Embed Size (px)
Citation preview

JOURNAL OF BACTERIOLOGY,0021-9193/99/$04.0010
June 1999, p. 3730–3742 Vol. 181, No. 12
Copyright © 1999, American Society for Microbiology. All Rights Reserved.
Bacterioferritin A Modulates Catalase A (KatA) Activity andResistance to Hydrogen Peroxide in Pseudomonas aeruginosa
JU-FANG MA,1 URS A. OCHSNER,2 MARTIN G. KLOTZ,3,4 VAGIRA K. NANAYAKKARA,1,5
MICHAEL L. HOWELL,1 ZAIGA JOHNSON,2 JAMES E. POSEY,6 MICHAEL L. VASIL,2
JOHN J. MONACO,1,5 AND DANIEL J. HASSETT1*
Department of Molecular Genetics, Biochemistry and Microbiology1 and Howard Hughes Medical Institute,5
University of Cincinnati College of Medicine, Cincinnati, Ohio 45267-0524; Department of Microbiologyand Immunology, University of Colorado Health Sciences Center, Denver, Colorado 802622;
Department of Biology3 and Center for Genetics and Molecular Medicine,4
University of Louisville, Louisville, Kentucky 40292; and Department ofMicrobiology, University of Georgia, Athens, Georgia 30609-40666
Received 16 September 1998/Accepted 8 April 1999
We have cloned a 3.6-kb genomic DNA fragment from Pseudomonas aeruginosa harboring the rpoA, rplQ,katA, and bfrA genes. These loci are predicted to encode, respectively, (i) the a subunit of RNA polymerase; (ii)the L17 ribosomal protein; (iii) the major catalase, KatA; and (iv) one of two iron storage proteins calledbacterioferritin A (BfrA; cytochrome b1 or b557). Our goal was to determine the contributions of KatA and BfrAto the resistance of P. aeruginosa to hydrogen peroxide (H2O2). When provided on a multicopy plasmid, the P.aeruginosa katA gene complemented a catalase-deficient strain of Escherichia coli. The katA gene was found tocontain two translational start codons encoding a heteromultimer of ;160 to 170 kDa and having an apparentKm for H2O2 of 44.7 mM. Isogenic katA and bfrA mutants were hypersusceptible to H2O2, while a katA bfrAdouble mutant demonstrated the greatest sensitivity. The katA and katA bfrA mutants possessed no detectablecatalase activity. Interestingly, a bfrA mutant expressed only ;47% the KatA activity of wild-type organisms,despite possessing wild-type katA transcription and translation. Plasmids harboring bfrA genes encoding BfrAaltered at critical amino acids essential for ferroxidase activity could not restore wild-type catalase activity inthe bfrA mutant. RNase protection assays revealed that katA and bfrA are on different transcripts, the levels ofwhich are increased by both iron and H2O2. Mass spectrometry analysis of whole cells revealed no significantdifference in total cellular iron levels in the bfrA, katA, and katA bfrA mutants relative to wild-type bacteria. Ourresults suggest that P. aeruginosa BfrA may be required as one source of iron for the heme prosthetic group ofKatA and thus for protection against H2O2.
Bacterial aerobic respiration involves a four-electron reduc-tion of molecular oxygen (O2) to water. Depending upon theenvironmental conditions, aerobic respiration can be ex-tremely dangerous to the cell. Such is the case when aberrantelectron flow from the electron transport chain or cellularredox enzymes to O2 leads to the production of reactive oxygenintermediates (ROIs). These include superoxide (O2
2), hydro-gen peroxide (H2O2), and hydroxyl radical (HO.). The un-checked production of each of these species can lead to celldamage, mutations, or death. The production of HO., the mostdestructive of the above compounds, is dependent in part uponthe presence of a transition metal, such as iron or copper, andeither O2
2 or H2O2. Relief from ROIs is provided by variousdefense systems, including antioxidant enzymes (superoxidedismutase [SOD]), catalase, and peroxidase), DNA repair en-zymes and binding protein (e.g., Dps [DNA binding proteinfrom starved cells] [33]), and free-radical-scavenging agents (6,24).
Pseudomonas aeruginosa is a gram-negative bacterium thatgains its greatest metabolic energy through aerobic respiration.To counter the production of ROIs, the organism possessestwo SODs, with either iron (Fe2; encoded by sodB [18, 20]) or
manganese (Mn2; encoded by sodA [18, 20]) as cofactor andwhose function is to disproportionate O2
2 to H2O2 and O2(34). To remove H2O2, P. aeruginosa possesses three catalases,KatA (10, 17), KatB (10), and KatC (40). KatA activity is themajor catalase activity detected in all phases of growth (10, 17).In contrast, KatB activity is detectable in bacteria exposed toH2O2 or paraquat, the latter of which generates a constant fluxof H2O2 through SOD-catalyzed dismutation of O2
2 (10). Un-like KatA and KatB, little is known of the biological role ofKatC in P. aeruginosa. In fact, the putative katC gene was onlyrecently discovered fortuitously via the Pseudomonas GenomeProject (40).
Most bacterial catalases are multimers (typically dimers, tet-ramers, or hexamers) that require heme b or heme d for cat-alytic activity. The final step of heme synthesis is catalyzed byferrochelatase, which condenses Fe21 into protoporphyrin IX.Little is known of the cellular source of iron required for hemeassembly. One protein that could provide iron for such a pro-cess is bacterioferritin A (BfrA, also known as cytochrome b1or b557), the major iron storage protein in P. aeruginosa (38).Actually, there is evidence in P. aeruginosa for two Bfr proteins(BfrA and BfrB), which differ in their N-terminal amino acidsequences (38, 38a). BfrA is a complex of 24 subunits capableof binding 700 iron atoms (38). It also binds 3 to 9 heme groupsper 24 subunits in vivo and 24 heme groups in vitro (25).Recently, Kim et al. (27) identified a bfr gene encoding abacterioferritin in the related organism P. putida; this gene waslocated downstream of a gene encoding a group III catalase,
* Corresponding author. Mailing address: Department of MolecularGenetics, Biochemistry and Microbiology, University of CincinnatiCollege of Medicine, 231 Bethesda Ave., Cincinnati, OH 45267-0524.Phone: (513) 558-1154. Fax: (513) 558-8474. E-mail: [email protected].
3730
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

CatA. However, the attractive hypothesis that one function ofP. putida Bfr is to provide iron for the heme prosthetic groupof CatA and thus to contribute to resistance to H2O2 was notpursued. A precedent for such a hypothesis stemmed fromresearch with Campylobacter jejuni, for which mutants deficientin ferritin, a protein related to bacterioferritin, were moresensitive to oxidative stress than wild-type organisms (50).
In this study, we have cloned and characterized the genesencoding KatA and BfrA in P. aeruginosa. Our studies suggest
a necessity for BfrA in the maintenance of optimal KatA ac-tivity. Hence, we propose that BfrA stores iron that is incor-porated into heme, a necessary prosthetic group for KatAactivity.
MATERIALS AND METHODS
Bacterial strains, plasmids, and growth conditions. All bacteria used in thisstudy are listed in Table 1 and were grown in either Luria (L) broth (10 g oftryptone, 5 g of yeast extract, and 5 g of NaCl per liter) or M9 minimal medium
TABLE 1. Strains and plasmids used in this study
Strain or plasmid Genotype or characteristicsa Source or reference
StrainsE. coli
DH5a F2 f80dlacZDM15 recA1 endA1 gyrA96 thi-1 relA1 supE44 hsdR17(rK2
mK1) D(lacZYA-argF)U169
Gibco-BRL
SM10 Mobilizer strain 47BL21(lDE3) F2 ompT hsdSB (rB
2 mB2) gal dcm (DE3) Novagen
CSH7 lacY rpsL thi-1 31UM1 As CSH7 plus katE1 katG14 31
P. aeruginosaPAO1 Wild type; prototroph 22PAO1 katA Gmr katA::Gm This studyPAO1 bfrA Gmr DbfrA::Gm This studyPAO1 katA bfrA Gmr DkatA bfrA::Gm This studyFRD2 katB algT18 katB::Gm 10
PlasmidspBluescript(KS)- or pBluescript (KS)1 Extended polylinker pUC derivative StratagenepKS-TA Apr; TA PCR cloning vector that uses EcoRV site for cloning purposes This studypCRII Apr; TA PCR cloning vector InVitrogenpCR2.1 Apr; TA PCR cloning vector InVitrogenpUCGM Apr Gmr; pUC19 1 850-bp Gmr cassette 45pPZ30 Apr; broad-host-range lacZ-based promoter probe vector 43pUCP19 Apr; broad-host-range expression vector 52pUCP21T Apr; broad-host-range expression vector 52pEX100T Apr Cbr; mobilizable oriT sacB vector for construction of mutants 46pRK2013 Kmr Ori (ColE1) OriT (Mob1) Tra1 14pET23a Apr; overexpression vector NovagenpJFM12 Apr; pKS2 with 3.6-kb EcoRI-EcoRV fragment containing
rpsD9-rpoA-rplQ-katA-bfrAThis study
pJFM13 Apr; pEX100T with blunted 3.6-kb EcoRI-EcoRV fragment containingrpsD9-rpoA-rplQ-katA-bfrA9 in the SmaI site of the vector
This study
pJFM14 Apr Gmr; pJFM13 with 850-bp aaC1 cassette within the SmaI site ofkatA
This study
pJFM15 Apr Gmr; pEX100T with blunted EcoRI fragment frompBFR1020DbfrA::Gm and with 850-bp aaC1 cassette within thedeleted 520-bp NdeI-SstII fragment, deleting bfrA
This study
pJFM16 Apr Gmr; pJFM13 with 850-bp aaC1 cassette within the deleted1,311-bp SmaI-NdeI site, deleting katA and bfrA
This study
pJFM17 Apr; pKS2 with 1,450-bp katA PCR product This studypJFM18 Apr; pET23a with NcoI-EagI katA This studypRP411 Apr; pKS1 containing 411-bp SalI-EcoRV fragment of katA-bfrA
intergenic regionThis study
pBFR1020 pCR2.1 harboring 1,020-bp PCR fragment containing bfrA This studypBFR1020DbfrA::Gm pBFR1020 with 520-bp NdeI-SstII deletion of bfrA replaced by aaC1
cassetteThis study
pPZ-katA Apr; pPZ30 containing the katA promoter on a 756-bp EcoRI-PstIfragment translationally fused to lacZ
This study
pPZ-bfrA Apr; pPZ30 containing the bfrA promoter on a 389-bp EcoRI-PstIfragment translationally fused to lacZ
This study
pBFR4 pUCP19 with wild-type bfrA gene This studypBFR18 pUCP19 with bfrA18 encoding Bfr18 (E18K) This studyPBFR25 pUCP19 with bfrA25 encoding Bfr25 (Y25I) This study
a Abbreviations used for genetic markers were those described by Holloway et al. (22). Mob1, mobilization site (ColE1); Tra1, conjugative phenotype; oriT, originof transfer (RK2); Apr, ampicillin resistance; Cmr, chloramphenicol resistance; TA, thymine-adenine; pKS2, pBluescript KS(2); pKS1, pBluescript (KS)1.
VOL. 181, 1999 BfrA MODULATES CATALASE A ACTIVITY IN P. AERUGINOSA 3731
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

(6 g of Na2HPO4, 3 g of KH2PO4, 1 g of NH4Cl, 0.5 g of NaCl, 3 mg of CaCl2,0.25 g of MgSO4 z 7H2O, and 2 g of glucose per liter). Suspensions were grownat 37°C with shaking at 300 rpm or on a roller wheel at 70 rpm. Culture volumeswere 1/10 the total flask volume to ensure maximum aeration. Media weresolidified with 1.5% Bacto Agar. Frozen stocks were stored indefinitely at 280°Cin a 1:1 mixture of 25% glycerol and stationary-phase suspension.
Construction of a P. aeruginosa katB genomic library, cloning steps, andsequence analysis. Genomic DNA (50 mg) from P. aeruginosa FRD2 katB (10)was digested with 10 U each of EcoRI and EcoRV at 37°C for 2 h. DNAfragments were separated on a 10 to 40% sucrose gradient (3). Purified 2- to 5-kbfragments were ligated into EcoRI-EcoRV-digested pBluescript KS(2) andscreened for the presence of P. aeruginosa katA with a heterologous catA geneprobe from P. putida (27). Plasmid DNA from positive clones was transformedinto catalase-deficient Escherichia coli UM1 (31). Bacterial colonies harboringthe P. aeruginosa katA gene bubbled vigorously when coated with 8.8 M H2O2. Aselected plasmid, pJFM12, that complemented for catalase activity was se-quenced on both strands with a PRISM Dye Deoxy Terminator cycle sequencingkit and analyzed on an ABI model 373A DNA sequencer. Oligonucleotides forsequencing and PCR analysis were synthesized at the DNA Core Facilities in theDepartment of Molecular Genetics, Biochemistry and Microbiology at the Uni-versity of Cincinnati College of Medicine or in the Department of Microbiologyand Immunology at the University of Colorado Health Sciences Center. Se-quence analysis was performed with MacVector 6.5 (Eastman Chemical Co.,New Haven, Conn.), Gene Runner (Hastings Software, Inc.), or Sequencer 3.0(GeneCodes, Madison, Wis.). Amino acid alignments were performed with ei-ther the BLASTP program provided by the National Center for BiotechnologyInformation (1) or the Align Plus 3.0 global alignment program (Sci-Ed Soft-ware, Durham, N.C.).
Manipulation of recombinant DNA and genetic techniques. Plasmid DNA wastransformed into either E. coli DH5a-MCR (Gibco-BRL, Gaithersburg, Md.) orE. coli SM10 (47). 5-Bromo-4-chloro-3-indolyl-b-D-galactopyranoside (X-Gal; 40mg/ml) was often added to agar medium to detect the presence of insert DNA.Restriction endonucleases, the Klenow fragment, T4 DNA polymerase, and T4DNA ligase were used as specified by the vendor (Gibco-BRL). Plasmid DNAwas isolated with plasmid mini-isolation kits (Qiagen Corp.). Restriction frag-ments were recovered from agarose gels with SeaPlaque low-melting-point aga-rose (FMC BioProducts, Rockland, Maine). PCRs were performed with TaqDNA polymerase (Gibco-BRL) and appropriate primers by use of a Perkin-Elmer Cetus thermal cycler with 30 cycles of denaturation (1 min, 94°C), an-nealing (1 min, 54°C), and extension (1 min, 72°C). Amplified DNA fragmentswere gel purified, cloned into pCRII or pCR2.1 (both from InVitrogen) or apBluescript KS(2)-based PCR vector (this study), and sequenced.
Phylogenetic analyses. The aligned amino acid sequences were processed byheuristic parsimonial analyses with PAUP version 3.1.1 (48). In order to mini-mize the possibility that the algorithm would detect local parsimony (potentialmonophyly of clusterings comprised of more than one species), 200 bootstrapreplicates were generated. A 50% majority-rule consensus tree was constructedfrom parsimony replicates by use of tree bisection-reconnection and nearest-neighbor branch-swapping methods with stepwise addition of the closest se-quence.
Overexpression of KatA in E. coli. To overexpress P. aeruginosa KatA, PCRprimers (sense, 59-CATATGGAAGAGAAGACCCGCCTGAC-39; antisense,59-CGGCGGCGTCCAGCTTCAGGCCGAGGG-39) were used to amplify a1,450-bp katA fragment with pJFM12 as a template. This fragment was clonedinto a pBluescript KS(2)-based PCR cloning vector, pKS-TA (Table 1), and thekatA fragment was excised with NdeI and EagI and ligated into pET23a (Nova-gen). After transformation into E. coli BL21(lDE3), bacteria were grown aero-bically to the mid-logarithmic phase (optical density at 600 nm, 0.6) and treatedwith 0.4 mM isopropyl-b-D-thiogalactopyranoside (IPTG) for 3 h at 37°C. Bac-teria were harvested by centrifugation at 10,000 3 g for 10 min at 4°C and washedin 0.9% saline, and the pellet was resuspended in 0.1 M NaH2PO4 (pH 8.0)containing 8 M urea. Six-His-tagged KatA was purified under denaturing con-ditions using the Qiagen Expressionist kit. The purity of recombinant KatA wasassessed after sodium dodecyl sulfate (SDS)-polyacrylamide gel electrophoresis(PAGE) with 10% acrylamide and staining with Coomassie blue.
Construction of P. aeruginosa katA, bfrA, and katA bfrA mutants. The strategyfor insertional inactivation of the katA and bfrA genes was facilitated by use ofthe gene replacement vector pEX100T (46), which allowed for the selection ofdouble-crossover events with 6% sucrose (44). To construct a katA mutant, a;3.6-kb EcoRI-EcoRV fragment from pJFM12 was filled in with the Klenowfragment and ligated into the unique SmaI site within pEX100T, formingpJFM13. This plasmid was cut with SmaI, a unique site within the katA locus, andligated to an 850-bp aaC1 (encoding gentamicin resistance [Gmr]) cassette ex-cised from pUCGM (45), forming pJFM14. For the construction of the DbfrAmutant, a 1,020-bp fragment containing the bfrA region was generated by PCRwith primers having the sequences 59-ACCGGGTGGACGACGACTACT-39and 59-GCCAACTGGCTGGTCAACCTC-39 and cloned into pCR2.1, yieldingpBFR1020. A 520-bp NdeI-SstII fragment of pBFR1020 comprising the entirebfrA coding sequence was replaced with the aaC1 cassette, resulting inpBFR1020DbfrA::Gm. The bfrA::Gm fragment was excised with EcoRI, filled inwith the Klenow fragment, and cloned into SmaI-cut pEX100T, formingpJFM15. A katA bfrA double mutant was constructed by replacing the SmaI-NdeI
katA9-bfrA9 fragment from pJFM13 with the aaC1 cassette, forming pJFM16.After biparental mating of E. coli SM10 harboring pJFM14, pJFM15, or pJFM16with recipient P. aeruginosa PAO1, plasmid integration into the genome byhomologous recombination was assessed by selection on Pseudomonas isolationagar-gentamicin (300 mg/ml) plates. Isolated Gmr colonies were picked andgrown in L broth until the mid-log phase, and serial dilutions were plated onPseudomonas isolation agar-gentamicin plates containing 6% sucrose. Candidatemutants were confirmed by Southern blot and catalase activity gel analyses (forkatA and katA bfrA mutants [19]).
Construction of altered BfrA proteins. Plasmid pBFR18 (E18K; see below)was constructed as follows. The 59 0.4-kb portion of bfrA (fragment A) wasamplified by PCR with primer 1 (59-ACCGGGTGGACGACGACTACT-39) andHindIII-containing primer 2 (HindIII sequence underlined: 59-AAGCTTGCCGGTCAACAGCGTATTG-39). The 39 0.63-kb portion of bfrA (fragment B) wasamplified with primer 3 (59-AAGCTTGCCGCGCGCGACCAGT-39) andprimer 4 (59-GCCAACTGGCTGGTCAACCTC-39). PCR fragments A and Bwere cloned into pCR2.1 and sequenced for verification and orientation. Frag-ment A was excised with EcoRI-HindIII and ligated into pUCP19 linearized withEcoRI-HindIII. The resulting plasmid was linearized with HindIII, and fragmentB was ligated into the HindIII site, yielding pBFR18. This plasmid contains theHindIII recognition sequence AAG CTT at codons 18 and 19 (the wild-typesequence at these positions is GAG CTG), resulting in a glutamate-to-lysinechange at position 18 (E18K). Plasmid pBFR25 (Y25I; see below) was con-structed as follows. The 59 0.42-kb portion of bfrA (fragment C) was amplified byPCR with primer 1 and SspI-containing primer 5 (SspI sequence underlined:59-AATATTTGGTGCCGCGCGGCCAGCTC-39). The 39 0.61-kb portion ofbfrA (fragment D) was amplified with primer 6 (59-AATATTCATCCACTCGCGCATGTAC-39) and primer 4. PCR fragments C and D were cloned intopCR2.1 and sequenced. Fragment D was excised with SspI-HindIII and ligatedinto pCR2.1 containing fragment C linearized with SspI-HindIII. The 1.03-kbfused fragments C and D were excised with EcoRI and ligated into pUCP19linearized with EcoRI, resulting in pBFR25. This plasmid contains the sequenceCAA ATA TTC at codons 24, 25, and 26 (the wild-type sequence at thesepositions is CAG TAC TTC), resulting in a tyrosine-to-isoleucine change atposition 25 (Y25I).
Construction of katA-lacZ and bfrA-lacZ fusions. The katA promoter wasisolated by PCR with primers having the sequences 59-AAGTGGTCGTCACCTGAGC-39 and 59-TCTCGAGGAACCACACGTC-39, cloned into pCR2.1, se-quenced, and directionally cloned as a 756-bp EcoRI-PstI fragment into pPZ30cut with EcoRI and PstI. The resulting pPZ-katA construct represents an in-frame katA-lacZ translational fusion after 33 codons. Similarly, the bfrA pro-moter was isolated by PCR with primers having the sequences 59-ACCGGGTGGACGACGACTACT-39 and 59-CTGCAGCGTATTGAGGTAATCG-39, cloned,and subsequently ligated into pPZ30 as a 389-bp EcoRI-PstI fragment. Theresulting construct, pPZ-bfrA, contains the first 14 codons of the bfrA gene fusedin frame to lacZ.
Purification of P. aeruginosa KatA. P. aeruginosa FRD2 katB (10), a nonmu-coid algT18 mutant of mucoid cystic fibrosis isolate FRD1 (13), was grown in 10liters of L broth containing 2 mM FeCl3 for 17 h at 37°C, followed by a 2-haerobic incubation in the presence of 350 mM paraquat and 10 mM H2O2 tostimulate katA transcription. The bacteria were pelleted by centrifugation at10,000 3 g for 15 min, washed in 0.9% saline, and resuspended in 50 mMTris-HCl (pH 7.4) containing lysozyme (0.02%) and the protease inhibitorsphenylmethylsulfonyl fluoride (0.5 mM), leupeptin (0.5 mM), and pepstatin (0.5mM). The suspension was subjected to three freeze-thaw (280°C-37°C) cycles toaid in breakage of the cells and further disrupted three times with a Frenchpressure cell at 12,000 lb/in2 and 4°C. Unbroken cells and cell debris wereclarified by ultracentrifugation at 100,000 3 g for 1 h at 4°C. The clarified extractwas brought to 80% saturation with ammonium sulfate and incubated at 4°C for17 h, and the precipitated protein was clarified by centrifugation at 10,000 3 g for20 min. The precipitate was dissolved in Tris-HCl (pH 7.4) and dialyzed againstsix 1-liter changes of the same buffer at 4°C. This solution was filtered through a0.22-mm-pore-size filter (Nalgene) and concentrated with an Amicon YM-100membrane. The retentate, containing KatA, was passed over a DE-52 column (2by 18 cm; Whatman International Ltd., Kent, England) and eluted with a 0 to 200mM NaCl gradient. After concentration of the catalase-positive fractions anddialysis against distilled water and then 50 mM potassium phosphate (pH 7.4),the sample was loaded on a hydroxyapatite column (2 by 13 cm) equilibrated withpotassium phosphate. KatA has previously been found not to bind hydroxyapa-tite (10). KatA-positive fractions were applied to a Phenyl-Sepharose column,and the enzyme was eluted with a decreasing gradient of ammonium sulfate.Purified KatA fractions were pooled, concentrated, and stored on ice at 0°C. Themolecular mass of purified native KatA was estimated by gel filtration withSephacryl S300 equilibrated with 50 mM Tris-HCl–100 mM NaCl (pH 7.4) andwith the known molecular mass standards b-amylase (200 kDa), yeast alcoholdehydrogenase (150 kDa), bovine serum albumin (65 kDa), and carbonic anhy-drase (29 kDa). The a- and b-subunit sizes of KatA were determined by dena-turing (boiled-sample) SDS-PAGE.
Hydrogen peroxide sensitivity assays. (i) Broth sensitivity. Bacteria weregrown aerobically for 17 h at 37°C in L broth. Suspensions were diluted 1:100 infresh, prewarmed L broth and grown until the bacteria reached the early loga-rithmic phase (optical density at 600 nm, 0.6). Organisms were diluted 1:10 in 3
3732 MA ET AL. J. BACTERIOL.
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

ml of prewarmed L broth and incubated with increasing concentrations of H2O2(Sigma Chemical Co.) for 15 min. The suspensions were serially diluted in 0.9%saline containing 10 mg of bovine liver catalase (Boehringer Mannehim Bio-chemicals) per ml, and aliquots were plated on L agar. CFU were enumeratedafter incubation at 37°C for 24 to 48 h.
(ii) Disk sensitivity. To assess the role of iron in sensitivity to H2O2, bacteriawere grown to the stationary phase in aerobic M9 broth with 0.5% glucose as thecarbon source and with or without 50 mM FeCl3. Samples of 100 ml were dilutedin 3 ml of molten (50°C) M9 top agar containing 0.6% agar and layered on M9agar plates. Sterile filter paper disks (7 mm) saturated with 10 ml of 8.8 M H2O2was determined by measuring the diameter of growth inhibition after aerobicincubation of the plates at 37°C for 24 h.
RNase protection assays. RNase protection assays were performed with theRiboprobe system (Promega). A 411-bp SalI-EcoRV fragment containing the 39end of katA and a portion of the 59 end of bfrA was cloned behind the T7promoter of pBluescript (KS)1 cut with SalI and EcoRV, resulting in pRP411.The antisense katA-bfrA riboprobe was generated and radiolabeled by in vitrorunoff transcription from the T7 promoter of SalI-linearized pRP411. Normal-ized (20 mg) sample of total RNA extracted from cells during the exponential (6h) or stationary (12 h) growth phase under low- or high-iron conditions werehybridized to excess katA-bfrA riboprobe. As a control for RNA integrity andloading accuracy, a constitutively expressed housekeeping gene, omlA, was alsoused as a riboprobe as previously described (49). RNA protected from single-strand-specific RNase was separated on a denaturing 5% polyacrylamide–8 Murea gel that was dried and analyzed by autoradiography.
Mass spectrometry analysis. (i) Sample preparation for MALDI analysis.SDS-polyacrylamide gels were stained with 200 mM imidazole for 15 min, fol-lowed by a 5-min incubation with 50 mM ZnCl2. The ;56- and 45-kDa KatAbands were excised from the gels as 1-mm2 sections and destained twice with 200ml of 50 mM citric acid for 20 min, followed by 300 ml of 10 mM NH4HCO3 in25% (vol/vol) acetonitrile for 30 min. Gel fragments were rinsed with 100 ml ofdeionized distilled water for 15 min and dried in a Speed Vac. The dried gelfragments were swollen with 6 ml of a 1-mg/ml solution of sequencing-gradetrypsin (Promega) dissolved in 50 mM acetic acid. The mixture was brought topH 8.0 by the addition of 44 ml of 25 mM NH4HCO3 digestion buffer, digestionwas allowed to proceed for 24 h at 37°C, and the reaction was stopped by theaddition of 75 to 100 ml of 0.1% trifluoroacetic acid (TFA). As controls, twoother samples, one with protein-free gel fragments and the other with trypsin anddigestion buffer, were processed in the same fashion. All samples were centri-fuged at 13,000 3 g for 5 min, and only the supernatant was removed. TFA (200ml; 0.1%) was added to the remaining gel particles, and the resulting solution wasincubated for 20 min with intermittent vortexing. The supernatant was removed,and the gel fragments were resuspended in 60% acetonitrile in 0.1% TFA. Afteradditional incubation and vortexing for 30 min, the supernatant was removed.The last step was repeated twice. All extracts were pooled and dried in a SpeedVac. To the dried extract was added 5 ml of 50% acetonitrile in 0.1% TFA, andthe mixture was spotted on a stainless steel target. When the sample was dry, 1ml of saturated matrix solution (4-hydroxy-a-cyanocinnamic acid in 0.1% TFA)was spotted on the sample and allowed to air dry.
(ii) MALDI analysis. Matrix assisted laser desorption ionization (MALDI)mass spectra were obtained on a MALDI TOFSPEC SE mass spectrometer inreflectron mode. The laser used for ionization was set at 337 nm with a pulsewidth of 4 ns and ;180 mJ per pulse. The spectra presented in this work areaverages of 20 to 30 laser shots. The ion acceleration voltage was set at 25 kV.The data were processed and stored on a DEC-3000 a-work station.
(iii) Cellular iron content. Bacteria were grown aerobically in 400 ml of Lbroth until the stationary phase. After centrifugation at 10,000 3 g for 10 min at4°C, organisms were washed twice in 200 ml of phosphate-buffered saline (PBS)with 1 mM EDTA (pH 7.4) and resuspended in 200 ml of PBS without EDTA.After centrifugation, the pellet was resuspended in 15 ml of PBS, 10 ml of whichwas used for iron analysis. Total viable cells and cell dry weight were estimatedwith the remaining 5-ml suspension. For iron analysis, pelleted bacteria wereresuspended in 2 ml of Ultrex II nitric acid (J. T. Baker, Phillipsburg, N.J.) andincubated at 80°C for 1 h, and the volume was brought to 20 ml with deionizeddistilled water. The samples were analyzed for iron content by inductively cou-pled plasma-optical emission spectroscopy with a model 965 Plasma Atomcompapparatus (Thermo Jarrell Ash, Franklin, Md.) at the Chemical Analysis Labo-ratory, University of Georgia, Athens. All buffers and nitric acid solutions wereanalyzed as described above to correct for background. The data were calculatedas both the number of iron atoms per cell and milligrams of iron per milligramof cell dry weight.
Cell extract preparation, nondenaturing gel electrophoresis, and biochemicalassays. Cell extracts were prepared from cultures harvested by centrifugation at10,000 3 g for 10 min at 4°C. Bacteria were washed twice in ice-cold 50 mMpotassium phosphate buffer (pH 7.0) and sonicated in an ice-water bath for 10 swith a model W-225 sonicator (Heat-Systems, Inc., Farmington, N.Y.) at setting5. The sonicate was clarified by centrifugation at 13,000 3 g for 10 min at 4°C.Cell extract preparation for native gel electrophoresis was performed as de-scribed above, except that 50 mM Tris-HCl (pH 7.8) was used as the diluent.Catalase activity was determined by monitoring the decomposition of 18 mMH2O2 at 240 nm (5, 10, 19). One unit of activity is that which decomposes 1 mmolof H2O2 min21 mg21. Determination of the Km value for purified KatA was
accomplished at 22°C with 1 to 80 mM H2O2 and 7 3 10210 M KatA. b-Galac-tosidase assays were performed on chloroform-SDS-treated bacteria with o-ni-trophenyl-b-D-galactopyranoside, and the results are expressed as Miller units(35). Protein concentrations were estimated by the method of Bradford (8) withbovine serum albumin fraction V (Sigma) as the standard.
Nucleotide sequence accession number. The DNA and amino acid sequencespresented in this work have been assigned GenBank accession no. AF047025.
RESULTSCloning and characterization of the katA gene of P. aerugi-
nosa: identification of adjacent loci rpoA, rplQ, and bfrA. DNAsequence analysis of the 3.6-kb genomic DNA fragment fromP. aeruginosa FRD2 that allowed for catalase expression incatalase-deficient E. coli UM1 revealed four open readingframes (ORFs), a map of which is depicted in Fig. 1. The firstORF comprised 999 bp and encoded a predicted protein of 333amino acids. A BLASTP GenBank search for this proteinrevealed similarity to the a subunit of the DNA-directed RNApolymerase family (data not shown). The second ORF com-prised 390 bp and encoded a putative protein of 129 aminoacids which was similar to 50S ribosomal subunit protein L17of E. coli (GenBank accession no. U18997). The third ORFcomprised 1,446 bp and encoded a predicted monomer of 482amino acids. This protein was 82% identical to the catalase ofProteus mirabilis (GenBank accession no. P42321). The fourthORF comprised 462 bp and encoded a putative protein of 154amino acids. This protein was 79% identical to a bacteriofer-ritin of the related organism P. putida (27).
Amino acid identity of KatA and BfrA with bacterial cata-lases and bacterioferritins. Since this study was focused on thepotential relationship between KatA and BfrA in protecting P.aeruginosa against H2O2, we thought it necessary to determinethe similarity of KatA and BfrA to other bacterial catalasesand bacterioferritins. A BLASTP homology search for KatAand other bacterial catalases revealed the greatest identity withcatalases from P. mirabilis (GenBank accession no. P42321;82% identity over 377 amino acids), Vibrio fischeri (GenBankaccession no. AF011784; 79% identity over 371 amino acids),Bordetella pertussis (GenBank accession no. P48062; 77% iden-tity over 370 amino acids), and Bacteroides fragilis (GenBankaccession no. U66717; 75% identity over 374 amino acids)(alignment not shown). A search for Bfr proteins revealed thegreatest identity with Bfr proteins from P. putida (GenBankaccession no. U66717; 79% identity over 154 amino acids),Neisseria gonorrhoeae (GenBank accession no. P72080; 62%identity over 154 amino acids), and Salmonella typhimurium(GenBank accession no. AF058449; 43% identity over 141amino acids) (Fig. 2).
FIG. 1. Genetic map of the 3.605-kb EcoRI-EcoRV fragment harboringrpoA, rplQ, katA, and bfrA of P. aeruginosa PAO1 and putative gene products.The large loopholes indicate transcriptional terminators. The flags pointingdownward indicate the promoter regions upstream of the katA and bfrA genes.The restriction sites from which single katA and bfrA mutants were generated viainsertional mutagenesis are given.
VOL. 181, 1999 BfrA MODULATES CATALASE A ACTIVITY IN P. AERUGINOSA 3733
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

The crystal structure (15), iron and heme binding capacities(12), and residues essential for ferroxidase activity of the E. coliBfr protein are known. P. aeruginosa BfrA possesses the samefour glutamate and single tyrosine and histidine residues es-sential for ferroxidase activity as E. coli Bfr (30). Interestingly,P. aeruginosa BfrA and the other aligned Bfr proteins in Fig.2A possess the same conserved residues, suggesting that theyalso possess ferroxidase activity.
Phylogenetic analyses of KatA and BfrA. Following thealignment of amino acid sequences of KatA and BfrA withsimilar proteins, unrooted phylogenetic trees were constructedby parsimony methods based on the amino acid sequences of94 catalases (data not shown) and 35 (bacterio)ferritins (Fig.2B). The phylogenetic tree generated for catalases was similarto the one constructed recently for 74 eukaryotic and prokary-otic catalases (29). P. aeruginosa KatA is a group III bacterialcatalase (29), as is P. putida CatA and Wolbachia sp. Cat, andis most closely related to the major catalase, KatA, of P. mira-bilis.
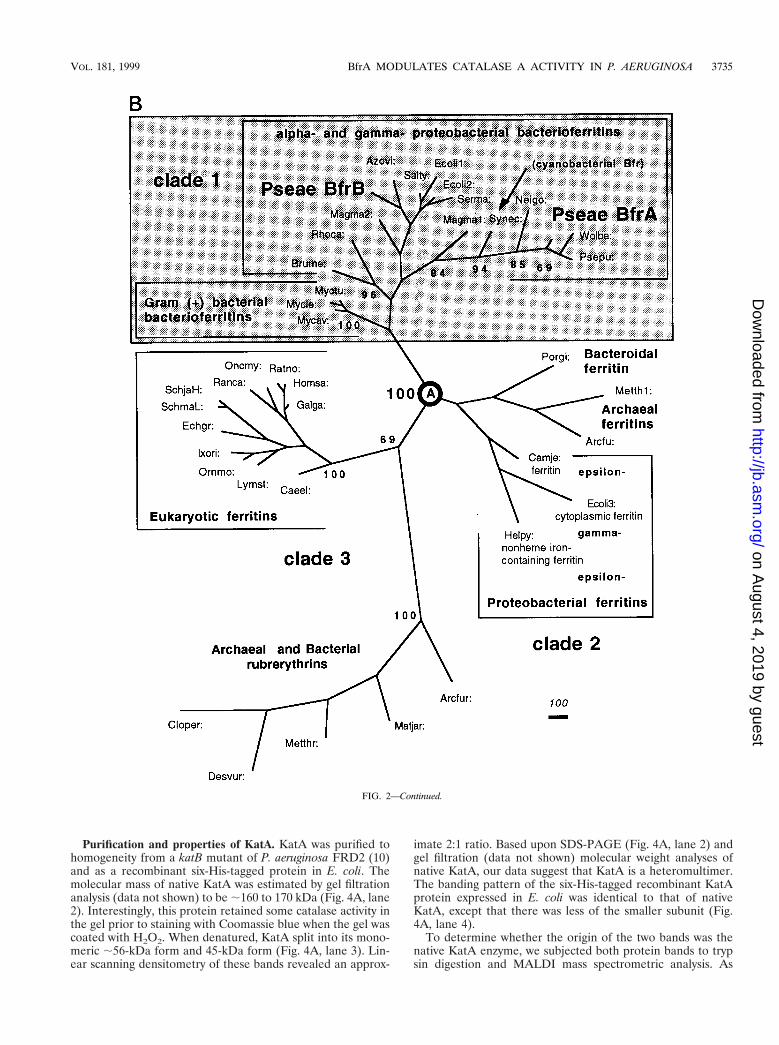
We selected 35 (bacterio)ferritin sequences from bacteria(all available sequences), archaea (3 sequences), and eukarya(12 sequences) for the analysis of the P. aeruginosa BfrA pro-tein. Because of their sequence relatedness (including the glu-tamate and tyrosine residues that are critical for ferroxidaseactivity), five archaeal and bacterial rubrerythrin sequenceswere used as the outgroup. The unrooted tree (Fig. 2B) con-sists of three clades that are separated at node A by the highestpossible confidence. P. aeruginosa BfrA groups in the bacteri-
oferritin-only clade 1, closest to the Bfr proteins from Wolba-chia sp. and P. putida. Interestingly, the archaeal and bacterialrubrerythrins group with eukaryal ferritins in clade 2, whileclade 3 contains a mixture of archaeal and unusual bacterialferritins. It is evident that the phylogenetic (bacterio)ferritintree obtained is not congruent with the tree reflective of thephylogenetic relationships based on 16S rRNA sequences (39).
Complementation of katA in catalase-deficient E. coli. Tofurther assess functional complementation by katA, cell ex-tracts were separated by nondenaturing PAGE and stained forcatalase activity. As shown in Fig. 3A, the primary catalaseactivity of P. aeruginosa, that of KatA, was detectable as asingle activity band (lane 1). Provision of plasmid pJFM12 tocatalase-deficient E. coli UM1 (Fig. 3A, lane 2) allowed for theexpression of KatA activity (lane 3).
We next assessed the contribution of P. aeruginosa KatA tothe resistance of catalase-deficient E. coli to exogenous H2O2.As shown in Fig. 3B, wild-type E. coli CSH7 was highly resis-tant to 1 to 10 mM H2O2 but was killed at 20 mM. As pre-dicted, catalase-deficient strain UM1 demonstrated slightlyhigher sensitivity than wild-type bacteria (31). We did notobserve the bimodal killing pattern of wild-type and DNA-repair-deficient strains of E. coli exposed to 1 to 5 mM H2O2(23). Still, catalase-deficient E. coli UM1 was predictably moresensitive to all tested concentrations of H2O2. The expressionof P. aeruginosa KatA in this strain allowed for a higher levelof protection against H2O2 than that seen for wild-type E. coli.
FIG. 2. Amino acid similarity of BfrA and other Bfr proteins. (A) Proteins were aligned with Align Plus 3.0. Dots indicate identical amino acids, while dashesindicate gaps in the protein sequence relative to P. aeruginosa BfrA. PA, P. aeruginosa BfrA; PP, P. putida Bfr; NG, N. gonorrhoeae Bfr; ST, S. typhimurium Bfr; EC,E. coli Bfr. The amino acids essential for ferroxidase activity in E. coli Bfr are conserved in each Bfr protein and are shown in boldface and marked with an asterisk.(B) Unrooted phylogenetic tree based on the amino acid sequences of 35 (bacterio)ferritins and 5 rubrerythrins and constructed by parsimony methods. The branchlengths (shown in italics) reflect the evolutionary distances calculated as the average number of amino acid changes per 1,000 residues. The numbers in bold in frontof the nodes represent the proportion of bootstrap samplings that support the topology shown. Two hundred bootstrap replicates were analyzed. For analysis of theBfrA protein, Bfr sequences were obtained from GenBank (accession no.) for the following organisms: Arcfu, Archaeoglobus fulgidus putative ferritin (AE001047);Arcfur, A. fulgidus rubrerythrin 1 (AE001047); Azovi, Azotobacter vinelandii Bfr (U83692); Brume, Brucella melitensis Bfr (U19760); Caeel, Caenorhabditis elegansferritin (AF106592); Camje, Campylobacter jejuni ferritin (D64082); Cloper, Clostridium perfringens rubreythrin (X92844); Desvur, Desulfovibrio vulgaris rubrerythrin(U82323); Echgr, Echinococcus granulosus ferritin (Z31712); Ecoli1, Escherichia coli ECOR30 Bfr (AF058450); Ecoli2, E. coli K-12 Bfr (M27176); Ecoli3, E. coli K-12MG1655 cytoplasmic ferritin (AF000335); Galga, Gallus gallus ferritin heavy (H) chain (Y14698); Helpy, Helicobacter pylori J99 non-heme iron-containing ferritin Pfr(AE00149); Homsa, Homo sapiens apoferritin H chain (X00318); Ixori, Ixodes ricinus ferritin (AF068224); Lymst, Lymnaea stagnalis snail soma ferritin (P42577/X56778); Magma1, Magnetospirillum magnetotacticum Bfr1 (AF001959); Magma2, M. magnetotacticum Bfr2 (AF001959); Metth1, Methanobacterium thermoautotro-phicum putative ferritin (AE000804); Metthr, M. thermoautotrophicum rubrerythrin (AE000854); Matjar, Methanococcus jannaschii rubrerythrin (U67520); Mycav,Mycobacterium avium Bfr (X76906); Mycle, Mycobacterium leprae Bfr (P43315); Myctu, Mycobacterium tuberculosis Bfr (Z97193); Neigo, Neisseria gonorrhoeae Bfr(U76633); Oncmy, Oncorhynchus mykiss ferritin-1 H chain (D86625); Ornmo, Ornithodoros moubata ferritin (AF068225); Porgi, Porphyromonas gingivalis ferritin(AB016086); Psepu: Pseudomonas putida Bfr (U66717); Pseae: Pseudomonas aeruginosa Bfr (AF047025); Ranca, Rana catesbeiana ferritin, middle subunit (J02724);Ratno, Rattus norvegicus ferritin H chain (P19132); Rhoca, Rhodobacter capsulatus Bfr (Z54247); Salty, Salmonella typhimurium LT2 Bfr (AF058449); SchjaH,Schistosoma japonicum putative ferritin-1 H chain (AF040385); SchmaL, Schistosoma mansoni ferritin light chain (M64538); Serma, Serratia marcescens Bfr(AF058451); Synec, Synechocystis sp. strain PCC6803 Bfr (D90905); and Wolba, Wolbachia sp. Bfr (21).
3734 MA ET AL. J. BACTERIOL.
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
Purification and properties of KatA. KatA was purified tohomogeneity from a katB mutant of P. aeruginosa FRD2 (10)and as a recombinant six-His-tagged protein in E. coli. Themolecular mass of native KatA was estimated by gel filtrationanalysis (data not shown) to be ;160 to 170 kDa (Fig. 4A, lane2). Interestingly, this protein retained some catalase activity inthe gel prior to staining with Coomassie blue when the gel wascoated with H2O2. When denatured, KatA split into its mono-meric ;56-kDa form and 45-kDa form (Fig. 4A, lane 3). Lin-ear scanning densitometry of these bands revealed an approx-
imate 2:1 ratio. Based upon SDS-PAGE (Fig. 4A, lane 2) andgel filtration (data not shown) molecular weight analyses ofnative KatA, our data suggest that KatA is a heteromultimer.The banding pattern of the six-His-tagged recombinant KatAprotein expressed in E. coli was identical to that of nativeKatA, except that there was less of the smaller subunit (Fig.4A, lane 4).
To determine whether the origin of the two bands was thenative KatA enzyme, we subjected both protein bands to trypsin digestion and MALDI mass spectrometric analysis. As
FIG. 2—Continued.
VOL. 181, 1999 BfrA MODULATES CATALASE A ACTIVITY IN P. AERUGINOSA 3735
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

shown in Fig. 4B, the molecular masses of the major peakswere identical for both the 56-kDa band (top panel) and the45-kDa band (bottom panel), with the exception of two pep-tides with molecular masses of 2,079.71 and 2,057.65 Da (ar-rows). Interestingly, the trypsin cleavage sites for these pep-tides correspond to amino acids 7 to 27 [(R)LTTAAGAPVVDNQNVQTAGPR(G)] and 58 to 76 [(K)GSAAYHTFTVTHDITPYTRAK(I)], respectively. These data suggest that thesmaller band is a second translational product encoded bykatA. The first translational start codon (ATG) after nucleo-tide 228 (76 amino acids) was in frame and located at position265, with two overlapping ribosome binding sites (253-AA-GAAGA-259) preceding it. From these data, we conclude thatKatA is a heteromultimer, possibly an a2b-heterotrimer. Gelfiltration and SDS-PAGE analyses of CatF from the relatedspecies P. syringae suggest that it, too, may be a heteromul-timer, although this notion has not yet been proven with massspectrometry or protein sequencing (28). This is the first dem-onstration of a heteromultimeric catalase among all three cata-lase groups (29).
Catalases are typically enzymes with low substrate affinities,with Km values for H2O2 of 2.07 mM for the catalase/peroxi-dase of Streptomyces cyaneus (36) and 78 mM for the catalaseof Bacillus subtilis (32) (the P. aeruginosa KatB Km is 10.6 mM[10]). After double-reciprocal Lineweaver-Burk analysis, wedetermined an apparent Km of 44.7 mM with H2O2 as thesubstrate for KatA purified from P. aeruginosa FRD2 katB (10)(Fig. 4C). This rather high apparent Km is similar to that forthe structurally similar but phylogenetically different catalaseCatF from P. syringae (Km, ;60 mM) (28).
Phenotypes of katA, bfrA, and katA bfrA mutants. The majorcatalase activity of P. aeruginosa is that of KatA (10, 17). Sinceone of our hypotheses was that iron bound to BfrA is necessaryfor the production of some heme, the prosthetic group ofKatA, we predicted that bfrA mutants would have reducedKatA activity. Catalase activity gel staining of cell extracts from
the wild type and isogenic bfrA, katA, and katA bfrA mutantsrevealed that wild-type bacteria produce only KatA (Fig. 5A,lane 1), with a specific activity of 2,018 U/mg (Fig. 5B, lane 1).This finding is consistent with our earlier observation that thesecond catalase, KatB, is not expressed unless organisms aretreated with H2O2 (10). Interestingly, the bfrA mutant pro-duced visibly less KatA than wild-type bacteria in catalaseactivity gels (Fig. 5A, lane 2); this result correlated with a 47%loss of catalase activity determined spectrophotometrically(Fig. 5B, lane 2). The katA and katA bfrA mutants produced nodetectable catalase, as monitored by native gel or spectropho-tometric assays (Fig. 5A and B, lanes 3 and 4).
Ferroxidase activity of BfrA is essential for optimal KatAactivity. The Bfr protein of E. coli possesses several aminoacids that are critical for the ferroxidase activity that oxidizesFe21 to Fe31 within the BfrA core (30). An alignment of P.aeruginosa BfrA with E. coli Bfr and other bacterioferritinsrevealed that each protein harbors these residues (Fig. 2A). Totest our hypothesis that the ferroxidase activity of BfrA isessential for optimal KatA activity, two bfrA mutant plasmidswere constructed. The first, pBFR18, possessed a glutamate-to-lysine change at amino acid 18 (E18K). The second,pBFR25, possessed a tyrosine-to-isoleucine change at aminoacid 25 (Y25I). As shown in Fig. 6, wild-type organisms (lane1) possessed nearly twice the catalase activity of the bfrA mu-tant (lane 2), consistent with the results shown in Fig. 5B.Provision of pBFR4 harboring the wild-type bfrA gene partiallyrestored catalase activity (Fig. 6, lane 3). In contrast, the cata-lase activity of the bfrA mutant harboring either pBFR18(E18K) or pBFR24 (Y25I) remained at bfrA mutant levels(Fig. 6, lanes 4 and 5).
BfrA and KatA are important in optimal resistance to H2O2:role of iron. Since catalase activity was reduced in the bfrAstrain and absent in the katA and katA bfrA strains, we pre-dicted that these mutants would demonstrate enhanced sensi-tivity to H2O2 and that cellular iron levels might influence
FIG. 3. (A) Complementation of P. aeruginosa katA in E. coli UM1. Cell extracts (20 mg) of aerobically grown, stationary-phase organisms were separated bynondenaturing PAGE and stained for catalase activity (51). Lane 1, P. aeruginosa PAO1; lane 2, E. coli UM1; lane 3, E. coli UM1(pJFM12). (B) Enhanced resistanceof E. coli UM1 harboring P. aeruginosa katA to H2O2. Mid-logarithmic-phase bacteria were exposed to various concentrations of H2O2 for 15 min at 37°C (23). Theresults are typical of three separate experiments. Symbols: h, E. coli CSH7; Œ, E. coli UM1; E, E. coli UM1(pJFM12).
3736 MA ET AL. J. BACTERIOL.
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

FIG. 4. Purification (A), mass spectrometric analysis (B), and Km measurement (C) of P. aeruginosa KatA. (A) Lane 1, protein molecular mass standards; lane 2,60 ng of purified unboiled KatA; lane 3, 120 ng of purified boiled KatA; lane 4, 160 ng of purified boiled recombinant six-His-tagged KatA. (B) Mass spectrometricanalysis of the 55-kDa subunit (top panel) and smaller subunits (bottom panel) of KatA. The arrows in the top panel indicate peptides that are absent in the bottompanel. (C) Double-reciprocal Lineweaver-Burk plot of KatA activity with various H2O2 concentrations. Experiments were performed as described in Materials andMethods.
VOL. 181, 1999 BfrA MODULATES CATALASE A ACTIVITY IN P. AERUGINOSA 3737
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

H2O2 sensitivity. As shown in Fig. 7A, the bfrA, katA, andespecially katA bfrA mutants demonstrated enhanced sensitiv-ity to H2O2 relative to wild-type organisms. The sensitivity ofthe katA and katA bfrA mutants was, in part, dependent uponcellular iron levels, since these organisms were less sensitive toH2O2 when grown in iron-limiting medium than when grown iniron-rich medium. Catalase activity was also highest when or-ganisms were grown in iron-replete medium (Fig. 7B).
Regulation of katA and bfrA in P. aeruginosa: role of iron andH2O2. Since the enhanced sensitivity of the bfrA, katA, andkatA bfrA mutants to H2O2 was greatest in the presence ofiron, we postulated that iron and H2O2 levels might control thetranscriptional activation of both katA and bfrA. First, growth-phase- and iron-dependent expression patterns were moni-tored by RNase protection analysis with a riboprobe that al-lowed for the simultaneous detection of katA and bfrAtranscripts (Fig. 8A). A loading control with a “housekeeping”gene (omlA) was included in Fig. 8B (middle panel). The levelof expression of katA was somewhat higher in iron-rich relativeto iron-poor medium during the exponential growth phase andwas upregulated at least fivefold during the stationary phase(Fig. 8B, left panel). The bfrA gene was expressed at high levelsunder both low- and high-iron conditions during the exponen-tial growth phase. However, during the stationary phase, thelevel of bfrA expression was maximal in iron-replete mediumbut was lower than that expressed in exponential phase. An
absence of BfrA had no effect on katA transcription (Fig. 8B,right panel).
A more detailed analysis of katA and bfrA expression withrespect to iron concentration and response to H2O2 was per-formed with translational fusions to the lacZ reporter gene(Fig. 8C). The level of KatA::LacZ expression was enhancedduring the stationary phase and was predictably higher in iron-rich versus iron-poor medium. Treatment with exogenousH2O2 caused an ;3-fold increase in KatA::LacZ activity. Theregulation and activity of KatA::LacZ in a DbfrA backgroundwere virtually identical to those in wild-type bacteria. The levelof expression of BfrA::LacZ was highest in the stationaryphase and was upregulated at least twofold upon H2O2 treat-ment. The high level of stationary-phase BfrA expression dif-fered from the pattern observed in the RNase protection anal-ysis. However, it is difficult to compare data from an RNaseprotection analysis with LacZ reporter data, since the formerdetects transcripts at a given instant and the latter measuresthe accumulation of a translated product over the entire timeperiod. Still, taken together, our data suggest that both KatAexpression and BfrA expression respond to growth phase, iron,and H2O2.
Iron levels in bfrA, katA, and katA bfrA strains. Since KatAis the predominant catalase in P. aeruginosa and the bfrA mu-tant possessed less KatA activity than wild-type organisms, wepostulated that iron would be redistributed in the cell, resultingin no net change in total cellular iron levels. To test thishypothesis, iron levels in wild-type and mutant strains weremeasured by ion-spray mass spectrometry. Not surprisingly,total iron levels varied little in wild-type, katA, bfrA, and katAbfrA strains, ranging from 2.8 3 105 to 4.3 3 105 iron atoms perviable cell and 0.99 3 1024 to 1.3 3 1024 mg of iron per mg ofcell dry weight. These data are supportive of the view thatcellular iron concentrations are regulated at the level of uptake(9).
DISCUSSION
The initial goal of this work was to clone the P. aeruginosakatA gene and determine the contribution of its product toH2O2 detoxification. However, when we discovered the bfrA
FIG. 6. Importance of ferroxidase-center amino acids in optimal KatA activ-ity in P. aeruginosa. P. aeruginosa harboring plasmids with wild-type or alteredbfrA genes was grown aerobically to the stationary phase in L broth at 37°C.Catalase activity of cell extracts was monitored in triplicate. 1, Wild type pluspUCP19) 2, bfrA plus pUCP19) 3, bfrA plus pBFR4; 4, bfrA plus pBFR18(E18K); 5, bfrA plus pBFR25 (Y25I).
FIG. 5. Catalase activity (A) and activity staining (B) of P. aeruginosa strains.(A) Catalase activity in cell extracts from stationary-phase organisms was mea-sured as described by Beers and Sizer (5); the values are means 6 standarderrors for three experiments. 1, P. aeruginosa PAO1; 2, bfrA; 3, katA; 4, katA bfrA.(B) Cell extracts (20 mg) from the above organisms were separated by nonde-naturing PAGE in triplicate and stained for catalase activity (51). Lane 1, P.aeruginosa PAO1; lane 2, bfrA; lane 3, katA; lane 4, katA bfrA. The KatA activityband is shown by an arrow.
3738 MA ET AL. J. BACTERIOL.
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

gene downstream of katA, we wondered whether their closeproximity could be extended to a functional relationship ofboth gene products. Our first attempt at understanding thepotential utility of katA and bfrA being so close together on theP. aeruginosa genome was through a phylogenetic analysis ofboth KatA and BfrA proteins. The phylogenetic analysis ofcatalases performed in this study will be discussed in moredetail elsewhere (31a). However, it is worth mentioning herethat P. aeruginosa KatA is a group III bacterial catalase (29)and thus is closely related to the CatA proteins found in P.putida and a Wolbachia sp., a nematode-endosymbiotic mem-ber of the Rickettsiales order (21). This fact is important be-cause the genes encoding these catalases are succeeded bybacterioferritin-encoding genes in each of the organisms. Sincethe Wolbachia sp. contains only one catalase, the proximity ofthe kat and bfr genes in the three bacteria is intriguing. Al-though katA and bfrA have their own promoters, which arepositively responsive to iron and H2O2, it appears that thesegenes have not evolved independently due to functional pres-sures on their expression products. In addition, the constructedferritin protein tree leads to the conclusion that a bacteriofer-ritin gene was already present in the common ancestor ofgram-positive bacteria, cyanobacteria, and proteobacteria,which diverged during further species evolution.
Further compelling evidence for a functional relationshipbetween KatA and BfrA surfaced when we demonstrated thata P. aeruginosa mutant lacking BfrA produces ;50% of wild-type KatA activity and shows greater H2O2 sensitivity despitepossessing wild-type katA transcription and translation. Theenhanced log-phase expression of BfrA relative to KatA mayserve to sequester labile iron before releasing it for distributioninto iron- and/or heme-containing proteins such as KatA.Thus, we believe that BfrA-bound iron is required for optimalKatA activity and, in turn, resistance to H2O2.
P. aeruginosa BfrA is a complex of 24 subunits capable ofbinding up to 24 heme moieties (25, 38). Because of the hy-drophobic nature of heme, it is not likely released from BfrAin vivo but is essential for the optimal release of reduced ironfrom its core in vitro (37). Because BfrA can also bind ;700 Featoms (11), we postulate that Fe21 released from BfrA can beincorporated into protoporphyrin IX by ferrochelatase-depen-
dent condensation, forming heme which, in turn, is incorpo-rated into the folding KatA enzyme. Since both iron and H2O2stimulate katA and bfrA transcription, we postulate that suchconditions may cause iron release from the BfrA core. Ironrelease from the related protein ferritin is mediated, in part, byO2
2 and not H2O2 (4, 7). However, H2O2 may indirectly pro-duce elevated levels of O2
2 through its reaction with HO., areaction proposed for the resistance of E. coli to mode IIkilling by 5 to 20 mM H2O2 (23, 24). The mechanism for thisevent is as follows. First, basal levels of O2
2 cause the releaseof Fe21 from the core of BfrA. In the presence of H2O2, someFe21 reacts with O2
2 in a Fenton reaction to form HO.. Inturn, the HO. can react with H2O2 to form even more O2
2,thereby reducing additional core BfrA Fe31 to Fe21. The re-lease of Fe21 from Bfr and ferritin in E. coli is also importantin the repair of O2
2-mediated damage to [4Fe-4S] clusterproteins (26).
Bfr proteins and ferritins possess ferroxidase activity, whichoxidizes Fe21 to Fe31 to form ferric-oxy-hydroxide-phosphatecomplexes within their cores (2). Mutagenesis of the conservedand required glutamate, histidine, or tyrosine residues (Fig.2B) present in P. aeruginosa BfrA was shown to abolish thisactivity (30). We have demonstrated that P. aeruginosa BfrAwith either an E18K or a Y25I substitution does not showwild-type KatA activity (Fig. 5). Ferroxidase activity could ben-efit the organism by limiting the amount of labile iron availableto undergo the Fenton reaction (Fe21 1 H2O23HO. 1 Fe31),thus restricting ensuing damage of biological molecules medi-ated by H2O2 and HO.. While not yet tested with P. aeruginosa,this hypothesis has been confirmed for murine erythroleuke-mia cells expressing the ferroxidase-center-containing subunitof the related iron storage protein ferritin; labile iron levelswere reduced 2.3-fold in H subunit-overexpressing cells (42).P. aeruginosa also possesses BfrB, and we postulated that it,too, could limit labile iron levels and thus assist in the protec-tion of organisms against oxidative stress. However, a bfrBmutant was not more susceptible to H2O2 and possessed wild-type catalase activity (38a). This result further supports a func-tional link between BfrA and KatA.
In addition to controlling the level of labile iron withinbacteria, BfrA may also indirectly control DNA damage. Re-
FIG. 7. H2O2 disk sensitivity (A) and catalase activity (B) of P. aeruginosa strains: influence of iron. (A) P. aeruginosa strains were grown aerobically in M9 minimalmedium with or without 50 mM FeCl3 to the stationary phase. Organisms were diluted 30-fold in 3 ml of molten 0.6% M9 top agar and layered on M9 agar plates. Sterilefilter paper disks (7 mm) were impregnated with 10 ml of 8.8 M H2O2 and placed in triplicate on the top-agar surface, and the plates were incubated at 37°C for 17 h.Zones of growth inhibition were measured. Shaded columns, M9 medium plus 50 mM FeCl3; open columns, M9 medium alone. The results are expressed as themeans 6 standard errors for nine different experiments. 1, P. aeruginosa PAO1; 2, bfrA; 3, katA; 4, katA bfrA. (B) Catalase activity (5) was measured in cell extractsfrom each strain and expressed as the means 6 standard errors for three different experiments. Columns are as in panel A.
VOL. 181, 1999 BfrA MODULATES CATALASE A ACTIVITY IN P. AERUGINOSA 3739
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

cently, it was found that the E. coli Bfr crystal structure con-tains a four-helix bundle that is nearly identical to the E. coliDps monomer (DNA binding protein from starved cells (16).Dps has been shown to bind and protect DNA from Fentonreaction-mediated oxidative DNA damage (33). Because ofthe remarkable structural identity between the E. coli Dps andBfr proteins, it is conceivable that one mechanism by which
Dps and possibly bacterioferritins protect DNA is throughtheir capacity to bind iron. Interestingly, the DpsA protein ofa Synechococcus sp. binds DNA, contains heme and catalaseactivity, and possesses a C-terminal domain that is 55% similarto that of the Azotobacter vinelandii bacterioferritin (41). Al-though there is no evidence that bacterioferritins bind DNA,they may protect it indirectly via their capacity to sequester
FIG. 8. Regulation of katA and bfrA: effect of growth phase, iron, and H2O2. (A) Genetic organization of katA and bfrA and location of the riboprobe used for thesimultaneous detection of the corresponding transcripts. The promoter regions upstream of katA and bfrA are depicted as shaded boxes. Loopholes indicatetranscriptional terminators. nt., nucleotides. (B) RNase protection analysis of katA and bfrA. P. aeruginosa was grown aerobically under low (2)- or high (1)-ironconditions. An RNA ladder, undigested probe (P), and detected transcripts for bfrA and katA are indicated (left panel), together with omlA as a constitutive and loadingcontrol (middle panel) and the katA transcript in the wild type and the bfrA mutant (right panel). (C) Translational katA-lacZ and bfrA-lacZ activities. All bacteria weregrown aerobically in M9 medium. Open columns, M9 low-iron medium (0.2 mM dipyridyl); shaded columns, M9 high-iron medium (50 mg of FeCl3 per ml). The threedata pairs in each panel reflect the measured activities during the stationary phase after overnight growth (o/n), during exponential growth (log), and after 1 h oftreatment with 1 mM H2O2 every 10 min (1H2O2). The values are the averages for quadruplicate cultures, and error bars (,10%) are omitted for clarity.
3740 MA ET AL. J. BACTERIOL.
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

reactive iron. Identification of cellular conditions triggeringiron and/or heme release from BfrA and determination ofwhether such conditions also increase KatA activity and oxi-dative DNA damage are studies currently being carried out.Finally, the trafficking of iron from BfrA to other molecules inbacteria, potentially by iron chaperones, is likely critical for avariety of cellular processes. Future studies will address themechanism(s) by which iron is released from BfrA and how itis conditionally and preferentially designated for different iron-and/or heme-containing proteins.
ACKNOWLEDGMENTS
We thank P. Loewen (University of Manitoba) for assistance withinterpretation of the KatA absorption spectra and for constructivecomments regarding KatA structure. We thank A. J. Anderson (UtahState University) for providing a plasmid containing the P. putida catAgene.
This work was supported in part by grants AI-40541 (to D.J.H.) andAI-15940 (to M.L.V.) from the National Institutes of Health (toD.J.H.), Cystic Fibrosis grant HASSET97PO (to D.J.H.), and start-upfunds from the Department of Molecular Genetics, Biochemistry andMicrobiology at the University of Cincinnati College of Medicine.
REFERENCES
1. Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990.Basic local alignment search tool. J. Mol. Biol. 215:403–410.
2. Andrews, S. C., J. M. Smith, S. J. Yewdall, J. R. Guest, and P. M. Harrison.1991. Bacterioferritins and ferritins are distantly related in evolution: con-servation of ferroxidase-centre residues. FEBS Lett. 293:164–168.
3. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. D. Seidman, andK. Struhl. 1993. Current protocols in molecular biology, p. 5.3.2–5.3.8. JohnWiley & Sons, Inc., New York, N.Y.
4. Bando, Y., and K. Aki. 1990. Superoxide-mediated release of iron fromferritin by some flavoenzymes. Biochem. Biophys. Res. Commun. 168:389–395.
5. Beers, R. F., Jr., and I. W. Sizer. 1952. A spectrophotometric method formeasuring the breakdown of hydrogen peroxide by catalase. J. Biol. Chem.195:133–140.
6. Beyer, W., J. Imlay, and I. Fridovich. 1991. Superoxide dismutases. Prog.Nucleic Acid Res. 40:221–253.
7. Bolann, B. J., and R. J. Ulvik. 1987. Release of iron from ferritin by xanthineoxidase: role of the superoxide radical. Biochem. J. 243:55–59.
8. Bradford, M. M. 1976. A rapid and sensitive method for the quantitation ofmicrogram quantities of protein utilizing the principle of protein-dye bind-ing. Anal. Biochem. 72:248–254.
9. Braun, V. 1997. Avoidance of iron toxicity through regulation of bacterialiron transport. Biol. Chem. 378:779–786.
10. Brown, S. M., M. L. Howell, M. L. Vasil, A. Anderson, and D. J. Hassett.1995. Cloning and characterization of the katB gene of Pseudomonas aerugi-nosa encoding a hydrogen peroxide-inducible catalase: purification of KatB,cellular localization, and demonstration that it is essential for optimal resis-tance to hydrogen peroxide. J. Bacteriol. 177:6536–6544.
11. Cheesman, M. R., F. H. A. Kadir, J. Al-Basseet, F. Al-Massad, J. Farrar, C.Greenwood, A. J. Thompson, and G. R. Moore. 1992. E.p.r. and magneticcircular dichroism spectroscopic characterization of bacterioferritin fromPseudomonas aeruginosa and Azotobacter vinelandii. Biochem. J. 286:361–367.
12. Cheesman, M. R., N. E. le Brun, F. H. Kadir, A. J. Thomson, G. R. Moore,S. C. Andrews, J. R. Guest, P. M. Harrison, J. M. Smith, and S. J. Yewdall.1993. Haem and non-haem iron sites in Escherichia coli bacterioferritin:spectroscopic and model building studies. Biochem. J. 292:47–56.
13. DeVries, C. A., and D. E. Ohman. 1994. Mucoid-to-nonmucoid conversion inalginate-producing Pseudomonas aeruginosa often results from spontaneousmutations in algT, encoding a putative alternate sigma factor, and showsevidence for autoregulation. J. Bacteriol. 176:6677–6687.
14. Figurski, D., and D. R. Helinski. 1979. Replication of an origin-containingderivative of plasmid RK2 dependent on a plasmid function provided intrans. Proc. Natl. Acad. Sci. USA 76:1648–1652.
15. Frolow, F., A. J. Kalb, and J. Yariv. 1994. Structure of a unique twofoldsymmetric haem-binding site. Nat. Struct. Biol. 1:453–460.
16. Grant, R. A., D. J. Filman, S. E. Finkel, R. Kolter, and J. M. Hogle. 1998. Thecrystal structure of Dps, a ferritin homolog that binds and protects DNA.Nat. Struct. Biol. 5:294–303.
17. Hassett, D. J., L. Charniga, K. A. Bean, D. E. Ohman, and M. S. Cohen.1992. Antioxidant defense mechanisms in Pseudomonas aeruginosa: resis-tance to the redox-active antibiotic pyocyanin and demonstration of a man-ganese-cofactored superoxide dismutase. Infect. Immun. 60:328–336.
18. Hassett, D. J., H. P. Schweizer, and D. E. Ohman. 1995. Pseudomonasaeruginosa sodA and sodB mutants defective in manganese- and iron-cofac-tored superoxide dismutase activity demonstrate the importance of the iron-cofactored form in aerobic metabolism. J. Bacteriol. 177:6330–6337.
19. Hassett, D. J., P. Sokol, M. L. Howell, J.-F. Ma, H. P. Schweizer, U. Ochsner,and M. L. Vasil. 1996. Ferric uptake regulator (Fur) mutants of Pseudomo-nas aeruginosa demonstrate defective siderophore-mediated iron uptake andaltered aerobic metabolism. J. Bacteriol. 178:3996–4003.
20. Hassett, D. J., W. A. Woodruff, D. J. Wozniak, M. L. Vasil, M. S. Cohen, andD. E. Ohman. 1993. Cloning of the sodA and sodB genes encoding manga-nese and iron superoxide dismutase in Pseudomonas aeruginosa: demonstra-tion of increased manganese superoxide dismutase activity in alginate-pro-ducing bacteria. J. Bacteriol. 175:7658–7665.
21. Henkle-Duhren, K., V. H. O. Eckelt, G. Wildenburg, M. Blaxter, and R. D.Walter. 1998. Gene structure, activity and localization of a catalase fromintracellular bacteria in Onchocerca volvolus. Mol. Biochem. Parasitol. 96:69–81.
22. Holloway, B. W., V. Krishnapillai, and A. F. Morgan. 1979. Chromosomalgenetics of Pseudomonas. Microbiol. Rev. 43:73–102.
23. Imlay, J. A., and S. Linn. 1986. Bimodal pattern of killing of DNA-repair-defective or anoxically grown Escherichia coli by hydrogen peroxide. J. Bac-teriol. 166:519–527.
24. Imlay, J. A., and S. Linn. 1987. Mutagenesis and stress responses induced inEscherichia coli by hydrogen peroxide. J. Bacteriol. 169:2967–2976.
25. Kadir, F. H. A., and G. R. Moore. 1990. Bacterial ferritin contains 24 haemgroups. FEBS Lett. 271:141–143.
26. Keyer, K., and J. A. Imlay. 1997. Inactivation of dehydratase [4Fe-4S] clus-ters and disruption of iron homeostasis upon cell exposure to peroxynitrite.J. Biol. Chem. 272:27652–27659.
27. Kim, Y. C., C. D. Miller, and A. J. Anderson. 1997. Identification of adjacentgenes encoding the major catalase and a bacterioferritin from the plant-beneficial bacterium Pseudomonas putida. Gene 199:219–224.
28. Klotz, M. G., Y. C. Kim, A. J. Katsuwon, and A. J. Anderson. 1995. Cloning,characterization, and phenotypic expression in Escherichia coli of catF, whichencodes the catalytic subunit of catalase isozyme CatF of Pseudomonassyringae. Appl. Environ. Biotechnol. 43:656–666.
29. Klotz, M. G., G. R. Klassen, and P. C. Loewen. 1997. Phylogenetic relation-ships among prokaryotic and eukaryotic catalases. Mol. Biol. Evol. 14:951–958.
30. Le Brun, N. E., S. C. Andrews, J. R. Guest, P. M. Harrison, G. R. Moore, andA. J. Thomson. 1995. Identification of the ferroxidase centre of Escherichiacoli bacterioferritin. Biochem. J. 312:385–392.
31. Loewen, P. C. 1984. Isolation of catalase-deficient Escherichia coli mutantsand genetic mapping of katE, a locus that affects catalase activity. J. Bacte-riol. 157:622–626.
31a.Loewen, P. C., M. G. Klotz, and D. J. Hassett. Catalase—an “old” enzymethat continues to surprise us. Submitted for publication.
32. Loewen, P. C., and J. Switala. 1988. Purification and characterization of aspore-specific catalase-2 from Bacillus subtilis. Biochem. Cell Biol. 66:707–714.
33. Martinez, A., and R. Kolter. 1997. Protection of DNA during oxidative stressby the nonspecific DNA-binding protein Dps. J. Bacteriol. 179:5188–5194.
34. McCord, J. M., and I. Fridovich. 1969. Superoxide dismutase: an enzymicfunction for erythrocuprein. J. Biol. Chem. 244:6049–6055.
35. Miller, J. H. 1992. Procedures for working with lac, p. 72–74. In A shortcourse in bacterial genetics: a laboratory manual and handbook for Esche-richia coli and related bacteria. Cold Spring Harbor Laboratory Press, Pla-inview, N.Y.
36. Mliki, A., and W. Zimmermann. 1992. Purification and characterization ofan intracellular peroxidase from Streptomyces cyaneus. Appl. Environ. Mi-crobiol. 58:916–919.
37. Moore, G. R., F. H. A. Kadir, and F. Al-Massad. 1992. Haem binding toferritin and possible mechanisms of physiological iron uptake and release byferritin. J. Inorg. Biochem. 47:175–181.
38. Moore, G. R., F. H. A. Kadir, F. K. Al-Massad, N. E. Le Brun, A. J. Thomson,C. Greenwood, J. N. Keen, and J. B. C. Findlay. 1994. Structural heteroge-neity of Pseudomonas aeruginosa bacterioferritin. Biochem. J. 304:493–497.
38a.Ochsner, U. A., et al. Unpublished data.39. Olsen, G. J., C. R. Woese, and R. Overbeek. 1994. The winds of (evolution-
ary) change: breathing new life into microbiology. J. Bacteriol. 176:1–6.40. Pathogenesis Corporation and the Cystic Fibrosis Foundation. 15 March
1999, posting date. Pseudomonas Genome Project. [Online.] http://www.pseudomonas.com. [April 1999, last date accessed.]
41. Pena, M. M., and G. S. Bullerjahn. 1995. The DpsA protein of Synechococ-cus sp. strain PCC7942 is a DNA-binding hemoprotein: linkage of the Dpsand bacterioferritin protein families. J. Biol. Chem. 270:22478–22482.
42. Picard, V., S. Epsztejn, P. Santambrogio, Z. I. Cabantchik, and C. Beau-mont. 1998. Role of ferritin in the control of the labile iron pool in murineerythroleukemia cells. J. Biol. Chem. 273:15382–15386.
43. Schweizer, H. P. 1991. Improved broad-host-range lac-based plasmid vectorsfor the isolation and characterization of protein fusions in Pseudomonasaeruginosa. Gene 103:87–92.
VOL. 181, 1999 BfrA MODULATES CATALASE A ACTIVITY IN P. AERUGINOSA 3741
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

44. Schweizer, H. P. 1992. Allelic exchange in Pseudomonas aeruginosa usingnovel ColE1-type vectors and a family of cassettes containing a portable oriTand the counter-selectable Bacillus subtilis sacB marker. Mol. Microbiol.6:1195–1204.
45. Schweizer, H. P. 1993. Small broad-host-range gentamicin resistance genecassettes for site-specific insertion and deletion mutagenesis. BioTechniques15:831–833.
46. Schweizer, H. P., and T. T. Hoang. 1995. An improved system for genereplacement and xylE fusion analysis in Pseudomonas aeruginosa. Gene 158:15–22.
47. Simon, R., U. Priefer, and A. Puehler. 1983. A broad host range mobilizationsystem for in vivo genetic engineering: transposon mutagenesis in gramnegative bacteria. Bio/Technology 1:784–791.
48. Swofford, D. L. 1993. PAUP: phylogenetic analysis using parsimony. Version3.1.1 for the Macintosh. Smithsonian Institute, Washington, D.C.
49. Vasil, M. L., U. A. Ochsner, Z. Johnson, J. A. Colmer, and A. N. Hamood.1998. The Fur-regulated gene encoding the alternative sigma factor PvdS isrequired for iron-dependent expression of the LysR-type regulator PtxR inPseudomonas aeruginosa. J. Bacteriol. 180:6784–6788.
50. Wai, S. N., K. Nakayama, K. Umene, T. Moriya, and K. Amako. 1996.Construction of a ferritin-deficient mutant of Campylobacter jejuni: contri-bution of ferritin to iron storage and protection against oxidative stress. Mol.Microbiol. 20:1127–1134.
51. Wayne, L. G., and G. A. Diaz. 1986. A double staining method for differen-tiating between two classes of mycobacterial catalase in polyacrylamide gels.Anal. Biochem. 157:89–92.
52. West, S. E. H., H. P. Schweizer, C. Dall, A. K. Sample, and L. J. Runyen-Janecky. 1994. Construction of improved Escherichia-Pseudomonas shuttlevectors derived from pUC18/19 and sequence of the region required for theirreplication in Pseudomonas aeruginosa. Gene 148:81–86.
3742 MA ET AL. J. BACTERIOL.
on August 4, 2019 by guest
http://jb.asm.org/
Dow
nloaded from





























