Embed Size (px)
Citation preview

JOURNAL OF BACTERIOLOGY, June 1974, p. 1059-1066Copyright 0 1974 American Societv for Microbiology
Vol. 118. No. :3Printed in U.S.A.
Bacterial Bioluminescence In Vivo: Control and Synthesis ofAldehyde Factor in Temperature-Conditional Luminescence
MutantsTHOMAS W. CLINE' AND J. W. HASTINGS
The Biological Laboratories, Harvard University, Cambridge, Massachusetts 02138
Received for publication 25 February 1974
Bioluminescent marine bacteria possess luciferase, which catalyzes the oxida-tion of reduced flavin mononucleotide and long-chain aldehyde to produce light.Temperature-sensitive mutants of these bacteria can be obtained which requireexogenous aldehyde for light production at higher temperatures. In Beneckeaharveyi. two classes of such mutants were found which differed with regard totheir response to temperature shifts. In one class, a shift from permissive tononpermissive temperature in liquid cultures resulted in a rapid (t,, a 3 min)loss of luminescence. In the other, there was no immediate decline in lumines-cence; it was the increase of luminescence that was blocked. Through studies ofthese and other effects of temperature shifts on the in vivo luminescence of thesemutants, we conclude that at least two genes are specifically involved in the invivo biosynthesis of aldehyde for the luminescence reaction and that both genes
are coordinately controlled with that for luciferase.
The in vitro bacterial bioluminescence reac-tion involves the oxidation of reduced flavinmononucleotide (FMNH2) and a long-chainaliphatic aldehyde by molecular oxygen, withthe concomitant emission of light (8, 9). Rogersand McElroy (17) first reported a class of darkmutants which can be stimulated to emit lightsimply by exposing the living cells to aldehydevapor. These "aldehyde" mutants are presumedto be somehow blocked in aldehyde synthesis oractivity. But in fact, the identity of the alde-hyde, or indeed its very existence in vivo, is notyet established.More recently we reported mutants of this
class which are conditional upon temperature(2). These "temperature-sensitive aldehyde-stimulable" (TSAS) mutants can be as brightas the wild type below 26 to 30 C, but severalorders of magnitude darker than the wild typeat the nonpermissive temperature, 36 C. At 36C, the cells may nevertheless synthesize wild-type levels of luciferase and will immediatelyluminesce in vivo at wild-type levels upon theaddition of decanal to the culture medium.Although these mutant cells were very dark
after growth at 36 C, the luminescence of aculture grown at 25 C was only very slowly lostafter warming to the higher, nonpermissive
I Present address: Center for Pathobiology, University ofCalifornia at Irvine, Irvine, Calif. 92664.
temperature. More recently, mutants of a dis-tinctly different type were found. In these, thelight emission is extinguished rapidly aftershifting from 26 C to 36 C. The experimentsdescribed here provide an analysis and compari-son of these two types of mutants.Aldehyde has now been shown to be a cosub-
strate in the in vitro luminescence reaction,with the corresponding acid as the product (5,12, 19). On the assumption that the in vivoaldehyde factor is itself not temperature sensi-tive nor subject to mutation which would makeit so, the most likely explanation for the alde-hyde mutants is that they fail at the nonpermis-sive temperature to synthesize aldehyde or afactor analogous to it. Although we cannot asyet map the genes involved in aldehyde-factorsynthesis nor identify the products of thesegenes, we have concluded that there are at leasttwo genes specifically involved with the synthe-sis of aldehyde factor for the in vivo biolumines-cence reaction, the expression of which appearsto be jointly controlled with that of the gene forluciferase.
MATERIALS AND METHODSThe mutants reported in this work were obtained
by mutagenesis of Beneckea harveyi (16) (previouslyreferred to [9] as strain MAV) with N-methyl-N'-nitro-N-nitrosoguanidine, according to the procedureoutlined by Cline and Hastings (3). Temperature-sen-
1059
on May 17, 2020 by guest
http://jb.asm.org/
Dow
nloaded from

CLINE AND HASTINGS
sitive luminescence mutants were identified as strainswhose colonies were bright at 22 C but dark at 36 C.Among these, strains were designated as TSAS whosecolonies grown and kept at 36 C luminesced brightlyonly when exposed to decanal vapors.
Another screen involved growth at the permissivetemperature followed by a shift to 36 C. Most TSASmutants in which the luminescent system was devel-oped at 22 C continued to luminesce brightly upon
shifting to 36 C. With 159 independently isolatedTSAS mutants, colonies grown at 22 C continued toluminesce brightly without decanal when observed 90min after shifting the temperature to 36 C. TSAS-1 isa mutant of this type. However, two TSAS mutantswere exceptions to this rule; colonies of these two were
extremely dim when observed, even shortly after theshift-up. TSAS-F1 is a mutant of this latter type.
Preliminary classification of the 161 TSAS mutantswas, as indicated above, based on their behavior on
solid medium; however, since luminescence behaviorfor the wild type as well as a number of mutant classesis not always strictly comparable on solid and liquidmedia, caution must be exercised in extrapolation ofresults from solid to liquid media. Of the TSASmutants, only TSAS-1 and TSAS-F1 have beenstudied extensively both on solid and in liquid media.For these two mutants, however, the behavior isqualitatively similar under these two culture condi-tions.
Cells were grown by using a complete mediumcontaining, per liter, tryptone (5 g; Difco), yeastextract (3 g; Difco), glycerol (2 ml), distilled water(200 ml), and sea water (800 ml). Solid mediumcontained in addition 14 g of agar (Difco). Cultureswere grown with 75 ml (initial volume) of medium in250-ml wide-mouth, fluted shake flasks (Bellco) andaerated by vigorous shaking in temperature-con-trolled shaking water baths (New Brunswick Aqua-therm). Cultures were inoculated with no more than0.2 ml of an overnight culture grown at 36 C.
Optical density (OD) of liquid cultures was deter-mined at 660 nm in the Coleman Jr. spectrophotome-ter in which one OD unit corresponded to approxi-mately 5 x 108 cells per ml at 32 C. Luminescence was
measured with a photomultiplier-photometer (13).Measurements on the cells during growth were madeon 1-ml samples removed from the flasks at the timesindicated, determined at 20 C within 15 s after beingwithdrawn. Where indicated, 200 gliters of an alde-hyde solution was added by injection from a syringe tothe sample during the actual light measurement. Thesolution was prepared by sonication of 10 gliters ofdecanal in 10 ml of distilled water, subsequentlydiluted 1:2 in fresh growth medium. The sampleswere discarded after use. Additional details are pro-
vided in the footnotes and figure legends.
RESULTS
Table 1 and Fig. 1 illustrate the behavior ofthe two types of TSAS mutants in liquid media.The two mutants were quite similar when grown
entirely at 26 or 36 C (Table 1), except thatTSAS-F1 was a bit more decanal stimulable atthe permissive temperature was than TSAS-1,and TSAS-1 was a bit darker in the absence ofdecanal at 36 C than was TSAS-F1.The important difference between the two
mutants was in their response to a temperatureshift-up (Fig. 1). The luminescence of TSAS-1increased a little after the shift-up and after 40min began to slowly decline; however, theluminescence in the presence of decanal in-creased after the shift in a fashion very similarto the luminescence increase for the wild typewithout decanal. The slow decline of TSAS-1luminescence occurred at a rate similar to thedecline in wild-type luminescence, which oc-
curred at a point considerably later on in growth(not shown), after peak luminescence had beenreached. In contrast, although the decanal-stimulable level of luminescence for theTSAS-F1 culture increased after the shift likethat of TSAS-1, TSAS-F1 luminescence in theabsence of decanal plummeted immediately
TABLE 1. Luminescence and extractable luciferase activity from TSAS mutants at the permissive andnonpermissive temperatures
Cells cultured at 26 C Cells cultured at 36 CDetermination
Wild type TSAS-1 TSAS-F1 Wild type TSAS-1 TSAS-F1
In vivo luminescence aNo aldehyde 3.0 2.2 1.9 1.4 0.0001 0.0009With aldehyde NS 2.4 2.7 NS 1.0 1.7
Extractable luciferase activityb 5.2 5.2 4.9 4.3 3.6 3.6
aExpressed as quanta per second per milliliter of culture x 10- 2. Measurements were made at peakluminescence of culture. Luminescence with decanal was measured as peak light intensity upon the addition of0.2 ml of aldehyde suspension to a 1-ml sample of culture. NS, No stimulation by decanal.
'Expressed as in a; 35 ml were harvested by centrifugation when culture reached OD 5.0 + 0.2 (this is thepoint of peak luminescence at 26 C, 15 to 50 min after peak at 36 C, depending on strain). Cell pellets werefrozen and then resuspended in 4 ml of distilled water at 4 C, lysed by sonication, and centrifuged to remove celldebris. Lysates were assayed for luciferase activity by the procedure in Hastings et al. (10) at 20 C, pH 7, withdecanal.
1060 J. BACTERIOL.
on May 17, 2020 by guest
http://jb.asm.org/
Dow
nloaded from
BACTERIAL BIOLUMINESCENCE
_ I
/0
W.T.
I I L I
I,'
,0 \
TSAS-I
I,vLo,o o I:
l TSAS-FI
WO-l I-0 50 100 0 50 100 0 50 100
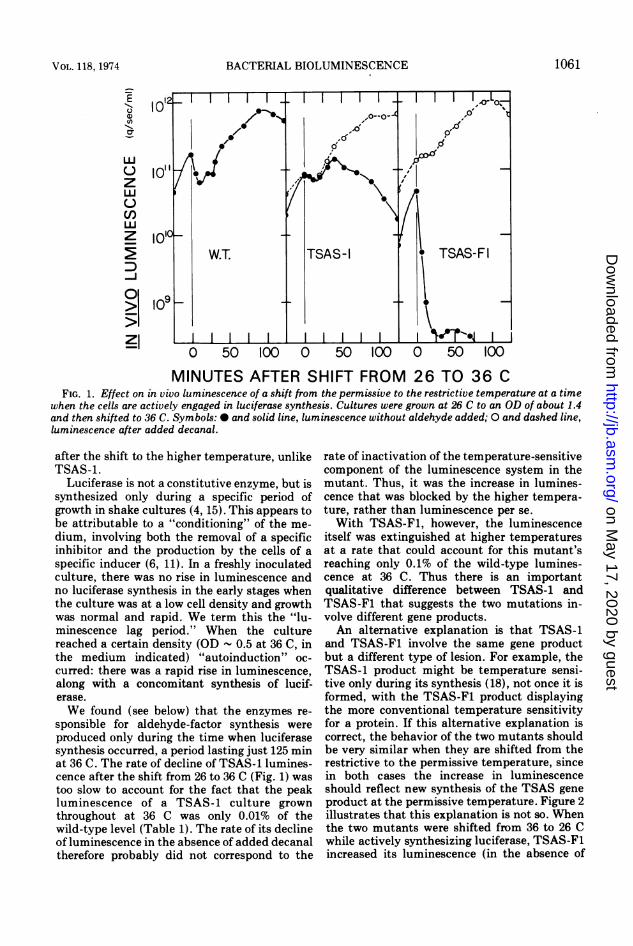
MINUTES AFTER SHIFT FROM 26 TO 36 CFIG. 1. Effect on in vivo luminescence of a shift from the permissive to the restrictive temperature at a time
when the cells are actively engaged in luciferase synthesis. Cultures were grown at 26 C to an OD of about 1.4and then shifted to 36 C. Symbols: 0 and solid line, luminescence without aldehyde added; 0 and dashed line,luminescence after added decanal.
after the shift to the higher temperature, unlikeTSAS-1.
Luciferase is not a constitutive enzyme, but issynthesized only during a specific period ofgrowth in shake cultures (4, 15). This appears tobe attributable to a "conditioning" of the me-
dium, involving both the removal of a specificinhibitor and the production by the cells of a
specific inducer (6, 11). In a freshly inoculatedculture, there was no rise in luminescence andno luciferase synthesis in the early stages whenthe culture was at a low cell density and growthwas normal and rapid. We term this the "lu-minescence lag period." When the culturereached a certain density (OD - 0.5 at 36 C, inthe medium indicated) "autoinduction" oc-
curred: there was a rapid rise in luminescence,along with a concomitant synthesis of lucif-erase.
We found (see below) that the enzymes re-sponsible for aldehyde-factor synthesis wereproduced only during the time when luciferasesynthesis occurred, a period lasting just 125 minat 36 C. The rate of decline of TSAS-1 lumines-cence after the shift from 26 to 36 C (Fig. 1) wastoo slow to account for the fact that the peakluminescence of a TSAS-1 culture grownthroughout at 36 C was only 0.01% of thewild-type level (Table 1). The rate of its declineof luminescence in the absence of added decanaltherefore probably did not correspond to the
rate of inactivation of the temperature-sensitivecomponent of the luminescence system in themutant. Thus, it was the increase in lumines-cence that was blocked by the higher tempera-ture, rather than luminescence per se.With TSAS-F1, however, the luminescence
itself was extinguished at higher temperaturesat a rate that could account for this mutant'sreaching only 0.1% of the wild-type lumines-cence at 36 C. Thus there is an importantqualitative difference between TSAS-1 andTSAS-F1 that suggests the two mutations in-volve different gene products.An alternative explanation is that TSAS-1
and TSAS-F1 involve the same gene productbut a different type of lesion. For example, theTSAS-1 product might be temperature sensi-tive only during its synthesis (18), not once it isformed, with the TSAS-F1 product displayingthe more conventional temperature sensitivityfor a protein. If this alternative explanation iscorrect, the behavior of the two mutants shouldbe very similar when they are shifted from therestrictive to the permissive temperature, sincein both cases the increase in luminescenceshould reflect new synthesis of the TSAS geneproduct at the permissive temperature. Figure 2illustrates that this explanation is not so. Whenthe two mutants were shifted from 36 to 26 Cwhile actively synthesizing luciferase, TSAS-F1increased its luminescence (in the absence of
EI 2
0 1(I'
-7
10"lwLLJ0z
cJ)z
D-j
0
ZI
1061VOL. 118, 1974
pId'd
i/J./
log9
on May 17, 2020 by guest
http://jb.asm.org/
Dow
nloaded from
background level of luminescence in the ab-I E . sence of added aldehyde can be reduced by
three or four orders of magnitude below the levelfor the wild type simply by culturing the inocu-
TSAS- F lum at 36 C. Consequently, even very low rates0s/ of synthesis of the aldehyde-factor enzymes
.* _ could be detected. When medium at 26 C was*/ / inoculated with TSAS cells grown at 36 C, there
A/ was no detectable increase in luminescence at26 C until the onset of luciferase synthesis,confirming that the aldehyde-factor-synthesiz-
* TSAS- A ing enzymes (or at least those corresponding tothe TSAS-1 or TSAS-F1 gene products) are in-deed not produced before initiation of luciferasesynthesis.
A. *_AThe luminescence during the initial lag phase
A / presumably reflects in part the aldehyde-factor+ ^ I Ar *A synthetic machinery synthesized before inocu-
0 5 10 15 lation. Figure 3 illustrates the TSAS mutants'behavior when they were allowed to produce
MINUTES AFTER SHIFT functional enzymes at 26 C but were thendiluted into fresh medium and shifted to 36 C
FROM 36 TO 23.5 C while still at very low culture densities. Sincevivo luminescence of TSAS cultures neither the mutants nor the wild type would be
thesizing luciferase after a shift from 36 to synthesizing additional aldehyde-factor en-ures were shifted down after growth at 36 zymes before autoinduction, stability of the2.3. Luminescence values are with no pre-existing mutant and wild-type lumines-Ided. cence machinery could be compared during
this period.ibstantially faster than TSAS-1. Five Although TSAS-1 developed aldehydeter the shift, the two mutants showed stimulability somewhat sooner than the wilddifference in their rates of lumines- type, perhaps reflecting a lower reserve of alde-ease. The possibility that the rapid hyde-factor-synthesizing activity even at 26 C,ease in TSAS-F1 luminescence is due the initial rate of decline of luminescence whereal of the temperature inactivation of decanal stimulability developed was nearlyhich accumulated before the shift is identical to the decline in the wild type. The)y the fact that chloramphenicol ef- difference between the two curves occurredblocked the luminescence increase when the wild type resumed synthesis of lucif-hift (95% or more when added 10 min erase and aldehyde-factor-synthesizing en-shift at a concentration of 100 ug/ml zymes.for both TSAS-1 and TSAS-F1 (not The behavior of TSAS-F1 during this period
contrasted dramatically with both the wild typeet al. (15) found that decanal can and TSAS-1. Luminescence activity was lostthe luminescence of the wild type in rapidly from the moment the cells were shiftedt a low cell density during growth to 36 C. Figure 3 confirms the conclusion frominitiation of luciferase synthesis, and Fig. 1 that it was only the increase in the rate ofthat aldehyde-factor synthesis is aldehyde-factor synthesis that was condition-
ntrolled with luciferase synthesis. ally blocked in TSAS-1, whereas with TSAS-F1this conclusion was somewhat inse- it was the capacity to synthesize aldehydeuse of the fact that the degree of factor per se that was temperature sensitive.;imulability of the wild type which This indicates that the products of the TSAS-1luring this period depends (among and TSAS-F1 genetic loci are different.gs) upon the history of the inoculum; We have already presented evidence that theere are some conditions under which synthesis of the TSAS-1 and TSAS-F1 genedo not become appreciably decanal products are jointly controlled with the synthe-The TSAS mutants allow a much sis of luciferase during the luminescence lag
itive test for the production of alde- period. The experiment illustrated in Fig. 4tr-synthesizing enzymes, since the shows that these two putative aldehyde-factor-
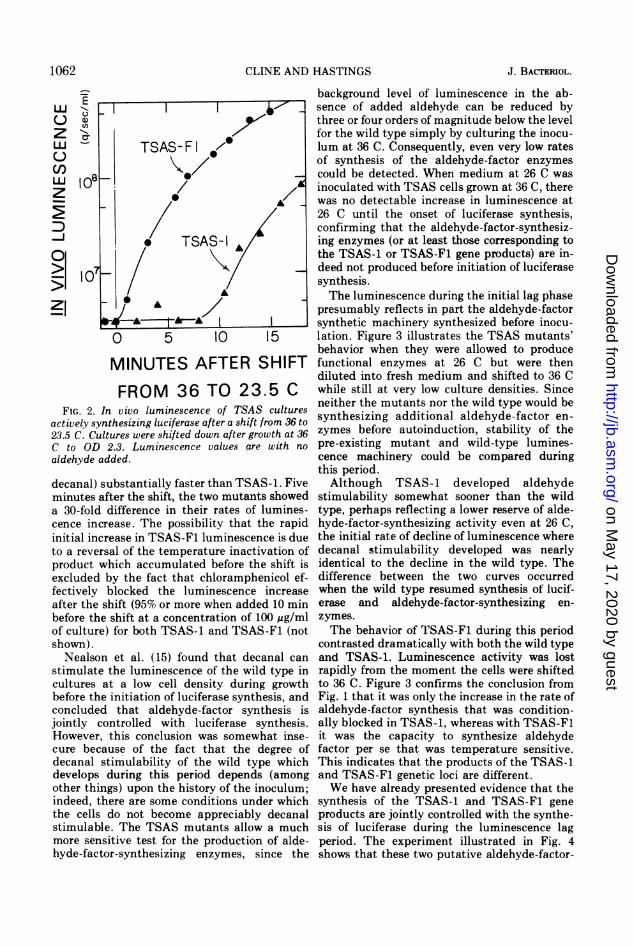
FIG. 2. Inactively synt23.5 C. CultC to ODaldehyde ad
decanal) s.minutes afia 30-fold ccence incr4initial incrito a revers
product wi
excluded bfectively I
after the si
before theof culture)shown).
Nealsonstimulate 1
cultures albefore theconcludedjointly co]However, Icure becaidecanal stdevelops cother thingindeed, ththe cellsstimulablemore sensihyde-facto:
E
1-
Lu
zlLU')ulz
D-j
0
5i
1062 CLINE AND HASTINGS J. BACTERIOL.
on May 17, 2020 by guest
http://jb.asm.org/
Dow
nloaded from
r--,N-o 10
Eo 107 p
50 10 500 100 "5 0100
z 'S~~~~0.0N0~ I.
C)~~~~~~~~~~~
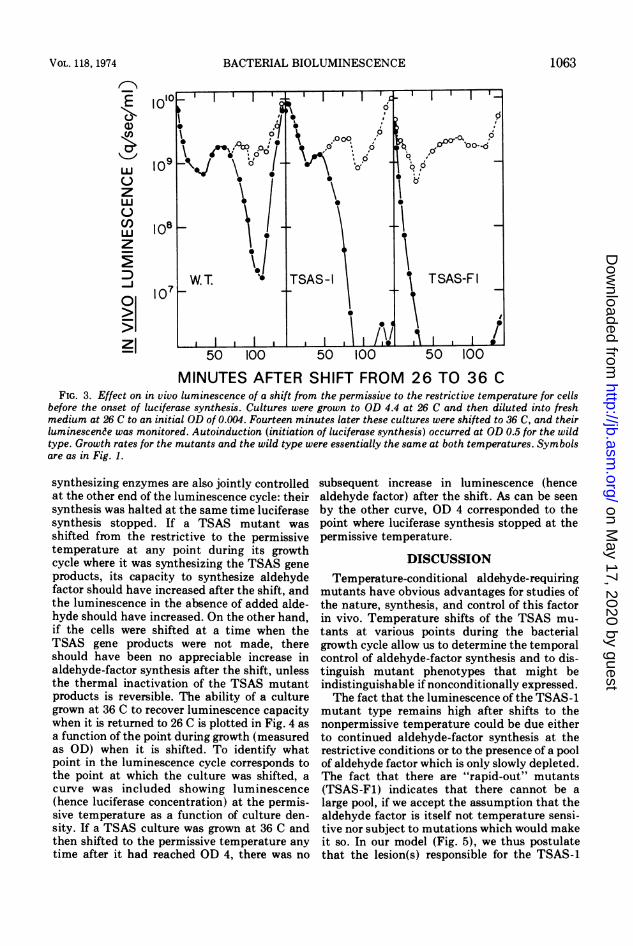
MINUTES AFTER SHIFT FROM 26 TO 36 CFIG. 3. Effect on in vivo luminescence of a shift from the permissive to the restrictive temperature for cells
before the onset of luciferase synthesis. Cultures were grown to OD 4.4 at 26 C and then diluted into freshmedium at 26 C to an initial OD of 0.004. Fourteen minutes later these cultures were shifted to 36 C, and theirluminescence was monitored. Autoinduction (initiation of luciferase synthesis) occurred at OD 0.5 for the wildtype. Growth rates for the mutants and the wild type were essentially the same at both temperatures. Symbolsare as in Fig. 1.
synthesizing enzymes are also jointly controlledat the other end of the luminescence cycle: theirsynthesis was halted at the same time luciferasesynthesis stopped. If a TSAS mutant wasshifted from the restrictive to the permissivetemperature at any point during its growthcycle where it was synthesizing the TSAS geneproducts, its capacity to synthesize aldehydefactor should have increased after the shift, andthe luminescence in the absence of added alde-hyde should have increased. On the other hand,if the cells were shifted at a time when theTSAS gene products were not made, thereshould have been no appreciable increase inaldehyde-factor synthesis after the shift, unlessthe thermal inactivation of the TSAS mutantproducts is reversible. The ability of a culturegrown at 36 C to recover luminescence capacitywhen it is retumed to 26 C is plotted in Fig. 4 asa function of the point during growth (measuredas OD) when it is shifted. To identify whatpoint in the luminescence cycle corresponds tothe point at which the culture was shifted, acurve was included showing luminescence(hence luciferase concentration) at the permis-sive temperature as a function of culture den-sity. If a TSAS culture was grown at 36 C andthen shifted to the permissive temperature anytime after it had reached OD 4, there was no
subsequent increase in luminescence (hencealdehyde factor) after the shift. As can be seenby the other curve, OD 4 corresponded to thepoint where luciferase synthesis stopped at thepermissive temperature.
DISCUSSIONTemperature-conditional aldehyde-requiring
mutants have obvious advantages for studies ofthe nature, synthesis, and control of this factorin vivo. Temperature shifts of the TSAS mu-tants at various points during the bacterialgrowth cycle allow us to determine the temporalcontrol of aldehyde-factor synthesis and to dis-tinguish mutant phenotypes that might beindistinguishable if nonconditionally expressed.The fact that the luminescence of the TSAS-1
mutant type remains high after shifts to thenonpermissive temperature could be due eitherto continued aldehyde-factor synthesis at therestrictive conditions or to the presence of a poolof aldehyde factor which is only slowly depleted.The fact that there are "rapid-out" mutants(TSAS-F1) indicates that there cannot be alarge pool, if we accept the assumption that thealdehyde factor is itself not temperature sensi-tive nor subject to mutations which would makeit so. In our model (Fig. 5), we thus postulatethat the lesion(s) responsible for the TSAS-1
VOL. 118, 1974 BACTERIAL BIOLUMINESCENCE 1063
on May 17, 2020 by guest
http://jb.asm.org/
Dow
nloaded from
CLINE AND HASTINGS
(]) lUU TSAS-1 \A
050A50LD A
100
>| 0 TSAS-Fl
z 50
l 111L.-1 L L0.5 1.0 2 3 6
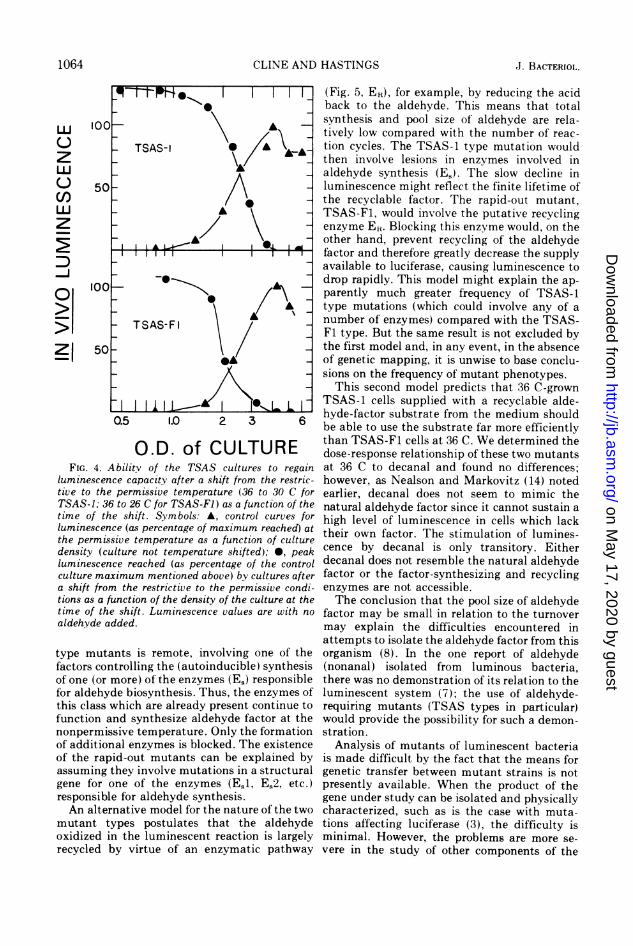
O.D. of CULTUREFIG. 4. Abilitv of the TSAS cultures to regain
luminescence capacity after a shift from the restric-tive to the permissive temperature (36 to 30 C forTSAS-1; 36 to 26 C for TSAS-F1) as a function of thetime of the shift. Symbols: A, control curves forluminescence (as percentage of maximum reached) atthe permissive temperature as a function of culturedensity (culture not temperature shifted); 0, peakluminescence reached (as percentage of the controlculture maximum mentioned above) by cultures aftera shift from the restrictive to the permissive condi-tions as a function of the density of the culture at thetime of the shift. Luminescence values are with no
aldehyde added.
type mutants is remote, involving one of thefactors controlling the (autoinducible) synthesisof one (or more) of the enzymes (E8) responsiblefor aldehyde biosynthesis. Thus, the enzymes ofthis class which are already present continue tofunction and synthesize aldehyde factor at thenonpermissive temperature. Only the formationof additional enzymes is blocked. The existenceof the rapid-out mutants can be explained byassuming they involve mutations in a structuralgene for one of the enzymes (ES1, ES2, etc.)responsible for aldehyde synthesis.An alternative model for the nature of the two
mutant types postulates that the aldehydeoxidized in the luminescent reaction is largelyrecycled by virtue of an enzymatic pathway
(Fig. 5, ER), for example, by reducing the acidback to the aldehyde. This means that totalsynthesis and pool size of aldehyde are rela-tively low compared with the number of reac-tion cycles. The TSAS-1 type mutation wouldthen involve lesions in enzymes involved inaldehyde synthesis (E,). The slow decline inluminescence might reflect the finite lifetime ofthe recyclable factor. The rapid-out mutant,TSAS-F1, would involve the putative recyclingenzyme ER. Blocking this enzyme would, on theother hand, prevent recycling of the aldehydefactor and therefore greatly decrease the supplyavailable to luciferase, causing luminescence todrop rapidly. This model might explain the ap-parently much greater frequency of TSAS-1type mutations (which could involve any of anumber of enzymes) compared with the TSAS-Fl type. But the same result is not excluded bythe first model and, in any event, in the absenceof genetic mapping, it is unwise to base conclu-sions on the frequency of mutant phenotypes.
This second model predicts that 36 C-grownTSAS-1 cells supplied with a recyclable alde-hyde-factor substrate from the medium shouldbe able to use the substrate far more efficientlythan TSAS-F1 cells at 36 C. We determined thedose-response relationship of these two mutantsat 36 C to decanal and found no differences;however, as Nealson and Markovitz (14) notedearlier, decanal does not seem to mimic thenatural aldehyde factor since it cannot sustain ahigh level of luminescence in cells which lacktheir own factor. The stimulation of lumines-cence by decanal is only transitory. Eitherdecanal does not resemble the natural aldehydefactor or the factor-synthesizing and recyclingenzymes are not accessible.The conclusion that the pool size of aldehyde
factor may be small in relation to the turnovermay explain the difficulties encountered inattempts to isolate the aldehyde factor from thisorganism (8). In the one report of aldehyde(nonanal) isolated from luminous bacteria,there was no demonstration of its relation to theluminescent system (7); the use of aldehyde-requiring mutants (TSAS types in particular)would provide the possibility for such a demon-stration.
Analysis of mutants of luminescent bacteriais made difficult by the fact that the means forgenetic transfer between mutant strains is notpresently available. When the product of thegene under study can be isolated and physicallycharacterized, such as is the case with muta-tions affecting luciferase (3), the difficulty isminimal. However, the problems are more se-vere in the study of other components of the
1064 J. BACTERIOL.
on May 17, 2020 by guest
http://jb.asm.org/
Dow
nloaded from

BACTERIAL BIOLUMINESCENCE
SYNTHESISOFr' k,7WlkAC
FMNH2+02
k LUCIFERASEt- NZYMLb INT I
ks-I
ION ZE/ \AC IDt LIGHT
ES- I E5-2 ES 3A -> B -> C -->ALDEHYDE-I
TSAS-FLESION
N ER-',
FIG. 5. Scheme for the mode of action in vivo of TSAS lesions in the bacterial bioluminescence system. Inthe TSAS-1 class, high temperature would block the specific synthesis of the enzyme or enzymes (Es) whichproduce aldehyde factor which reacts with intermediate II as shown to produce light. The TSAS-Fl typemutants would involve a temperature-sensitive lesion in the aldehyde-factor enzvmes themselves. Analternative hypothesis is also discussed in the text which involves the pathwav indicated by a dashed line for therecycling of aldehyde factor via enzyme ER.
luminescence system whose identity and modeof synthesis are unknown. We hope that in vivocharacterization of the aldehyde-factor-synthe-sizing enzymes, as revealed by studies on TSASmutants, will help in the eventual identificationof these enzymes in vitro. For example, theTSAS-1 and TSAS-F1 gene products may beidentifiable as altered proteins co-induced withluciferase. Direct identification of these pro-
teins based on their enzymatic function is madedifficult by the fact that aldehydes such as
decanal will not mimic the aldehyde factor invivo, and by the fact that attempts to identifyand isolate aldehyde from this strain have beenunsuccessful. The recent demonstration that a
long-chain nitrite can replace aldehyde (1) is ofspecial interest in this connection. In additionto its activity in the in vitro assay, it was shownto stimulate light emission in dark aldehydemutants and, unlike aldehyde, to do so in a
long-term nontransitory fashion. Analysis of itseffect on the different classes of aldehyde mu-
tants will be of considerable interest.
ACKNOWLEDGMENTSThis work was supported in part by National Science
Foundation research grant GB 31977X. T. W. C. was a PublicHealth Service trainee under grant GM 00138-13 from theNational Institute of General Medical Sciences.We thank Miriam Nicoli for helpful discussions.
LITERATURE CITED
1. Bentley, D., A. Eberhard, and R. Solsky. 1974. Decylnitrite: an aldehyde analog in the bacterial biolumines-cence reaction. Biochem. Biophys. Res. Commun.
56:865-868.2. Cline, T. W., and J. W. Hastings. 1971. Temperature-sen-
sitive mutants in the bacterial bioluminescence sys-
tem. Proc. Nat. Acad. Sci. U.S.A. 68:500-504.3. Cline, T. W., and J. W. Hastings. 1972. Mutationally
altered bacterial luciferase. Implications for subunitfunctions. Biochemistry 11:3359-3370.
4. Coffey, J. J. 1967. Inducible synthesis of bacterial lucif-erase: specificity and kinetics of induction. J. Bac-teriol. 94:1638-1647.
5. Dunn, D. K., G. A. Michaliszyn, I. G. Bogacki, and E. A.Meighen. 1973. Conversion of aldehyde to acid in thebacterial bioluminescent reaction. Biochemistry12:4911-4918.
6. Eberhard, A. 1972. Inhibition and activation of bacterialluciferase synthesis. J. Bacteriol. 109:1101-1105.
7. Ferrell, W. J., R. J. Kessler, and M. Drouillard. 1971.Identification of nonaldehyde in P. fischeri. Chem.Phys. Lipids 6:131-134.
8. Hastings, J. W. 1968. Bioluminescence. Annu. Rev.Biochem. 37:597-630.
9. Hastings, J. W., A. Eberhard, T. 0. Baldwin, M. Z.Nicoli, T. W. Cline, and K. H. Nealson. 1973. Bacterialbioluminescence. Mechanistic implications of activecenter chemistry of luciferase, p. 369-380. In M. J.Cormier, D. M. Hercules, and J. Lee (ed.), Chemilumi-nescence and bioluminescence. Plenum PublishingCo., New York.
10. Hastings, J. W., K. Weber, J. Friedland, A. Eberhard, G.W. Mitchell, and A. Gunsalus. 1969. Structurallydistinct bacterial luciferases. Biochemistry8:4681-4689.
11. Kempner, E. S., and F. E. Hanson. 1968. Aspects of lightproduction by Photobacterium fischeri. J. Bacteriol.95:975-979.
12. McCapra, F., and D. W. Hysert. 1973. Bacterial bio-luminescence-identification of fatty acid as product,its quantum yield and suggested mechanism. Biochem.Biophys. Res. Commun. 52:298-304.
13. Mitchell, G., and J. W. Hastings. 1971. A stable inexpen-sive solid state photomultiplier photometer. Anal.Biochem. 39:243-250.
14. Nealson, K. H., and A. Markovitz. 1970. Mutant analysis
TSALES
1065VOL. 118, 1974
on May 17, 2020 by guest
http://jb.asm.org/
Dow
nloaded from

CLINE AND HASTINGS
and enzyme subunit complementation in bacterialbioluminescence in Photobacterium fischeri. J. Bacte-riol. 104:300-312.
15. Nealson, K. H., T. Platt, and J. W. Hastings. 1970.Cellular control of the synthesis and activity of thebacterial luminescent system. J. Bacteriol.104:313-322.
16. Reichelt, J. L., and P. Baumann. 1973. Taxonomy of themarine, luminous bacteria. Arch. Mikrobiol.94:283-330.
17. Rogers, P., and W. D. McElroy. 1955. Biochemicalcharacteristics of aldehyde and luciferase mutants of
luminous bacteria. Proc. Nat. Acad. Sci. U.S.A.41:67-70.
18. Roodman, S. T., and G. R. Greenberg. 1971. Conditionsallowing synthesis of thymidylate synthesis at thenonpermissive temperature in a temperature-sensitivethy mutant normally blocked in its translation. J. Biol.Chem. 246:4853-4858.
19. Shimomura, O., F. H. Johnson, and Y. Kohama. 1972.Reactions involved in bioluminescence systems of lim-pet (Latia neritoides) and luminous bacteria. Proc. Nat.Acad. Sci. U.S.A. 69:2086-2089.
1066 J. BACTERIOL.
on May 17, 2020 by guest
http://jb.asm.org/
Dow
nloaded from