Embed Size (px)
Citation preview

Asf1 Is Required for Viability and Chromatin Assembly duringDNA Replication in Vertebrate Cells*
Received for publication, October 26, 2005, and in revised form, March 14, 2006 Published, JBC Papers in Press, March 14, 2006, DOI 10.1074/jbc.M511590200
Fumiyuki Sanematsu‡, Yasunari Takami‡, Hirak Kumar Barman‡, Tatsuo Fukagawa§, Tatsuya Ono¶,Kei-ichi Shibahara¶, and Tatsuo Nakayama‡�1
From the ‡Section of Biochemistry and Molecular Biology, Department of Medical Sciences, Miyazaki Medical College,University of Miyazaki, 5200 Kihara, Kiyotake, Miyazaki 889-1692, Japan, the ¶Department of Integrated Genetics,National Institute of Genetics, 1111 Yata, Mishima, Shizuoka 411-8540, Japan, the §Department of Molecular Genetics,National Institute of Genetics, 1111 Yata, Mishima, Shizuoka 411-8540, Japan, and the �Department of Life Science,Frontier Science Research Center, University of Miyazaki, 5200, Kihara, Kiyotake, Miyazaki 889-1692, Japan
Asf1 (anti-silencing function 1), a well conserved protein fromyeast to humans, acts as a histone chaperone and is predicted toparticipate in a variety of chromatin-mediated cellular processes.To investigate the physiological role of vertebrate Asf1 in vivo, wegenerated a conditional Asf1-deficient mutant from chicken DT40cells. Induction of Asf1 depletion resulted in the accumulation ofcells in S phase with decreased DNA replication and increasedmitoticaberrancy formingmultipolar spindles, leading tocelldeath. Inaddition, nascent chromatin in Asf1-depleted cells showed increasednuclease sensitivity, indicating impaired nucleosome assembly dur-ing DNA replication. Complementation analyses revealed that thefunctional domain of Asf1 for cell viability was confined to theN-terminal core domain (amino acids 1–155) that is a binding plat-form for histones H3/H4, CAF-1p60, and HIRA, whereas Asf1mutant proteins, abolishing binding abilities with both p60 andHIRA, exhibit no effect on viability. These results together indicatethat the vertebrate Asf1 plays a crucial role in replication-coupledchromatin assembly, cell cycle progression, and cellular viabilityand provide a clue of a possible role in a CAF-1- and HIRA-inde-pendent chromatin-modulating process for cell proliferation.
During S phase, newly synthesized histones H3-H4 are assembledbehind the replication fork, followed by loading of the H2A-H2B dimerto complete de novo nucleosome formation on newly replicated DNA(1–3). Besides such a replication-coupled chromatin assembly process,a DNA synthesis-independent chromatin assembly process also existsto operate histone deposition during transcription and DNA repair (4).These processes aremediated by several specialized histone chaperones(1). CAF-1 (chromatin assembly factor-1), a trimeric protein complexconsisting of p150, p60, and p48 subunits, is the most characterizedchaperone responsible for loading of histones H3 and H4 onto replicat-ing DNA through an interaction with proliferating cell nuclear antigen(PCNA),2 the DNA polymerase sliding clamp (5). In fact, loss of CAF-1
by a small interfering RNA-mediated knock-down system results inboth impaired S phase progression and cell viability accompanied withdefect of nascent chromatin assembly (6, 7), suggesting its essentialityfor coordinated DNA synthesis and chromatin assembly (6, 8). How-ever, the yeast CAF-1 mutant exhibits no growth retardation, implyingthe availability of other histone H3-H4 deposition factors (9).Asf1 was originally identified in yeast as its anti-silencing function in
silentmating loci (10). Biochemically identifiedDrosophilaRCAFcomplex,containingAsf1 and newly synthesized acetylated histonesH3 andH4,wasshown to facilitate efficient replication-coupled nucleosome assembly onlyin the presence of a limited amount of CAF-1 (11). Yeast Asf1 and twohuman forms, Asf1a and Asf1b, can interact with CAF-1p60 and promotereplication or repair-dependent chromatin assembly synergistically withCAF-1 (12–14). Two biochemically purified histone H3.1 and H3.3 com-plexes, containing CAF-1 and HIRA, possess the replication-coupled and-independent chromatin assembly activities, respectively (15, 16). Asf1 isretained in these twoH3 variant complexes, suggesting the involvement ofit in both chromatin assembly pathways (15). These together enforced aspeculation about its role as histone donor chaperone effectively supplyinghistones to either the CAF-1- or HIRA-mediated chromatin assemblypathway (2). Recently, it was reported that RNAi-mediated knockdown ofthe human or Drosophila Asf1 leads to impaired S phase progression (17,18), asobserved inCAF-1-depletedhumancells (6). Inyeast, the lossofAsf1results in impaired cell proliferation, increased sensitivity to methyl meth-anesulfonate (11), andminordefectsof gene silencingat telomereandsilentmating loci. These effects are enhanced by incorporating additional muta-tions either of CAF-1 or Hir (11–13). Thus, both genetic and biochemicaldata suggest that Asf1 synergistically acts with CAF-1 or HIRA in the reg-ulation of chromatin assembly that has impact on DNA replication, cellcycle progression, and formation/maintenance of heterochromatin struc-ture (2, 19, 20).In addition, Asf1 exhibits several distinct features in addition to a
synergistic role with CAF-1. For example, recent findings in yeast indi-cate that, in contrast to CAF-1, Asf1 functions as a disassembly factorfor histone deposition on overall genome (21) and facilitates chromatindisassembly at PHO5 promoter loci so as to allow transcription activa-tion (22), suggesting that it acts as a histone acceptor. The nature of Asf1as an interactor with the TFIID subunit also suggests its participation intranscription control at various RNA polymerase II-dependent geneloci (23, 24). Furthermore, yeast Asf1 is involved in genomic stability in
* This work was supported in part by the 21st Century Center of Excellence Program (LifeScience); a grant-in-aid for scientific research from the Ministry of Education, Culture,Sports, Science, and Technology; and grants from the Japan Society for the Promo-tion of Science and CREST from Japan Science and Technology of Japan. The costs ofpublication of this article were defrayed in part by the payment of page charges. Thisarticle must therefore be hereby marked “advertisement” in accordance with 18 U.S.C.Section 1734 solely to indicate this fact.
1 To whom correspondence should be addressed: Section of Biochemistry and MolecularBiology, Dept. of Medical Sciences, Miyazaki Medical College, University of Miyazaki,5200, Kihara, Kiyotake, Miyazaki 889-1692, Japan. Tel.: 81-985-85-3127; Fax: 81-985-85-6503; E-mail: [email protected].
2 The abbreviations used are: PCNA, proliferating cell nuclear antigen; aa, amino acid(s);tet, tetracycline; HA, hemagglutinin; FACS, fluorescence-activated cell sorting;
BrdUrd, 5-bromodeoxyuridine; FITC, fluorescein isothiocyanate; PI, propidium iodide;GST, glutathione S-transferase; WT, wild type; PBS, phosphate-buffered saline;MNase, micrococcal nuclease; DAPI, 4�,6-diamidino-2-phenylindole; EST, expressedsequence tag; RNAi, RNA interference; TUNEL, terminal deoxynucleotidyltransferase-mediated dUTP nick end labeling.
THE JOURNAL OF BIOLOGICAL CHEMISTRY VOL. 281, NO. 19, pp. 13817–13827, May 12, 2006© 2006 by The American Society for Biochemistry and Molecular Biology, Inc. Printed in the U.S.A.
MAY 12, 2006 • VOLUME 281 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 13817
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

conjunction with the checkpoint machinery (25) and interacts with theDNA damage checkpoint kinase Rad53 (26, 27), and human Asf1 isphosphorylated by tousled-like kinase (Tlk), which is inhibited byATM/ATR/Chk1 kinases in response to DNA damage, suggesting itsrole as mediator of checkpoint signal to chromatin (28, 29). Geneticanalyses inDrosophila suggest a linkage between cell proliferation and aTlk-Asf1-mediated chromatin assembly pathway (30).Asf1 is a highly conserved protein among eukaryotes and shares com-
mon features associatedwith a variety of chromatin-mediated processesas described above. Although the inactivation of Asf1 leads to a broadrange of cellular defects based on altering chromatin structure in yeast(2, 3, 19), its role in vertebrates has not yet been fully explored. In thispaper, to investigate the physiological role of Asf1 in vertebrates, wegenerated a conditional Asf1-deficient DT40 mutant cell, using thegene targeting technique. Analyses of the mutant cells indicate thatvertebrate Asf1 plays a crucial role in replication-coupled nucleosomeassembly linked to DNA replication, S phase progression, and viability.
EXPERIMENTAL PROCEDURES
Cell Culture—DT40 cells and mutants were cultured in RPMI 1640medium supplemented with 10% fetal calf serum, 1% chicken serum, 2mM L-glutamine, 10�5 M �-mercaptoethanol, and penicillin and strep-tomycin at 39 °C in a 5%CO2 incubator. Transfections and selections ofdrug-resistant cells were conducted as described (31). At the indicatedtimes, cells were counted to determine the growth rate. COS-7 cellswere grown in Dulbecco’s modified Eagle’s medium supplemented with10% fetal calf serum at 37 °C in a 5% CO2 incubator. Transfection wasconducted as described (32).
Cloning of Genomic DNA, Gene Constructs, Transformation, andMutant Isolation—Chicken full-length Asf1 cDNA was PCR-amplifiedfrom DT40 cDNA on the basis of the sequence from the chicken WebBursal EST data base (available on the World Wide Web at www.chick.umist.ac.uk/index.html). The nucleotide sequence of obtainedcDNA was verified by the dye terminator method (Applied BiosystemsDivision, PerkinElmer Life Sciences). Genomic Asf1 DNA clones wereisolated by screening the DT40 � FIX II genomic library (33), using Asf1cDNA as a probe. The Asf1 disruption constructs were made in pBlue-script vector by subcloning 2.0-kb upstream and 2.5-kb downstreamfragments ofAsf1 genomeDNA. The neomycin cassette flanked by loxPsite or bsr/cre-ER drug resistance cassette (34) was inserted betweenupstream and downstream arms. Gene targeting with these constructswas expected to disrupt 131 amino acids (aa) in exon 3 of the Asf1 gene.The tetracycline (tet)-responsible Asf1 expression vector was con-structed by inserting the HA-tagged full-length chicken Asf1 cDNAinto the pUHD10-3 plasmid (35). To obtain the ptTA-bleo construct, acassette of the bleomycin-resistant gene driven by the�-actin promoterwas inserted into the pUHD15-1 plasmid, which contains the tet-re-sponsive transactivator gene controlled by the cytomegalovirus pro-moter. For FLAG-taggedwild-type andmutantAsf1 expression vectors,the respective Asf1 cDNA fragments generated by PCR amplification orthe QuikChange method using Pfu turbo (Stratagene) were insertedinto pApuro, carrying the chicken �-actin promoter upstream of thecloning site and a marker gene, the puro-resistant gene, under the con-trol of the SV40 promoter (36) or into the pCite vector (Invitrogen).Knock-in type targeting construct for CENP-H-GFP was the same asdescribed (37). pCiteHA-HIRA was also the same as described (38).
Flow Cytometric Analysis of Cell Cycle—Flow cytometric analyseswere carried out using a FACSCalibur (BD Biosciences) as described(39). For synchronization into mitotic phase, cells were cultured in theabsence or presence of tet for 24 h and treated with nocodazole (200
ng/ml) for 8 h, released from the block by washing with medium threetimes, and further culturedwith orwithout tet. At 2-h intervals, the cellswere collected, fixed in 70% ethanol, stainedwith propidium iodide (PI),and then analyzed. For two-dimensional cell cycle analysis, cells werecultured in the presence of 20 �M BrdUrd at 39 °C for 10 min, fixed in70% ethanol, and stained with FITC-labeled anti-BrdUrd antibody(1:400; Chemicon) and PI.
Analysis of DNA Synthesis and Chromatin Structure—To monitortheDNA synthesis rate during Asf1 deletion, cells were culturedwith orwithout tet (1 �g/ml) for the indicated times and then pulse-labeled bythe addition of 2 �Ci/ml [3H]thymidine (PerkinElmer Life Sciences) for10 min. Collected cells were lysed by NaOH, followed by precipitationwith trichloroacetic acid, and passed through a filter, and then incorpo-rated radioactivities were counted using a liquid scintillation counter.To examine nascent chromatin structure, cells were pulse-labeled
with [3H]thymidine (20 �Ci/ml) for 10 min, and labeled nuclei wereprepared as follows. Cells were washed with cold PBS and incubated inNB (15 mM Tris-HCl, pH 8.0, 0.5 mM EDTA, 2 mMmagnesium acetate,2mMCaCl2, 1mM dithiothreitol, protease inhibitormixture (Sigma)) inthe presence of 0.1% Nonidet P-40. The resultant nuclei were washedtwicewithNB, suspended at 20A260/ml (A260 wasmeasured in 2MNaCland 5 M urea) in NB, and digested at 37 °C for 8 min with 0.0025–0.2units/ml micrococcal nuclease (MNase) (Sigma). The reactions werestopped by adding 10 mM EDTA and 0.5% SDS, and then DNA waspurified by incubation with 100 �g/ml proteinase K for 2 h at 37 °C,followed by phenol-chloroform extraction and ethanol precipitation.DNA was electrophoresed in a 1.2% agarose gel followed by EtBr stain-ing and then transferred to a Hybond N� membrane. To detect 3H-la-beled DNA, blots were directly exposed on a BAS screen (TR2040)specified for tritium and visualized using a Mac BAS-1000 Bio ImagingAnalyzer (Fuji Film). Densitometric tracing of the nucleosomal ladderwas carried out by using Image Gauge version 3.3 (Fuji Film).
Western Blotting and Co-immunoprecipitation—For production ofanti-chicken Asf1, anti-chicken CAF-1p150, and anti-chicken CAF-1p60 antibodies, GST fusion proteins (GST-Asf1-(1–204), GST-p150-(720–938), andGST-p60-(1–256)) were expressed in bacteria, and thenpurified fusion proteins were injected into rabbits. Affinity purificationof antibodies was performed using standard procedures.DT40 cells (1 � 107) were lysed in 200 �l of SDS buffer, and then
aliquots (10–20�l) of the resultant cell extracts were separated by SDS-PAGE, followed by electroblotting onto a polyvinylidene difluoridemembrane. The filter was probed with a 1:1000 dilution of antibodiesagainst Asf1, p150, p60, p48 (Pharmingen), PCNA (PC10; Sigma),�-H2AX (Upstate Biotechnology, Inc.), �-tubulin (Sigma), HA (12CA5;Roche Applied Science), or FLAG M2 (Sigma), followed by incubationwith a 1:1000 dilution of secondary antibodies (horseradish peroxidase-conjugated anti-mouse or anti-rabbit IgG antibodies (Dako)). The sig-nal was developed using a Super Signal West Pico ChemiluminescentSubstrate (Pierce) and visualized with a LAS-1000 (Fuji Film).For co-immunoprecipitation analysis, cells (5 � 107) were lysed in
radioimmune precipitation buffer (20 mM Tris-HCl, pH 7.8, 150 mM
NaCl, 0.1 mM dithiothreitol, 1% Nonidet P-40) containing proteaseinhibitor mixture (Sigma) for 30 min on ice, and then clarified lysateswere immunoprecipitated with anti-FLAG M2 immobilized beads(Sigma) at 4 °C for 2 h. After washing by radioimmune precipitationbuffer three times, beads were suspended in SDS sample buffer, fol-lowed by Western blotting using antibodies against FLAG M2, p60,histone H3 (Cell signaling), and acetylated histone H4 (K12) (UpstateBiotechnology). To examine the phosphorylation states of Asf1, beadscontaining Asf1-FLAG protein were untreated or treated with 10 units
Asf1 Is Essential for Vertebrate Cells
13818 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 19 • MAY 12, 2006
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from
of alkaline phosphatase in 25 �l of buffer (50 mM Tris-HCl, pH 9.0, 10mM MgCl2) at 37 °C for 1 h. After elution with SDS sample buffer,mobility of Asf1-FLAG in SDS-gel was analyzed by Western blottingusing anti-FLAG antibody.For in vitro immunoprecipitation analysis, 35S-labeled proteins, FLAG-
tagged Asf1 and its derivatives, HA-tagged HIRA, and HA-tagged Tip60were produced by an in vitro translation kit STP3 (Novagen) using pCitevectors containing cognate regionsofAsf1 andHIRAorTip60.The follow-ing procedures for the in vitro translation and immunoprecipitation werecarried out as described (38).
ImmunofluorescenceMicroscopy—DT40 cells were attached on poly-ethyleneimine-coated slides, fixed by 4% paraformaldehyde in PBS for10 min, permeabilized by 0.15% Triton X-100 in PBS, and rinsed withPBS. After blocking with 5% bovine serum albumin plus PBS for 20min,cells were incubated with primary and secondary antibodies in 3%bovine serum albumin plus PBS for 1 h at 37 °C. The following antibod-ies were used: rat anti-HAmonoclonal (1:1000; RocheApplied Science),mouse anti-PCNA (1:400; PC10; Sigma), rabbit anti-p150 (1:1000), rab-bit anti-p60 (1:1000), and FITC-conjugated anti-�-tubulin monoclonal(1:100; Sigma). Primary antibodies were detected with AlexaFluor 488-conjugated anti-mouse IgG or AlexaFluor 594-conjugated anti-rabbitand rat IgG antibodies (1:400; Molecular Probes, Inc.). DNA was coun-terstained with 4�,6-diamidino-2-phenylindole (DAPI) (Sigma) at 0.1�g/ml.
For Triton extraction, attached cells on slides were incubated withCSK (100mMNaCl, 300mM sucrose, 3 mMMgCl2, 10mM PIPES-KOH,pH 6.8) containing 0.2% Triton X-100 for 5 min at 4 °C followed byfixation with 2% paraformaldehyde for 10 min and absolute methanolfor 20 min at �20 °C as described (40). To examine co-localization ofBrdUrd with PCNA, cells were pulse-labeled with 20 �M BrdUrd at39 °C for 10 min. After staining of PCNA with AlexaFluor 594-conju-
gated anti-mouse IgG as described above, cells were fixed with 4%paraformaldehyde and then treated with 2 M HCl containing 0.5% Tri-ton X-100 for 20 min. After washing three times, cells were incubatedwith FITC-labeled anti-BrdUrd antibody (1:400; Chemicon). Fluores-cence images were visualized with a cooled CCD camera (ORCA ER;Hamamatsu Photonics) using Axiovert M-200 (Zeiss).
TUNEL Assay—TUNEL assay was performed using a MEBSTAINApoptosis Kit Direct (MBL). Briefly, cells were collected, fixed in 4%paraformaldehyde for 10min at 4 °C, and permeabilized in PBS contain-ing 0.5% Tween 20 for 15 min at room temperature. DNA break endswere labeled using terminal deoxynucleotidyl transferase and FITC-dUTP, and then DNA was counterstained with PI and visualized byfluorescence microscopy.
Complementation Assay—To determine the ability of Asf1 and itsderivatives to rescue the tet-induced lethality of the Asf1-deficient cells,a complementation assay was carried out as described (39).Asf1-condi-tional knock-out cells were transfectedwith pApuro-Asf1-FLAGand itsderivatives and split into two portions. A half-portion was incubated on96-well plates inmediumcontaining 0.4�g/ml puromycin to determinethe frequency of transfection, and the other was incubated on 96-wellplates in medium containing 0.4 �g/ml puromycin plus 1 �g/ml tet.After incubation for 10 days, the numbers of surviving colonies werecounted.
RESULTS
Generation of Conditional Asf1 Knock-out DT40 Cells—To designgene distribution constructs, chicken Asf1 cDNA was isolated by com-bination of EST searching (available on the World Wide Web at swal-low.gsf.de/dt40Est.html) and reverse transcription-PCR.We found sev-eral EST sequences that have significant homology to human Asf1a.Using reverse transcription-PCR based on these sequences, we cloned
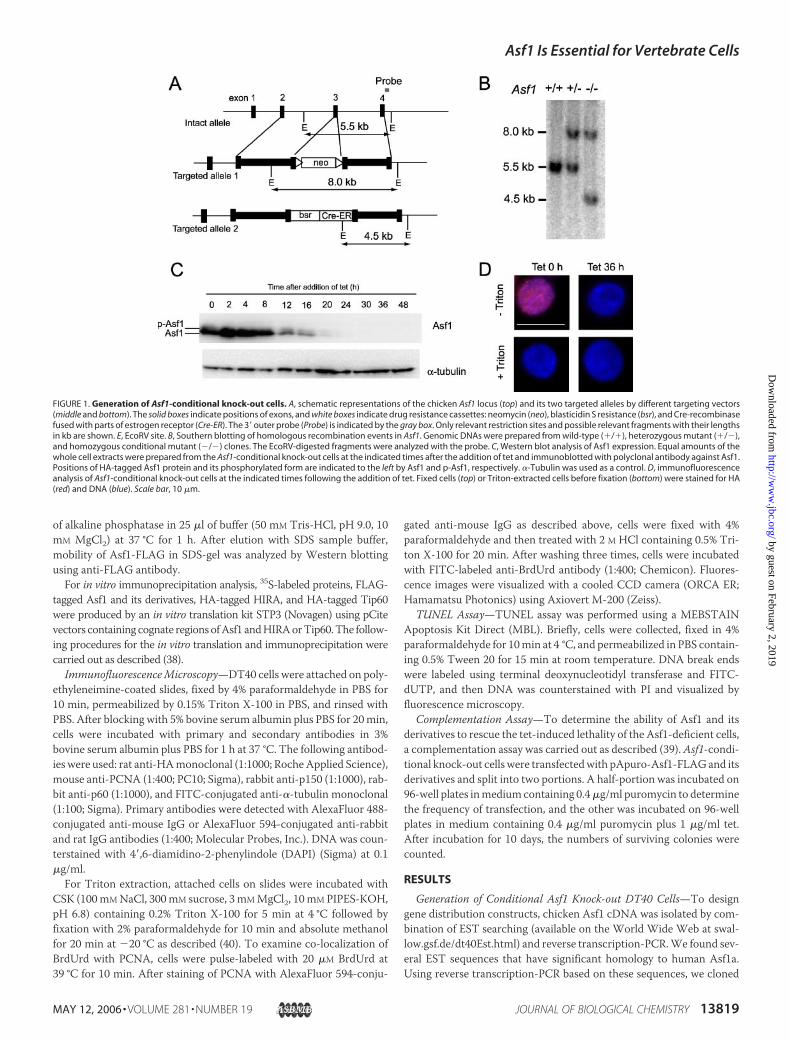
FIGURE 1. Generation of Asf1-conditional knock-out cells. A, schematic representations of the chicken Asf1 locus (top) and its two targeted alleles by different targeting vectors(middle and bottom). The solid boxes indicate positions of exons, and white boxes indicate drug resistance cassettes: neomycin (neo), blasticidin S resistance (bsr), and Cre-recombinasefused with parts of estrogen receptor (Cre-ER). The 3� outer probe (Probe) is indicated by the gray box. Only relevant restriction sites and possible relevant fragments with their lengthsin kb are shown. E, EcoRV site. B, Southern blotting of homologous recombination events in Asf1. Genomic DNAs were prepared from wild-type (�/�), heterozygous mutant (�/�),and homozygous conditional mutant (�/�) clones. The EcoRV-digested fragments were analyzed with the probe. C, Western blot analysis of Asf1 expression. Equal amounts of thewhole cell extracts were prepared from the Asf1-conditional knock-out cells at the indicated times after the addition of tet and immunoblotted with polyclonal antibody against Asf1.Positions of HA-tagged Asf1 protein and its phosphorylated form are indicated to the left by Asf1 and p-Asf1, respectively. �-Tubulin was used as a control. D, immunofluorescenceanalysis of Asf1-conditional knock-out cells at the indicated times following the addition of tet. Fixed cells (top) or Triton-extracted cells before fixation (bottom) were stained for HA(red) and DNA (blue). Scale bar, 10 �m.
Asf1 Is Essential for Vertebrate Cells
MAY 12, 2006 • VOLUME 281 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 13819
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from
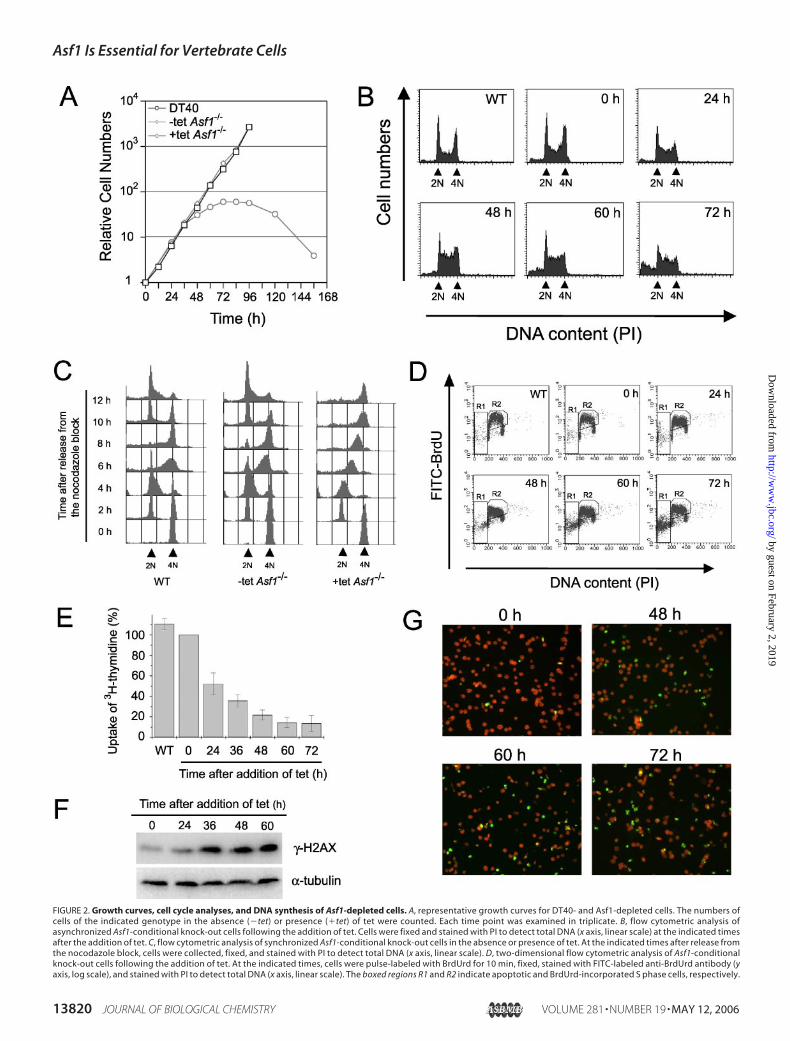
FIGURE 2. Growth curves, cell cycle analyses, and DNA synthesis of Asf1-depleted cells. A, representative growth curves for DT40- and Asf1-depleted cells. The numbers ofcells of the indicated genotype in the absence (�tet) or presence (�tet) of tet were counted. Each time point was examined in triplicate. B, flow cytometric analysis ofasynchronized Asf1-conditional knock-out cells following the addition of tet. Cells were fixed and stained with PI to detect total DNA (x axis, linear scale) at the indicated timesafter the addition of tet. C, flow cytometric analysis of synchronized Asf1-conditional knock-out cells in the absence or presence of tet. At the indicated times after release fromthe nocodazole block, cells were collected, fixed, and stained with PI to detect total DNA (x axis, linear scale). D, two-dimensional flow cytometric analysis of Asf1-conditionalknock-out cells following the addition of tet. At the indicated times, cells were pulse-labeled with BrdUrd for 10 min, fixed, stained with FITC-labeled anti-BrdUrd antibody (yaxis, log scale), and stained with PI to detect total DNA (x axis, linear scale). The boxed regions R1 and R2 indicate apoptotic and BrdUrd-incorporated S phase cells, respectively.
Asf1 Is Essential for Vertebrate Cells
13820 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 19 • MAY 12, 2006
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

chicken full-length Asf1a cDNA. The deduced aa sequences of chickenAsf1a completelymatched those of human andmouse Asf1a, indicatingthat it is a highly conserved protein throughout eukaryotes. Althoughtwo forms of Asf1a andAsf1b, sharing 69% aa sequence homology, werefound in humans and mice, we could not find a chicken Asf1b homologin either the EST or genome databases. Consistent with this, Southernhybridization under low stringency conditions using the chicken Asf1acDNA as a probe failed to detect any signal other than the Asf1a hom-olog in DT40 genomic DNA. Western blotting also failed to detectAsf1b signal in DT40 cells using our raised antibody against Asf1a,which could recognize both Asf1a and Asf1b proteins in HeLa cells(data not shown). These suggest that chicken had only the Asf1a gene,and Asf1bmight be divergent from Asf1a throughout vertebrate evolu-tion. Hereafter, we referred the Asf1a homolog as chicken Asf1 (acces-sion number AB238225).We isolated chicken genomic Asf1 DNA by library screening and
designed gene targeting constructs (Fig. 1A).We could obtain heterozy-gous knock-out clones (�/�) but never obtain homozygous knock-outclones (�/�), even after several attempts. Therefore, we tried to obtaintet-responsive conditional null mutants of Asf1, since this system hasbeen applied successfully to generate conditional homozygous mutantsfor other essential genes (39). After we obtained Asf1�/� clones (Fig. 1,A and B) where one allele had been disrupted by the neo disruptionconstruct, we produced several Asf1�/�� tetHA-Asf1 cell lines, inwhichN-terminal HA-tagged Asf1 was expressed under the control of atet-responsive promoter. After confirmation of tet-repressive expres-sion of HA-Asf1, one of the clones was transfected for the second rounddisruption of the remaining allele of Asf1 by the bsr/cre-ER disruptionconstruct to obtain three independent Asf1�/��tetHA-Asf1 clones(Fig. 1, A and B). Hereafter, we referred to these cell lines as Asf1-conditional knock-out cell lines. In these cells, levels of HA-Asf1 pro-teins were slightly higher than that of the endogenous Asf1 protein inDT40 cells (data not shown), but upon the addition of tet, HA-Asf1protein level decreased gradually and became undetectable under ourblotting conditions after 24 h of treatment (Fig. 1C). Of note, doubletAsf1 bands in SDS-PAGEmay represent phosphorylated (upper bands)and unphosphorylated (lower bands) forms (28), which were confirmedas described below (see Fig. 5D). Progressive decrease of Asf-1 proteinlevel was also confirmed by indirect immunofluorescence. Asf1 stainingsignal was distributedwithin the nucleus, but by 36 h, this staining signalwas no longer detectable (Fig. 1D, top). It should be noted that treatmentof Triton-X100 prior to fixation of cells led to diffusion of theAsf1 signalout of the nucleus (Fig. 1D, bottom), suggesting that themajority of Asf1protein exists in a soluble nuclear fraction that is not tightly bound withchromatin. This observation was consistent with a previous report thatsubnuclear localization of human Asf1 is sensitive to Triton extraction(14). Since all of theAsf1-conditional knock-out clones exhibited essen-tially the same phenotypic properties following the treatment with tet,we describe phenotypes of one of these clones hereafter.
Growth and Cell Cycle Analysis of Asf1-conditional Knock-out Cells—We next monitored proliferative properties of Asf1-conditional knock-out cells (Fig. 2A). Upon the addition of tet, the growth rate began toslow down from day 2 with a drastic reduction during 3–4 days, fol-lowed by complete cessation. A portion of dead cells appeared at day 2,and finally almost all of themutants died by day 5, indicating that Asf1 isessential for cell viability.
FACS analysis in terms of DNA content indicated that Asf1 defi-ciency led to the accumulation ofmid-S phase cells (Fig. 2B). In addition,a population of sub-G1 content also became apparent at 48 h, represent-ing apoptotic cell populations. To verify more closely the effect of Asf1deficiency for S phase progression, Asf1 null mutants were synchro-nized to mitotic phase by nocodazole treatment, the cell populationbeing �90% at M phase. After removal of the drug, the DNA contentwas monitored by FACS at 2-h intervals. In the absence of tet (�tetAsf1), the mutant cells, like DT40 cells (WT), had proceeded to G1 andG2/M phases by 2 and 8 h, respectively, after release from the nocoda-zole block (Fig. 2C,middle and left). When Asf1-conditional knock-outcells had been treated with tet for 24 h (�tet Asf1), the cell cycle pro-gression fromM to G1 phase was not affected, but delayed progressionthrough S phase, especially middle to late S phase, was observed (see 6and 8 h in Fig. 2C, right).With defects in S phase progression, DNA replication was also
impaired in Asf1-depleted cells. Two-dimensional FACS analyses forDNAcontent andBrdUrd uptake showed that the proportion of S phasecells incorporating BrdUrd appeared to increase upon Asf1 depletion,but the amount of BrdUrd incorporation was slightly reduced at 24 hand was greatly reduced by 48–72 h (see the level of the y axis, andcompare the shapes of the BrdUrd arc), indicating an impaired DNAsynthesis in S phase cells (Fig. 2D). Gradual reduction of DNA synthesisrate following Asf1 depletion was also confirmed by a [3H]thymidineuptake assay (Fig. 2E). Taken together, we concluded that Asf1 isrequired for efficient DNA replication that is linked to proper S phaseprogression.Following the accumulation of S phase cells, a proportion of dead
cells, indicated by the sub-G1 fraction of FACS, was increased by 48 hupon depletion of Asf1. To evaluate whether this cell death pathway isrelated to the induction of double-stranded DNA breaks and apoptosis,we first examined phosphorylated form of histone H2AX (�-H2AX),one of the earliest responses to sites of double-stranded DNA breaks(41), following loss of Asf1. The signal for �-H2AX was increased sig-nificantly at 36 h after the addition of tet (Fig. 2F), suggesting that dou-ble-stranded DNA breaks are accumulated upon the depletion of Asf1.To confirm this finding by another means, we performed the TUNELassay to ask whether cell death after Asf1 depletion occurs throughapoptosis. As shown in Fig. 2G, the population of TUNEL-positive cells,representing the cells with fragmented genomic DNA, was progres-sively increased to 5.6, 5.2, 15.5, 21.7, and 29.3% at 0, 36, 48, 60, and 72 hafter the addition of tet, respectively. Taken together, we conclude thatdepletion of Asf1 leads to an induction of double-stranded DNA breaksand the activation of programmed cell death.
Asf1 Contributes to Replication-coupled Nucleosome Assembly inVivo—Asf1 is predicted to facilitate replication-coupled nucleosomeassembly in concert withCAF-1 (11). To examinewhether there are anyroles of Asf1 associated with the events related to replication-couplednucleosome assembly, we compared the stability of pulse-labeled nas-cent chromatin structure in the presence and absence of it. The Asf1-conditional knock-out cells untreated and pretreated with tet for 24 and30 h were pulse-labeled with [3H]thymidine for 10 min, and then thelabeled DNAs were isolated for the MNase digestion assay. After gelelectrophoresis of MNase-digested samples, DNA was stained by EtBrto evaluate bulk chromatin structure, and pulse-labeled DNA on theblot was detected using radioimaging (Fig. 3, A and B). We could not
E, relative DNA synthesis rate following Asf1 depletion. The [3H]thymidine incorporations at the indicated times after the addition of tet were normalized by dividing the total cellnumber at each time point by that of at 0 h, defined as 100%. The data are representative results from three separate experiments. F, phosphorylation of H2AX upon the loss of Asf1.At the indicated times after the addition of tet, cells were collected, and equal amounts of total cell extracts were subjected to Western blotting using anti-�-H2AX antibody. �-Tubulinwas used as a control. G, TUNEL assay. At the indicated times after the addition of tet, cells were collected, and DNA breaks were labeled with FITC-dUTP (green) by using terminaltransferase (see “Experimental Procedures”). DNA was counterstained with PI (red).
Asf1 Is Essential for Vertebrate Cells
MAY 12, 2006 • VOLUME 281 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 13821
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from
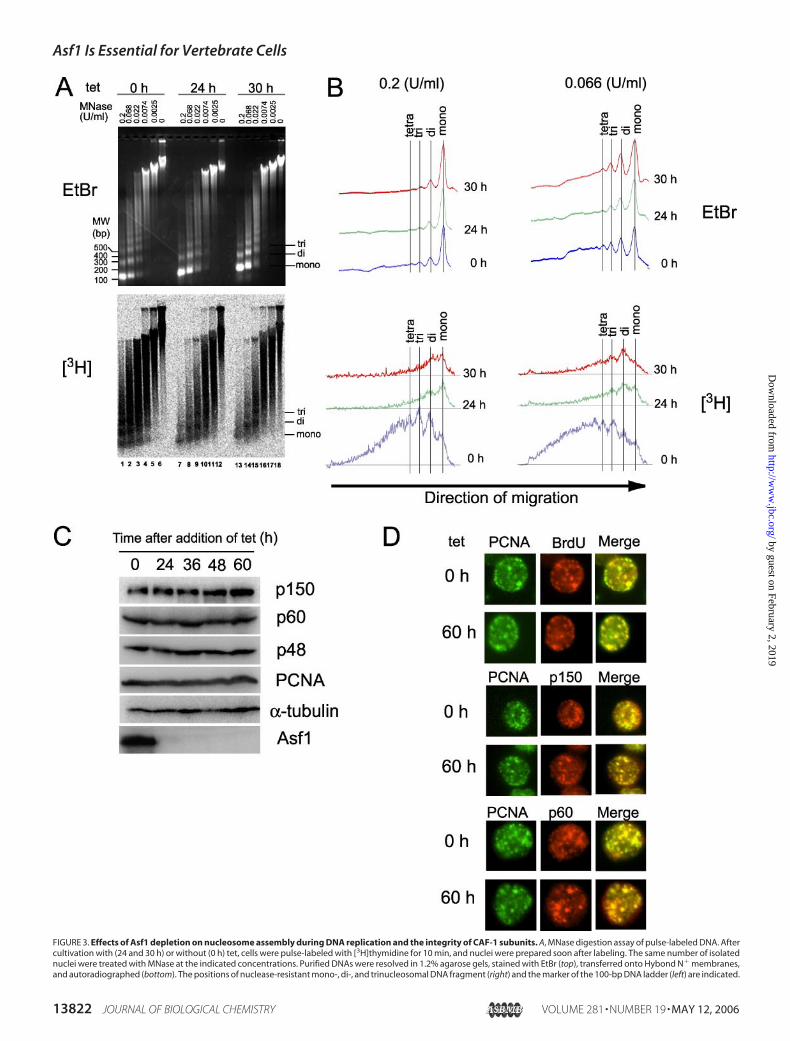
FIGURE 3. Effects of Asf1 depletion on nucleosome assembly during DNA replication and the integrity of CAF-1 subunits. A, MNase digestion assay of pulse-labeled DNA. Aftercultivation with (24 and 30 h) or without (0 h) tet, cells were pulse-labeled with [3H]thymidine for 10 min, and nuclei were prepared soon after labeling. The same number of isolatednuclei were treated with MNase at the indicated concentrations. Purified DNAs were resolved in 1.2% agarose gels, stained with EtBr (top), transferred onto Hybond N� membranes,and autoradiographed (bottom). The positions of nuclease-resistant mono-, di-, and trinucleosomal DNA fragment (right) and the marker of the 100-bp DNA ladder (left) are indicated.
Asf1 Is Essential for Vertebrate Cells
13822 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 19 • MAY 12, 2006
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from
detect any apoptotically degraded DNA at least at 24 and 30 h after tettreatment (lanes 6, 12, and 18 in Fig. 3A, top), consistent with previousresults (Fig. 2,D and F). BulkDNA fromAsf1-depleted cells treatedwithtet for 24 and 30 h showed MNase resistance similar to that from non-depleted cells (compare lanes 1–5with lanes 7–11 and 13–17 in Fig. 3,A(top) and B (top)). This indicates that the global chromatin structure isnot so impaired at least until 30 h of tet treatment. When we examinedpulse-labeled chromatin, MNase digestion of Asf1-proficient chroma-tin (tet 0 h) gave characteristic ladders indicative of mono-, di-, andtrinucleosomes (lanes 1–5 in Fig. 3A (bottom) and 0 h in Fig. 3B (bot-
tom)), indicating that oligonucleosomes were formed onto newly repli-cated DNA within 10 min of DNA replication. On the other hand,Asf1-depleted chromatin (tet 24 h and 30 h) showed greatly increasedMNase digestion sensitivity, indicating that chromatin structure ontonewly replicated DNA was severely impaired (lanes 7–11 and 13–17 inFig. 3, A (bottom) and B (bottom)). These results strongly indicate thatAsf1 is involved in the replication-coupled nucleosome assembly proc-ess in vivo.Since CAF-1 is a primary factor for nucleosome assembly during repli-
cation, it was possible that the observed defective nascent nucleosome
B, densitometry tracing of nucleosome ladders. MNase digestion patterns were quantified by densitometry tracing. The tracing data of bulk DNA (EtBr) and newly synthesized DNA(3H) are shown at the top and bottom, respectively, at the indicated MNase concentrations and times after tet addition. C, protein levels of CAF-1 subunits during Asf1 depletion. Atthe indicated times after the addition of tet, cells were collected, and equal amounts of total cell extracts were subjected to Western blotting using anti-chicken CAF-1p150, p60, p48,PCNA, and Asf1 antibodies. As a control, �-tubulin was used. D, co-localization of CAF-1p150 and p60 with PCNA. Cultured cells with (60 h) or without (0 h) tet were doubly stainedwith antibodies for appropriate proteins and BrdUrd.
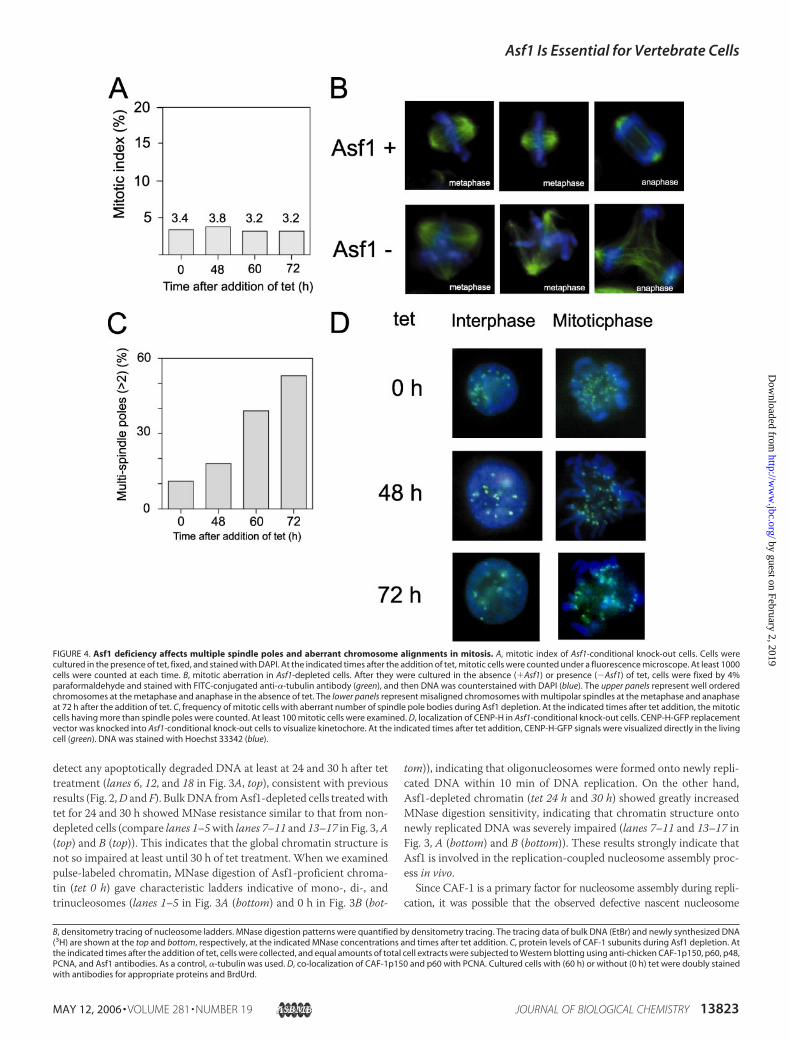
FIGURE 4. Asf1 deficiency affects multiple spindle poles and aberrant chromosome alignments in mitosis. A, mitotic index of Asf1-conditional knock-out cells. Cells werecultured in the presence of tet, fixed, and stained with DAPI. At the indicated times after the addition of tet, mitotic cells were counted under a fluorescence microscope. At least 1000cells were counted at each time. B, mitotic aberration in Asf1-depleted cells. After they were cultured in the absence (�Asf1) or presence (�Asf1) of tet, cells were fixed by 4%paraformaldehyde and stained with FITC-conjugated anti-�-tubulin antibody (green), and then DNA was counterstained with DAPI (blue). The upper panels represent well orderedchromosomes at the metaphase and anaphase in the absence of tet. The lower panels represent misaligned chromosomes with multipolar spindles at the metaphase and anaphaseat 72 h after the addition of tet. C, frequency of mitotic cells with aberrant number of spindle pole bodies during Asf1 depletion. At the indicated times after tet addition, the mitoticcells having more than spindle poles were counted. At least 100 mitotic cells were examined. D, localization of CENP-H in Asf1-conditional knock-out cells. CENP-H-GFP replacementvector was knocked into Asf1-conditional knock-out cells to visualize kinetochore. At the indicated times after tet addition, CENP-H-GFP signals were visualized directly in the livingcell (green). DNA was stained with Hoechst 33342 (blue).
Asf1 Is Essential for Vertebrate Cells
MAY 12, 2006 • VOLUME 281 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 13823
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from
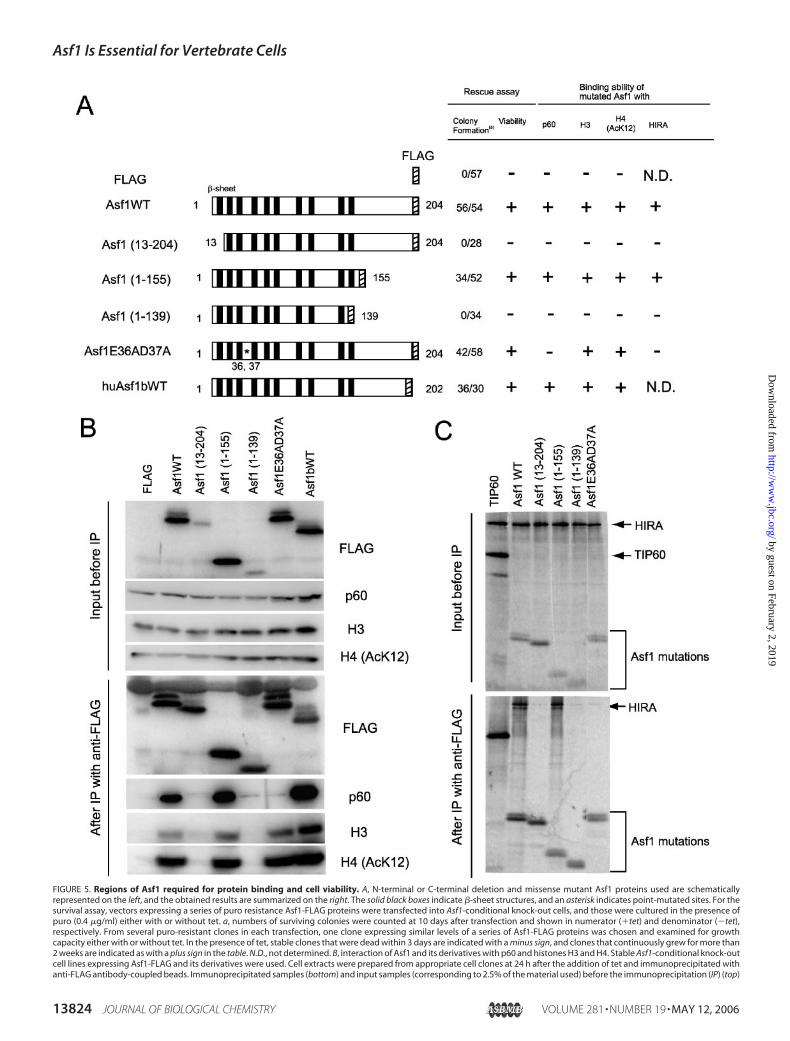
FIGURE 5. Regions of Asf1 required for protein binding and cell viability. A, N-terminal or C-terminal deletion and missense mutant Asf1 proteins used are schematicallyrepresented on the left, and the obtained results are summarized on the right. The solid black boxes indicate �-sheet structures, and an asterisk indicates point-mutated sites. For thesurvival assay, vectors expressing a series of puro resistance Asf1-FLAG proteins were transfected into Asf1-conditional knock-out cells, and those were cultured in the presence ofpuro (0.4 �g/ml) either with or without tet. a, numbers of surviving colonies were counted at 10 days after transfection and shown in numerator (�tet) and denominator (�tet),respectively. From several puro-resistant clones in each transfection, one clone expressing similar levels of a series of Asf1-FLAG proteins was chosen and examined for growthcapacity either with or without tet. In the presence of tet, stable clones that were dead within 3 days are indicated with a minus sign, and clones that continuously grew for more than2 weeks are indicated as with a plus sign in the table. N.D., not determined. B, interaction of Asf1 and its derivatives with p60 and histones H3 and H4. Stable Asf1-conditional knock-outcell lines expressing Asf1-FLAG and its derivatives were used. Cell extracts were prepared from appropriate cell clones at 24 h after the addition of tet and immunoprecipitated withanti-FLAG antibody-coupled beads. Immunoprecipitated samples (bottom) and input samples (corresponding to 2.5% of the material used) before the immunoprecipitation (IP) (top)
Asf1 Is Essential for Vertebrate Cells
13824 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 19 • MAY 12, 2006
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from
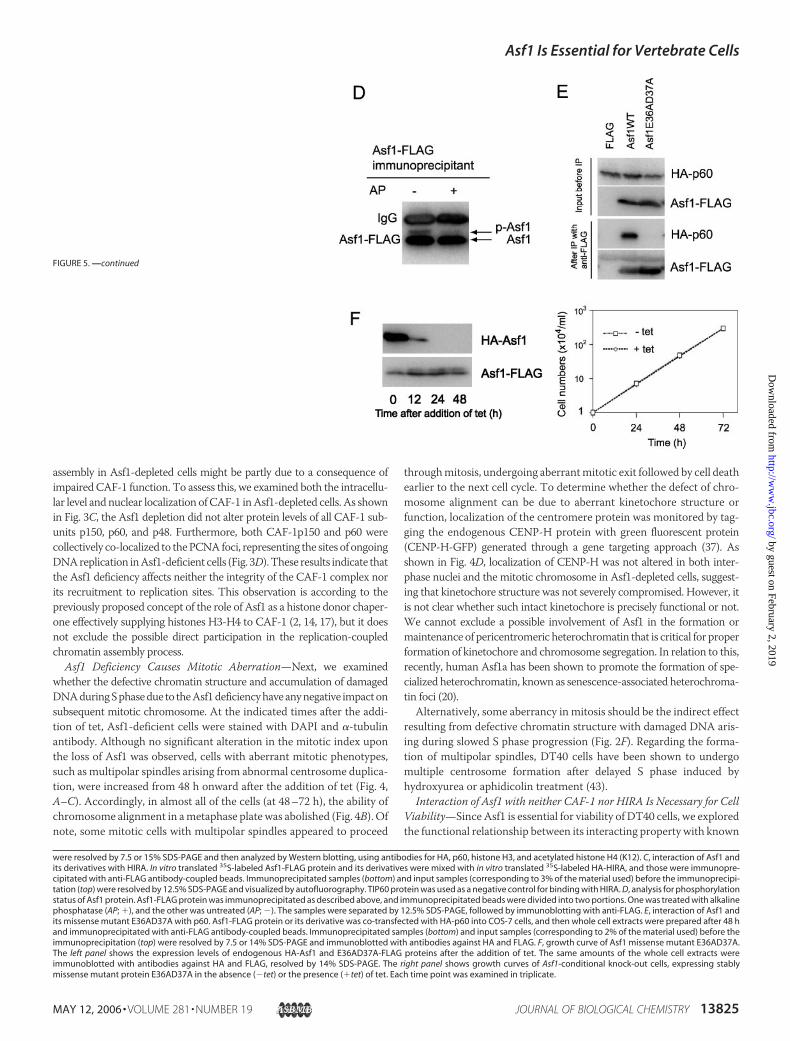
assembly in Asf1-depleted cells might be partly due to a consequence ofimpaired CAF-1 function. To assess this, we examined both the intracellu-lar level and nuclear localization ofCAF-1 inAsf1-depleted cells. As shownin Fig. 3C, the Asf1 depletion did not alter protein levels of all CAF-1 sub-units p150, p60, and p48. Furthermore, both CAF-1p150 and p60 werecollectively co-localized to thePCNAfoci, representing the sites of ongoingDNAreplication inAsf1-deficient cells (Fig. 3D).These results indicate thatthe Asf1 deficiency affects neither the integrity of the CAF-1 complex norits recruitment to replication sites. This observation is according to thepreviously proposed concept of the role of Asf1 as a histone donor chaper-one effectively supplying histones H3-H4 to CAF-1 (2, 14, 17), but it doesnot exclude the possible direct participation in the replication-coupledchromatin assembly process.
Asf1 Deficiency Causes Mitotic Aberration—Next, we examinedwhether the defective chromatin structure and accumulation of damagedDNAduringSphasedue to theAsf1deficiencyhaveanynegative impactonsubsequent mitotic chromosome. At the indicated times after the addi-tion of tet, Asf1-deficient cells were stained with DAPI and �-tubulinantibody. Although no significant alteration in the mitotic index uponthe loss of Asf1 was observed, cells with aberrant mitotic phenotypes,such as multipolar spindles arising from abnormal centrosome duplica-tion, were increased from 48 h onward after the addition of tet (Fig. 4,A–C). Accordingly, in almost all of the cells (at 48–72 h), the ability ofchromosome alignment in ametaphase plate was abolished (Fig. 4B). Ofnote, some mitotic cells with multipolar spindles appeared to proceed
throughmitosis, undergoing aberrantmitotic exit followed by cell deathearlier to the next cell cycle. To determine whether the defect of chro-mosome alignment can be due to aberrant kinetochore structure orfunction, localization of the centromere protein was monitored by tag-ging the endogenous CENP-H protein with green fluorescent protein(CENP-H-GFP) generated through a gene targeting approach (37). Asshown in Fig. 4D, localization of CENP-H was not altered in both inter-phase nuclei and the mitotic chromosome in Asf1-depleted cells, suggest-ing that kinetochore structure was not severely compromised. However, itis not clear whether such intact kinetochore is precisely functional or not.We cannot exclude a possible involvement of Asf1 in the formation ormaintenance of pericentromeric heterochromatin that is critical for properformation of kinetochore and chromosome segregation. In relation to this,recently, human Asf1a has been shown to promote the formation of spe-cialized heterochromatin, known as senescence-associated heterochroma-tin foci (20).Alternatively, some aberrancy in mitosis should be the indirect effect
resulting from defective chromatin structure with damaged DNA aris-ing during slowed S phase progression (Fig. 2F). Regarding the forma-tion of multipolar spindles, DT40 cells have been shown to undergomultiple centrosome formation after delayed S phase induced byhydroxyurea or aphidicolin treatment (43).
Interaction of Asf1 with neither CAF-1 nor HIRA Is Necessary for CellViability—Since Asf1 is essential for viability of DT40 cells, we exploredthe functional relationship between its interacting property with known
were resolved by 7.5 or 15% SDS-PAGE and then analyzed by Western blotting, using antibodies for HA, p60, histone H3, and acetylated histone H4 (K12). C, interaction of Asf1 andits derivatives with HIRA. In vitro translated 35S-labeled Asf1-FLAG protein and its derivatives were mixed with in vitro translated 35S-labeled HA-HIRA, and those were immunopre-cipitated with anti-FLAG antibody-coupled beads. Immunoprecipitated samples (bottom) and input samples (corresponding to 3% of the material used) before the immunoprecipi-tation (top) were resolved by 12.5% SDS-PAGE and visualized by autofluorography. TIP60 protein was used as a negative control for binding with HIRA. D, analysis for phosphorylationstatus of Asf1 protein. Asf1-FLAG protein was immunoprecipitated as described above, and immunoprecipitated beads were divided into two portions. One was treated with alkalinephosphatase (AP; �), and the other was untreated (AP; �). The samples were separated by 12.5% SDS-PAGE, followed by immunoblotting with anti-FLAG. E, interaction of Asf1 andits missense mutant E36AD37A with p60. Asf1-FLAG protein or its derivative was co-transfected with HA-p60 into COS-7 cells, and then whole cell extracts were prepared after 48 hand immunoprecipitated with anti-FLAG antibody-coupled beads. Immunoprecipitated samples (bottom) and input samples (corresponding to 2% of the material used) before theimmunoprecipitation (top) were resolved by 7.5 or 14% SDS-PAGE and immunoblotted with antibodies against HA and FLAG. F, growth curve of Asf1 missense mutant E36AD37A.The left panel shows the expression levels of endogenous HA-Asf1 and E36AD37A-FLAG proteins after the addition of tet. The same amounts of the whole cell extracts wereimmunoblotted with antibodies against HA and FLAG, resolved by 14% SDS-PAGE. The right panel shows growth curves of Asf1-conditional knock-out cells, expressing stablymissense mutant protein E36AD37A in the absence (�tet) or the presence (�tet) of tet. Each time point was examined in triplicate.
FIGURE 5. —continued
Asf1 Is Essential for Vertebrate Cells
MAY 12, 2006 • VOLUME 281 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 13825
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

binding partners such as histones H3/H4, CAF-1p60, and HIRA. First,to determine the binding region(s) of Asf1 to these proteins, we gener-ated Asf1-conditional knock-out cells, expressing stably C-terminalFLAG-tagged Asf1 and its mutant proteins as depicted schematically inFig. 5A (left). Then we performed co-immunoprecipitation assays forH3, H4, and CAF-1p60 proteins, using anti-FLAG antibody and lysatesfrom corresponding mutant cells that had been treated with tet for 24 hto eliminate the effect of HA-tagged Asf1. Each immunoprecipitatedcomplex was analyzed by Western blotting using anti-p60, histone H3,and acetylated histoneH4 (K12) antibodies (Fig. 5B). On the other hand,the binding region(s) of Asf1 with HIRA was assessed by co-immuno-precipitation assays using in vitro translated HA-tagged HIRA andFLAG-tagged Asf1 cognate proteins (Fig. 5C). As expected, the wildtype Asf1 exhibited binding partnership with all of these proteins exam-ined (Fig. 5, B and C). Deletion mutational analyses revealed that theminimum binding domain of Asf1 for p60, histones H3/H4, and HIRAwas confined to 1–155 aa, which lacks a Ser- and Thr-rich region of theC-terminal tail. This N-terminal domain, containing 10 �-sheets, hasbeen well defined to be structurally conserved (44). Two other deletionmutant proteins (aa 13–204 and aa 1–139) that failed to bind with all ofthese proteins are likely to be unstable, since the detected levels of theseconstantly remained low in clones tested, compared with other mutantproteins, probably due to a lack of complete �-sheet structures. Asshown in Fig. 1C, the mobility shifting of Asf1 proteins in SDS-PAGEcould be seen as doublet bands in several mutant proteins (Fig. 5B). Theresult that treatment with alkaline phosphatase of Asf1-FLAG-contain-ing beads caused a loss of the upper band of Asf1 (Fig. 5D) revealed thatthis was due to phosphorylation of Asf1 protein. Since mobility-shiftedbands were not detected for C-terminal deleted mutant proteins 1–155and 1–139 (Fig. 5B), it is suggested that the region of aa 156–204 con-tains major phosphorylation sites. Consistent with the previous report(44), the missense mutant protein E36AD37A could not bind to HIRAin vitro (Fig. 5C). Interestingly, this mutant protein also abolished bind-ing ability as to p60 but retained binding ability to histones H3/H4 (Fig.5B). These findings were also confirmed in COS-7 cells, in which thosewere transiently expressed (Fig. 5E). In addition, human Asf1b couldbind to p60 and histones H3 and H4 (Fig. 5B).Next, we determined the region of Asf1 responsible for viability of
DT40 cells. The complementing criterion of Asf1 mutant proteins wasexamined based on the appearance of surviving colonies in the presenceof both tet and puromycin after transfection with expression vectorsencoding FLAG-tagged Asf1 and its mutants (Fig. 5A, right). As expected,the wild-type Asf1 protein could efficiently form the surviving colonies,whereas introduction of empty construct could not. Deletion muta-tional analyses showed that the minimum essential region for viabilitywas located within aa 1–155 of Asf1. Interestingly, the missense mutantprotein E36AD37A could rescue the lethality caused by the depletion ofHA-tagged Asf1 completely. To confirm this, we examined the growthrate of conditional Asf1-depleted cells, expressing stably the FLAG-tagged mutant Asf1 protein E36AD37A. As expected, FLAG-taggedE36AD37A of Asf1 was constantly expressed through a period of timeexamined, whereas HA-tagged Asf1 protein disappeared completely by24 h after the addition of tet (Fig. 5F, left). Even in the absence of HA-tagged wild-type Asf1 protein, the stable expression of the E36AD37Amutant protein could compensate for the loss of function of the wild-type Asf1 in the normal growth of the DT40 cell line (Fig. 5F, right).Together, these results revealed no requirement of stable association ofAsf1 with p60 and HIRA for cell growth ability. In addition, humanAsf1b also could do, suggesting that Asf1a and Asf1b act in a comple-mentary fashion for cell viability (Fig. 5A), consistentwith the result that
the synergetic role for cell proliferation was observed in human cells,using an RNAi-mediated knockdown system for Asf1a and Asf1b (17).Taken together, these results indicate not only that the N-terminal
core domain (aa 1–155) of Asf1 is necessary and sufficient for both cellviability and association with CAF-1p60, HIRA, and histones H3-H4but also that its interactionwithCAF-1 andHIRA is not required for cellviability.
DISCUSSION
Because DNA replication and chromatin assembly are tightly cou-pled processes having a tremendous impact on cell growth of verte-brates, a slight deviation in these would definitely affect cell prolifera-tion (3, 6, 7, 8, 19). RNAi-mediated knockdown of CAF-1, the primaryfactor for histone H3-H4 deposition during DNA replication, leads toretarded replication-coupled chromatin assembly, slowed DNA repli-cation, slowed S phase progression, and eventually apoptotic cell death(6, 7).On the other hand, the in vivo role ofAsf1, another histoneH3-H4chaperone, has not been fully elucidated yet, except that RNAi-medi-ated knockdown of Asf1 impairs S phase progression in human andDrosophila cell lines (17, 18). To better understand the physiologicalfunctions of Asf1 and to dissect its functional domains, we geneticallycreated the Asf1-conditional knock-out DT40 cell line.Using this system, we clearly established that Asf1 is essential for
viability of DT40 cells. The depletion of Asf1 caused defects in S phaseprogression accompanied by slowed DNA replication, and then thesedefective phenotypic properties were similar to those seen in CAF-1-depleted DT40 cells.3 Furthermore, as in the case with the CAF-1depletion (7),3 newly synthesized DNA was highly sensitive toMNase digestion (Fig. 3, A and B), indicating that CAF-1 and Asf1are individually required for replication-coupled nucleosome assembly.Thus, the in vivo replication-coupled nucleosome assembly depends onnot only CAF-1 function but also Asf1 function, in agreement with theproposed synergistic role of Asf1 with CAF-1 in the in vitro replication-coupled nucleosome assembly (11, 14). The defect in replication-couplednucleosome assembly could be a cause for slowedDNA replication and Sphase progression, as proposed for CAF-1-depleted cells (6, 8). Addi-tionally, this defective chromatin structure possibly triggered other cel-lular defects, such as accumulation of damaged DNA (Fig. 2, F and G),which was also evident in yeast (45, 46), and aberrant mitotic process(Fig. 4).In a variety of chromatin-modulating processes, it is difficult to assess
which deviated chromatin modulation is associated with cognate cellu-lar aberrancy caused by the Asf1 depletion. Several lines of evidence areavailable demonstrating its association with histones H3-H4, CAF-1p60, and HIRA (11, 13, 14, 15, 44) (Fig. 5), probably facilitating nucleo-some assembly. In addition, Asf1 has been reported to be involved inproviding a buffering system to preserve excess H3-H4 pool (17). Theseresults strongly suggested the role of Asf1 as a histone donor in bothCAF-1-mediated replication-coupled and HIRA-mediated replication-independent chromatin assembly processes (2). The similar aberrantphenotypes observed between CAF-1-depleted and Asf1-depletedDT40 cells were in line with this theory, suggesting that Asf1-mediatedCAF-1 function might be responsible for cellular aberrancy to a greaterextent as described above, but the Asf1-mediated HIRA function maynot be involved in it, because such aberrancywas not observed inHIRA-deficient DT40 cells (48). Alternatively, since Asf1 interacts with TFIID(23) and regulates expressions of a broad array of genes in yeast (24),some of the cellular abnormalities might be accounted for by the lack of
3 Y. Takami, submitted for publication.
Asf1 Is Essential for Vertebrate Cells
13826 JOURNAL OF BIOLOGICAL CHEMISTRY VOLUME 281 • NUMBER 19 • MAY 12, 2006
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

the Asf1 function as a transcription regulator by disturbing the expres-sion of specific genes.ChickenAsf1 protein, like those in other organisms, is composed of two
structural distinct domains, the N-terminal evolutionarily conserveddomain (aa 1–155) and the C-terminal divergent one (aa 156–204).Recently, a three-dimensional structural model of yeast Asf1 protein, inwhich theN-terminal coredomain adopts a compact immunoglobulin foldstructure consisting of 10�-sheets, has been proposed (44, 49). In addition,the N-terminal domain is sufficient not only for stimulating the in vitroreplication-dependentnucleosomeassemblybutalso forconferringmethylmethanesulfonate resistance and gene silencing (44). Consistent with this,our complementation analyses revealed that the N-terminal aa 1–155domain is the minimum essential region for cell proliferation and is suffi-cient for binding to histones H3-H4, CAF-1p60, and HIRA (Fig. 5). How-ever, of our findings, the most intriguing one is that the binding abilities ofAsf1 as to both CAF-1p60 and HIRA proteins are not connected with itscell viability function, implying the possible existence of an alternativemediating role of Asf1, other than H3-H4 deposition, that is distinct fromthose concerning these twoproteins for cell proliferation. In relation to this,a direct involvement of Asf1 in the DNA replication machinery wasrecently evaluated in yeast (i.e.maintaining a subset of replication elonga-tion factors by directly interacting with the RFC during stress) (42). If thismechanism is also operating in vertebrates, loss of this function should leadto more severe cellular defects in normally proliferating DT40 cells thanyeast, since the spontaneously occurring DNA damages during replicationare more evident in higher eukaryotes than in yeast because of its largergenome size. On the other hand, the possible function of the C-terminalregion of Asf1 remains to be resolved. This C-terminal region exhibits arelativedivergence in lengthbut similarity among species. For example, thisregion is rich in acidic amino acid residues in yeast, but in vertebrates it isenriched with Ser and Thr residues that are considered to be kinase-sub-strate sites (28). Interestingly, thephosphorylated formofAsf1disappeared,when theC-terminal regionwas deleted (Fig. 5B), indicating that it is beingphosphorylated in DT40 cells. The deletion of this region exhibited noeffects on either the cell growth or binding capability as to histonesH3-H4,p60, andHIRA (Fig. 5), consistentwith the previous findings that phospho-rylation statusdoesnot affect either the in vitrochromatinassembly activity(14) orhistone flow (17).Thus, theC-terminal regionofAsf1 is predicted tohave an additional regulatory role. Since Asf1 phosphorylation is regulatedby Tlk kinase, which is inhibited by ATR/ATM kinase triggered by DNAdamage and replication stress (29), the phosphorylation status might beinvolved in signal transduction in response to DNA damage.In conclusion, our results revealed that the vertebrate Asf1 is an inte-
gral player for the nucleosome assembly process during DNA replica-tion, including being essential in normal DNA replication and S phaseprogression, and provides a possible clue of its participation in cell pro-liferation distinct from CAF-1- and HIRA-mediated chromatin assem-bly pathways. Our established cell lines, expressing exclusively Asf1mutant proteins lacking the phosphorylation site or binding platformfor CAF1-p60 and HIRA, would be of great help in elucidating addi-tional roles of Asf1 for many chromatin-modulating processes main-taining chromosomal integrity and cell viability.
Acknowledgments—We thank N. Nagamatsu-Yamamoto for technical assist-ance and Dr. H. Kikuchi for helpful support.
REFERENCES1. Tyler, J. K. (2002) Eur. J. Biochem. 269, 2268–22742. Loyola, A., and Almouzni, G. (2004) Biochim. Biophys. Acta 1677, 3–113. Polo, S. E., and Almouzni, G. (2005) Cancer Lett. 220, 1–9
4. Ahmad, K., and Henikoff, S. (2002) Proc. Natl. Acad. Sci. U. S. A. 99, Suppl. 4,16477–16484
5. Shibahara, K., and Stillman, B. (1999) Cell 96, 575–5856. Hoek, M., and Stillman, B. (2003) Proc. Natl. Acad. Sci. U. S. A. 100, 12183–121887. Nabatiyan, A., and Krude, T. (2004)Mol. Cell Biol. 24, 2853–28628. Ye, X., Franco, A. A., Santos, H., Nelson, D. M., Kaufman, P. D., and Adams, P. D.
(2003)Mol. Cell 11, 341–3519. Kaufman, P. D., Kobayashi, R., and Stillman, B. (1997) Genes Dev. 11, 345–35710. Le, S., Davis, C., Konopka, J. B., and Sternglanz, R. (1997) Yeast 13, 1029–104211. Tyler, J. K., Adams, C. R., Chen, S. R., Kobayashi, R., Kamakaka, R. T., and Kadonaga,
J. T. (1999) Nature 402, 555–56012. Sharp, J. A., Fouts, E. T., Krawitz, D. C., and Kaufman, P. D. (2001) Curr. Biol. 11,
463–47313. Krawitz, D. C., Kama, T., and Kaufman, P. D. (2002)Mol. Cell Biol. 22, 614–62514. Mello, J. A., Sillje, H. H., Roche, D.M., Kirschner, D. B., Nigg, E. A., and Almouzni, G.
(2002) EMBO Rep. 3, 329–33415. Tagami, H., Ray-Gallet, D., Almouzni, G., and Nakatani, Y. (2004) Cell 116, 51–6116. Ray-Gallet, D., Quivy, J. P., Scamps, C., Martini, E.M., Lipinski, M., and Almouzni, G.
(2002)Mol. Cell 9, 1091–110017. Groth, A., Ray-Gallet, D., Quivy, J. P., Lukas, J., Bartek, J., and Almouzni, G. (2005)
Mol. Cell 17, 301–31118. Schulz, L. L., and Tyler, J. K. (2006) FASEB J. 20, 488–49019. Gunjan, A., Paik, J., and Verreault, A. (2005) Biochimie (Paris) 87, 625–63520. Zhang, R., Poustovoitov, M. V., Ye, X., Santos, H. A., Chen, W., Daganzo, S. M.,
Erzberger, J. P., Serebriiskii, I. G., Canutescu, A. A., Dunbrack, R. L., Pehrson, J. R.,Berger, J. M., Kaufman, P. D., and Adams, P. D. (2005) Dev. Cell 8, 19–30
21. Adkins, M. W., and Tyler, J. K. (2004) J. Biol. Chem. 279, 52069–5207422. Adkins, M. W., Howar, S. R., and Tyler, J. K. (2004)Mol. Cell 14, 657–66623. Chimura, T., Kuzuhara, T., andHorikoshi,M. (2002) Proc. Natl. Acad. Sci. U. S. A. 99,
9334–933924. Zabaronick, S. R., and Tyler, J. K. (2005)Mol. Cell Biol. 25, 652–66025. Myung, K., Pennaneach, V., Kats, E. S., and Kolodner, R. D. (2003) Proc. Natl. Acad.
Sci. U. S. A. 100, 6640–664526. Hu, F., Alcasabas, A. A., and Elledge, S. J. (2001) Genes Dev. 15, 1061–106627. Emili, A., Schieltz, D.M., Yates, J. R., III, andHartwell, L. H. (2001)Mol. Cell 7, 13–2028. Sillje, H. H., and Nigg, E. A. (2001) Curr. Biol. 11, 1068–107329. Groth, A., Lukas, J., Nigg, E. A., Sillje, H. H., Wernstedt, C., Bartek, J., and Hansen, K.
(2003) EMBO J. 22, 1676–168730. Carrera, P., Moshkin, Y.M., Gronke, S., Sillje, H. H., Nigg, E. A., Jackle, H., and Karch,
F. (2003) Genes Dev. 17, 2578–259031. Takami, Y., and Nakayama, T. (1997) Genes Cells 2, 711–72332. Ahmad, A., Takami, Y., and Nakayama, T. (2003) Biochem. Biophys. Res. Commun.
312, 1266–127233. Takami, Y., Kikuchi, H., and Nakayama, T. (1999) J. Biol. Chem. 274, 23977–2399034. Fukagawa, T., Hayward,N., Yang, J., Azzalin, C., Griffin, D., Stewart, A. F., and Brown,
W. (1999) Nucleic Acids Res. 27, 1966–196935. Gossen, M., and Bujard, H. (1992) Proc. Natl. Acad. Sci. U. S. A. 89, 5547–555136. Takata, M., Sabe, H., Hata, A., Inazu, T., Homma, Y., Nukada, T., Yamamura, H., and
Kurosaki, T. (1994) EMBO J. 13, 1341–134937. Fukagawa, T., Mikami, Y., Nishihashi, A., Regnier, V., Haraguchi, T., Hiraoka, Y.,
Sugata, N., Todokoro, K., Brown, W., and Ikemura, T. (2001) EMBO J. 20,4603–4617
38. Ahmad, A., Takami, Y., and Nakayama, T. (2004) Gene (Amst.) 342, 125–13639. Takami, Y., and Nakayama, T. (2000) J. Biol. Chem. 275, 16191–1620140. Martini, E., Roche, D. M., Marheineke, K., Verreault, A., and Almouzni, G. (1998)
J. Cell Biol. 143, 563–57541. Rogakou, E. P., Boon, C., Redon, C., and Bonner,W.M. (1999) J. Cell Biol. 146, 905–91642. Franco, A. A., Lam, W. M., Burgers, P. M., and Kaufman, P. D. (2005) Genes Dev. 19,
1365–137543. Dodson, H., Bourke, E., Jeffers, L. J., Vagnarelli, P., Sonoda, E., Takeda, S., Earnshaw,
W. C., Merdes, A., and Morrison, C. (2004) EMBO J. 23, 3864–387344. Daganzo, S.M., Erzberger, J. P., Lam,W.M., Skordalakes, E., Zhang, R., Franco, A. A.,
Brill, S. J., Adams, P. D., Berger, J. M., and Kaufman, P. D. (2003) Curr. Biol. 13,2148–2158
45. Ramey, C. J., Howar, S., Adkins, M., Linger, J., Spicer, J., and Tyler, J. K. (2004) Mol.Cell Biol. 24, 10313–10327
46. Prado, F., Cortes-Ledesma, F., and Aguilera, A. (2004) EMBO Rep. 5, 497–50247. Sutton, A., Bucaria, J., Osley, M. A., and Sternglanz, R. (2001) Genetics 158,
587–59648. Ahmad, A., Kikuchi, H., Takami, Y., and Nakayama, T. (2005) J. Biol. Chem. 280,
32090–3210049. Mousson, F., Lautrette, A., Thuret, J. Y., Agez, M., Courbeyrette, R., Amigues, B.,
Becker, E., Neumann, J. M., Guerois, R., Mann, C., and Ochsenbein, F. (2005) Proc.Natl. Acad. Sci. U. S. A. 102, 5975–5980
Asf1 Is Essential for Vertebrate Cells
MAY 12, 2006 • VOLUME 281 • NUMBER 19 JOURNAL OF BIOLOGICAL CHEMISTRY 13827
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from

Tatsuya Ono, Kei-ichi Shibahara and Tatsuo NakayamaFumiyuki Sanematsu, Yasunari Takami, Hirak Kumar Barman, Tatsuo Fukagawa,
in Vertebrate CellsAsf1 Is Required for Viability and Chromatin Assembly during DNA Replication
doi: 10.1074/jbc.M511590200 originally published online March 14, 20062006, 281:13817-13827.J. Biol. Chem.
10.1074/jbc.M511590200Access the most updated version of this article at doi:
Alerts:
When a correction for this article is posted•
When this article is cited•
to choose from all of JBC's e-mail alertsClick here
http://www.jbc.org/content/281/19/13817.full.html#ref-list-1
This article cites 49 references, 24 of which can be accessed free at
by guest on February 2, 2019http://w
ww
.jbc.org/D
ownloaded from





























