Embed Size (px)
Citation preview

www.elsevier.com/locate/apcata
Applied Catalysis A: General 312 (2006) 86–94
Baeyer–Villiger oxidation of cyclohexanone with hydrogen
peroxide/benzonitrile over hydrotalcites as catalysts
Cesar Jimenez-Sanchidrian *, Julia Marıa Hidalgo, Rafael Llamas, Jose Rafael Ruiz **
Departamento de Quımica Organica,Universidad de Cordoba, Campus de Rabanales, Edificio Marie Curie,
Carretera Nacional IV-A, km. 396, 14014 Cordoba, Spain
Received 7 March 2006; received in revised form 20 June 2006; accepted 20 June 2006
Available online 25 July 2006
Abstract
Various Mg/Al, Mg/Al/Sn and Mg/Al/Zr hydrotalcite-like compounds (HTs) were prepared for use as catalysts in the Baeyer–Villiger (BV)
oxidation of cyclohexanone with H2O2/benzonitrile as oxidant and benzonitrile itself as the solvent. The reaction gave water and benzamide as
byproducts, the latter can be recycled in an appropiate process. Analysis of the solids revealed that tin and zirconium are in fact incorporated into
the HT structure. The solids containing Mg/Al and Mg/Al/Sn and their calcination products (viz. Mg/Al and Mg/Al/Sn mixed oxides) were found
to be effective catalysts for the BVoxidation of cyclohexanone. The Sn solids exhibited a higher activity than a more conventional catalyst obtained
from an HT containing Mg and Al only (17% in the best case). Also, the catalyst containing tin was active when we used H2O2/acetonitrile system
as oxidant. The solids containing Zr were found to promote the decomposition of the hydrogen peroxide and hence to adversely influence the
oxidation reaction. A mechanism accounting for the experimental results is proposed. The reaction was conducted under very mild conditions (viz.
atmospheric pressure and a temperature of 70 8C), and conversions obtained were higher than 80% in some cases and 100% selectivity after 6 h.
The most active catalyst, which was that containing the largest amount of tin, retained full activity after three reuses.
# 2006 Elsevier B.V. All rights reserved.
Keywords: Hydrotalcite; LDH; Baeyer–Villiger oxidation; Cyclohexanone; Tin; Zirconium
1. Introduction
Hydrotalcite-like compounds (HTs), also known as layered
double hydroxides, have aroused much debate over their
potential uses in various scientific areas including catalysed
organic synthesis [1–3]. The parent compound, a natural mineral
called hydrotalcite [4], is a magnesium-aluminium hydroxy-
carbonate of formula Mg6Al2(OH)16CO3�4H2O the structure of
which is similar to that of brucite except that some Mg2+ ions are
substituted by Al3+; this substitution produces layers bearing
positive charges that are neutralized by carbonate ions present in
the interlayer spacings. By replacing the magnesium, aluminium
or both cations, and the carbonate, by other ions, a large family of
compounds known as hydrotalcite-like (HT) or layered double
hydroxides (LDHs) of general formula [M(II)1�xM(III)x(OH)2]x+
(An�)x/n�mH2O can be obtained where M(II) is a divalent cation
* Corresponding author.
** Corresponding author. Tel.: +34 957 218638; fax: +34 957 212066.
E-mail address: [email protected] (J.R. Ruiz).
0926-860X/$ – see front matter # 2006 Elsevier B.V. All rights reserved.
doi:10.1016/j.apcata.2006.06.031
such as Mg, Cu, Ni, Co or Zn [3], M(III) a trivalent cation such as
Al, Fe, Cr, V, Mn, Ga or In [3,5,6] and An� the charge balancing
anion, which can be an inorganic or organic species of widely
variable nature [3].
There are two main types of HTs. One consists of divalent
and trivalent cations plus various interlayer anions; the other,
contains lithium and aluminium (i.e. a univalent metal and a
trivalent one) [7,8]. However, some HTs reported in the last
decade contain tetravalent metals such as tin [9], titanium [10]
or zirconium [11]; in any case, they require the presence of a
trivalent metal in addition to the divalent one as high
concentrations of the tetravalent metal cause the formation
of various phases (particularly mixed hydroxides of the di- and
tetravalent metal) in addition to the HT.
HTs containing tetravalent metals are finding interesting
uses in Catalysis. This led us to prepare HTs containing Mg, Al
and Sn or Mg, Al and Zr in variable Mg/Al/M(IV) proportions
that were characterized by using a variety of instrumental
techniques including X-ray diffraction, IR and NMR spectro-
scopies, and nitrogen porosimetry. Once synthesized and
characterized, the different solids were used as catalysts in the

C. Jimenez-Sanchidrian et al. / Applied Catalysis A: General 312 (2006) 86–94 87
Scheme 1. General mechanism for the Baeyer–Villiger oxidation of ketones with organic peracids.
Baeyer–Villiger oxidation of cyclohexanone. This reaction was
first reported by Baeyer and Villiger in 1899 and provides a
method for converting ketones into esters [12]; since then, it has
become a very important organic synthetic pathway, as
reflected in a vast number of books and papers including not
only specific reviews [13–17], but also articles dealing with
organic oxidation reactions at large [18,19]. At present, the
Baeyer–Villiger reaction involves the oxidation of a ketone
with an organic peroxyacid, hydrogen peroxide or an alkyl
hydroperoxide to give an ester or lactone (or an alcohol or acid
derivative). Similar reactions involving the oxidation of
aldehydes to formates or their hydrolysis products are also
included in this designation. The general mechanism for the
process involves two steps (see Scheme 1). In the first, the
peroxyacid adds to the carbonyl compound to form a so-called
Criegee adduct (1), which undergoes rearrangement to the end-
product (2) in the second. The migration of group R0 is
concerted with the cleavage of the O–O bond. Bases favour
rearrangement of the adduct by facilitating the release of the
hydroxyl proton in the intermediate. The Baeyer–Villiger
oxidation of ketones provides a number of advantages
including (a) a high tolerance of a variety of functional groups
(e.g. when the double bond is conjugated with the carbonyl
group the selectivity is towards BV oxidation, but when the
C C bond is not conjugated the selectivity depends on the
substitution of this double bond [14]), (b) the regiochemistry
can usually be controlled via the migration ability of the
different groups – in some cases (e.g. with bicyclic systems),
however, the regioselectivity of the oxygen insertion process
can be influenced by various stereoelectronic factors – , and (c)
the reaction allows the use of a wide variety of oxidants.
Usually, the BV reaction is conducted with homogeneous
catalysts; however, an increasing number of heterogeneous
catalysts has been used for this purpose over the past decade.
Also, the growing environmental concern of recent years has
led to the replacement of many organic oxidants with more
environmentally benign agents such as hydrogen peroxide. This
has indeed been the case with the Baeyer–Villiger reaction,
where a number of heterogeneous catalysts have been
effectively used in conjunction with hydrogen peroxide.
Ti-Silicalite (TS-1) was one of the catalysts that revolutio-
nized the field of organic oxidations. This solid consists of a
zeolite structure (silicalite) into which titanium is incorporated
[20]. Its high oxidizing power lies in the ability of titanium
metal sites to form Ti-peroxy species that can activate hydrogen
peroxide in various oxidation reactions including epoxidations,
ammonoxidations and CH oxidations [21,22]. These catalysts
have been further developed by inserting new oxidizing metals
into zeolite structures, albeit with poorer results than those
originally obtained with titanium. However, Corma et al.
developed a catalyst consisting of Sn incorporated into a beta
zeolite [23]. In subsequent work [24–29], Corma et al.
conducted extensive research into Baeyer–Villiger oxidation
reactions using their Sn-beta zeolite catalysts. Recently, they
proposed a mechanism for the underlying process from a
combination of theoretical and experimental data [30].
Asnotedearlier, in this workwe used HT-based heterogeneous
catalysts containing metals such as Sn and Zr in the Baeyer–
Villiger reaction of cyclohexanone with hydrogen peroxide and
benzonitrile as oxidant. Our group previously found HTs to be
effective catalysts for reactions requiring the assistance of a base
(e.g. the epoxidation of limonenewith hydrogen peroxide [31,32]
where the peroxycarboximidic acid is also the intermediate or the
Meerwein–Ponndorf–Verley reaction [33–36]). Hydrotalcite-
like compounds have also been explored by others authors in the
BV reaction, but using a mixture of molecular oxygen and
benzaldehyde [37–39] or peroxyacid as oxidant [40]. Also, a
hydrotalcite-supported SnO2 catalyst was found to effect the BV
reaction with hydrogen peroxide as oxidant in the presence of a
nitrile as oxygen transfer agent [41]. However, these authors used
Mg/Al hydrotalcite as a support for SnO2, which was the actual
catalyst, and obtained zero conversion with hydrotalcite contain-
ing no SnO2. In previous work [32], we showed the formation of
the peroxycarboximidic intermediate to require the presence of a
nitrile containing no a protons (e.g. benzonitrile). Otherwise (e.g.
with acetonitrile or butyronitrile), the epoxidation of limonene –
the first step of which (viz. the formation of a peroxycarbox-
ymidic intermediate) is similar to that for the Baeyer–Villiger
oxidation – developed to a smaller extent than it did in the
presence of benzonitrile (the conversion at 8 h was 90% with
benzonitrile, and only 20% and 22% with acetonitrile and
butyronitrile, respectively). We ascribed this result to the
formation of carbanionic species from the nitriles possessing a
protons and to the retention of such species on the hydrotalcite
surface hindering further reaction development.
2. Experimental
2.1. Preparation of hydrotalcite-like compounds
The HT containing only magnesium and aluminium
was prepared from solutions of Mg(NO3)2�6H2O and

C. Jimenez-Sanchidrian et al. / Applied Catalysis A: General 312 (2006) 86–9488
Table 1
Metal ratios and chemical formulae of the HTs
Catalyst Mg/[Al + M(IV)]a Al/M(IV)a Chemical formulaeb
HT-Mg/Al 1.8 – Mg0.631Al0.369(OH)2(CO3)0.185�0.65H2O
HT-Sn-1 1.8 30.8 Mg0.650Al0.339Sn0.011(OH)2(CO3)0.125�0.66H2O
HT-Sn-2 1.8 13.0 Mg0.650Al0.325Sn0.025(OH)2(CO3)0.115�0.65H2O
HT-Sn3 – – Hydrotalcite + magnesium hydroxystannate
HT-Zr1 2.2 7.7 Mg0.684Al0.280Zr0.036(OH)2(CO3)0.160�0.80H2O
HT-Zr2 2.4 3.0 Mg0.710Al0.217Zr0.073(OH)2(CO3)0.145�1.20H2O
HT-Zr3 – – Hydrotalcite + brucite
a Metal ratios as determined by ICP-MS.b Crystallization water was determined thermogravimetrically.
Al(NO3)3�9H2O in Mg(II)/Al(III) ratio = 2, using a coprecipi-
tation method described elsewhere [5]. In a typical synthetic
run, a solution containing 0.3 mol of Mg(NO3)2�6H2O and
0.15 mol of Al(NO3)3�9H2O in 250 mL of de-ionized water was
used. This solution was slowly dropped over 500 mL of a
Na2CO3 solution at pH 10 at 60 8C under vigorous stirring. The
pH was kept constant by adding appropriate volumes of 1 M
NaOH during precipitation. The suspension thus obtained was
kept at 80 8C for 24 h, after which the solid was filtered and
washed with 2 L of de-ionized water. The magnesium,
aluminium and tin or zirconium HTs with Mg(II)/[Al(III) + -
M(IV)] = 2 was obtained by following the same procedure,
using appropriate amounts of the Mg(II) and Al(III) nitrates and
Sn(IV) chloride or Zr(IV) oxychloride.
The HTs thus prepared were ion-exchanged with
carbonate to remove ions intercalated between layers. The
procedure involved suspending the solids in a solution
containing 0.345 g of Na2CO3 in 50 mL of bidistilled, de-
ionized water per gram of HT at 100 8C for 2 h. Then, each
solid was filtered off in vacuo and washed with 200 mL of
bidistilled, de-ionized water. The HTs thus obtained were
subjected to a second ion-exchange operation under the same
conditions. The exchanged Mg/Al solid was named HT-Mg/
Al and its Mg/Al/Sn or Mg/Al/Zr counterparts HT-Sn1, HT-
Sn2, HT-Sn3 and HT-Zr1, HT-Zr2 and HT-Zr3 (the former
being that with the lower Sn or Zr content) (see Table 1).
These solids were calcined at 450 8C in the air for 8 h, using
a temperature gradient of 1 8C/min.
2.2. Experimental techniques
The HTs and their calcination products obtained from them
were characterized by using various instrumental techniques.
Thus, the metal contents in the catalysts were determined by
inductively coupled plasma-mass spectrometry on a Perkin-
Elmer ICP-MS instrument under standard conditions.
X-ray diffraction (XRD) analysis was performed in all
catalysts in order to check for crystallinity. Powder patterns
were recorded on a Siemens D-5000 diffractometer using Cu
Ka radiation. Scans were performed over the 2u ranges from 58to 708, using a resolution of 0.028 and a count time of 2 s at each
point.
Fourier transform infrared (FT-IR) spectra for the HTs were
recorded over the wavenumber range from 400 to 4000 cm�1
on a Bomen MB-100 FT-IR spectrophotometer. Samples were
prepared by mixing the powdered solids with KBr (the blank).
Solid-state 27Al MAS NMR spectra were recorded at room
temperature on a Bruker ACP-400 spectrometer. A resonance
frequency of 104.21 MHz, a recycle delay of 0.1 s, and short
3 ms pulses (458 pulses of 3 ms) and a spin rate of 12 kHz were
applied with an accumulation of 3500 scans. Signals were
referred to Al(H2O)63+ (0 ppm).
Thermogravimetric analysis were performed on a Setaram
Setsys 12 instrument by heating in an argon atmosphere from
25 to 800 8C at a 10 8C/min rate.
BET surface areas, pore radii and pore volumes were
calculated from nitrogen adsorption–desorption isotherms that
were obtained at �196 8C using a Micromeritics ASAP 2010
instrument. Samples were outgassed in vacuo at 100 8C for 12 h
prior to use.
2.3. Reaction conditions
Baeyer–Villiger oxidations runs were performed at 70 8C in
a two-necks flask containing 0.012 mol of carbonyl compound,
0.098 mol of benzonitrile, 0.10 mol of hydrogen peroxide (30%
(v/v) aqueous solution) and 0.25 g of catalyst. One of the flask
necks was fitted with a reflux condenser and the other was used
for sampling at regular intervals. The system was stirred
throughout the process. Products were identified from their
retention times as measured by GC–MS analysis on an HP 5890
GC instrument furnished with a Supelcowax 30 m � 0.32 mm
column and an HP 5971 MSD instrument.
3. Results and discussion
3.1. Characterization of catalysts
3.1.1. Elemental analysis
Table 1 shows the chemical composition and empirical
formula of each solid as determined by ICP-MS. The
differences between the theoretical and experimental values
were all within the range of experimental error.
3.1.2. XRD results
Fig. 1 shows the XRD pattern for solid HT-Mg/Al. The
signals are typical of hydrotalcite [42], with tall, narrow,
symmetric peaks in the planes (0 0 3), (0 0 6), (1 1 0) and
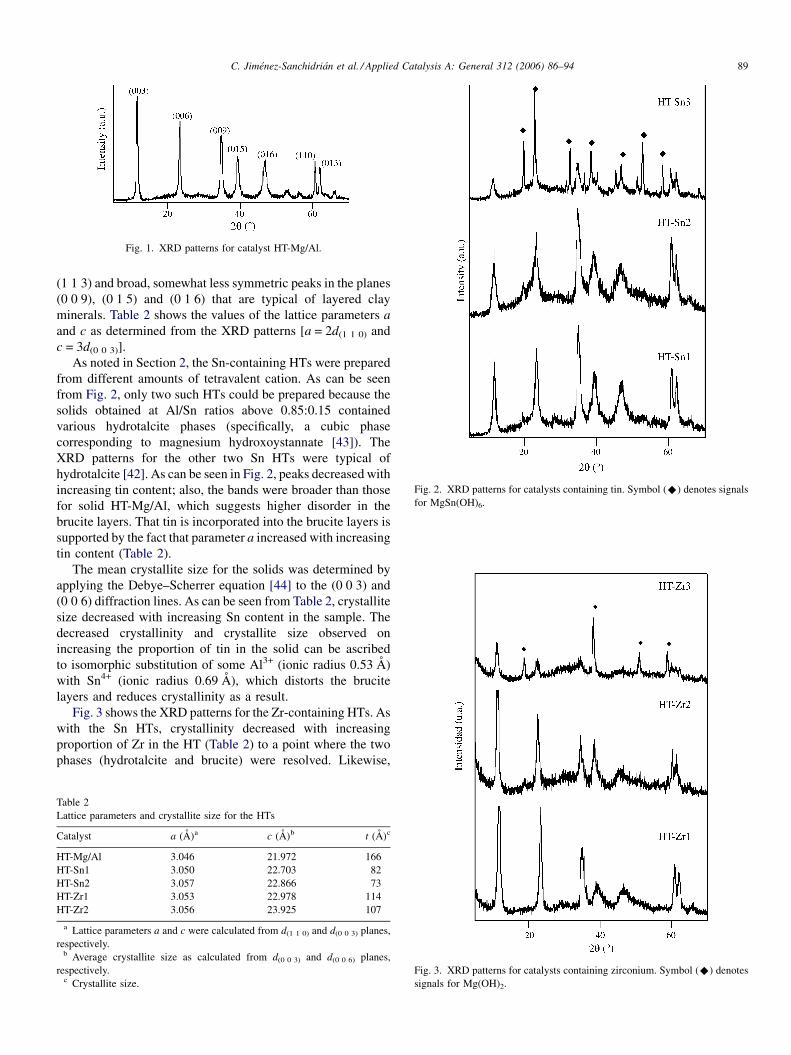
C. Jimenez-Sanchidrian et al. / Applied Catalysis A: General 312 (2006) 86–94 89
Fig. 1. XRD patterns for catalyst HT-Mg/Al.
Fig. 2. XRD patterns for catalysts containing tin. Symbol (^) denotes signals
for MgSn(OH)6.
(1 1 3) and broad, somewhat less symmetric peaks in the planes
(0 0 9), (0 1 5) and (0 1 6) that are typical of layered clay
minerals. Table 2 shows the values of the lattice parameters a
and c as determined from the XRD patterns [a = 2d(1 1 0) and
c = 3d(0 0 3)].
As noted in Section 2, the Sn-containing HTs were prepared
from different amounts of tetravalent cation. As can be seen
from Fig. 2, only two such HTs could be prepared because the
solids obtained at Al/Sn ratios above 0.85:0.15 contained
various hydrotalcite phases (specifically, a cubic phase
corresponding to magnesium hydroxoystannate [43]). The
XRD patterns for the other two Sn HTs were typical of
hydrotalcite [42]. As can be seen in Fig. 2, peaks decreased with
increasing tin content; also, the bands were broader than those
for solid HT-Mg/Al, which suggests higher disorder in the
brucite layers. That tin is incorporated into the brucite layers is
supported by the fact that parameter a increased with increasing
tin content (Table 2).
The mean crystallite size for the solids was determined by
applying the Debye–Scherrer equation [44] to the (0 0 3) and
(0 0 6) diffraction lines. As can be seen from Table 2, crystallite
size decreased with increasing Sn content in the sample. The
decreased crystallinity and crystallite size observed on
increasing the proportion of tin in the solid can be ascribed
to isomorphic substitution of some Al3+ (ionic radius 0.53 A)
with Sn4+ (ionic radius 0.69 A), which distorts the brucite
layers and reduces crystallinity as a result.
Fig. 3 shows the XRD patterns for the Zr-containing HTs. As
with the Sn HTs, crystallinity decreased with increasing
proportion of Zr in the HT (Table 2) to a point where the two
phases (hydrotalcite and brucite) were resolved. Likewise,
Table 2
Lattice parameters and crystallite size for the HTs
Catalyst a (A)a c (A)b t (A)c
HT-Mg/Al 3.046 21.972 166
HT-Sn1 3.050 22.703 82
HT-Sn2 3.057 22.866 73
HT-Zr1 3.053 22.978 114
HT-Zr2 3.056 23.925 107
a Lattice parameters a and c were calculated from d(1 1 0) and d(0 0 3) planes,
respectively.b Average crystallite size as calculated from d(0 0 3) and d(0 0 6) planes,
respectively.c Crystallite size.
Fig. 3. XRD patterns for catalysts containing zirconium. Symbol (^) denotes
signals for Mg(OH)2.
C. Jimenez-Sanchidrian et al. / Applied Catalysis A: General 312 (2006) 86–9490
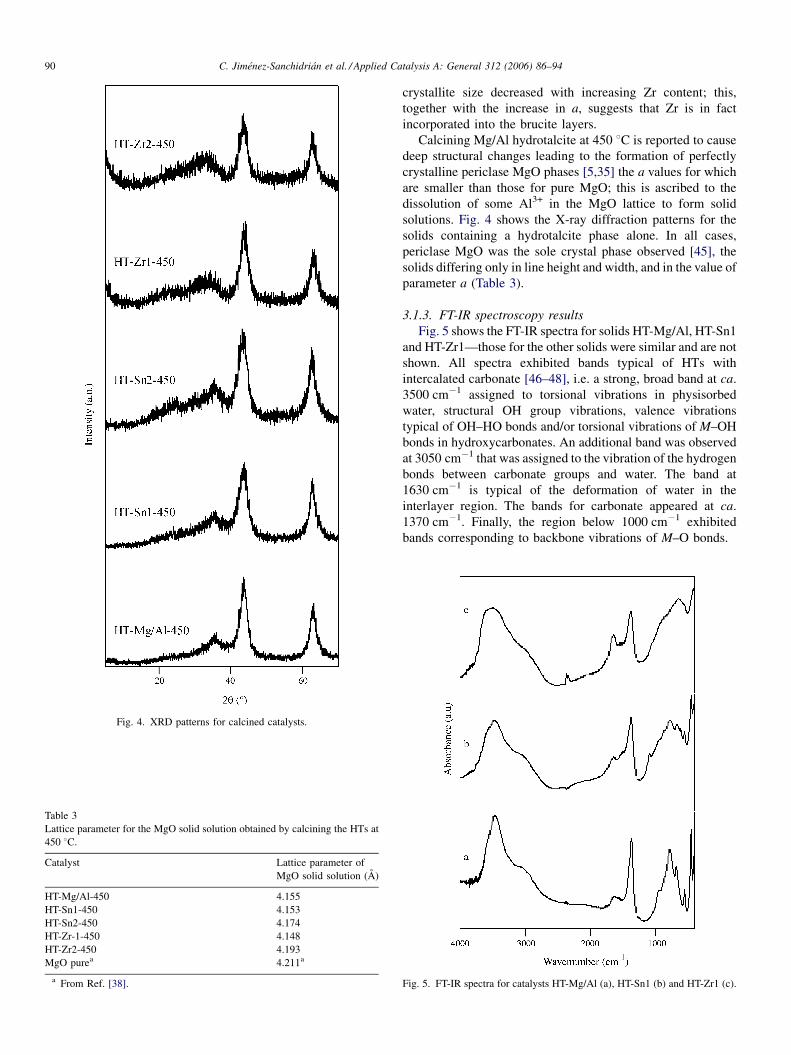
Fig. 4. XRD patterns for calcined catalysts.
Table 3
Lattice parameter for the MgO solid solution obtained by calcining the HTs at
450 8C.
Catalyst Lattice parameter of
MgO solid solution (A)
HT-Mg/Al-450 4.155
HT-Sn1-450 4.153
HT-Sn2-450 4.174
HT-Zr-1-450 4.148
HT-Zr2-450 4.193
MgO purea 4.211a
a From Ref. [38].
crystallite size decreased with increasing Zr content; this,
together with the increase in a, suggests that Zr is in fact
incorporated into the brucite layers.
Calcining Mg/Al hydrotalcite at 450 8C is reported to cause
deep structural changes leading to the formation of perfectly
crystalline periclase MgO phases [5,35] the a values for which
are smaller than those for pure MgO; this is ascribed to the
dissolution of some Al3+ in the MgO lattice to form solid
solutions. Fig. 4 shows the X-ray diffraction patterns for the
solids containing a hydrotalcite phase alone. In all cases,
periclase MgO was the sole crystal phase observed [45], the
solids differing only in line height and width, and in the value of
parameter a (Table 3).
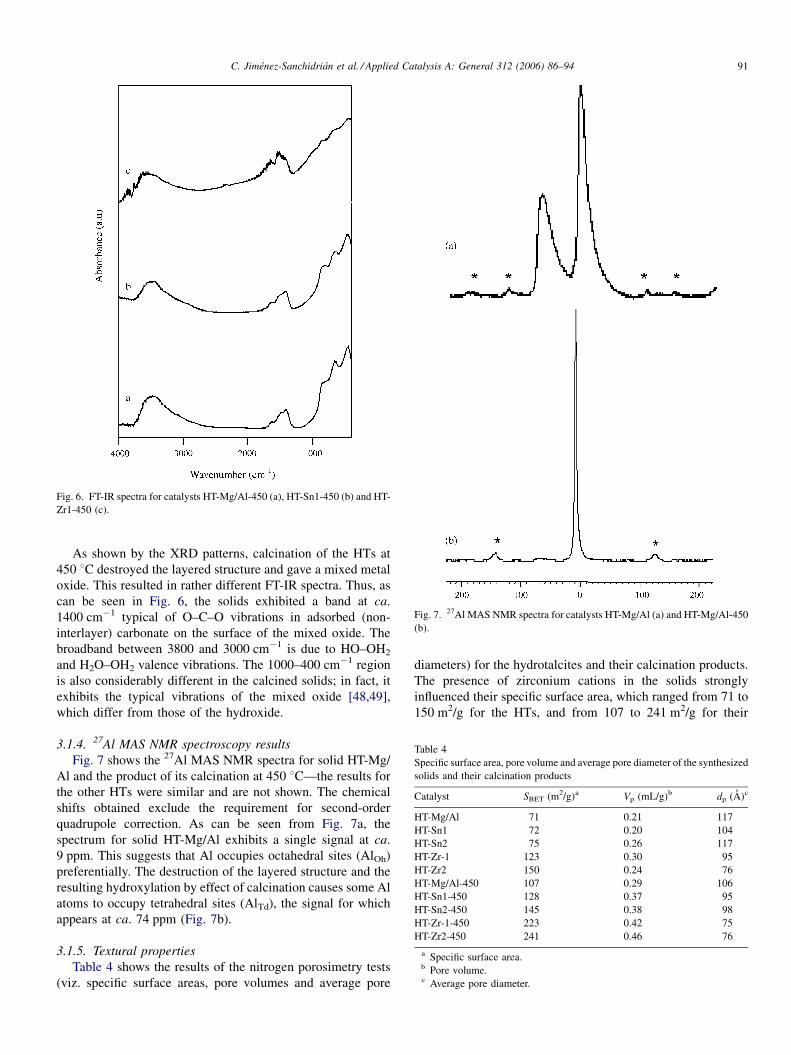
3.1.3. FT-IR spectroscopy results
Fig. 5 shows the FT-IR spectra for solids HT-Mg/Al, HT-Sn1
and HT-Zr1—those for the other solids were similar and are not
shown. All spectra exhibited bands typical of HTs with
intercalated carbonate [46–48], i.e. a strong, broad band at ca.
3500 cm�1 assigned to torsional vibrations in physisorbed
water, structural OH group vibrations, valence vibrations
typical of OH–HO bonds and/or torsional vibrations of M–OH
bonds in hydroxycarbonates. An additional band was observed
at 3050 cm�1 that was assigned to the vibration of the hydrogen
bonds between carbonate groups and water. The band at
1630 cm�1 is typical of the deformation of water in the
interlayer region. The bands for carbonate appeared at ca.
1370 cm�1. Finally, the region below 1000 cm�1 exhibited
bands corresponding to backbone vibrations of M–O bonds.
Fig. 5. FT-IR spectra for catalysts HT-Mg/Al (a), HT-Sn1 (b) and HT-Zr1 (c).
C. Jimenez-Sanchidrian et al. / Applied Catalysis A: General 312 (2006) 86–94 91
Fig. 6. FT-IR spectra for catalysts HT-Mg/Al-450 (a), HT-Sn1-450 (b) and HT-
Zr1-450 (c).
Fig. 7. 27Al MAS NMR spectra for catalysts HT-Mg/Al (a) and HT-Mg/Al-450
(b).
Table 4
Specific surface area, pore volume and average pore diameter of the synthesized
solids and their calcination products
Catalyst SBET (m2/g)a Vp (mL/g)b dp (A)c
HT-Mg/Al 71 0.21 117
HT-Sn1 72 0.20 104
HT-Sn2 75 0.26 117
HT-Zr-1 123 0.30 95
HT-Zr2 150 0.24 76
HT-Mg/Al-450 107 0.29 106
HT-Sn1-450 128 0.37 95
HT-Sn2-450 145 0.38 98
HT-Zr-1-450 223 0.42 75
HT-Zr2-450 241 0.46 76
a Specific surface area.b Pore volume.c Average pore diameter.
As shown by the XRD patterns, calcination of the HTs at
450 8C destroyed the layered structure and gave a mixed metal
oxide. This resulted in rather different FT-IR spectra. Thus, as
can be seen in Fig. 6, the solids exhibited a band at ca.
1400 cm�1 typical of O–C–O vibrations in adsorbed (non-
interlayer) carbonate on the surface of the mixed oxide. The
broadband between 3800 and 3000 cm�1 is due to HO–OH2
and H2O–OH2 valence vibrations. The 1000–400 cm�1 region
is also considerably different in the calcined solids; in fact, it
exhibits the typical vibrations of the mixed oxide [48,49],
which differ from those of the hydroxide.
3.1.4. 27Al MAS NMR spectroscopy results
Fig. 7 shows the 27Al MAS NMR spectra for solid HT-Mg/
Al and the product of its calcination at 450 8C—the results for
the other HTs were similar and are not shown. The chemical
shifts obtained exclude the requirement for second-order
quadrupole correction. As can be seen from Fig. 7a, the
spectrum for solid HT-Mg/Al exhibits a single signal at ca.
9 ppm. This suggests that Al occupies octahedral sites (AlOh)
preferentially. The destruction of the layered structure and the
resulting hydroxylation by effect of calcination causes some Al
atoms to occupy tetrahedral sites (AlTd), the signal for which
appears at ca. 74 ppm (Fig. 7b).
3.1.5. Textural properties
Table 4 shows the results of the nitrogen porosimetry tests
(viz. specific surface areas, pore volumes and average pore
diameters) for the hydrotalcites and their calcination products.
The presence of zirconium cations in the solids strongly
influenced their specific surface area, which ranged from 71 to
150 m2/g for the HTs, and from 107 to 241 m2/g for their
C. Jimenez-Sanchidrian et al. / Applied Catalysis A: General 312 (2006) 86–9492
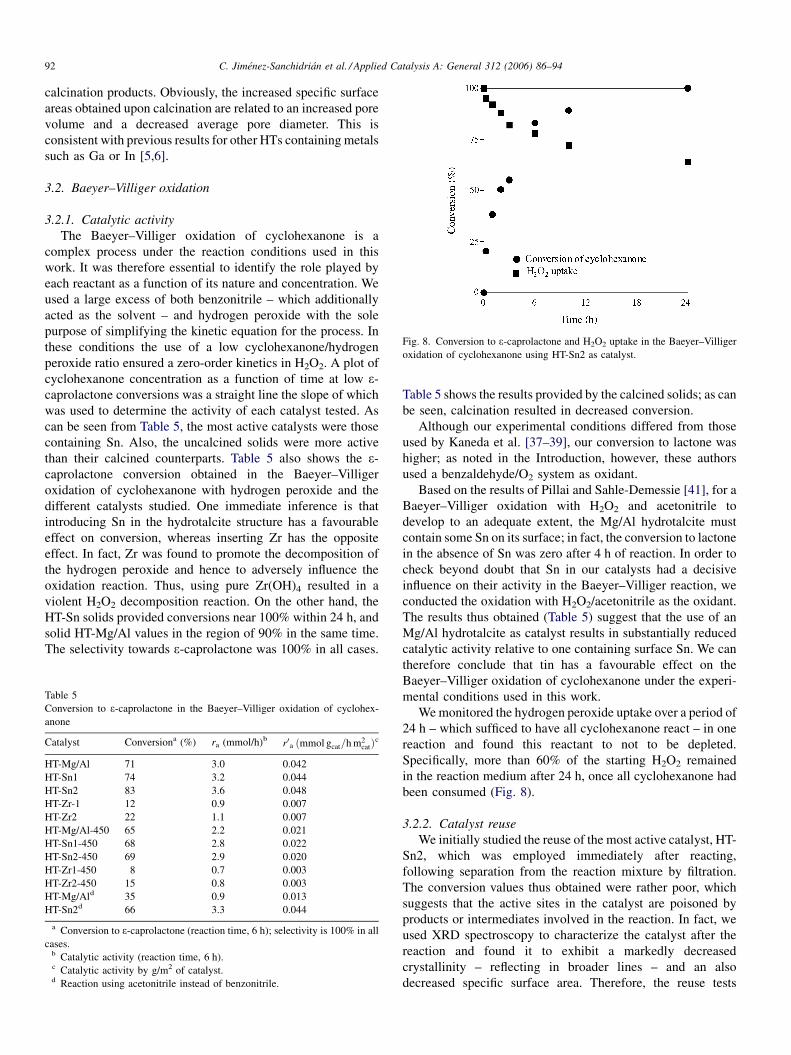
Fig. 8. Conversion to e-caprolactone and H2O2 uptake in the Baeyer–Villiger
oxidation of cyclohexanone using HT-Sn2 as catalyst.
calcination products. Obviously, the increased specific surface
areas obtained upon calcination are related to an increased pore
volume and a decreased average pore diameter. This is
consistent with previous results for other HTs containing metals
such as Ga or In [5,6].
3.2. Baeyer–Villiger oxidation
3.2.1. Catalytic activity
The Baeyer–Villiger oxidation of cyclohexanone is a
complex process under the reaction conditions used in this
work. It was therefore essential to identify the role played by
each reactant as a function of its nature and concentration. We
used a large excess of both benzonitrile – which additionally
acted as the solvent – and hydrogen peroxide with the sole
purpose of simplifying the kinetic equation for the process. In
these conditions the use of a low cyclohexanone/hydrogen
peroxide ratio ensured a zero-order kinetics in H2O2. A plot of
cyclohexanone concentration as a function of time at low e-
caprolactone conversions was a straight line the slope of which
was used to determine the activity of each catalyst tested. As
can be seen from Table 5, the most active catalysts were those
containing Sn. Also, the uncalcined solids were more active
than their calcined counterparts. Table 5 also shows the e-
caprolactone conversion obtained in the Baeyer–Villiger
oxidation of cyclohexanone with hydrogen peroxide and the
different catalysts studied. One immediate inference is that
introducing Sn in the hydrotalcite structure has a favourable
effect on conversion, whereas inserting Zr has the opposite
effect. In fact, Zr was found to promote the decomposition of
the hydrogen peroxide and hence to adversely influence the
oxidation reaction. Thus, using pure Zr(OH)4 resulted in a
violent H2O2 decomposition reaction. On the other hand, the
HT-Sn solids provided conversions near 100% within 24 h, and
solid HT-Mg/Al values in the region of 90% in the same time.
The selectivity towards e-caprolactone was 100% in all cases.
Table 5
Conversion to e-caprolactone in the Baeyer–Villiger oxidation of cyclohex-
anone
Catalyst Conversiona (%) ra (mmol/h)br0a ðmmol gcat=h m2
catÞc
HT-Mg/Al 71 3.0 0.042
HT-Sn1 74 3.2 0.044
HT-Sn2 83 3.6 0.048
HT-Zr-1 12 0.9 0.007
HT-Zr2 22 1.1 0.007
HT-Mg/Al-450 65 2.2 0.021
HT-Sn1-450 68 2.8 0.022
HT-Sn2-450 69 2.9 0.020
HT-Zr1-450 8 0.7 0.003
HT-Zr2-450 15 0.8 0.003
HT-Mg/Ald 35 0.9 0.013
HT-Sn2d 66 3.3 0.044
a Conversion to e-caprolactone (reaction time, 6 h); selectivity is 100% in all
cases.b Catalytic activity (reaction time, 6 h).c Catalytic activity by g/m2 of catalyst.d Reaction using acetonitrile instead of benzonitrile.
Table 5 shows the results provided by the calcined solids; as can
be seen, calcination resulted in decreased conversion.
Although our experimental conditions differed from those
used by Kaneda et al. [37–39], our conversion to lactone was
higher; as noted in the Introduction, however, these authors
used a benzaldehyde/O2 system as oxidant.
Based on the results of Pillai and Sahle-Demessie [41], for a
Baeyer–Villiger oxidation with H2O2 and acetonitrile to
develop to an adequate extent, the Mg/Al hydrotalcite must
contain some Sn on its surface; in fact, the conversion to lactone
in the absence of Sn was zero after 4 h of reaction. In order to
check beyond doubt that Sn in our catalysts had a decisive
influence on their activity in the Baeyer–Villiger reaction, we
conducted the oxidation with H2O2/acetonitrile as the oxidant.
The results thus obtained (Table 5) suggest that the use of an
Mg/Al hydrotalcite as catalyst results in substantially reduced
catalytic activity relative to one containing surface Sn. We can
therefore conclude that tin has a favourable effect on the
Baeyer–Villiger oxidation of cyclohexanone under the experi-
mental conditions used in this work.
We monitored the hydrogen peroxide uptake over a period of
24 h – which sufficed to have all cyclohexanone react – in one
reaction and found this reactant to not to be depleted.
Specifically, more than 60% of the starting H2O2 remained
in the reaction medium after 24 h, once all cyclohexanone had
been consumed (Fig. 8).
3.2.2. Catalyst reuse
We initially studied the reuse of the most active catalyst, HT-
Sn2, which was employed immediately after reacting,
following separation from the reaction mixture by filtration.
The conversion values thus obtained were rather poor, which
suggests that the active sites in the catalyst are poisoned by
products or intermediates involved in the reaction. In fact, we
used XRD spectroscopy to characterize the catalyst after the
reaction and found it to exhibit a markedly decreased
crystallinity – reflecting in broader lines – and an also
decreased specific surface area. Therefore, the reuse tests

C. Jimenez-Sanchidrian et al. / Applied Catalysis A: General 312 (2006) 86–94 93
Table 6
Recycling of the HT-Mg/Al and HT-Sn2 catalysts in the Baeyer–Villiger
oxidation of cyclohexanone
Recycle number Conversion (%)a
HT-Sn2 fresh 83
1b 12
1 81
2 80
3 81
a e-Caprolactone conversion (reaction time, 6 h); selectivity is 100% in all
cases.b Unactivated catalyst.
Scheme 2. Proposed mechanism for the Baeyer–Villiger reaction of cyclohexanone over HTs.
Table 7
Influence of the H2O2:cyclohexanone molar ratio in the Baeyer–Villiger
oxidation of cyclohexanone with HT-Sn2 as catalyst
H2O2:cyclohexanonea Conversion (%)b ra (mmol/h)c
8.3 83 3.6
5 48 3.0
4 45 2.7
3 41 2.6
2 37 2.0
a Molar ratio.b Conversion to e-caprolactone (reaction time, 6 h); selectivity is 100% in all
cases.c Catalytic activity (reaction time, 6 h).
required previously separating the catalyst from the reaction
mass. Once isolated, the catalyst was activated by calcination at
450 8C and subsequent rehydration in a 5% Na2CO3 solution
prior to recycling. This process converted the hydrotalcite into a
mixture of oxides that regained the hydrotalcite structure – as
checked by XRD spectroscopy – upon subjection to an aqueous
treatment exploiting their memory effect. Table 6 shows the
results obtained in the three reuse cycles; as can be seen, the
catalyst lost no activity after them.
3.2.3. Mechanism of the process
Scheme 2 depicts a plausible mechanism for the reaction.
Initially, hydrogen peroxide attacks a Bronsted basic site at the
catalyst surface to form a hydroperoxide species that
subsequently attacks benzonitrile to give a peroxycarboximidic
acid intermediate [50]. In the second step of the process, the
previous intermediate attacked cyclohexanone adsorbed at an
acid site of the catalyst to form an intermediate equivalent to the
Criegee adduct in homogeneous catalysis processes; finally, the
intermediate undergoes rearrangement to e-caprolactone, and
the peroxycarboximidic acid intermediate is transformed in
benzamide. This benzamide can be recycled to benzonitrile
using an appropriate procedure [51–54]. This mechanism
explains why the catalyst containing Sn4+ provided higher
catalytic activity and conversion values; in fact, this cation is
more acidic than Mg2+ and Al3+, so it must favour adsorption of
cyclohexanone and hence result in increased conversion values
relative to the solid containing the latter two cations.
Once the mechanism for the process was elucidated, the
Baeyer–Villiger was examined under more ‘‘realistic’’ condi-
tions in order to extrapolate the results to a large-scale scenario.
To this end, the H2O2:cyclohexanone ratio was decreased to a
level were, as can be seen from Table 7, the initial catalytic
activity was not significantly decreased. However, conversion
levelled off through exhaustion of peroxide after some time
under these conditions. We thus adopted a H2O2:cyclohexanone
ratio of 4 as optimal for the reaction.
4. Conclusion
The method used in this work to prepare tin- and zirconium-
containing hydrotalcite-like compounds allows the metal to be
incorporated into the structure as confirmed by the XRD
patterns for the solids. Thermal treatment of the resulting
LDHs at 450 8C for several hours destroys their layered
structure and produces mixed oxides of the constituent metals.
All uncalcined and calcined solids thus obtained exhibit
acceptable conversion and selectivity towards e-caprolactone
in the Baeyer–Villiger oxidation of cyclohexanone; however,
the best catalysts were found to be the Mg/Al/Sn hydrotalcites,
the activity of which increased with increasing tin content.
These results are consistent with a mechanism involving the
adsorption of hydrogen peroxide at a Bronsted basic site of the
catalyst to attack benzonitrile and form a peroxycarboximidic
acid intermediate that transfers oxygen to cyclohexanone

C. Jimenez-Sanchidrian et al. / Applied Catalysis A: General 312 (2006) 86–9494
adsorbed at a Lewis acid site of the catalyst. The most active
catalyst was found to remain active after three reuses.
Acknowledgments
The authors gratefully acknowledge funding by Spain’s
Ministerio de Educacion y Ciencia, Feder Funds and to the
Consejerıa de Innovacion, Ciencia y Empresa de la Junta de
Andalucıa.
References
[1] A. Vaccari, Appl. Clay Sci. 14 (1999) 161.
[2] B.F. Sels, D.E. De Vos, P.A. Jacobs, Catal. Rev. Sci. Eng. 43 (2001) 443.
[3] F. Cavanni, F. Trifiro, A. Vaccari, Catal. Today 2 (1991) 11.
[4] S. Miyata, Clay Clay Miner. 23 (1975) 369.
[5] M.A. Aramendia, Y. Aviles, V. Borau, J.M. Luque, J.M. Marinas, J.R.
Ruiz, F.J. Urbano, J. Mater. Chem. 9 (1999) 1603.
[6] M.A. Aramendia, V. Borau, C. Jimenez, J.M. Luque, J.M. Marinas, J.R.
Ruiz, F.J. Urbano, Mater. Lett. 43 (2000) 118.
[7] C.A. Drewien, D.R. Tallant, M.O. Eatough, J. Mater. Sci. 31 (1996) 4321.
[8] L. Anicai, A.C. Manea, T. Visan, Mol. Cryst. Liq. Cryst. 418 (2004) 41.
[9] O. Saber, H. Tagaya, J. Porous Mater. 10 (2003) 83.
[10] O. Saber, H. Tagaya, J. Inclusion Phenom. Macro. Chem. 45 (2003) 109.
[11] M. Intissar, S. Holler, F. Malherbe, J.P. Besse, F. Leroux, J. Phys. Chem.
Solids 65 (2004) 453.
[12] A. Baeyer, V. Villiger, Ber. Deut. Chem. Ges. 32 (1899) 3625.
[13] C.H. Hassal, Org. React. 9 (1957) 73.
[14] G.R. Krow, Org. React. 42 (1993) 251.
[15] G.R. Krow, Tetrahedron 37 (1981) 2697.
[16] G. Strukul, Angew. Chem. Int. Ed. 37 (1998) 1198.
[17] M. Renz, B. Meunier, Eur. J. Org. Chem. (1999) 737.
[18] B. Plesnicar, in: W.S. Trahanosky (Ed.), Oxidation in Organic Chemistry,
Academic Press, New York, 1978, p. 254.
[19] M. Hudlicky, Oxidations in Organic Chemistry, American Chemical
Society, Washington, 1990, p. 186.
[20] M. Taramasso, G. Perego, B. Notari, US Patent 4,410,501 (1983).
[21] A. Corma, H. Garcıa, Chem. Rev. 102 (2002) 3837.
[22] A. Corma, H. Garcıa, Chem. Rev. 103 (2003) 4307.
[23] A. Corma, L.T. Nemeth, M. Renz, S. Valencia, Nature 412 (2001) 423.
[24] M. Renz, T. Blasco, A. Corma, V. Fornes, R. Jensen, L. Nemeth, Chem.
Eur. J. 8 (2002) 4708.
[25] A. Corma, M.T. Navarro, M. Renz, J. Catal. 219 (2003) 242.
[26] A. Corma, V. Fornes, S. Iborra, M. Mifsud, M. Renz, J. Catal. 221 (2004)
67.
[27] A. Corma, S. Iborra, M. Mifsud, M. Renz, M. Susarte, Adv. Synth. Catal.
346 (2004) 257.
[28] M. Boronat, P. Concepcion, A. Corma, M. Renz, S. Valencia, J. Catal. 234
(2005) 111.
[29] A. Corma, S. Iborra, M. Mifsud, M. Renz, J. Catal. 234 (2005) 96.
[30] M. Boronat, A. Corma, M. Renz, G. Sastre, P.M. Viruela, Chem. Eur. J. 11
(2005) 6905.
[31] M.A. Aramendia, V. Borau, C. Jimenez, J.M. Luque, J.M. Marinas, J.R.
Ruiz, F.J. Urbano, Appl. Catal. A: Gen. 216 (2001) 257.
[32] M.A. Aramendia, V. Borau, C. Jimenez, J.M. Luque, J.M. Marinas, F.J.
Romero, J.R. Ruiz, F.J. Urbano, Stud. Surf. Sci. Catal. 130 (2000) 1667.
[33] M.A. Aramendia, V. Borau, C. Jimenez, J.M. Marinas, J.R. Ruiz, F.J.
Urbano, Appl. Catal. A: Gen. 206 (2001) 95.
[34] M.A. Aramendia, V. Borau, C. Jimenez, J.M. Marinas, J.R. Ruiz, F.J.
Urbano, J. Mol. Catal. A: Chem. 171 (2001) 153.
[35] M.A. Aramendia, V. Borau, C. Jimenez, J.M. Marinas, J.R. Ruiz, F.J.
Urbano, Appl. Catal. A: Gen. 255 (2003) 301.
[36] J.R. Ruiz, C. Jimenez-Sanchidrian, J. Hidalgo, J.M. Marinas, J. Mol.
Catal. A: Chem. 246 (2006) 190.
[37] K. Kaneda, S. Ueno, T. Imanaka, J. Chem. Soc., Chem. Commun. (1994)
797.
[38] K. Kaneda, S. Ueno, ACS Symp. Ser. 638 (1996) 300.
[39] S. Ueno, K. Ebitani, A. Ookubo, K. Kaneda, Appl. Surf. Sci. 121/122
(1997) 366.
[40] K. Kaneda, T. Yamashita, Tetrahedron Lett. 37 (1996) 4555.
[41] U.R. Pillai, E. Sahle-Demessie, J. Mol. Catal. A: Chem. 191 (2003) 93.
[42] JCPDS X-ray Powder Diffraction File no. 22-700, 1986.
[43] JCPDS X-ray Powder Diffraction File no. 9-27, 1986.
[44] J. Kannan, S. Velu, V. Ramkumar, C.S. Swamy, J. Mater. Sci. 30 (1995)
1462.
[45] JCPDS X-ray Powder Diffraction file no. 45-946.
[46] F. Rey, V. Fornes, J.M. Rojo, J. Chem. Soc., Faraday Trans. 88 (1992)
2233.
[47] S. Narayanan, K. Krishna, Appl. Catal. A: Gen. 174 (1998) 221.
[48] M.A. Aramendıa, Y. Aviles, J.A. Benıtez, V. Borau, C. Jimenez, J.M.
Marinas, J.R. Ruiz, F.J. Urbano, Micropor. Mesopor. Mater. 29 (1999) 319.
[49] S. Miyata, W. Wakamiya, I. Kobakawa, J. Catal. 34 (1974) 117.
[50] R. Prihod’ko, M. Sychev, I. Kolomitsyn, P.J. Stobbelaar, E.J.M. Hensen,
R.A. Van Santen, Micropor. Mesopor. Mater. 56 (2002) 241.
[51] D.S. Bose, A.V. Narsaiah, Synthesis (2001) 373.
[52] D.S. Bose, P.R. Goud, Tetrahedron Lett. 40 (1999) 747.
[53] C.R. Stephens, E.J. Bianco, F.J. Pilgrim, J. Am. Chem. Soc. 77 (1955)
1701.
[54] G.E. Ficken, H. France, R.P. Linstead, J. Chem. Soc. (1954) 3730.





![Intercalation of hydrotalcites with hexacyanoferrate(II) and (III) – … · 2010. 6. 9. · effect the d(003) spacing of the hydrotalcite [31]. The potential application of hydrotalcites](https://img.pdfslide.us/doc/110x75/60e8d3340c17be51b86a281e/intercalation-of-hydrotalcites-with-hexacyanoferrateii-and-iii-a-2010-6.jpg)























