Embed Size (px)
Citation preview

Arf tumor suppressor and miR-205 regulate celladhesion and formation of extraembryonic endodermfrom pluripotent stem cellsChunliang Lia,b, David Finkelsteinc, and Charles J. Sherra,b,1
aHoward Hughes Medical Institute, and Departments of bTumor Cell Biology and cComputational Biology, St. Jude Children’s Research Hospital, Memphis,TN 38105
Contributed by Charles J. Sherr, February 4, 2013 (sent for review December 22, 2012)
Induction of theArf tumor suppressor (encoded by the alternate read-ing frameof theCdkn2a locus) followingoncogeneactivationengagesa p53-dependent transcriptional program that limits the expansion ofincipient cancer cells. Although the p19Arf protein is not detected inmost tissues of fetal or young adult mice, it is physiologically ex-pressed in the fetal yolk sac, a tissue derived from the extraembryonicendoderm (ExEn). Expression of the mouse p19Arf protein marks latestages of ExEn differentiation in cultured embryoid bodies (EBs) de-rived from either embryonic stem cells or induced pluripotent stemcells.Arf inactivation delays differentiation of the ExEn lineagewithinEBs, butnot the formationofothergermcell lineages frompluripotentprogenitors. Arf is required for the timely induction of ExEn cells inresponse to Ras/Erk signaling and, in turn, acts through p53 to ensurethe development, but not maintenance, of the ExEn lineage. Remark-ably, a significant temporal delay in ExEn differentiation detected dur-ing thematurationofArf-null EBs is rescuedby enforced expressionofmouse microRNA-205 (miR-205), a microRNA up-regulated by p19Arf
and p53 that controls ExEn cell migration and adhesion. The nonca-nonical and canonical roles of Arf in ExEn development and tumorsuppression, respectively, may be conceptually linked through mech-anisms that govern cell attachment and migration.
p53 tumor suppressor | epithelial to mesenchymal transition
The Ink4–Arf (Cdkn2a,b) locus, which is only 50 kb in length,encodes three intimately linked tumor suppressor genes. The
Ink4a and Ink4b genes encode polypeptides (p16Ink4a andp15Ink4b) that inhibit cyclin D-dependent kinases to maintain theretinoblastoma protein (Rb) in its active inhibitory state, therebylimiting cell proliferation. In contrast, the Arf protein (p19Arf inthe mouse, p14ARF in humans) inhibits the Mdm2 E3 ubiquitinligase to activate and stabilize p53, a transcription factor thatcoordinates a complex gene expression program that potentlyguards against tumor formation (1, 2). The p19Arf and p16Ink4a
proteins are encoded in part by unique first exons, whose productsare spliced to a second shared exon that is translated in alternativereading frames, yielding proteins that bear no shared amino acidsequences and that are functionally distinct. The Ink4a–Arf locus isgenerally not expressed under normal physiological circumstancesbut is induced by aberrant mitogenic signals that result from on-cogene activation. By engaging Rb- and p53-dependent tran-scriptional programs, the Ink4–Arf proteins counter tumor cellprogression by eliciting cell cycle arrest, apoptosis, or cellular se-nescence. Deletion of this small gene cluster incapacitates thefunctional Rb/p53 tumor-suppressive network and is one of themost common events observed in human cancers.The Ink4a–Arf locus is silenced in stem cells—whether of
embryonic, fetal, or adult somatic tissue origin—thereby facili-tating their capacity for continuous cellular self-renewal. Incontrast, the locus is epigenetically remodeled in more differ-entiated cell types to allow its engagement in response to on-cogenic stress signals. Despite the risk of its deletion in cancer,the evolutionary conservation of the Ink4–Arf locus in mammalsmay provide a mechanism for limiting the numbers of stem and
progenitor cells (2). In agreement with the idea that epigeneticsilencing of the locus is necessary to maintain cellular self-renewal,reprogramming of somatic cells to yield induced pluripotentstem (iPS) cells is accompanied by Ink4–Arf repression (see be-low) and facilitated by Ink4–Arf deletion (3).Paradoxically, the p19Arf protein is physiologically expressed in a
few disparate tissues during mouse development, including peri-vascular cells within the hyaloid vasculature of the eye (4–6), mi-totically dividing spermatogonia within seminiferous tubules (6, 7),and the fetal yolk sac (8). Inactivation ofArf results in blindness andreduced sperm production, but effects of Arf deletion on yolk sacdevelopment have not been investigated. Whether these diversephysiological roles of Arf can be explained through a commonmechanism and whether they reflect the canonical role of Arf asa potent tumor suppressor remain a mystery. We demonstrate thata signaling pathway involving Ras/Erk, p19Arf, p53, and microRNA205 (miR-205) regulates a cell motility and adhesion program thatfacilitates formation of extraembryonic endoderm (ExEn) cellsfrom pluripotent embryonic stem (ES) or iPS cell progenitors.
ResultsExpression of Arf in ExEn. Blastocysts harvested from mouse em-bryos at embryonic day (E) 4.5 exhibit pluripotent Oct4-positivecells in the inner cell mass surrounded by Gata4-marked primitiveendoderm (PrE) cells in a generally mutually exclusive pattern(Fig. 1A Upper). By using a particularly sensitive and specificmonoclonal antibody (9), p19Arf expression was not detected inthe early PrE lineage at E4.5 (Fig. 1A Lower), whereas embryosrecovered at E7.5 revealed p19Arf expression in ExEn tissues
Significance
The Arf tumor suppressor gene is not expressed in most normaltissues but when activated by oncogenic stress signals engagesa p53-dependent transcriptional program that prevents tumorformation. Surprisingly, expression of the p19Arf protein inmouseembryoid bodies is required for the timely formation of extra-embryonic endoderm (ExEn). Inactivation of Arf down-regulatesa singlemicroRNA,miR-205,which can “rescue” ExEn formation inArf-null embryonic or induced pluripotent stem cells. During ExEnformation, miR-205 regulates a suite of genes that govern cellmigration and adhesion, suggesting a conceptual basis for linkingthe roles of Arf in ExEn differentiation and tumor metastasis.
Author contributions: C.L. and C.J.S. designed research; C.L. and D.F. performed research;C.L. and C.J.S. contributed new reagents/analytic tools; C.L., D.F., and C.J.S. analyzed data;and C.L. and C.J.S. wrote the paper.
The authors declare no conflict of interest.
Freely available online through the PNAS open access option.
Data deposition: The data reported in this paper have been deposited in the Gene Ex-pression Omnibus (GEO) database, www.ncbi.nlm.nih.gov/geo (accession no. GSE42210).1To whom correspondence should be addressed. E-mail: [email protected].
This article contains supporting information online at www.pnas.org/lookup/suppl/doi:10.1073/pnas.1302184110/-/DCSupplemental.
www.pnas.org/cgi/doi/10.1073/pnas.1302184110 PNAS Early Edition | 1 of 10
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US

surrounding Oct4-positive cells (Fig. 1B). Inter-crossing Arf+/−
mice yields Arf−/− offspring at the expected Mendelian ratio, andArf-null females produce equivalent numbers of blastocysts com-pared with those of wild-type (WT) mice and generate normal-sized litters (10). Examination of 10 Arf−/− and 12 Arf+/+ embryosrecovered at E7.5 revealed as much morphological variation withinindividual cohorts as between them.Given difficulties in defining any overt developmental de-
ficiency in Arf-null embryos, we attempted to model the earlieststages of ExEn formation in embryoid bodies (EBs) derived fromcultured ES cells. When undifferentiated ES cells are suspendedin hanging drops and transferred to ultralow attachment dishesin medium lacking leukemia-inhibitory factor (LIF), the well-organized EBs that develop are rimmed by a single ExEn layer,which surrounds the pluripotent epiblast and other cells withinthe inner mass that are differentiating to form the three othergerm cell lineages. Cryosections of EBs that arose after 4 d of
culture clearly revealed p19Arf expression restricted to the outerExEn layer, which was marked by expression of the Polycombgroup protein Bmi1 (11) (Fig. 1C). Bmi1 is essential for silencingof the entire Ink4–Arf locus in adult hematopoietic and neuralstem cells and is required for formation of the early ExEn lineage(12), where, in contrast, it does not interfere with p19Arf ex-pression (Fig. 1C).At early times of embryonic development (E4.5) when p19Arf
is not detected (Fig. 1A), some cells within the inner cell massexpress the ExEn markers Gata6, Dab2, and Lrp2 and exhibitdiminished expression of the pluripotency markers Nanog andOct4 (13). PrE cells then migrate to the EB periphery to sur-round pluripotent cells. Because we never detected p19Arf in theinner cell mass, we assumed that the emergence of p19Arf-positivecells only occurs after the migration of PrE cells to form the outerEB layer. We crossed females of a knock-in strain that expressesCre recombinase under the control of the endogenous cellular Arfpromoter (6) to reporter male mice that conditionally express Cre-dependent LacZ from the Rosa-26 locus. Although there was noexpression of LacZ in pluripotent ES cells due to the silenced Arfpromoter, the Arf–Cre allele was activated in EBs as expected, andβ-galactosidase expression was largely limited to the EB periphery(Fig. 1D).
Retardation of ExEn Differentiation in Cultured Arf-Null ES Cells.Unlike WT EBs that spontaneously develop in cultures de-prived of LIF for 4 d, Arf-null EBs expressed the pluripotencymarker Oct4 in lieu of Dab2 at their periphery (Fig. 2 A and B).Independently derived Arf-null ES cell clones that underwentdifferentiation after LIF withdrawal exhibited highly robustNanog protein expression, but reduced levels of the ExEnmarkers Dab2 and Lrp2, compared with those in WT EBs (Fig.2C), consistent with the failure to detect ExEn cells on the sur-face of Arf-null EBs. Given that an inability of Arf-null progen-itors to form ExEn cells might be compensated during earlyembryonic development in the mouse, the duration of EB cellculture was extended to determine whether ExEn cell devel-opment was temporally delayed. EB cells derived from Arf-nullES clones recovered Dab2 expression after 10 d of suspensionculture and produced Dab2 at levels equivalent to those detectedin differentiating WT cells cultured for only 4 d (Fig. 2D). Byusing Arf-null ES cells in which a cDNA encoding green fluo-rescent protein (GFP) was substituted for Arf exon-1β sequencesunder the control of the cellular Arf promoter (14), ArfGfp/Gfp
ES clones produced GFP- and Gata4-positive ExEn cells after10 d of prolonged EB culture (Fig. 2E). Hence, the functionallynull ArfGfp alleles were induced as Gata4-positive ExEn cellseventually emerged.
Arf-null, but Not Ink4a-Deficient, iPS Cells Exhibit Defective ExEnDifferentiation. The creation of iPS cells by introduction of fourtranscription factors, Oct4, Sox2, Klf4, and c-Myc, into mouseembryo fibroblasts (MEFs) is facilitated by inactivation of p53(15–18). Because elevated Myc signaling engages Arf to inducep53 (19), Arf-null MEFs might be as susceptible to four-factoriPS reprogramming as their p53-null counterparts. We derived2 WT, 3 Arf+/−, and 14 Arf-null iPS cell lines, the latter origi-nating from Arf−/− or ArfGfp/Gfp strains (10, 14). All iPS cell linesselected for subsequent studies maintained normal karyotypesand expressed the pluripotency markers Oct4, Sox2, Nanog, andCD15/SSEA1; all could generate cell types representing threeembryonic germ layers; and all formed teratomas in immuno-deficient mice (Fig. S1 A–D). Like p53-null MEFs, strains lackingArf generated iPS cells at ∼50-fold greater efficiency than cellscontaining a single Arf allele (Fig. S1E).When allowed to differentiate for 4 d in the absence of LIF,
Arf+/− iPS cells generated EBs that expressed p19Arf and Gata4at the periphery (Fig. S1 F and G). However, Arf-null iPS clones
Fig. 1. p19Arf protein expression in ExEn. (A) WT E4.5 embryos were fixedand stained with the indicated antibodies and visualized for immunofluo-rescence with a confocal microscope. Pluripotent cells (red) in the inner cellmass and differentiated primitive endoderm cells (green) were visualizedwith antibodies to Oct4 and Gata4, respectively; p19Arf protein was notdetected. DAPI was used to visualize cell nuclei. (Scale bars, 25 μm.) (B)Section of WT embryo recovered from the decidua of the uterus at E7.5stained as above. p19Arf-positive ExEn cells (green, asterisks) surround theOct4-positive epiblast (red). (Scale bars, 200 μm, Upper; 100 μm, Lower.) (C)Cryosectioned WT EBs were stained and visualized as above. Expression ofp19Arf (green) and Bmi1 (red) colocalize (yellow) in a single ExEn cell layer atthe EB periphery. (Scale bar, 50 μm.) (D) Lineage tracing. Knock-in mice withCre under the regulatory control of the cellular Arf promoter were crossedto an indicator strain that expresses LacZ in response to Cre-mediated exci-sion of a “lox–stop–lox” cassette. ES cells obtained from these blastocystswere induced to differentiate to EBs. β-galactosidase was detected at theperiphery of EBs expressing Arf–Cre (Lower; magnification: 50×).
2 of 10 | www.pnas.org/cgi/doi/10.1073/pnas.1302184110 Li et al.
mimicked their ES cell counterparts and expressed much lowerlevels of the ExEn markers Dab2 and Lrp2 than Arf+/− iPS cells(Fig. S1H) and were defective in producing fully differentiatedExEn cells in EBs (Fig. S1 I and J). Two iPS cell lines derivedfrom p16Ink4a-null MEFs generated EBs that expressed the ExEnmarker proteins Gata4 and Lrp2 at levels equivalent to those oftheir WT counterparts (Fig. S1K) and exhibited comparativelyhigher levels of Dab2 and Gata4 mRNAs (Fig. S1L). Thus, al-though the entire mouse Ink4a–Arf locus is silenced during thereprogramming of iPS cells, and although p19Arf and p16Ink4a areboth reexpressed as iPS cells differentiate, onlyArf loss of functiondelays ExEn differentiation. Given that Arf-null ES and iPS cellsexhibited identical phenotypes, both were exploited subsequently.
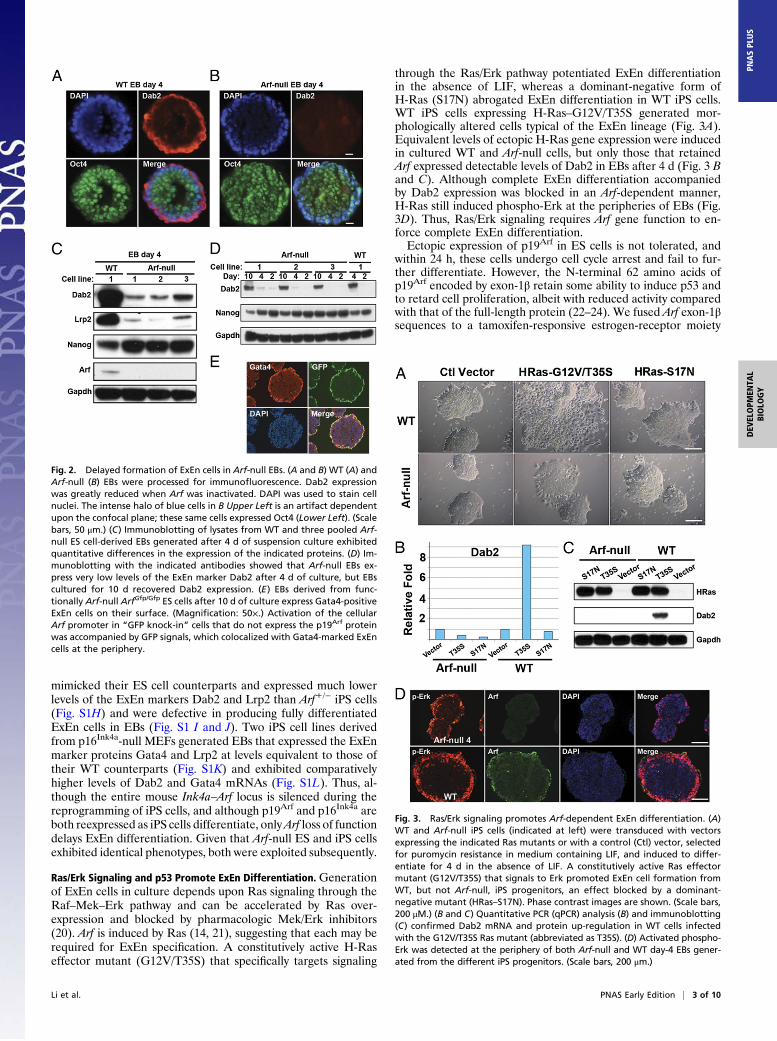
Ras/Erk Signaling and p53 Promote ExEn Differentiation. Generationof ExEn cells in culture depends upon Ras signaling through theRaf–Mek–Erk pathway and can be accelerated by Ras over-expression and blocked by pharmacologic Mek/Erk inhibitors(20). Arf is induced by Ras (14, 21), suggesting that each may berequired for ExEn specification. A constitutively active H-Raseffector mutant (G12V/T35S) that specifically targets signaling
through the Ras/Erk pathway potentiated ExEn differentiationin the absence of LIF, whereas a dominant-negative form ofH-Ras (S17N) abrogated ExEn differentiation in WT iPS cells.WT iPS cells expressing H-Ras–G12V/T35S generated mor-phologically altered cells typical of the ExEn lineage (Fig. 3A).Equivalent levels of ectopic H-Ras gene expression were inducedin cultured WT and Arf-null cells, but only those that retainedArf expressed detectable levels of Dab2 in EBs after 4 d (Fig. 3 Band C). Although complete ExEn differentiation accompaniedby Dab2 expression was blocked in an Arf-dependent manner,H-Ras still induced phospho-Erk at the peripheries of EBs (Fig.3D). Thus, Ras/Erk signaling requires Arf gene function to en-force complete ExEn differentiation.Ectopic expression of p19Arf in ES cells is not tolerated, and
within 24 h, these cells undergo cell cycle arrest and fail to fur-ther differentiate. However, the N-terminal 62 amino acids ofp19Arf encoded by exon-1β retain some ability to induce p53 andto retard cell proliferation, albeit with reduced activity comparedwith that of the full-length protein (22–24). We fused Arf exon-1βsequences to a tamoxifen-responsive estrogen-receptor moiety
Fig. 2. Delayed formation of ExEn cells in Arf-null EBs. (A and B) WT (A) andArf-null (B) EBs were processed for immunofluorescence. Dab2 expressionwas greatly reduced when Arf was inactivated. DAPI was used to stain cellnuclei. The intense halo of blue cells in B Upper Left is an artifact dependentupon the confocal plane; these same cells expressed Oct4 (Lower Left). (Scalebars, 50 μm.) (C) Immunoblotting of lysates from WT and three pooled Arf-null ES cell-derived EBs generated after 4 d of suspension culture exhibitedquantitative differences in the expression of the indicated proteins. (D) Im-munoblotting with the indicated antibodies showed that Arf-null EBs ex-press very low levels of the ExEn marker Dab2 after 4 d of culture, but EBscultured for 10 d recovered Dab2 expression. (E) EBs derived from func-tionally Arf-null ArfGfp/Gfp ES cells after 10 d of culture express Gata4-positiveExEn cells on their surface. (Magnification: 50×.) Activation of the cellularArf promoter in “GFP knock-in” cells that do not express the p19Arf proteinwas accompanied by GFP signals, which colocalized with Gata4-marked ExEncells at the periphery.
Fig. 3. Ras/Erk signaling promotes Arf-dependent ExEn differentiation. (A)WT and Arf-null iPS cells (indicated at left) were transduced with vectorsexpressing the indicated Ras mutants or with a control (Ctl) vector, selectedfor puromycin resistance in medium containing LIF, and induced to differ-entiate for 4 d in the absence of LIF. A constitutively active Ras effectormutant (G12V/T35S) that signals to Erk promoted ExEn cell formation fromWT, but not Arf-null, iPS progenitors, an effect blocked by a dominant-negative mutant (HRas–S17N). Phase contrast images are shown. (Scale bars,200 μM.) (B and C) Quantitative PCR (qPCR) analysis (B) and immunoblotting(C) confirmed Dab2 mRNA and protein up-regulation in WT cells infectedwith the G12V/T35S Ras mutant (abbreviated as T35S). (D) Activated phospho-Erk was detected at the periphery of both Arf-null and WT day-4 EBs gener-ated from the different iPS progenitors. (Scale bars, 200 μm.)
Li et al. PNAS Early Edition | 3 of 10
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US

(ERTAM) and separated these cassettes by a 16-amino acid linkerpeptide (Arf residues 63–78) containing the epitope recognizedby the 5C3-1 monoclonal antibody to p19Arf (9) (SI Materials andMethods). Following its transduction into WT MEFs, the ∼50-kDa fusion protein was detected with antibodies to either p19Arf
or ER (Fig. 4A). Arf-null NIH 3T3 cells, MEFs, and iPS cellsexpressing the uninduced fusion protein continued to proliferate.After introduction of the minigene into Arf-null iPS cells, whoseRNA expression was detected by using PCR primers directed tothe ERTAM cassette (Fig. 4B Upper Left), tamoxifen treatmentinduced modestly but significantly increased expression of Dab2and Gata4 mRNAs and decreased expression of Nanog (Fig. 4BUpper Right, Lower Left, and Lower Right). Hence, induction ofArf–exon1β–ERTAM was capable of driving elements of the ExEndifferentiation program.Wedetermined that EBs derived from p53-null iPS cells failed to
exhibit a distinct halo of ExEn cells on their surface (Fig. 4C), andDab2 levels in pooled day-4 EBs were significantly reduced, similarto that seen in the Arf-null setting (Fig. 4D). Despite the absenceof p53, the p19Arf protein was expressed on the EB periphery, butDab2 and Gata4 expression could not be detected (Fig. 4E). Oct4-positive cells localized on the surface instead (Fig. 4E). Thus, Arfenforces p53-dependent differentiation of ExEn cells.EBs derived from Arf-null iPS cells transduced with H-Ras–
G12V/T35S expressed increased phospho-Erk activity at theirperiphery (Fig. 3D), but like p53-null EBs, these cells did notform Dab2-positive ExEn cells in the absence of Arf (Fig. 3 B andC). We expressed p53–ERTAM in Arf-null iPS cells either aloneor in those engineered to stably express H-Ras–G12V/T35S.Conditional induction of Dab2 was achieved following tamoxifenexposure during 4 d of EB development only when Ras was over-expressed (Fig. 4F), indicating that the Ras/Erk pathway comple-ments p19Arf
–p53 signaling to enforce ExEn differentiation.
WT ExEn Cells Form Chimeric EBs When Admixed with Arf-Null ESCells. ExEn cell lines, also established from mouse blastocysts(25, 26), exhibit a morphology distinct from cultured ES cells(Fig. 5A) and express a different repertoire of ExEn proteins andmRNAs (Fig. 5 B and C). Because we did not succeed in gener-ating such cultures from Arf-null ES cells, we established ExEncells from blastocysts containing “floxed” Arf alleles (ArfFL/FL) (6)and deleted Arf exon-1β with a retrovirus encoding Cre recom-binase. These Arf-null ExEn cells continued to proliferate con-tinuously, and expression of ExEnmarkers was maintained, soArfis not required for ExEn cell line viability and maintenance.We infected cultured WT ExEn cells with a retrovirus
encoding GFP and admixed them in a 1:1 ratio with Arf-null EScells to form chimeric EBs (26). Two days later, ExEn cells hadlocalized to the EB outer layer, spontaneously segregating fromDAPI-stained ES cells that comprised the inner pluripotentcompartment (Fig. 5D). Because Arf-null ES cells cannot con-tribute to ExEn formation under these conditions, the exclusionof GFP-marked ExEn cells from the inner pluripotent cells andtheir localization and retention at the periphery is an inherentproperty of differentiated ExEn cells themselves.
miR-205 Can Rescue ExEn Differentiation in Arf-Null Cells. Arf isexpressed in mitotically dividing spermatogonia, but not in pri-mary spermatocyctes derived from them or in other cells withinthe testis (6, 7). In unrelated studies, we determined that thelevels of a single microRNA, miR-205, were selectively de-creased by >15-fold in the Arf-null testes of postnatal day-18mice, a time when p19Arf levels in spermatogonia are maximaland are synchronously expressed throughout the developingseminiferous tubules. miR-205 is up-regulated by p53 (27), andits promoter is directly bound and induced by other p53-relatedfamily members, p63 and p73 (28, 29).
Motivated to test whether Arf–p53 signaling and miR-205expression are directly correlated in ES cells, we obtained an EScell line (designated Arf154) in which a miR-30–based shRNAthat targets Arf mRNA (but not sequences shared with Ink4a) is
Fig. 4. Arf and p53–ER promote expression of ExEn marker proteins. (A)Lysates of WT MEFs (left lane) or from cells transduced with a vectorencoding Arf exon-1β sequences (center lane) or with a naked control vector(right lane) were immunoblotted with antibodies directed to p19Arf (Upper)or to the ER cassette (Lower). The endogenous p19Arf protein was detectedin all three samples, whereas the 50-kDa minigene-coded fusion protein wasdetected only in the center lane. The asterisk indicates a staining artifact. (B)Arf-null iPS cells transduced with the vector encoding the Arf–exon-1βminigene were subjected to qPCR analysis. Primers that amplified the ERmoiety confirmed generally equivalent levels of vector-coded RNA expres-sion in cells treated with different concentrations of tamoxifen (TAM;see legends). Dab2 and Gata4 mRNAs in transduced Arf-null iPS cells wereinduced, and Nanog expression was decreased in response to minigene ex-pression and tamoxifen treatment. Error bars, mean ± SEM (n = 3 experi-ments). (C) Phase contrast micrographs reveal that EBs derived from p53-nulliPS cells (Right) fail to express a halo of ExEn cells visualized in WT EBs (Left).(D) Like day-4 EBs derived from Arf-null iPS cells, p53-null iPS cells exhibitedreduced Dab2 protein levels compared with WT iPS controls. (E) Althoughp19Arf protein expression was detected at the periphery of day-4 EBs derivedfrom p53-null iPS cells, neither Gata4 nor Dab2 were detected. Instead, Oct4-positive cells were observed at the periphery. (Scale bars, 200 μm.) (F) Arf-nulliPS cells were transduced with a retroviral vector expressing the HRas–G12V/T35 mutant. Cells engineered to coexpress p53–ERTAM (Center andRight) were treated with tamoxifen (Right) or not (Center), and Dab2 ex-pression was quantified by flow cytometric analysis. The percentages of cellsthat exhibited increased Dab2 expression are noted.
4 of 10 | www.pnas.org/cgi/doi/10.1073/pnas.1302184110 Li et al.

induced by doxycycline (Dox) treatment (30). Arf154 ES cellswere suspended for EB formation and treated with Dox for 4 d.Expression of miR-205 RNA was decreased following Doxtreatment, which was without effect in WT ES cells (Fig. 6ALeft). Conversely, introduction of the Arf–exon-1β–ERTAM fusionprotein into Arf-null iPS cells triggered a conditional increase inmiR-205 levels following tamoxifen treatment (Fig. 6A Right).WT ES cells were transduced with miR-205 vectors coex-
pressing GFP (Fig. 6B) or with a control vector expressing GFPalone, and GFP-positive cells were sorted 2 d later and replatedin the presence of LIF to preserve their undifferentiated state.Cells expressing GFP alone maintained their characteristic ESmorphology, but those expressing threefold more miR-205 un-derwent partial differentiation in 2 d, even in the presence ofLIF, to yield ExEn-like progeny (Fig. 6C) that exhibited in-creased Dab2 and decreased Nanog expression (Fig. 6D). Fourdays after plating these cells in the absence of LIF, expression ofGata4 and Sox7 mRNAs, as well as p19Arf, p53, and p21Cip1,were up-regulated as expected, but without effects on the relativeexpression of markers of ES-derived ectoderm (Fgf5), mesoderm(T/Brachyury), or trophectoderm (Cdx2) (Fig. 6E, left two col-umns). In contrast, enforced miR-205 expression in established,fully differentiated WT ExEn cell lines did not affect the ex-pression of these RNAs (Fig. 6E, right two columns). Thus,overexpression of miR-205 could up-regulate the Arf–p53–p21signaling axis and preferentially drive pluripotent WT ES cells toassume an ExEn fate but did not affect the same target genesonce ExEn differentiation was established. Given that p19Arf caninduce miR-205 (Fig. 6A) and vice versa (Fig. 6E), a feed-forward
mechanism appears to drive ExEn formation, after which miR-205 is without effect.We used the same strategy to introduce miR-205 into Arf-null
ES cells and promoted their spontaneous differentiation bywithdrawing LIF. Cells exhibiting a significant (P < 0.0032) 3.2-fold increase in miR-205 expression differentiated within 4 d toform typical ExEn cells (Fig. 7A). Dab2-positive cells were nowidentified at the EB periphery (Fig. 7B), and other ExEn markerswere further up-regulated (Fig. 7C). Strikingly, then, enforcedmiR-205 expression not only potentiated ExEn differentiation inWT ES cells (Fig. 6) but induced fully differentiated ExEn cellson the EB periphery even when Arf function was completelydisabled (Fig. 7).
ExEn Gene Expression Program Regulated by miR-205. Two in-dependently derived Arf-null ES cell lines infected in duplicatewith control or miR-205–encoding vectors were sorted 2 d laterfor GFP expression and cultured for an additional 4 d in theabsence of LIF to form EBs. Gene expression was profiled byusing Affymetrix Mouse Gene Chips (Version 1.0), which in-clude 33,283 probe sets. We identified 632 genes that were dif-ferentially up- or down-regulated at least twofold in response toenforced miR-205 expression vs. their levels in cells infected withthe control vector (Fig. 8A). Of 144 probe sets down-regulatedin ES cells 2 d after introduction of miR-205 but before LIFwithdrawal (Fig. 8A, lane 2 vs. 1), none corresponded to highlypredicted miR-205 targets (SI Materials and Methods). ExEnmarkers (Dab2, Gata4, Gata6, Sox17, and Afp) were further in-duced in response to miR-205, whereas expression of pluri-potency markers (such as Klf4 and Tbx3) was reduced (Fig. 8 Band C). Gene Ontology (Database for Annotation, Visualization,and Integrated Discovery) analysis indicated that the most sig-nificantly up-regulated genes encode glycoproteins (2.4E-17;4.2% of the total genes), affect in utero development (4.29E-05;0.74% of total genes), and govern cell motility and migration(4.64–8.71E-04; 0.74% of total genes) and cell adhesion (2.37E-04; 0.82% of total genes) (Fig. 8D).The cell-autonomous ability of WT ExEn cells to properly
form chimeric EBs when cocultured with Arf-null ES cells (Fig.5) mimics the classical sorting behavior of cells that expressvariable levels of cadherins (31, 32). The E-cadherin (Cdh1) re-pressor Snail (33) was expressed at higher levels in ExEn cellscompared with their ES cell progenitors with concomitant Cdh1down-regulation (Fig. 5). Affymetrix chip gene profiling (Fig.8D) and PCR analysis (Fig. S2A) confirmed the up-regulationof Snail2 in EBs responding to miR-205, but down-regulationof Cdh1 was not observed when day-4 EBs expressing a controlvector and miR-205 were directly compared by microarrayanalysis. However, introduction of miR-205 into Arf-null ES cellsresulted in a very rapid reduction in Cdh1 levels, which occurredeven before LIF was withdrawn (Fig. S2A). Although the fewreported direct targets of miR-205 include Zeb proteins that alsoact as Cdh1 corepressors (34), Zeb2 levels were elevated in re-sponse to transduction of the miR-205 vs. control vector in Arf-null EBs (Fig. 8D and Fig. S2A). We used imaging techniques tolook at the topological distribution of E-cadherin in EBs formedfrom Arf-null ES cells that had been infected either with thecontrol vector or that encoding miR-205 (Fig. 9). Strikingly,E-cadherin was no longer detected in relative abundance at theperiphery of EBs overexpressing miR-205, and this result wasaccompanied by induction of vimentin (Fig. 8D) and its prefer-ential distribution to the ExEn layer at the EB surface (Fig. 9).Significantly increased expression of candidate genes that reg-
ulate cell migration and adhesion, including Cdh2 (N-Cadherin),Cdh5, Cdh6, and Cdh11, was observed (Fig. 8D and Fig. S2A).Many other genes reported to be essential for cell migration,invasion, and adhesion also exhibited the same pattern of expres-sion as the latter cadherins (Fig. 8D). Wnt pathway components,
Fig. 5. Self-sorting of ExEn cells in chimeric EBs. (A) Morphology of ExEncells visualized by phase contrast microscopy. (Magnification: 100×.) (B andC) Immunoblotting of proteins (B) and PCR analysis of transcripts (C) in WTES or ExEn cell lines. (D) WT ExEn cells expressing p19Arf, Gata4, and Dab2were transduced with a vector expressing GFP, mixed with an equal numberof Arf-null ES cells, and suspended in the absence of LIF to form chimeric EBs.Two days after suspension, EBs were harvested, cryosectioned, and stainedwith antibodies to the indicated proteins. Marked ExEn cells spontaneouslysorted to the periphery of the chimeric EBs and completely segregated fromDAPI-stained ES cells in the inner mass. (Scale bars, 100 μm.)
Li et al. PNAS Early Edition | 5 of 10
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US

including Wnt3, Wnt5a, and Lef1, were enriched in the up-regulated group of genes (Fig. S2B), consistent with previousobservations that activation of the β-catenin/Tcf–Lef signalingpathway is an obligatory step required for the retinoic acid-induced differentiation of pluripotent cells into ExEn cells (35).Collectively, these results imply that miR-205–dependent generegulation can substitute for p19Arf signaling in altering a pro-gram of gene expression required for the proper migration andsorting of differentiating ExEn cells to their final location on theEB periphery.
DiscussionThe p53-dependent role of Arf as a tumor suppressor is widelyappreciated, but the functions of Arf in other seemingly arcanephysiological settings are poorly understood. The p19Arf proteinwas not detected in mouse embryos at E4.5 but is expressed inthe ExEn lineage by E7.5, consistent with its detection in the yolksac later during fetal development (8). We modeled the earlieststages of ExEn cellular differentiation and development bystudying pluripotent ES and iPS cells of various genotypes thatwere induced to differentiate in culture into EBs in which cells of
Fig. 6. miR-205 is regulated by Arf and enhances ExEn formation fromWT ES progenitors. (A Left) Arf154 ES cells expressing a Dox-inducible Arf shRNA werecultured to form EBs in the absence of LIF and presence of Dox, resulting in a small decrease (P < 0.01) in miR-205 mRNA expression, quantified by PCR. WT EScells were unaffected by Dox. Error bars, mean ± SEM (n = 3 experiments). (Right) Arf-null ES cells transduced with Arf–exon-1β–ERTAM were induced to formEBs and simultaneously treated with tamoxifen (TAM) for 4 d. (B) The V1 vector transcribes miR-205 (hairpin) embedded in a miR-30 backbone, whereas theV2 vector expresses the complete pre-miRNA sequence (gray rectangle). Vectors included a PGK promoter-driven cassette and encoded neomycin resistance(Neor); GFP was translated from an internal ribosome entry site (IRES). Long terminal repeat (LTR) sequences required for viral integration and viral promoter-driven mRNA expression and a psi-2 (φ) virion packaging sequence are indicated. (C) WT ES cells infected with a control vector (Ctl GFP; Left) or with miR-205vectors (Center and Right) were maintained in LIF to retard differentiation. Nonetheless, enforced miR-205 expression generated cells with altered mor-phology reminiscent of ExEn cells. (Magnification: 100×.) (D) Cells shown in C down-regulated the pluripotency marker Nanog and up-regulated the ExEnmarker Dab2. (E) A more comprehensive quantitative RT-PCR analysis of infected WT iPS cells (left two bars) revealed up-regulation of other ExEn markers,Gata4 and Sox7, but insignificant alterations in the expression of markers of other germ-cell layers (mesoderm T, ectoderm Fgf5, and trophectoderm Cdx2). Asexpected, p19Arf, p53, and the p53-responsive gene p21Cip1 were induced as cells assumed the ExEn fate. By contrast, enforced expression of miR-205 inestablished ExEn cell lines (right two bars) did not affect the mRNA expression of the above genes.
6 of 10 | www.pnas.org/cgi/doi/10.1073/pnas.1302184110 Li et al.

the ExEn lineage, born internally, differentiate and migrate tothe periphery. The Ink4–Arf locus is silenced in pluripotent ESand iPS cells, and, like p53, Arf inactivation increases the fre-quency of successful four-factor reprogramming by >50-fold. WTiPS cells mimicked ES cells in reengaging Arf expression duringExEn development.Unlike other ExEn marker proteins—such as Gata4 and
Dab2, which are first detected in select cells within the inner cell
mass as they lose expression of pluripotency factors—the in-duction of p19Arf occurs only after primitive endoderm cells havemigrated to form the single EB outer layer. Notably, the ap-pearance of differentiated ExEn cells in Arf-null EBs (but not inInk4a-null EBs) was significantly delayed, requiring 10 d of exvivo culture instead of the 4-d period normally sufficient for Arf+/+
ES cells to generate mature ExEn derivatives. Given that Arf-nullpups are born in appropriate numbers at the expected Mendelianratio, compensation for Arf loss-of-function might occur duringearly mouse development.Development of the ExEn lineage is enforced by Ras and
inhibited by Myc. Ras/Erk signaling is central to these effects, anddrugs that inhibit either Mek or Erk interfere with this process(20). Although Ras potently elicits phospho-Erk expression at theperiphery of Arf-null EBs, other markers of the ExEn lineage,including Dab2, Lrp2, and Gata4, were not detected, implyingthat p19Arf acts “downstream” of, or parallel to, Ras/Erk signalingto facilitate later stages of ExEn differentiation. p53-deficient iPScells also failed to generate EBs that exhibited mature ExEn cellson their surface. However, in this setting, p19Arf was expressed atthe EB periphery, but again without the appearance of Dab2- orGata4-positive cells, implying that the ability of p19Arf to induceExEn differentiation is p53-dependent. In turn, conditional acti-vation of p53–ERTAM in Arf-null iPS cells engineered to expressH-Ras–G12V-T35S bypassed the Arf requirement and restoredthe appearance of Dab2- and Gata4-positive ExEn cells.We successfully generated ExEn cell lines from WT and ArfFl/Fl
embryos but were unable to derive them from Arf-null embryos.Conditional Cre recombinase-mediated deletion of ArfFL allelesfrom established ExEn lines had no effect on their viability orability to be continuously passaged in culture; hence, Arf playsa role in late stages of ExEn lineage differentiation but not inmaintaining ExEn cell lines once they have been established.When suspended Arf-null ES and WT ExEn cells were admixedand cocultured to form EBs, GFP-marked ExEn cells segregatedfrom Oct4-positive ES cells and rapidly localized to the periph-ery of chimeric EBs. Because ExEn cells cannot arise from Arf-null EBs under these conditions, their ability to sort properly isan inherent property.Adhesion in tissues is predominantly mediated by cadherins,
whose quantitative differences in expression are sufficient to gov-ern the spontaneous self-sorting behavior of two cell populations,rendering them immiscible (32). E-cadherin is required for ag-gregation of ES cells and for the proper formation of EBs (36).In model systems analogous to the formation of chimeric EBs,differences in the levels of cellular surface tension arising fromcadherin expression are sufficient to enable two populations tosort out, allowing cells with the lower level of cadherin to en-velop the others (37). Consistent with this view, ExEn cells ex-press lower Cdh1 levels than their ES progenitors. We thereforeconsidered the idea that Arf may regulate a p53-dependentprogram of gene expression that affects ExEn cell migration andadhesion within differentiating EBs.Arf plays a salutary role in male germ-cell development where
its transient expression, restricted to basement membrane-boundspermatogonial progenitors in seminiferous tubules, ensures thesurvival of detached spermatocytes that have extinguished Arfexpression and entered meiotic prophase I (7). Detachment ofArf-null progenitors from the tubular lining triggers DNA damageand p53-dependent apoptosis (anoikis) of primary spermatocytes,resulting in reduced numbers of mature sperm. Sequencingof microRNAs extracted from whole testes, in which relativelysynchronous and maximal expression of p19Arf is achieved atpostnatal day 18, revealed that miR-205 was singularly down-regulated by >15-fold when Arf was inactivated. Analysis of EBs inwhich Arf expression could be conditionally up- or down-regulatedprovided direct evidence for positively correlated Arf and miR-205 expression. In turn, miR-205 expression is induced by p53,
Fig. 7. miR-205 induces ExEn formation from Arf-null ES progenitors. (A)Arf-null ES cells infected with a naked control (Ctl) GFP vector or oneencoding miR-205 from a miR-30 backbone together with GFP were sorted2 d after infection, placed in culture, and visualized 2 d later by phasecontrast microscopy at magnification of 50× (Left) or 100× (Right). Cells withthe characteristic morphology of ExEn cells appeared in response to miR-205.(B) EBs derived from WT or Arf-null ES cells and infected with control or miR-205–encoding vectors (indicated at the top) were stained for Dab2 proteinexpression. WT cells (Left) and Arf-null EBs transduced with miR-205 (Right)expressed Dab2 at their periphery, whereas Arf-null cells, whether un-infected or transduced with a control vector, did not (second and third fromleft). (Scale bars, 50 μm.) (C) Quantitative RT-PCR was used to quantify mRNAexpression of five ExEn markers (indicated at the top) in vector-transducedArf-null ES cells grown in the presence of LIF (day 0) and in EBs derived fromthem (day 4). Cells were transduced with a control virus (Ctl) or a vectorencoding miR-205 (205) as indicated in the legend. Error bars, mean ± SEM(n = 3 experiments).
Li et al. PNAS Early Edition | 7 of 10
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US

and its promoter is directly regulated by other p53 family mem-bers (27–29).Threefold overexpression of miR-205 in WT ES cells accel-
erated ExEn formation without affecting differentiation of othergerm-cell lineages. Notably, our goal was not to try to identifymRNA targets regulated by miR-205 in ES cells, but, instead, totest whether miR-205, acting as a p19Arf
–p53 signaling responseelement, might “rescue” Arf deficiency and promote ExEn dif-ferentiation. Strikingly, introduction of miR-205 into Arf-null EScells was sufficient to induce complete ExEn differentiation inEBs after LIF was withdrawn. Given that p19Arf regulates miR-205 levels and vice versa, a feed-forward mechanism likely drivesExEn formation, after which miR-205 is without observed effectsin mature ExEn cells. Unlike the vast majority of microRNAs,inactivation of miR-205 results in early embryonic lethality (38),whereas physiological up-regulation of miR-205 might compen-sate for Arf loss in the embryo proper.PCR analysis, immunofluorescence imaging, and gene profiling
pointed to widespreadmiR-205–induced effects on the expressionof genes that govern cell migration and adhesion. Introductionof miR-205 into Arf-null ES cells acutely triggered Cdh1 down-regulation even before LIF was withdrawn, and elevated expres-sion of the Cdh1 corepressors Snail and Zeb2 was observed asmiR-205-expressing Arf-null EBs emerged and formed ExEn
cells. Vimentin was up-regulated in response to enforced miR-205synthesis, and the protein preferentially localized to the EB outerlayer in lieu of E-cadherin. In contrast, another family of Cdhgenes (Cdh4, 5, 6, and 11) was induced by miR-205, as were manyother genes implicated in regulating cell motility and adherence.Given that qualitative differences in cadherin signaling dictatepatterns of mutual adhesive binding and help to define mechan-ical polarization boundaries (39), the segregation of externalExEn tissue from internal ES cells within EBs likely dependson many such factors, and not simply one. In agreement withobservations that activation of β-catenin signaling is requiredfor retinoic acid-induced differentiation of ES cells into ExEncells (35), Wnt pathway components, includingWnt3, Wnt5a, andLef1, were also induced. Together, these findings indicate thatregulation of miR-205 by p19Arf
–p53 signaling affects the ex-pression of genes required for late stages of ExEn differentiation,including many that govern cell migration and sorting within EBs.Differential adhesion plays well-documented roles not only in
embryogenesis but also in malignancy. Notably, some of the out-comes observed during ExEn formation in EBs were inconsistentwith the reported role of miR-205 in maintaining epithelial fatesand E-cadherin expression and in forestalling the epithelial tomesenchymal transition (EMT) during tumor progression (29, 33,34). The few reported targets of miR-205 in tumor cells include
Fig. 8. Gene expression profiling of Arf-null ES cells and EBs transduced with miR-205. Arf-null ES cells infected with a retroviral vector expressing GFP alone(Ctl) or with a vector coexpressing miR-205 and GFP were sorted for GFP expression, plated in the presence of LIF (designated ES), or induced for 4 d in theabsence of LIF to form EBs (designated EB). Isolated RNAs labeled with fluorochromes were used to probe Affymetrix Mouse Gene Chips (Version 1.0). Thedata were normalized (Z score transformed) across samples so that the heat intensity scales shown in A, C, and D are in units of SD. (A) Indicated in the heatmap are 632 probe sets that were significantly up-regulated (red) or down-regulated (green) with twofold or more changes. Control ES cells, ES cellsexpressing miR-205, control EBs, and EBs expressing miR-205 are indicated and are designated 1–4, respectively, at the top of the heat map. Although theseexperiments were not designed to identify genes directly targeted by miR-205, we selected the most probable miR-205 mouse target genes in each of threepublic databases (miRDB.org, TargetScan.org, and microRNA.org) and chose 26 genes for further analysis that were concordantly ranked in the “top 60.”None of the 144 probe sets corresponding to selected candidates exhibited >1.5-fold down-regulation in miR-205–expressing ES cells taken for comparativemicroarray analysis before LIF withdrawal. Six genes [Cdh11 (see also Fig. S2A), Cldn11, Plcb1, Ralyl, Zfp558, and Zfp758] were not expressed at significantlevels in control GFP+ ES cells. The remaining candidates included Acsl1, Akna, BC030336, Cadm1, Ccny, Chn1, Ctps2, Dmxl2, Ezr, Lrch3, Lrp1, Mgrn1, Mllt4,Nacc2, Nfat5, Rfk, Sbf2, Sdha, Sorbs1, and Spata13. (B) qPCR analysis confirmed up-regulation of two ExEn markers in EBs transduced with miR-205. Error bars,mean ± SEM (n = 3 experiments). (C) Heat map of a subset of genes associated with pluripotency and differentiation toward germ layers other than ExEn(terms indicated at the right). Of note, Klf4, Tbx3, Lefty2, Tdgf1, Ncam1, and Fgf5 revealed less than twofold changes. (D) Heat map of genes involved in celladhesion (i) and cell motility and migration (ii). Designation of lanes in C and D is identical to that in A. The data have been deposited in the Gene ExpressionOmnibus (GEO) under accession no. GSE42210.
8 of 10 | www.pnas.org/cgi/doi/10.1073/pnas.1302184110 Li et al.

Zeb1/2 corepressors, which down-regulate Cdh1. However, whenmiR-205 expression was enforced in Arf-null ES progenitors thatwere induced to form EBs, Zeb2 expression was unexpectedlyincreased, and Cdh1 levels fell. We can only presume that otheras-yet-undefined mechanisms intercede in preventing Zeb2mRNA down-modulation by miR-205 during EB formation.Despite these differences, our data argue that miR-205 ex-
pression is positively regulated by Arf–p53 signaling and that Arfloss compromises miR-205 expression in at least two differentphysiological settings—ExEn differentiation and spermatogen-esis. Down-regulation of miR-205 in tumor cells promotes theEMT and metastasis in several forms of cancer (29, 33, 34, 40,41). A parsimonious hypothesis is that a role for Arf as a tumorsuppressor in somatic cells is mediated, at least in part, by itsregulation of EMT in response to oncogene activation. Thisfinding is consistent with a plethora of studies indicating that Arfexpression is induced by activated oncogenes and that sub-sequent Arf inactivation facilitates late stages of tumor pro-gression and metastasis.
Materials and MethodsAnimals and Cell Lines. Work with mice was performed under establishedguidelines and supervision by the St. Jude Children’s Research Hospital’s(SJCRH) Institutional Animal Care and Use Committee, as required by the USAnimal Welfare Act and National Institutes of Health policy to ensure propercare and use of laboratory animals for research. We previously generatedArf-null (10), Arf–GFP (14), Arf–Flox, and Arf–Cre mice (6). Mouse strainsdeficient for Ink4a (42) and Ink4a–Arf (43) were derived by others. Geneti-cally engineered mice were back-crossed nine or more times onto a C57BL/6background to create isogenic strains. C57BL/6 mice deficient for p53 werepurchased from Jackson Laboratories (stock no. 2101), and immunodeficientnude mice (stock no. 086) were from Charles River Laboratories.
ES cell lines (44) and ExEn cells (25) from E3.5 embryos were derived asdescribed. E3.5 blastocysts were flushed and cultured in suspension untilE4.5 before analysis by immunofluorescence. ES cells (Arf154) expressing anshRNA directed to Arf mRNA (30) were generously provided by Prem Pre-msrirut (Cold Spring Harbor Laboratory, Cold Spring Harbor, NY; Mirimus,Cold Spring Harbor, NY). Early passage primary MEF strains were generatedas described (10).
A single inducible lentiviral vector containing a Dox-responsive elementcoregulating Oct4, Klf4, Sox2, and cMyc (STEMCCA-tetO-4F) and anothervector expressing the reverse tetracycline activator (rtTA) were used toproduce iPS cells (45, 46). Approximately 1 × 105 MEF cells were seeded in60-mm culture plates and infected with 2.5 mL of medium containing the
rtTA vector plus 2.5 mL of medium containing the STEMCCA-tetO-4F vectorin the presence of polybrene (5 μg/mL) (Sigma-Aldrich). The medium wasreplaced after 24 h with ES cell medium and changed every 2–3 d. Dox(Sigma-Aldrich) was added 24 h after infection at a final concentration of1 μg/mL and then removed at day 14. iPS colonies, picked 20–25 d after in-fection on the basis of morphology, were expanded on gelatin-coated cul-ture dishes in ES cell medium. Then, 4 × 106 undifferentiated iPS and ES cellswere injected into the hind leg muscles of 6- to 8-wk-old nude mice, andteratomas were analyzed 4 wk later.
Plasmid Vectors. Construction of the Arf–exon1β–ERTAM plasmid is describedin SI Materials and Methods. The p53ERTAM vector (47) was obtained withpermission of Gerard Evan from Douglas Green (SJCRH), and miR-205 vec-tors were provided by Gregory Hannon (Cold Spring Harbor Laboratory).Vectors encoding H-Ras–G12V/T35S, H-Ras–S17N were provided by HiroshiKoide (Kanazawa University, Kanazawa, Ishikawa, Japan) (48).
EB Formation. EBs were generated by trypsinizing undifferentiated mouse ESor iPS cells and placing 300 cells into 50-μL hanging drops. Two days later,aggregates were collected and cultured in ultralow attachment Petri disheswith EB culture medium, consisting of 90% DMEM, 10% FBS (both vol/vol),100 U/mL penicillin, 100 μg/mL streptomycin, 2 mM L-glutamine (all fromInvitrogen), and 0.1 mM mercaptoethanol (Gibco). The medium was changedevery other day. To generate chimeric EBs, ES and ExEn cells were trypsinized,mixed 1:1, and dropped onto ultralow attachment Petri dishes at a density of300 cells per 50 μL. Two days later, chimeric EBs were collected, embedded intissue freezing medium (Triangle Biomedical Sciences), and sectioned into (10μm) slices for immunofluorescence analysis.
RT-PCR. Total RNA was extracted from cells by using TRIzol (Invitrogen) andtranscribed into cDNA by using a high-capacity reverse-transcription kit (ABI),which included oligo(dT)15 and ReverTra Ace reverse transcriptase. FormiRNA reverse transcription, universal PCR Master Mix, No AmpErase UNG(Applied Biosystems) was used. For quantitative RT-PCR, raw data wereobtained on PRISM 7900 (ABI) by using SYBR Green I double-stranded DNA-binding dye chemistry (Applied Biosystems) as described (44). For detection ofmiR-205 and endogenous miRNA RNU6B, TaqMan MicroRNA Assay kits(Applied Biosystems) were used. See Table S1 for primers.
Immunofluorescence and Immunoblotting. Staining was performed as de-scribed (49). Primary antibodies listed in Table S2 were diluted in blockingbuffer and incubated with sections overnight at 4 °C. Fluorescently coupledsecondary antibodies, including Alexa anti-rat 488, anti-mouse 488, anti-rabbit 488, anti-rat 555, anti-mouse 555, and anti-rabbit 555 (all from Invi-trogen), were incubated for 1 h at room temperature. Representative imageswere captured by confocal microscopy (Zeiss LSM 510 NLO Meta). Immu-noblotting was performed as described (50).
Affymetrix Microarray Analysis. RNA from Arf-null iPS cells and EBs infectedwith a control vector expressing GFP alone or with another vector coex-pressing GFP and miR-205 were collected and subjected to hybridization byusing Affymetrix Mouse Gene Chips (Version 1.0). Data presentation andanalysis are described in SI Materials and Methods. Expression of selectedgenes was validated by qPCR using the PCR primers listed in Table S1.
RNA sequencing experiments performed on whole mouse testes of WTand Arf-null mice by Kaja Wasik and Gregory Hannon (Cold Spring HarborLaboratory) first revealed down-regulation of miR-205 in response to Arfinactivation. These insights provided the impetus for studying miR-205 ex-pression during extraembryonic development. Relevant RNA sequencingdata were generously provided for our own use.
ACKNOWLEDGMENTS. Relevant RNA sequencing data were generouslyprovided by Kasa Wasik and Gregory Hannon. We thank Gregory Hannonand Scott Lowe for providing vectors encoding miR-205; Konrad Hoched-linger for lentiviruses encoding four reprogramming factors and rtTA;Gerard Evan for p53–ERTAM; Prem Premsrirut for Arf154 ES cells; Ronald A.DePinho for Ink4a-null mice; Hiroshi Koide for H-Ras mutants; NadineHachouche and Rebecca Singleterry for excellent technical assistance; Fred-erique Zindy for providing mice and cell lines; core resources of St. JudeComprehensive Cancer Center CA-21765 for imaging, flow cytometric anal-yses, synthesis of oligonucleotides, and gene profiling analysis; and membersof the C.J.S./Martine F. Roussel laboratory for critical comments and encour-agement. This work was supported in part by ALSAC of SJCRH. C.J.S. is anInvestigator of the Howard Hughes Medical Institute.
Fig. 9. miR-205 drives vimentin synthesis and relocalization of E-cadherinwithin Arf-null EBs. Immunofluorescence staining of Arf-null day-4 EBs de-rived from cells infected with a control vector (Ctl; Left) and a vectorexpressing miR-205 (205; Right) is shown. Confocal images of EBs stainedwith antibodies to E-cadherin or vimentin and with DAPI to visualize nucleiare merged. (Scale bars, 100 μm.)
Li et al. PNAS Early Edition | 9 of 10
DEV
ELOPM
ENTA
LBIOLO
GY
PNASPL
US

1. Kim WY, Sharpless NE (2006) The regulation of INK4/ARF in cancer and aging. Cell127(2):265–275.
2. Sherr CJ (2012) Ink4-Arf locus in cancer and aging. Wiley Interdiscip Rev Dev Biol 1(5):731–741.
3. Li H, et al. (2009) The Ink4/Arf locus is a barrier for iPS cell reprogramming. Nature460(7259):1136–1139.
4. McKeller RN, et al. (2002) The Arf tumor suppressor gene promotes hyaloid vascularregression during mouse eye development. Proc Natl Acad Sci USA 99(6):3848–3853.
5. Silva RL, et al. (2005) Arf-dependent regulation of Pdgf signaling in perivascular cellsin the developing mouse eye. EMBO J 24(15):2803–2814.
6. Gromley A, Churchman ML, Zindy F, Sherr CJ (2009) Transient expression of the Arftumor suppressor during male germ cell and eye development in Arf-Cre reportermice. Proc Natl Acad Sci USA 106(15):6285–6290.
7. Churchman ML, Roig I, Jasin M, Keeney S, Sherr CJ (2011) Expression of arf tumorsuppressor in spermatogonia facilitates meiotic progression in male germ cells. PLoSGenet 7(7):e1002157.
8. Jerome-Majewska LA, et al. (2005) Tbx3, the ulnar-mammary syndrome gene, andTbx2 interact in mammary gland development through a p19Arf/p53-independentpathway. Dev Dyn 234(4):922–933.
9. Bertwistle D, Zindy F, Sherr CJ, Roussel MF (2004) Monoclonal antibodies to the mousep19(Arf) tumor suppressor protein. Hybrid Hybridomics 23(5):293–300.
10. Kamijo T, et al. (1997) Tumor suppression at the mouse INK4a locus mediated by thealternative reading frame product p19ARF. Cell 91(5):649–659.
11. Sparmann A, van Lohuizen M (2006) Polycomb silencers control cell fate,development and cancer. Nat Rev Cancer 6(11):846–856.
12. Lavial F, et al. (2012) Bmi1 facilitates primitive endoderm formation by stabilizingGata6 during early mouse development. Genes Dev 26(13):1445–1458.
13. Chazaud C, Yamanaka Y, Pawson T, Rossant J (2006) Early lineage segregationbetween epiblast and primitive endoderm in mouse blastocysts through the Grb2-MAPK pathway. Dev Cell 10(5):615–624.
14. Zindy F, et al. (2003) Arf tumor suppressor promoter monitors latent oncogenicsignals in vivo. Proc Natl Acad Sci USA 100(26):15930–15935.
15. Banito A, et al. (2009) Senescence impairs successful reprogramming to pluripotentstem cells. Genes Dev 23(18):2134–2139.
16. Kawamura T, et al. (2009) Linking the p53 tumour suppressor pathway to somatic cellreprogramming. Nature 460(7259):1140–1144.
17. Marión RM, et al. (2009) A p53-mediated DNA damage response limitsreprogramming to ensure iPS cell genomic integrity. Nature 460(7259):1149–1153.
18. Utikal J, et al. (2009) Immortalization eliminates a roadblock during cellularreprogramming into iPS cells. Nature 460(7259):1145–1148.
19. Zindy F, et al. (1998) Myc signaling via the ARF tumor suppressor regulates p53-dependent apoptosis and immortalization. Genes Dev 12(15):2424–2433.
20. Li LJ, et al. (2010) Stk40 links the pluripotency factor Oct4 to the Erk/MAPK pathwayand controls extraembryonic endoderm differentiation. Proc Natl Acad Sci USA107(4):1402–1407.
21. Palmero I, Pantoja C, Serrano M (1998) p19ARF links the tumour suppressor p53 toRas. Nature 395(6698):125–126.
22. Quelle DE, Cheng M, Ashmun RA, Sherr CJ (1997) Cancer-associated mutations at theINK4a locus cancel cell cycle arrest by p16INK4a but not by the alternative readingframe protein p19ARF. Proc Natl Acad Sci USA 94(2):669–673.
23. Weber JD, Taylor LJ, Roussel MF, Sherr CJ, Bar-Sagi D (1999) Nucleolar Arf sequestersMdm2 and activates p53. Nat Cell Biol 1(1):20–26.
24. Weber JD, et al. (2000) Cooperative signals governing ARF-mdm2 interaction andnucleolar localization of the complex. Mol Cell Biol 20(7):2517–2528.
25. Kunath T, et al. (2005) Imprinted X-inactivation in extra-embryonic endoderm celllines from mouse blastocysts. Development 132(7):1649–1661.
26. Rula ME, et al. (2007) Cell autonomous sorting and surface positioning in theformation of primitive endoderm in embryoid bodies. Genesis 45(6):327–338.
27. Piovan C, et al. (2012) Oncosuppressive role of p53-induced miR-205 in triple negativebreast cancer. Mol Oncol 6(4):458–472.
28. Alla V, et al. (2012) E2F1 confers anticancer drug resistance by targeting ABCtransporter family members and Bcl-2 via the p73/DNp73-miR-205 circuitry. Cell Cycle11(16):3067–3078.
29. Tucci P, et al. (2012) Loss of p63 and its microRNA-205 target results in enhanced cellmigration and metastasis in prostate cancer. Proc Natl Acad Sci USA 109(38):15312–15317.
30. Premsrirut PK, et al. (2011) A rapid and scalable system for studying gene function inmice using conditional RNA interference. Cell 145(1):145–158.
31. Alberts B, et al., eds (2008) The Molecular Biology of the Cell (Garland Science, NewYork), pp 1140–1142.
32. Steinberg MS (2007) Differential adhesion in morphogenesis: A modern view. CurrOpin Genet Dev 17(4):281–286.
33. Cano A, et al. (2000) The transcription factor snail controls epithelial-mesenchymaltransitions by repressing E-cadherin expression. Nat Cell Biol 2(2):76–83.
34. Gregory PA, et al. (2008) The miR-200 family and miR-205 regulate epithelial tomesenchymal transition by targeting ZEB1 and SIP1. Nat Cell Biol 10(5):593–601.
35. Sato N, Meijer L, Skaltsounis L, Greengard P, Brivanlou AH (2004) Maintenance ofpluripotency in human and mouse embryonic stem cells through activation of Wntsignaling by a pharmacological GSK-3-specific inhibitor. Nat Med 10(1):55–63.
36. Larue L, et al. (1996) A role for cadherins in tissue formation. Development 122(10):3185–3194.
37. Foty RA, Steinberg MS (2005) The differential adhesion hypothesis: A directevaluation. Dev Biol 278(1):255–263.
38. Park CY, et al. (2012) A resource for the conditional ablation of microRNAs in themouse. Cell Rep 1(4):385–391.
39. Amack JD, Manning ML (2012) Knowing the boundaries: Extending the differentialadhesion hypothesis in embryonic cell sorting. Science 338(6104):212–215.
40. Baffa R, et al. (2009) MicroRNA expression profiling of human metastatic cancersidentifies cancer gene targets. J Pathol 219(2):214–221.
41. Majid S, et al. (2010) MicroRNA-205-directed transcriptional activation of tumorsuppressor genes in prostate cancer. Cancer 116(24):5637–5649.
42. Sharpless NE, et al. (2001) Loss of p16Ink4a with retention of p19Arf predisposes miceto tumorigenesis. Nature 413(6851):86–91.
43. Serrano M, et al. (1996) Role of the INK4a locus in tumor suppression and cellmortality. Cell 85(1):27–37.
44. Li CL, et al. (2009) Germline-competent mouse-induced pluripotent stem cell linesgenerated on human fibroblasts without exogenous leukemia inhibitory factor. PLoSONE 4(8):e6724.
45. Wernig M, et al. (2008) A drug-inducible transgenic system for direct reprogrammingof multiple somatic cell types. Nat Biotechnol 26(8):916–924.
46. Sommer CA, et al. (2009) Induced pluripotent stem cell generation using a singlelentiviral stem cell cassette. Stem Cells 27(3):543–549.
47. Christophorou MA, et al. (2005) Temporal dissection of p53 function in vitro and invivo. Nat Genet 37(7):718–726.
48. Yoshida-Koide U, et al. (2004) Involvement of Ras in extraembryonic endodermdifferentiation of embryonic stem cells. Biochem Biophys Res Commun 313(3):475–481.
49. Li CL, et al. (2009) Pluripotency can be rapidly and efficiently induced in humanamniotic fluid-derived cells. Hum Mol Genet 18(22):4340–4349.
50. Zindy F, et al. (2007) Genetic alterations in mouse medulloblastomas and generationof tumors de novo from primary cerebellar granule neuron precursors. Cancer Res67(6):2676–2684.
10 of 10 | www.pnas.org/cgi/doi/10.1073/pnas.1302184110 Li et al.





























