Embed Size (px)
Citation preview

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
Audited firm Annex of the directive Standards Type of consideration
Annex ll Annex V Annex Vl
ISO 9001 / 2 EN 46001 / 2 FDA QSR’s
certification audit pre-audit surveillance-audit
follow up-audit re-audit
Telephone-Nr. Documentation review/report from Reference document of the firm
Telefax-Nr. On-site audit carried out on (date)
Lead auditor Co-auditor / technical expert Co-auditor
Audited section/area (area of validation)
Products:
SUMMARY STATEMENTS:
Scope and Aim of the Quality Audit
The audit performed on __________ at __________________ located in _________________ is de-signed to evaluate the quality system implemented according to the FDA Quality System Require-ments (QSR’s) as given in 21 CFR § 820, ISO 9001/2, and EN 46001/2. There was a particular em-phasis on the use of the ISO 9001/2, EN 46001/2 audit check list provided by this author of this re-port.
The scope and aim of the audit is for the qualification of this Company (at this one facility only). Any other facility must be evaluated on its own merits.
Critical Observations: __Major Observations: __ (None have an impact on Safety, Form, Fit, Function)Minor Observations : __Recommendations: __
Date / Signature of Auditor(s)
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 1 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
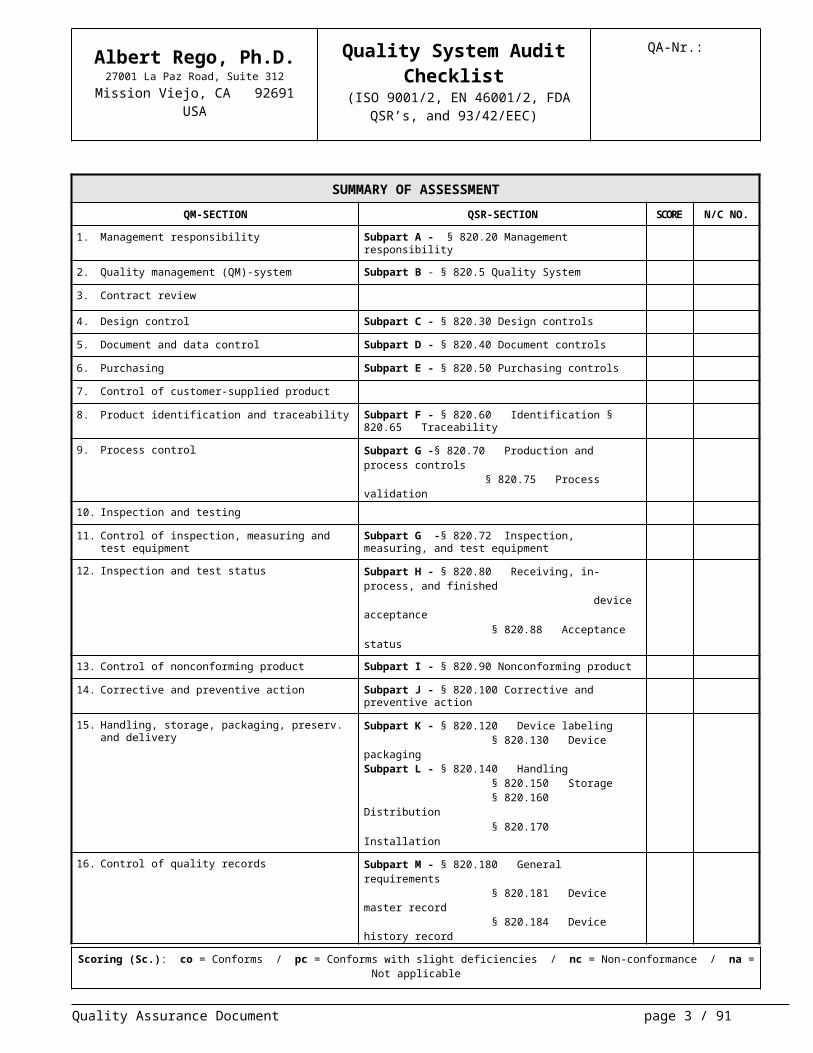
SUMMARY OF ASSESSMENTQM-SECTION QSR-SECTION SCORE N/C NO.
1. Management responsibility Subpart A - § 820.20 Management responsibility
2. Quality management (QM)-system Subpart B - § 820.5 Quality System
3. Contract review
4. Design control Subpart C - § 820.30 Design controls
5. Document and data control Subpart D - § 820.40 Document controls
6. Purchasing Subpart E - § 820.50 Purchasing controls
7. Control of customer-supplied product
8. Product identification and traceability Subpart F - § 820.60 Identification § 820.65 Traceability
9. Process control Subpart G -§ 820.70 Production and process controls § 820.75 Process validation
10. Inspection and testing
11. Control of inspection, measuring and test equipment Subpart G -§ 820.72 Inspection, measuring, and test equipment
12. Inspection and test status Subpart H - § 820.80 Receiving, in-process, and finished device acceptance § 820.88 Acceptance status
13. Control of nonconforming product Subpart I - § 820.90 Nonconforming product
14. Corrective and preventive action Subpart J - § 820.100 Corrective and preventive action
15. Handling, storage, packaging, preserv. and delivery Subpart K - § 820.120 Device labeling § 820.130 Device packagingSubpart L - § 820.140 Handling § 820.150 Storage § 820.160 Distribution § 820.170 Installation
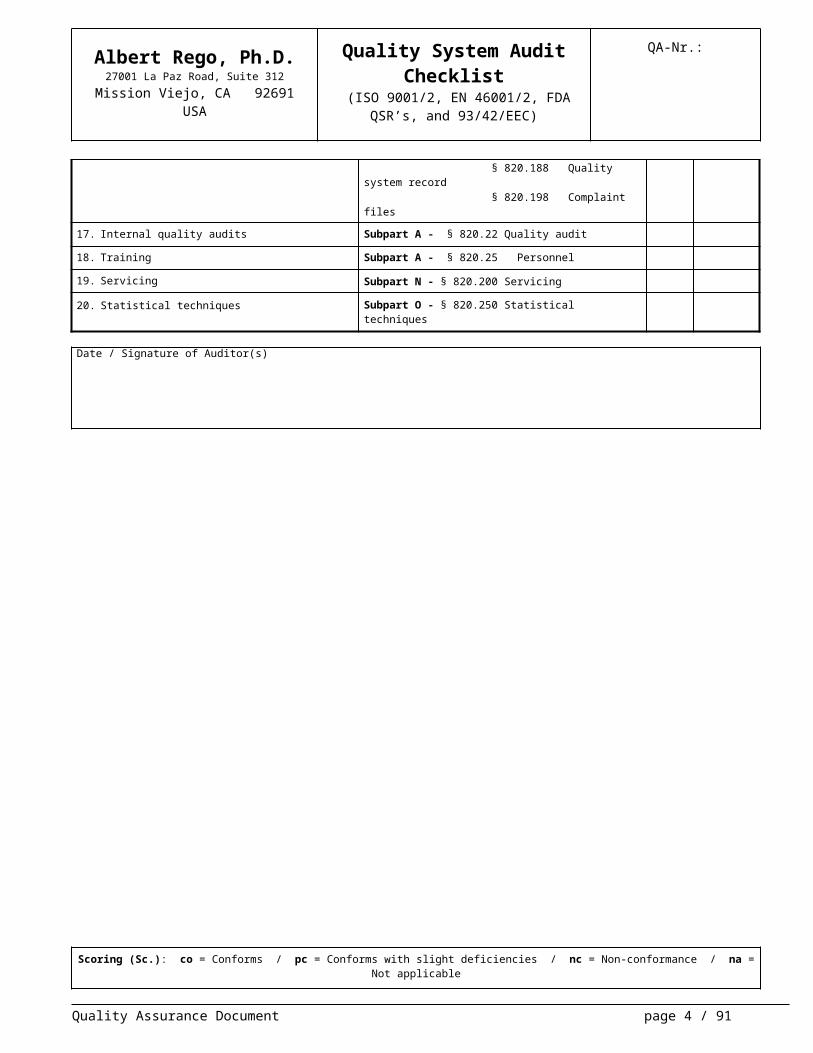
16. Control of quality records Subpart M - § 820.180 General requirements § 820.181 Device master record § 820.184 Device history record § 820.188 Quality system record § 820.198 Complaint files
17. Internal quality audits Subpart A - § 820.22 Quality audit
18. Training Subpart A - § 820.25 Personnel
19. Servicing Subpart N - § 820.200 Servicing
20. Statistical techniques Subpart O - § 820.250 Statistical techniques
Date / Signature of Auditor(s)
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 2 / 57
Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.1 - Management responsibility / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.1.1 Quality policy
Is this defined and documented?
Is there understanding at all lev-els of the organization? (e.g. through a circular letter, management assem-bly, notices, training programs, semi-nars)
§ 820.20 Management Responsibility(a)Quality Policy 1.Management with executive re-
sponsibility has established a policy demonstrating its objectives for and commitment to quality.
2.Management with executive re-sponsibility has ensured that the pol-icy is understood, implemented, and maintained at all levels of the organi-zation.
Reviews are documented.
4.1.1 Quality Goals
Does this quality policy provide clear directions on quality goals and commitment?Is there surveillance of these goals?Are these goals adapted or adjusted?
4.1.2.1 Organization /Responsibility/Au-thority
Is the responsibility, authority and the interrelation of personnel who manage, perform and verify work af-fecting quality (clearly) defined and documented?
For example in the organizational structure, job description, matrices, workplace description.
§ 820.20 Management Responsibility(b)Organization The manufacturer has established
and maintained an adequate organi-zational structure.
(1)Responsibility and authority 1.The manufacturer has established
the appropriate responsibility, author-ity, and interrelation of all personnel who manage, perform, and assess work affecting quality.
2.The manufacturer has provided the independence and authority neces-sary to perform these tasks.
4.1.2.2 Resources
Is there identification of resource requirements?
Is there provision of adequate resources?
Is there provision of trained per-sonnel (e.g. internal audits) ?
820.25- Personnel(a)General There are sufficient personnel with
the necessary education, back-ground, training, and experience to assure that all activities required by this part are correctly performed.
§ 820.20 Management Responsibility(2)Resources The manufacturer has provided ade-
quate resources, including the as-signment of trained personnel, for management, performance of work, and assessment activities, including internal quality audits.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 3 / 57
Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.1 - MANAGEMENT RESPONSIBILITY / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.1.2.3 QM representative
Is there a QM representative ap-pointed?
Has the QM representative the necessary authority and compe-tence?
§ 820.20 Management Responsibility (3)Management representative
Management with executive respon-sibility has appointed and docu-mented a member of management who, irrespective of other responsibil-ities, has established authority over and responsibility for: Ensuring that quality system re-
quirements are effectively estab-lished and maintained in accor-dance with this part;
Reporting on the performance of the quality system to manage-ment with executive responsibil-ity for review,
4.1.3 Management review
Is there revision/review of the quality system, by the suppliers man-agement, at defined intervals?
Is this review documented?
Were records of such QM re-views maintained?
Please refer to records in Section 4.16
§ 820.20 Management Responsibility(c)Management review
1.Management with executive re-sponsibility has reviewed the suitabil-ity and effectiveness of the quality system at defined intervals and with sufficient frequency according to es-tablished procedures to ensure that the quality system satisfies the re-quirements and the established qual-ity policy and objectives.
2.The dates and results of quality systems reviews are documented.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 4 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.2 - QUALITY MANAGEMENT SYSTEM / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.2.1 QM-manual
Is a QM-manual available? Does this include or make refer-
ence to the quality system proce-dures?
Does this outline the structure of the documentation used in the quality system?(eventual reference to ISO 10013)
§ 820.5 Quality System
The manufacturer has established and maintained an appropriate quality system.
Particular requirements for all medical devices (EN 46001/2)
Are the specified requirements estab-lished and documented?
Note: When the quality system is used, the relevant requirements of the regula-tions should be included in the specified requirements.
4.2.2 Quality system procedure
Does the QM manual contain documented procedures?
When no, are references made to document procedures?
In the documented procedures is reference made to work instruc-tions that define how an activity is performed?
§ 820.20 Management ResponsibilityQuality system procedures
1.The manufacturer has established quality system procedures and in-structions.
2.An outline of the structure of the documentation used in the quality system has been established.
Particular requirements for all medical devices (EN 46001/2)
Does there exist for each product/product group a file (technical document/product file) containing, at least, the following: product specifications classification of the medical
products manufacturing specifications quality assurance specifications.
The records that are not in a central ar-chive shall, within these files, refer to the location of these records.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 5 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.2 - Quality management System / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.2.3 Quality planning
(QM Plan: document, in which are the specific quality procedures and means of assistance and even task/activity descrip-tion Is there description, in the quality
plan, for customers products and ser-vice requirements?
§ 820.20 Management Responsibility(d)Quality planning
1.The manufacturer has established a quality plan which defines the qual-ity practices, resources, and activities relevant to devices that are designed and manufactured.
2.The manufacturer has established how the requirements for quality will be met.
Is there consideration, in the quality plan, given to the following sections:
Report on the managerial and testing process.
Identification of suitable verifica-tion at appropriate stages in the real-ization process.
Development of new instrumen-tation
Identification of any measure-ment requirement involving capabil-ity that exceeds the known state of the art.
Clarification of standards of ac-ceptability for all features and re-quirements , inclusive of those which contain a subjective section.
Identification and preparation of quality records.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 6 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.3 contract review
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.3.1 General
Is the procedure for contract re-view established? Are there established and maintained documented procedures for contract review and for the coordination of these activities?
4.3.2 Review
Is the statement of requirements adequately defined and documented?
Does a review take place be-fore making an offer in respect to acceptance of a contract/order?
Are requirements adequately defined and documented?
Will differences between the contract or the order requirements and those in the tender be resolved?
Is it ensured that requirements can be fulfilled/kept?
In the instance of an order re-ceived by verbal means (word of mouth/telephone), does there exist an insurance of requirements agreed before acceptance?
4.3.3 Amendment to a contract
Are all concerned functions in-formed?
4.3.4 Records
Are the quality records in Section 4.16 taken in consideration?
Are channels for communication and interfaces with the customer’s or-ganization in these contract matters established? (see comment 9 of stan-dard)
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 7 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.4 - DESIGN CONTROL / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.4.1 General
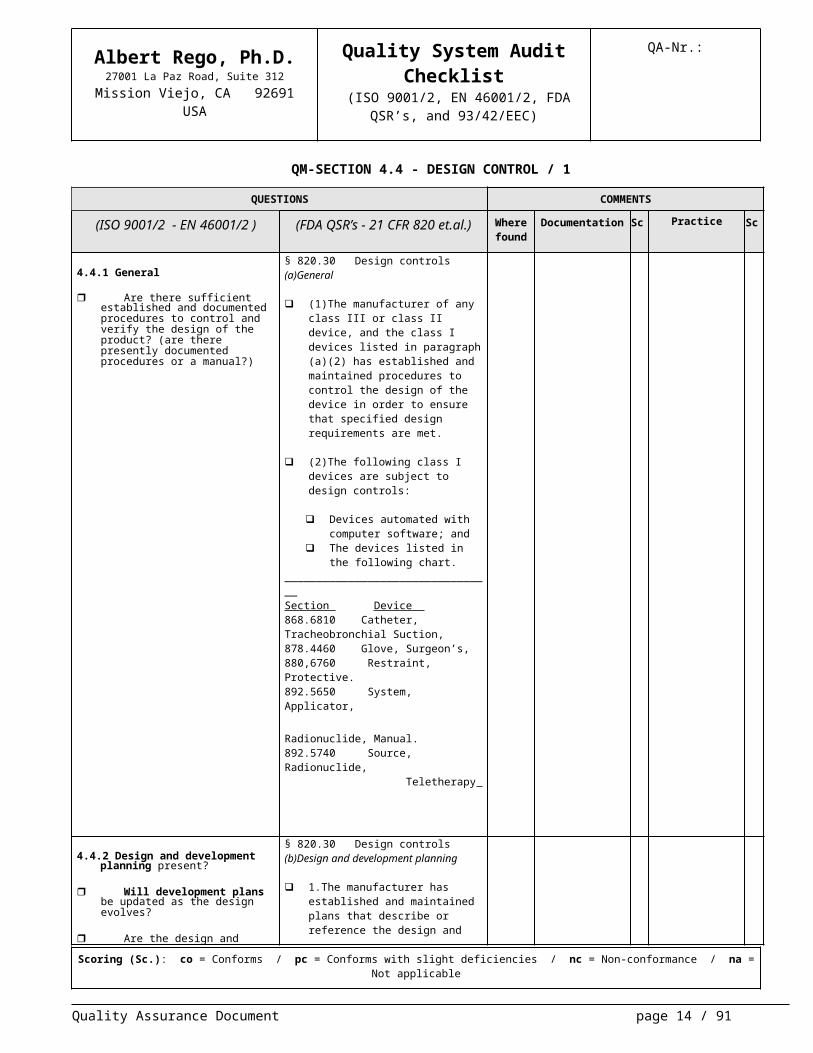
Are there sufficient established and documented procedures to con-trol and verify the design of the prod-uct? (are there presently documented procedures or a manual?)
§ 820.30 Design controls(a)General
(1)The manufacturer of any class III or class II device, and the class I de-vices listed in paragraph (a)(2) has established and maintained proce-dures to control the design of the de-vice in order to ensure that specified design requirements are met.
(2)The following class I devices are subject to design controls:
Devices automated with com-puter software; and
The devices listed in the follow-ing chart.
_________________________________Section Device 868.6810 Catheter, Tracheobronchial Suction,878.4460 Glove, Surgeon’s,880,6760 Restraint, Protective.892.5650 System, Applicator, Radionuclide, Manual.892.5740 Source, Radionuclide, Teletherapy
4.4.2 Design and development planning present?
Will development plans be up-dated as the design evolves?
Are the design and development activities assigned to qualified per-sonnel?
Are adequate resources avail-able for the development process?
§ 820.30 Design controls (b)Design and development planning
1.The manufacturer has established and maintained plans that describe or reference the design and develop-ment activities
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 8 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.4 - DESIGN CONTROL / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.4.3 Organizational and technical inter-faces
Are the organizational and tech-nical interfaces between different groups which input into the design process defined?
Is necessary written informa-tion documented, transmitted and regularly reviewed?
§ 820.30 Design controls 2.The plans identify and describe the
interfaces with different groups or ac-tivities that provide, or result in, input to the design and development process.
3.The plans are reviewed, updated, and approved as design and develop-ment evolves.
4.4.4 Design input
Are design input requirements defined and documented?
Are inputs from the contract testing considered?
Are the requirements complete and unambiguous?
§ 820.30 Design controls (c)Design input
1.The manufacturer has established and maintained procedures to ensure that the design requirements relating to a device are appropriate and ad-dress the intended use of a device, including the needs of the user and patient.
2.The procedures include a mecha-nism for addressing incomplete, am-biguous, or conflicting requirements.
3.The design input requirements are documented and reviewed and ap-proved by a designated individual(s).
4.The approval, including the date and signature of the individual(s) ap-proving the requirements, is docu-mented.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 9 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.4 - Design control / 3
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.4.4 CONTINUATION
Particular requirements for all medical devices (EN 46001)
Is there identification of requirements that are related to the safety of the medical device and include such re-quirements as design input data?
DIRECTIVE 93/42/EEC
ANNEX I / essential requirements
Is there a record of how the essential requirements are to be fulfilled?
Does there exist a record/list of rele-vant standards?
ANNEX lX / classification criteria
Is classification stipulated according to the annex IX of the directive?
Are the definitions for the classifica-tion rules applied?
ANNEX X / clinical evaluation
Is clinical evaluation necessary?
If yes, is there clinical evaluation ac-cording to EN 540?
Will a clinical evaluation be under-taken?
Does the evaluation adhere to: market experience, scientific literature and their
evaluation
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 10 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.4 - Design control / 4
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.4.4 CONTINUATION
ANNEX 1, 13 / Information supplied by the manufacturer Are the requirements for the
symbol considered (13.3 a to m, e.g. Symbols w.r.t. EN 980)?
Is the intended purpose (13.4) obvious or is this clearly stated?
Are all the necessary instructions covered (13.6 a to p)?
4.4.5 Design output
Is the design output documented and expressed in terms that can be verified and validated against the de-sign input requirements? Will the design input require-
ments be meet? Are acceptance criteria defined?
Are characteristics of the design that are crucial to the safe and proper functioning identified?
§ 820.30 Design controls(d)Design output
1.The manufacturer has established and maintained procedures for defin-ing and documenting design output in terms that allow an adequate evalua-tion of conformance to design input requirements.
2.Design output procedures contain or make reference to acceptable cri-teria and ensure that those design outputs that are essential for the proper functioning of the device are identified.
3.Design output is documented, re-viewed, and approved before release.
4.The approval, including the date and signature of the individual(s) ap-proving the output is identified.
4.4.6 Design review
At appropriate stages of design are reviews of the design results planned and conducted?
Will these reviews be documented?
Are the participants, at each de-sign review, including representatives of all functions concerned with the de-sign stage, been reviewed?
Are specialist personnel neces-sary?
§ 820.30 Design controls(e)Design review
1.The manufacturer has established and maintained procedures to ensure that formal documented reviews of the design results are planned and conducted at appropriate stages of the device’s design development.
2.The procedure ensures that partici-pants at each design review include representatives of all functions con-cerned with the design stage being reviewed and an individual(s) who does not have direct responsibility for the design stage being reviewed, as well as any specialists needed.
3.The results of a design review, in-cluding identification of the design, the date, and the individual(s) per-forming the review, are documented in the design history file (DHF).
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 11 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.4 - DESIGN CONTROL / 5
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.4.7 Design verification must at appropri-ate stages of design be performed.
Through the design verification, how will the developmental results and corresponding design phase of the requirements be fulfilled : Performing alternative calcula-
tions Comparing the new design with a
similar proven design, if avail-able
Undertaking tests and demon-strations
Reviewing the design stage doc-uments before release
§ 820.30 Design controls(f)Design verification
1.The manufacturer has established and maintained procedures for verify-ing the device design.
2.Design verification confirms that the design output meets the design input requirements.
3.The results of the design verifica-tion, including identification of the de-sign, method(s), the date, and the in-dividual(s) performing the verification , are documented in the DHF.
Particular requirements for all medical devices (EN 46001)
Are there documented and main-tained records (see Section 4.16) of all design verification activities includ-ing those where clinical investigation was involved?
4.4.8 Design validation
Will a conclusive validation be performed?
For example. : After successful verification; Under defined operating condi-
tions; Necessary in earlier stages prior
to product completion
Will this ensure that the product conforms to defined user needs and/or requirements?
§ 820.30 Design controls(g)Design validation 1.The manufacturer has established
and maintained procedures for vali-dating the device design.
2.Design validation is performed un-der defined operating conditions on initial production units, lots, or batches, or their equivalents.
3.Design validation ensures that de-vices conform to defined user needs and intended uses and includes test-ing of production lots under actual and simulated use conditions.
4.Design validation includes software validation and risk analysis, where appropriate.
5.The results of the design validation, including identification of the design, method(s), the date, and the individ-ual(s) performing the validation, is documented in the DHF.
QM-SECTION 4.4 - DESIGN CONTROL / 6
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 12 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s – 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.4.9 Design changes
Are all design changes and mod-ifications identified and documented?
Will all design changes be re-viewed and approved by authorized personnel before their implementa-tion?
Have the records in Section 4.16 been considered?
§ 820.30 Design controls (h)Design transfer The manufacturer has established
and maintained procedures to ensure that the device design is correctly translated into production specifica-tions.
(i)Design changes The manufacturer has established
and maintained procedures for the identification, documentation, valida-tion or where appropriate, verification, review, and approval of the design changes before their implementation.
(j)Design history file 1.Each manufacturer has established
and maintained a DHF for each type of device.
2.The DHF contains or references the records necessary to demon-strate that the design was developed in accordance with the approved de-sign plan and the requirements of this part.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 13 / 57
Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.5 DOCUMENT AND DATA CONTROL / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s – 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.5.1 General/documented procedures
Are there established and main-tained document procedures to con-trol all documents and data?
Are documents of external ori-gin (to the extent applicable) in data control systems integrated?
For example Standards, Customer drawings, Laws,
Subpart D - § 820.40 Document controls
The manufacturer has established and maintained procedures to control all documents that are required by this part. The procedures provide for the following:
4.5.2 Document and data approval and issue
Is there a master list or equiva-lent control procedure identifying the current revision status and is available to preclude the use of in-valid and/or obsolete documents?
Are there authorized personnel that review and approve for ade-quacy, documents and data prior to issue?
The control shall ensure that:
the pertinent issues of appropri-ate documents are available at all relevant locations?
invalid and/or obsolete docu-ments are promptly removed from all points of issue or use? E.g. changing of collecting list
the current revision status is made known?
any obsolete documents re-tained for legal and/or knowl-edge-preservation purposes are suitably identified?
§ 820.40 Document controls(a)Documentation approval and distribu-tion
1.The manufacturer has designated an individual(s) to review for ade-quacy and approve prior to issuance all documents established to meet the requirements of this part.
2.The approval, including the date and signature of the individual(s) ap-proving the document, is docu-mented.
3.Documents established to meet the requirements of this part are available at all locations for which they are des-ignated, used, or otherwise docu-ments are promptly removed from all points of use or otherwise prevented from unintended use.
Particular requirements for all medical devices (EN 46001/2)
Is there a defined period where obsolete documents shall be re-tained?
Does this period insure that specifications are available for at least the lifetime of the medical de-vice?
Please refer to Section 4.16?
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 14 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.5 DOCUMENT AND DATA CONTROL / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s – 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.5.3 Document and data changes
Are changes to documents and data reviewed and approved by the same functions/organizations that performed the original review and approval?
Is the nature of the change identified in the document or the appropriate at-tachments (Where is this feasible)?
§ 820.40 Document controls(b)Document changes
1.Changes to documents are re-viewed and approved by an individ-ual(s) that performed the original re-view and approval, unless specifically designated otherwise.
2.Approved changes are communi-cated to the appropriate personnel in a timely manner.
3.The manufacturer maintains records of changes to documents.
4. Change records include a descrip-tion of the change, identification of the affected documents, the signature of the approving individual(s), the ap-proval date, and when the change be-comes effective.
DIRECTIVE 93/42/EEC
ANNEX II 6., V 5., VI 5.
The following administrative provisions must, at least, after the manufacture of the last product be controlled: declaration of conformity, documentation concerning the QM sys-
tem alterations/changes to the QM system, documentation for the product con-
struction (only w.r.t. annex II), technical documentation, all decisions and reports from the noti-
fied body
Note: Documents are input, where they serve as a revision to a standard. Data is defined as digitized documents or records.
The standard makes explicit reference to the least quality record in a QM system (see Section 4.16).
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 15 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.6 PURCHASING / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s – 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.6.1 General/documented procedures
Are there established and maintained documented proce-dures to ensure that purchased prod-ucts conform to specified require-ments? (Is the procedure sufficiently detailed?)
Subpart E – Purchasing Controls§ 820.50 Purchasing controls
The manufacturer has established and maintained procedures to ensure that all purchased or otherwise re-ceived product and services conform to specified requirements.
4.6.2 Evaluation of subcontractors
Will the evaluation and selection of subcontractors be based on their ability to meet subcontract require-ments?
§ 820.50 Purchasing controls
(a)Evaluation of suppliers, contractors, and consultants
1.The manufacturer has established and maintained the requirements, in-cluding quality requirements, that must be met by suppliers, contrac-tors, and consultants.
2.The manufacturer has: Evaluated and selected potential
suppliers, contractors, and con-sultants on the basis of their ability to meet specified require-ments, including quality require-ments. This evaluation is docu-mented.
Define the type and extent of control to be exercised over the product, services, suppliers, contractors, and consultants, based on the evaluation results.
Establish and maintain records of acceptable suppliers, contrac-tors, and consultants.
Current evaluation?
Who will evaluate and select subcon-tractors on their ability to meet require-ments including quality systems and any specific quality assurance require-ments?
What other aspects and requirements are taken into evaluation ?
Is the type and extent of control exer-cised by the supplier over subcontrac-tors defined?
E.g. quality audit reports and/or quality records, performance of subcontrac-tors.
QM-SECTION 4.6 PURCHASING / 2
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 16 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.6.3 Purchasing data
Do the purchasing documents contain data that clearly describes the product ordered?
In so far as applicable the purchas-ing data shall contain the follow-ing: Precise identification of the prod-
uct Title or other positive identifica-
tion, and application issues of specifications, drawings?),
Requirements for the product process;
Release procedures; Requirements for personal; Requirements for approval or
qualification of product.
§ 820.50 Purchasing controls (b)Purchasing data
1.The manufacturer has established and maintained data that clearly de-scribe or reference the specified re-quirements, including quality require-ments, for purchased or otherwise re-ceived product and services.
2.Purchasing documents include, where possible, an agreement that the suppliers, contractors, and con-sultants agree to notify the manufac-turer of changes in the product or ser-vice so that the manufacturer may de-termine whether the changes may af-fect the quality of a finished device.
3.Purchasing data is approved in ac-cordance with § 820.40.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 17 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.6 PURCHASING / 3
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.6.3 CONTINUED
Particular requirements for all medical devices (EN 46001/2)
With reference to traceability, is there retention of copies of relevant pur-chasing documents (Section 4.16)?
4.6.4 Verification of purchased product
4.6.4.1 Supplier verification at subcon-tractor’s premises (denoted in the standard as the supplier)
Is the purchased product verified at the subcontractor’s premises?
If yes, are the verification ar-rangements and the method of prod-uct release in the purchasing docu-ments specified?
4.6.4.2 Customer verification of subcon-tracted product
Where specified in the contract, is the supplier’s customer or the customer’s representative afforded the right to verify at the subcontractor’s premises and the supplier’s premises that the subcontracted product conforms to the specified requirements?
If yes, how has the customer right to verification?
Note: Verification by the customer shall not ab-solve the supplier of the responsibility to provide acceptable product, nor shall it pre-clude subsequent rejection by the cus-tomer.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 18 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.7 - control of customer supplied product
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.7 Is there established and documented procedures for control of
verification
storage
maintenance
of customer supplied products?(Products that are provided for incor-poration into the supplies or of related activities)
Is there consideration how products are recorded and re-ported to the customer in the case of loss or damage?
Is such a process documented?
Is the filing from this documenta-tion systematic and ordered? (see Section 4.16)
Are there contractual agreements (Section 4.3/contract review), where by the handling of the products are docu-mented?
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 19 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4. 8 - product identification and traceability / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.8 Product identification and traceability
Are procedures for identifying the product established and docu-mented (where is this appropriate?)
Is the identification clearly de-fined?
Is the traceability a stipulated require-ment?
If yes, are the responsibilities stipu-lated for identification of the respective products?
Are these in accordance with the re-quirements?
Is the identification clearly stipulated?
Will records be kept of such?
please refer to Section 4.16
DIRECTIVE 93/42/EEC
Annex ll 3.2.d
Does there exist adequate product identification procedures?
Can it be stipulated that the product identification procedures are kept up to date (from drawings, specifications) at every stage of construction?
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 20 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4. 8 - Product Identification and traceability / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
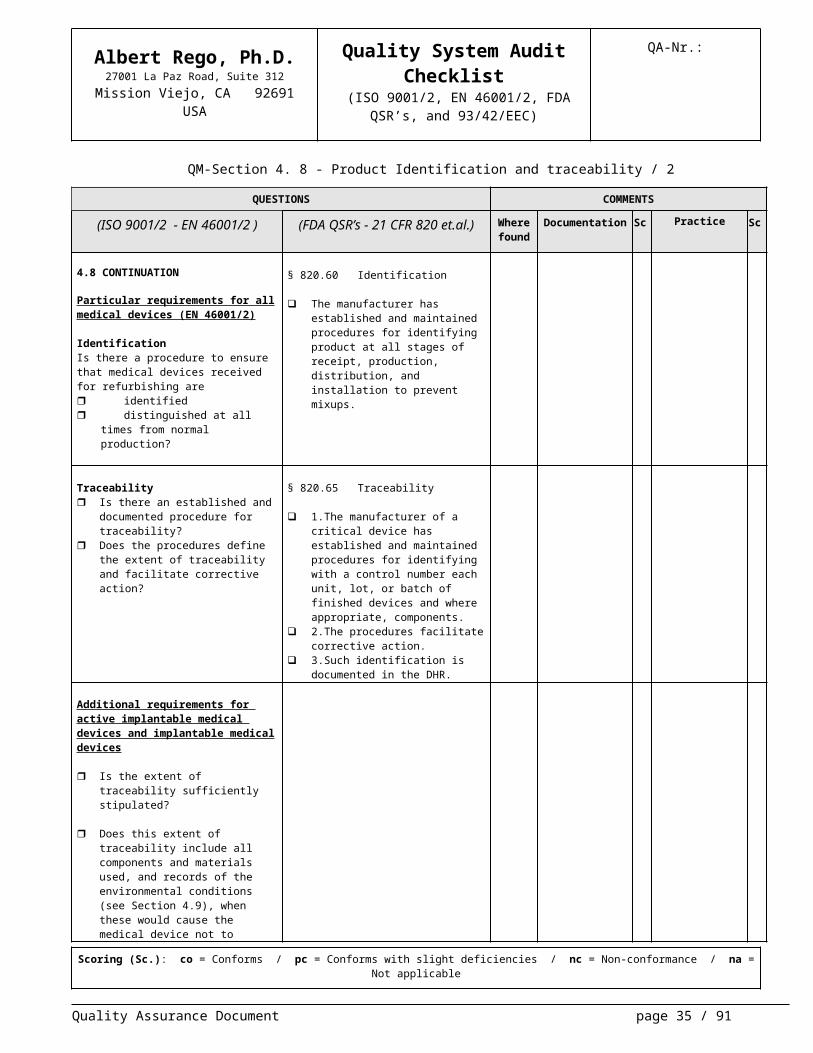
4.8 CONTINUATION
Particular requirements for all medical devices (EN 46001/2)
IdentificationIs there a procedure to ensure that medical devices received for refurbishing are identified distinguished at all times from
normal production?
§ 820.60 Identification
The manufacturer has established and maintained procedures for identi-fying product at all stages of receipt, production, distribution, and installa-tion to prevent mixups.
Traceability Is there an established and docu-
mented procedure for traceability? Does the procedures define the extent
of traceability and facilitate corrective action?
§ 820.65 Traceability
1.The manufacturer of a critical de-vice has established and maintained procedures for identifying with a con-trol number each unit, lot, or batch of finished devices and where appropri-ate, components.
2.The procedures facilitate corrective action.
3.Such identification is documented in the DHR.
Additional requirements for active im-plantable medical devices and im-plantable medical devices
Is the extent of traceability sufficiently stipulated?
Does this extent of traceability include all components and materials used, and records of the environmental con-ditions (see Section 4.9), when these would cause the medical device not to satisfy specified requirements?
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 21 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4. 9 - Process control / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.9 Production, installation, and servic-ing processes
Are the production, installa-tion, and servicing processes, which directly affect quality, identified and planned?
Are these processes carried out un-der controlled conditions?
Are the following points addressed?
Are the production, installation, and servicing processes defined in documented procedures?
Is there use of suitable produc-tion, installation and servicing processes?
Is the work environment suit-able?
Is there compliance with refer-ence standards/codes, quality plans and/or documented proce-dures?
Is there monitoring and control of suitable process parameters and production characteristics?
Is there inclusion of approval of processes and equipment, as appropriate?
How are the criteria for work-manship documented?
Is there a clear and unambigu-ous manner/criteria stipulated? (e.g. written standards, represen-tative samples or illustrations?)
Is there a suitable maintenance process documented?
Subpart G - § 820.70 Production and process controls(a)General 1.The manufacturer has developed,
conducted, controlled, and monitored the production processes to ensure that a device conforms to its specifi-cations.
2.Where deviations from device specifications could occur as a result of the manufacturing process, the manufacturer has established and maintained process control proce-dures that describe any process con-trols necessary to ensure confor-mance to specifications.
3.Where process controls are needed, they include: Documented instructions, stan-
dard operating procedures (SOP’s), and methods that de-fine and control the manner of production;
Monitoring and control of process parameters and compo-nent and device characteristics during production;
Compliance with reference stan-dards or codes;
The approval of processes and process equipment; and
Criteria for workmanship which is expressed in documented standards or by means of identi-fied and approved representa-tive samples.
(b)Production and process changes 1.The manufacturer has established
and maintained procedures for changes to a specification, method, process, or procedure.
2.Such changes are verified or where appropriate validated according to § 820.75, before implementation and these activities are documented.
3.Changes are approved in accor-dance with § 820.40.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 22 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.9 Process control / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.9 CONTINUED
Processes with beforehand qualification (also referred to as „special process“)
Will the processes be undertaken by qualified operators?
When no, will the processes be con-tinually monitored and controlled?
Are records maintained for qualified processes, equipment and personnel?
Has there been consideration of Sec-tion 4.16/control of quality records?
§ 820.70 Production and process controls (3)Adjustment The manufacturer ensures that any
inherent limitations or allowable toler-ances are visibly posted on or near equipment requiring periodic adjust-ments or are readily available to per-sonnel performing these adjustments.
(h)Manufacturing material 1.Where a manufacturing material
could reasonably be expected to have an adverse effect on product quality, the manufacturer has estab-lished and maintained procedures for the use and removal of such manu-facturing material to ensure that it is removed or limited to amount that does not adversely affect the device’s quality.
2.The removal or reduction of such manufacturing material is docu-mented.
(i)Automated processes 1.When computers or automated data
processing systems are used as part of production of the quality system, the manufacturer has validated com-puter software for its intended use ac-cording to an established protocol.
2.All software changes are validated before approval and issuance.
3.These validation activities are docu-mented.
Particular requirements for all medical devices (EN 46001/2)
A) Personnel Are there established, documented and
maintained requirements for health cleanliness clothing
of personnel if contact between such personnel and product or environment could adversely affect the quality of product?
§ 820.70 Production and process controls(d)Personnel
1.The manufacturer has established and maintained requirements for the health, cleanliness, personal prac-tices, and clothing of personnel if contact between such personnel and product or environment could have an adverse effect on product quality.
2.The manufacturer ensures that maintenance and other personnel who are required to work temporarily under special environmental condi-tions are appropriately trained or su-pervised by a trained individual.
QM-SECTION 4.9 PROCESS CONTROL / 3
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 23 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
B) Environmental control in manufac-ture
Are there environmental controls es-tablished and documented for medical devices:a) that are supplied sterile?b) that are supplied non-sterile and
intended for sterilization before use?
c) where the microbiological and/or particulate cleanliness or other environmental conditions are of significance in their use?
d) where the environmental condi-tions are of significance in their manufacture?
If appropriate, are the environmental conditions controlled and/or moni-tored?
Subpart G - § 820.70 Production and process controls
(c)Environmental control
1.Where environmental conditions could reasonably be expected to have an effect on product quality, the manufacturer has established and maintained procedures to adequately control these environmental condi-tions.
2.Environmental control system(s) are periodically inspected to verify that the system, including necessary equipment, is adequate and function-ing properly.
3.These activities are documented and reviewed.
C) Cleanliness of product Are there established, documented
and maintained requirements for cleanliness of the product if: a) (a) the product is cleaned by the
supplier prior to sterilization and/or its use?
b) (b) the product is supplied non-sterile to be subjected to a clean-ing process prior to sterilization and/or its use?
c) (c) the product is supplied to be used non-sterile and its cleanli-ness is of significance in use?
d) (d) process agents are to be re-moved from the product during manufacture?Important: If appropriate, prod-uct cleaned in accordance with a) or b) above need not to be subject to the preceding particu-lar requirements, i.e. A) person-nel and B) environmental control in manufacture, prior to the cleaning procedure.
§ 820.70 Production and process controls (e) Contamination control
The manufacturer has established and maintained procedures to pre-vent contamination of equipment or product by substances that could rea-sonably be expected to have an ad-verse effect on product quality.
(f)Buildings
Buildings are of suitable design and contain sufficient space to perform necessary operations, prevent mix-ups, and assure orderly handling.
(g)Equipment
The manufacturer ensures that all equipment used in the manufacturing process meets specified require-ments and is appropriately designed, constructed, placed, and installed to facilitate maintenance, adjustment, cleaning, and use.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 24 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.9 Process control / 4
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.9 CONTINUATION
D) MaintenanceAre there established and docu-mented requirements for maintenance activities if such activities possibly af-fect product quality?
Are records of such maintenance kept (see Section 4.16)?
§ 820.70 Production and process controls
(1)Maintenance schedule 1.The manufacturer has established
and maintained schedules for the ad-justment, cleaning, and other mainte-nance of equipment to ensure that manufacturing specifications are met.
2.Maintenance activities, including the date and individual(s) performing the maintenance activities, are docu-mented.
(2)Inspection 1.The manufacturer conducts peri-
odic inspections in accordance with established procedures to ensure ad-herence to applicable equipment maintenance schedules.
2.The inspections, including the date and individual(s) conducting the in-spections, are documented.
.
E) Installation If appropriate, are there estab-
lished and documented, both in-structions and acceptance crite-ria for :
installing and, checking the medical device?
Will records of such be retained see Section 4.16?
§ 820.170 Installation
1.The manufacturer of a device re-quiring installation has established and maintained adequate installation and inspection instructions, and where appropriate, test procedures.
2.Instructions and procedures include directions for ensuring proper installa-tion so that the device will perform as intended after installation.
3.The manufacturer distributes the in-structions and procedures with the device or otherwise make them avail-able to the person(s) installing the de-vice.
(b)The person installing the device ensures that the installation, inspec-tion, and any required testing are per-formed in accordance with the manu-facturer’s instructions and procedures and document the inspection and any test results to demonstrate proper in-stallation.
QM-Section 4.9 Process control / 5
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 25 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
F) Special processes If, when necessary, the quality tests
can not be reviewed in complete scope/comprehensively, will these processes be performed through qual-ity personnel and/or the process pa-rameters be constantly reviewed?
Do the quality records identify:a) the work instructions used?,b) the date the special process was
performed?,c) the identity of the operator of the
special process?
§ 820.75 Process validation
1.Where the results of a process can-not be fully verified by subsequent in-spection and test, the process is vali-dated with a high degree of assur-ance and approved according to es-tablished procedures.
2.The validation activities and results, including the date and signature of the individual(s) approving the valida-tion and where appropriate the major equipment validated, are docu-mented.
(b) The manufacturer has established and maintained procedures for moni-toring and control of process parame-ters for validated processes to ensure that the specified requirements con-tinue to be met.
(1)The manufacturer has ensured that the validated processes are per-formed by qualified individual(s).
(2)For validated processes, the moni-toring and control methods and data, the date performed, and where ap-propriate, the individual(s) performing the process or the major equipment used is documented.
(c) 1.When changes or process devi-ations occur, the manufacturer re-views and evaluates the process and performs revalidations where appro-priate..
2. These activities are documented.
Particular requirements for all medical devices (EN 46001/2)
Is the sterilization process validated?
Are all the control parameters of the sterilization process stipulated? ( see Section 4.16)
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 26 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.10 - inspection and testing / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.10.1 General
Are the inspection and testing procedures sufficiently detailed in writ-ing?
Are the required inspection and testing, and the records to be estab-lished, defined in a quality plan or documented procedures?
4.10.2 Receiving inspection and testing
4.10.2.1 Is it ensured that incoming prod-
ucts have been inspected or other-wise verified as conforming to specific requirements?
4.10.2.2 Will consideration be given to the
amount of control exercised at the subcontractor’s premises and the recorded evidence of conformance provided?
4.10.2.3 Does there exist an incoming
product released for urgent pro-duction purposes prior to verifica-tion?
If yes, is there identification in or-der to permit immediate recall ?
Subpart H - Acceptance Activities§ 820.80 Receiving, in-process, and fin-ished device acceptance
(a)General
The manufacturer has established and maintained procedures for ac-ceptance activities.
(b)Receiving acceptance activities 1.The manufacturer has established
and maintained procedures for ac-ceptance of each incoming product.
2.Incoming product is inspected, tested, or otherwise verified as con-forming to specified requirements.
3.Acceptance or rejection is docu-mented.
4.10.3 In process inspection and testing
Are there in-process inspection and testing as required by the quality plan and/or documented procedures?
Will untested products be held back until the required inspection and tests have been completed or necessary re-ports have been verified?
When no, has the product been re-leased under positive-recall proce-dures ( see Section 4.10.2.3)?
(Note, release under positive-recall proce-dures shall not preclude the activities out-lines in Section 4.10.3a above)
Subpart H - Acceptance Activities§ 820.80 Receiving, in-process, and fin-ished device acceptance
(c)In-process acceptance activities
1.The manufacturer has established and maintained acceptance proce-dures, where appropriate, to ensure that specified requirements for in-process product are met.
2.Such procedures ensure that in-process product is controlled until the required inspection and tests or other verification activities have been com-pleted, or necessary approvals are received, and are documented.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 27 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.10 inspection and testing / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.10.4 Final inspection and testing
Is all final inspection and test-ing carried out in accordance with the quality plan and/or documented proce-dures?
Has the final inspection and test-ing been fulfilled?
Is there evidence of confor-mance of the finished product to the specified requirements in accordance with the quality plan or documented procedures?
Subpart H - Acceptance Activities§ 820.80 Receiving, in-process, and fin-ished device acceptance
(d)Final acceptance activities 1.The manufacturer has established
and maintained procedures for fin-ished device acceptance to ensure that each production run, lot, or batch of finished devices meets acceptance criteria.
2.Finished devices are held in quar-antine or otherwise adequately con-trolled until released.
3.Finished devices are not released for distribution until: The activities required in the
DMR are completed; The associated data and docu-
mentation is reviewed; The release is authorized by the
signature of designated individ-ual(s);
The authorization is dated.
4.10.5 Inspection and test records
Please refer to Section 4.16.
Is it evident from the records, where the responsible testing
place is? that the products have fulfilled
the necessary criteria? that whereby non-conforming
products, that failed to pass any inspection and/or test, the pro-cedures for control of non-con-forming product apply?
Subpart H - Acceptance Activities§ 820.80 Receiving, in-process, and fin-ished device acceptance
(e)Acceptance records 1.The manufacturer documents the
acceptance activities. 2.These records include:
The acceptance activities per-formed;
The dates the acceptance activi-ties are performed;
The results; The signatures of the
individual(s) conducting the ac-ceptance activities;
Where appropriate, the equip-ment used.
3.These records are part of the DHR.
Particular requirements for active medi-cal devices and implantable medical de-vices (EN 46001/2)
Where are the identity records of personnel performing inspections or testing?
Is there a requirement for storage/keeping of identity records (see Section 4.16, Identification)?
QM-Section 4. 11 - control inspection, measuring and test equipment / 1
QUESTIONS COMMENTS
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 28 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.11.1 General Are there documented proce-
dures for the control calibration maintenance of inspectionin the measuring and test equipment?
820.70 Inspection, measuring, and test equipment(a)Control of inspection, measuring, and test equipment1.The manufacturer ensures that all inspec-tion, measuring, and test equipment, in-cluding mechanical, automated, or elec-tronic inspection and test equipment, is suitable for its intended purposes and is capable of producing valid results.3.The procedures include provisions for han-dling, preservation, and storage of equip-ment so that its accuracy and fitness for use are maintained.4.These activities are all documented.
In the control, inspection, mea-suring and testing are the following used: test software comparative references
When yes, will they be checked prior to release for use
during production? rechecked at prescribed inter-
vals? establishment of the extent and
frequency of such tests?
4.11.2 Control procedure Is there determination of the measurements to be undertaken? What is the accuracy required? Is there selection of the appro-
priate inspection, measuring and test equipment?
How is the selection of such ap-propriate inspection, measuring and test equipment?
Is this selection of the appropri-ate inspection, measuring and test equipment capable of the necessary accuracy and preci-sion?
QM-Section 4. 11 - control inspection, measuring and test equipment / 2
QUESTIONS COMMENTS
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 29 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
Are all inspection, measuring and test equipment identified, calibrated or adjusted?
Is this undertaken at prescribed intervals or prior to use?
Is the inspection, measuring and test equipment calibrated and adjusted against certified equipment having a known valid relationship? (With re-spect to internationally or nationally recognized standards!).
In the case where no such stan-dards exist, is the basis used for cali-bration documented?
820.70 Inspection, measuring, and test equipment (b)
Calibration1.Calibration procedures include specific di-rections and limits for accuracy and preci-sion.2.When accuracy and precision limits are not met, there are provisions for remedial action to reestablish the limits and to eval-uate whether there was any adverse effect on the device’s quality.3.These activities are documented.(1)Calibration standardsCalibration standards used for inspection, measuring, and test equipment are trace-able to national or international standards. If national or international standards are not practical or available, the manufacturer uses an independent, reproducible stan-dard. If no applicable standard exists, the manufacturer has established and main-tained an in-house standard.
Is the calibration process defined? (in-cluding details of equipment type, unique identification, location, fre-quency of checks, check method, ac-ceptance criteria and the action to be taken when results are unsatisfactory)
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 30 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4. 11 - CONTROL OF INSPECTION, MEASURING AND TEST EQUIPMENT / 3
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.11.2 CONTINUATION
How is the process which is em-ployed for the calibration of inspec-tion, measuring and test equipment defined? (e.g. equipment type, color code, unique identification )
820.70 Inspection, measuring, and test equipment
(2)Calibration records (b)1.The equipment identification, cal-
ibration dates, the individual perform-ing each calibration, and the next cali-bration date are documented.
Where are the calibration records for inspection, measuring and test equip-ment maintained?
Please refer to Section 4.16?
820.70 Inspection, measuring, and test equipment
2.These records are displayed on or near each piece of equipment or are readily available to the personnel us-ing such equipment and to the indi-viduals responsible for calibrating the equipment.
Will the validity of previous inspection and test results when inspection, mea-suring and test equipment, is found to be out of calibration, be accessed and documented? (if necessary new tests)
820.70 Inspection, measuring, and test equipment
(b)Calibration 2.When accuracy and precision limits
are not met, there are provisions for remedial action to reestablish the lim-its and to evaluate whether there was any adverse effect on the device’s quality.
3.These activities are documented.
Are the environmental conditions suitable for the calibrations, inspec-tions, measurements and tests be-ing carried out?(e.g. is a climatizied room necessary or practical?)
Is it ensured that the handling, preser-vation and storage of inspection, mea-suring and test equipment (inclusive of test hardware and software) is such that the accuracy and fitness for use are maintained?
How is the inspection, measuring and test facilities, including both the test hardware and software, safe-guarded from adjustments that would invalidate the calibration set-ting?
820.70 Inspection, measuring, and test equipment(a)Control of inspection, measuring, and test equipment 2.The manufacturer has established
and maintained procedures to ensure that equipment is routinely calibrated, inspected, checked, and maintained.
3.The procedures include provisions for handling, preservation, and stor-age of equipment so that its accuracy and fitness for use are maintained.
4.These activities are all docu-mented.
QM-Section 4.12 inspection and test status
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where Documentation Sc Practice Sc
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 31 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
found
4.12 Inspection and test status
How is the identification and test status maintained?
Subpart H - § 820.80 Receiving, in-process, and finished device acceptance§ 820.88 Acceptance status
1.The manufacturer has identified by suitable means the acceptance sta-tus of product.
2.The identification of acceptance status is maintained throughout man-ufacturing, packaging, installation, and servicing of the product to ensure that only product which has passed the required acceptance activities is distributed, used, or installed.
Is the identification of inspection and test status maintained throughout production, installation and servicing of the product? (e.g. in the quality plan and/or documented procedures)
Is such identification defined in the quality plan and/or documented procedures?
How is it ensured, in the case of prod-ucts (produced, installed or serviced) that are released under an authorized concession (see Section 4.13.2)?
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 32 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.13 - control of nonconforming product / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.13.1 General
How is it ensured that products that do not conform to specified require-ments are prevented from unin-tended use or installation? (e.g. in documented procedures)
How does this control provide for . identification, documentation, evaluation, segregation (when practical),disposition of nonconforming prod-uct and for notification to the func-tions concerned?
Subpart I – Nonconforming Product - 820.90
(a)Control of nonconforming product 1.The manufacturer has established
and maintained procedures to control product that does not conform to specified requirements.
2.The procedures address the identi-fication, documentation, evaluation, segregation, and disposition of non-conforming product.
3.The evaluation of nonconformance includes a determination of the need for the persons or organizations re-sponsible for the nonconformance.
4.The evaluation and any investiga-tion is documented.
4.13.2 Review and disposition of non-conforming product
Is the responsibility for review and authority for the deposition of nonconforming product defined?
(b)Nonconformity review and disposition 1.The manufacturer has established
and maintained procedures that de-fine the responsibility for review and the authority for the disposition of nonconforming product.
Is it ensured (in accordance with doc-umented procedures) how a noncon-forming product is reviewed?This review maybe reworked accepted with or without repair
by concession regarded for alternative applica-
tion rejected or scrapped
2.The procedures set forth the review
and disposition process.
3.Disposition of nonconforming prod-uct is documented.
Shall repaired/reworked product con-ditions be newly tested?
5.The manufacturer has established
and maintained procedures for re-work, to include retesting and reeval-uation of the nonconforming product after rework, to ensure that the prod-uct meets its current approved speci-fications.
Where required by the contract, shall the proposed use or repair of product, which does not conform to specified requirements, be reported for concession to the customer or cus-tomer’s representative?
How is the accepted description of nonconformity, and of repairs, recorded to denote the actual condi-tion?
(see Section 4.16?)
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 33 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.13 - control of nonconforming product / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.13.2 CONTINUATION
Particular requirements for all medical devices (EN 46001/2)
How is it stipulated that a nonconform-ing product is accepted by concession only if regulatory requirements are met?
Where is recorded the identity of the person authorizing the concession?
4.Documentation includes the justifi-cation for use of nonconforming prod-uct and the signature of the individ-ual(s) authorizing the use.
Is the reworking of a product in a work instruction documented?
Has this rework, of a product, in a work instruction undergone the same authorization and approval pro-cedure as the original work instruc-tion?
6.Rework and reevaluation activities,
including a determination of any ad-verse effect from the rework upon the product, is documented in the DHR.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 34 / 57
Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.14 - corrective and preventive action / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.14.1 General
Is there an established and main-tained documented procedure for im-plementing corrective and preventive action?
1. The manufacturer has established and maintained procedures for imple-menting corrective and preventive ac-tion.
4.14.2 Corrective action Do the procedures for corrective
action include the following aspects: an effective handling of customer
complaints and reports of prod-uct non-conformities?
Failure analysis w.r.t. product, process and QM-system also?
Will results of such investigations be recorded (Section 4.6)?
Is there determination of corrective action needed to eliminate the cause of non-conformities?
Is there application of controls to en-sure that corrective action is taken and that it is effective?
2.The procedures include require-ments for: Analyzing processes, work op-
erations, concessions, quality audit reports, quality records, service records, complaints, re-turned product, and other sources of quality data to iden-tify existing and potential causes of nonconforming product, or other quality problems;
Investigating the cause of non-conformities relating to product, processes, and the quality sys-tem;
Identifying the action(s) needed to correct and prevent recur-rence of nonconforming product and other quality problems;
Verifying or validating the cor-rective and preventive action to ensure that such action is effec-tive and does not adversely af-fect the finished device;
Implementing and recording changes in methods and proce-dures needed to correct and prevent identified quality prob-lems;
Ensuring that information related to quality problems or noncon-forming product is disseminated to those directly responsible for assuring the quality of such product or the prevention of such problems;
Submitting relevant information on identified quality problems, as well as corrective and pre-ventive actions, for management review.
(b) All activities required under this section, and their results, are docu-mented.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 35 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.14 - corrective and preventive action / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
Particular requirements for all medical devices (EN 46001/2)
Is there an established and main-tained documented feedback system to provide early warning of quality problems and for the input into the corrective action system
Is this corrective action system in compliance with the regulatory re-quirements of the authorities in the dif-ferent countries
DIRECTIVE 93/42/EEC (ARTICLE 10)
Can also characteristics/marks and instructions be recalled/withdrawn?
(a)1.The manufacturer maintains complaint files.
2.The manufacturer has established and maintained procedures for re-ceiving, reviewing, and evaluating complaints by a formally designated unit.
3.Such procedures ensure that: All complaints are processed in
a uniform and timely manner; Oral complaints are documented
upon receipt; and Complaints are evaluated to de-
termine whether the complaint represents an event which is re-quired to be reported to FDA un-der part 803 or 804, MDR.
(b) 1.The manufacturer reviews and evaluates all complaints to determine whether an investigation is neces-sary. When no investigation is made, the manufacturer maintains a record which includes the reason no investi-gation was made and the name of the individual responsible for the decision not to investigate.
(c) Any complaint involving the possi-ble failure of a device, labeling, or packaging to meet any of its specifi-cations is reviewed, evaluated, and investigated, unless such investiga-tion has already been performed for a similar complaint and another investi-gation is not necessary.
QM-SECTION 4.14 - CORRECTIVE AND PREVENTATIVE ACTION / 3
QUESTIONS COMMENTS
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 36 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.14.3 Preventive action
Do the procedures for preventive ac-tion include the following aspects:
use of appropriate sources of infor-mation, such as:
processes and work operationsconcessionsservice reportsaudit resultsquality recordscustomer complaints to detect , analyze and eliminate potential causes of non-conformities?
Are steps determined to deal with any problems requiring preventive action?
(d) 1.Any complaint that represents an event which must be reported to FDA under part 803 or 804 of this chapter is promptly reviewed, evalu-ated, and investigated by a desig-nated individual(s) and is maintained in a separate portion of the complaint files or otherwise clearly identified.
2.In addition to the information re-quired by § 820.198(c), records of in-vestigation under this paragraph in-clude a determination of: Whether the device failed to
meet specifications; Whether the device was being
used for treatment or diagnosis; and
The relationship, if any, of the device to the reported incident or adverse event.
(e) 1.When an investigation is made under this section, a record of the in-vestigation is maintained by the for-mally designated unit identified in paragraph (a) of this section.
QM-SECTION 4.14 - CORRECTIVE AND PREVENTATIVE ACTION / 4
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 37 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
820.198 Complaint files
(a)1.The manufacturer maintains complaint files.
2.The manufacturer has established and maintained procedures for re-ceiving, reviewing, and evaluating complaints by a formally designated unit.
3.Such procedures ensure that:
All complaints are processed in a uniform and timely manner;
Oral complaints are documented upon receipt; and
Complaints are evaluated to de-termine whether the complaint represents an event which is re-quired to be reported to FDA un-der part 803 or 804, MDR.
(b) 1. The manufacturer reviews and evaluates all complaints to determine whether an investigation is neces-sary. When no investigation is made, the manufacturer maintains a record which includes the reason no investi-gation was made and the name of the individual responsible for the decision not to investigate.
(c) (1) Any complaint involving the possible failure of a device, labeling, or packaging to meet any of its speci-fications is reviewed, evaluated, and investigated, unlesssuch investiga-tion has already been performed for a similar complaint and another investi-gation is not necessary.
QM-SECTION 4.14 - CORRECTIVE AND PREVENTATIVE ACTION / 5
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
How is it ensured that relevant infor-mation on actions taken is submitted for management review (4.1.3)?
2.The record of investigation in-cludes: The name of the device; The date the complaint was re-
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 38 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
ceived; Any device identification(s) and
control number(s) used; The name, address, and phone
number of the complainant; The nature and details of the
complaint; The dates and results of the in-
vestigation; Any corrective action taken; and Any reply to the complainant.
(f) When the manufacturer’s formally designated complaint unit is located at a site separate from the manufac-turing establishment, the investigated complaint(s) and the record(s) of in-vestigation are reasonably accessible to the manufacturing establishment.
(g)If a manufacturer ‘s formally desig-nated complaint unit is located out-side of the United States, records re-quired by this section are reasonably accessible in the United States at ei-ther:
A location in the U.S. where the manufacturer’s records are reg-ularly kept; or
The location of the initial distrib-utor.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 39 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.15 - handling, storage, packaging, preservation and delivery / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.15.1 General
Are there established and main-tained documented procedures for handling, storage, packaging, preser-vation and delivery of product?
Particular requirements for all medical devices (EN 46001/2)
Are there documented procedures for the control of product or products with a limited shelf life or requiring special storage conditions?
Are such special storage conditions controlled and recorded?
Are there special provision made for the handling of used products in order to prevent contamination of other product, the manufacturing environ-ment or personnel (in so far as achievable)?
4.15.2 Handling
Are the methods such to prevent product handling and deterioration?
§ 820.140 Handling
The manufacturer has established and maintained procedures to ensure that mixups, damage, deterioration, contamination, or other adverse ef-fects to product do not occur during handling.
4.15.3 Storage
Are there designated storage areas or stock rooms to prevent damage or de-terioration of product, pending use or delivery?
Are the storage areas or stock rooms sufficiently clearly identified?
Is there appropriate methods stipu-lated for authorizing:
receipt? store? dispatch?
Are the intervals of stock assessment appropriate.
§ 820.150 Storage
1.The manufacturer has established and maintained procedures for the control of storage areas and stock rooms for product to prevent mixups, damage, deterioration, contamination, or other adverse effects pending use or distribution and to ensure that no obsolete, rejected, or deteriorated product is used or distributed.
2.When the quality of product deterio-rates over time, it is stored in a man-ner to facilitate proper stock rotation, and its condition is assessed as ap-propriate.
(b)The manufacturer has established and maintained procedures that de-scribe the methods for authorizing re-ceipt from and dispatch to storage ar-eas and stock rooms.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 40 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.15 - Handling, storage, packaging, preservation and delivery / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.15.4 Packaging
During the processes of: packing, packaging and marking processes, including materials used
is necessary control exercised?
Is this control to such an extent to en-sure necessary conformance to speci-fied requirements?
§ 820.130 Device packaging
The manufacturer ensures that de-vice packaging and shipping contain-ers are designed and constructed to protect the device from alteration or damage during the customary condi-tions of processing, storage, handling, and distribution.
Particular requirements for all medical devices (EN 46001/2)
How is it assured that; the medical device is presented in a
container which maintains the sterility of the medical device?
the medical device is capable of being presented in an aseptic manner (if its use so requires)?
the package, or medical device if only the inner surface is sterile, clearly re-veals that it has been opened?
Subpart K - § 820.120 Device labeling
The manufacturer has established and maintained procedures to control labeling activities.
(a)Label integrity Labels are printed and applied so as
to remain legible and affixed during the customary conditions of process-ing, storage, handling, distribution, and where appropriate use.
(b)Labeling inspection 1.Labeling is not released for storage
or use until a designated individual(s) has examined the labeling for accu-racy, including, where applicable, the correct expiration date, control num-ber, storage instructions, handling in-structions, and any additional pro-cessing instructions.
2.The release, including the date and signature of the individual(s) perform-ing the examination, are documented in the DHR.
(c)Labeling storage The manufacturer stores labeling in a
manner that provides proper identifi-cation and is designed to prevent mix-ups.
(d)Labeling operations 1.The manufacturer controls labeling
and packaging operations to prevent labeling mixups.
2.The label and labeling used for each production unit, lot, or batch is documented in the DHR.
(e)Control number Where a control number is required
by § 820.65, that control number is on or accompanies the device through distribution.
QM-Section 4.15 - Handling, storage, packaging, preservation and delivery / 3
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 41 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
Additional requirements for active im-plantable medical devices and im-plantable medical devices (EN 46001/2)
Where is recorded the identity of per-sons who perform the final labeling operation?
DIRECTIVE 93/42/EEC
ANNEX l / Sterile products or products for prior sterilization:
Does the packaging ensure mainte-nance of the medical devices until use (8.3)?
Is the packaging of non-sterile de-vices suitable to ensure a suitable level of cleanliness until prior steriliza-tion (8.6)?
Does the packaging/label distinguish between identical and similar products in both sterile and non-sterile condi-tions (8.7)?
Does the label indicate the recom-mended word STERILE according to EN 980 (13.3)?
4.15.5 Preservation
Are methods appropriate? Is there segregation (where neces-
sary) of the products?
4.15.6 Delivery
Is there an arrangement for the protec-tion of the quality of product after final inspection and test?
Is it contractually specified, that this protection shall be extended to include delivery to destination?
§ 820.160 Distribution
1.The manufacturer has established and maintained procedures for control and distribution of finished devices to ensure that only those devices ap-proved for release are distributed and that purchase orders are reviewed to ensure that ambiguities and errors are resolved before devices are re-leased for distribution.
2.Where a device’s fitness for use or quality deteriorates over time, the pro-cedures ensure that expired devices or devices deteriorated beyond ac-ceptable fitness for use are not dis-tributed.
(b)The manufacturer maintains distri-bution records which include or refer to the location of: The name and address of the
initial consignee; The identification and quantity of
devices shipped; The date shipped; and Any control number(s) used.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 42 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.15 - Handling, storage, packaging, preservation and delivery / 4
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
Additional requirements for active im-plantable medical devices and im-plantable medical devices (EN 46001/2)
Where is recorded the name and ad-dress of the shipping package con-signee?
Is this record a section of the quality record (see Section 4.16)?
Is the audited firm required that any unauthorized representative maintains records of distribution of medical de-vices and that such records are avail-able for inspection?
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 43 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.16 control of quality records / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.16 Controf quality records
Is there an established and main-tained documented procedure for quality records?
Does the contents of this documented procedure include identification? collection? indexing? access? filing? storage? maintenance? disposition? (e.g. matrix or list for quality symbols)
Through the quality records are the following demonstrated conformance to specified require-
ments and effective operation of the quality
system?
Is it ensured that quality records are legibleand readily retrievable?
Are the facilities and conditions for filing and storage, such to provide a suitable environment to prevent damage or deterioration and to pre-vent loss?
Where agreed contractually, are qual-ity records made available for evalua-tion by the customer or the customer’s representative for an agreed period?
Subpart M - Records
820.180 General requirements
1. All records required by this part are maintained at the manufacturing es-tablishment or other location that is reasonably accessible to responsible officials of the manufacturer and to employees of FDA designated to per-form inspections.
2. Such records are legible and are stored to minimize deterioration and to prevent loss.
3. Those records stored in automated data processing systems are backed up.
Confidentiality
(b)Record retention period; All records required by this part are re-tained for a period of time equivalent to the design and expected life of the device, but in no case less than 2 years from the date of release for commercial distribution by the manu-facturer.
(c) Exceptions: Upon request of a designated employee of FDA, an em-ployee in management with executive responsibility certifies in writing that the management reviews, quality au-dits, and supplier audits have been performed and documented, the dates on which they were performed, and that any required corrective ac-tion has been undertaken.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 44 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.16 control of quality records / 2
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.16 CONTINUATION
Particular requirements for all medical devices (EN 46001/2)
Are the quality records retained for a time at least equivalent to the lifetime of the medical device defined by the supplier, but not less than 2 years from the date of dispatch from the supplier?
Is it stipulated, which records - corre-sponding to the requirement of Sec-tion 4.8/traceability - for each product, each appointment/post, each share/portion,which must be kept?
Do the records show the manufac-tured and released amount?
820.181 Device master record
1.The manufacturer maintains device master records (DMR’s).
2.The manufacturer ensures that each DMR is prepared and approved in accordance with § 820.40.
3.The DMR for each type of device includes or refers to the location of the following information:
Device specification including appropriate drawings, composi-tion, formulation, component specifications, and software specifications;
Production process specifica-tions including the appropriate equipment specifications, pro-duction methods, production procedures, and production en-vironment specifications;
Quality assurance procedures and specifications including ac-ceptance criteria and the quality assurance equipment to be used;
Packaging and labeling specifi-cations, including methods and processes used; and
Installation, maintenance, ser-vicing procedures & methods.
QM-Section 4.16 control of quality records / 3
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 45 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
Will the records be verified and signed (authorized)?Important: The standard explicitly sets down, in each element the least quality record standard in a QM sys-tem.
Are there respective standards w.r.t.: QM-valuation (4.1.3) contract review (4.3.4) design control (4.4.6) design verification (4.4.7) evaluation of subcontractors
(4.6.2) control of customer-supplied
product (4.7) identification - where necessary -
(4.8) process control (4.9) Preliminary release in incoming
products (4.10.2.3) test records (4.10.5) control of inspection, measuring
and test equipment (4.11.1) calibration (4.11.2 e) concession (4.13.2) corrective action (4.14.2) results of internal quality audits
(4.17) corrective action in following au-
dits (4.17) training (4.18)
820.184 Device history record
1.The manufacturer maintains device history records (DHR’s).
2.The manufacturer has established and maintained procedures to ensure that DHR’s for each batch, lot, or unit are maintained to demonstrate that the device is manufactured in accor-dance with the DMR and the require-ments of this part.
3.The DHR includes, or refers to the location of, the following information:
The dates of manufacture; The quantity manufactured; The quantity released for distri-
bution; The acceptance records which
demonstrate the device is man-ufactured in accordance with the DMR;
The primary identification label and labeling used for each pro-duction unit; and
Any device identification(s) and control number(s) used.
Are these the minimal considerations to be observed w.r.t. the quality con-trol process? 820.188 Quality system record
1.The manufacturer maintains a qual-ity system record (QSR).
2.The QSR includes, or refers to the location of, procedures and the docu-mentation of activities required by this part that are not specific to a particu-lar type of devices(s), including, but not limited to, the records required by § 820.20.
3.The manufacturer ensures that the QSR is prepared and approved in ac-cordance with § 820.40.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 46 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.17 Internal Quality audits / 1
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.17 Internal Qulaity Audits
Are there documented procedures for planning and implementing in-ternal quality audits?
Do these procedures comply with planned arrangements/ schedules?
Subpart B – Quality System Require-ments820.22 Quality Audit
1.The manufacturer has established procedures for quality audits and has conducted such audits.
Are the internal audits carried out by personnel independent of those having direct responsibility for the activity being audited?
2.The quality audits are conducted by individuals who do not have direct re-sponsibility for the matters being au-dited.
Are the internal audits carried out by sufficiently qualified personnel (see paragraph 4.1.2.2)?
Will the results of the internal audits be recorded? brought to the attention of the
personnel having responsibility in the audited area?
brought to the attention of the highest management personnel ( also for the QM assess-ment?)
5.The dates and results of quality au-dits and reaudits are documented.
Do the responsible management per-sonnel take timely corrective action on deficiencies found during the audit?
4.A report of each quality audit, and reaudits, are made and reviewed by management having responsibility for the matters being audited.
Do follow-up audit activities verify and record the implementation and effectiveness of the corrective ac-tion taken?
Will the corresponding results of the audits and corrective actionbe registered? ( 4.16?)
3.Corrective action(s), including a reaudit of deficient matters, are taken when necessary.
QM-Section 4.17 Internal Quality audits / 2Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 47 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.17 CONTINUATION
DIRECTIVE 93/42/EEC
ANNEX ll, V, VI 3.2 b)
Will the functioning of the QM system be regularly investigated?
In the frame of the internal audit, is the QM system investigated to see if it is suited for the quality lay out and prod-uct?
Will it be invetsigated in internal audits if the QM system is suited for identifi-cation of non-conforming products?
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 48 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.18 TRAINING
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.18 Training
Are there documented procedures for identifying training needs?
Is this in an established manner?
Subpart B – Quality System Require-ments820.25 Personnel
1.There are procedures for identifying training needs and ensuring that all personnel are trained to adequately perform their assigned responsibili-ties.
2.This training is documented. 3.As part of their training, personnel
are made aware of device defects which may occur from the improper performance of their specific jobs.
4.Personnel who perform verification and validation activities are made aware of defects and errors that may be encountered as part of their job functions.
Are the personnel performing activi-ties affecting quality trained?
Will the means of training be as-sessed?
3.As part of their training, personnel are made aware of device defects which may occur from the improper performance of their specific jobs.
Are the personnel performing spe-cific assigned tasks qualified on the basis of appropriate education?, appropriate training? and/or experience?
4.Personnel who perform verification and validation activities are made aware of defects and errors that may be encountered as part of their job functions.
Particular requirements for all medical devices (EN 46001/2)
Are personnel that work under special environment
conditions, who performs special processes or
functions, appropriately trained?
or is it the case that: they are supervised by a trained per-
son?
training certificates are recorded and put in file?
Is this record a section of the quality records (see Section 4.16)?
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 49 / 57
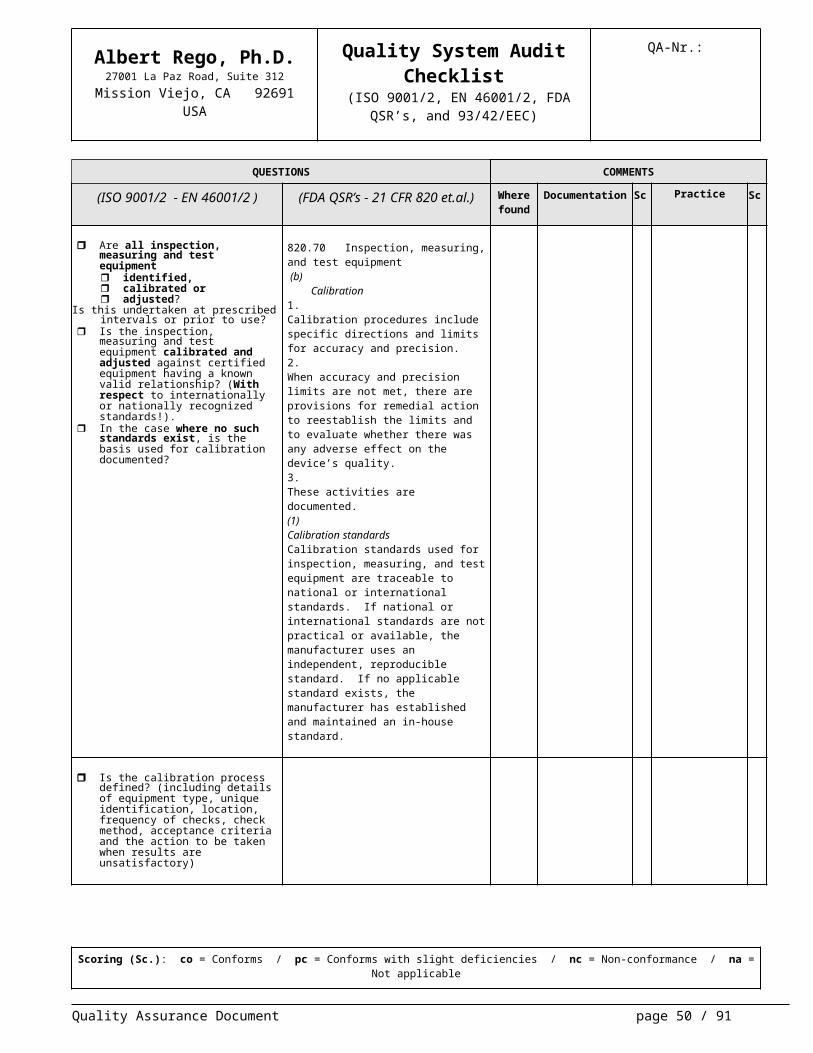
Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-SECTION 4.19 SERVICING
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.19 Servicing
Is servicing a specified require-ment?
If yes, are the procedures through performance and testing stipulated?
Are such documented in a procedure?
In the description of the docu-mented procedures are the following considered
performance of the activity?
verification of the activity?
reporting that the servicing meets the specified requirements?
Subpart N - 820.200
(a)Where servicing is a specified re-quirement, the manufacturer has es-tablished and maintained instructions and procedures for performing and verifying that the servicing meets the specified requirements.
(b)The manufacturer analyzes ser-vice reports with appropriate statisti-cal methodology in accordance with § 820.100.
(c)The manufacturer who receives a service report that represents an event which must be reported to FDA under part 803 or 804 of this chapter automatically considers the report a complaint and processes it in accor-dance with the requirements of § 820.198.
(d)Service reports are documented and include: The name of the device ser-
vices; Any device identification(s) and
control number(s) used; The date of service; The individual(s) servicing the
device; The service performed; and The test and inspection data.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 50 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
QM-Section 4.20 Statistical techniques
QUESTIONS COMMENTS
(ISO 9001/2 - EN 46001/2 ) (FDA QSR’s - 21 CFR 820 et.al.) Where found
Documentation Sc Practice Sc
4.20.1 Identification of need
Is the identification of statistical techniques a need?
How is need for statistical tecnique required identified w.r.t.:
establishing, controlling, verifyingprocess capability and product char-acteristics?
Subpart O - 820.250
(a) Where appropriate, the manufac-turer has established and maintained procedures for identifying valid statis-tical techniques required for estab-lishing, controlling, and verifying the acceptability of process capability and product characteristics.
Particular requirements for all medical devices (EN 46001/2)
Are there established and main-tained procedures to ensure that sam-pling methods are regularly reviewed in the light of;
the occurrence of non-conforming products
quality audit reports (see Section 4.17),
feedback information (see Section 4.14),
and other appropriate considerations?
4.20.2 Procedures
Are there documented procedures to
implement control
the application of the statistical proce-dures?
Is it stipulated where the respective methods for application originate?
Are the concerned personnel suffi-ciently trained/qualified?
Subpart O - 820.250
(b) 1.Sampling plans, when used, are written and based on a valid statisti-cal rationale.
(b) 2.The manufacturer has estab-lished and maintained procedures to ensure that sampling methods are adequate for their intended use and to ensure that when changes occur the sampling plans are reviewed.
(b) 3.The activities are documented.
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 51 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
AUDIT - SUMMARY POSITIVE ASPECTS OF THE QM-SYSTEMS
COMMENTS / POTENTIAL FOR IMPROVEMENTS
DEFICIENCIES
GENERAL COMMENTS
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 52 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
Scoring (Sc.): co = Conforms / pc = Conforms with slight deficiencies / nc = Non-conformance / na = Not applicable
Quality Assurance Document page 53 / 57

Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
Deviation Nr. of
Examined firm Date Lead Auditor
QM-Standard Aspect (Section of standard/ Council Directive 93/42/EEC)
Stipulated deviations
Corrective procedures from audited firm
Corrective procedures will be carried out until Responsible signature of the audited firm
Corrective procedures carried out on Responsible signature of the audited firm
Endorsement of consideration
Date Signature
Quality Assurance Document Page 54 of 57
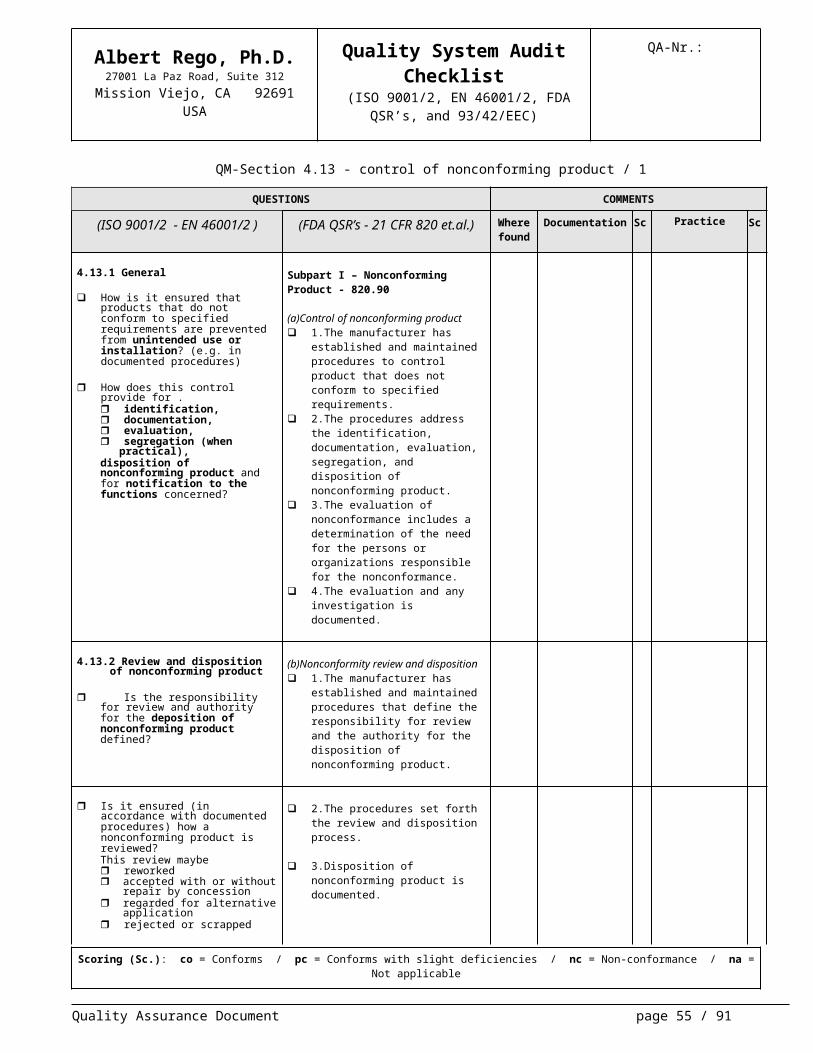
Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
List of viewed/copied documents and records
Document / record (title, nr.) Rel.-edition / date
department/section location of plant/firm
V viewed
C copy
Quality Assurance Document Page 55 of 57
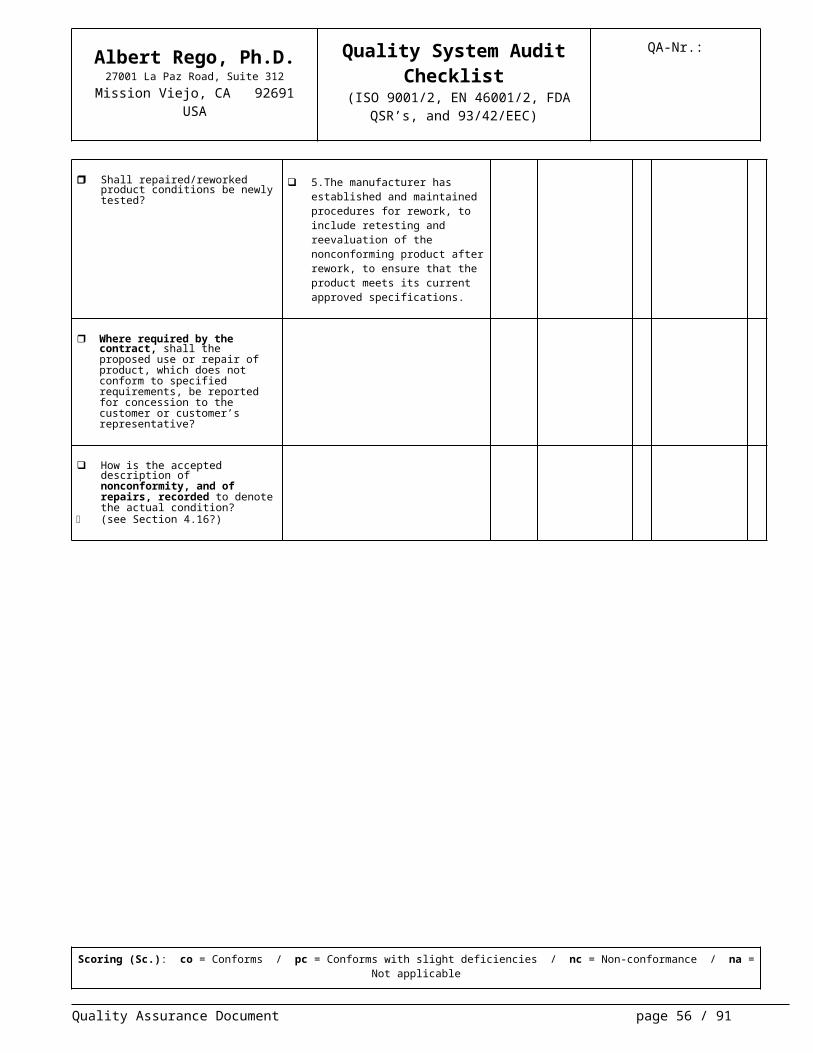
Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
List of the participants
Name Department/section/function Comments
Quality Assurance Document Page 56 of 57
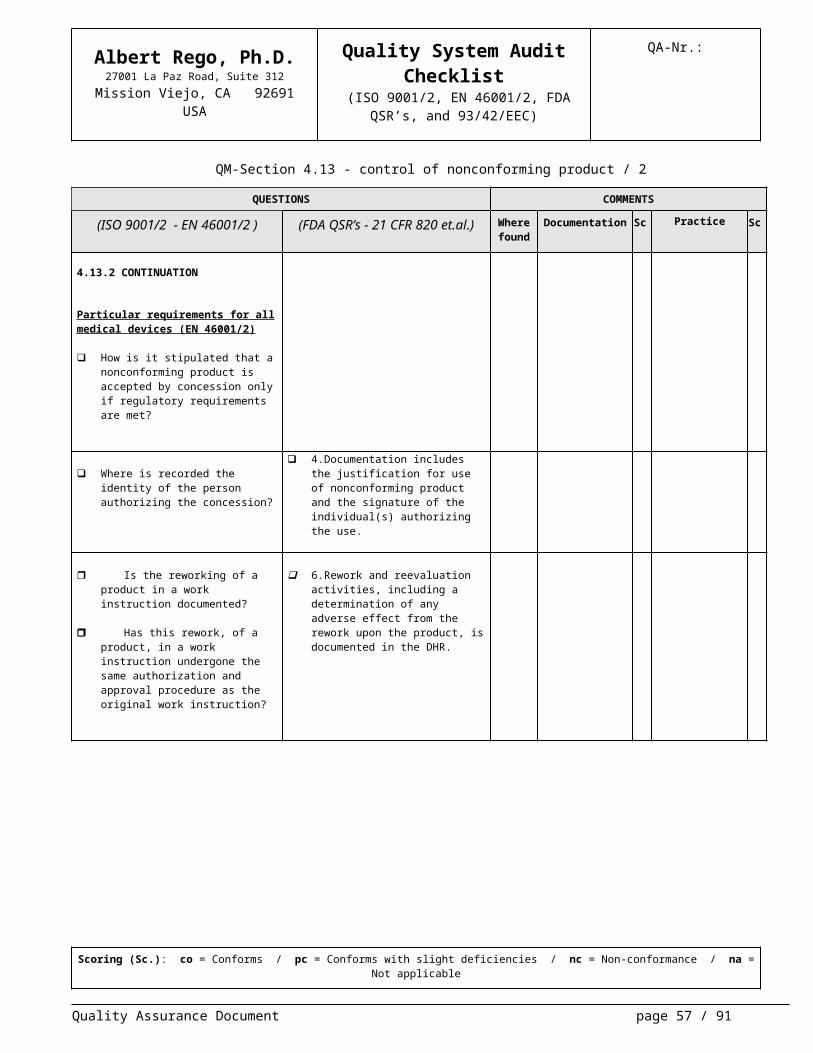
Albert Rego, Ph.D.27001 La Paz Road, Suite 312
Mission Viejo, CA 92691USA
Quality System Audit Checklist
(ISO 9001/2, EN 46001/2, FDA QSR’s, and 93/42/EEC)
QA-Nr.:
Quality Services Audit Report Summary
_________________________________________
_________________________
Date of the Audit: ______________Representatives: ______________ Lead Auditor
Scope and Aim of the Quality Audit
The audit performed on __________ at ______________ located in ___________ was designed to evaluate the quality system implemented according to the FDA Quality System Requirements (QSR’s) as given in 21 CFR § 820, ISO 9001/2, and EN 46001/2. There was a particular emphasis on the use of the Linvatec Supplier Selection Review form, SOP 06204.
The scope and aim of the audit is for the qualification of this supplier for ______________.
Critical Observations: 0Major Observations: 0 (Major items marked as Form/ Fit Function/ Safety must be corrected)Minor Observations: 0Recommendations: 0
City, StateDate
____________________Lead Auditor and Expert
Quality Assurance Document Page 57 of 57


![AR01...2012/03/06 · Annual Return, Mar. 06, 2012. Companies House.] AR01 Annual Return (For returns made up to a date on or after 1 October 2011) w Company typeo Please confirm](https://img.pdfslide.us/doc/110x75/5f565ca97a6540408437eb8d/ar01-20120306-annual-return-mar-06-2012-companies-house-ar01-annual.jpg)


























