Embed Size (px)
Citation preview

APPLICATIONS OF MODERN NMR METHODS TO STRUCTURAL STUDIES OF NATURAL AND
SYNTHETIC ORGANIC COMPOUNDS
Suzanne Monk B issada
A Thesis submitted in confomity with the requirements for the degree of Master of Science Graduate Department of Chemistry
University of Toronto
Q Siislanne M. Bissada, 1999.

National Library 1+1 of Canada Bibliothèque nationale du Canada
Acquisitions and Acquisitions et Bibliographie Services services bibliographiques 395 Wellington Street 395, rue Wellington OttawaON K1AON4 OttawaON K1AON4 Canada Canada
The author bas granted a non- L'auteur a accordé une licence non exclusive Licence allowing the exclusive permettant à la National Library of Canada to Bibliothèque nationale du Canada de reproduce, loan, disiribute or sel1 reproduire, prêter, distribuer ou copies of this thesis in rnicroform, vendre des copies de cette thèse sous paper or electronic formats. la forme de microfiche/film, de
reproduction sur papier ou sur format électronique.
The author retains ownership of the L'auteur conserve la propriété du copyright in this thesis. Neither the droit d'auteur qui protège cette thèse. thesis nor substantial extracts fiom it Ni la thèse ni des extraits substantiels may be printed or otherwise de celle-ci ne doivent être imprimés reproduced without the author's ou autrement reproduits sans son permission. autorisation.

Abstract
The study of natural and synthetic organic compounds has been investigated.
The use of one-dimensional and two-dimensional NMR methods as a method for rapidly
assigning structures was demonstrated. Structurai elucidation was perfomed on
unknown side-products in synthetic sequences to conclusively determine what reactions
had taken place. Steps toward a biomimetic synthesis of the alkaloid, dihydrocadambine,
were carried out. The isolation of the nahu;il product precursor to this alkaloid,
secologanin, was first performed. in the course of isolating secologanin, two other
natural products, loganin and sweroside were also isolated and structurally elucidated.
The intermediates in the atternpted synthesis of dihydrocadambine were also examined.
Finally, NMR methods were applied to characterization of diterpene and sesquiterpene
naturai products isolated in the Caribbean. Eleven of the natural products investigated
were determined to be novel.

Ac knowledeemen ts
1 wish to express my sincere gratitude to my supenisors, Professor Stewart
McLean and Professor William F. Reynolds. The opporhmity to work with two such
experienced, knowledgeable. worldly and genuine people will always be appreciated. 1
am thankful for the guidance and patience that was readily provided to me.
The expenence of working with the Caribbean affiliates from the University of
West Indies and the Mexican affiliates at Universidad Nacional Autonoma de México
was truly a pleasure. Working in such an international comrnunity provided more than
just a scientific learning experience
1 would like to thank Dr. Timothy Burrows for al1 the extra help he gave me on
instrumentation in the NMR facilities at the University of Toronto. Thanks are also due
to Ms. Nura Hagi-Nur for her contribution in isolating the natural products fiom the
honeysuc kle plant.
1 cannot forget the people that have been a constant support throughout my life. 1
can never be thankful enough to al1 my parents, Faiza Fahim Bissada, Mo& Giryagos
Bissada, Samir Magar, Feria1 Magar and Sarnia Bissada, and my brothers, Freddy Farid
Tengberg, Yousry Bissada and Ramses Bissada for their undying support and patience.

For my mother
Faiza Fahim Bissada
iii

Table of Contents --
Abstract Acknowledgement Dedica tion Table of Contents List of Ta b1es List of Abbreviations
Chapter 1 - Description of NMR Methods
1 . 1 Introduction
1.2. 2D NMR Expenments
1.3. Chemical Shifts
1.4. Multiplicity and Coupling
1 S. Instrumental Experimental
Chapter 2- Structural Elucidation of a Synthetic Intermediate
2.1. Background
2.2. NMR Data for Structure 3
2.3. Structural Determination of 3
2.4. Interpretation of Data
Chapter 3 - Synthetic Plan
3.1. Previous Synthesis of 3a and 3P- dihydrocadarnbine
3.2. Synthetic Plan
Page i ii iii iv vi vii

Chapter 4 - Isolates from Lonicera tartarica
4.1 . Iridoids of Lonicera tartarica
4.2. General Experimental
4.3. Isolation of Secologanin fiom Lonicera tartarica
4+4. Products Isolated fiom Extract
4.5. Forming dimethyl acetal O f 9
Chapter 5 - Steps toward Synthesis of Dihydrocadambine
5.1. mCPBA oxidation of 8
5.2. Dihydroxylation of STAMA
5.3. Formation of Key Epoxide
5.4. Pictet-Spengler Reaction
5 S. Deprotection of Dimethyl Acetal
5.6. Coupling of Epoxide 28 to tryptarnine
5.7. Discussion of Results
Chapter 6 - NMR Analysis of Natural Products
6.1. Terpenes
6.2. Determination of Structures of Two Sesquiterpenes fiom Capraria
6.3. Elucidation of Unknown Caesalpinia bonduc Root Extracts
6.4. Structural Elucidation of Awiellamines A and B
References

List of Tables --
Table
2-1
4-1
4-2
5- 1
5-2
5-3
5-4
5-5
6-1
6-2
6-3
6-4
6-5
6-6
6-7
Titfe
500 MHz NMR data for 3
400 MHz NMR data for 22
500 MHz NMR data for 21
400 MHz NMR data for glycol 24
500 MHz NMR data for mono-epoxide 26
500 MHz I3c. ' H and HMBC data for 18a
500 MHz "c. 'H, COSY and HMBC for 27
500 MHz 13c, 'H md HMBC data for 29
500 MHz 13c. 'H and HMBC data for 30
500 MHz 13c, 'H and HMBC data for 31
500 MHz 'H and "C data for 32 and 33
500 MHz 'H and l3c data for 34 and 35
500 MHz 'H and "C data for 36 and 37
500 MHz 'H and I3c data for 38 and 39
500 MHz 'H and 13c data for 40 and 41
Page
12
27
3 1
37
39
43
46
52
58
60
64
66
69

List of Abbreviations --
COSY: Correlation Spectroscopy
EtOAc: ethyl acetate
HMBC: ' ~de t ec t ed multiple-bond heteronuclear multiple-quantum coherence)
HMQC: '~Ddetected heteronuclear multiple-quantum coherence
HRMS : high-resolution mass spectrometry
HSQC: 'H-detected heteronuclear single-quantum coherence
mCPB A: me tu-c hloro peroxy benzoic acid
n.0.e.: nuclear Overhauser effect or enhancement
NMO : N-methy lmo rpholine Nsxide
NMR: Nuclear Magnetic Resonance
NOESY: homonuclear Overhauser effect spectroscopy
pTSA: para-toluene sulfonic acid
ROESY: rotating frame NOESY
STA: secologanin tetraacetate
STAMA: secologanin tetraacetate dimethyl acetal
TBAF: tetnibutylammonium fluoride
TLC: Thin Layer C hromatography
TMSOTfi Trimethyl silyl tnfluoro methane sulfonate
Ts : p-toluenesulfony 1
vii

Chapter 1. Descriplon of NMR Methods
1.1. Introduction
Nuclear magnetic resonance (NMR) spectroscopic data provides chemists with
information that is indispensable for unraveling the structure of organic compounds. The
interface of synthetic chemistry and NMR spectroscopy is becoming more closely
entwined as both develop. The utility of NMR to elucidate the structure of both expected
and unexpected products is of vital importance. Previously, when a synthetic sequence
was desired, characteristics for the desired product were usually the only ones looked for.
A method for rapidly assigning the structures of synthetic intermediates would provide a
secure way of ensuring that the proposed synthetic route was, in fact, being followed.
Identifjing the structure and charactenstics of an unknown side-product can result in
interesting chemistry, and modem methods of NMR spectroscopy in combination with
other instrumentation techniques, such as mass spectrometry and infrared spectroscopy,
are ideal for this purpose. The use of NMR techniques by means of one-dimensional and
two-dimensional nuclear magnetic resonance (2D NMR) for structure elucidation of both
naturai products and synthetic products were examined in the course of this thesis
researc h.
1.2. 2D NMR Exiieriments 1 2 3 4
Multi-dimensional NMR experiments have revolutionized the use of NMR
spectroscopy for structure elucidation of al1 molecules varying fiom small molecules to
large complex proteins. 2D NMR experiments are designed to generate different kinds of

fiequency information dong the two axes. The preparation time is a relaxation delay,
followed by one or more pulses to start the experiment.
The two distinct mechanisms for magnetization transfer are scalar coupling and
cross-relaxation. Scalar coupling yields information on the proximity of spins through
the covalent framework of the molecule and the latter provides information on spatial
proximity and molecular motion. In COSY (correlated spectroscopy), the tl period is
initiated with a 90" 'H pulse. Magnetization transfer arnong the transitions of a coupled
system is mediated by a second 90" 'H pulse before the t2 period. As a result of net
magnetization transfer, cross-peaks appear between al1 transitions belonging to the same
coupled spin system. Heteronuclear 2-D experiments cm also be perfonned for exarnple
when 'H magnetization can be transferred to a 13c nuclei by means of a 90" 'H pulse
followed by a delay and a 90" 13c puise. By employing proper delays in the preparation
period, HMQC ('H-detected heteronuclear multiple-quantum coherence) or HSQC ('H-
detected heteronuclear single quantum coherence) provide correlation for directly bonded
'H and I3c pairs of nuclei. HSQC differs fiom HMQC in that 'H multiplet structure
5 appears alongfr in the latter sequence but not in the former. HSQC was found, in our
lab, to provide better 13c resolution and sensitivit$ and consequently was used in place
of HMQC throughout these investigations. Similarly, W C (I~~detected multiple-bond
heteronuclear multiple-quantum coherence) provides 'H-'~c correlation via long-range
coupling. In both HSQC and HMBC experiments, another 90" "C pulse prior to the tz
period transfers the magnetization back to the 'H nuclei for detection.
Overhauser effect spectroscopy is based on cross-relaxation which is an
interaction in which the spins exchange magnetization by fluctuating through-space

dipole-dipole couplings. As a result of the distance dependence of the dipolar interaction,
cross-peaks in the 2D spectra may indicate spatially proximate partners. NOESY
(homonuclear Overhauser effect spectroscopy) has a pulse sequence similar to that of
COSY, but with an extra delay and a 90" 'H pulse before the t2 penod. During this
delay, nOe (nuclear Overhauser effect or enhancement) develops dong the z-axis. The
nOe causes the change in intensity (increase or decrease) during decoupling experiments.
The nuclear Overhauser measurements can also be performed in the rotating fiame and
under these conditions nOe factors are always positive. ROESY (rotating fiame
NOESY) experiments are used for molecules tumbling at a rate where nOe crosses over
from positive to negative. ROESY experiments are particularly important for
intermediate molecular weight molecules (ca. 500- 1 500).
The real usefûlness of multi-dimensional NMR in general is not in the
information provided by a single expenment, but rather in the synergy provided by
canying out several different experiments on the sarne molecule. Combining al1 these
experiments together, aid in identifjing each nucleus in a molecule, and their relative and
spatial positioning. Thus the experiments can be used either for complete NMR spectral
assignments of known molecules or mapping the structure of unknowns. A schematic
illustration for the various 2D NMR experiments w d for structural elucidation is shown .
in Figure 1-1.

1. COSY
2. HSQC
3. HMBC
Figure 1-1 - Schematic ülustration of the procedures used for structural assignment. 1 .) COSY assigns sequences of coupled protons 2.) HSQC assigns carbons for the same sequences. 3.) HMBC assigns non-protonated carbons and ties together molecular fragments separated by non-protonated carbons or heteroatoms.
1.3. Chernical Shifts 'g8?
Chernical shifts reflect the extent to which the basic Larmor fiequency of a
nucleus is altered by its electronic environment in the molecule. Magnetic field-induced
circulation of electrons in a molecule induces a shielding field, which opposes the
extemal magnetic field. The induced field arkes from both electrons associated with the
atom and electmnr in neighbouring atoms and bonds. As a result of this shielding field, a
particular nucleus in a molecule will give a NMR signal at a lower fiequency than for an
isolated nucleus with no surrounding electrom. For historical reasons, a nucleus giving a

signal at relatively low fiequency is said to be shielded, while one giWig a signal at
relatively higher fiequency is said to be deshielded. This reflects the fact that early NMR
spectrometers operated at constant fiequency while varying the magnetic field. By
contrat, modem spectrometers operate at constant field and mesure fiequency
differences.
Because the Larmor fiequency is proportional to the strength of the magnetic
field, there is no absolute scale of chemical shiR Thus, a freguency difference (Hz) is
measured fiom the resonance of a reference compound, normally tetramethylsilane
(TMS) for 'H and I3c NMR, and divided by the absolute value of the Larmor fiequency
of the reference compound, which itself is proportional to the strength of the magnetic
field. TMS is generally useful because it is chernically inert, symmeaical, soluble in most
organic solvents, and absorbs at higher field (shielded) than almost al1 protons of organic
molecules. The chemical shifi is therefore given in the dimensionless scale of parts per
million (ppm, 6 scale), because a fiequency difference in Hz is divided by a fiequency in
MHz, these values are in a proportion of 1 : 1 06.
The local electronic environment normally dominates the chemical shift of a
nucleus. However, the anisotropic magnetic susceptibility of an adjacent bond may
influence the chemical shift of a nucleus as well. For example, the carbon-carbon bond is
the axis of a deshielding cone. Single bonds expenence shielding as in the case of
cyclohexane (See Figure 1-2). Here, a difference of about 0.5 ppm exists between the
resonance frequencies of the axial and equatorial protons. The equatorial 'H, which is in
the deshielding cone, is ofien more deshielded than the axial 'H on the sarne carbon. This

differential influence on the resonance is an important aid in confornational analysis of
compounds.
Figure 1-2 - Effect on equatorial (eq) and axial (ax) protons caused by a desbielding cone in six membered rings
This magnetic anisotropic effect can occur in any chemical group such as a
carbonyl (See Fijyre 1-3), where the circulation of electrons is less restricted about one
molecular axis than the others. The diamagnetic shielding of a proton by its 1s electron
density is relatively small compared with the shielding of nuclei of heavier atoms that
have filled inner shells. The proton near the functional groups experiences both high-
field and low-field shifts depending on theu position with respect to the anisotropic
group. Bemene protons have a chemical shift of -7.3 ppm, which is caused by
deshielding by a ring current of x electrons.

Figure 1-3 - The anisotropic shielding cones for a carbonyl group. Anisotropy causes deshielding of protons lying in thc cone whose a h is along the C=O bond and sheildiog outside this cone
1.4. Multiolicitv and Cou~ling 10,ll
Indirect or scalar coupling of nuclear spins through covalent bonds causes the
splitting of NMR signais into multiplets in high-resolution NMR spectroscopy in the
solution state. The signai multiplicity is the extent to which a NMR signal is split as a
result of spin-spin coupling. Signals that show no splitting are known as singlets (s).
Those with two, three, four, five, six or seven lines are known as doublets (d), triplets (t),
quartets (q, Figure 1-4), quintets (qui), sextets (sxt) and septets (sep) respectively. These
multiplet lines, however, must be equal distance apart to ensure that the one coupling
constant is shared by al1 of them. When m o or three different coupling constants
produce a multiplet, this is referred to as a two or three-fold multiplet, respectively [e.g. a
doublet of doublets (dd) or a doublet of doublets of doublets (ddd), Figure 1-41.

Qu artet ( q) Doublet of doublets (dd) Pseudotriplet Threefold doublet (ddd)
Figure 1-4 - Quartet, doublet of doublets, pseudotriplet and three-fold doublet (doublet of doublets of doublets)
The coupling constant is the frequency difference J i n Hz between two multiplet
lines. Unlike chernicd shift, the fiequency value of a coupling constant does not depend
on the strength of the rnagnetic field. In high-resolution NMR a distinction is made
between coupling through one bond ('JCH), and coupling through several bonds, e.g. two
bonds ( 2 ~ C ~ , gerninal couplings), ihree bonds (3~CH, vicinal couplings) or four or five
1 bonds (4~cH or 5.JCH, long-range couplings). The one-bond coupiing constant, JCHi is
proportional to the s character of the hybrid bonding orbital of the coupling carbon atom
(Le. '.JCH =500xs, where s = 0.5, 0.33 and 0.25 for sp, sp2, and sp3 hybridized carbon
atoms respectively). ' J ~ ~ increases with the electronegativity of the substituent bound to
the coupled carbon atoms. Two bond couplings, 2 ~ C H , across carbons of sp2 or sp3
hybridization are usually small with the exception of coupling across a carbonyl group.
Vicinal CH coupling constants, ' J ~ , resemble '.JHH in their dependence on the dihedral
angle between the C-C bond to the coupled C atom and the CH bond to the coupled
proton. An electronegative substituent on the coupled C raises the 3 ~ C H while one on the
coupling path lowers it.

1 .S. Instrumentation Ex~erimental
Mass spectral data (hi& resolution and low-resolution electron, EI) were
detemined at an ionizing voltage of 70 eV at 250°C on a Micromass 70-250 S (double
focusing) mass spectrometer. Optical rotations were measured on a Perkin-Elmer 341
polarimeter in CHC13 solutions. Fourier transfomed infrared (FTIR) spectra were
recorded on a Perkin Elmer Pentagon 500 instrument, using KBr discs. Melting points
were determined on Thomas-Kofler micro hot stage and are corrected.
Routine 'H and I3c NMR spectra were obtained on a Gemini-300 or Varian
üNITY-400 spectrometer equipped with a four-nucleus computer switchable probe while
2D experiments were carried out on a Varian UNITY-500 spectrometer equipped with a
5-mm inverse detection probe. The sarnples were dissolved in 0.8 ml of deuterated
solvent (-2-1 5 rnd0.8 ml solvent). COSY, NOESY, ROESY and HMBC experiments
were carried out using standard Varian software. HSQC spectra were obtained using
12 software developed in our laboratories. NOESY, ROESY and HSQC spectra were
acquired in phase sensitive mode, while HMBC was processed using mixed mode (phase
sensitive alongfi, absolute value alongfr). A mixing tirne of 0.1 - 1.25 s were used for
ROESY experiments. Linear prediction was used for al1 2D spectra except absolute
value COSY spectra.

Chaprer 2. Structural Elucidation of a Svnthetic Intermediate
2.1. Backeround
The first NMR investigation was used to elucidate the structure of an unknown
side product obtained in a previous attempted synthesis of aspidospermidine in our
laboratory. l 3 NMR methods and mass spectrometry were used to elucidate the structure.
Scheme 2-1 shows the synthetic steps taken to form the aspidospermidine precursor, S.
The key step, the photocyclization of azide 4 failed, but a small amount of an apparent
byproduct was obtained fiom one attempt. Elucidation of this product was of interest,
because understanding what intermediate was produced may explain what mechanism
had actually taken place in this photosynthetic reaction. Examination of this byproduct
eliminated the possibility that it was the desired product, 5. However, instrumental
limitations prevented a complete elucidation of this product. The structure of this
cornpound remained a mystery and this appeared to be an excellent subject for study
using our present NMR methods.
Figure ; C-1- Structure of aspidospermine

2 aq. acetone 1 NaN,
"J3
5 Desired Product
Scheme 2-1 - Synthetic steps peiiormed in attempt to synthesize precursor to Aspidospermidine, 5.
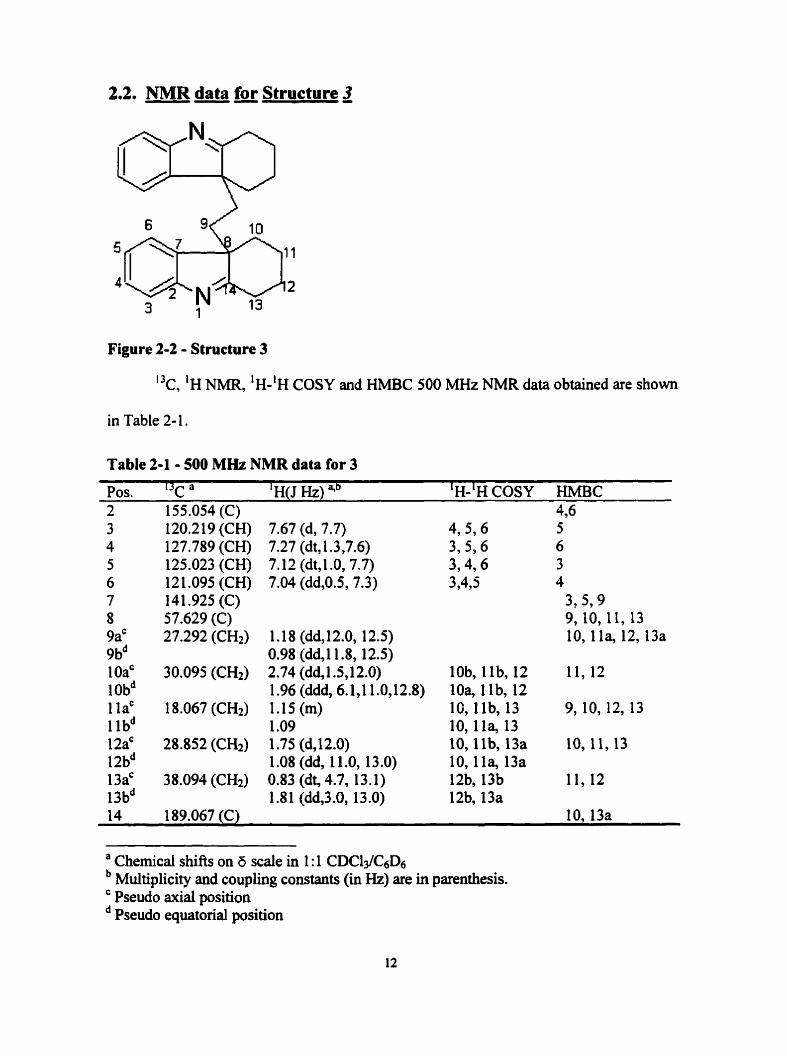
2.2. NMR data for Structure 3
Figure 2-2 - Structure 3
I3c, 'H NMR, 'H-'H COSY and HMBC 500 MHz NMR data obtained are shown
in Table 2- 1.
Table 2-1 - 500 MHz NMR data for 3
Pos. I3c a ' H(J HZ) qb 'H-'H COSY HMBC 2 155.054 (C) 4,6
120.219 (CH) 127.789 (CH) 125.023 (CH) 121 .O95 (CH) 141.925 (C) 57.629 (C) 27.292 (CH2)
7.67 (d, 7.7) 7.27 (dt, t .3,7.6) 7.12 (dt, 1 .O, 7.7) 7.04 (dd,0.5, 7.3)
1.18 (dd, 12.0, 12.5) 0.98 (ddJ1.8, 12.5) 2.74 (dd, 1.5,12.0) 1.96 (ddd, 6.1,l 1 .O,l2.8) 1.15 (m) 1 .O9 1.75 (dJ2.0) 1 .O8 (dd, 1 1 .O, 13 .O) 0.83 (dt, 4.7, 13.1) 1.8 1 (dd,3.0, 13.0)
4,5,6 3,596 3,4,6 3,435
lob, l lb, 12 10% llb, 12 16, l lb, 13 10,1la, 13 10, l lb , 13a 10,l la, 13a 12b, 13b 12b, 13a
5 6 3 4
3,5? 9 9, 10, 11, 13 10,l la, 12,13a
14 189.067 (C) 10,13a
a Chernicai shifts on 6 scaîe in 1 : 1 CDC13/c6D6 Multiplicity and coupling constants (in Hz) are in parenthesis. Pseudo axial position Pseudo equatorial position

2.4. Structural Determination of 3
Cornpound 3 was a white crystalline solid with a melting point of 243-245 OC. The
molecular formula, C20H28N2, was confimed by HRMS with a calculated molecular
weight of 368.2254 and experimental of 368.226849. This provided the fist piece of
evidence that the product was actually some form of a dimer. The 'H and I3c NMR
experiments in CDC13 gave evidence for 14 carbons and the correspondhg protons,
M e r providing evidence that the product was actually a symmetrical dimer. Since
there was overlapping of peaks of the 'H NMR in the aliphatic region, the expenment
was performed with increasing amounts of CsDs to resolve these peaks and determine
their coupling constants. Optimum resolution was found using a 1: 1 mixture of the two
deuterated solvents. COSY, HSQC and HMBC experiments were also performed using a
1: 1 mixture of CDCll and C6D6. These experiments were used to map together how this
molecule was attached.
Due to the molecular symmetry of the rnolecule, each side gave identical
chemical shifts and only half the nurnber of carbons and protons were evident. Carefùl
analysis indicated a cyclic ring attached to an indolenine. However, five distinct
methylene groups were present. The breakdown of the electron impact mass spectra with
molecular ion at 368 gave the next strong peak at 198, which would be the ion for 1 with
two CH2 attached. Additional breakdown peaks were at 184 and 170, which are due to
the loss of each of the methylenes. This suggested the existence of two six-membered
rings attached by two methylene groups at position 6, instead of two attached seven-

membered rings. Mapping of the 2-D NMR experiments determined the structure to be
3.
2.3 Iater~retation of Data
Since structure 3 is now established, it becomes clear that it is very unlikely that it
was produced in the photochemical reaction. Retracing the steps in the original synthesis
showed that 3 was actually an intemediate impurity fonned during the synthesis of 4. It
is almost certainly a byproduct fkom an earlier step.
In the first step of the synthetic sequence, carbazole 1 was treated first with ethyl
magnesium bromide in an anisole solution, and then with dibromoethane in a Grignard
reaction. Precautions were taken to minimize the possibility that both halogens of the 1,2
dibromoethane would react with 2 molecules of 1 to form 3. To form 4, the bromo
compound (2) was refluxed in an aqueous acetone solution mixture containing an excess
of sodium azide. Isolation and structural elucidation of this product, however, shows that
in fact some of the dibromoethane in the Grignard reaction did undergo a reaction where
each reacted with two molecules of 1. Despite purification, some of this product must
have been carried through to the photolytic step where it would not have been expected to
undergo m e r reactions. As a result, the structural question was resolved and the
possibility that an unknown photochemical reaction had taken place was conclusively
s h o w not to have taken place.
The stereochemical question, however, has not been resolved. The starthg
material was achiral, and there were no chiral constraints used to introduce the two stereo
centers at position 8 at each side of the dimer. Since only one set of chemical shifis was

observed, 3 should be either the racemic modification (RR, SS) or the meso modification
(W.

Chap fer 3 - Synthetic Plan of D ihydrocadambine
3.1. Previous svnthesis of 3a and 3B- dihvdrocadambine
3.1.1. Cadambine, 3a- and 3 0-~ihvdrocadambine'~ An extract of the heartwood of Anthocephalus cadamba first led to the isolation
of the indole glycosides, cadambine (7, and 3a-(8a) and Jfi-dihydrocadambine (8b).
These alkaloids are tryptamine derivatives with seven-membered rings. The structure
and relative configuration of 8a was assigned by McLean et al. as an alkaioid of Nauclea
diderrichds and later, by Brown and Fraser as an isolate fiom Anthocephalus ~adarnba'~.
Figure 3-1 - 7) cadambine; 8a) 3a-dihydrocadambine where 3H=a; 8b) 3p- dihydrocadambine where 3H=P
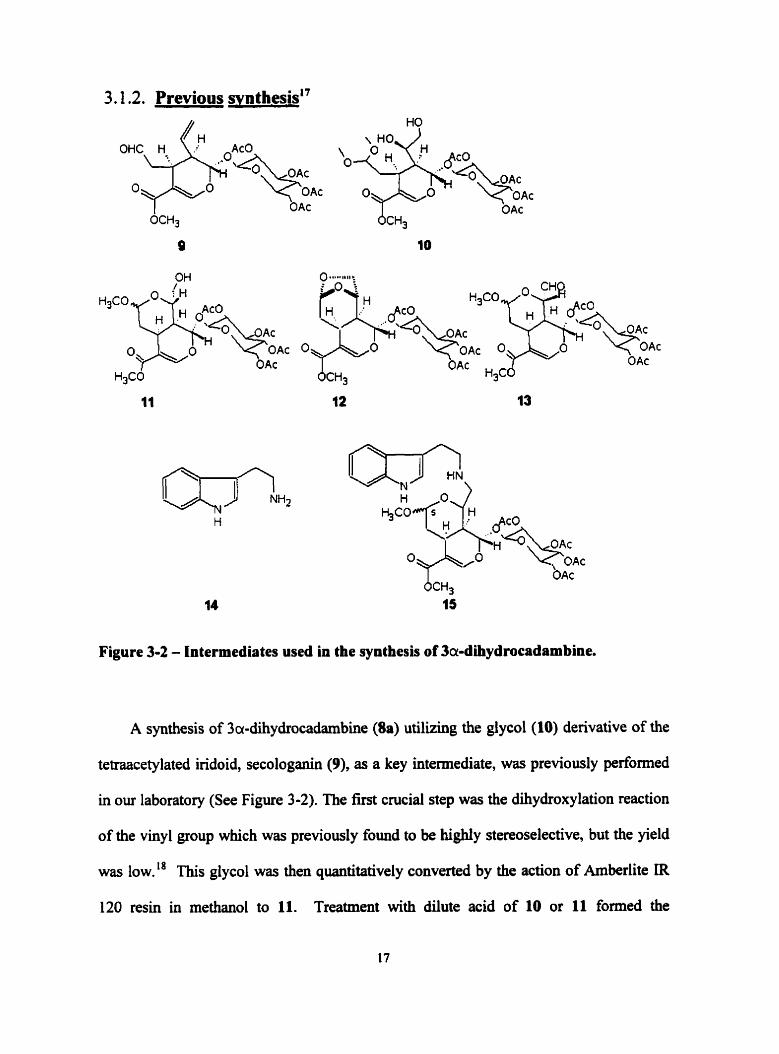
3.1.2. Previous synthesis"
OAc
OAc
Figure 3-2 - Intermediates used in the synthesis of 3a-dihydrocadrmbine.
A synthesis of 3a-dihydrocadarnbine (Ba) utilizing the glycol (10) denvative of the
tetraacetylated iridoid, secologanin (9), as a key intemediate, was previously performed
in our laboratory (See Figure 3-2). The first crucial step was the dihydroxylation reaction
of the vinyl group which was previously found to be highly stereoselective, but the yield
was low." This glycol was then quantitatively converted by the action of Amberlite IR
120 resin in methanol to 11. Treatment with dilute acid of 10 or 11 formed the

undesired-bridged bicyclic acetal 11. When instead, 11 was selectively oxidized with
pyridinium chlorochrornate at the primary alcohol it provided the aldehyde, 13. This key
intemediate locked the protective fiuiction of the cyclic acetal of both the secondary
hydroxyl group and the aldehyde. 13 was then reductively coupled with tryptarnine (14)
hydrochloride in the presence of sodium cyanoborohydride to form 15.
The completion of the synthesis of the alkaloidal framework then required
deprotection of the aldehyde funciion at C-5 of 15 and subsequent coupling with the
tryptamine moiety in an intramolecular Pictet-Spengler reaction. This proved difficult
due to acidic conditions needed and the susceptibility of the many cryptoaldehydes of the
iridoid structure. In addition, it was known that hydrolysis of the acetal was diflicult in
the presence of the neighbouring basic nitrogen. M e r extensive investigations, it was
found finally, that upon treatment with 90% formic acid, the target alkaloid was formed
as a mixture of 8a and 8b at high yield. The overall yield, however, was poor due to
instability of some key intermediates and the low yield obtained in the dihydroxylation.
This proved the Pictet-Spengler reaction could form a seven-membered ring in good yield
to form this alkaioid, but with M e stereoselectivity.
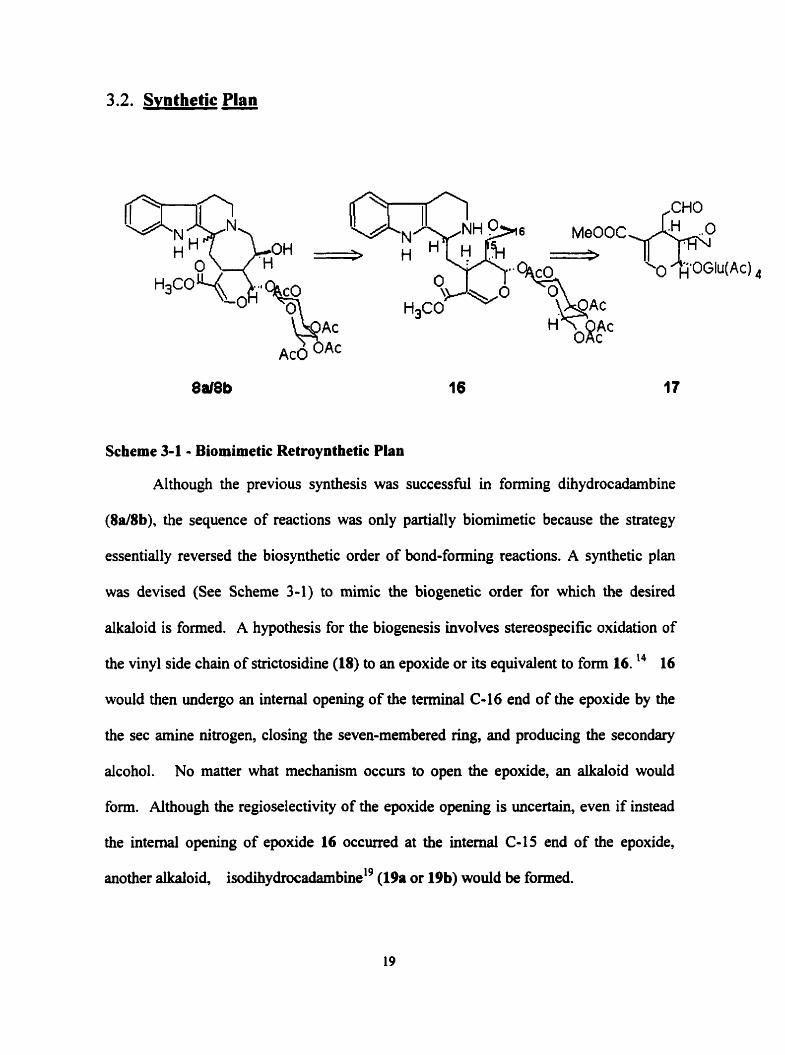
3.2. Svnthetic Plan
8 CHO
Scheme 3-1 - Biomimetic Retroynthetic Plan
Although the previous synthesis was successful in forming dihydrocadambine
(8a/8b), the sequence of reactions was only partiaily biomimetic because the strategy
essentially reversed the biosynthetic order of bond-forming reactions. A synthetic plan
was devised (See Scheme 3-1) to mimic the biogenetic order for which the desired
alkaloid is formed. A hypothesis for the biogenesis involves stereospecific oxidation of
the vinyl side chah of strictosidine (18) to an epoxide or its equivalent to form 16. l4 16
would then undergo an intemal opening of the terminal C-16 end of the epoxide by the
the sec amine nitrogen, closing the seven-membered Nig, and producing the secondary
alcohol. No matter what mechanism occurs to open the epoxide, an alkaloid would
form. Although the regioselectivity of the epoxide opening is uncertain, even if instead
the intemal opening of epoxide 16 occurred at the intemal C-15 end of the epoxide,
another alkaloid, is~dih~drocadambiw'~ (19a or 19b) would be formed.

Figure 3-3 - Structures of 18.) Strictosidine, where 3H=a 18b) Vincosidine, where 3H43 19a) 3a-Isodihydrocadambine, where 3H=a and 19b) 3 P- Isodihydrocadambine, where 3H=P
Strictosidine (180) itself, is a well-known monoterpene indole alkaloid glycoside
that is a precursor for over 2200 monoterpene indole and related alkaloids. 18a was first
isolated h m Rharya stricta by G.N. ~rnith.~' It was earlier thought that oniy vincoside
(18b) serves as a precursor for H-3a and H-3P indole alkaloid representatives of the
Corynanthe, Aspidosperma and Zboga fiameworks in Vinca ~ o s e a . ~ ' Strong proof of the
configuration of 18a had not been presented since it cannot be obtained in crystalline
f ~ r m . ' ~ It was found that only strictosidine and not vincoside is differentially
incorporated into several indole alkaioids such as those belonging to Corynanthe (3a and
3P series) Aspidospema and ~ b o ~ a ? Recent NMR studies have shown that the C-3
chiral center is in the S configuration, and thus it is strictosidine that serves as the major
precursor.24 In vivo, strictosidine is a biosynthetic equivalent of a Pictet-Spengler
condensation of a tryptarnine unit and secologanin (9), a reaction catalyzed by the
enzyme strictosidine synthase, which has been i~ola ted .~~

To form 16, a Pictet-Spengler condensation of the mono-epoxide of secologanin
(17) and the cornmercially available tryptamine hydrochloride could be perfomed. The
Pictet-Spengler reaction has long been an important procedure for the synthesis of both
indole and isoquinoline a lka l~ids~~, and in ideal conditions few side reactions are
reported. In the coupling reaction of tryptamine and secologanin, a new chiral center is
formed with complete stereoselectivity in the presence of the enzyme to form
strictosidine. In this synthetic sequence, it was hoped that the small amount of acid
present in the tryptamine hydrochloride would have three hctions. First, it could be
used to hydrolyze the dimethyl acetai to reveal the aldehyde group. Second, it could
catalyze the reaction of the aldehyde with the secondary amine of tryptamine to form an
imine, which is the start of the Pictet-Spengler reaction. Finally, it could also function to
protonate the imine to make it ~ ~ c i e n t l y electrophüic to do a substitution on the indole
(position 2) and complete the Pictet-Spengler.
A key step to this synthetic sequence is also the formation of the mono-epoxide,
17. The correct configuration of the epoxide is essential, first, to form 8a or 8b. In
addition, the configuration of the epoxide may have some infiuence on directing the
opening of the epoxide. Forming the correct isomer is known to be problernatic. Some
studies of the dihydroxylation and epoxidation of the vinyl side chah of the aldehyde-
protected form of secologanin (21) have previously been performed.27 Another iridoid,
sweroside (20), was used as a mode1 for the studies, but it was found that the two
derivatives behaved very differently in the oxidations carried out. 20 reacted site-
selectively with Os04 and m-chiorobenzoic acid at the vinyl side chah, leading to an
epimeric pair of products (di01 and epoxide, respectively), but with little

stereoselectively. 21 reacted at both the endocyclic double bond (a P-allcoxyacrylate) and
the vinyl side chah with little apparent site selectivity, low yield, but with very high
stereoselectivity to fom the desired configuration for this ~ynthesis.*~
Figure 3-4 - 20) tetraacetylated Sweroside and 21) aldebyde protected dimethyl acetal of 9 (secologaain)
It is hypothesized that either stenc factors or the conformation of the vinyl side-
chain are responsible for the reactivity of the Palkoxyacrylate of 21. The carboxyl
function of 20 is part of a lactone, and the carbonyl is locked in a conformation where its
n orbital interacts with that of the C=C, withdrawing electron density and deactivating it
to electrophilic attack. In the case of the 21, the ester can rotate so that the carbonyl x
orbital is not lined up with that of the C=C. With 21, the ring is more flexible since it is
not fused to the lactone ring. It readily places the side chain in an equatorial
environment, where there is considerably more stenc congestion, and the two faces of the
carbon double bond are quite different in the stenc himirance experienced by an
approaching reagent.
The low degree of stereoselectivity at the vinyl side chai. of 20 can be explained
by postulathg that this side chah is held in an axial coaformation in al1 derivatives of 20
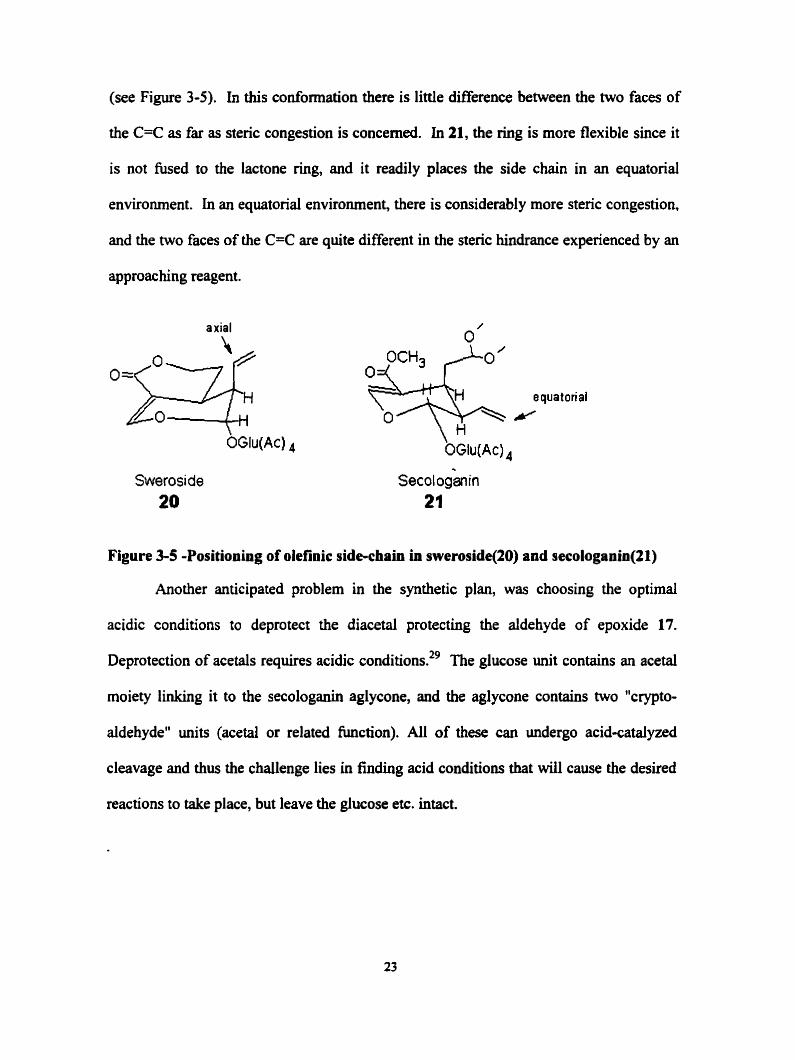
(see Figure 3-5). In this conformation there is little difference beiween the two faces of
the C=C as far as stenc congestion is concemed. In 21, the ring is more flexible since it
is not fused to the lactone ring, and it readily places the side chah in an equatorial
environment. In an equatorial environment, there is considerably more steric congestion,
and the two faces of the C=C are quite differed in the steric hindrance experienced by an
approac hing reagent.
axial
Swerosi de 20
Figure 3-5 -Positioning of olefinic side-chah in sweroside(20) and secologanin(21)
Another anticipated problem in the synthetic plan, was choosing the optimal
acidic conditions to deprotect the diacetal protecting the aldehyde of epoxide 17.
Deprotection of acetals requires acidic c0nditions.2~ The glucose unit contains an acetal
rnoiety linking it to the secologanh aglycone, and the aglycone contains two "crypto-
aldehyde" units (acetai or related function). Al1 of these cm undergo acid-catalyzed
cleavage and thus the challenge lies in finding acid conditions that will cause the desired
reactions to take place, but leave the glucose etc. intact.

Chapter 4. Isolations fronr Lonicera tartarica
The synthesis of 3a- or 3P-dihydrocadambine requires the use of the natural
product, secologanin (9), as a synthetic precursor. It was known that secologanin cm be
isolated fiom the common honeysuckle plant, Lonicera tartarica. The first task was to
isolate this natural product from its known naturai source.
4.1. Iridoids of Lonicera tartarica
Secologanin, a monoterpenoid glycoside, is a precursor of more than 2200
alkaloids. It has been used as an insect repellant and is a nototious stimulator of
The compound is exceptionally rich in functional groups and chernical reactions can be
carried out at al1 carbon atoms of the aglycone skeleton.
Secologanin was first theoretically elucidated as a result of biosynthetic studies on
the rnonoterpenoid indole alkaloids3', which first established loganin (22) as the key
intermediate and it was then predicted that secologanin should be the last non-
nitrogenous key intexmediate to form the protoalkaloids.32 Secologanin is the ultimate
precursor for the C-9lC-10 non-tryptamhe carbon skeleton common to the majority of
indole alkaloids. 33
A related irîdoid, sweroside (20) can also be isolated fiom Lonicera tartarica.
Sweroside is a close relative of secologanin, but with the aidehyde function of
secologanin reduced to the level of an alcohol and incorporated into a lactone.
Secologanin and sweroside are cailed secoîridoids because they are biogeneticaily formed
from loganin, an iridoid glucoside, by cleavage of its cyclopentane ring?4

Column chromatography was performed using BDH silica gel (40-63 pm).
Charcoal and celite were obtained fiom Fisher Chernical Co. Pre-made preparative thin
layer chromatography (thickness of 0.25mm, lm) containhg a fluorescent indicator
(254 nm) were obtained fiom Toronto Research Chemicals. Reagent grade solvents were
used for al1 preparative chrornatography. Analytical thin layer chromatography (TLC)
analyses were c h e d out using Whatman polyester or aluminum backed plated precoated
with silica gel UV2s4 (0.25mm). TLC were viewed under ultraviolet light (254 and 366
nm) before spraying with phosphomolybdic acidkeric sulphate spray made up with
phosphomolybdic acid (4g), cenc sulphate (0.5 g) in 100 ml of a 20% &SO4 solution.
4.3. Isolation of Secologanin from Lonicera tartarica
Shoots of Lonicera tarturica (the common honeysuckle plant), bearing both pink
and yellow flowers were gathered from Su~ybrook hospitai by permission. The plants
were gathered the first week of June of 1997, while plant flowers were just begiming to
die.
The shoots (-5 kg) were broken manually Uito smaller pieces. The broken shoots
were left to soak in rnethanol approximately 2 hours. A Waring blender (2-litre capacity)
was then used at low speed to blend the mixture. A total of Idlitres of methanol was
used for soaiing and blending. The aqueoudmethanol extract was filtered through

Whatman filter paper and concentrated by mtaevaporation. The remaining filter cake
was saved for fûrther extraction, repeating the previous steps of soaking, blending and
filtering. Al1 extracts were collected together to form a thick greenhlack liquid.
The concentrate was extracted with 5 x petroleum ether or hexanes. The aqueous
solution was once again concentrated by rotaevaporation to collect approximately Mitre.
The aqueous solution was extracted successively with chloroform and ethyl acetate. The
ethyl acetate-soluble residue contained the most products by mas, with one compound
corresponding to the Rfof the desired product, secologanin, by TLC. This solution was
run through a celite/activated coconut charcoal column (300g/700g) which was set with
methanol. The solution obtained was an amber brown colour. The crude mixture was
acetylated by treatment with acetic anhydride in pyridine to protect the free hydroxyl
groups. Silica column chromatography was perfonned using various eluents to puri@ the .
products. Three main products 9,20 and 22 were obtained and structurally examined.

4.4. Products Isolated from Extract
Figure 4-1 - Structures 9,20 and 22
4.4.1. Identification of 22
5 mg of compound 22 was isolated as oil and examined by 'H, I3c, I H - I H COSY
NMR and HSQC expenments. NMR data is show in Table 4-1.
Table 4-1 - 400 MHz NMR data for 22
Pos. 13C a 1 ' H - I H COSY
1 97.7 (CH) 5.1 1 (d, 4.6) 2 7 - 43.8 (CH) 2.5 13 (m) 1 .3 ,7
3 42.7 (CH) 1.81 (ddd, 4.6,6.8,9.3 ) 2,4,5
4 13.4 (CH3) 1 .O7 (d, 6.8) 3
5 75.1 (CH) 4.02 (m) 3,6a, 6b
6a 42.2 (CH2) 2.23 (ddd, 1.6, 7.9, 14.2) 5,6b, 7
6b - 3.94 (dd, 1.3, 10.1) 5,6a, 7 7 46.5 (CH) 3.10 (m) 6a, 6b
8 114.1 (C) - - 9 152.2 (CH) 7.38 (d, 1.3) 1
' Chernical shif€s on 6 scale in CD30D In brackets, mdtiplicities and coupling constants (Hz)

11 51.5 (CH3) 3.68 (s)
1 ' 100.2 (CH) 4.72 (d, 7.6)
2 ' 74.6 (CH) 3.30 (dd, 7.6,9.7)
3 ' 78.2 (CH) 3.55 (t, 9.7) 2'' 4'
4' 7 1.3 (CH) 3.18 (t, 9.7) 3'' 5'
5 ' 77.8 (CH) 3.25 (m) 4'' d'a, 6'b
6'a 62.1 (CH2) 3.52 (dd, 9.6, 10.1) 5'' 6'b
6'b - 4.15 (dd, 4.5, 10.1) 5', 6'a
Compound 22 had a molecular ion peak at 600, and the high-resolution mass
spectrurn confirmed the molecular formula as C2,H350 Compound 22 was identified
as loganin pentaacetate fiom examination of NMR data and the structure and
configurations were confvmed by cornparison of data with reported values.35 Although
loganin is the known precursor to secologanin, which is abundant in Lonicera tartarica,
to the best of our knowledge, this is the fist time loganin has been isolated fiom this
plant.
a Note: The NMR experiments performed could not determine exact positions of acetates

4.4.2 - Identification ef structure a 530 mg of compound 20 was isolated as a gurn fkom the extract. 'H, 13c and 'H-
'H COSY experiments were perfonned. The NMR data are as follows. 'H NMR (400
MHz, CDCb) 6: 1.68(rn, H-6a and H-6b), 1.97(s, Ac), 2.02(s, Ac), 2.04(s, Ac), 2.1 1 (s,
Ac), 2.70 (ddd, 1.5, 5.5, 9.7), 2.86(m, H-7), 3.75(ddd, 2.1, 4.0, 9.5, H-57, 4.14 (dd, 2.2,
12.4, H-6'a), 4.3 1 (dd, 4.8, 12.4, H-6'b), 4.32(m, H-Sb), 4.46(dt, 2.8, 10.8, H-5a), 4.92(d,
8.1, H-1'), 5.01(dd, 8.1, 9.7, H-2'), 5.10(t, 9.7, H-47, 5.25(t, 9.7, H-37, 5.28(m, H-4a),
5.3 1(m, H-4b), 5.46(ddd, 9.5, 10, 17.6, H-3), 7.55(d, 2.4, H-9). 13c NMR (125 MHz,
CDC13) 6: 20.6(CH3 for acetates), 24.7(C-6), 27.7(C-7), 42.2(C-2), 61.7(C-6'), 68.1(C-
4'), 68.5(C-5), 70.3(C-2'), 72.O(C-3 '), 72.5(C-S'), 96.O(C- 1 '), W.O(C- 1 ), 105 S(C-8),
i2OS(C-4), 13 1 S(C-3), 15 1.6(C-9), 165.7(C-1 O), 169.1, 169.5, 1 70.0, 1 70.6(C=O for
acetates). Compound 20 was confirmed to be the tetraacetate of sweroside. The positions
and configuration of protons and carbons were determined by comparing to previously
reported values.36 Compound 20 had a molecular ion peak at 526, and the high-
resohtion mass spectnun confimied the molecdar formula as C2,$H3()OI3.
4.4.3 - Determination structure 2 2.2 g of compound 9 was isolated as oil fiom the extract. 'H, "c, HSQC and
W C experiments were performed. The NMR data obtained are as follows. 'H NMR
(400 MHz, CDC13) 8: 9.70(t, 1.5, H-S), 7.42(d, 1.8, H-9), 5.46(ddd, 9.3, 10.1, 1 7.9,
5.28(dd, 2.1, 10.7, H-4a), 5.30(ddd, 2.1, 5.6, 17.1, H-4b), 5.23(t, 9.5, H-37, 5.10(t, 9.6),
5.02(dd, 8.1, 9.5, H-2'),4.89(d, 8.0, H-l'), 4.29(dd, 4.2, 12.4, H-6'a), 4.17(dd7 2.2, 12.4,
H-6'b), 3.74(m, H-S), 3.69(~, H-Il), 3.32(~, 0CH3), 3.30(s, OCHa), 2.94(ddd7 1.5, 6.2,

17.0, H-7), 2.8 1 (m, H-2), 2.42(dddY 1.2, 7.6, 18.0, H-6a), 1.54 (rn, H-6b), 2.1 1 (s, 6'-
Ac), 2.04(sy4'-AC), 2.01(s,3'-Ac), 1.91(s, 2'-Ac). 13c Nh4R (125 MHz, CDCli) 8:
20.6(CH3 for AC), 20.6(CH3 for 3', 4' and AC), 26.7(C-7), 42.2(C-2), 43.6(Cœ6)M,
5 1.3(C-Il), 5 1 .4(C-0CH3), 53.7(C-0CH3), 68.2(C-2'), 70.5(C-4'), 72.2(C-5')' 72.8(C-
3'), 95.7(C- 1 ), 95.9(C- 1 '), 96.1 (C-6'), 102.4(C-5), 1 1 1.4(C-8)' l2O.2(C-4), 132.2(C-3),
15 1.3(C-9), I69.O(C=O for AC), 169.5(C=O for A AC), 170.3(C=O for AC),
170.8(C=O for 6'Ac). The positions of carbons and protons were assigned fiom
HSQC and HMBC data. Compound 9 was confïmed to be the tetraacetate of
secologanin.
4.5. Forming dimethvl acetal of 9
The aldehyde function at position 5 of secologanin (9) was protected to prevent
oxidation and as a protective group against attack by various reagents such as strong or
moderately strong nucleophiles in subsequent synthetic reactions. This was done by
adding 9 (1.8g) to a solution of methanol and Arnberlite IR 120 resin as the source of acid
catalyst. The reaction mixture was then filtcred through celite and concentrated by
rotaevaporation to obtain 23 at quantitative yield.
More detailed structural elucidation was performed on the tetmacetylated
dimethyl acetal of secologanin (STAMA, 21) since this would be the precursor for most
of the synthetic reactions, and thus more knowledge was needed of the coupling constants
for each proton.

\
b I o 8
4 OAc
Figure 4-2 -Structure of 21 a) Stereo projection of STAMA; b) Chair representation of STAMA
The proton, carbon and 'H-'H COSY NMR assignments for structure 21 are
shown in Table 4-2. 'H, 13c, 'H-'H COSY, HSQC and NOESY experiments were
Table 4-20 500 M H z NMR data for 21
POS. I3c a 'H ab Coupling (Hz) 1 ~ - 1 ~ COSY 1 96.149 (CH) 5.30 (d) J12 = 4.4 2 2 43.222 (CH) 2.71 1 (m) 1,3,7 3 133.194 (CH) 5.608 (ddd) = 17.1 2,4a, 4b
JpCn = 10.7 J33 = 9.2
a Chernical shifis on 6 scde in CDC13 In brackets, multiplicities

Pos. I3c a 'H a,b Coupling (Hz) 'H-'H COSY 120.153 (CH2) 5.260 (ddd) 3,4b 4a
4b
5
6a
6b 7 8 9 1 ' 2'
3 '
4'
5'
6'a
6'b
2' Ac Acetate 3' Ac Acetate 4'-AC Acetate 6'Ac Acetate Acetal Acetal
102.442 (CH)
- 27.304 (CH) 1 1 1.406 (C) 150.327 (CH) 95.755 (CH) 70.576 (CH)
72.499 (CH)
68.143 (CH)
72.166 (CH)
5.297 (ddd)
4.479 (dd)
1.540 (ddd)
2.170 (m) 2.787 (m)
7.320 (d) 4.873 (d) 5.019 (dd)
5.210 (dd)
5.1 O7 (dd)
3.726 (ddd)
4.138 (dd)
4.287 (dd)
2.029 (s)
2.003 (s)
1.926 (s)
2.104 (s)
3.309 (s) 3.299 (s)
3,4a
da, 6b
5,6b, 7
5'6% 7 2,7,6a, 60
7 2' l' , 3'
2'' 4'
3'' 5'
4', 6'a, 6'b
5', 6'b
5'' 6'a
(C=O)OCH3 66.983 (CH3) 3.700 (s)
The molecular formula of 23, C27&00L7, was confinned by HRMS. IR confïrmed
the presence of ester carbonyl at 1756 cm-' also the vinyl ether at 1228 cm'. The 'H-'H

COSY spectrum confmed the relative positioning of al1 protons. H d a and H-6b showed
different chernical shifis, but the COSY cross-section shows H-6a and H-6b showed cross
peaks with each other, H-5 and H-7, while H-5 and H-7 showed no cross peaks with each
other. Examination of HSQC spectra provided the correlation for directly bonded proton
and carbon pairs.

The reactions performed in an attempt to accomplish the synthesis of
dihydrocadambine (8a or Sb) are discussed in this chapter.
5.1. mCPBA oxidation of 8
A peracid oxidation of STAMA (21) was performed to see if conditions could be
optimized to form the mono-epoxide at the vinyl position.
Figure 5-1 - Structure of bis-epoxide 23
5.1.2. General ~rocedure for ~eracid oxidation of STAMA (21)
To a solution of olefin 21 (C27H380is; 150 mg, 0.25mmol) in dichloromethane
(hl) at 0°C was added mCPBA (150mg, 3 equiv.). AAer an hou, the ice bath was
removed and the mixture was shed at room temperature for 72 hours. 5% NaHC03
(20rnl) was added and the mixture was extracted with dichloromethane ( 2 x ) . The organic

phase was then washed with brine, dried over MgS04 and concentrated by
rotaevaporation. 2 products were visible by TLC. Chromatography on silica using 9: 1
CHC13/acetone was performed. The main product was obtained at 50% yield as the bis-
epoxide 23, with a mp of 153-1 55.5 OC. 'H NMR (300 MHz, CDC13) 65.082 (H-1, d,
1 .a), 1 -623 (H-2, ddd, 10.0, 7.8, 1.8), 3.272 (H-3, 6.76, m), 2.47 (H-4a, 2.777 (H-4b, dd,
4.5,3.6) 4.672 (H-5, t, 5.5) 1.89 1 (H-6% m) 1.801 (H-6b, m) 3.172 (H-7, m), 4.853 (H-
9, s), 3.776 (CH30-IO , s, 3), 3.301 (CH30-acetal, s, 3), 3.340 (CH30-acetal, s, 3),
4.921 (H-l', d, 8.1), 5.013 (H-2', t, 9.3), 5.203 (H-3', dd, 9.4, 9.7), 5.105 (H-4', t,
9.7), 3.862 (H-5', m), 4.167 (H-6'a, dd, l2.5,2.3), 4.820 (H-6'b, dd, l2.5,4.6), 2.1034
(Ac, s, 3H), 2.033 (Ac, s, 3H),2.010(Ac, s, 3H), 1.954 (Ac, s, 3H).
Another product was obtained at 30% yield, which appeared to be oxidized at the
P-alkoxy acrylate bond and not the vinyl bond, because of the disappearance of the H-9
proton by NMR while the H-3 and H-4 vinyl protons were retained. Prolonged mixing
and heating of this product only resulted in the production of more of the bis-epoxide
(23) indicating that the 8-alkoxy acrylate bond was actually the first to be oxidized.
5.2. Dihvdroxvlation of STAMA 37,38
The oxidation of olefms is known to selectively perform dihydroxylation
reactions at high yields using osmium tetraoxide (0~0~). Although 0 ~ 0 4 , like the
peracids, is selective for less polarized double bonds with more electron donating
substituents, the selectivity has been shown to be greate?9 and the influence of steric
factors seems to be stronger than in the peracid oxidations.

To prepare the 1% 0s04 solution in pyridine, reagent grade Os04 (99+% pure)
was obtained in 1 g glass ampoules which could be scored with a file, cooled in Dry Ice
and broken open. The orange solid was quickly transferred to an amber vial that had
been pre-cooled, with septum under argon gas. The &O4 was dissolved in 10 ml of
pyridine .
A catalytic amount of osmium tetraoxide ushg N-methylmorpholine N-oxide
hydrate (NM0.H20) as a CO-catalyst was used. Reactions were carried using Os04
dissolved in pyridine or tert-butyl alcohol.
To a solution of STAMA (21) (C27H3(015; I.Og, 1.7 rnrnol) and N-
methylmorpholine N-oxide hydrate (NMO-H20, 0.48g, 2.1 equivalents) in acetone (12
ml) and water (1.2 ml) at room temperature was added a solution of Os04 (1 -2 ml of a 1 %
solution in pyridine. The mixture was stirred at room temperature for 48 hours. A
saturated solution of Na2S03 (20 ml) was then added to the resulting mixture, and
continued stimng for 2 hours. The mixture was extracted with ethyl acetate ( 2 x ) and the
organic phase was then washed successively with saturated Na2S03. water, and brine and
then dned over MgS04 and concentrated d o m by rotaevaporation. Flash
chromatography using 4: 1 CHC13/acetone was performed to isolate the product. A solid
was obtained at 79% yield.
5.2.1. NMR Examination a glvcol24 The proton and deduced carbon NMR assignments for glycol 24 are shown in
Table 5-1. 'H, HSQC, COSY and ROESY experiments were performed in CDCl,.

Table 5-1 - 400 MHz "C and 'H NMR Data for glycol 24
Pos. J values 9 7.326 (d) 1.5 151.2
3.001 (m)
1.885 (m)
1 S60 (m)
4.576 (dd)
3.560 (bs)
3.860 (bs)
2.810 (m)
5.302 (d)
4.923 (d)
5.024 (t)
5.259 (d)
5.107 (t)
3.716 (m)
4.265 (dd)
4.199 (dd)
Acetd 3.410 (s,3)
Acetal 3.264 (s,3)
Acetates 2.1 OS(s,3), 2.049(~,3), 2.025(~,3) and 2.046 (s,3)
Glycol 24 was obtained as a white crystalline solid with a melting point of 84-
86°C. The carbon NMR assignments shown in Table 5-1 were al1 deduced fiom an
a Chernical shifts on 6 scale in CDC13 Al1 carbon shifts were determined by HSQC

HSQC experiment. COSY data was used to confimi positions of ail protons.
5.3. Formation of kev epoxide (27)
Scheme 5-10 Conversion of glycol 24 to epoùde 26
5.3.1. Conversion glwoi 24 to e~oxide 26
nie glycol (24) could now be used to form the monospoxide of 21, using the
steps outlined in Scheme 5-1. This sequence of reactions was perforrned in two steps,
without intermediate cation of the tosylate (25).

Glycol 24 (C27&0017; 400mg, 0.6 rnrnol) was dissolved in dry pyridine (20 ml)
with freshly regenerated 4A molecular sieves, and cooled to 0°C. pToluenesulfony1
chloride (1 30 mg, 1 .1 equiv.) was added as a solid and the resulting mixture was stirred at
0°C for an additional 2 hours and then gradually warmed to room temperature. After
stimng at room temperature for 24 ~ O W S , 5% HCI was added and the resulting mixture
was extractcd with EtOAc (2x). The organic layer was washed with brine, dried over
MgS04 and concentrated. This residual oil was mixed with K2C03 (100 mg, 2.1 equiv.)
in methanol (70 ml) and was stirred at room temperature for 48 minutes, and then
saturated N h C l was added. The mixture was extracted with EtOAc (2x) and the organic
layers were washed with brine, dried over MgS04 and concentrated. The oil was purified
by silica chromatography using 9:l CHC13/acetone to obtain a solid at an overall yield of
70%.
5.3.2. NMR Examination of eooxide 26
Proton, 'H-IH-COSY, HSQC and ROESY experiments were performed and NMR
data are shown in Table 5-2.
Table 5-2 -500 MHz NMR of mono-epoxide 26
Labeled Proton #
J values
9 7.413 (d) 1.8 150.1 7 3.053 (ddd) 17.9,6.1, 5.4 42.4 6a 1.99 (m) - 43.10 6b 2.05 (m) - - 5 4.572 (dd) 3.6.7.6 105.1
a Chernical shifts on 6 scale in CDC13 MUltiplicities in parentheses Obtained fiom HSQC

Labeled IHB.~ J values 13cqc Proton # 4a 2.568 (dd) 4.8,2.6 47.9
5' 6'a 6'b (C=O)Oc& Acetal
Acetates
2.844 (m) 2.746 (m) 1.752 (m) 5.358 (d) 4.878 (d) 4.991 (t) 5.207 (t) 5.098 (t) 3.735 (ddd) 4.261 (dd) 4.162 (dd) 3.709 (s,3) 3.327 (s,3) 3.281 (s,3) 2.109,2.034,2.013 and 1.934
NMR analysis showed the main product to have the desired, R configuration.
5.4. Pictet-Spengler reaction
5.4.1. Cou~ling a STAMA J21) and trwtamine
Before attempting to perform the Pictet-Spengler condensation on mono-epoxide
26 and tryptamine hydrochloride, conditions needed to be optimized to see if the acid
present in the tryptamine could hydrolyze the protected aldehyde and immediately
cataiyze the Pictet-Spengler reaction. As a model, the reaction was performed on
STAMA (21) instead of epoxide 26 so that conditions could first be optimized. The
reaction was fist carried out using the method described by Battersby that uses
tryptamine hydrochloride.40 We were disappointed to find there was no hydrolysis of the
aldehyde and no reaction had taken place. Many conditions were then explored, using a

variety of acids, solvents and temperatures, but al1 of these were unsuccessN in
deprotection. No reaction was also obtained in one attempt to view if the use of epoxide
26 would yield diflerent results.
While exploring the various acidic environments, some side-products products
were obtained, and analyzed by NMR. The loss of the glucose residue was evident in
some cases, which was easily established fiom the losses of the acetates and sugar
protons by 'H NMR, and the concomitant isolation of pure acetylated glucose residue.
5.4.2. Coupling reaction of STA (9) and trv~tamine
In light of the problems deprotecting the dimethyl acetal of the aldehyde in situ, it
became obvious that epoxide 26 would fust have to be deprotected and then coupled with
tryptamine in two steps. Optimization of conditions to couple tryptamine was first
explored using the free aldehyde, secologanin (9) as a model. It was found that an
additional amount of a mild acid to that present in the tryptamine hydrochloride was
required for the reaction to proceed. Many conditions were explored, including catalytic
amounts of mineral acids, pTSA and acid resin.
5.4.2.1. General orocedure for cou~ling of 9 and trv~tamine
To a solution of 9 (C25H32014; 100 mg, 0.18mmol) in benzene (20 ml), water (1
ml) and acetic acid (0.5 mi) was added tryptamine hydrochloride (2.1 equiv., 75 mg).
The solution was heated at 50°C for 24 hours. The solution was then quenched in
ammonium acetate and extracted with EtOAc. The organic phase was then washed with
brine, dned over MgS04 and concentrated down by rotaevaporation. Flash
chromatography using 9:l CHC13/acetone was performed to isolate the product. Two
products, 18a and 27 were obtained.
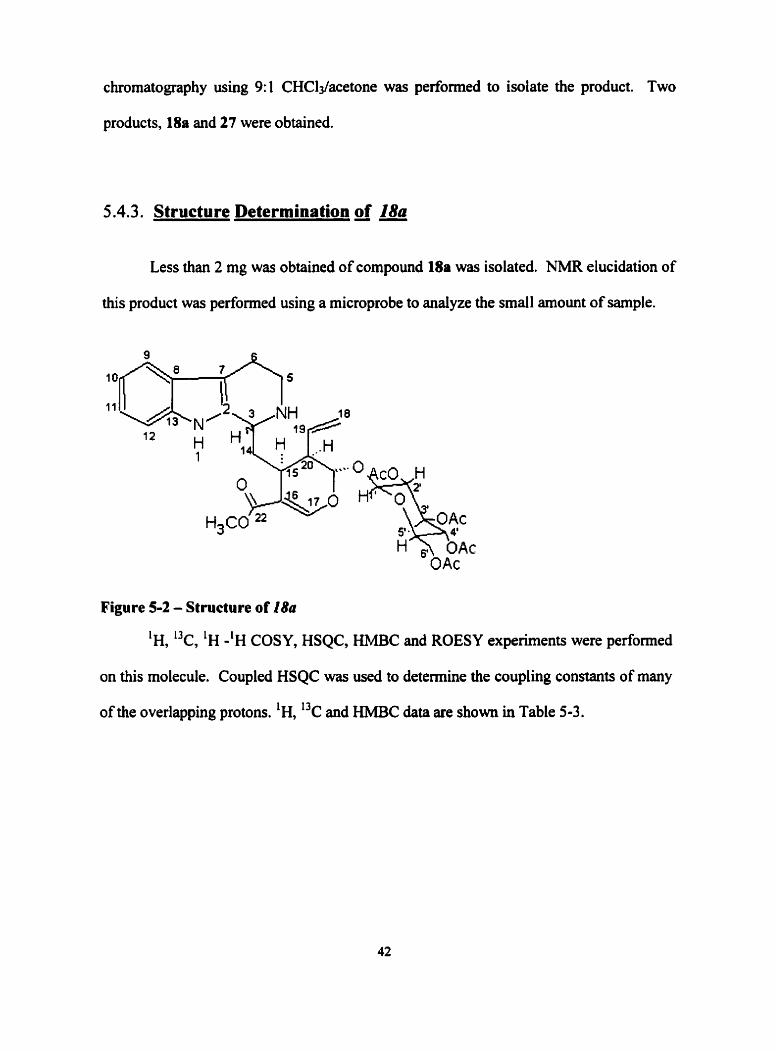
5.4.3. Structure Determination of 18a
Less than 2 mg was obtained of compound 18a was isolated. NMR elucidation of
this product was performed using a microprobe to analyze the small amount of sample.
Figure 5-2 - Structure of 18a
'H, 13c, 'H -'H COSY, HSQC, HMBC and ROESY experiments were performed
on this molecule. Coupled HSQC was used to determine the coupling constants of many
of the overlapping protons. 'H, "C and HMBC data are shown in Table 5-3.

Table 5-3 -500 MHz "c, 'H and HMBC data for I8a
Pos. I3c a 1 ~ 8 . b
HMBC
0 133.1(C) 52.0 (CH) (NI 42.8 (CH2) - 20.9 (CH2) - 107.5 (C) 126.9 (C) 1 18.5 (CH) 120.1 (CH) 122.5 (CH) 11 1.8 (CH) 137.8 (C) 35.5 (CH2) - 32.4 (CH) 109.7 (C) 156.3 (CH) 1 19.4 (CH2) - 135.6 (CH) 45.2 (CH) 97.7 (CH) 171.1 (C=O) 52.3 (CH3) 93.9 (CH) 73.4 (CH) 72.5 (CH) 68.9 (CH) 71.7 (CH) 62.8 (CH2)
20.61 7 (CH3) 20.61 8 (CH3) 20.6 19 (CH3) 20.787 (CH3)
7.80 - 4.30 (ddd, 1.3,3.0, 11.4) 3.01 (s) 3.17 (ddd, 5.3,8.8, 12.3) 3.48 (ddd, 4.2,5.3, 12.3) 2.84 (ddd, 4.2,5.3,15.7) 2.96 (ddd, 5.3,8.7, 15.7) O
O
7.44 (d, 7.8) 6.99 (dt, 1.3, 7.4) 7.05 (dt, 1.1, 7.9) 7.30 (d, 7.7) - 2.09 (dd, 3.9, 14.7) 2.21 (dd, 1 1.5, 14.7) 3.05 (m) - 7.73 (d, 2.4) 5.24 (ddd, 1.0,2.1, 10.6) 5.35 (ddd, 1.0,2.1, 17.4) 5.79 (ddd, 7.6, 10.6, 17.4) 2.70 (ddd, 1.7,5.8,9.7) 5.73 (d, 4.4) - 3.78 4.90 (d, 8.1) 5.03 (dd, 8.1,9.7) 5.25 (t, 9.6) 5.11 (dd, 9.6,9.7) 3.77 (ddd, 2.1,6.1,9.7) 4.33 (dd, 6.1,ll.g) 4.1 1 (dd, 2.l,ll.g) 2.05 (s, 3) 2.07 (s, 3) 1.97 (s, 3) 2.13 (s, 3) -
Sa, 5b -
a Chernical shifls on 6 scale in CD30D In brackets, multiplicities and coupling constants in Hz

Pos. 13c a ' H " ~ HMBC
3' Ac 168.8(C=O) - 3 ' 4' Ac 167.6 (GO) - 4' 6' Ac 166.5 (GO) - 6 '
Not enough product was obtained to determine melting point or to obtain an IR
spectra. The results of the HSQC and HMBC experiments were used to detennine the
relative positioning of al1 carbons, nitrogens and protons. The results of the ROESY
experiment showed n.0.e. between H-15 and H-14a. H-3, however, only showed n.0.e.
with H- 14b and none with H-15. The stereochemical assignment for H-3 as the S isomer
was assigned on this basis. Bases on these results, the product was determined to be the
alkaloid, strictosidine.
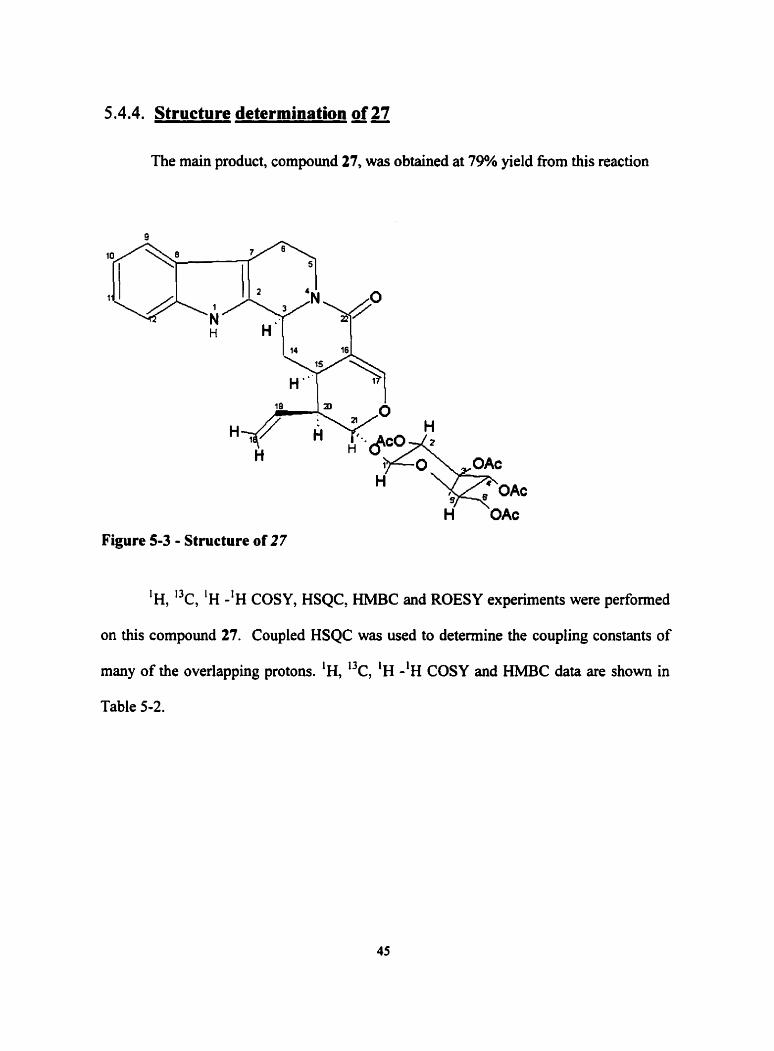
5.4.4. Structure determination of 27
The main product, compound 27, was obtained at 79% yield fiom this reaction
9
H H' 14 16
H"
H
OAc
Figure 5 3 - Structure of 27
'H, 'k, 'H -'H COSY, HSQC, HMBC and ROESY experiments were performed
on this compound 27. Coupled HSQC was used to determine the coupling constants of
many of the overlapping protons. 'H, 13c, 'H -'H COSY and HMBC data are shown in
Table 5-2.

Table 5-4 500 MHz I3c, 'H, COSY and HMBC data for 27
Pos. 13c a ' H ' ~ ' H - ~ H COSY HMBC
(NI 132.693 (C) 52.853 (CH) 39.562 (CH2) - 20.948 (CH2) - 109.9 (C) 126.752 (C) 1 1 8.444 (CH) 1 19.905 (CH) 122.292 (CH) 1 10.884 (CH) 136.230 (C) 3 1.607 - 26.288 (CH) 108.306 (C) 146.835 (CH) 120.687 (CH2) - 13 1.754 (CH) 42.690 (CH) 96.064 (CH) 170.056 (C=O) 95.872 (CH) 70.461 (CH) 72.261 (CH) 68.133 (CH) 72.229 (CH) 61.723 (CH*) - 20.6 1 3 20.6 13 20.6 13 20.787
7.788 (br s) - 4.871 (d, 10.7) 5.165 (d, 11.9) 2.866 (dt, 4.5, 1 1.9) 2.805 (m) 2.805 (m) - - 7.509 (d, 7.7) 7.126 (td, 1.1, 7.9) 7.192 (td, 1.3, 7.4) 7.335 (d, 7.8) - 1.58 (ddd, 12, 13, 13) 2.18 (dddO, 4'4, 13) 2.94 (m) - 7.48 (d, 2.4) 5.18 (dd,1.7,10.1) 5.26 (dd, 1.7, 16.9) 5.47 (ddd (**) 2.70 (ddd, 1.8,5.8,9.7) 5.29 (d,4.4) - 4.91 (d, 8.9) 5.03 (dd, 8.9'9.7) 5.25 (t, 9.5) 5.11 (dd, 9.5,9.7) 3.77 (ddd, 2.4,4.4,9.7) 4.33 (dd, 4.4J2.8) 4.15 (dd, 2.4,12.8) 2.022 (s, 3) 2.044 (s, 3) 1.997 (s, 3) 2.111 (s, 3) -
- - 14% 14b 5b, 6 5% 6 5% 5b Sa, 5b - - IO, I l , 12 9, 11, 12 9, 10, 12 9, 10, 11 - 3, 14b, 15 3,14a, 15 14% 14b, 21 - - 18b, 19 18a, 19 18a,18b, 20 19,21 20 - 2' 1', 3' 2', 4' 3', 5' 4', 6'a, 6'b 5', 6'b 5', 6'a - - - -
' Chemicai shifts on 6 scaie in CDCll in brackets, multiplicities and coupling constants in HZ Pseudo axial position Pseudo equatorial position

Pos. 13c a ' H " ~ WH COSY HMBC
5.4.4.1. Structural Determination of 27
The product was obtained as an orange-brown solid with an [a], = -45 O . High-
resolution mass spectral anaiysis confirmed the molecular formula of C34H38N2012,
which has a calculated mass of 666.24254 and an experimental mass of 666.243061. The
infrared spectmn confirmed absorption due to primary amine (3378 cm-'), carbonyl
(1 755 cm-'), vinyl ether (1 228 cm-') and disubstituted aromatic (755 cm").
The 'H NMR resonance peak at 6 7.788 (N-1) showed evidence that the indole
NH remained intact. There was no amine peak indicated for position 4 in the 1 H-NMR,
indicating that this nitrogen had no attached protons as strictosidine (18a) did. In
addition, there were no HMBC correlation's of 639.562 (C-5) with any protons except
those at C-6. No geminal coupling exists for 6a and 6b since they essentially have the
same chemical shift. The "C resonance's for the lactam carbonyl at 5170.056 and the
sugar carbonyls 6 170.03 1 (2') 169.742 (3') 169.506 (4') 170.622 (6') and the lactam
were similar, however HMBC clearly distinguished the positioning of dl.
Interestingly, the two-gerninal protons at position 5 had two very different
chemical shifts of 2.886 and 5.165 ppm. The ciifferences in sh ih present here are due to
the anisotropic effects caused by the amide. The positioning of these protons was evident
by the HSQC contours and the ')c cross-section of the coupled HSQC spectrurn, which
showed thwe two protons, attached to C-5. Further evidence that both these protons were

attached to C-5 came fiom the COSY spectnun, which showed cross-peaks between H-Sa
and H-Sb were present with Hda and H-6b.
The configuration at C-3 was deteminrd by a NOESY experiment, which
showed that the proton was on the same face as H-14b and H-15. The product obtained
had 3R-configuration. The NOESY enhancements are shown in Figure 5-4.
Figure 5-1 - nOe enhancements observed for Compound 27
The designation of the 3R-configuration is the reason it was assigned as
vincosamide and not stricto~amide.~~ Vincosamide has been show as a precursor in the
biogenetic route to the synthesis of the N d e a akaloid, nauclefidine" and naucleudinal.
(Figure S-S) .~~~" Nauclefidine has been found to exhibit potent andgesic and antibacterial
activities.

Nauclefidine Naucleidinai
Figure 5-5 - Structure of Nauclefidine and naucleudinal
5.5. Deprotection of dimethvl acetal of 26
Since the coupling to tryptamine had to take place in two steps, epoxide 26 had to
be converted to its deprotected ddehyde. Before deprotecting the diacetal of epoxide 26,
STAMA (21) was used as a mode1 to explore optimum conditions. The problem proved
to be more problematic than anticipated. It was first hoped that mixing of 23 in a 1: 1
benzenelwater mixture using acid resin as a cataiyst would be suficient for the
deprotection. The results were unsatisfactory and other routes to deprotection were
investigated. In one attempt to optimize the coupling of tryptamine to 23, trimethylsilyl
trifluoro methane sulfonate (TMSOTf) was used as a ~ a t a l ~ s t . ~ ~ It was found instead that
these reaction conditions successfully deprotected the dirnethyl acetal of 23. These
conditions were then applied to epoxide 26.
To a solution of epoxide 26 (CZ7&&; 100 mg, 0.18mmol) in 10 ml dry
tetrahydrofuran -20°C under argon gas, was added TMSOTf (2 equiv., 120 kL). The
reaction was stirred for 45 min and 1 ml of water was added. The reaction mixture was
then quenched in ammonium acetate and extracted (3x) with ethyl acetate. The reaction

mixture was dned over MgSOd and concentrated. Silica chromatography using 9:l
CHC13/acetone was perforrned to isolate epoxide 28 at 80% yield.
Figure 5-6 - Structure and proton 400 MIIz NMR assignments for epoxide 28.
The proton assignments for 28 are show in Figure 5-6. Proton and 'H-'H COSY
NMR experiments confirmed that the desired fiee aidehyde epoxide derivative of
secologanin (28) needed to couple to tryptamine in the biomimetic synthesis of
dihydrocadambine (8d8b) was successfully formed.
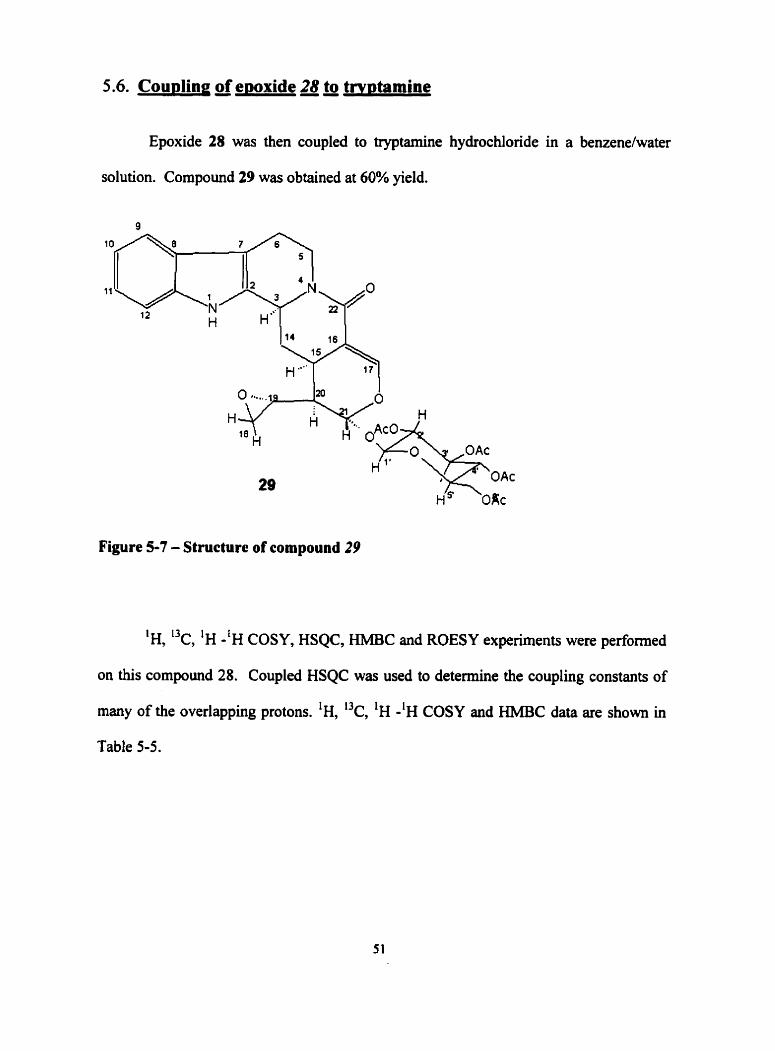
5.6. Cou~ling of ewxide 28 to tw~tamine
Epoxide 28 was then coupled to tryptamine hydrochloride in a benzene/water
solution. Compound 29 was obtained at 60% yield.
Figure 5-7 - Structure of compound 29
'H, 13c, 'H -'H COSY, HSQC, W C and ROESY experiments were performed
on this compound 28. Coupled HSQC was used to determine the coupling constants of
many of the overlapping protons. 'H, "c, 'H - l ~ COSY and HMBC data are shown in
Table 5-5.

Table 5-5 - 500 MHz NMR 'k, 'H and HMBC data for 29
Pos. 13c a ' H " ~ HMBC
(NI 131.8(C) 53.4(CH) 39.6 (CH2) - 20.9 (CH2) - 1 10.1 (C) 126.5 (C) 131.1 (CH) 122.1 (CH) 1 19.7 (CH) 1 18.5 (CH) 137.2 (C) 31.91 (CH2) - 26.92 (CH) 109.7 (C) 156.3 (CH) 40.9 (CH2) - 48.82 (CH) 45.2 (CH) 90.25 (CH) 171.1 (C=O) 95.76 (CH) 72.3 (CH) 70.7 (CH) 69.1 (CH) 67.9 (CH) 62.8 (CH2) - 20.61 (CH3) 20.62 (CH3) 20.62 (CH3) 20.8 (CH3)
8.05 (s) - 4.756 (d, 11.4) 5.13 (d, 12.5) 2.86 (m) 2.79 (d4,6.8,12.7) 2.10 (m) - - 7.51 (d, 7.8) 7.138 (dt, 1.3, 7.4) 7.1 1 (dt, 1.3,7.9) 7.14 (d, 7.7) - 2.44 (d, 13.6) 1.80 (m) 3.10 (m) - 7.73 (d, 2.4) 2.34 (d, 6.8) 2.58 (m) 2.88 (m) 2.70 (ddd, 1.7, 5.8,9.7) 5.47 (d, 4.4) - 4.87 (d, 8.3) 5.26 (dd, 8.3,9.7) 5.25 (dd, 9.6'9.7) 5.10 (dd, 9.6'9.7) 3.78 (ddd, 2.1,6.1,9.7) 4.33 (dd, 6.1,11.9) 4.11 (dd, 2.1,11.9) 2.04 (s, 3) 2.07 (s, 3) 1.97 (s, 3) 2.12 (s, 3) -
-
' Chernicd shifts on b scale in CDC13 In brackets, multiplicities and coupling constants in Hz

Pos. 'Y a ' H ' ~ HMBC
The resuits of HSQC and HMBC experirnents for compound 29 were used to determine
relative position of atoms. The configuration around H-3 was determined to be R upon
examination of ROESY expenments. The results show that lactamization occmed under
these conditions also to f o n this epoxide denvative of vincosamide.

5.6. Discussion of results
The lactarns 27 and 29 were the major products formed in the Pictet-Spengler
reaction attempts. This demonstrates that in acidic conditions, the nucleophilic nitrogen
preferentially attacks the sp2 carbon of the ester over the sp3 of the epoxide. Once it was
realized that acid media caused attack of the methyl ester in the coupling of tt-yptarnine to
secologanin derivatives, attempts were made to perform a Pictet-Spengler reaction in
aprotic media using methods described by Cook et without success. The
formation of an epoxide derivative of strictosidine (16) was more problematic than
anticipated. Conditions need to be found to successfully complete the Pictet-Spengler
reaction of epoxide 27 to tryptamine in conditions that will prevent the lactarnization of
the ester. The reaction may also be successful if the ester in epoxide 27 could be
converted to its acid denvative to make this carbonyl Iess electrophilic and successfully
couple this derivative to tryptamine.

Chapter 6. NMR Analysis of Natuai Products
NMR methods were also used to elucidate the structure of unknown naturd
products. Al1 of the natural products analyzed in this chapter were done in collaboration
with scientists from the University of West Indies campuses in Barbados and Jamaica.
The samples exarnined in this chapter were isolated and purified in the West Indies, and
the NMR data were performed at the University of Toronto.
6.1. Terpenes 48
Terpenes (also referred to as isoprenoids) are naturally occurring compounds
having isoprene (C5Hs) skeletal units (see figure 2-1). Terpenes rnay contain two, three,
or more isoprene units. Their molecules may be open chah or cyclic. The number of
isoprene units they contain classifies terpenes as follows:
Monoterpene 2 isoprene units 1 10 carbons
Sesquiterpene 3 isoprene units / 15 carbons
Diterpene 4 isoprene units / 20 carbons
Triterpene 6 isoprene units / 30 carbons
Tetraterpene 8 isoprene units 1 40 carbons
Most of the compounds discussed in this chapter are either sesquiterpenes or diterpenes.

Figure 6-1 - Isopreae unit and a terpene
6.2. Determination a Structures of Two Sesauiterwnes from Carrraria
The IWO compounds were collecied and provided by Dr. Paul Reese of the
University of West Indies, Mona campus in Kingston, Jamaica. They are isolated from
the plant Capraria biflora, a member of the Scrophulariaceae family. The common name
for this vascular plant is Goatweed. These plants are found in south Florida, the Keys
and tropical Arnerica. The traditional medicinal uses for extracts of this plant include
treatrnent for promoting labor, earache, indigestion, dianhea, hemorrhoids, cough, fever,
post-childbirth recover, painful menstruation, for swollen body parts, vaginal douche,
tiredness, rheumatism and kidney problems.49
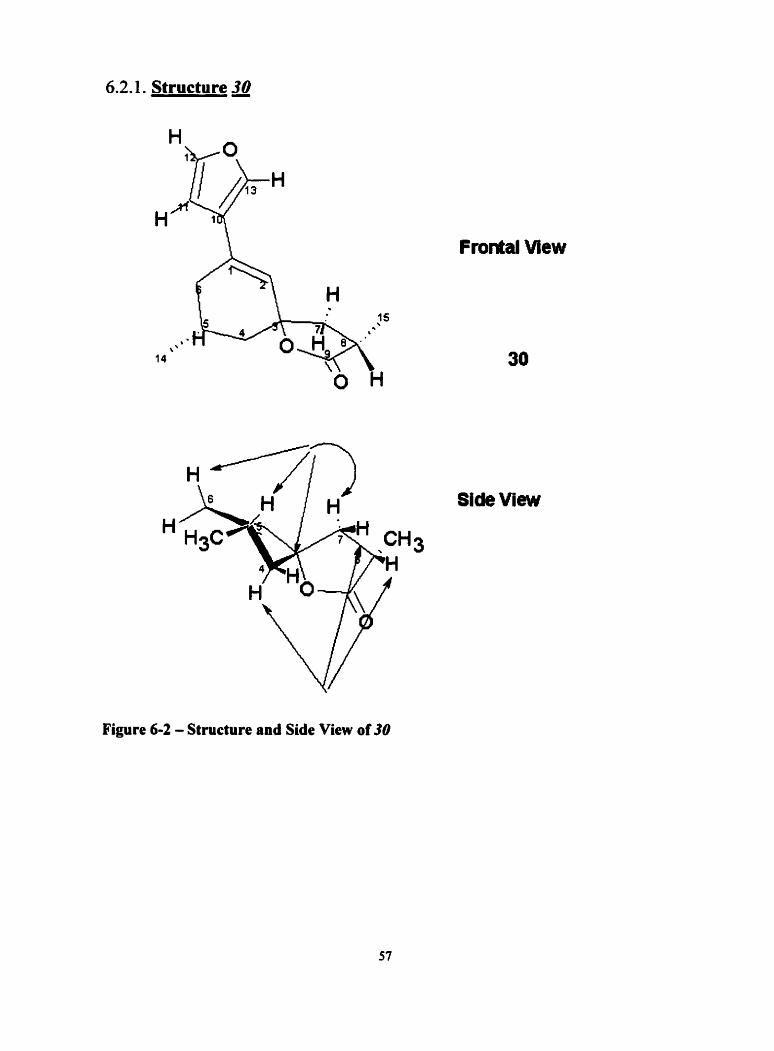
6.2.1. Structure a
Frontal View
Side View
Figure 6 3 - Structure and Side View of 30

The "c, 'H and HMBC assignments for structure 30 are provided in Table 6-2.
Table 6-1 - 500 MHz 'k, 'H and HMBC data for JO
POS 1 3 ~ a 'H HlMBC
H, HO 1 143.90 (C) 7P, 6% 6P, 2 2 1 14.32 (CH) 6.3 1 (3.1,S.O) 6~~ 6P 3 90.31(C) 7P, 6P, 4a, 4P, 2 4 45.16 (CH2) 1.65 (1 1.5, 11.5, 1.4)' 2.09 (1 1.6,6.8,3.0)~ 7P, 6P, 5 5 30.52 (CH) 2.10 (m) 6a, 6P, 4a 6 38.37 (CH2) 2.19 (17.3,9.5,3.1)' 2.74 (17.3,7.3,2.0,2.0)~ 2,5 7 44.51 (CH2) 2.45 (12.5,8.3) 1.85 (12.5, 12.0, 1.4) 8a, 4a 8 35.59 (CH) 2.82 (m) 7a, 7P, 15 9 179.21 (C) 7c1,8c1 10 122.92 (C) 13, 12, 11 i l 110.19(CH) 6.48 (2.0,O.g) 13, 12,2 12 143.02 (CH) 7.40 (2.0,2.0) 13, 11 13 141.05 (CH) 7.46 (m) 12, 11,2 14 19.91(CH) 1 .16 (6.3) 6a, 401 15 15.07 (CH) 1.32 (6.1) 8 ~ , 7P
6.2.1.1. Structural Determination of a
'H, 13c, COSY, HSQC, W C and ROESY NMR experiments were performed
in CDC13. Electron impact mass spectral analysis of the product showed the molecular
ion at 246. The calculated high-resolution mass spectrum for Ci sH sOa was 246.1256
and was found experimentally to be 246.1267. The presence of the substituted furan
moiety attributed to the 'H NMR resonances at 8 6.48 (H-1 1, dd, J = 2.0,0.8 Hz), 6 7.40
(H- 12, dd, 2.0,2.0) and 8 7.46 (H- 13, m ). The conjugated olefîn showed a peak at 6 6.3 1
a Chernical shifts on 6 scaie in CDC13 In brackets, coupling constants in Hz Axial hydmgen Pseudo equatorial hydrogen
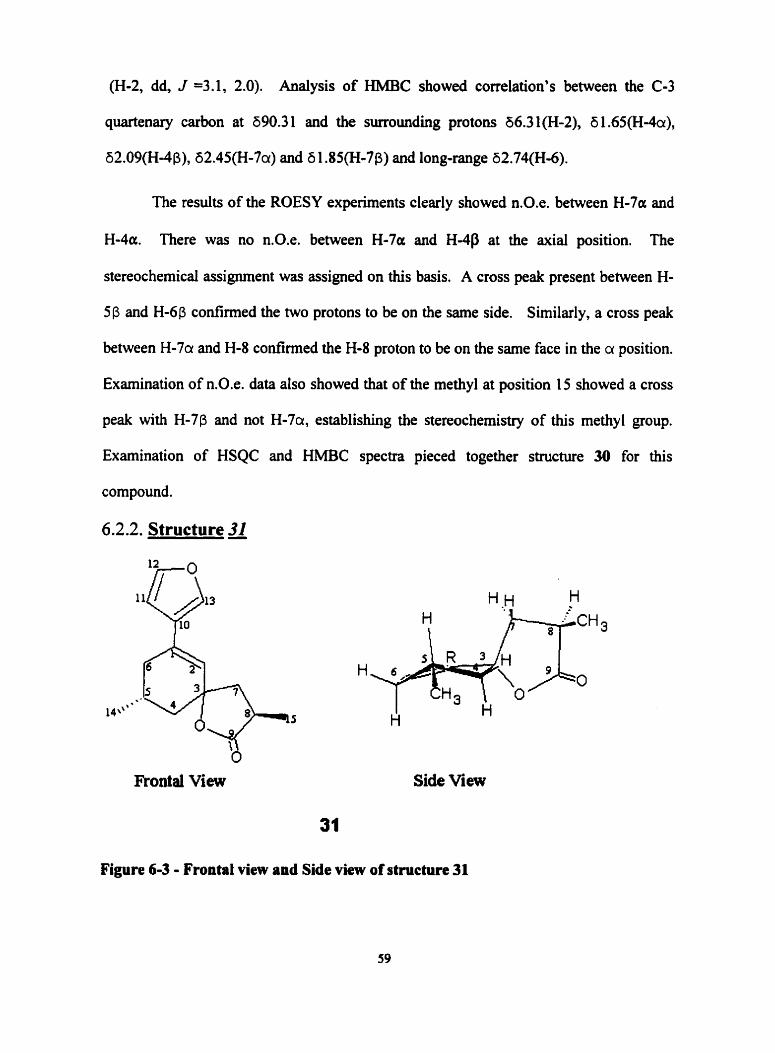
(H-2, dd, J =3.1, 2.0). Analysis of HMBC showed correlation's between the C-3
quartenas, carbon at 690.3 1 and the surroundhg protons 66.3 1 (H-2), 6 1.65(H-4a),
62.09(Ho4P), 62.45(H-7a) and 6 1.8S(HJp) and long-range 62.74(H-6).
The results of the ROESY experiments clearly showed n.0.e. between H-7a and
H-4a. There was no n.0.e. between H-7a and H-4P at the axial position. The
stereochemical assignment was assigned on this basis. A cross peak present between H-
5p and H-6P ~ o ~ r m e d the two protons to be on the same side. Similarly, a cross peak
between H-7ir and H-8 confirmed the H-8 proton to be on the same face in the a position.
Exarnination of n.0.e. data also showed that of the methyl at position 15 showed a cross
peak with H-7p and not H-7a, establishing the stereochemistry of this methyl group.
Exarnination of HSQC and HMBC spectra pieced together structure 30 for this
compound.
6.2.2. Structure a
Frontal View Side View
Figure 6-3 - Frontal view and Side view of structure 31

The 13c, 'H and HMBC assignments for structure 31 are provided in Table 6-2.
Table 6-2 - 500 MHz NMR "c, 'H and HMBC data for 31
Pos. I3c a 'H 4b EIMBC H a Ha
1 143.54 (C) 7a, 6% 613, 4P, 2a, 2 1 1 1.85 (CH) 6.10(7.5,2.8) 6% 613 3 89.84(C) 713,613, 4P, 2 4 47.28 (CH2) 1.88 (1 1.5, 1 1.5)' 1.98 (1 1.5, 5.8)d*C 14,7a, 6P 5 29.00(CH) 2.20 (m)' 14,7cx, 6a, 6P, 4P, 2 6 37.50 (CH2) 2.18 (17.5,8.7,2.8)' 2.75 (17.5,8.5, 14,4P,
2.51~ 7 42.87 (CH2) 1.87 (12.5, 1.08)' 2.55 (12.5,9.0)~ 15,8,4p, 8 34.32 (CH) 2.94 (m) 15,7a, 713 9 179.83 (C) 15,8,7P 10 122.64(C) 13, 12, 11 1 1 1 10.24 (CH) 6.45 (1.8,0.9) 13, 12,2 12 143.00(CH) 7.39 (1.8, 1.6) 13,11 13 140.94 (CH) 7.43 (m) 12, 11,2 14 20.55 (CH) 1.16 (6.7) 6% 15 15.69(CH) 1.32 (6.2) 8, 7a
6.2.2.1. Structural Determination of a 'H, 13c, COSY, HSQC, HMBC and ROESY NMR experiments were performed.
The mass of this produci was the same as that for 31 and the HRMS also confirmed that
the molecular formula was also identical. A 70:30 CsD6:CDC13 mixture was necessary
for this compound to resolve overlapping peaks, which were more problematic than 31.
The presence of the substituted îùran moiety attiibuted to the 'H NMR resonances at O
6.45 (H-11, dd, J = 1.8, 0.9 Hz), 6 7.39 (H-12, dd, 1.8, 1.6) and 6 7.43 (H-13,m ). The
' Chernical shifts on 6 scale in CDCb In brackets, coupling constants in Hz ' Pseudo axial hydrogen
Pseudo equatorial hydrogen ' Peaks are slightly broadened, due to m e r unresolved coupling. The COSY spec tm shows weak cross peaks between protons at 8 1.98 and 6 2.75

conjugated olefui showed a peak at 6 6.10 (H-2, dd, J =7.5, 2.8). Analysis of HMBC
showed correlation's between the C-3 quartenary carbon at 690.3 1 and the surrounding
protons at positions 56.10(H-2), 6 1.98(H-4) and 52.55(H-7).
Cross peaks present between H-5P and H-6P confirmed the two protons to be on
the same side. H-4P was established because it showed an n.0.e. with H-5P and
because of the axial-equatorial coupling it exhibited. H-7a showed an n.0.e. with the
equatorial H-4a proton and none with H-4p establishing its position. Examination of
n.0.e. data also showed that of the H-15 methyl showed a cross peak with H-7u, and not
H-7p showing this methyl group to be the inverted isomer to 30. Examination of HSQC
and HMBC spectra pieced together structure 31 for this compound.
6.2.3. Inter~retation of Data
The two products were determined to be sesquiterpenes of structures 30 and 31.
Configurations were successfûlly established with n.0.e. data. The products were found
to be identical and ody differed in configuration around position 15. The two
sesquiterpenes (C-15 terpenes) were found to be new compounds, which was confirmed
by a CAS online search .

6.3. Elucidation un known CaesalDnia bonduc root extracts.
Caesalpinia is a pantropical genus with approxirnateiy 150 species. The species,
Caesalpinia bonduc is a medicinai plant of wide distribution throughout the tropics and
s u b t r ~ ~ i c s . ~ ~ Many of the compounds present in the root extract of CaesaZpinia bonduc
are still unknown. Investigation of the root extracts of C. bonduc collected and provided
by Professor W.F. Tinto of the University of West Indies in Barbados, were carried out
and elucidation of the structures were performed using NMR rnethods. Previously in our
labs, chemicai investigations of Caesaipinia bonduc lead to the isolation of a new
reamnged cassane furanoditerpene, caesalpinin (Figure 6-4), being the first report of a 19
(4+3)-abeo-cassane diterpene. Further investigations of the root extracts lead to the
isolation of several new rearranged cassane furanoditerpenes.
Figure 6-4 - Structure of Caesalpinin

6.3.1 - Structures 32 and 33
- - - -
Figure 6-5 - Structures of compounds 32 and 33
The proton and carbon NMR assignments for structures 32 and 33 are shown in
Table 6-3. 'H, I3c, COSY, HSQC, HMBC and NOESY experiments were performed in
CDC13.
Table 6 3 - 500 MHz NMR 'H and 13c data for structures 32 and 33
Structure 32 Pos. 13c 'H 1 70.9(CH) 3.89(m)
2 39.1 (CH*) 2.53 (br d, 19.1) 2.20 (br d, 19.1)
3 127.7 (C) - 4 127.7 (C) - 5 76.6 (C) - 6 76.0 (CH) 5.38 (d, 8.3) 7 74.2 (CH) 5.63 (dd, 10.2,8.3)
Structure 33 'Y a 'H 34.6 (CHi) 1.44 (m)
1.53 (m) 18.2 (CH2) 1.5 1 (m)
1.71 (m) 38.1 (CH2) 1.14 (m)
1.71 (m) 38.9 (C) - 76.2 (C) - 72.3 (CH) 5.23 (t, 3.3) 3 1.4 (CH2) 1.5 1 (m)
2.19 (ddd, 13.4, I3.4,3.3)
a Chernical shifts on 6 scaie in CDC13 In brackets, coupling constants in Hz

Stnicture 32 Pos. 13c a 'H 8 48.1 (CH) 2.30 (dd, 1 1.9. 10.2)
33.2 (CH) 43.3 (C) 23.2 (CHÎ)
148.4 (C) 125.6 (C) 72.9 (C) 107.5 (CH) 141.9 (CH) 24.8 (CH3) 20.5 (CH3) 14.7 (CH3) 15.8 (CH3) - - 170.9(C=O) 21.8 (CH3) 171.1(C=O)
2.95 (ddd, 11.9. 11.2, 5.6)
2.80 (dd, 15.6,5.6) 2.48 (dd, 1 5.6, 1 1.2)
6.39 (d, 1.9) 7.25 (d, 1.9) 1.56 (s) 1.68 (br s) 1.74 (br s) 1 .O4 (s) 3.02 (d, 6.4) 3.44 (br s) - 2.1 O (s) -
2 1.7 (CH3) 1.99 (s)
6.3.1.1. Structural Determination of32
Structure 33 I3c a 'H 4b
30.4 (CH) 1.26 (m) 37.9 (CH) 2.34 (m) 4 1.4 (C) - 2 1.7 (CH2) 2.47 (dd, 18.0, 10.0)
2.5 1 (dd, 18.0, 8.0) 149.s (C) - 122.3 (C) - 31.1 (CH) 2.57 (dq, 6.8,5.7) 109.4 (CH) 6.19 (d, 1.7) 140.3 (CH) 7.22 (d, 1.7) 1 7.6 (CH3) 0.99 (d, 6.8) 27.6 (CH3) 0.98 (s) 25.7 (CH3) 1.25 (s) 16.5 (CH3) 1.34 (s)
The compound was isolated as a white solid with a melting point range between
163-165 OC and [alD +7.2 O . The molecular formula, C24H3208, was c o n f i e d by hi&-
resolution mass spectrornetry. The IR spectrum had absorption due to hydroxyl (3429
cm"), ester carbonyl (1735 cm") and furan (757 c d ' ) functionalities respectively. The
presence of the 1,t-disubstituted furan moiety attributed to the 'H NMR peaks at 6 6.39
(1 H, d, J = 1.9 Hz, H-15) and 6 7.25 (1 H, d, J = 1.9 Hz, H-16). The 'H NMR spectnim
also had resonance's for two olefinic methyl's at 6 1.68 (H-19) and 6 1.74 (H-19), two
acetoxy methyl's at 6 1.99 (7-OAc) and 62.10 (6-OAc) and two tertiary methyl singlets at
6 1 .O4 (H-20) and 6 1.56 (H-17). In addition, there were three oxymethine resonances at
6 3.89 (m, H-l), 6 5.38 (d, J = 8.3 Hz, H-6) and 6 (dd,+ 10.2, 8.3, Hz, H-7). The latter

two oxymethines were associated with the acetoxy groups attached to C-6 (6 76.0) and C-
7 (6 74.2) on the basis of 'H-'H COSY, HMQC and HMBC experiments. The third
oxymethine proton was assigned to C-1 since the C-20 methyl group at 6 1.04 showed
HMBC correlation's to the C-1 carbon at 6 70.9 as well as C-5 (876.6), C-9(633.2) and
C- 1 O(643 .3). In the ' H - ~ H COSY spectnun, H- 1 has cross peaks with methylene protons
at 62.53 and 62.20; these protons showed direct connectivity to C-2 (639.1) in the
HMQC spectrum. The methylene group at C-2 also had 'H-'H COSY cross peaks with
the methyl protons at 6 1.68(H-18) and 51.74(H-19), respectively, while these methyl
protons had cross peaks with each other, establishing their geminal disposition.
Stereochemistry was determined f'rorn vicinal coupling constants and NOESY cross-
peaks.
6.3.1.2 Structural Determination of 33
The molecular formula of C22H3204 was confhned by H R M S . The IR spectrum
had absorption's typical of hydroxyl (3447 cm"), carbonyl ester (1732 cm") and furan
(758 cm-') functionalities. The 'H NMR spectrum showed resonances indicating the
presence of the three tertiary methyl groups at 80.98 (H- 1 8), 5 1.25 (H-19) and 6 1.34 (H-
20); a secondary methyl group at 60.99 (H-17, d J = 6.8 Hz), and a more deshielded
acetoxyl group at 62.06 (attached at 16). The presence of the 1,2-disubstitued furan was
evident fiom resonances at 67.22 (H-16, d, J = 1.7 Hz) and 66.19 (H-15, d, J = 1.7 Hz).
An oxyrnethine was observed at 65.23 (H-6). Examination of HSQC and HMBC spectra
pieced together structure 33 for this compound.
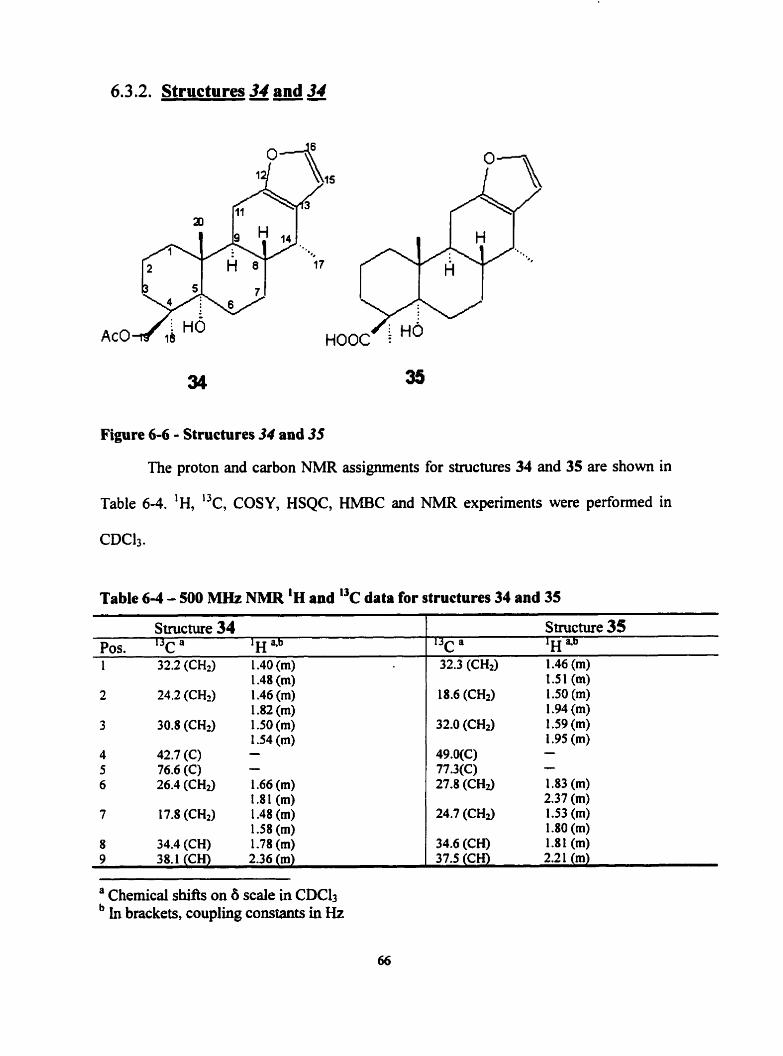
6.3.2. Structures 34 and 34
Figure 6-6 - Structures 34 and 35
The proton and carbon NMR assignrnents for structures 34 and 35 are shown in
Table 6-4. 'H, 'k, COSY, HSQC, HMBC and NMR experiments were performed in
CDC13.
Table 6-4 - 500 MHz NMR 'H and I3c data for structures 34 and 35
1 -48 (m) 2 24.2 (CH2) 1.46 (m)
1.82 (m) 3 30.8 (CH3 1.50 (m)
1.54 (m) 4 42.7 (C) - 5 76.6 (C) - 6 26.4 (CH3 1.66 (m)
1.81 (m) 7 17.8 (CH2) 1.48 (m)
1.58 (m) 8 34.4 (CH) 1.78 (m) 9 38.1 (CH) 2.36 (m)
Structure 34 Pos. I3c a 'H 1 32.2 (CH2) 1.40 (m)
1.51 (m) 18.6 (CHz) 1.50 (m)
1.94 (m) 32.0 (CHt) 1.59 (m)
1.95 (m) 49.WC) - 77.3(C) - 27.8 (CH3 1 .83 (m)
2.37 (m) 24.7 (CH2) 1.53 (m)
1.80 (m) 34.6 (CH) 1.81 (m) 37.5 (CH) 2.21 (m)
Structure 35 I3c a 'H 32.3 (CH2) 1.46 (m)
a Chernical shifis on 6 scale in CDC13 In brackets, coupling constants in Hz

Structure 34 Pos. 13c a 'H "b 1 O 4 1.4 (C) -
22.2 (CH2)
149.8 (C) l22.8(C)
3 t .4 (CH) t 09.6 (CH) 140.6 (CH) 17.5 (CH3) 21.1 (CH3) 67.5 (CH3)
16.9 (CH3) 17 1.5(C=O)
2.36 (m) 2.48 (dd, 15.9,9.1) - - 2.60 (m) 6.18 (d, 2.4) 7.22 (d, 2.4) t .O l (d, 6.8) 1 .O0 (s) 4.05 (d, 10.7) 4.43 (d, 10.7) 1 .O2 (s) -
Structure 35 ' j ~ a 'H "b 42.0 (C) - 22.5 (CH2) 2.34 (dd, 16.7, 1 1.1)
2.50 (dd, l6.7,6.l) 149.4 (C) - 122.3 (C) -
3 1.6(CH) 2.62 (m) 109.5 (CH) 6.19 (d, 1.9) 140.3 (CH) 7.22 (d, 1.9) 17.5 (CH3) 1 .O2 (d. 6.8) 24.1 (CH3) 1.26 (s) 18 1.3 (CH3)
15.3 (CH3) 0.94 (s)
6.3.2.1. Structural Determination of 34
The molecular formula of CuHa04 was confirmed HRMS. The IR spectnim had
absorption's typical of hydroxyl (3487 cm''), carbonyl ester (1741 cm") and furan (757
cm-') functionaiities. The 'H NMR spectnun showed resonances indicating the presence
of the two tertiary methyl groups at 6 1 .O2 (H-20) 6 1 .O0 (H-18). At position 19, an
oxymethylene was present, displaying different resonances for each proton at 6 4.43 (d, J
= 10.7 Hz) and 64.05 (d, J = 10.7 Hz). The presence of the h a n ring was also indicated
with sirnilar resonances to compound 33 at 67.22 and 66.1 8.
6.3.2.2. Structural Determination of 35
The molecular formula of C20H2804 was cotlfinned HRMS. The IR spectnim had
absorption's typical of hydroxyl (3550 cm-'), carboxylic acid -OH and carbonyl (3300-
2506 cm-' and 1732 cm-' respectively) and furan (758 cm-') functionaiities. The 'H NMR
spectnim showed resonances indicating the presence of the two tertiary methyl groups at

60.94 (H-20), 6 1.26 (FI- 18) and a secondary methyl group at 6 1 .O2 (H-17, d J = 6.8 Hz).
The presence of the furan ring was indicated with resonances at 67.22 (H-16) and
66.19(H-15). Examination of the HMBC spectrum indicated that the tedary methyl
group at 6 1.26 (H-18) showed long-range correlation's to the carboxylic acid carbon at
6 18 1.3 (C-19), the quartenary carbon at 649.0 (C-4), the rnethylene carbon at 632.0 (C-
3) and the oxygenated carbon at 677.3 (C-5).
6.3.3. Structures 36and 37
Figure 6-7 - Structures 36 and 37
The proton and carbon NMR assignments for structures 36 and 37 are shown in
Table 6-5. 'H, 13c, COSY, HSQC, HMBC and NMR experiments were performed in
CDC13.

Table 6-5 - 500 MHz NMR 'H and data for structures 36 and 37
Structure 36 Pos. I3c a 'H "'
32.0 (CH2)
17.8 (CH2)
30.5 (CH2)
42.6 (C) 77.3 (C) 32.3 (CH2)
71.7 (CH)
39.5 (CH) 36.9 (CH) 40.9 (C) 22.4 (CHr)
149.0 (C) 12 1.9(C) 27.5 (CH) 109.6 (CH) 140.7 (CH) 17.2 (CH3) 2 1 .O (CH3) 67.0 (CH2)
17.2 (CH3) 1 70.7(C=O) 2 1.3 (CH3) 170.7(C=O) 2 1 .O (CH3)
1.42 (rn) 1.51 (rn) 1.49 (m) 1.51 (m) 1.48 (rn) 1.48 (m) - - 1.69 (dd, 13.8, 10.5) 2.16 (dd, 13.8,5.9) 5.28 (ddd, 10.4, 10.4,5.9)
1.93 (ddd, 11.0, 10.5,4.7) 2.55 (dd, 1 1 .O, 4.7) - 2.44 (dd, 15.1,6.3) 2.53 (dd, 15.1, 1 1 .O) - - 2.83 (dq, 7.3,4.7) 6.19 (d, 1.7) 7.25 (d, 1.7) 1 .O0 (d, 7.3) 1 .O1 (s) 4.05 (d, 10.5) 4.40 (d, 10.5) 1 .O8 (s)
2.07 (s)
2.07 (s)
Structure 37 ' 3 ~ a lu 0 u L 1
32.9 (CH3 1.96 (m)
19.4 (CH9
3 1.6 (CH2)
48.7 (C) 75.5 (C) 25.5 (CH-)
23.7 (CH2)
126.6 (C) 143.0 (C) 43.9 (C) 105.8 (CH)
153.8 (C) l25.7(C) 128.3 (C) 104.9 (CH) 144.3 (CH) 15.9 (CH3) 23.9 (CH3) 177.5 (C=O)
27.5 (CH3)
2. I O (rn) 1.69 (m) 2.09 (m) 1.76 (ddd, 12.6, 12.6,5.0) 2.02 (rn) - - 2.28 (ddd, 13.7,7.9,4.1) 2.62 (ddd, 13.7,8.1,2.7) 2.87(ddd, 17.8,8.1,4.1) 2.62 (ddd, 13.7,8.1,2.7) - - 7.35 (s)
- 6.73 (d, 2.1) 7.54 (d, 3.1) 2.39 (s) 1.34 (s) - 1.15 (s)
6.3.3.1. Structural determination of 36
The molecular formula, C24HM06, was confirmed HRMS. The IR spectrum had
absorption due to hydroxyl (3440 cm-'), ester carbonyl (1732 cm") and furan (758 cm-')
fùnctionalities respectively. The presence of the 1,2-disubstituted furan moiety attributed
to the 'H NMR peaks at &7.25(H-16) and 66. W(H-15). The two tertiary methyl groups
' Chemical shih on 6 scale in CDClt
69

at C- 18 and C-20 showed resonances at 6 1 .O1 and 6 1 .O8 respectively, the secondary
methyl at C-17 peak was at 6 1 .O0 (d, J = 7.3 Hz) and the two acetoxyl groups attached to
Cd and C-7 showed one peak at 62.07 (6H). The oxymethine proton had a signal at
65.28 (H-7, ddd, J = 10.4, 10.4, 5.9 Hz) and the oxymethylene group at C-19 showed
two peaks at 64.40 and 64.05(5 = 1 0.5 Hz). The secondary methyl at 6 1 .O0 (H- 1 7) was
assigned as attached to C-14 by HMBC, because it showed long-range correlation's to
carbons at 6 12 1.9(C-13) and 639.5 (C-8). In the 'H-~H COSY spectrum the oxymethine
proton at 5.28 had cross peaks to H-8 and C-6 methylene protons at 62.16 (dd, J = 13 -8,
5.9 Hz) and 6 1.69 (dd, J = 13.8, 10.5 Hz), thus supporting its assignment at (2-7.
6.3.3.2. Structural determination of 37
The molecular formula, C2 H2604, was confirmed by high-resolution mass
spectrometry. The IR s p e c t m had absorption due to hydroxyl (3565 cmd1), ester
carbonyl (1724 cm-') and furan (772 cm") functiondities respectively. The presence of
the 1,2-disubstituted h a n moiety attributed to the 'H NMR peaks at 67.54(H-16) and
66.73(H-15). The deshielded proton in the aromatic region at ti7.35(H-1 l), dong with
an aromatic methyl group at 62.39(H-17), confirmed the presence of a trisubstituted
benzofuran moiety. The I3c NMR spectnim had a resonance at 675.5 due to the
presence of an oxygenated quartenary carbon. Position 11 was confirmed when HSQC
showed the carbon attached the aromatic proton (67.35) was 6 105.8 and this, in turn,
showed correlation's to the aromatic carbons at 6 153.8(C- 12), 6 125.7(C- 13), 6 l26.6(C-
8) and long-range with the quartenary carbon at 643.9(C- 1 0).
' In brackets, coupling constants in Hz
70

6.3.4. Structures 38 and 39
Figure 6-8 - Structures of 38 and 39.
The proton and carbon NMR assignments for structures compounds 38 and 39 are
shown in Table 66. 'H, "c, COSY, HSQC, HMBC and NMR experiments were
performed in CDCIi.
Table 6-6 - 500 MHz NMR 'H and "C data for structures 38 and 39
Structure 38 Pos. '32 a 'H 4b 1 32.2 (CH2) 1.4 1 (m)
1.50 (m) 2 18.7 (CH?) 1.49 (m)
1.89 (m) 3 32.0 (CHI) 1.57 (m)
1.95 (m) 4 49.0 (C) - 5 76.8 (C) - 6 27.7 (CH2) 1.85 (m)
2.24 (m) 7 20.0 (CH) 1.68 (m)
1.8 1 (m) 8 36.qCH) 1.96 (m) 9 40.6 (CH) 2.23 (m) 10 42.0 (C) -
Structure 39 1 3 r a lu 4b
36.2 (CH2) 1.71 (m) 1.68 (m)
23.1 (CH2) 1.65 (m) 1.82 (m)
73.9 (CH) 4.76 (dd, 1 1.9,4.8)
40.4 (C) - 43.8 (CH) 1.49 (m) 26.6 (CH2) 1.48 (m)
1.89 (m) 75.7 (CH) 4.99 (m)
46.6 (CU) 2.02 (m) 46.0 (CH) 1.26 (m) 36.5 (C) -
" Chernical shifts on b scale in CDC13 In brackets, coupling constants in Hz

Structure 38 Pos. 13c a 'H 1 1 22.3 (CH2) 2.38 (dd, 14.8,8.2)
2.48 (dd, 14.8,6.8) 12 151.0(C) - 13 122.1(C) - 14 77.3 (C) - 15 107.7 (CH) 6.28 (d, 1.8) 16 140.9 (CH) 7.23(d, 1.8)
17 25.5 (CH3) 1.28 (s) 18 23.9 (CH3) 1.20 (s) 19 177.3(C=O) -
20 27.5 (CH3) 0.86 (s) 2 1 57.5 (CH2) 3.14 (dq, 15.0, 7.0)
3.29 (dq, 1 5.0, 7.0) 22 16.1 (CH,) 1.14 (t, 7.0) 3-AC
Structure 39 13c a 'H 4b 24.5 (CHZ) 2.03 (m)
2.09 (m) 122.4 (CH) 5.79 (t, 4.8) 14 1.1(C) - 73.7 (C) - 134.7 (CH) 6.40 (dd, 17.9, 1 1.9) 114.2 (CH2) "5.09 (dd, 1 1.9,2.3)
b5.45 (dd, 17.9,2.3) 23.8 (CH3) 1 -29 (s) 13.6 (CH3) 0.88 (s) 64.9 (CH2) 3.68 (d, 12.7)
3.82 (d, 12.7) 14.8 (CH3) 0.99 (s)
169.4 (C=O) - 20.9 (CH3) 2.12 (s) 170.6 (C=O) - 2 1.2 (CH3) 2.03 (s) 17 1 .O (C=O) - 2 1.6 (CH3) 2.09 (s)
6.3.4.1. Structural determination of 38
The molecular formula, CuH340s, was confirmed by high-resolution mass
spectrometry. The IR spectrum had absorption's characteristic of hydroxyl, (3528 cm"),
carbonyl ester (1718 cm"), and furan (756 cm-') functionalities. The 'H NMR spectrum
has resonances for three tertiary methyl groups at 60.86 (H-20), 6 1.20 (H-18), and 6 1.28
(H- 1 7) and a methyl ester at 63.69. The presence of the ethoxy group was evident frorn
1 H NMR resonances at 63.14 (dq, 15.0, 7.0), 63.29 (dq, 15.0, 7.0) and 61.14 (1, 7.0).
The tertiary methyl group at 61.20(H-18) had HMBC correlation's to the
methoxycarbonyl at 6 177.3 (C- lg), the methylene carbon at 632.0 (C-3), the quartenary
carbon at 677.3(C-4) and the oxygenated carbon at 676.8 (C-5).

6.3.4.1. Structural determination of 39
The molecular formula, C&13807, was confirmed by high-resolution mass
spectrometry. The 'H NMR spectrum showed resonances indicating the presence of the
three tertiary methyl groups at 80.88 (H-18), 60.99 (H-20) and 6 1.29 (H-17) and three
acetoxyl methyl groups at 62.03 (H-6Ac), 62.09 (Fi-7Ac) and 62.12 (H-3Ac). For this
compound, the presence of the k a n ring was not indicated. Instead, signals for olefinic
protons typical of a vinyl moiety were observed at 56.40 (H-14, dd, J = 17.9, 1 1.9) 65.45
(H-16b, dd, J = 17.9,2.3 Hz) and 65.09 (H-16% dd, J = 11.9,2.3 Hz).
6.3.5. Interpretation of Data
Compound 32 was detennined to be a new product and labeled caesalpinin B.^'
Caesalpinin B (32) represents only the second example of a cassane hanoditerpene with
a rearranged skeleton.'* Compound 33 was found to be the previously reported
caesaldekarin A." The other minor cassane diterpenoids, compounds 34-39, have not
been previously reported. Compound 36 was found to be a benzofuran, and it is known
that cassane fùranoditerpenes bearing a C-14 hydroxyl group cm be transformed into
benzohirans by the action of mild acid treatmerd4 It was presumed, however, that 36
was a natural product because no acid was used in its isolation process. Compounds 34,
35, 36, 37, 38, and 39 are known as Caesaldekarin H, Demethylcaesaldekarin C,
Caesaldekarh 1, Caesaideka~ J, Caesaldekarin K, Caesaldekarin J, Caesaldekarin K and
Caesaldekarin L re~pec t ive l~ .~~

6.4. Structural elucidation of Axinellamines A and B
A number of compounds with interesting biological properties have been isolated
fiom the marine sponges of the genus Axinella belonging to the class ~ e r n o s ~ o n ~ i a e . ~ ~
Many of these compounds contain nitrogen and have been found to possess cytotoxic and
antheoplastic a ~ t i v i t ~ . ~ ~ * ~ ~ TWO pyrrole alkaloid compounds were isolated fiom
Caribbean Axinella sp. and provided by Professor W.F. Tinto of the University of the
West Indies in Barbados, and their structures were examined by NMR spectroscopy
methods. The sponge was collected around Nelson Island, of Trinidad's Western
peninsula in July 1996 and identified by Mr. Richard Hubbard, Institute of Marine
Affairs, Trinidad and Tobago.
6.4.1 - Structures 40 and 41
Figure 6-9 - Stuctures 40 and 41

'H , "c, COSY, HSQC, and HMBC experiments were performed. The proton and
carbon NMR assignments for structures 40 and 41 are shown in Table 6-7.
Table 6-7 - 500 MHz NMR and 'H data for 40 and 41 -- - -
40 Position I3c a 'H 4b
- 130.1 (C) 105.6 (CH) 108.4 (CH) 1 16.7 (CH) 3 1.1 (CH2) 128.3 (CH) 132.3 (CH) 128.0 (CH) 139.8 (CH) 38.4 (CH) 29.7 (CH2) 1 1.8 (CH3) 20.1 (CH3) - -
7.98 (br s) - 5.95 (m) 6.14 (m) 6.68 (m) 3.40 (br d, 7.2) 5.68 (dt, 15.0, 7.5) 6.09 (dd, 15.0, 10.4) 6.00 (dd, 15.2, 10.4) 5.52 (dd, 15.2, 7.5) 2.05 (m) 1.32 (m) 0.86 (t, 7.0) 0.95 (d, 6.9) - -
- 7.51 (br s) 129.5(C) - 104.9(CH) 5.81(m) 103.6(CH) 5.94(m) 138.1 (C) - 3 1.1 (CH3 3 -28 (br d, 7.0) 128.2 (CH) 5.62 (dt, 15.0, 7.0) 132.0 (CH) 6.05 (dd, 15.0,9.8) 128.0 (CH) 6.00 (dd, 14.3,9.8) 139.5 (CH) 5.48 (dd, 14.3, 7.1) 38.5 (CH) 2.05 (m) 29.7 (CH2) 1.33 (m) 1 1.8 (CH3) 0.85 (t, 7.2) 20.1 (CH3) 0.98 (d, 6.8) 35.4 (CH3) - 29.3 (CH3) 1.61 (s)
6.4.1.1. - Structural elucidatioa of 40
The product was isolated as a pale yellow gurn with [alD 45.4". The molecular
formula of 40, Ci3HisN was connmied by hi&-resolution mass spectrornetry with an
experimental mass of 1 89.1525 and calculated mass of 189.1 5 18. The IR spectrum had
absorptions at 3391 cm-' due to the amine functionality and at 1662 cm-' from C=C
stretches. The 'H-NMR resonances for the two-methyl groups occurred at 60.86(H-13)
and 60.95(H-14). The peaks at 65.95, 66.14 and 66.68 are attributed to the 2-substituted
a Chernical shifts on 8 scale in CDC13 In brackets, coupling constants in Hz; m = unresolved multiplet

pyrrole. The resonances at 65.52 (H-10, dd, J = 15.2, 7.5 Hz), 65.68 (H-7, dt, J = 15.0,
7.2 Hz), 66.0O(H-9, dd, J = 15.2, 10.4 Hz) and d 6.09(H-8, dd, J = 15.0, 10.4 Hz) gave
evidence of the presence of a 1,4-disubstituted butadiene system. The stereochemistry of
the double bonds was detemined to be E due to the sizes of the vicinal 'H-'H coupling
constants. The structure of the 6-methyl-2,4-octadienyl side chah was established by
analysis of a *H-'H COSY spectrum, while the proton-carbon correlation's were
established by an HMQC expenment.
6.4.1.2. - Structural elucidation of 41
The product was isolated as a pale yellow gum and [alD -8.9'. The IR spectrum
had absorption's at 3401 cm" due to the amine fùnctionality and a stretch at 1652 cm-'
fiom C=C olefin stretch. The molecular formula of 41, C2~h2N2 was codirrned by high-
resolution mass spectrometry with an expeiimental mass of 41 8.3329 and calculated mass
of 4 1 8.3348. The molecular formula suggested that 40 was a dimerized form of 41, with
an extra three-carbon unit. Further evidence for this was provided by the 13c spectnun,
which only showed resonances for fifieen carbon atoms, suggesting a symmetrical dimer
similar to structure 3 (Chapter 2). The 'H-NMR spectrum had resonances due to the
presence of six methyl groups showing only three resonance peaks because ail were
chernicaily equivalent. Two occurred as singlets at 6 1.6 1 (H-l6/17), two occurred as a
triplet at 60.85 (H-13, J = 7.2 Hz) while the other two occurred as a doublet at 60.98 (H-

14, J = 6.8 Hz). The 2,5dibubstituted pyrrole had resonances at 65.8 1(H-3) and 65.94
(H-4).
6.4.2 - Interpretation of Data
Both products were detemined to be new pyrrole alkaloids named Axinellamines
A and B.'^ Axinellamine A (40) was detennined to be 2-((EE)-6-methyl-2,4-
octadieny1)pyrrole. Axinellamine B (41) was conclusively detemined to be a related
dimer of Axinellamine A (40), but containing an additional (CH3)2C unit between the C-
5 and C-5' carbons of the two pyrrole rings.

References
1 D. Neuhaus and M.P. Williamson. The nuclear Overhauser effect in structurai and conformation analvsis. VCH, New York, (1 989).
2 R.R. Ernst, G. Bodenhausen and A. Wokaun. Princioles of nuclear mametic resonance in one and two dimensions. Clarendon, Oxford, (1987).
3 H. Gunther. NMR S~ectroscopy, 2" edition. John Wiley, Chichester, (1995).
4 W .F. Reynolds. NMR Pulse Sequences in Encvclowdia of Soectroscoov and Spectromeûy. E h . J.C. Lindon, G. Tranter, J. Hohes, Academic Press, London (1999).
5 G. Bodenhausen and H. Ruben, Chem. Phys. Lett., 69, 185, (1980).
6 W.F. Reynolds, S. McLean, L. Tay, M. Yu, R.G. E ~ q u e z , D.M. Estaick and K.O. Pascoe. Mag. Res. 35,455, (1997).
7 R.J. Abraham, J. Fisher and P. Loftus. Introduction to NMR S~ectroscoov, John Wiley and Sons, New York, (1988).
8 E. Breitrnaier. Structure elucidation bv NMR in oraanic chemistry-A practical guide. John Wiley and Sons, New York, 1993.
9 J.K. M. Sanders and B.K. Hunter. Modem NMR Swctrosco~y. Oxford University Press, Toronto, (1 987)
10 R.M. Silverstein, G.C. Bassler, T.C. Momll. Swctromeaic Identification of Ornanic Compounds, 5' edition, John Wiley and Sons, New York, (1 99 1).
1 1 A.E. Derome, Modem NMR techniaues for chemistrv research Pergamon, New York, (1 987).
12 L. Tay, Imoroved ~rocedure for settine up. runnine and intemreting 2D-NMR snectra used for structurai elucidation. [Thesis], University of Toronto, (1 997).
1 3 A. L. Fallas, Structural and Synthetic Studies in Aspidosperma AIkdoidr [Thesis], University of Toronto, (1965).
14 R.S. Kapil and R.T. Brown. The Alkaloids Chemistw and Phvsiolom Volume XVII, Ed. R.H. F. Manske and R. Rodrigo, Academic Press Inc., New York, (1979).
15 G.I. Dmitrienko, D.G. Murray and S. McLean. Tetrahedron Lett., 196 1, (1 974); S. McLean, G.I. Dirnitrienko and A. Szakolcd Can. J. Chem., 54,1262, (1 976).
16 R.T. Brown and S.B. Fraser. Tetrahedron Lett., 1957, (1 976).
17 R.G. Hamilton, G.N. Saunders and S. McLean. Can. J. Chem., 61,2,284, (1983).
18 J.R. Purdy, R.G. Hamilton, L. Akhter and S. McLean, Con. J Chenr, 59,2,2 10, (1 98 1).
19 R.T. Brown and S.B. Fraser, Tetrahedron Lett., 3853, (1 974).

20 G.N. Smith Chem Commun., 219, (1968).
21 A.R. Battersby, A.R. Burnett, E.S. Hall, and P.G. Parsons, Chern. Commun., 1282, (1 968).
22 Atta-ur-Rahman, A. Basha. Biosynthesis of Indole Alkaloids, Clarendon Press, Oxford, pp. 45-93., (1 983).
23 N. Nagakura, M. Reffer and M.H. Zenk. J. Chem. Soc. P e r h Tram 1,1979,2308.
24 A. Pathy-Lukats, L. Karolyhazy, L.F. Szabo and B. Podanyi. J. Nat. Prod., 1997,60, 69.
25 L A Stevens, J. Schripsema, E.J. Mirand and R. Verpoorte. Plant Physiol. Biochem. 30,674, (1 992).
26 E.D. Cox and J.M. Cook. Chem. Rev., 1797, (1 995).
27.J.~. Purdy, R.G. Hamilton, L. Akhter and S. McLean, Cm. J Chem., 59,210, (1981).
28 G.N. Saunders, J.R. Purdy and S. McLean. Can. J Chem., 61,2,276, (1 983).
29 T. W. Greene. Protective Grouos in Organic Synthesis. John Wiley and Sons, New York pp. 1 15-135, (1981).
3 0 ~ . Schwartz, L.F. Scabo, B. Podanyi. Tetrahedron, 53,30, 10489, (1 997).
3 1 A. R. Battersby, A.R. Burnett and P.G. Parsons. A Chem.Soc., 1 1 87, (1 969).
32 R. T. Brown and S.B. Fraser, Tetrahedron Lett., 84 1, (1 978).
33 A. R. Battersby, A.R. Burnett and P.G. Parsons. J.Chem.Soc., 1 193, (1969).
34 J. Mann, R.S.Davidson, J.B. Hobbs, D.V. Banthorpe and J.B. Harbome. Natural Products: their chemistry and biologicai simificame. Clarendon Press: Oxford, pp. 300- 309, (1 992).
35 H. Kawai, M. Kuroyanagi and A. Ueno. Chem. Pharm. Bull., 36,364, (1 988).
36 R.G. Hamilton. Secoindoids as svnthetic mecursors to 3a-dihvdrocadambine. [Thesis], University of Toronto, (1 98 1).
37 K.B. Sharpless and K. Akashi. A Am. Chem. Soc., 98, 1986, (1 976).
38 H.C. Kolb, M.S. VanNieuwenze and K.B. Sharpless. Chem. Rev., 994,2483, (1994).
39 R.G. Hamilton and S. McLean. Can. J. Chem., 59,2,215, (198 1).
40 A.R. Battersby, AR. Bumett and P.G. Parsons. J Chem. Soc. (C), 1 193, (1 969).
41 H. Achenback and M. Benirschke. Phytochemistry. 44,7,1387, (1997).
42 H. Takayama, Y. Miyabe, T. Shito, M. Kitajima and N. Aimi. Chem. Pharm. Bull., 44,11,2 192, (1996).

43 H. Takayama, R. Yamamoto, M. Kurihara, M. Kitajirna, N. Aimi, L. Mao and S. Sakai. Tetrahedron Lett., 35,88 13, (1 994).
44 H. Takayama, R. Yamamoto, M. Kurihara, M. Kitajirna, N. Aimi, L. Mao and S. Sakai. Tetrahedron Let fers, 35,4 7,88, (1 994).
45 A. Haudrechy, W. Picoul and Y. Langlois. Tetrahedron: Asymmetry, 8,1, 139. (1 997).
46 J.Sandrin, S.P. Hollinshead and J.M. Cook, J Org. Chem., 54,5436, (1 989).
47 M.L. Trudell, D. Soerans, R.W.Weber, L. Hutchins, D. Grubisha, D. Bennett and J.M. Cook. Tetrahedron, 48,10, 1 805, (1 992).
48 R.J. Fessenden and J.S. Fessenden, Organic Chemistn, bh Edition, Brooks Cole Publishing Company: Monterey, pp. 804-8 1 1 (1 990).
49 D. Scofield. Native Plants of South Florida, John Wiley and Sons: New York, p. 65 (1 998)
50 S. Carrington. Wild Plants of Barbados, Macmillan: London, p.37, and (1993).
51 D.L. Lyder, W.F. Tinto, S. M. Bissada, S. McLean and W.F. Reynolds. Heterocycles, 48, 7, 1465, (1 998).
52 S.R. Peter, W.F. Tinto, S. McLean, W.F. Reynolds, and L.-L. Tay. Tetruhedron Lett., 38,5767, (1997).
53 1. Kitagawa, P. Simanjuntak, T. Watano, H. Shibuya, S. Fujii, Y. Yamagata, and M. Kobayashi. Chem. Pharm. Bull., 42, 1798, (1 994).
54 K.O. Pascoe, B.A. Burke and W.R. Chan. J. Nat. Prod 49,913, (1 986).
55 D.L. Lyder, S.R. Peter, W.F. Tinto, S.M. Bissada, S. McLean and W.F. Reynolds. J. Nat. Prod., 61, 1462, (1998).
56 P.Ciminiello, E. Fattorusso, S. Magno and L. Mayol. Con. J. Chem., 67,5 18, (1 987).
57 R. Herb, A.R. Carroll, W.Y. Yoshida, P.J. Scheuer, and V.J. Paul, Tetrhedron, 46, 3089, (1 990).
58 G. R. Penit, F. Fao, and R. Cerny, Heterocycles, 35,7 1 1, (1 993).
59 K.C. Bascombe, S.R. Peter, W.F. Tinto, S.M. Bissada, S.McLean and W.F. Reynolds. HeterocycZes, 48, 7, 146 1, (1 998).





























