[2,3]–ζ basis sets. Only the functional (Efctl) component of the energy is extrapolated. For the B97M-V215
functional, where Efctl contains the non-local correlation (Enl) term, this term is subtracted from the216
11

Efctl value to be directly comparable to the HelFEM results.217
0.5
0.5
1.0
1.0
1.5
1.5
2.0
2.0
M06L-D3M052X-D3
B97M-VdlDF+D10
SCAN-D3(BJ)B97-D3(BJ)PBE-D3(BJ)
BLYP-D3(BJ)revPBE-D3(BJ)B3LYP-D3(BJ)
H-F
AeX
+/-
0.66+/-0.220.78+/-0.081.10+/-0.271.05+/-0.141.23+/-0.251.31+/-0.211.32+/-0.231.33+/-0.231.31+/-0.341.41+/-0.191.66+/-0.09
Figure 4: Box plots of α of the studied DFAs with cc-pvXz-pp basis sets, for the reference dataset of9 closed-shell diatomics of Halkier et al. The symbols are means (N) and medians (|). The medians ofeach DFA are also listed on the right y-axis.
Box plots of the αs for each studied DFA are shown in Fig. 4, using an exponential extrapolation218
with the cc-pvXz-pp family of basis sets. The H-F result of α = 1.61 (α = 1.66) agrees well with the219
reported data of Halkier et al. obtained with the same dataset, [5] validating the current methodology.220
The two Minnesota DFAs are clear outliers, with α below 1.0. This can be partially attributed to the221
non-negligible amount of dispersion trained into both M06L and M052X, especially as the dispersionless222
dlDF, which uses a functional form similar to M052X, is much closer to the overall mean. The overall223
averages (〈α〉) over all but Minnesota DFAs are 1.26± 0.11 for exponential, 4.06± 0.34 for power, and224
4.58 ± 0.40 for expsqrt extrapolation functions in a “global” extrapolation; for a “[d,t]” extrapolation225
these values decrease to 1.26 ± 0.07, 3.12 ± 0.17, and 3.97 ± 0.22, respectively. Results for other basis226
set families are in the Supplemental material.227
The effect of increasing amount of (Hatree-)Fock exchange (Fx) in the DFA on α is shown in Fig. 5,228
for a series of PBE-related functionals. With the exception of H-F which contains no correlation, only229
the percentages of Fock and PBE exchanges are modified, and the PBE correlation is always fully230
included. There is a clear trend of increasing α and α with increasing proportion of Fock exchange.231
The σα decreases significantly with a higher percentage of Fx. A qualitatively similar trend can be seen232
between the BLYP and B3LYP data, and M06L and M052X data in Figure 4. The four GGA functionals233
investigated in this study (PBE, revPBE, BLYP, and B97-D) have a very similar α ≈ 1.28, and the234
increase in α between B3LYP (20% Fx) and BLYP, ∆α = 0.08, is comparable to the difference between235
12
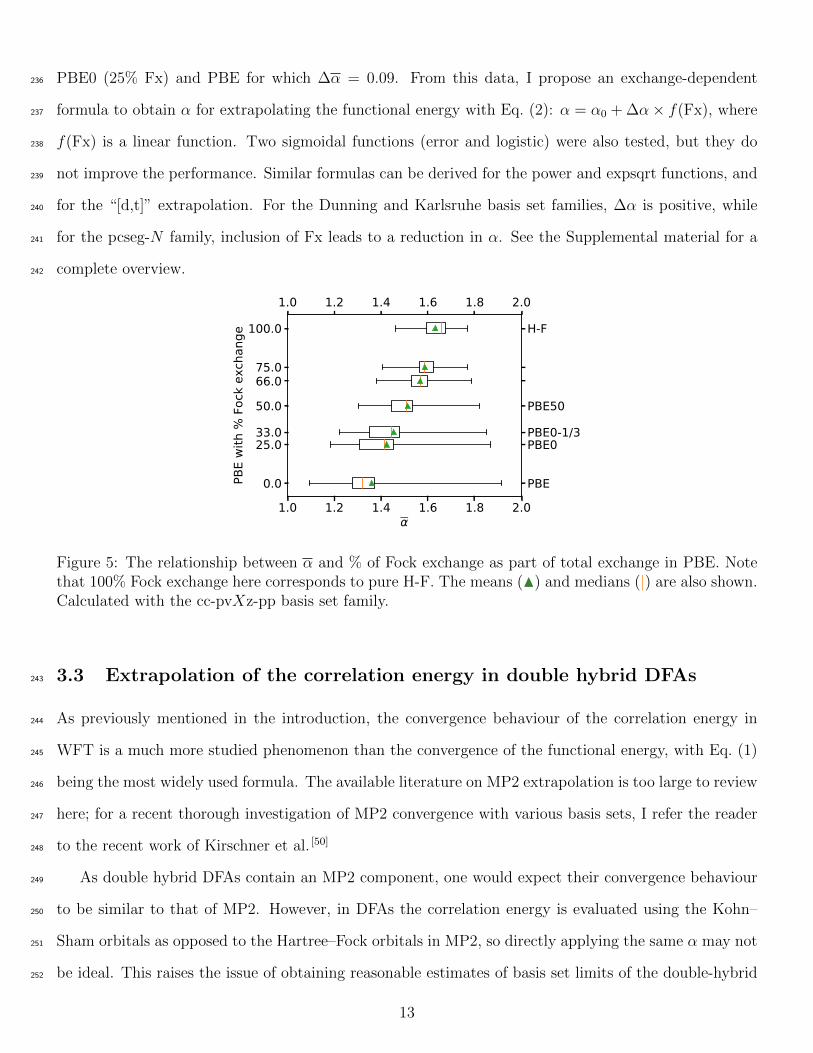
PBE0 (25% Fx) and PBE for which ∆α = 0.09. From this data, I propose an exchange-dependent236
formula to obtain α for extrapolating the functional energy with Eq. (2): α = α0 + ∆α× f(Fx), where237
f(Fx) is a linear function. Two sigmoidal functions (error and logistic) were also tested, but they do238
not improve the performance. Similar formulas can be derived for the power and expsqrt functions, and239
for the “[d,t]” extrapolation. For the Dunning and Karlsruhe basis set families, ∆α is positive, while240
for the pcseg-N family, inclusion of Fx leads to a reduction in α. See the Supplemental material for a241
complete overview.242
1.0
1.0
1.2
1.2
1.4
1.4
1.6
1.6
1.8
1.8
2.0
2.0
0.0
25.033.0
50.066.075.0
100.0
PBE
with
% F
ock
exch
ange
PBE
PBE0PBE0-1/3
PBE50
H-F
Figure 5: The relationship between α and % of Fock exchange as part of total exchange in PBE. Notethat 100% Fock exchange here corresponds to pure H-F. The means (N) and medians (|) are also shown.Calculated with the cc-pvXz-pp basis set family.
3.3 Extrapolation of the correlation energy in double hybrid DFAs243
As previously mentioned in the introduction, the convergence behaviour of the correlation energy in244
WFT is a much more studied phenomenon than the convergence of the functional energy, with Eq. (1)245
being the most widely used formula. The available literature on MP2 extrapolation is too large to review246
here; for a recent thorough investigation of MP2 convergence with various basis sets, I refer the reader247
to the recent work of Kirschner et al. [50]248
As double hybrid DFAs contain an MP2 component, one would expect their convergence behaviour249
to be similar to that of MP2. However, in DFAs the correlation energy is evaluated using the Kohn–250
Sham orbitals as opposed to the Hartree–Fock orbitals in MP2, so directly applying the same α may not251
be ideal. This raises the issue of obtaining reasonable estimates of basis set limits of the double-hybrid252
13

correlation (∆Edh∞ ). Numerical estimates of the MP2 limit (∆EMP2
∞ ) are not as common as for the H-F253
limit. [44] Fortunately, Ranasinghe et al. [18] have collated accurate literature values of the MP2 core-core254
and core-valence contributions from finite element calculations of atoms, [51–54] and explicitly correlated255
calculations of molecules, [55] and proposed a simple linear extrapolation method to obtain near-reference256
results from [3,4,5]–ζ calculations (Eq. (5)).257
∆EMP2∞ ≈ ∆EMP2
5ZaPa−CV + λ× (∆EMP24ZaPa−CV −∆EMP2
3ZaPa−CV), λ = 0.4307 (5)
To prepare a set of ∆Edh∞ data for each of the four functionals studied, I have collated the total MP2258
correlation energies from the same dataset (C, N, O, F, Ne, Ar, F−, Mg, Be, CH2, HF, NH3, F2, N2259
and H2O). The valence-only MP2 correlation is then compared to the extrapolated values using frozen260
core calculations and the above extrapolation approach. [18] The root mean square deviation (RMSD)261
of the two sets is 17.7 mEh, which is higher than the RMSD for core-core + core-valence correlation262
(0.5 mEh), but sufficiently accurate for extrapolation purposes. For comparison, a two point cubic263
power extrapolation from [4,5]ZaPa-CV data has an RMSD of 18.1 mEh. Therefore, the estimated264
∆Edh∞ values were calculated using Eq. (5) (see Supplemental information for the raw data).265
2.2
2.2
2.4
2.4
2.6
2.6
2.8
2.8
B2PLYP-D3(BJ)
DSD-BLYP-D3(BJ)
PBE0DH-D3(BJ)
PWPB95-NL
MP2
AX
+/-
2.59+/-0.16
2.55+/-0.14
2.58+/-0.16
2.48+/-0.15
2.53+/-0.12
Figure 6: Box plots of α for the studied double-hybrid DFAs and MP2 with cc-pvXz-pp basis sets, forthe reference dataset of atoms and small molecules of Ranasinghe et al. The symbols are means (N)and medians (|). The medians of each DFA are also listed on the right y-axis.
The four double-hybrid DFAs and MP2 have a very similar α, as shown in Fig. 6. The convergence266
of MP2, with a global αMP2 = 2.53 is significantly slower than the ideal cubic scaling. For the Dunning267
and Karlsruhe basis set families, the “[d,t]” fits of αdh are ∼ 0.3 lower than the global fits; the Jensen268
14

basis sets are again an outlier for which the global fit is smaller. The “[d,t]” αMP2 values for Ne, HF,269
H2O, and N2 are 2.18, 2.21, 2.37, and 2.28, which is in a good agreement with Truhlar’s αMP2 = 2.2 for270
the same species and basis sets, [4] validating the current method.271
3.4 Performance of [2,3]-ζ extrapolation in GMTKN55272
To significantly reduce the number of calculations required for the benchmarking below, I resort to the273
use of the diet100 subset [45] of the GMTKN55 dataset. [19] The diet100 subset has been validated for274
all of the functionals studied by Goerigk et al., [19] yielding good quantitative agreement between the275
diet100 and full GMTKN55 WTMADs. Here, I re-validate the use of diet100 as a proxy for GMTKN55276
by considering the PBE-D3(BJ) functional and the def2-Xzvpd basis set family. The results presented277
in Table 5 show the discrepancy between the two benchmarks is small, with the largest deviation of278
3 kJ/mol occuring in the def2-qzvpd data. An agreement of the two sets to within 0.2 kJ/mol in the279
extrapolated data validates the use of diet100 subset for this purpose.280
Table 5: Comparison of WTMAD values of the diet100 subset and the full GMTKN55 database for thePBE-D3(BJ) functional, def2-Xzvpd basis sets, and selected extrapolation methods.
Basisdiet100 GMTKN55
WTMAD [kJ/mol]def2-svpd 64.3 62.8def2-tzvpd 43.5 43.6def2-qzvpd 42.6 45.6[s, t]zvpd ORCA [8] 43.1 43.2[s, t]zvpd expsqrt 42.5 42.7
As comparing the extrapolation results of all methods with all studied functionals for all four basis281
set families would be tedious, an overview of the performance of 13 extrapolation methods with the282
cc-pvXz-pp basis sets and a smaller set of functionals is shown in Table 6. Results for other basis283
set families are included in Supplemental material. The extrapolated results are listed along non-284
extrapolated values calculated with 3– and 4–ζ basis sets. A well performing [2,3]–ζ extrapolation285
should approach 4–ζ results. The exponential function performs rather poorly in DFT extrapolations,286
with the extrapolated BLYP-D3(BJ) and B3LYP-D3(BJ) results barely beating 3–ζ data, while for the287
double-hybrid B2PLYP-D3(BJ) it performs even worse. Scaling α based on Fx also shows no benefit,288
15
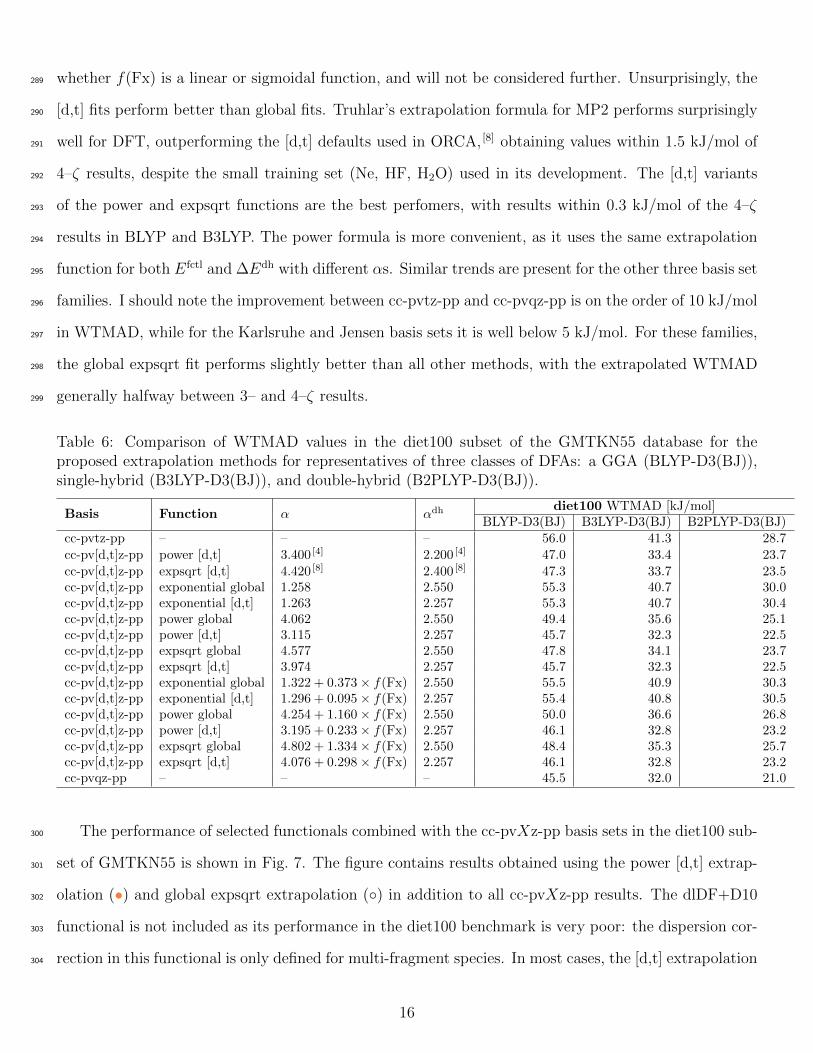
whether f(Fx) is a linear or sigmoidal function, and will not be considered further. Unsurprisingly, the289
[d,t] fits perform better than global fits. Truhlar’s extrapolation formula for MP2 performs surprisingly290
well for DFT, outperforming the [d,t] defaults used in ORCA, [8] obtaining values within 1.5 kJ/mol of291
4–ζ results, despite the small training set (Ne, HF, H2O) used in its development. The [d,t] variants292
of the power and expsqrt functions are the best perfomers, with results within 0.3 kJ/mol of the 4–ζ293
results in BLYP and B3LYP. The power formula is more convenient, as it uses the same extrapolation294
function for both Efctl and ∆Edh with different αs. Similar trends are present for the other three basis set295
families. I should note the improvement between cc-pvtz-pp and cc-pvqz-pp is on the order of 10 kJ/mol296
in WTMAD, while for the Karlsruhe and Jensen basis sets it is well below 5 kJ/mol. For these families,297
the global expsqrt fit performs slightly better than all other methods, with the extrapolated WTMAD298
generally halfway between 3– and 4–ζ results.299
Table 6: Comparison of WTMAD values in the diet100 subset of the GMTKN55 database for theproposed extrapolation methods for representatives of three classes of DFAs: a GGA (BLYP-D3(BJ)),single-hybrid (B3LYP-D3(BJ)), and double-hybrid (B2PLYP-D3(BJ)).
Basis Function α αdh diet100 WTMAD [kJ/mol]BLYP-D3(BJ) B3LYP-D3(BJ) B2PLYP-D3(BJ)
cc-pvtz-pp – – – 56.0 41.3 28.7cc-pv[d,t]z-pp power [d,t] 3.400 [4] 2.200 [4] 47.0 33.4 23.7cc-pv[d,t]z-pp expsqrt [d,t] 4.420 [8] 2.400 [8] 47.3 33.7 23.5cc-pv[d,t]z-pp exponential global 1.258 2.550 55.3 40.7 30.0cc-pv[d,t]z-pp exponential [d,t] 1.263 2.257 55.3 40.7 30.4cc-pv[d,t]z-pp power global 4.062 2.550 49.4 35.6 25.1cc-pv[d,t]z-pp power [d,t] 3.115 2.257 45.7 32.3 22.5cc-pv[d,t]z-pp expsqrt global 4.577 2.550 47.8 34.1 23.7cc-pv[d,t]z-pp expsqrt [d,t] 3.974 2.257 45.7 32.3 22.5cc-pv[d,t]z-pp exponential global 1.322 + 0.373× f(Fx) 2.550 55.5 40.9 30.3cc-pv[d,t]z-pp exponential [d,t] 1.296 + 0.095× f(Fx) 2.257 55.4 40.8 30.5cc-pv[d,t]z-pp power global 4.254 + 1.160× f(Fx) 2.550 50.0 36.6 26.8cc-pv[d,t]z-pp power [d,t] 3.195 + 0.233× f(Fx) 2.257 46.1 32.8 23.2cc-pv[d,t]z-pp expsqrt global 4.802 + 1.334× f(Fx) 2.550 48.4 35.3 25.7cc-pv[d,t]z-pp expsqrt [d,t] 4.076 + 0.298× f(Fx) 2.257 46.1 32.8 23.2cc-pvqz-pp – – – 45.5 32.0 21.0
The performance of selected functionals combined with the cc-pvXz-pp basis sets in the diet100 sub-300
set of GMTKN55 is shown in Fig. 7. The figure contains results obtained using the power [d,t] extrap-301
olation (•) and global expsqrt extrapolation () in addition to all cc-pvXz-pp results. The dlDF+D10302
functional is not included as its performance in the diet100 benchmark is very poor: the dispersion cor-303
rection in this functional is only defined for multi-fragment species. In most cases, the [d,t] extrapolation304
16
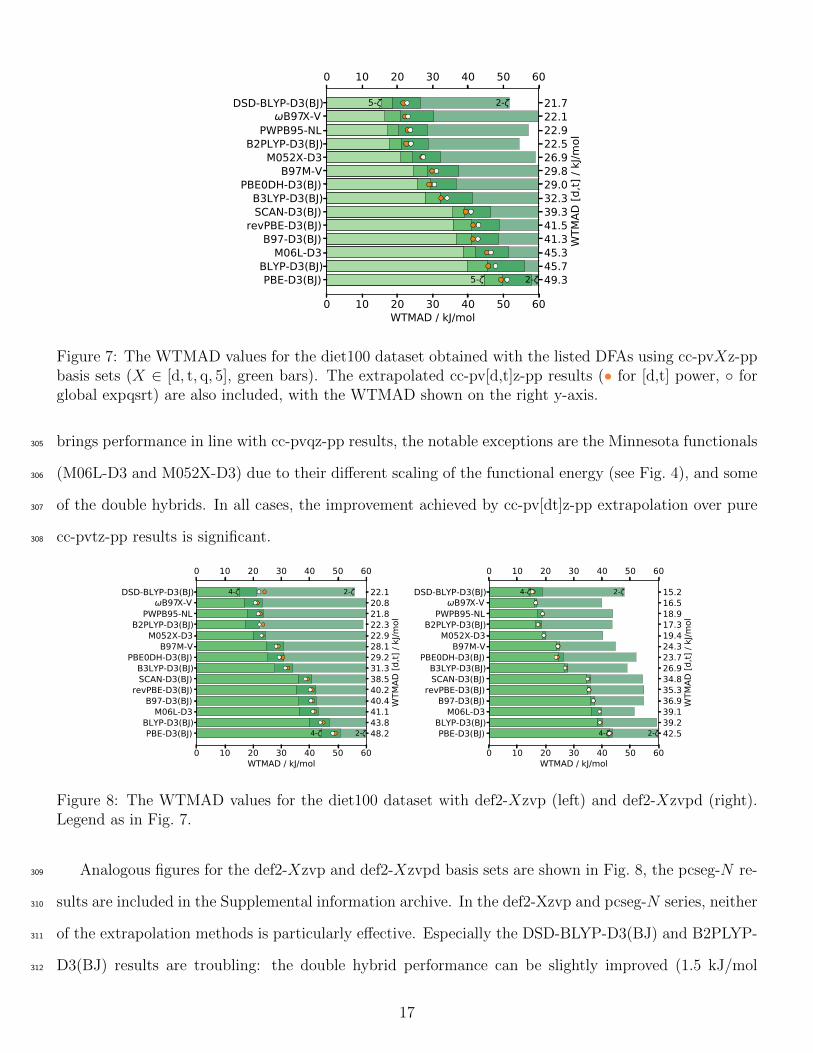
0
0
10
10
20
20
30
30
40
40
50
50
60
60
WTMAD / kJ/mol
PBE-D3(BJ)BLYP-D3(BJ)
M06L-D3B97-D3(BJ)
revPBE-D3(BJ)SCAN-D3(BJ)B3LYP-D3(BJ)
PBE0DH-D3(BJ)B97M-V
M052X-D3B2PLYP-D3(BJ)
PWPB95-NLB97X-V
DSD-BLYP-D3(BJ) 5- 2-
5- 2- 49.345.745.341.341.539.332.329.029.826.922.522.922.121.7
WTM
AD [d
,t] /
kJ/m
ol
Figure 7: The WTMAD values for the diet100 dataset obtained with the listed DFAs using cc-pvXz-ppbasis sets (X ∈ [d, t, q, 5], green bars). The extrapolated cc-pv[d,t]z-pp results (• for [d,t] power, forglobal expqsrt) are also included, with the WTMAD shown on the right y-axis.
brings performance in line with cc-pvqz-pp results, the notable exceptions are the Minnesota functionals305
(M06L-D3 and M052X-D3) due to their different scaling of the functional energy (see Fig. 4), and some306
of the double hybrids. In all cases, the improvement achieved by cc-pv[dt]z-pp extrapolation over pure307
cc-pvtz-pp results is significant.308
0
0
10
10
20
20
30
30
40
40
50
50
60
60
WTMAD / kJ/mol
PBE-D3(BJ)BLYP-D3(BJ)
M06L-D3B97-D3(BJ)
revPBE-D3(BJ)SCAN-D3(BJ)B3LYP-D3(BJ)
PBE0DH-D3(BJ)B97M-V
M052X-D3B2PLYP-D3(BJ)
PWPB95-NLB97X-V
DSD-BLYP-D3(BJ) 4- 2-
4- 2- 48.243.841.140.440.238.531.329.228.122.922.321.820.822.1
WTM
AD [d
,t] /
kJ/m
ol
0
0
10
10
20
20
30
30
40
40
50
50
60
60
WTMAD / kJ/mol
PBE-D3(BJ)BLYP-D3(BJ)
M06L-D3B97-D3(BJ)
revPBE-D3(BJ)SCAN-D3(BJ)B3LYP-D3(BJ)
PBE0DH-D3(BJ)B97M-V
M052X-D3B2PLYP-D3(BJ)
PWPB95-NLB97X-V
DSD-BLYP-D3(BJ) 4- 2-
4- 2- 42.539.239.136.935.334.826.923.724.319.417.318.916.515.2
WTM
AD [d
,t] /
kJ/m
ol
Figure 8: The WTMAD values for the diet100 dataset with def2-Xzvp (left) and def2-Xzvpd (right).Legend as in Fig. 7.
Analogous figures for the def2-Xzvp and def2-Xzvpd basis sets are shown in Fig. 8, the pcseg-N re-309
sults are included in the Supplemental information archive. In the def2-Xzvp and pcseg-N series, neither310
of the extrapolation methods is particularly effective. Especially the DSD-BLYP-D3(BJ) and B2PLYP-311
D3(BJ) results are troubling: the double hybrid performance can be slightly improved (1.5 kJ/mol312
17

reduction in WTMAD) by increasing αdh from 2.382 to 3.0, but even then the extrapolation barely313
outperforms def2-tzvp results. Extrapolation of def2-[s,t]zvp or pcseg-[1,2] results therefore cannot be314
recommended. Upon diffuse augmentation, the difference between 3–ζ and 4–ζ results shrinks further.315
However, this time the global expsqrt extrapolation consistently improves upon def2-tzvpd results, with316
the exception of PWPB95-NL which remains near def2-tzvpd performance.317
3.5 Automated [2,3]–ζ extrapolation of non-covalent interaction energies318
While the GMTKN55 database contains several subsets of non-covalent interaction energies, I have319
decided to use an independent dataset to check the performance for non-covalent energies and also320
gather timing data in an automated fashion. For this purpose, I have modified the cbs() routine of321
Psi4 to accept DFAs, decompose them into fctl, dh, and disp stages consistent with Eq. (4), and allow322
independent extrapolation of these components.323
Table 7: Comparison of RMSD values in the NCDT dataset of non-covalent energies, with cc-pvXz-ppbasis sets. Extrapolation with [d,t] power function, α = 3.115, αdh = 2.257. Timing is the ratio of totalexecution times of cc-pv[dt]z-pp versus cc-pvqz-pp.
FunctionalNCDT RMSD [kJ/mol]
Timingcc-pvtz-pp cc-pvqz-pp cc-pv[dt]z-pp
BLYP-D3(BJ) 7.01 4.84 4.81 83.8%PBE-D3(BJ) 8.86 6.73 6.78 81.3%revPBE-D3(BJ) 5.92 4.44 4.56 81.5%B97-D3(BJ) 5.61 4.14 4.17 83.2%SCAN-D3(BJ) 9.06 7.53 7.62 67.0%M06L-D3 4.29 3.18 3.54 66.1%B97M-V 5.36 3.48 4.07 138.7%B3LYP-D3(BJ) 6.96 4.87 4.81 81.5%M052X-D3 6.02 4.30 4.33 46.6%dlDF+D10 8.28 9.42 9.28 66.1%ωB97X-V 5.03 2.84 3.21 152.4%B2PLYP-D3(BJ) 5.13 2.96 3.60 81.9%DSD-BLYP-D3(BJ) 5.31 3.31 4.31 88.5%PBE0DH-D3(BJ) 7.20 5.60 5.84 94.5%PWPB95-NL 4.05 2.00 2.56 151.1%
Table 7 presents the results for cc-pvXz-pp basis sets, with the cc-pv[d,t]z-pp extrapolation performed324
using the [d,t] power formula from Table 6. For most GGAs, meta-GGAs, and single hybrids, the325
RMSD of the extrapolated results is almost identical to the RMSD of cc-pvqz-pp data at ∼ 80% of the326
18

computational cost. The exceptions are: i) extrapolated M06L-D3, which would benefit from a lower α327
(see Fig. 4); ii) dlDF+D10, where cc-pvtz-pp performs better than both cc-pvqz-pp and cc-pv[dt]z-pp;328
and iii) extrapolated B97M-V and ωB97X-V, which are 50% slower than cc-pvqz-pp due to the cost of329
evaluation of the VV10 contribution as part of the self-consistent cycle. This issue also affects PWPB95-330
NL. However, the Enl contribution can be evaluated as an additive correction at no loss to accuracy. [56]331
Further speedups may be possible, if for example the converged wavefunction at the smaller basis set was332
used as an initial guess for the larger basis set. The improvement of the [d,t] extrapolation over cc-pvtz-333
pp results in double hybrids is generally worse than for the other DFAs, and can be only recommended for334
larger systems, where the cost of cc-pvqz-pp calculations would be prohibitively expensive: the largest335
system in the NCDT dataset is DMS–SO2, for which the computational time of B2PLYP-D3(BJ)/cc-336
pv[dt]z-pp is 56% of B2PLYP-D3(BJ)/cc-pvqz-pp.337
The performance of def2-[s,t]zvp extrapolation (global expsqrt fit) for the NCDT dataset is in line338
with the GMTKN55 results above: the RMSDs of def2-[s,t]zvp are not worse than for def2-tzvp, but def2-339
qzvp performance is not achieved. In any case, the use of large basis sets without diffuse augmentation340
is questionable when accurate non-covalent interaction energies are required. [57] For the same reason,341
I will omit the analysis of pcseg-N with the NCDT dataset; the results for both basis set families are342
tabulated in the Supplemental material. Upon augmentation with diffuse functions, the performance343
of def2-[s,t]zvpd extrapolation (also global expsqrt fit) for non-covalent energies is a positive surprise:344
the RMSDs of def2-qzvpd are matched, if not outperformed, by the extrapolated results (see Table 8).345
dlDF+D10 is again an outlier, as its performance decreases when going from 3–ζ to a 4–ζ basis set.346
3.6 Note on non-local correlation correction347
As the -NL (or -V) non-local corrections are included in the Efctl component, they are extrapolated348
using the same formula and α as the rest of the functional energy. Results for B97M-V presented in349
Fig. 4 show a comparably large spread and a lower α than all other non-Minnesota functionals. The350
Enl term seems to scale almost linearly in the cc-pvXz-pp basis set family, regardless of the integration351
grid used. The DFAs including the VV10 term are some of the best performing functionals, but they do352
not seem to benefit from basis set extrapolation nearly as much as other functionals. The most obvious353
19

Table 8: Comparison of RMSD values in the NCDT dataset of non-covalent energies, with def2-Xzvpdbasis sets. Extrapolation with a global expsqrt function, α = 7.886, αdh = 2.267. Timing is the ratio oftotal execution times of def2-[s,t]zvpd versus def2-qzvpd.
FunctionalNCDT RMSD [kJ/mol]
Timingdef2-tzvpd def2-qzvpd def2-[st]zvpd
BLYP-D3(BJ) 4.09 4.04 3.98 75.8%PBE-D3(BJ) 5.15 5.08 4.89 78.0%revPBE-D3(BJ) 4.30 4.29 4.22 80.6%B97-D3(BJ) 3.91 3.90 3.82 77.8%SCAN-D3(BJ) 6.57 6.45 6.32 62.4%M06L-D3 2.86 2.95 2.75 61.4%B97M-V 2.71 2.68 2.52 132.5%B3LYP-D3(BJ) 3.74 3.66 3.52 76.5%M052X-D3 3.61 3.46 3.39 66.2%dlDF+D10 9.88 10.16 10.08 65.6%ωB97X-V 1.37 1.16 1.20 150.7%B2PLYP-D3(BJ) 1.82 1.85 1.53 77.1%DSD-BLYP-D3(BJ) 2.41 2.48 1.90 77.6%PBE0DH-D3(BJ) 4.65 4.66 4.30 77.5%PWPB95-NL 1.40 1.28 1.38 136.7%
case is in the NCDT results of PWPB95-NL and ωB97X-V with def2-Xzvpd basis sets. It may well be354
better to scale the Enl term separately from Efctl, however, further research in this area is necessary.355
4 Conclusion356
There is no universal recipe for basis set extrapolation in density functional theory. In selected cases, such357
as for cc-pvXz-pp or def2-Xzvpd basis set families, a [2,3]–ζ extrapolation is a cost-efficient approach of358
obtaining results of nearly 4–ζ quality, and I can recommend its use for cases where a 4–ζ calculation is359
prohibitively expensive. However, the recipes are specific to each basis set family, with the cc-pvXz-pp360
sets performing better with a power function fitted to [d,t] data with α = 3.115, and the def2-Xzvpd sets361
with an exponential–square-root function with α = 7.886. I cannot recommend extrapolating pcseg-N362
or def2-Xzvp data.363
The MP2-like correction present in double hybrid density functionals can be extrapolated using a364
power function with αdh = 2.257 for cc-pvXz-pp, and 2.267 for def2-Xzvpd. Despite the use of different365
orbitals, the ∆Edh scales comparably to ∆EMP2. A keen reader will notice the similarity of these results366
20

with Truhlar’s proposed MP2 scaling of 2.2. [4]367
The optimal extrapolation method for Hartree-Fock in [2,3]–ζ basis sets is not exponential, despite368
the exponential convergence of Hartree-Fock towards the complete basis set limit. The power function369
of Truhlar [4] tailored for cc-pv[d,t]z extrapolations with an α = 3.4 remains the best performer given this370
set of requirements. The ideal global α for extrapolation of Hartree-Fock energies of diatomic molecules371
correlates with system charge, but is independent of system multiplicity.372
The development of optimal extrapolation parameters for other basis set families can be considerably373
accelerated by fitting to the basis set limit data, calculated for several functionals in this work. I have374
shown that the dataset of 9 diatomics used by Halkier et al. [5] is more than sufficient for extrapolation375
purposes. While there is a correlation between the amount of Fock exchange in the density functional376
approximation and the optimal extrapolation α, scaling formulas that take the amount of Fock exchange377
into account do not perform as well as functional-independent αs. The modified cbs() routine in Psi4378
allows for an intuitive and automated use of extrapolated density functional theory in calculations of379
energies, gradients, and Hessians.380
5 Acknowledgements381
I would like to thank Susi Lehtola for his help with HelFEM, Julian Gale for comments and feedback,382
and the Forrest Research Foundation for funding. This work was supported by resources provided by the383
Pawsey Supercomputing Centre (project f97) and the National Computational Infrastructure (project384
f97), with funding from the Australian Government and the Government of Western Australia.385
References386
[1] T. Helgaker, W. Klopper, H. Koch, and J. Noga, “Basis-set convergence of correlated calculations387
on water,” The Journal of Chemical Physics, vol. 106, pp. 9639–9646, June 1997.388
[2] D. Feller, “Application of systematic sequences of wave functions to the water dimer,” The Journal389
of Chemical Physics, vol. 96, pp. 6104–6114, Apr. 1992.390
21

[3] C. E. Warden, D. G. A. Smith, L. A. Burns, U. Bozkaya, and C. D. Sherrill, “Efficient and auto-391
mated computation of accurate molecular geometries using focal-point approximations to large-basis392
coupled-cluster theory,” J. Chem. Phys., vol. 152, p. 124109, Mar. 2020.393
[4] D. G. Truhlar, “Basis-set extrapolation,” Chemical Physics Letters, vol. 294, pp. 45–48, Sept. 1998.394
[5] A. Halkier, T. Helgaker, P. Jørgensen, W. Klopper, and J. Olsen, “Basis-set convergence of the395
energy in molecular Hartree–Fock calculations,” Chemical Physics Letters, vol. 302, pp. 437–446,396
Mar. 1999.397
[6] D. G. A. Smith, L. A. Burns, A. C. Simmonett, R. M. Parrish, M. C. Schieber, R. Galvelis, P. Kraus,398
H. Kruse, R. Di Remigio, A. Alenaizan, A. M. James, S. Lehtola, J. P. Misiewicz, M. Scheurer, R. A.399
Shaw, J. B. Schriber, Y. Xie, Z. L. Glick, D. A. Sirianni, J. S. O’Brien, J. M. Waldrop, A. Kumar,400
E. G. Hohenstein, B. P. Pritchard, B. R. Brooks, H. F. Schaefer, A. Y. Sokolov, K. Patkowski,401
A. E. DePrince, U. Bozkaya, R. A. King, F. A. Evangelista, J. M. Turney, T. D. Crawford, and402
C. D. Sherrill, “Psi4 1.4: Open-source software for high-throughput quantum chemistry,” J. Chem.403
Phys., vol. 152, p. 184108, May 2020.404
[7] J. F. Stanton, J. Gauss, L. Cheng, M. E. Harding, D. A. Matthews, and P. G. Szalay, “CFOUR,405
coupled-cluster techniques for computational chemistry, a quantum-chemical program package. Ver-406
sion 2.1,” July 2019.407
[8] F. Neese, “Software update: The ORCA program system, version 4.0,” WIREs Computational408
Molecular Science, vol. 8, no. 1, p. e1327, 2018.409
[9] A. J. Varandas, “Straightening the hierarchical staircase for basis set extrapolations: A low-cost410
approach to high-accuracy computational chemistry,” Annu. Rev. Phys. Chem., vol. 69, pp. 177–411
203, Apr. 2018.412
[10] K. S. Raymond and R. A. Wheeler, “Compatibility of correlation-consistent basis sets with a hybrid413
Hartree-Fock / density functional method,” Journal of Computational Chemistry, vol. 20, no. 2,414
pp. 207–216, 1999.415
22

[11] F. Jensen, “Polarization consistent basis sets. II. Estimating the Kohn–Sham basis set limit,” The416
Journal of Chemical Physics, vol. 116, pp. 7372–7379, May 2002.417
[12] B. C. Cabral and S. Canuto, “The enthalpy of the O–H bond homolytic dissociation: Basis-set418
extrapolated density functional theory and coupled cluster calculations,” Chemical Physics Letters,419
vol. 406, pp. 300–305, May 2005.420
[13] N. Mardirossian and M. Head-Gordon, “Characterizing and understanding the remarkably slow ba-421
sis set convergence of several Minnesota density functionals for intermolecular interaction energies,”422
J. Chem. Theory Comput., vol. 9, pp. 4453–4461, Oct. 2013.423
[14] Y.-Y. Chuang and S.-M. Chen, “Infinite basis set extrapolation for double hybrid density functional424
theory 1: Effect of applying various extrapolation functions,” J. Comput. Chem., vol. 32, pp. 1671–425
1679, June 2011.426
[15] Y.-Y. Chuang and S.-M. Chen, “Infinite basis set extrapolation for double hybrid density functional427
theory 2: Effect of adding diffuse basis functions,” Jnl Chinese Chemical Soc, vol. 59, pp. 1094–1103,428
Sept. 2012.429
[16] F. Jensen, “How large is the elephant in the density functional theory room?,” J. Phys. Chem. A,430
vol. 121, pp. 6104–6107, Aug. 2017.431
[17] D. Feller and D. A. Dixon, “Density functional theory and the basis set truncation problem with432
correlation consistent basis sets: Elephant in the room or mouse in the closet?,” J. Phys. Chem. A,433
vol. 122, pp. 2598–2603, Mar. 2018.434
[18] D. S. Ranasinghe, M. J. Frisch, and G. A. Petersson, “Core-core and core-valence correlation energy435
atomic and molecular benchmarks for Li through Ar,” The Journal of Chemical Physics, vol. 143,436
p. 214110, Dec. 2015.437
[19] L. Goerigk, A. Hansen, C. Bauer, S. Ehrlich, A. Najibi, and S. Grimme, “A look at the density func-438
tional theory zoo with the advanced GMTKN55 database for general main group thermochemistry,439
kinetics and noncovalent interactions,” Phys. Chem. Chem. Phys., vol. 19, no. 48, pp. 32184–32215,440
2017.441
23

[20] A. D. Becke, “Density-functional exchange-energy approximation with correct asymptotic behav-442
ior,” Phys. Rev. A, vol. 38, pp. 3098–3100, Sept. 1988.443
[21] C. Lee, W. Yang, and R. G. Parr, “Development of the Colle-Salvetti correlation-energy formula444
into a functional of the electron density,” Phys. Rev. B, vol. 37, pp. 785–789, Jan. 1988.445
[22] S. Grimme, S. Ehrlich, and L. Goerigk, “Effect of the damping function in dispersion corrected446
density functional theory,” J. Comput. Chem., vol. 32, pp. 1456–1465, May 2011.447
[23] J. P. Perdew, K. Burke, and M. Ernzerhof, “Generalized gradient approximation made simple,”448
Phys. Rev. Lett., vol. 77, pp. 3865–3868, Oct. 1996.449
[24] Y. Zhang and W. Yang, “Comment on “Generalized gradient approximation made simple”,” Phys.450
Rev. Lett., vol. 80, pp. 890–890, Jan. 1998.451
[25] S. Grimme, “Semiempirical GGA-type density functional constructed with a long-range dispersion452
correction,” Journal of computational chemistry, vol. 27, no. 15, pp. 1787–1799, 2006.453
[26] J. Sun, A. Ruzsinszky, and J. P. Perdew, “Strongly constrained and appropriately normed semilocal454
density functional,” Phys. Rev. Lett., vol. 115, p. 036402, July 2015.455
[27] J. G. Brandenburg, J. E. Bates, J. Sun, and J. P. Perdew, “Benchmark tests of a strongly constrained456
semilocal functional with a long-range dispersion correction,” Phys. Rev. B, vol. 94, p. 115144, Sept.457
2016.458
[28] Y. Zhao and D. G. Truhlar, “A new local density functional for main-group thermochemistry,459
transition metal bonding, thermochemical kinetics, and noncovalent interactions,” The Journal of460
Chemical Physics, vol. 125, p. 194101, Nov. 2006.461
[29] L. Goerigk and S. Grimme, “A thorough benchmark of density functional methods for general main462
group thermochemistry, kinetics, and noncovalent interactions,” Phys. Chem. Chem. Phys., vol. 13,463
no. 14, p. 6670, 2011.464
24

[30] N. Mardirossian and M. Head-Gordon, “ΩB97M-V: A combinatorially optimized, range-separated465
hybrid, meta-GGA density functional with VV10 nonlocal correlation,” The Journal of Chemical466
Physics, vol. 144, p. 214110, June 2016.467
[31] A. D. Becke, “A new mixing of Hartree–Fock and local density-functional theories,” The Journal468
of Chemical Physics, vol. 98, pp. 1372–1377, Jan. 1993.469
[32] P. J. Stephens, F. J. Devlin, C. F. Chabalowski, and M. J. Frisch, “Ab initio calculation of vi-470
brational absorption and circular dichroism spectra using density functional force fields,” J. Phys.471
Chem., vol. 98, pp. 11623–11627, Nov. 1994.472
[33] N. Mardirossian and M. Head-Gordon, “ωB97X-V: A 10-parameter, range-separated hybrid, gener-473
alized gradient approximation density functional with nonlocal correlation, designed by a survival-474
of-the-fittest strategy,” Phys. Chem. Chem. Phys., vol. 16, no. 21, p. 9904, 2014.475
[34] Y. Zhao, N. E. Schultz, and D. G. Truhlar, “Design of density functionals by combining the method476
of constraint satisfaction with parametrization for thermochemistry, thermochemical kinetics, and477
noncovalent interactions,” J. Chem. Theory Comput., vol. 2, pp. 364–382, Mar. 2006.478
[35] K. Pernal, R. Podeszwa, K. Patkowski, and K. Szalewicz, “Dispersionless density functional theory,”479
Phys. Rev. Lett., vol. 103, p. 263201, Dec. 2009.480
[36] R. Podeszwa, K. Pernal, K. Patkowski, and K. Szalewicz, “Extension of the Hartree-Fock plus481
dispersion method by first-order correlation effects,” J. Phys. Chem. Lett., vol. 1, pp. 550–555, Jan.482
2010.483
[37] S. Kozuch and J. M. L. Martin, “Spin-component-scaled double hybrids: An extensive search484
for the best fifth-rung functionals blending DFT and perturbation theory,” J. Comput. Chem.,485
pp. 2327–2344, July 2013.486
[38] S. Grimme, “Semiempirical hybrid density functional with perturbative second-order correlation,”487
Journal of Chemical Physics, vol. 124, no. 3, pp. 034108–034108, 2006.488
25

[39] L. Goerigk and S. Grimme, “Efficient and accurate double-hybrid-meta-GGA density function-489
als—Evaluation with the extended GMTKN30 database for general main group thermochemistry,490
kinetics, and noncovalent interactions,” J. Chem. Theory Comput., vol. 7, pp. 291–309, Feb. 2011.491
[40] W. Hujo and S. Grimme, “Performance of the van der waals density functional VV10 and (hy-492
brid)GGA variants for thermochemistry and noncovalent interactions,” Journal of Chemical Theory493
and Computation, vol. 7, no. 12, pp. 3866–3871, 2011.494
[41] F. Yu, “Spin-component-scaled double-hybrid density functionals with nonlocal van der Waals495
correlations for noncovalent interactions,” J. Chem. Theory Comput., vol. 10, pp. 4400–4407, Oct.496
2014.497
[42] E. Bremond and C. Adamo, “Seeking for parameter-free double-hybrid functionals: The PBE0-DH498
model,” The Journal of Chemical Physics, vol. 135, p. 024106, July 2011.499
[43] D. Bousquet, E. Bremond, J. C. Sancho-Garcıa, I. Ciofini, and C. Adamo, “Non-parametrized500
functionals with empirical dispersion corrections: A happy match?,” Theor Chem Acc, vol. 134,501
p. 1602, Jan. 2015.502
[44] S. Lehtola, “Fully numerical Hartree-Fock and density functional calculations. II. Diatomic503
molecules,” Int J Quantum Chem, vol. 119, Oct. 2019.504
[45] T. Gould, “‘Diet GMTKN55’ offers accelerated benchmarking through a representative subset ap-505
proach,” Phys. Chem. Chem. Phys., vol. 20, no. 44, pp. 27735–27739, 2018.506
[46] P. Kraus, D. A. Obenchain, and I. Frank, “Benchmark-quality semiexperimental structural param-507
eters of van der Waals complexes,” J. Phys. Chem. A, vol. 122, pp. 1077–1087, Feb. 2018.508
[47] P. Kraus, “Non-covalent dimers and trimers (NCDT) database version 2.0,” Feb. 2020.509
[48] A. J. C. Varandas, “CBS extrapolation of Hartree–Fock energy: Pople and Dunning basis sets510
hand-to-hand on the endeavour,” Phys. Chem. Chem. Phys., vol. 21, no. 15, pp. 8022–8034, 2019.511
[49] S. Lehtola, “A review on non-relativistic, fully numerical electronic structure calculations on atoms512
and diatomic molecules,” Int J Quantum Chem, vol. 119, p. e25968, Oct. 2019.513
26

[50] K. N. Kirschner, D. Reith, and W. Heiden, “The performance of Dunning, Jensen, and Karlsruhe514
basis sets on computing relative energies and geometries,” Soft Materials, pp. 1–15, Feb. 2020.515
[51] K. Jankowski, P. Malinowski, and M. Polasik, “Second-order correlation energies for F1-, Na1+,516
Mg2+, and Ar8+: Z dependence of irreducible-pair energies,” Phys. Rev. A, vol. 22, pp. 51–60, July517
1980.518
[52] J. R. Flores, “Hierarchic computation of atomic correlation energies using a p-version finite element519
method,” Chemical Physics Letters, vol. 195, pp. 377–382, July 1992.520
[53] J. R. Flores and P. Redondo, “Accurate second-order correlation energies for Mg and Ar,” Int. J.521
Quantum Chem., vol. 45, no. 6, pp. 563–572, 1993.522
[54] J. R. Flores, “New benchmarks for the second-order correlation energies of Ne and Ar through the523
finite element MP2 method,” Int. J. Quantum Chem., vol. 108, pp. 2172–2177, May 2008.524
[55] J. Noga, P. Valiron, and W. Klopper, “The accuracy of atomization energies from explicitly cor-525
related coupled-cluster calculations,” The Journal of Chemical Physics, vol. 115, pp. 2022–2032,526
Aug. 2001.527
[56] A. Najibi and L. Goerigk, “The nonlocal kernel in van der Waals density functionals as an additive528
correction: An extensive analysis with special emphasis on the B97M-V and ωB97M-V approaches,”529
J. Chem. Theory Comput., vol. 14, pp. 5725–5738, Nov. 2018.530
[57] P. Kraus, D. A. Obenchain, S. Herbers, D. Wachsmuth, I. Frank, and J.-U. Grabow, “Xe· · ·OCS:531
Relatively straightforward?,” Phys. Chem. Chem. Phys., vol. 22, no. 10, pp. 5615–5624, 2020.532
27

download fileview on ChemRxivmain_v1.0.pdf (408.61 KiB)

Basis set extrapolations for density functional theory
P. Kraus∗
Supplementary Material
Additional supplemental files and archives in computer-readable form are available onZenodo, see DOI: 10.5281/zenodo.3904120. An executable version of this archive is availableon Binder.
∗E-Mail: [email protected]
S1
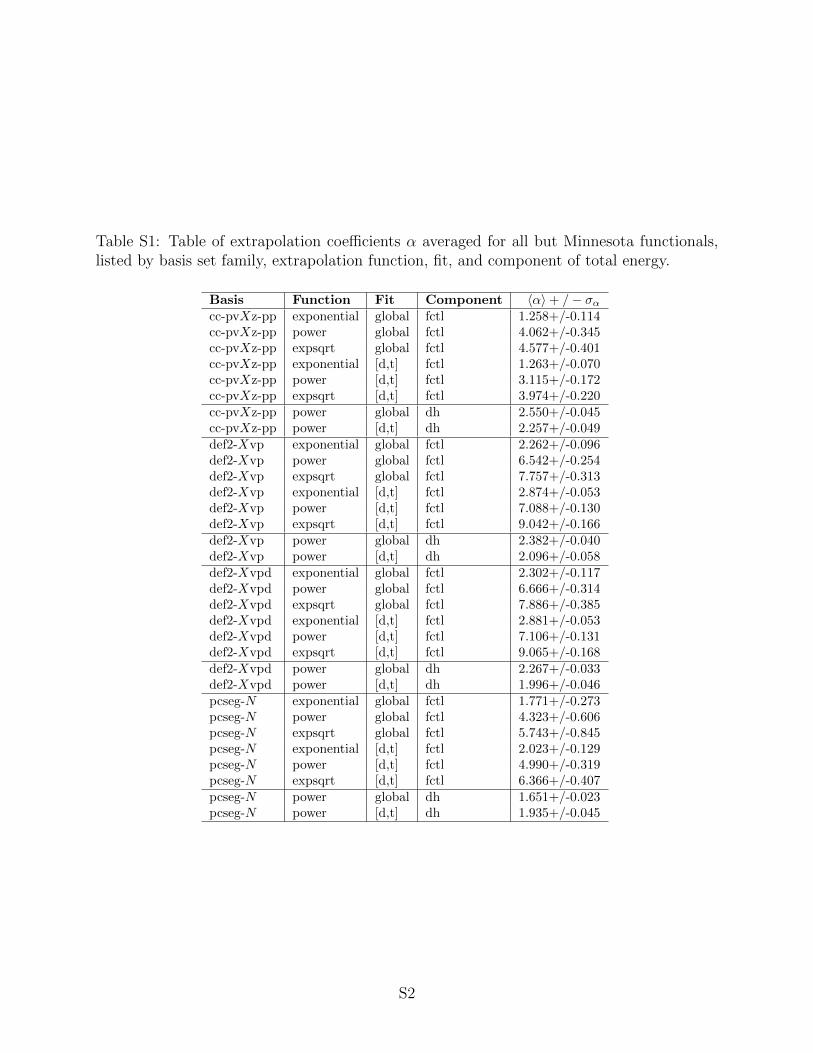
Table S1: Table of extrapolation coefficients α averaged for all but Minnesota functionals,listed by basis set family, extrapolation function, fit, and component of total energy.
Basis Function Fit Component 〈α〉+ /− σαcc-pvXz-pp exponential global fctl 1.258+/-0.114cc-pvXz-pp power global fctl 4.062+/-0.345cc-pvXz-pp expsqrt global fctl 4.577+/-0.401cc-pvXz-pp exponential [d,t] fctl 1.263+/-0.070cc-pvXz-pp power [d,t] fctl 3.115+/-0.172cc-pvXz-pp expsqrt [d,t] fctl 3.974+/-0.220cc-pvXz-pp power global dh 2.550+/-0.045cc-pvXz-pp power [d,t] dh 2.257+/-0.049def2-Xvp exponential global fctl 2.262+/-0.096def2-Xvp power global fctl 6.542+/-0.254def2-Xvp expsqrt global fctl 7.757+/-0.313def2-Xvp exponential [d,t] fctl 2.874+/-0.053def2-Xvp power [d,t] fctl 7.088+/-0.130def2-Xvp expsqrt [d,t] fctl 9.042+/-0.166def2-Xvp power global dh 2.382+/-0.040def2-Xvp power [d,t] dh 2.096+/-0.058def2-Xvpd exponential global fctl 2.302+/-0.117def2-Xvpd power global fctl 6.666+/-0.314def2-Xvpd expsqrt global fctl 7.886+/-0.385def2-Xvpd exponential [d,t] fctl 2.881+/-0.053def2-Xvpd power [d,t] fctl 7.106+/-0.131def2-Xvpd expsqrt [d,t] fctl 9.065+/-0.168def2-Xvpd power global dh 2.267+/-0.033def2-Xvpd power [d,t] dh 1.996+/-0.046pcseg-N exponential global fctl 1.771+/-0.273pcseg-N power global fctl 4.323+/-0.606pcseg-N expsqrt global fctl 5.743+/-0.845pcseg-N exponential [d,t] fctl 2.023+/-0.129pcseg-N power [d,t] fctl 4.990+/-0.319pcseg-N expsqrt [d,t] fctl 6.366+/-0.407pcseg-N power global dh 1.651+/-0.023pcseg-N power [d,t] dh 1.935+/-0.045
S2

Table S2: Table of Fock exchange dependent extrapolation coefficients α + ∆α, listed bybasis set family, extrapolation function, and fit. f(Fx) is a linear function.
Basis Function Fit α+ ∆α× f(Fx)cc-pvXz-pp exponential global 1.322 + 0.373 f(Fx)cc-pvXz-pp exponential [d,t] 1.296 + 0.095 f(Fx)cc-pvXz-pp power global 4.254 + 1.160 f(Fx)cc-pvXz-pp power [d,t] 3.195 + 0.233 f(Fx)cc-pvXz-pp expsqrt global 4.802 + 1.334 f(Fx)cc-pvXz-pp expsqrt [d,t] 4.076 + 0.298 f(Fx)def2-Xvp exponential global 2.286 + 0.481 f(Fx)def2-Xvp exponential [d,t] 2.847 + 0.564 f(Fx)def2-Xvp power global 6.582 + 1.451 f(Fx)def2-Xvp power [d,t] 7.022 + 1.390 f(Fx)def2-Xvp expsqrt global 7.830 + 1.639 f(Fx)def2-Xvp expsqrt [d,t] 8.958 + 1.773 f(Fx)def2-Xvpd exponential global 2.347 + 0.423 f(Fx)def2-Xvpd exponential [d,t] 2.857 + 0.535 f(Fx)def2-Xvpd power global 6.780 + 1.256 f(Fx)def2-Xvpd power [d,t] 7.045 + 1.320 f(Fx)def2-Xvpd expsqrt global 8.033 + 1.473 f(Fx)def2-Xvpd expsqrt [d,t] 8.988 + 1.683 f(Fx)pcseg-N exponential global 1.961 - 0.232 f(Fx)pcseg-N exponential [d,t] 2.102 - 0.096 f(Fx)pcseg-N power global 4.757 - 0.561 f(Fx)pcseg-N power [d,t] 5.185 - 0.238 f(Fx)pcseg-N expsqrt global 6.339 - 0.750 f(Fx)pcseg-N expsqrt [d,t] 6.615 - 0.303 f(Fx)
S3

Tab
leS3:
Tab
leof
lite
ratu
rean
des
tim
ated
vale
nce
-only
∆E
MP2
∞an
d∆E
dh ∞.
Lin
ear
corr
esp
onds
toth
elinea
rex
trap
olat
ion
usi
ng
Eq.
(5),
cubic
corr
esp
onds
toa
cubic
extr
apol
atio
nusi
ng
Eq.
(1).
Molecule
MP2
DFA
Refere
nce
Linear
Cubic
B2P
LY
P-D
3(B
J)
DS
D-B
LY
P-D
3(B
J)
PB
E0D
H-D
3(B
J)
PW
PB
95-N
LC
O-0
.403
74-0
.357
46-0
.35673
-0.1
1004
-0.1
7529
-0.0
5126
-0.0
8491
F2
-0.6
1140
-0.5
8799
-0.5
8691
-0.1
7934
-0.2
8531
-0.0
8327
-0.1
3293
N-0
.113
77-0
.135
52-0
.13554
-0.0
4214
-0.0
6653
-0.0
1970
-0.0
3630
HF
-0.3
1958
-0.3
0152
-0.3
0091
-0.0
9192
-0.1
4696
-0.0
4266
-0.0
7044
CH
2-0
.155
81-0
.168
84-0
.16845
-0.0
5420
-0.0
8528
-0.0
2538
-0.0
4614
Ar
-0.2
5462
-0.2
5331
-0.2
5274
-0.0
8013
-0.1
2627
-0.0
3744
-0.0
6251
Ne
-0.3
2022
-0.3
1960
-0.3
1939
-0.0
9714
-0.1
5530
-0.0
4505
-0.0
7372
C-0
.081
66-0
.098
28-0
.09836
-0.0
3293
-0.0
5160
-0.0
1549
-0.0
3073
N2
-0.4
2078
-0.3
5799
-0.3
5728
-0.1
0977
-0.1
7513
-0.0
5114
-0.0
8462
H2O
-0.3
0035
-0.2
8028
-0.2
7957
-0.0
8553
-0.1
3688
-0.0
3973
-0.0
6631
F-0
.246
20-0
.246
04-0
.24550
-0.0
7401
-0.1
1806
-0.0
3435
-0.0
5758
NH
3-0
.264
37-0
.258
90-0
.25814
-0.0
7905
-0.1
2669
-0.0
3679
-0.0
6217
F−
-0.3
3576
-0.3
3636
-0.3
3611
-0.1
0750
-0.1
6784
-0.0
4980
-0.0
8095
O-0
.177
29-0
.195
87-0
.19561
-0.0
6223
-0.0
9857
-0.0
2906
-0.0
5232
S4

Tab
leS4:
Com
par
ison
ofW
TM
AD
valu
esin
the
die
t100
subse
tof
the
GM
TK
N55
dat
abas
efo
rth
epro
pos
edex
trap
olat
ion
met
hods
wit
hdef
2-X
zvp
bas
isse
tfa
mily.
Basis
Function
ααdh
diet1
00
WT
MA
D[k
J/m
ol]
BLY
P-D
3(B
J)
B3LY
P-D
3(B
J)
B2P
LY
P-D
3(B
J)
def
2-tz
vp
––
–47.1
34.0
22.3
def
2-[s
,t]z
vp
exp
sqrt
[d,t
]10.3
90[1]
2.400[1]
45.7
32.9
23.4
def
2-[s
,t]z
vp
exp
onen
tial
glob
al2.2
62
2.382
47.0
34.0
24.6
def
2-[s
,t]z
vp
exp
onen
tial
[d,t
]2.8
74
2.096
47.0
34.0
25.2
def
2-[s
,t]z
vp
pow
ergl
obal
6.5
42
2.382
44.4
31.8
22.6
def
2-[s
,t]z
vp
pow
er[d
,t]
7.0
88
2.096
44.9
32.3
23.5
def
2-[s
,t]z
vp
exp
sqrt
glob
al7.7
57
2.382
43.8
31.3
22.3
def
2-[s
,t]z
vp
exp
sqrt
[d,t
]9.0
42
2.096
44.9
32.3
23.5
def
2-[s
,t]z
vp
exp
onen
tial
glob
al2.2
86
+0.4
81×f
(Fx)
2.382
47.0
34.0
24.6
def
2-[s
,t]z
vp
exp
onen
tial
[d,t
]2.8
47
+0.5
64×f
(Fx)
2.096
47.0
34.0
25.2
def
2-[s
,t]z
vp
pow
ergl
obal
6.5
82
+1.4
51×f
(Fx)
2.382
44.4
32.1
23.1
def
2-[s
,t]z
vp
pow
er[d
,t]
7.0
22
+1.3
90×f
(Fx)
2.096
44.9
32.4
23.8
def
2-[s
,t]z
vp
exp
sqrt
glob
al7.8
30
+1.6
39×f
(Fx)
2.382
43.9
31.7
22.8
def
2-[s
,t]z
vp
exp
sqrt
[d,t
]8.9
58
+1.7
73×f
(Fx)
2.096
44.9
32.4
23.8
def
2-qzv
p–
––
39.8
27.7
17.3
S5

Tab
leS5:
Com
par
ison
ofW
TM
AD
valu
esin
the
die
t100
subse
tof
the
GM
TK
N55
dat
abas
efo
rth
epro
pos
edex
trap
olat
ion
met
hods
wit
hdef
2-X
zvp
dbas
isse
tfa
mily.
Basis
Function
ααdh
diet1
00
WT
MA
D[k
J/m
ol]
BLY
P-D
3(B
J)
B3LY
P-D
3(B
J)
B2P
LY
P-D
3(B
J)
def
2-tz
vp
d–
––
40.0
27.8
18.6
def
2-[s
,t]z
vp
dex
psq
rt[d
,t]
10.3
90[1]
2.400[1]
39.6
27.3
17.5
def
2-[s
,t]z
vp
dex
pon
enti
algl
obal
2.302
2.267
40.0
27.8
17.9
def
2-[s
,t]z
vp
dex
pon
enti
al[d
,t]
2.881
1.996
40.0
27.8
17.9
def
2-[s
,t]z
vp
dp
ower
glob
al6.
666
2.267
39.3
27.0
17.4
def
2-[s
,t]z
vp
dp
ower
[d,t
]7.
106
1.996
39.4
27.1
17.6
def
2-[s
,t]z
vp
dex
psq
rtgl
obal
7.886
2.267
39.2
26.9
17.3
def
2-[s
,t]z
vp
dex
psq
rt[d
,t]
9.065
1.996
39.4
27.1
17.6
def
2-[s
,t]z
vp
dex
pon
enti
algl
obal
2.347
+0.4
23×f
(Fx)
2.267
40.0
27.8
17.9
def
2-[s
,t]z
vp
dex
pon
enti
al[d
,t]
2.857
+0.5
35×f
(Fx)
1.996
40.0
27.8
17.9
def
2-[s
,t]z
vp
dp
ower
glob
al6.
780
+1.2
56×f
(Fx)
2.267
39.3
27.1
17.5
def
2-[s
,t]z
vp
dp
ower
[d,t
]7.
045
+1.3
20×f
(Fx)
1.996
39.3
27.2
17.6
def
2-[s
,t]z
vp
dex
psq
rtgl
obal
8.033
+1.4
73×f
(Fx)
2.267
39.2
27.0
17.4
def
2-[s
,t]z
vp
dex
psq
rt[d
,t]
8.988
+1.6
83×f
(Fx)
1.996
39.3
27.2
17.6
def
2-qzv
pd
––
–38.3
26.6
16.5
S6

Tab
leS6:
Com
par
ison
ofW
TM
AD
valu
esin
the
die
t100
subse
tof
the
GM
TK
N55
dat
abas
efo
rth
epro
pos
edex
trap
olat
ion
met
hods
wit
hp
cseg
-Nbas
isse
tfa
mily.
Basis
Function
ααdh
diet1
00
WT
MA
D[k
J/m
ol]
BLY
P-D
3(B
J)
B3LY
P-D
3(B
J)
B2P
LY
P-D
3(B
J)
pcs
eg-2
––
–40.5
28.6
18.6
pcs
eg-[
1,2]
exp
sqrt
[d,t
]7.0
20[1]
2.010[1]
38.7
26.8
19.4
pcs
eg-[
1,2]
exp
onen
tial
glob
al1.7
71
1.651
40.4
28.5
22.3
pcs
eg-[
1,2]
exp
onen
tial
[d,t
]2.0
23
1.935
40.4
28.5
21.2
pcs
eg-[
1,2]
pow
ergl
obal
4.3
23
1.651
38.0
26.3
20.2
pcs
eg-[
1,2]
pow
er[d
,t]
4.9
90
1.935
38.3
26.6
19.4
pcs
eg-[
1,2]
exp
sqrt
glob
al5.7
43
1.651
38.1
26.3
20.3
pcs
eg-[
1,2]
exp
sqrt
[d,t
]6.3
66
1.935
38.3
26.6
19.4
pcs
eg-[
1,2]
exp
onen
tial
glob
al1.9
61−
0.2
32×f
(Fx)
1.651
40.4
28.5
22.3
pcs
eg-[
1,2]
exp
onen
tial
[d,t
]2.1
02−
0.0
96×f
(Fx)
1.935
40.4
28.5
21.2
pcs
eg-[
1,2]
pow
ergl
obal
4.7
57−
0.5
61×f
(Fx)
1.651
38.2
26.4
20.3
pcs
eg-[
1,2]
pow
er[d
,t]
5.1
85−
0.2
38×f
(Fx)
1.935
38.5
26.7
19.4
pcs
eg-[
1,2]
exp
sqrt
glob
al6.3
39−
0.7
50×f
(Fx)
1.651
38.3
26.5
20.4
pcs
eg-[
1,2]
exp
sqrt
[d,t
]6.6
15−
0.3
03×f
(Fx)
1.935
38.5
26.7
19.4
pcs
eg-3
––
–36.7
25.0
15.7
S7
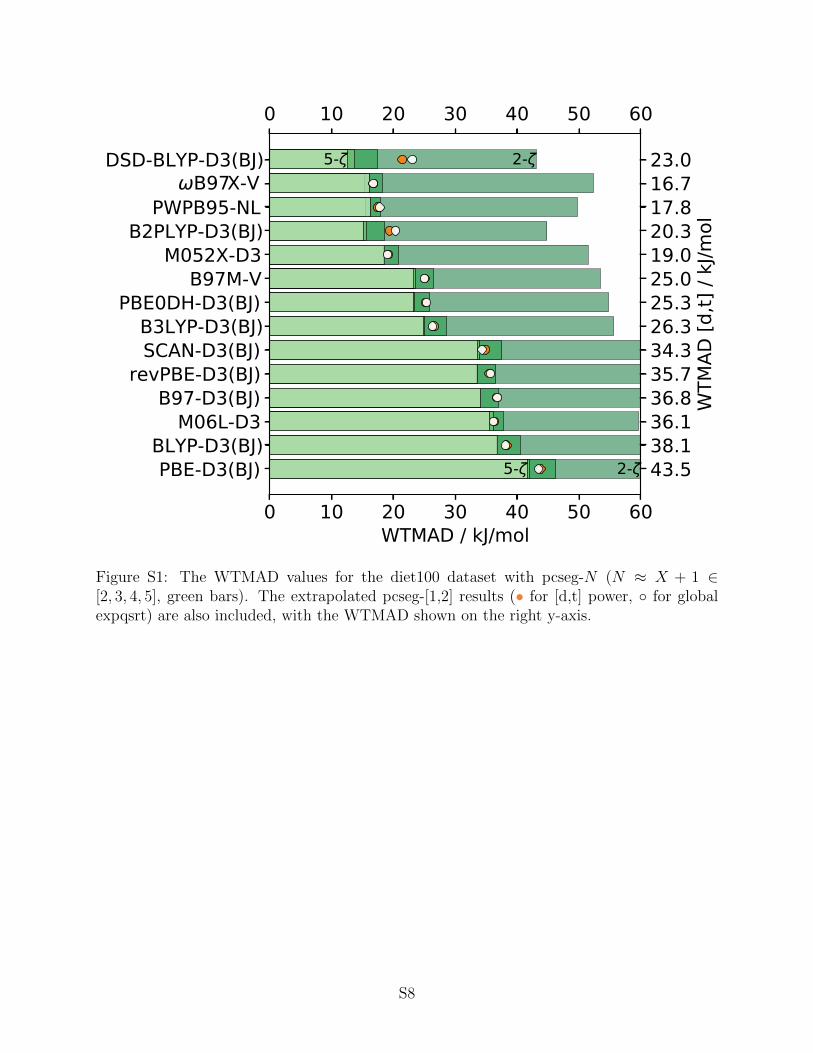
0
0
10
10
20
20
30
30
40
40
50
50
60
60
WTMAD / kJ/mol
PBE-D3(BJ)BLYP-D3(BJ)
M06L-D3B97-D3(BJ)
revPBE-D3(BJ)SCAN-D3(BJ)B3LYP-D3(BJ)
PBE0DH-D3(BJ)B97M-V
M052X-D3B2PLYP-D3(BJ)
PWPB95-NLB97X-V
DSD-BLYP-D3(BJ) 5- 2-
5- 2- 43.538.136.136.835.734.326.325.325.019.020.317.816.723.0
WTM
AD [d
,t] /
kJ/m
ol
Figure S1: The WTMAD values for the diet100 dataset with pcseg-N (N ≈ X + 1 ∈[2, 3, 4, 5], green bars). The extrapolated pcseg-[1,2] results (• for [d,t] power, for globalexpqsrt) are also included, with the WTMAD shown on the right y-axis.
S8
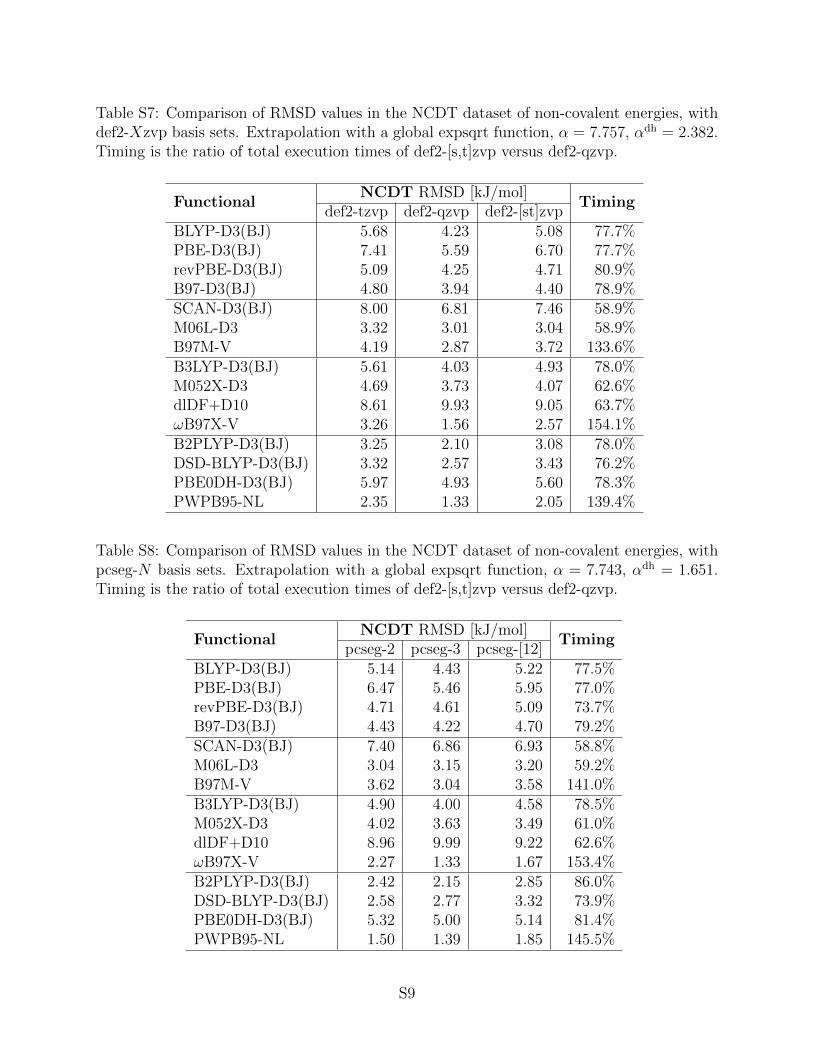
Table S7: Comparison of RMSD values in the NCDT dataset of non-covalent energies, withdef2-Xzvp basis sets. Extrapolation with a global expsqrt function, α = 7.757, αdh = 2.382.Timing is the ratio of total execution times of def2-[s,t]zvp versus def2-qzvp.
FunctionalNCDT RMSD [kJ/mol]
Timingdef2-tzvp def2-qzvp def2-[st]zvp
BLYP-D3(BJ) 5.68 4.23 5.08 77.7%PBE-D3(BJ) 7.41 5.59 6.70 77.7%revPBE-D3(BJ) 5.09 4.25 4.71 80.9%B97-D3(BJ) 4.80 3.94 4.40 78.9%SCAN-D3(BJ) 8.00 6.81 7.46 58.9%M06L-D3 3.32 3.01 3.04 58.9%B97M-V 4.19 2.87 3.72 133.6%B3LYP-D3(BJ) 5.61 4.03 4.93 78.0%M052X-D3 4.69 3.73 4.07 62.6%dlDF+D10 8.61 9.93 9.05 63.7%ωB97X-V 3.26 1.56 2.57 154.1%B2PLYP-D3(BJ) 3.25 2.10 3.08 78.0%DSD-BLYP-D3(BJ) 3.32 2.57 3.43 76.2%PBE0DH-D3(BJ) 5.97 4.93 5.60 78.3%PWPB95-NL 2.35 1.33 2.05 139.4%
Table S8: Comparison of RMSD values in the NCDT dataset of non-covalent energies, withpcseg-N basis sets. Extrapolation with a global expsqrt function, α = 7.743, αdh = 1.651.Timing is the ratio of total execution times of def2-[s,t]zvp versus def2-qzvp.
FunctionalNCDT RMSD [kJ/mol]
Timingpcseg-2 pcseg-3 pcseg-[12]
BLYP-D3(BJ) 5.14 4.43 5.22 77.5%PBE-D3(BJ) 6.47 5.46 5.95 77.0%revPBE-D3(BJ) 4.71 4.61 5.09 73.7%B97-D3(BJ) 4.43 4.22 4.70 79.2%SCAN-D3(BJ) 7.40 6.86 6.93 58.8%M06L-D3 3.04 3.15 3.20 59.2%B97M-V 3.62 3.04 3.58 141.0%B3LYP-D3(BJ) 4.90 4.00 4.58 78.5%M052X-D3 4.02 3.63 3.49 61.0%dlDF+D10 8.96 9.99 9.22 62.6%ωB97X-V 2.27 1.33 1.67 153.4%B2PLYP-D3(BJ) 2.42 2.15 2.85 86.0%DSD-BLYP-D3(BJ) 2.58 2.77 3.32 73.9%PBE0DH-D3(BJ) 5.32 5.00 5.14 81.4%PWPB95-NL 1.50 1.39 1.85 145.5%
S9

References
[1] F. Neese, “Software update: The ORCA program system, version 4.0,” WIREs Compu-tational Molecular Science, vol. 8, no. 1, p. e1327, 2018.
S10

download fileview on ChemRxivsi_v1.0.pdf (201.81 KiB)




![1 FRONT PAGES · 2019-04-14 · with the best physical basis is the modern version of the Pulay method [9-11]. 2.2.4. Density functional theory Density functional theory is a quantum](https://img.pdfslide.us/doc/110x75/5f57670e233ccd5aca5975d5/1-front-pages-2019-04-14-with-the-best-physical-basis-is-the-modern-version-of.jpg)
























