Embed Size (px)
Citation preview

Survival of Staphylococcus epidermidis in Fibroblasts andOsteoblasts
Kimberly Perez,a Robin Patelb,c
aDepartment of Immunology, Mayo Clinic, Rochester, Minnesota, USAbDivision of Clinical Microbiology, Department of Laboratory Medicine and Pathology, Mayo Clinic, Rochester,Minnesota, USA
cDivision of Infectious Diseases, Department of Medicine, Mayo Clinic, Rochester, Minnesota, USA
ABSTRACT Staphylococcus epidermidis is a leading cause of infections associatedwith indwelling medical devices, including prosthetic joint infection. While biofilmformation is assumed to be the main mechanism underlying the chronic infectionsS. epidermidis causes, we hypothesized that S. epidermidis also evades immune kill-ing, contributing to its pathogenesis. Here, we show that prosthetic joint-associatedS. epidermidis isolates can persist intracellularly within human fibroblasts and insidehuman and mouse osteoblasts. We also show that the intracellularly persisting bac-teria reside primarily within acidic phagolysosomes and that over the course of in-fection, small-colony variants are selected for. Moreover, upon eukaryotic cell death,these bacteria, which can outlive their host, can escape into the extracellular envi-ronment, providing them an opportunity to form biofilms on implant surfaces at de-layed time points in implant-associated infection. In summary, the acidic phagolyso-somes of fibroblasts and osteoblasts serve as reservoirs for chronic or delayed S.epidermidis infection.
KEYWORDS fibroblast, intracellular infection, osteoblast, Staphylococcus epidermidis
Staphylococcus epidermidis is a generally nonvirulent Gram-positive bacteriumwhich is a member of the normal human skin microbiota. Similar to many gut
bacteria, this commensal organism typically serves a beneficial role, educating theimmune system and allowing immune cells to keep the potentially harmful micro-biota in check while not mounting an inflammatory response (1–5). As an example,S. epidermidis produces phenol-soluble modulin (PSM) peptides, present on thesurface of the human skin (1), which have antimicrobial activity against somepathogenic bacteria, including Staphylococcus aureus and Streptococcus pyogenes(6). In addition, S. epidermidis stimulates keratinocytes to produce antimicrobialpeptides which inhibit the growth of S. aureus and S. pyogenes (2). Despite itsnormally constructive role, with the advent of implantation of medical devices, S.epidermidis has emerged as an important foreign body-associated pathogen and isnow, for example, one of the leading causes of prosthetic joint infection (PJI) (7–11).Although S. epidermidis infection does not frequently lead to death, the chronicinfection it causes contributes to a high economic burden and morbidity (8–12). In2015, an estimated 1.3 million hip and knee replacement procedures were per-formed in the United States, with numbers estimated to double by 2020 (13).Infection rates have held steady at 2 to 2.4% and lead to an annual cost projectedto reach 1.62 billion dollars by 2020 (13). Together, S. aureus and S. epidermidis arethe causative agents for more than half of all PJI cases. Given the steadily increasingnumbers of joint arthroplasty surgeries being performed, and accordingly thenumber of associated infections, it is important to define their pathogenesis.
Received 30 March 2018 Returned formodification 31 May 2018 Accepted 20 July2018
Accepted manuscript posted online 30 July2018
Citation Perez K, Patel R. 2018. Survival ofStaphylococcus epidermidis in fibroblasts andosteoblasts. Infect Immun 86:e00237-18.https://doi.org/10.1128/IAI.00237-18.
Editor Nancy E. Freitag, University of Illinois atChicago
Copyright © 2018 American Society forMicrobiology. All Rights Reserved.
Address correspondence to Robin Patel,[email protected].
BACTERIAL INFECTIONS
crossm
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 1Infection and Immunity
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

Many specific mechanisms of immune evasion have been described for S. aureus,including the production of several factors that inhibit complement activation, com-promise the activity of neutrophils and macrophages, lyse neutrophils, neutralizeantimicrobial peptides (AMPs), and allow for both survival in and escape from phago-somes (12, 14–23). In contrast, there is less known about S. epidermidis pathogenicity.S. epidermidis does not express many of the virulence factors that S. aureus does, andbiofilm formation has been traditionally considered its primary mechanism of patho-genesis when it establishes implant-associated infection (10, 24, 25). In addition toserving as a barrier, biofilm formation has also been shown to decrease deposition ofC3b and IgG, both of which help facilitate phagocytosis and killing by polymorphonu-clear leukocytes (PMNs), on bacterial surfaces (26). Moreover, polysaccharide intercel-lular adhesin (PIA), extracellular matrix-binding protein (Embp), and accumulation-associated protein (Aap), found on the surface of S. epidermidis bacteria, play roles inintercellular adhesion (27–29) and protect biofilms from macrophage phagocytosis (30).
Although existing in a biofilm may enable S. epidermidis to escape the host immunesystem and remain relatively undetected, this is unlikely to be the sole reason that thisorganism is such a prominent cause of implant-associated infections, because manybacteria are capable of biofilm formation. In addition, if biofilm formation were the onlymechanism involved in infection, implant removal might be expected to be universallycurative. In this regard, immune evasion may be operative. Factors found to play rolesin immune evasion by S. epidermidis include its aps and sepA loci. The former senses thepresence of human AMPs, while the latter destroys them (31). Further, the staphylo-coccal quorum sensing system, accessory gene regulator (agr), has been suggested toplay a role in resistance to PMN killing and resistance to the PMN antimicrobial products(26, 32). agr regulates many colonization and virulence factors; one category ofvirulence factors upregulated by agr is PSMs. PSMs are amphipathic peptides producedby most staphylococci that have been demonstrated to play important roles in S. aureusinfection (33) by lysing neutrophils, altering dendritic cell function, and skewing T cellpriming (1, 14, 17, 31, 34, 35). Although S. epidermidis has the capacity to producecytolytic PSMs, it produces such low levels of these peptides that it has little to noability to lyse PMNs (31).
We hypothesized there to be an additional mechanism that S. epidermidis uses tocause chronic infection. Based on the demonstration that S. aureus can invade andpersist in a myriad of cell types (36–47), we hypothesized that S. epidermidis also evadesimmune killing by persisting within nonimmune cells located around implants. Previ-ously it has been reported that a reference strain not associated with human diseaseand two S. epidermidis strains isolated from peritonitis and osteomyelitis cases can beinternalized in MG63 bone cells (48). In addition, we have shown that clinical S.epidermidis PJI isolates can be internalized by and persist within human fibroblasts (49).
The aim of this study was to further investigate intracellular persistence as anothermeans, besides biofilm formation, that S. epidermidis may employ to escape innateimmune killing and cause chronic infection. As several cell types, including fibroblastsand osteoblasts, can be exposed to pathogens in device-related infections, we studiedthe interactions of these cells with S. epidermidis. We evaluated the ability of clinical S.epidermidis PJI isolates to persist intracellularly and to escape host cells using in vitrocell culture infection models, flow cytometry, fluorescence microscopy, and spectrom-etry.
RESULTSS. epidermidis invasion of and persistence in fibroblasts and osteoblasts in
vitro. Planktonic S. epidermidis clinical isolates are readily killed by human neutrophils(see Fig. S1 in the supplemental material), suggesting that some form of evasion of thiskilling is needed to allow for chronic infection. To test the hypothesis that S. epidermidiscan invade and persist within nonprofessional phagocytic cells normally found at thesite of joint arthroplasty, a lysostaphin/daptomycin protection assay was used to assayintracellular infection. Fibroblasts, commonly found at the site of implants due to their
Perez and Patel Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 2
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from
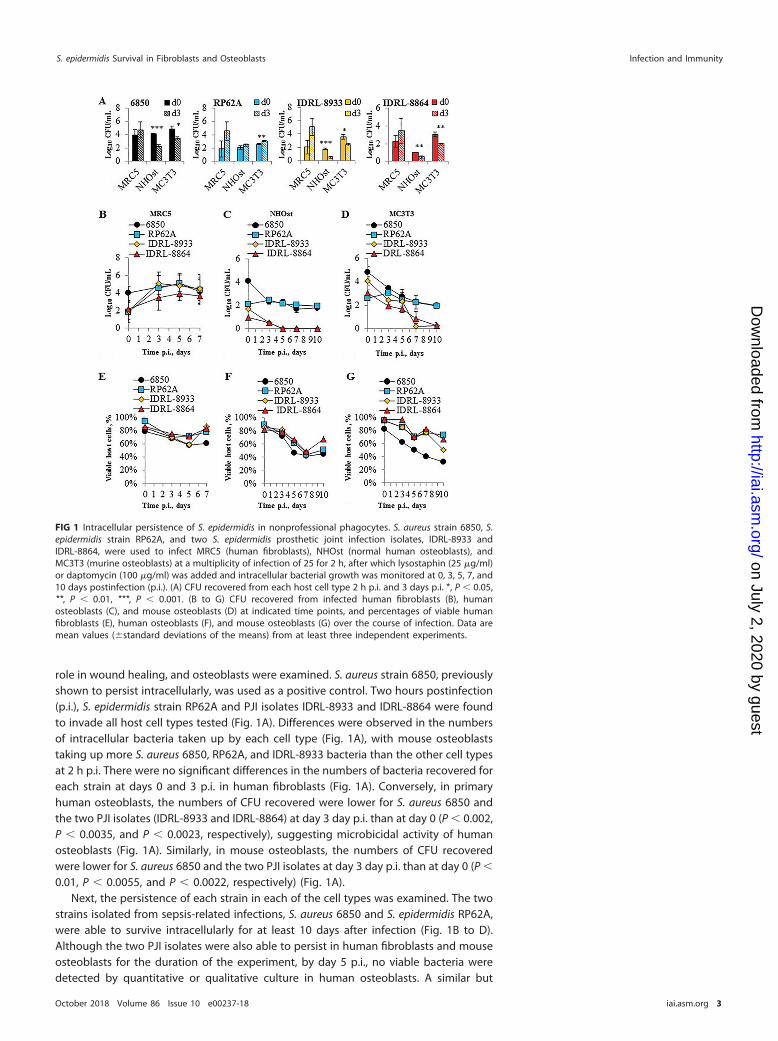
role in wound healing, and osteoblasts were examined. S. aureus strain 6850, previouslyshown to persist intracellularly, was used as a positive control. Two hours postinfection(p.i.), S. epidermidis strain RP62A and PJI isolates IDRL-8933 and IDRL-8864 were foundto invade all host cell types tested (Fig. 1A). Differences were observed in the numbersof intracellular bacteria taken up by each cell type (Fig. 1A), with mouse osteoblaststaking up more S. aureus 6850, RP62A, and IDRL-8933 bacteria than the other cell typesat 2 h p.i. There were no significant differences in the numbers of bacteria recovered foreach strain at days 0 and 3 p.i. in human fibroblasts (Fig. 1A). Conversely, in primaryhuman osteoblasts, the numbers of CFU recovered were lower for S. aureus 6850 andthe two PJI isolates (IDRL-8933 and IDRL-8864) at day 3 day p.i. than at day 0 (P � 0.002,P � 0.0035, and P � 0.0023, respectively), suggesting microbicidal activity of humanosteoblasts (Fig. 1A). Similarly, in mouse osteoblasts, the numbers of CFU recoveredwere lower for S. aureus 6850 and the two PJI isolates at day 3 day p.i. than at day 0 (P �
0.01, P � 0.0055, and P � 0.0022, respectively) (Fig. 1A).Next, the persistence of each strain in each of the cell types was examined. The two
strains isolated from sepsis-related infections, S. aureus 6850 and S. epidermidis RP62A,were able to survive intracellularly for at least 10 days after infection (Fig. 1B to D).Although the two PJI isolates were also able to persist in human fibroblasts and mouseosteoblasts for the duration of the experiment, by day 5 p.i., no viable bacteria weredetected by quantitative or qualitative culture in human osteoblasts. A similar but
FIG 1 Intracellular persistence of S. epidermidis in nonprofessional phagocytes. S. aureus strain 6850, S.epidermidis strain RP62A, and two S. epidermidis prosthetic joint infection isolates, IDRL-8933 andIDRL-8864, were used to infect MRC5 (human fibroblasts), NHOst (normal human osteoblasts), andMC3T3 (murine osteoblasts) at a multiplicity of infection of 25 for 2 h, after which lysostaphin (25 �g/ml)or daptomycin (100 �g/ml) was added and intracellular bacterial growth was monitored at 0, 3, 5, 7, and10 days postinfection (p.i.). (A) CFU recovered from each host cell type 2 h p.i. and 3 days p.i. *, P � 0.05,**, P � 0.01, ***, P � 0.001. (B to G) CFU recovered from infected human fibroblasts (B), humanosteoblasts (C), and mouse osteoblasts (D) at indicated time points, and percentages of viable humanfibroblasts (E), human osteoblasts (F), and mouse osteoblasts (G) over the course of infection. Data aremean values (�standard deviations of the means) from at least three independent experiments.
S. epidermidis Survival in Fibroblasts and Osteoblasts Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 3
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

delayed trend was also seen during infection of mouse osteoblasts. To evaluate fordifferences in cytotoxicity among the strains tested, trypan blue exclusion was used toassess host cell death. At 2 h p.i., each strain tested induced less than 20% cell death(Fig. 1E to G), with at least 50% of the population persisting throughout the course ofinfection.
Neither agr nor PSMs are required for invasion or survival during intracellularinfection. To assess whether agr or PSMs are required for invasion and intracellularpersistence, a PSM� deletion mutant and an agr deletion mutant, and their correspond-ing wild-type (wt) strains, 1457 and Tü3298, respectively, were used to infect humanfibroblasts (Fig. S2A). Each of the S. epidermidis strains was able to invade and persistfor at least 7 days p.i., and there were no significant differences in the numbers of viableCFU recovered between the wt and deletion mutant pairs over the course of infection.There were also no differences in associated host cell death (Fig. S2B).
Lack of difference in uptake and intracellular persistence of S. aureus acute-and chronic-infection PJI isolates. Resch et al. have shown that there are differencesin gene expression between S. aureus planktonic and biofilm cells. They demonstratedupregulation of genes encoding toxins in planktonic cells and upregulation of genesassociated with the stress response and cell surface proteins in biofilm cells (50). As ithas been suggested that planktonic S. aureus cells are associated with acute infections,whereas biofilm cells are associated with chronic infections, we studied one S. aureusisolate each associated with chronic and acute PJI to examine for differences ininvasiveness between the two isolates upon infection of mouse osteoblasts. IDRL-9072(chronic PJI associated) and IDRL-11537 (acute PJI associated) invaded and persisted inosteoblasts for at least 7 days p.i., at similar levels as S. aureus 6850, inducing �40% cellhost death over the course of infection (Fig. S2C and D).
Intracellular persistence is accompanied by increased numbers of SCVs. Small-colony variants (SCVs) are slow-growing subpopulations that naturally arise in responseto various environmental pressures (51–53). They normally comprise a minor fraction ofthe source population and are commonly described in chronic S. aureus infections(54–58). Some SCV observations have also been described for S. epidermidis, mostly inassociation with foreign-body-related infections, such as PJI (8, 59–64). Previously, wehave shown that intracellular localization induces S. epidermidis SCV formation inhuman lung fibroblasts (49). To determine if these findings extend to intracellularpersistence in other cell types, we assessed colony phenotypes of viable bacteria afteran overnight incubation on sheep blood agar (SBA) at 37°C, followed by an additionalincubation overnight at room temperature. SCVs were identified on the basis of theirsize (Fig. 2A). At 2 h p.i., an average of 3, 24, and 7% of all viable intracellularly localizedbacteria had a SCV phenotype after infection in human fibroblasts, human osteoblasts,and mouse osteoblasts, respectively (Fig. 2B to D). While the number of viable intra-cellularly persisting bacteria remained fairly constant over the course of infection, thefrequency of SCVs increased in the intracellular environment, reaching an average of24% after 7 days in the human fibroblasts (Fig. 2B). Although the number of viablepersisting bacteria declined over time in the human and mouse osteoblasts, thefrequency of SCVs continued to increase over time, reaching an average of 19 and 25%after 10 days in the human osteoblasts and mouse osteoblasts, respectively (Fig. 2C andD). The drop observed in the average percentage of SCVs in the human osteoblasts wasdue to the death of the two PJI isolates.
Next, to assess whether SCVs have an advantage in intracellular invasiveness, wecompared the numbers of CFU recovered after infection with two normal-colony-phenotype (NCP) isolates and their SCV counterparts recovered from chronic knee andshoulder PJI. There were no statistically significant differences in the numbers ofrecovered bacteria between the NCP and SCV pairs at 2 h p.i. (Fig. S2C), suggesting thatSCVs do not have an increased ability to invade cells in comparison to NCP isolates, aspreviously shown for one S. aureus NCP and SCV pair (65).
Intracellularly persisting S. epidermidis bacteria colocalize with host cellphagolysosomes. Previously, we have shown that S. epidermidis RP62A is located
Perez and Patel Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 4
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

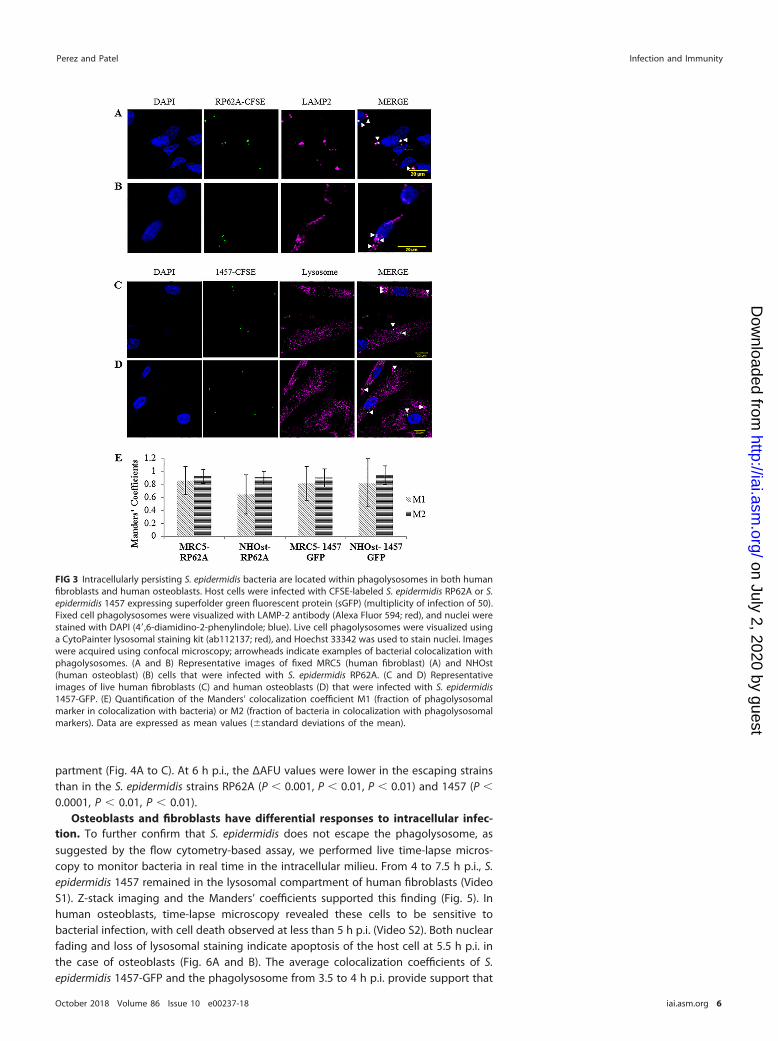
within the phagolysosomes of human fibroblasts (49). Here, we aimed to confirm thatthese findings are not limited to a single strain and to evaluate whether intracellularlypersisting S. epidermidis bacteria are also located in phagolysosomes within humanosteoblasts. CFSE (carboxyfluorescein succinimidyl ester)-labeled RP62A colocalizedwith LAMP-2� vesicles in both fixed human fibroblasts and osteoblasts (Fig. 3A and B).Similarly, S. epidermidis 1457-green fluorescent protein (GFP) colocalized with phagoly-sosomes in live cells (Fig. 3C and D). Colocalizations were supported by analysis ofManders’ coefficient. Manders’ coefficient M1 denotes the fraction of phagolysosomesthat colocalize with bacteria, whereas Manders’ coefficient M2 denotes the fraction ofbacteria that colocalize with phagolysosomes. Because there is an excess number ofphagolysosomes compared to bacteria, Manders’ coefficient M1 may be skewed, falselyindicating low colocalization. Thus, we ascribed increased significance to Manders’coefficient M2 in terms of quantifying colocalization. The average Manders’ coefficientM2 values for the two S. epidermidis isolates with the two different phagolysosomalstains were all �0.9, suggesting that intracellularly persisting bacteria are locatedwithin phagolysosomes in both human fibroblasts and osteoblasts (Fig. 3E). Thesefindings were similar for live cells infected with the USA300 strain LAC expressing GFP(LAC-GFP) 3 h p.i. (Fig. S3).
S. epidermidis RP62A and 1457 do not escape the phagolysosome of infectedcells. It has previously been shown that some S. aureus strains are capable of escapingfrom the phagosomal compartment to avoid phagolysosomal killing (15, 40, 44, 66–68).To address the question as to whether escape from the phagolysosome is a mechanismS. epidermidis uses to persist in nonprofessional phagocytes, we used a flow cytometry-based pH monitoring assay described by Lâm et al. (40). At 2 h p.i., all examined strainswere located within an acidic environment (Fig. 4A to C). Differences in arbitraryfluorescent units (ΔAFU) at 6 h p.i. indicated translocation of the two S. aureus strainstested (Fig. 4A to C), previously shown to escape the phagosome in HeLa, 293, andTHP-1 cells (40, 66). The escape of S. aureus strains 6850 and LAC-GFP was indicated bysignificant changes in ΔAFU between the 2- and 6-h time points (for 6850, P � 0.007,P � 0.006, P � 0.007; for LAC-GFP, P � 0.02, P � 0.005, P � 0.0007 [for MRC5 {humanfibroblasts}, NHOst {normal human osteoblasts}, and MC3T3 {murine osteoblasts},respectively]). However, both RP62A and 1457-GFP remained in the endosomal com-
FIG 2 Small-colony variant (SCV) formation increases over time during intracellular persistence. Colonyphenotypes of viable bacteria were assessed after an overnight incubation on sheep blood agar at 37°C,followed by an additional incubation overnight at room temperature. SCVs were identified on the basisof their size. (A) Representative images of colony phenotype of each strain; SCVs are denoted byarrowheads. (B to D) Percentages of viable intracellularly persisting bacteria with SCV phenotype inhuman fibroblasts (B), human osteoblasts (C), and mouse osteoblasts (D) over the course of infection.Data are mean values (�standard deviations of the means) from two independent experiments per-formed in triplicate.
S. epidermidis Survival in Fibroblasts and Osteoblasts Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 5
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from
partment (Fig. 4A to C). At 6 h p.i., the ΔAFU values were lower in the escaping strainsthan in the S. epidermidis strains RP62A (P � 0.001, P � 0.01, P � 0.01) and 1457 (P �
0.0001, P � 0.01, P � 0.01).Osteoblasts and fibroblasts have differential responses to intracellular infec-
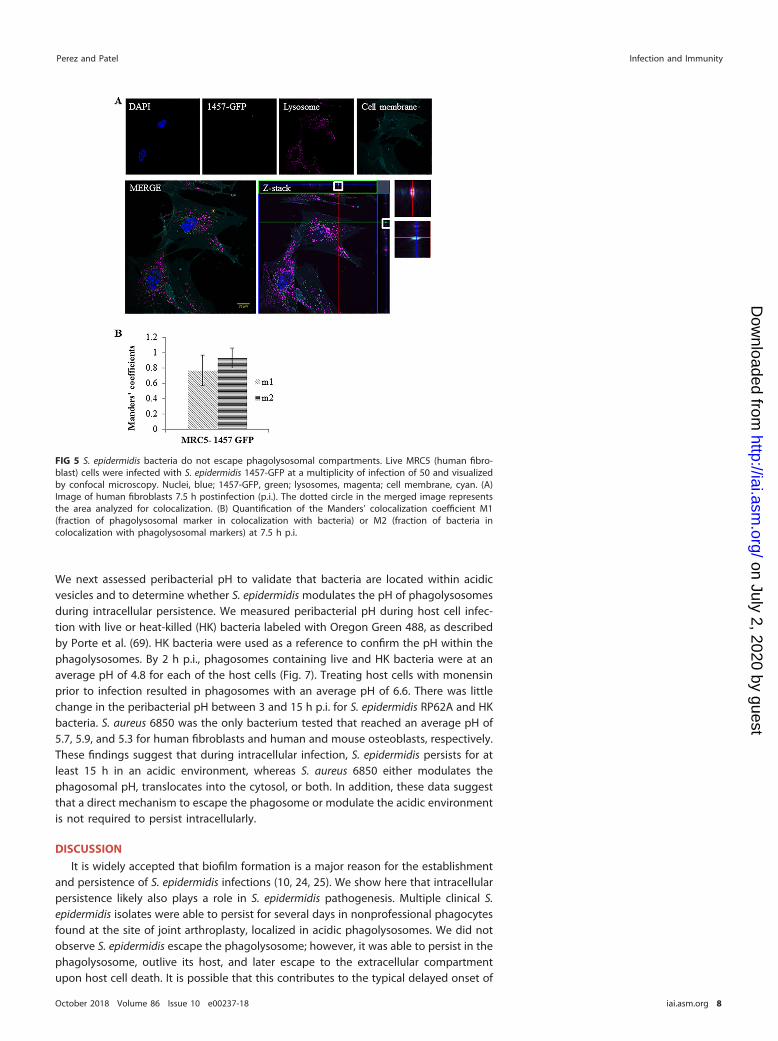
tion. To further confirm that S. epidermidis does not escape the phagolysosome, assuggested by the flow cytometry-based assay, we performed live time-lapse micros-copy to monitor bacteria in real time in the intracellular milieu. From 4 to 7.5 h p.i., S.epidermidis 1457 remained in the lysosomal compartment of human fibroblasts (VideoS1). Z-stack imaging and the Manders’ coefficients supported this finding (Fig. 5). Inhuman osteoblasts, time-lapse microscopy revealed these cells to be sensitive tobacterial infection, with cell death observed at less than 5 h p.i. (Video S2). Both nuclearfading and loss of lysosomal staining indicate apoptosis of the host cell at 5.5 h p.i. inthe case of osteoblasts (Fig. 6A and B). The average colocalization coefficients of S.epidermidis 1457-GFP and the phagolysosome from 3.5 to 4 h p.i. provide support that
FIG 3 Intracellularly persisting S. epidermidis bacteria are located within phagolysosomes in both humanfibroblasts and human osteoblasts. Host cells were infected with CFSE-labeled S. epidermidis RP62A or S.epidermidis 1457 expressing superfolder green fluorescent protein (sGFP) (multiplicity of infection of 50).Fixed cell phagolysosomes were visualized with LAMP-2 antibody (Alexa Fluor 594; red), and nuclei werestained with DAPI (4=,6-diamidino-2-phenylindole; blue). Live cell phagolysosomes were visualized usinga CytoPainter lysosomal staining kit (ab112137; red), and Hoechst 33342 was used to stain nuclei. Imageswere acquired using confocal microscopy; arrowheads indicate examples of bacterial colocalization withphagolysosomes. (A and B) Representative images of fixed MRC5 (human fibroblast) (A) and NHOst(human osteoblast) (B) cells that were infected with S. epidermidis RP62A. (C and D) Representativeimages of live human fibroblasts (C) and human osteoblasts (D) that were infected with S. epidermidis1457-GFP. (E) Quantification of the Manders’ colocalization coefficient M1 (fraction of phagolysosomalmarker in colocalization with bacteria) or M2 (fraction of bacteria in colocalization with phagolysosomalmarkers). Data are expressed as mean values (�standard deviations of the mean).
Perez and Patel Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 6
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

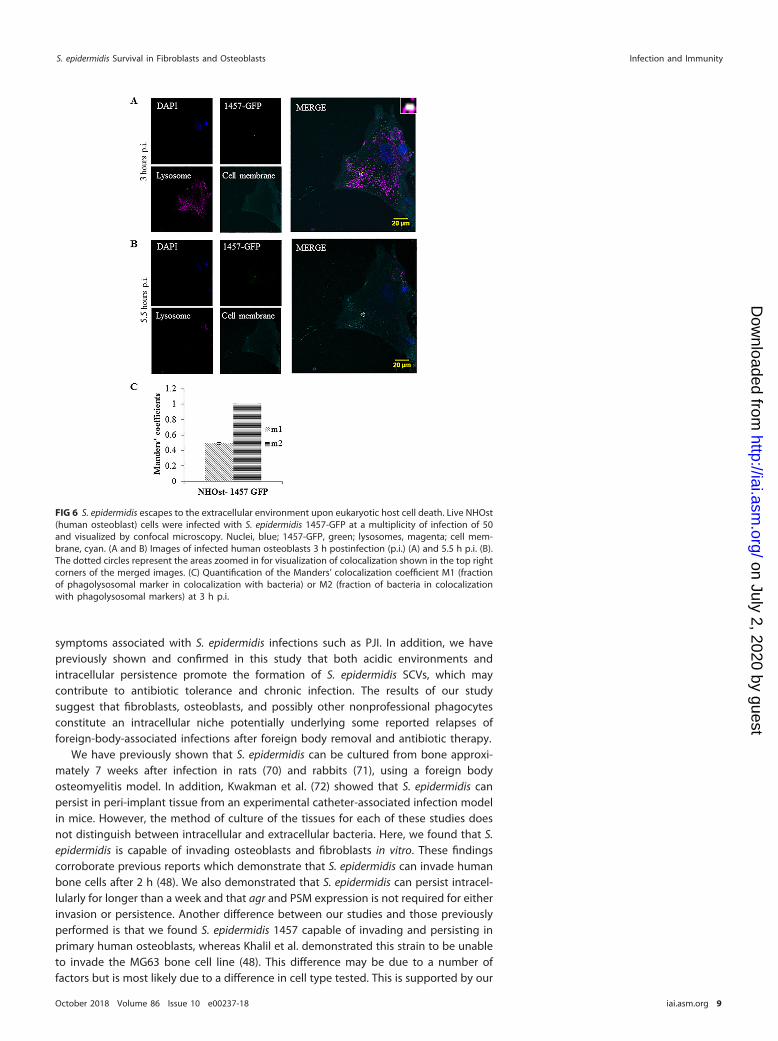
the bacteria are located within the phagosome (Fig. 6C). There are no coefficients for5 h p.i. due to the lack of colocalization found, but the continual GFP expressionindicates that S. epidermidis 1457-GFP outlived its captor. Together, the time-lapseexperiments provide evidence that S. epidermidis cannot actively escape the phagoly-sosome. However, we show that this organism can remain viable and be released upondeath of the host cell.
To compare the nature of infection between S. aureus and S. epidermidis, S. aureusLAC-GFP was also visualized during intracellular persistence in human fibroblasts andosteoblasts. LAC-GFP was found to escape lysosomal compartments in fibroblasts priorto 6 h p.i. and to replicate in the cytosol in a relatively short period of time (Fig. S4 andVideo S3). This corroborates the previous findings of Grosz et al. that S. aureus replicateswithin the host cell cytoplasm (66). The high standard deviation (SD) seen in the M2coefficient likely reflects the escape of some LAC-GFP bacteria and the replication ofthose bacteria that have escaped (Fig. S4D). Conversely, upon infection with osteo-blasts, some LAC-GFP bacteria were killed and the death of the host cells was alsovisualized (Fig. S5A and B and Video S4). The decrease in and high SD of both the M1and M2 coefficients from 4 to 8 h p.i. are indicative of both a decrease in GFP expression(death of bacteria) and a decrease in colocalization (death of host cell) (Fig. S5C).Together, the time-lapse experiments reveal a difference in sensitivity of fibroblasts andosteoblasts to bacterial infection in regard to microbicidal activity and death.
S. epidermidis persists during localization within acidic phagosomes. Despitebeing unable to escape the phagolysosome, S. epidermidis is able to persist in host cells.
FIG 4 S. epidermidis does not escape the phagolysosome by 6 h. Host cells were infected withFITC-labeled S. aureus 6850, FITC-labeled S. epidermidis RP62A, S. aureus LAC-GFP, or S. epidermidis1457-GFP at a multiplicity of infection of 50. The difference in arbitrary fluorescence units (ΔAFU) in thepresence and absence of monensin, normalized to the invasion rate {ΔAFU � [(AFU�monensin �AFU�monensin)/AFU�monensin] � 105}, was used as the readout for pH. Larger negative values correspondto more-acidic (i.e., lower) pH values. Staphylococci localization in infected MRC5 (human fibroblast) (A),NHOst (human osteoblast) (B), and MC3T3 (mouse osteoblast) (C) cells is shown. Data are mean values(�standard deviations of the means) from two independent experiments done in triplicate. *, P � 0.05,**, P � 0.01, ***, P � 0.001, ****, P � 0.0001. Significance indicated near strain designations was gatheredfrom a comparison of results at 2 and 6 h p.i.; significance indicated near data points was gathered froma comparison of results at 6 h p.i. between the corresponding S. aureus and S. epidermidis strains.
S. epidermidis Survival in Fibroblasts and Osteoblasts Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 7
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from
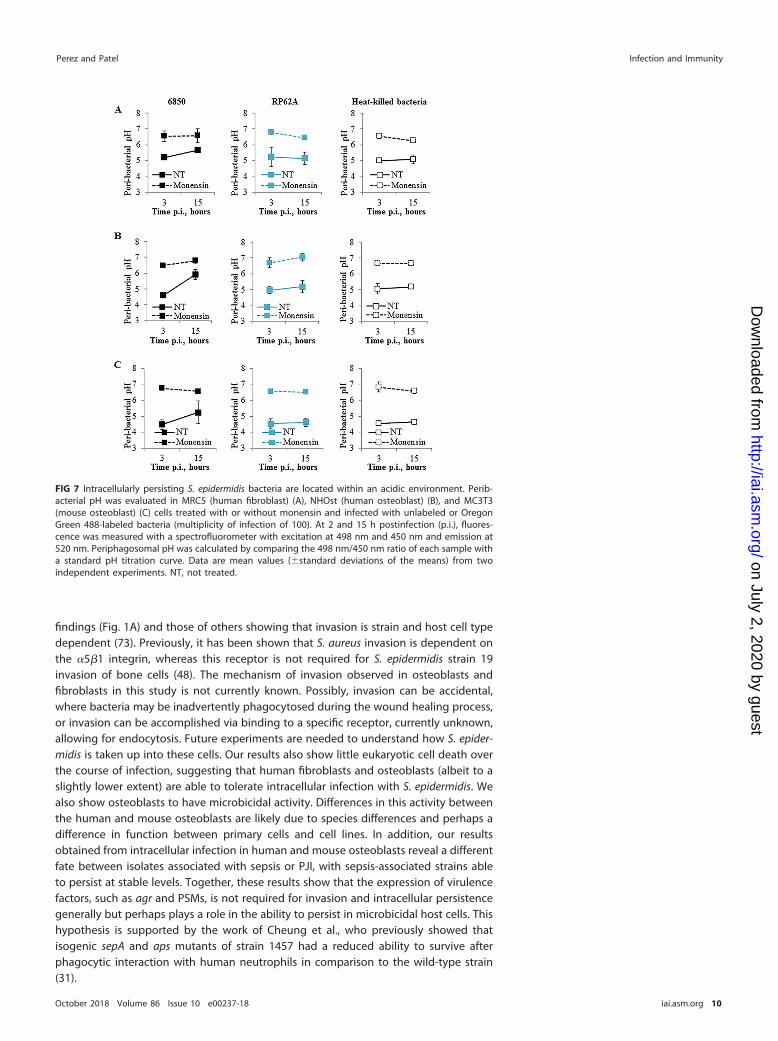
We next assessed peribacterial pH to validate that bacteria are located within acidicvesicles and to determine whether S. epidermidis modulates the pH of phagolysosomesduring intracellular persistence. We measured peribacterial pH during host cell infec-tion with live or heat-killed (HK) bacteria labeled with Oregon Green 488, as describedby Porte et al. (69). HK bacteria were used as a reference to confirm the pH within thephagolysosomes. By 2 h p.i., phagosomes containing live and HK bacteria were at anaverage pH of 4.8 for each of the host cells (Fig. 7). Treating host cells with monensinprior to infection resulted in phagosomes with an average pH of 6.6. There was littlechange in the peribacterial pH between 3 and 15 h p.i. for S. epidermidis RP62A and HKbacteria. S. aureus 6850 was the only bacterium tested that reached an average pH of5.7, 5.9, and 5.3 for human fibroblasts and human and mouse osteoblasts, respectively.These findings suggest that during intracellular infection, S. epidermidis persists for atleast 15 h in an acidic environment, whereas S. aureus 6850 either modulates thephagosomal pH, translocates into the cytosol, or both. In addition, these data suggestthat a direct mechanism to escape the phagosome or modulate the acidic environmentis not required to persist intracellularly.
DISCUSSION
It is widely accepted that biofilm formation is a major reason for the establishmentand persistence of S. epidermidis infections (10, 24, 25). We show here that intracellularpersistence likely also plays a role in S. epidermidis pathogenesis. Multiple clinical S.epidermidis isolates were able to persist for several days in nonprofessional phagocytesfound at the site of joint arthroplasty, localized in acidic phagolysosomes. We did notobserve S. epidermidis escape the phagolysosome; however, it was able to persist in thephagolysosome, outlive its host, and later escape to the extracellular compartmentupon host cell death. It is possible that this contributes to the typical delayed onset of
FIG 5 S. epidermidis bacteria do not escape phagolysosomal compartments. Live MRC5 (human fibro-blast) cells were infected with S. epidermidis 1457-GFP at a multiplicity of infection of 50 and visualizedby confocal microscopy. Nuclei, blue; 1457-GFP, green; lysosomes, magenta; cell membrane, cyan. (A)Image of human fibroblasts 7.5 h postinfection (p.i.). The dotted circle in the merged image representsthe area analyzed for colocalization. (B) Quantification of the Manders’ colocalization coefficient M1(fraction of phagolysosomal marker in colocalization with bacteria) or M2 (fraction of bacteria incolocalization with phagolysosomal markers) at 7.5 h p.i.
Perez and Patel Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 8
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from
symptoms associated with S. epidermidis infections such as PJI. In addition, we havepreviously shown and confirmed in this study that both acidic environments andintracellular persistence promote the formation of S. epidermidis SCVs, which maycontribute to antibiotic tolerance and chronic infection. The results of our studysuggest that fibroblasts, osteoblasts, and possibly other nonprofessional phagocytesconstitute an intracellular niche potentially underlying some reported relapses offoreign-body-associated infections after foreign body removal and antibiotic therapy.
We have previously shown that S. epidermidis can be cultured from bone approxi-mately 7 weeks after infection in rats (70) and rabbits (71), using a foreign bodyosteomyelitis model. In addition, Kwakman et al. (72) showed that S. epidermidis canpersist in peri-implant tissue from an experimental catheter-associated infection modelin mice. However, the method of culture of the tissues for each of these studies doesnot distinguish between intracellular and extracellular bacteria. Here, we found that S.epidermidis is capable of invading osteoblasts and fibroblasts in vitro. These findingscorroborate previous reports which demonstrate that S. epidermidis can invade humanbone cells after 2 h (48). We also demonstrated that S. epidermidis can persist intracel-lularly for longer than a week and that agr and PSM expression is not required for eitherinvasion or persistence. Another difference between our studies and those previouslyperformed is that we found S. epidermidis 1457 capable of invading and persisting inprimary human osteoblasts, whereas Khalil et al. demonstrated this strain to be unableto invade the MG63 bone cell line (48). This difference may be due to a number offactors but is most likely due to a difference in cell type tested. This is supported by our
FIG 6 S. epidermidis escapes to the extracellular environment upon eukaryotic host cell death. Live NHOst(human osteoblast) cells were infected with S. epidermidis 1457-GFP at a multiplicity of infection of 50and visualized by confocal microscopy. Nuclei, blue; 1457-GFP, green; lysosomes, magenta; cell mem-brane, cyan. (A and B) Images of infected human osteoblasts 3 h postinfection (p.i.) (A) and 5.5 h p.i. (B).The dotted circles represent the areas zoomed in for visualization of colocalization shown in the top rightcorners of the merged images. (C) Quantification of the Manders’ colocalization coefficient M1 (fractionof phagolysosomal marker in colocalization with bacteria) or M2 (fraction of bacteria in colocalizationwith phagolysosomal markers) at 3 h p.i.
S. epidermidis Survival in Fibroblasts and Osteoblasts Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 9
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from
findings (Fig. 1A) and those of others showing that invasion is strain and host cell typedependent (73). Previously, it has been shown that S. aureus invasion is dependent onthe �5�1 integrin, whereas this receptor is not required for S. epidermidis strain 19invasion of bone cells (48). The mechanism of invasion observed in osteoblasts andfibroblasts in this study is not currently known. Possibly, invasion can be accidental,where bacteria may be inadvertently phagocytosed during the wound healing process,or invasion can be accomplished via binding to a specific receptor, currently unknown,allowing for endocytosis. Future experiments are needed to understand how S. epider-midis is taken up into these cells. Our results also show little eukaryotic cell death overthe course of infection, suggesting that human fibroblasts and osteoblasts (albeit to aslightly lower extent) are able to tolerate intracellular infection with S. epidermidis. Wealso show osteoblasts to have microbicidal activity. Differences in this activity betweenthe human and mouse osteoblasts are likely due to species differences and perhaps adifference in function between primary cells and cell lines. In addition, our resultsobtained from intracellular infection in human and mouse osteoblasts reveal a differentfate between isolates associated with sepsis or PJI, with sepsis-associated strains ableto persist at stable levels. Together, these results show that the expression of virulencefactors, such as agr and PSMs, is not required for invasion and intracellular persistencegenerally but perhaps plays a role in the ability to persist in microbicidal host cells. Thishypothesis is supported by the work of Cheung et al., who previously showed thatisogenic sepA and aps mutants of strain 1457 had a reduced ability to survive afterphagocytic interaction with human neutrophils in comparison to the wild-type strain(31).
FIG 7 Intracellularly persisting S. epidermidis bacteria are located within an acidic environment. Perib-acterial pH was evaluated in MRC5 (human fibroblast) (A), NHOst (human osteoblast) (B), and MC3T3(mouse osteoblast) (C) cells treated with or without monensin and infected with unlabeled or OregonGreen 488-labeled bacteria (multiplicity of infection of 100). At 2 and 15 h postinfection (p.i.), fluores-cence was measured with a spectrofluorometer with excitation at 498 nm and 450 nm and emission at520 nm. Periphagosomal pH was calculated by comparing the 498 nm/450 nm ratio of each sample witha standard pH titration curve. Data are mean values (�standard deviations of the means) from twoindependent experiments. NT, not treated.
Perez and Patel Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 10
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

It has been reported that there are differences in gene expression between free-floating and adherent bacterial cells. For example, in S. aureus planktonic cells, agr andgenes encoding toxins are upregulated, while no toxin gene was expressed at highlevels in the S. aureus biofilm cells (50). In addition to changes in metabolism, biofilmcells also had an increase in the expression of ica genes in comparison to that ofplanktonic bacteria (50). Overall, it has been suggested that planktonic bacteria ex-pressing toxins are generally more virulent and have a greater ability to cause acuteinfection than biofilm cells, whereas biofilm cells tend to employ defensive molecularmechanisms to promote their survival, at times resulting in chronic infection (50, 74,75). Here we compared the invasion and persistence of one chronic-infection and oneacute-infection S. aureus PJI isolate and showed that both isolates are successfully ableto invade and persist within mouse osteoblasts. As seen for the S. epidermidis strains,the expression of virulence factors may not be required for invasion. However, furthercharacterization of these two isolates (and other chronic and acute infection-associatedisolates) and further time points are needed to distinguish any changes in the ability topersist past 7 days and to identify genes responsible for this persistence.
SCV formation is induced in response to diverse environmental pressures (51–53).SCVs typically comprise a minor proportion of the source population and are commonin chronic infections (54–58). S. epidermidis SCVs are common in PJI (8). Here weshowed there to be no difference in ability to invade cells between pairs of an isogenicSCV or closely related SCV and an NCP S. epidermidis isolate (see Fig. S2E and F in thesupplemental material), as previously shown for S. aureus (65). In addition, we showeda positive correlation between SCV formation and time of intracellular persistence inhuman fibroblasts with multiple staphylococcal strains and extended these findings toinfection in osteoblasts. Previously, we have shown that the number of SCVs can bereduced by treatment with lysosomotropic alkalinizing agents. For at least the firstweek, SCVs do not significantly contribute to survival, as there is no change in the totalnumber of CFU recovered from cells treated with these drugs (49). Although theincreasing numbers of SCVs suggest adaptation to the acidic environment, there maybe another role this subpopulation plays during infection. Recently, two groups haveimplicated SCVs in proinflammatory cytokine suppression. Ou et al. showed that after24 h, intracellular infection with NCP S. aureus in a human bronchial epithelial cell lineinduced a robust proinflammatory response evidenced by increased Toll-like receptor2 (TLR2), interleukin 1 (IL-1), IL-6, and IL-12 expression, whereas SCV infection inducedonly TLR2 expression (65). Magrys et al. showed that 4 h after infection of THP-1 cellswith S. epidermidis SCVs isolated from a hip PJI, there was an increase in AKT phos-phorylation compared to that in NCP-infected and noninfected cells (76). This group didnot show direct evidence for proinflammatory cytokine suppression, but because thephosphatidylinositol 3-kinase (PI3K)-AKT pathway has been shown to be involved withcytokine suppression, they suggested this role. Thus, together with our findings, thissuggests that intracellular persistence and tolerance to this persistence are maintainedby SCVs via mechanisms currently unknown, possibly by modulating signaling path-ways.
Classified as nonprofessional phagocytes, both fibroblasts and osteoblasts usemechanisms similar to those used by macrophages to phagocytose and degrade thecontents within their phagosomes (77–79). Using live time-lapse fluorescence micros-copy, we showed that within 2 h of infection, bacteria are taken up by osteoblasts andfibroblasts into acidic phagosomes, where some but not all S. epidermidis cells are killed.In addition, our results indicate that S. epidermidis is unable to escape the phagosomesor modulate the acidic environment after 15 h. This is different from results generatedwith S. aureus, both in our study and in those by others, describing phagosomal escapeand replication in the cytosol (15, 40, 66, 80). However, as not all S. aureus isolates areable to escape and some escape at later time points, our findings do not definitivelyprove that S. epidermidis cannot escape the phagolysosome. Because phagosomalescape for S. aureus is dependent upon PSM� expression (66), whose homologue isexpressed in much lower concentrations by S. epidermidis (31), it is plausible that this
S. epidermidis Survival in Fibroblasts and Osteoblasts Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 11
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

organism simply requires more time for phagosomal escape than does S. aureus. Mostimportantly, we were able to show that viable S. epidermidis is capable of escaping intothe extracellular environment upon host cell death. Together, the results suggest thatS. epidermidis is able to tolerate the acidic phagosomal environment for several days.
In conclusion, our findings show that intracellular infection of osteoblasts, fibro-blasts, and possibly other nonprofessional phagocytes comprise a second and equallyimportant mechanism, beyond biofilm formation, enabling evasion from host immunedefense and permitting bacteria to persist during infection and possibly treatment.Given that current treatments may not kill intracellularly persisting bacteria, targetingthis population may contribute to increased treatment success. Additionally, SCVformation may contribute to the subtle and silent nature of infection through as-yet-undescribed processes.
MATERIALS AND METHODSMicroorganisms. Bacterial strains used in this study are detailed in Table S1 in the supplemental
material. Prior to assays, staphylococci were grown overnight in 10 ml of tryptic soy broth (TSB) at 37°Cwith rotation. Bacteria expressing superfolder green fluorescent protein (sGFP) were grown in 10 ml TSBsupplemented with 10 �g/ml chloramphenicol. Bacteria were harvested by centrifugation, washed twicewith phosphate-buffered saline (PBS), and resuspended in assay medium. Bacterial cell numbers wereestimated spectrophotometrically at 600 nm and/or using McFarland standards. Heat-killed (HK) bacteriawere prepared by heating the bacteria for 10 to 15 min at 65°C. Lack of viability was confirmed by nogrowth after plating (data not shown). Prior to experimentation, S. epidermidis and S. aureus stockcultures were confirmed for agr expression via the production of PSM� and/or PSM�2 via reversetranscription PCR or ELISA (data not shown). SCVs and NCP isolates from the same joint were previouslyassessed for relatedness by pulsed-field gel electrophoresis (PFGE), as described previously (81). IDRL-8933 and IDRL-8934 had a 3-band difference, whereas IDRL-8864 and IRL-8866 were indistinguishable(81). To confirm that both SCV and NCP isolates originated from the same clone, whole-genomesequencing (WGS) had been previously conducted at the Department of Infection Control Science atJuntendo University Graduate School of Medicine (Tokyo, Japan) (82). WGS results were insufficient toconfirm that IDRL-8933 and IDRL-8934 were clonally related. However, WGS revealed approximately 10mutations between IDRL-8866 and IDRL-8864, supporting the theory that these two strains are clonal(82).
Cell culture. Human lung fibroblasts (MRC5 cells) were maintained in RPMI 1640, supplemented with10% heat-inactivated fetal bovine serum (Sigma). Murine osteoblasts (MC3T3-E1) were maintained inminimum essential medium alpha (MEM�) supplemented with 10% heat-inactivated fetal bovine serum.Mouse osteoblast cell authentication was performed by IDEXX BioResearch using short tandem repeat(STR) DNA profiling. Primary normal human osteoblast (NHOst) cells were maintained in osteoblast basalmedium supplemented with OGM SingleQuots (Lonza). Human osteoblast cell authentication wasperformed by the Mayo Clinic Genotyping Core using STR DNA profiling. Osteoblasts were alsoconfirmed by alkaline phosphatase and bone mineralization staining (data not shown).
Intracellular persistence. (i) Lysostaphin-daptomycin protection assay. Host cells were infectedat a multiplicity of infection (MOI) of 25 for intracellular assay. Host cells were washed daily with PBS andlysostaphin (Sigma) or daptomycin (to kill extracellular bacteria) and provided fresh medium supple-mented with 10% fetal bovine serum. At 0, 3, 5, and 7 days p.i., host cells were lysed and serial 10-folddilutions of the cell lysates were plated onto sheep blood agar (SBA) to detect and quantitate intracellularbacteria. CFU were enumerated after overnight incubation at 37°C in air. The SCV phenotype wasdetermined by size after a second overnight incubation at room temperature. Only intracellularlypersisting bacteria were monitored over time, as no colonies were found in the washing medium afterthe lysostaphin/daptomycin treatment (data not shown).
(ii) Viability. Host cells were detached from wells with trypsin-EDTA (0.25%) (Thermo Fisher), and a100-�l aliquot was diluted 1:1 in 0.4% trypan blue solution. The percentage of live cells was calculatedusing a Countess II FL automated cell counter (Thermo Fisher); two independent samples were analyzedper time point.
Phagolysosomal localization. (i) Confocal microscopy of fixed cells. S. epidermidis RP62A wasgrown and harvested as described above. Bacterial cells were washed and stained with 5 mM CFSE(eBioscience) in the dark for 10 min at room temperature. Host cells grown on glass coverslips were theninfected at an MOI of 50 with the CFSE-labeled bacteria for 2 h at 37°C in 5% CO2. Following the 2-hincubation, host cells were washed and incubated with 20 �g/ml lysostaphin to kill extracellular adherentbacteria. Host cells were then further incubated for 1 h at 37°C in 5% CO2. The cells were washed, fixed,and permeabilized. After blocking, the cells were incubated with primary antibodies overnight at 4°C,followed by three washes with PBS and incubation with secondary antibodies for 1 h and then threemore washes. Antibodies to human lysosomal-associated membrane protein-2 (LAMP-2) (H4B4; Abcam)were used at 1:100. LAMP-2 is abundantly and ubiquitously expressed on lysosomal membranes and isused to mark mature endosomes and lysosomes. The fluorophore-conjugated donkey anti-mousesecondary antibody was used at 1:500. Confocal microscopy was performed using an LSM 780 micro-scope (Carl Zeiss, Germany) with a 40� objective.
Perez and Patel Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 12
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

(ii) Confocal microscopy of live cells. S. epidermidis 1457 expressing sGFP was grown and harvestedas described above. Host cells were infected at an MOI of 50 for 1 h at 37°C in 5% CO2. Host cells werethen washed as described above, and a CytoPainter lysosomal staining kit (Abcam) was used to stain forlysosomes. The lysotropic indicator from the kit is designed to selectively accumulate in the lysosomesof live cells. Cells were then incubated for 30 min at 37°C in 5% CO2, washed, and stained with NucBlue(Invitrogen) for 5 min, followed by staining with the CellMask deep red plasma membrane stain (ThermoFisher) for 5 to 10 min, both at 37°C in 5% CO2. The cells were washed, and phenol-red free medium wasadded. Confocal microscopy was performed with an LSM 780 microscope (Carl Zeiss, Germany) using a40� objective in a controlled chamber providing incubation at 37°C in 5% CO2.
(iii) Colocalization. Colocalization of bacteria and phagolysosomes was determined by selecting atleast 15 randomly chosen regions of interest (ROI) containing intracellular bacteria using ImageJ Coloc2 software to determine the Manders’ coefficients. There is one Manders’ coefficient per channel, andeach coefficient is proportional to the amount of fluorescence in that channel that colocalizes with thefluorescence in the other channel over its total intensity. The coefficient varies from 0 to 1, with 0indicating no colocalization and 1 indicating perfect colocalization (83). The colocalization in livetime-lapse images was determined by averaging the coefficients of each image within respective timeframes.
(iv) Flow cytometry phagolysosome escape assay. The flow cytometry phagolysosome escapeassay, which monitors the pH of the environment of intracellular staphylococci, was performed asdescribed by Lâm et al. (40), with slight modification. Bacterial cultures were grown and harvested asdescribed above. Washed bacterial pellets were then resuspended in 0.1 M sodium bicarbonate buffer,and bacteria were labeled with fluorescein isothiocyanate (FITC) (Invitrogen) in the dark with rotation for30 min at room temperature. Bacterial cells were washed with Hanks balanced salt solution (HBSS) toremove unbound dye and then resuspended in HBSS. Prior to infection, host cells were seeded at 3 �105 cells per well and grown overnight in a 24-well culture disk to subconfluence. Host cells were placedon ice for 10 min to synchronize phagocytosis, and cells were then infected at an MOI of 50. Followingincubation on ice for 15 min, the cells were moved to 37°C in 5% CO2 for 55 min. The cells were washedand incubated with medium supplemented with lysostaphin (20 �g/ml) at 37°C for 30 min. The cellswere returned to the incubator for an additional 55 min or 4 h and 55 min, resulting in an overallinfection time of 2 or 6 h, respectively. Cells were washed with PBS, trypsinized, and transferred to 5-mlround-bottom tubes. Samples were incubated with or without 50 �M monensin for 20 min at 37°C,followed by propidium iodide treatment for live/dead staining. Samples were analyzed using FACSCantoX. For each cell type, a preset fixed gating and a fixed amplification were used. The invasion rate withmonensin (total internalized bacteria), expressed as numbers of arbitrary fluorescence units (AFU), wasdetermined according to the formula AFU � [(mean fluorescence intensity of cells in M) � (percentageof cells in M)]/1,000 and normalized to the mean fluorescence intensity of each corresponding bacterialpreparation. (The FITC-positive gate is denoted M.) The difference in AFU (ΔAFU) in the presence orabsence of monensin, normalized to the invasion rate {ΔAFU � [(AFU�monensin � AFU�monensin)/AFU�monensin] � 105}, was used as the readout for pH. Larger negative values correspond to more-acidic(i.e., lower) pH values.
Measurement of peribacterial pH. Live (overnight) or HK (prepared as described above) bacteriawere labeled with the pH reporter dye Oregon Green 488 (OG-488) (Invitrogen), which can assess pHvalues between 3.5 and 7 (69). The experimental setup for this assay was as described by Tranchemon-tagne et al. (18). Live and HK cultures were washed with Dulbecco’s phosphate-buffered saline (D-PBS),resuspended in OG-488 (10 mg/ml), and incubated for 30 min at 4°C in the dark. Labeled bacteria werecentrifuged, and the labeling reaction was stopped by adding Tris-HCl (pH 8.3) to a final concentrationof 100 mM. Bacteria were incubated at 4°C in the dark for 15 min, washed, and used for infection.Labeling with OG-488 did not affect the viability of live bacteria as measured by plating on bovine serumalbumin (BSA) (data not shown).
To determine the pH of the environment surrounding S. epidermidis, host cells were grown in phenolred-free RPMI 1640 in tissue culture-treated, black-walled 24-well microplates (PerkinElmer). Cells weretreated with or without 50 �M monensin and infected with Oregon Green 488-labeled or unlabeledbacteria at an MOI of 100, according to the procedure described above. At 2 and 15 h p.i., 0.1% trypanblue was added to infected cells for 1 min to quench the fluorescence of the remaining extracellularbacteria, and the cells were then washed three times with cell medium. With the emission wavelengthat 520 nm, the ratio of fluorescence with excitation at 498 to that at 450 nm was measured using aFluoroskan Ascent (Thermo Scientific). As described by Levitz et al. (84), a standard curve was generatedfor each condition by obtaining excitation ratios at 498 and 450 nm in infected cells where thephagosomal and extracellular pH values were equilibrated in nigericin buffers of defined pH (0.028 mMnigericin, 10 �M monensin, 150 mM sodium chloride, 80 mM potassium chloride, 1 mM magnesiumchloride, 5.5 mM glucose, 2 mM calcium chloride, and 10 mM HEPES) (data not shown). Backgroundvalues for excitation at 498 and 450 nm with emission at 520 nm for cells infected with unlabeledbacteria were subtracted from readings obtained with labeled bacteria. To calculate the averagephagosomal pH of the samples, the ratios of excitation at 498 to that at 450 nm were compared with thestandard pH titration curve generated from nigericin-permeabilized cells. HK bacteria were used as areference to confirm the pH over time in the phagolysosomes, as dead bacteria would not activelymodulate or escape the phagolysosome.
Statistical methods. Statistical analysis was performed using a two-tailed t test. We used an alphavalue of 0.05, so P values of �0.05 were considered significant (*, P � 0.05, **, P � 0.01, ***, P � 0.001,and ****, P � 0.0001). All error bars represent standard deviations (SD).
S. epidermidis Survival in Fibroblasts and Osteoblasts Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 13
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

SUPPLEMENTAL MATERIAL
Supplemental material for this article may be found at https://doi.org/10.1128/IAI.00237-18.
SUPPLEMENTAL FILE 1, PDF file, 0.6 MB.SUPPLEMENTAL FILE 2, AVI file, 13.5 MB.SUPPLEMENTAL FILE 3, AVI file, 6.0 MB.SUPPLEMENTAL FILE 4, AVI file, 18.8 MB.SUPPLEMENTAL FILE 5, AVI file, 15.8 MB.
ACKNOWLEDGMENTSS. aureus strain 6850 was kindly provided by Bettina Löffler (Institute of Medical
Microbiology, Jena, Germany). S. epidermidis strains 1457, 1457ΔPSM�, Tü3298, andTüF38 were kindly provided by Michael Otto (NIAID NIH, Bethesda, MD). S. epidermidisstrain 1457 expressing GFP was kindly provided by Alexander Horswill (University ofColorado Department of Immunology and Microbiology, Denver, CO). MRC5 cells werekindly provided by Kelli Black (Mayo Clinic Virology/Parasitology Laboratory, Rochester,MN), and MC3T3 cells were kindly provided by Jennifer Westendorf (Mayo ClinicDepartment of Orthopedic Surgery, Rochester, MN). Primary normal human osteoblastswere kindly provided by Paul German Norambuena Morales (Mayo Clinic Departmentof Orthopedic Surgery, Rochester, MN).
This work was supported by NIH grant GM055252, a Ph.D. Training Grant in BasicImmunology (T32 AI07425-21), NIH grant R01 AR056647, and NIH grant R01 AI91594.
K.P. performed the experiments. R.P. supervised K.P. and helped edit and revise themanuscript.
REFERENCES1. Cogen AL, Yamasaki K, Muto J, Sanchez KM, Crotty Alexander L, Tanios
J, Lai Y, Kim JE, Nizet V, Gallo RL. 2010. Staphylococcus epidermidisantimicrobial delta-toxin (phenol-soluble modulin-gamma) cooperateswith host antimicrobial peptides to kill group A Streptococcus. PLoS One5:e8557. https://doi.org/10.1371/journal.pone.0008557.
2. Lai Y, Cogen AL, Radek KA, Park HJ, Macleod DT, Leichtle A, Ryan AF, DiNardo A, Gallo RL. 2010. Activation of TLR2 by a small molecule pro-duced by Staphylococcus epidermidis increases antimicrobial defenseagainst bacterial skin infections. J Invest Dermatol 130:2211–2221.https://doi.org/10.1038/jid.2010.123.
3. Lai Y, Di Nardo A, Nakatsuji T, Leichtle A, Yang Y, Cogen AL, Wu ZR,Hooper LV, Schmidt RR, von Aulock S, Radek KA, Huang CM, Ryan AF,Gallo RL. 2009. Commensal bacteria regulate Toll-like receptor3-dependent inflammation after skin injury. Nat Med 15:1377–1382.https://doi.org/10.1038/nm.2062.
4. Wanke I, Steffen H, Christ C, Krismer B, Gotz F, Peschel A, Schaller M,Schittek B. 2011. Skin commensals amplify the innate immune responseto pathogens by activation of distinct signaling pathways. J InvestDermatol 131:382–390. https://doi.org/10.1038/jid.2010.328.
5. Scharschmidt TC, Vasquez KS, Truong H-A, Gearty SV, Pauli ML, NosbaumA, Gratz IK, Otto M, Moon JJ, Liese J, Abbas AK, Fischbach MA, Rosen-blum MD. 2015. A wave of regulatory T cells into neonatal skin mediatestolerance to commensal microbes. Immunity 43:1011–1021. https://doi.org/10.1016/j.immuni.2015.10.016.
6. Cogen AL, Yamasaki K, Sanchez KM, Dorschner RA, Lai Y, MacLeod DT,Torpey JW, Otto M, Nizet V, Kim JE, Gallo RL. 2010. Selective antimicro-bial action is provided by phenol-soluble modulins derived from Staph-ylococcus epidermidis, a normal resident of the skin. J Invest Dermatol130:192–200. https://doi.org/10.1038/jid.2009.243.
7. Lamagni T. 2014. Epidemiology and burden of prosthetic joint infec-tions. J Antimicrob Chemother 69(Suppl 1):i5–10. https://doi.org/10.1093/jac/dku247.
8. Tande AJ, Osmon DR, Greenwood-Quaintance KE, Mabry TM, HanssenAD, Patel R. 2014. Clinical characteristics and outcomes of prostheticjoint infection caused by small colony variant staphylococci. mBio5:e01910-14. https://doi.org/10.1128/mBio.01910-14.
9. Tande AJ, Patel R. 2014. Prosthetic joint infection. Clin Microbiol Rev27:302–345. https://doi.org/10.1128/CMR.00111-13.
10. Otto M. 2009. Staphylococcus epidermidis—the ‘accidental’ pathogen.Nat Rev Microbiol 7:555–567. https://doi.org/10.1038/nrmicro2182.
11. Vuong C, Otto M. 2002. Staphylococcus epidermidis infections. MicrobInfect 4:481– 489. https://doi.org/10.1016/S1286-4579(02)01563-0.
12. Cheung GY, Joo HS, Chatterjee SS, Otto M. 2014. Phenol-soluble modu-lins— critical determinants of staphylococcal virulence. FEMS MicrobiolRev 38:698 –719. https://doi.org/10.1111/1574-6976.12057.
13. Kurtz SM, Ong KL, Lau E, Bozic KJ. 2014. Impact of the economicdownturn on total joint replacement demand in the United States:updated projections to 2021. J Bone Joint Surg Am 96:624 – 630. https://doi.org/10.2106/JBJS.M.00285.
14. Armbruster NS RJ, Schreiner J, Klenk J, Manina G, Kretschmer D,Poschel S, Schenke-Layland K, Kalbacher H, Clark K, Autnrieth SE.2016. PSM peptides of Staphylococcus aureus activate the p38-CREBpathway in dendritic cells, thereby modulating cytokine productionand T cell priming. J Immunol 196:1284 –1292. https://doi.org/10.4049/jimmunol.1502232.
15. Blattner S, Das S, Paprotka K, Eilers U, Krischke M, Kretschmer D, Rem-mele CW, Dittrich M, Muller T, Schuelein-Voelk C, Hertlein T, Mueller MJ,Huettel B, Reinhardt R, Ohlsen K, Rudel T, Fraunholz MJ. 2016. Staphy-lococcus aureus exploits a non-ribosomal cyclic dipeptide to modulatesurvival within epithelial cells and phagocytes. PLoS Pathog 12:e1005857. https://doi.org/10.1371/journal.ppat.1005857.
16. Rasigade JP, Trouillet-Assant S, Ferry T, Diep BA, Sapin A, Lhoste Y,Ranfaing J, Badiou C, Benito Y, Bes M, Couzon F, Tigaud S, Lina G, EtienneJ, Vandenesch F, Laurent F. 2013. PSMs of hypervirulent Staphylococcusaureus act as intracellular toxins that kill infected osteoblasts. PLoS One8:e63176. https://doi.org/10.1371/journal.pone.0063176.
17. Schreiner J, Kretschmer D, Klenk J, Otto M, Buhring HJ, Stevanovic S,Wang JM, Beer-Hammer S, Peschel A, Autenrieth SE. 2013. Staphylococ-cus aureus phenol-soluble modulin peptides modulate dendritic cellfunctions and increase in vitro priming of regulatory T cells. J Immunol190:3417–3426. https://doi.org/10.4049/jimmunol.1202563.
18. Tranchemontagne ZR, Camire RB, O’Donnell VJ, Baugh J, BurkholderKM. 2016. Staphylococcus aureus strain USA300 perturbs acquisitionof lysosomal enzymes and requires phagosomal acidification forsurvival inside macrophages. Infect Immun 84:241–253. https://doi.org/10.1128/IAI.00704-15.
Perez and Patel Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 14
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

19. Jin T, Bokarewa M, Foster T, Mitchell J, Higgins J, Tarkowski A. 2004.Staphylococcus aureus resists human defensins by production of staphy-lokinase, a novel bacterial evasion mechanism. J Immunol 172:1169 –1176. https://doi.org/10.4049/jimmunol.172.2.1169.
20. Kim HK, Thammavongsa V, Schneewind O, Missiakas D. 2012. Recurrentinfections and immune evasion strategies of Staphylococcus aureus. CurrOpin Microbiol 15:92–99. https://doi.org/10.1016/j.mib.2011.10.012.
21. Koch TK, Reuter M, Barthel D, Bohm S, van den Elsen J, Kraiczy P, ZipfelPF, Skerka C. 2012. Staphylococcus aureus proteins Sbi and Efb recruithuman plasmin to degrade complement C3 and C3b. PLoS One7:e47638. https://doi.org/10.1371/journal.pone.0047638.
22. Laarman AJ, Ruyken M, Malone CL, van Strijp JA, Horswill AR, RooijakkersSH. 2011. Staphylococcus aureus metalloprotease aureolysin cleavescomplement C3 to mediate immune evasion. J Immunol 186:6445– 6453. https://doi.org/10.4049/jimmunol.1002948.
23. McGuinness WA, Kobayashi SD, DeLeo FR. 2016. Evasion of neutrophilkilling by Staphylococcus aureus. Pathogens 5:32. https://doi.org/10.3390/pathogens5010032.
24. Vuong C, Kocianova S, Yao Y, Carmody AB, Otto M. 2004. Increasedcolonization of indwelling medical devices by quorum-sensing mutantsof Staphylococcus epidermidis in vivo. J Infect Dis 190:1498 –1505. https://doi.org/10.1086/424487.
25. Yao Y, Sturdevant DE, Otto M. 2005. Genomewide analysis of geneexpression in Staphylococcus epidermidis biofilms: insights into thepathophysiology of S. epidermidis biofilms and the role of phenol-soluble modulins in formation of biofilms. J Infect Dis 191:289 –298.https://doi.org/10.1086/426945.
26. Kristian SA, Birkenstock TA, Sauder U, Mack D, Gotz F, Landmann R. 2008.Biofilm formation induces C3a release and protects Staphylococcus epi-dermidis from IgG and complement deposition and from neutrophil-dependent killing. J Infect Dis 197:1028 –1035. https://doi.org/10.1086/528992.
27. Vuong C, Voyich JM, Fischer ER, Braughton KR, Whitney AR, DeLeo FR,Otto M. 2004. Polysaccharide intercellular adhesin (PIA) protects Staph-ylococcus epidermidis against major components of the human innateimmune system. Cell Microbiol 6:269 –275. https://doi.org/10.1046/j.1462-5822.2004.00367.x.
28. Rohde H, Burdelski C, Bartscht K, Hussain M, Buck F, Horstkotte MA,Knobloch JK, Heilmann C, Herrmann M, Mack D. 2005. Induction ofStaphylococcus epidermidis biofilm formation via proteolytic processingof the accumulation-associated protein by staphylococcal and hostproteases. Mol Microbiol 55:1883–1895. https://doi.org/10.1111/j.1365-2958.2005.04515.x.
29. Christner M, Franke GC, Schommer NN, Wendt U, Wegert K, Pehle P, KrollG, Schulze C, Buck F, Mack D, Aepfelbacher M, Rohde H. 2010. The giantextracellular matrix-binding protein of Staphylococcus epidermidis medi-ates biofilm accumulation and attachment to fibronectin. Mol Microbiol75:187–207. https://doi.org/10.1111/j.1365-2958.2009.06981.x.
30. Schommer NN, Christner M, Hentschke M, Ruckdeschel K, AepfelbacherM, Rohde H. 2011. Staphylococcus epidermidis uses distinct mechanismsof biofilm formation to interfere with phagocytosis and activation ofmouse macrophage-like cells 774A.1. Infect Immun 79:2267–2276.https://doi.org/10.1128/IAI.01142-10.
31. Cheung GY, Rigby K, Wang R, Queck SY, Braughton KR, Whitney AR,Teintze M, DeLeo FR, Otto M. 2010. Staphylococcus epidermidis strategiesto avoid killing by human neutrophils. PLoS Pathog 6:e1001133. https://doi.org/10.1371/journal.ppat.1001133.
32. Yao Y, Vuong C, Kocianova S, Villaruz AE, Lai Y, Sturdevant DE, Otto M.2006. Characterization of the Staphylococcus epidermidis accessory-generegulator response: quorum-sensing regulation of resistance to humaninnate host defense. J Infect Dis 193:841– 848. https://doi.org/10.1086/500246.
33. Otto M. 2014. Phenol-soluble modulins. Int J Med Microbiol 304:164 –169. https://doi.org/10.1016/j.ijmm.2013.11.019.
34. Laabei M, Jamieson WD, Yang Y, van den Elsen J, Jenkins AT. 2014.Investigating the lytic activity and structural properties of Staphylococ-cus aureus phenol soluble modulin (PSM) peptide toxins. Biochim Bio-phys Acta 1838:3153–3161. https://doi.org/10.1016/j.bbamem.2014.08.026.
35. Wang R, Braughton KR, Kretschmer D, Bach TH, Queck SY, Li M, KennedyAD, Dorward DW, Klebanoff SJ, Peschel A, DeLeo FR, Otto M. 2007.Identification of novel cytolytic peptides as key virulence determinantsfor community-associated MRSA. Nat Med 13:1510 –1514. https://doi.org/10.1038/nm1656.
36. Fowler T, Wann ER, Joh D, Johansson S, Foster TJ, Hook M. 2000.Cellular invasion by Staphylococcus aureus involves a fibronectinbridge between the bacterial fibronectin-binding MSCRAMMs andhost cell beta1 integrins. Eur J Cell Biol 79:672– 679. https://doi.org/10.1078/0171-9335-00104.
37. Jevon M, Guo C, Ma B, Mordan N, Nair SP, Harris M, Henderson B, BentleyG, Meghji S. 1999. Mechanisms of internalization of Staphylococcusaureus by cultured human osteoblasts. Infect Immun 67:2677–2681.
38. Dziewanowska K, Patti JM, Deobald CF, Bayles KW, Trumble WR, BohachGA. 1999. Fibronectin binding protein and host cell tyrosine kinase arerequired for internalization of Staphylococcus aureus by epithelial cells.Infect Immun 67:4673– 4678.
39. Kalinka J, Hachmeister M, Geraci J, Sordelli D, Hansen U, Niemann S,Oetermann S, Peters G, Loffler B, Tuchscherr L. 2014. Staphylococcusaureus isolates from chronic osteomyelitis are characterized by high hostcell invasion and intracellular adaptation, but still induce inflammation.Int J Med Microbiol 304:1038 –1049. https://doi.org/10.1016/j.ijmm.2014.07.013.
40. Lâm T-T, Giese B, Chikkaballi D, Kühn A, Wolber W, Pané-Farré J, SchäferD, Engelmann S, Fraunholz M, Sinha B. 2010. Phagolysosomal integrity isgenerally maintained after Staphylococcus aureus invasion of nonprofes-sional phagocytes but is modulated by strain 6850. Infect Immun 78:3392–3403. https://doi.org/10.1128/IAI.00012-10.
41. Mohamed W, Sommer U, Sethi S, Domann E, Thormann U, Schutz I, LipsKS, Chakraborty T, Schnettler R, Alt V. 2014. Intracellular proliferation ofS. aureus in osteoblasts and effects of rifampicin and gentamicin on S.aureus intracellular proliferation and survival. Eur Cell Mater 28:258 –268.https://doi.org/10.22203/eCM.v028a18.
42. Hamill RJ, Vann JM, Proctor RA. 1986. Phagocytosis of Staphylococcusaureus by cultured bovine aortic endothelial cells: model for postadher-ence events in endovascular infections. Infect Immun 54:833– 836.
43. Clement S, Vaudaux P, Francois P, Schrenzel J, Huggler E, Kampf S,Chaponnier C, Lew D, Lacroix JS. 2005. Evidence of an intracellularreservoir in the nasal mucosa of patients with recurrent Staphylococ-cus aureus rhinosinusitis. J Infect Dis 192:1023–1028. https://doi.org/10.1086/432735.
44. Bayles KW, Wesson CA, Liou LE, Fox LK, Bohach GA, Trumble WR. 1998.Intracellular Staphylococcus aureus escapes the endosome and inducesapoptosis in epithelial cells. Infect Immun 66:336 –342.
45. Almeida RA, Matthews KR, Cifrian E, Guidry AJ, Oliver SP. 1996. Staphy-lococcus aureus invasion of bovine mammary epithelial cells. J Dairy Sci79:1021–1026. https://doi.org/10.3168/jds.S0022-0302(96)76454-8.
46. Hebert A, Sayasith K, Senechal S, Dubreuil P, Lagace J. 2000. Dem-onstration of intracellular Staphylococcus aureus in bovine mastitisalveolar cells and macrophages isolated from naturally infected cowmilk. FEMS Microbiol Lett 193:57– 62. https://doi.org/10.1111/j.1574-6968.2000.tb09402.x.
47. Ellington JK, Harris M, Webb L, Smith B, Smith T, Tan K, Hudson M. 2003.Intracellular Staphylococcus aureus. A mechanism for the indolence ofosteomyelitis. J Bone Joint Surg Br 85:918 –921. https://doi.org/10.1302/0301-620X.85B6.13509.
48. Khalil H, Williams RJ, Stenbeck G, Henderson B, Meghji S, Nair SP. 2007.Invasion of bone cells by Staphylococcus epidermidis. Microb Infect9:460 – 465. https://doi.org/10.1016/j.micinf.2007.01.002.
49. Perez K, Patel R. 2017. Staphylococcus epidermidis small-colony variantsare induced by low pH and their frequency reduced by lysosomalalkalinization. J Infect Dis 215:488 – 490. https://doi.org/10.1093/infdis/jiw503.
50. Resch A, Rosenstein R, Nerz C, Gotz F. 2005. Differential gene expressionprofiling of Staphylococcus aureus cultivated under biofilm and plank-tonic conditions. Appl Environ Microbiol 71:2663–2676. https://doi.org/10.1128/AEM.71.5.2663-2676.2005.
51. Bui LM, Turnidge JD, Kidd SP. 2015. The induction of Staphylococcusaureus biofilm formation or small colony variants is a strain-specificresponse to host-generated chemical stresses. Microbes Infect 17:77– 82.https://doi.org/10.1016/j.micinf.2014.09.009.
52. Kahl BC. 2014. Small colony variants (SCVs) of Staphylococcus aureus—abacterial survival strategy. Infect Genet Evol 21:515–522. https://doi.org/10.1016/j.meegid.2013.05.016.
53. Melter O, Radojevic B. 2010. Small colony variants of Staphylococcusaureus—review. Folia Microbiol (Praha) 55:548 –558. https://doi.org/10.1007/s12223-010-0089-3.
54. Johns BE, Purdy KJ, Tucker NP, Maddocks SE. 2015. Phenotypic andgenotypic characteristics of small colony variants and their role in
S. epidermidis Survival in Fibroblasts and Osteoblasts Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 15
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from

chronic infection. Microbiol Insights 8:15–23. https://doi.org/10.4137/MBI.S25800.
55. Proctor RA, von Eiff C, Kahl BC, Becker K, McNamara P, Herrmann M,Peters G. 2006. Small colony variants: a pathogenic form of bacteria thatfacilitates persistent and recurrent infections. Nat Rev Microbiol4:295–305. https://doi.org/10.1038/nrmicro1384.
56. Sendi P, Proctor RA. 2009. Staphylococcus aureus as an intracellularpathogen: the role of small colony variants. Trends Microbiol 17:54 –58.https://doi.org/10.1016/j.tim.2008.11.004.
57. Tuchscherr L, Kreis CA, Hoerr V, Flint L, Hachmeister M, Geraci J, Bremer-Streck S, Kiehntopf M, Medina E, Kribus M, Raschke M, Pletz M, Peters G,Loffler B. 2016. Staphylococcus aureus develops increased resistance toantibiotics by forming dynamic small colony variants during chronicosteomyelitis. J Antimicrob Chemother 71:438 – 448. https://doi.org/10.1093/jac/dkv371.
58. von Eiff C, Peters G, Becker K. 2006. The small colony variant (SCV)concept—the role of staphylococcal SCVs in persistent infections. Injury37(Suppl 2):S26 –33. https://doi.org/10.1016/j.injury.2006.04.006.
59. Bogut A, Niedzwiadek J, Koziol-Montewka M, Strzelec-Nowak D, BlachaJ, Mazurkiewicz T, Marczynski W, Plewik D. 2014. Characterization ofStaphylococcus epidermidis and Staphyloccocus warneri small-colonyvariants associated with prosthetic-joint infections. J Med Microbiol63:176 –185. https://doi.org/10.1099/jmm.0.066068-0.
60. Sander G, Borner T, Kriegeskorte A, von Eiff C, Becker K, Mahabir E. 2012.Catheter colonization and abscess formation due to Staphylococcusepidermidis with normal and small-colony-variant phenotype is mousestrain dependent. PLoS One 7:e36602. https://doi.org/10.1371/journal.pone.0036602.
61. von Eiff C, Proctor RA, Peters G. 2000. Small colony variants ofstaphylococci: a link to persistent infections. Berl Munch TierarztlWochenschr 113:321–325.
62. Baddour LM, Barker LP, Christensen GD, Parisi JT, Simpson WA. 1990.Phenotypic variation of Staphylococcus epidermidis in infection of trans-venous endocardial pacemaker electrodes. J Clin Microbiol 28:676 – 679.
63. Baddour LM, Simpson WA, Weems JJ, Jr, Hill MM, Christensen GD. 1988.Phenotypic selection of small-colony variant forms of Staphylococcusepidermidis in the rat model of endocarditis. J Infect Dis 157:757–763.https://doi.org/10.1093/infdis/157.4.757.
64. Adler H, Widmer A, Frei R. 2003. Emergence of a teicoplanin-resistantsmall colony variant of Staphylococcus epidermidis during vancomycintherapy. Eur J Clin Microbiol Infect Dis 22:746 –748. https://doi.org/10.1007/s10096-003-1029-9.
65. Ou JJJ, Drilling AJ, Cooksley C, Bassiouni A, Kidd SP, Psaltis AJ, WormaldPJ, Vreugde S. 2016. Reduced innate immune response to a Staphylo-coccus aureus small colony variant compared to its wild-type parentstrain. Front Cell Infect Microbiol 6:187. https://doi.org/10.3389/fcimb.2016.00187.
66. Grosz M, Kolter J, Paprotka K, Winkler AC, Schäfer D, Chatterjee SS,Geiger T, Wolz C, Ohlsen K, Otto M, Rudel T, Sinha B, Fraunholz M. 2014.Cytoplasmic replication of Staphylococcus aureus upon phagosomal es-cape triggered by phenol-soluble modulin �. Cell Microbiol 16:451– 465.https://doi.org/10.1111/cmi.12233.
67. Giese B, Glowinski F, Paprotka K, Dittmann S, Steiner T, Sinha B, Fraun-holz MJ. 2011. Expression of delta-toxin by Staphylococcus aureus me-diates escape from phago-endosomes of human epithelial and endo-thelial cells in the presence of beta-toxin. Cell Microbiol 13:316 –329.https://doi.org/10.1111/j.1462-5822.2010.01538.x.
68. Jarry TM, Cheung AL. 2006. Staphylococcus aureus escapes more effi-ciently from the phagosome of a cystic fibrosis bronchial epithelial cellline than from its normal counterpart. Infect Immun 74:2568 –2577.https://doi.org/10.1128/IAI.74.5.2568-2577.2006.
69. Porte F, Liautard J-P, Köhler S. 1999. Early acidification of phagosomescontaining Brucella suis is essential for intracellular survival in murinemacrophages. Infect Immun 67:4041– 4047.
70. Park KH, Greenwood-Quaintance KE, Mandrekar J, Patel R. 2016. Activityof tedizolid in methicillin-resistant Staphylococcus aureus experimentalforeign body-associated osteomyelitis. Antimicrob Agents Chemother60:6568 – 6572. https://doi.org/10.1128/AAC.01248-16.
71. Del Pozo JL, Rouse MS, Euba G, Kang CI, Mandrekar JN, Steckelberg JM,Patel R. 2009. The electricidal effect is active in an experimental modelof Staphylococcus epidermidis chronic foreign body osteomyelitis. Anti-microb Agents Chemother 53:4064 – 4068. https://doi.org/10.1128/AAC.00432-09.
72. Kwakman PH, te Velde AA, Vandenbroucke-Grauls CM, van Deventer SJ,Zaat SA. 2006. Treatment and prevention of Staphylococcus epidermidisexperimental biomaterial-associated infection by bactericidal peptide 2.Antimicrob Agents Chemother 50:3977–3983. https://doi.org/10.1128/AAC.00575-06.
73. Strobel M, Pförtner H, Tuchscherr L, Völker U, Schmidt F, Kramko N,Schnittler HJ, Fraunholz MJ, Löffler B, Peters G, Niemann S. 2016. Post-invasion events after infection with Staphylococcus aureus are stronglydependent on both the host cell type and the infecting S. aureus strain.Clin Microbiol Infect 22:799 – 809. https://doi.org/10.1016/j.cmi.2016.06.020.
74. Phillips PL, Schultz GS. 2012. Molecular mechanisms of biofilm infection:biofilm virulence factors. Adv Wound Care (New Rochelle) 1:109 –114.https://doi.org/10.1089/wound.2011.0301.
75. Garcia-Betancur JC, Goni-Moreno A, Horger T, Schott M, Sharan M,Eikmeier J, Wohlmuth B, Zernecke A, Ohlsen K, Kuttler C, Lopez D.2017. Cell differentiation defines acute and chronic infection celltypes in Staphylococcus aureus. Elife 6:e28023. https://doi.org/10.7554/eLife.28023.
76. Magrys A, Bogut A, Kiełbus M, Olender A. 2018. The role of the PI3K/mTOR signaling pathway in Staphylococcus epidermidis small colonyvariants intracellular survival. Immunol Invest 47:251–263. https://doi.org/10.1080/08820139.20181423569:1-13.
77. Rabinovitch M. 1995. Professional and non-professional phagocytes: anintroduction. Trends Cell Biol 5:85– 87. https://doi.org/10.1016/S0962-8924(00)88955-2.
78. Cerri PS. 2005. Osteoblasts engulf apoptotic bodies during alveolar boneformation in the rat maxilla. Anat Rec A Discov Mol Cell Evol Biol286:833– 840. https://doi.org/10.1002/ar.a.20220.
79. Arlein WJ, Shearer JD, Caldwell MD. 1998. Continuity between woundmacrophage and fibroblast phenotype: analysis of wound fibroblastphagocytosis. Am J Physiol Regul Integr Comp Physiol 275:R1041– 8.https://doi.org/10.1152/ajpregu.1998.275.4.R1041.
80. Rollin G, Tan X, Tros F, Dupuis M, Nassif X, Charbit A, Coureuil M. 2017.Intracellular survival of Staphylococcus aureus in endothelial ells: a mat-ter of growth or persistence. Front Microbiol 8:1354. https://doi.org/10.3389/fmicb.2017.01354.
81. Maduka-Ezeh AN, Greenwood-Quaintance KE, Karau MJ, Berbari EF,Osmon DR, Hanssen AD, Steckelberg JM, Patel R. 2012. Antimicrobialsusceptibility and biofilm formation of Staphylococcus epidermidis smallcolony variants associated with prosthetic joint infection. Diagn Micro-biol Infect Dis 74:224 –229. https://doi.org/10.1016/j.diagmicrobio.2012.06.029.
82. Uehara Y, Sasaki T, Hiramatsu K, Tande A, Greenwood-Quaintance K,Perez K, Patel R. 2016. Identification of mutations in Staphylococcusepidermidis small-colony variants associated with prosthetic joint infec-tion by direct whole genome sequencing from colonies. Open ForumInfect Dis 3:2215–2215. https://doi.org/10.1093/ofid/ofw172.1763.
83. Manders EMM, Verbeek FJ, Aten JA. 1993. Measurement of co-localization of objects in dual-colour confocal images. J Microsc 169:375–382. https://doi.org/10.1111/j.1365-2818.1993.tb03313.x.
84. Levitz SM, Nong SH, Seetoo KF, Harrison TS, Speizer RA, Simons ER. 1999.Cryptococcus neoformans resides in an acidic phagolysosome of humanmacrophages. Infect Immun 67:885– 890.
Perez and Patel Infection and Immunity
October 2018 Volume 86 Issue 10 e00237-18 iai.asm.org 16
on July 2, 2020 by guesthttp://iai.asm
.org/D
ownloaded from





























