Embed Size (px)
Citation preview

Aplastic Anemia and Paroxysmal Nocturnal Hemoglobinuria: Search for a Pathogenetic Link
By A. Griscelli-Bennaceur, E. Gluckman, M.L. Scrobohaci, P. Jonveaux, T. Vu, A. Bazarbachi, E.D. Carosella, F. Sigaux, and G. Socie
The association of paroxysmal nocturnal hemoglobinuria (PNH) and aplastic anemia (AA) raises the yet unresolved questions as to whether these two disorders are different forms of the same disease. We compared two groups of patients with respect to cytogenetic features, glycosylphos- phatidylinositol (GPII-linked protein expression, protein C/ protein Slthrombomodulinlantithrombin 111 activity, and PIG-A gene expression. The first group consisted of eight patients with PNH (defined as positive Ham and sucrose tests at diagnosis), and the second, 37 patients with AA. Twelve patients with AA later developed a PNH clone. Mono- clonal antibodies used to study GPI-linked protein expres- sion (CD14 [on monocytesl, CD16 [on neutrophils], CD48 [on lymphocytes and monocytes], CD67 [on neutrophils and eo- sinophils], and, more recently, CD55, 0 5 8 , and CD59 [on erythrocytesl) were also tested on a cohort of 20 normal subjects and five patients with constitutional AA. Ham and sucrose tests were performed on the same day as flow-cyto- metric analysis. Six of 12 patients with AA, who secondarily developed a PNH clone, had clinical symptoms, while all eight patients with PNH had pancytopenia and/or thrombo- sis andlor hemolytic anemia. Cytogenetic features were nor- mal in all but two patients. Proteins C and S, thrombomod- ulin, and antithrombin 111 levels were within the normal range in patients with PNH and in those with AA (with or without a PNH clone). In patients with PNH, CD16 and CD67 expression were deficient in 78% to 98% of the cells and CD14 in 7696 t o 100Y0. By comparison, a GPI-linked defect was detected in 13 patients with AA, affecting a mean of 32% and 33% of CD16/CD67 and CD14 cell populations, re-
CQUIRED APLASTIC ANEMIA (AA) is a heteroge- neous disease, in which several pathophysiologic fac-
tors are involved.' In contrast to patients who undergo bone marrow transplantation (BMT), those who are successfully treated with immunosuppressive therapy (IST) are at risk for subsequently developing paroxysmal nocturnal hemoglobin- uria (PNH), myelodysplastic syndromes, and acute myeloid l e ~ k e m i a . ~ . ~ D e novo PNHh-' is an acquired clonal disorderY"" characterized by complement-mediated hemolysis and the expansion of affected cells of various hematopoietic lin- eages. The most typical manifestation of PNH is intravascu- lar hemolysis due to abnormal sensitivity of red blood cells
A
From the Laboratoire Central d'Himatologie, and the Unite de Recheche sur la Biologie des Cellules Souches and Service de Greffr de Moelle, Hdpital Saint-Louis, Paris, France.
Submitted March 9, 1994; accepted October 21, 1994. Address reprint requests to G. Sori&, MD, PhD, Unit6 de Recher-
che sur la Biologie des Cellules Souches (LIRBKEA. DSV) et Service De Gre& de Moelle, Centre Hayem, Hfipitul Saint-Louis, I Avenue Claude Vellefaux, 7547.5 Paris Cedex IO, France.
The publication costs of this article were defrayed in parr by page charge payment. This article must therefore be hereby marked "advertisement" in accordance with 18 U.S.C. section 1734 solely to indicate this fact. 0 199.5 by The American Society of Hematology. 0006-4971/9.5/8505-0008$3.00/0
1354
spectively. Two of three tested patients with PNH and l of 12 patients with AA had a defect in the CD48 lymphocyte population. In a follow-up study of our patient cohort, we used the GPI-linked molecules on granulocytes and mono- cytes investigated earlier and added the study of CD55, CD58, and CD59 on erythrocytes. Two patients with PNH and 14 with AA were studied for 6 to 13 months after the initial study. Among patients with AA, four in whom no GPI- anchoring defect was detected in the first study had no de- fect in follow-up studies of all blood-cell subsets (including erythrocytes). Analysis of granulocytes, monocytes, and erythrocytes was performed in 7 of 13 AA patients in whom affected monocytes and granulocytes were previously de- tected. A GPI-anchoring defect was detected on erythrocytes in five of six. However, in one patient, who had a GPI-anchor- ing defect on leukocytes, we did not observe a defect on erythrocytes. In three patients without a GPI-anchoring de- fect after the initial study, a defect was detected on leuko- cytes only, with persistent negative Ham and sucrose tests in one patient, in all cell subsets in another, and on erythro- cytes only in the remaining patient. Finally, PIG-A gene ex- pression, studied by Northern blot analysis, showed that the PIG-A full-length transcript was barely detectable or absent in four patients with PNH and abnormal in length in one. All nine patients with AA who secondarily developed a PNH clone had a normal-sized PIG-A gene transcript. In conclusion, this study failed to show any significant difference, except for the size of the PNH clone, between PNH arising in patients with previous AA and in patients with de novo PNH. 0 1995 by The American Society of Hematology.
to autologous serum at either low ionic strength (sucrose test)" or low pH (acid Ham test).'* Venous thrombosis is another feature of the disease." While PNH is commonly associated with bone marrow aplasia, and while the appear- ance of a PNH clone is a recognized complication in patients treated with E T , the question as to whether these two disor- ders are different forms of the same disease is unresolved.""" One study has strongly suggested that AA patients have a PNH-like defect at the hematopoietic stem-cell level," and some investigators have postulated that marrow failure is the primary mechanism in both AA and PNH." In PNH, affected blood cells are deficient in glycosylphosphatidylinositol (GP1)-anchored proteins.17 A number of studies have demon- strated the usefulness of flow cytometry in the diagnosis of a GP1 defect."^" However, in these studies, details concern- ing the primitive disease (PNH or AA) are generally not reported. The elucidation of the GPI-anchor biosynthetic pathway has been facilitated by the availability of a panel of T-cell mutants." The mutants of classes A, C, and H cannot transfer N-acetylglucosamine to an inositol phospho- lipid acceptor, which suggests that the first step of GP1 syn- thesis is regulated by at least three genes.2x Two of these three genes coding for class and class H" gene prod- ucts have recently been cloned and sequenced.
In the present study, our aim was to compare clinical and biologic characteristics of patients with de novo PNH (positive Ham and sucrose tests at diagnosis) with those of
Blood, Vol 85, No 5 (March l), 1995: pp 1354-1363
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom

PNH-APLASTIC ANEMIA SYNDROME 1355
AA patients who only secondarily developed PNH (negative Ham and sucrose tests at diagnosis) to search for a pathoge- netic link between these two syndromes.
MATERIALS AND METHODS
Patients
Patients Patients were aware of the objectives of the study, and specimens
were collected in accordance with the requirements established by the local ethics committee of the Hdpital Saint-Louis. The patients' main characteristics are listed in Table 1.
Thirty-seven consecutive patients with acquired AA were included in this study. Among these 37 patients with negative Ham/sucrose tests at diagnosis, 31 patients had an idiopathic form of AA, three had drug-induced AA, two had AA associated with an autoimmune disease, and one had viral hepatitis before AA. Thirteen patients met the international criteria for the severity of the disease.34 These patients received cyclosporine (CSA) or antithymocyte globulin (ATG) plus prednisone, with or without androgens, as first-line ther- apy. As second-line therapy, 14 patients received either a second course of ATG, CSA, and/or recombinant human growth factor (in- terleukin-3 [IL-31 or granulocyte colony-stimulating factor [G-CSF]) (Table 1). One patient (no. 40) underwent BMT, one (no. 33) re- ceived prednisone, and one (no. 32) was given transfusion support as sole therapy. All patients had regular Ham and sucrose tests performed to detect the emergence of a PNH clone. Twelve of 37 patients with AA later developed PNH (positive Ham andlor sucrose tests) and one patient (no. 49) developed a myelodysplastic syn- drome.
Eight patients were diagnosed with PNH. Presenting manifesta- tions included pancytopenia in six, hemolytic anemia in six, and thrombosis (Budd Chiarri syndrome) in three (Table I ) .
Controls Twenty healthy blood donors and five patients with congenital
cytopenia (two cases of Fanconi's anemia, one of Schwachman- Diamond's syndrome, one of amegakaryocytic thrombocytopenia, and one of an unclassified familial AA) were used as controls in the flow-cytometry and Northern blot analyses.
Methods
Immunocytornetric Study of GPI-Anchored Molecules Cell preparation for immunofluorescence studies. Cells were
isolated from heparinized peripheral blood. Mononuclear cells and granulocytes were separated by Ficoll gradient centrifugation (ficoll Isopaque, d = 1.077; Eurobio, Paris, France). Mononuclear cells were tested by indirect immunofluorescence after lysing contaminat- ing erythrocytes with FACS lysing solution 1 X (Becton Dickinson, Pont de Claire, France). An aliquot of mononuclear cells was cryo- preserved in RPM1 1 W medium containing 10% DMSO (Sigma, Eragry, France) and 50% fetal calf serum (GIBCO-BRL, St Quentin, France). Erythrocytes were separated from granulocytes by sedimen- tation using high-molecular weight dextran and then tested by indi- rect immunofluorescence. Blood samples collected on EDTA were used to test granulocytes and monocytes by direct immunofluores- cence and for routine blood cell count.
Monoclonal antibodies. Monoclonal antibodies recognizing GPI-anchored surface molecules were as follows: CD14 conjugated to fluorescein isothiocyanate (FITC) (RM052, Immunotech, Mar- seille, France; and My4 nonconjugated, Coultronics, Margency, France); CD16 (3G8-FITC, Immunotech; and Leu-llb, Becton Dickinson); CD48 (TCT.1, kindly provided by F. Mami-Chouaib,
Institut Gustave Roussy, Villejuif, France); CD55 (anti-DAF, NaM16 4D3), CD58 (anti-LFA3, NaM20 5D12), and CD59 (anti- MIRL, NaM77 1E5) (kindly provided by D. Blanchard, CRTS, Nantes, France); and CD67 (80H3-FITC, Valbiotech, Paris, France).
For lymphocyte selection, a CD2/CD4 double-positive control was used (TII, Coultronics; Leu-3a, Becton Dickinson). We used a CD64 (32-2-FITC, Valbiotech) to identify monocytes and a CD13 (WM-47-PE [phycoerythrin; PE]-conjugated, Dako, France) to gate granulocytes and monocytes. Unspecific irrelevant FITCPE-conju- gated mouse IgG (MsIgG) and unlabeled isotypic unspecific MsIgG (Coultronics) were used as negative controls. A FTTC-conjugated goat anti-mouse (GAM-FITC) IgG (Fab')2 (Flobio Cappel, France) was used for indirect immunofluorescence. All of these antibodies were used at saturating concentrations, previously determined by flow cytometry.
Flow-cyfometry analysis. Samples were analyzed by flow cy- tometry (F'rofil 11; Coulter, Coultronics). Using forward and side- scatter gating, it was possible to analyze lymphocytes and monocytes separately from the peripheral blood mononuclear cell (PBMNC) fraction. Granulocytes were separated from monocytes in whole- blood samples, and 10,OOO cells were analyzed in each sample through a granulocyte live-scatter gate. A minimum of 1 ,OOO events was analyzed in a monocyte gate. The mean fluorescence intensity (MFI) was expressed on a logarithmic scale.
Ham Acid Lysis and Sucrose Tests Ham and sucrose tests were performed on the same day as part
of the routine work-up according to Dacie and Lewis. As a rule, the results of the lytic tests are given in pluses rather than as a percentage of lysis in our institution. However, results were quantitated by spectrophotometry in 12 patients (whose lytic tests and respective spectrometry results are used as references for the laboratory; Y. Bretagne, CTS HBpital St Antoine, Paris, France, unpublished re- sults). For the sucrose test, (+) corresponds to less than 5% lysis (2% to 5%). + to 10% lysis (8% to Is%), and ++ to greater than 30% lysis. For the Ham test, (+) corresponds to 8% lysis (8% to 12%). + to 15% lysis (10% to 20%), and ++ to greater than 30% lysis (25% to 40%).
Cytogenetic Studies Cytogenetic studies were performed after bone marrow culture in
vitro for 24 and/or 48 hours. RHG and GTG-bands were obtained as usual, and chromosomes were classified according to the interna- tional n~menclature.~~."
Coagulation Tests Proteins S and C-related antigens and thrombomodulin were mea-
sured using enzyme-linked immunosorbent assay (ELISA) meth- o d ~ . ~ ' Proteins S and C and antithrombin 111 activity were measured using functional chromogenic techniques3'
Study of PIG-A Gene Expression by Northern Blot Analysis
Total RNA was isolated from PBMNC using the guanidine iso- thiocyanate method. Northern blots were prepared using formalde- hyde-containing gels. The blots were probed with PIG-A cDNA after labeling by random priming (Amersham, Les Ullis, France; Megaprime DNA labeling system). The probe corresponds to the full-length PIG-A cDNA and was a generous gift of Dr J. Takeda.30 The same filters were subsequently hybridized with a &actin probe and used as a control. A semiquantitative estimation of PIG-A ex- pression was performed by densitometry using Quantiscan software (The Imager, Appligtne, Strasbourg, France).
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom

1356 GRISCELLI-BENNACEUR ET AL
Table 1. Clinical Characteristics of the Patients
Patient Age Interval No. (yr)/Sex Diagnosis Diagnosis/Study Etiology Treatment
Ham & Sucrose Tests Symptoms at Inclusion
1 2 3 4 5 6 7 8 9
10 11 12 13 14 15 16 18 19 20 21 22 23 24 25 26 27 28 29
31 32 33 34 35 36 37 40
43lF 22lF 28lF 45lM 57lF 47lM 451F 191M 3OlF 22lF 45lF 39lM 64lM 1 ?/M 1 OIF 13/F 27lF 30lM 59lM 24lF 38lM 49lF 17/F 58lM 191M 47lM 18/M 28lM
15lM 78lM 31lF 2 4lM 581M 36lM 7 OIM 34/M
SAA AA AA AA SAA AA AA SAA AA SAA AA AA AA AA SAA AA AA AA SAA SAA AA AA SAA SAA SAA AA AA SAA
AA AA AA SAA AA AA AA SAA
4 yr 5 vr
10 yr
3 Y' 6 vr
l ? yr
7 yr 2 yr 3 vr 9 yr
3 v 2 v 8 yr 2.5 yr 2.5 yr 6 v 1 vr
16 yr
6 vr 25 yr
7 v 8 mo 3 vr 3 m0
14 yr 5 m0
8 yr
12 yr 2 vr
10 yr 7 v 2 yr 5 vr 4 m0 6 m0
18 yr
T I I I I l I I I I i I I I l I I l I I T l I I I I ai l
ai I I I T post-hep I I
ATGlCSNIL3 ATG ATGlCSAIandrollL3 ATG ATG andro ATGICSA ATGICSNG-CSF CSA ATGICSA CSA CSA andro ATG ATGICSNG-CSF CSA ATGICSA ATGICSA ATG ATGICSA ATGIandro ATG ATGICSA ATGICSA ATGICSA ATGIandro ATG ATGICSNandro
ATGICSNandro /(transfusions support, prednisone andro CSNandro CSA andro studied before
/G-CSFIIL-3
H H None None None H H None None H H + inf None None None None None None None None None None None None None None None None None
None only) None
None None None None None None
-l +l 1
(+) l - (+) l+ (+ ) l (+ )
+l+ +l+ -l+
(+)l+ ++It+
(+)l- +/+ -l(+) -1- -1- -I- "/- -l" -1- -1- -1 -
- I - -/- -1- -1- -1".
-1- -1-
-1- -1- "/- -l" -l- -1- -1- -1-
transplantation 49 13lM AA 2 v I ATGICSAJandro None -1-
/G-CSF 42 53lF PNH 10 mo l ATGICSA H-inf-AA +I+ 44 22lM PNH 11 yr l andro Th-H-AA +l+ 45 271F PNH 12 yr l ATGICSA Th-H-AA + +/+ +
47 24lM PNH 2 v / l Th-H +/+ 46 58lF PNH 8 yr l andro H ++/+ +
t +/+ + ++/++
48 23lF PNH 5 mo I I AA 50 58/F PNH 30 yr I l AA-H 51 36lM PNH 1 mo l I AA-H +I+
Abbreviations: M, male; F, female; S W , severe AA; T, toxic; l, idiopathic, ai, autoimmune; post-hep. posthepatitis; ATG, antithvmocyte globulin; CSA, cyclosporine A; IL-~, interleukin-3; G-CSF; granulocyte colony-stimulating factor, andro, androgens; H, hemolysis; inf, infections;
Th, thrombosis.
RESULTS
Clinical Manifestations
during the study. This complication was asymptomatic in six, whereas five patients had intravascular hemolysis and one had both intravascular hemolysis and recurrent infec-
Twelve of 37 Patients With acquired AA secondarily de- tions. None had thrombosis. Among eight patients with PNH, veloped PNH (as defined earlier), with a mean time from pancytopenia was present at the time of the analysis in six, diagnosis tO positive Ham and sucrose tests Of 7.25 months hemolytic anemia in six, and thrombosis (Budd Chimi syn- (range, 5 to 19). None of these 37 patients were on therapy drome) in three (Table l ) .
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom
PNH-APLASTIC ANEMIA SYNDROME
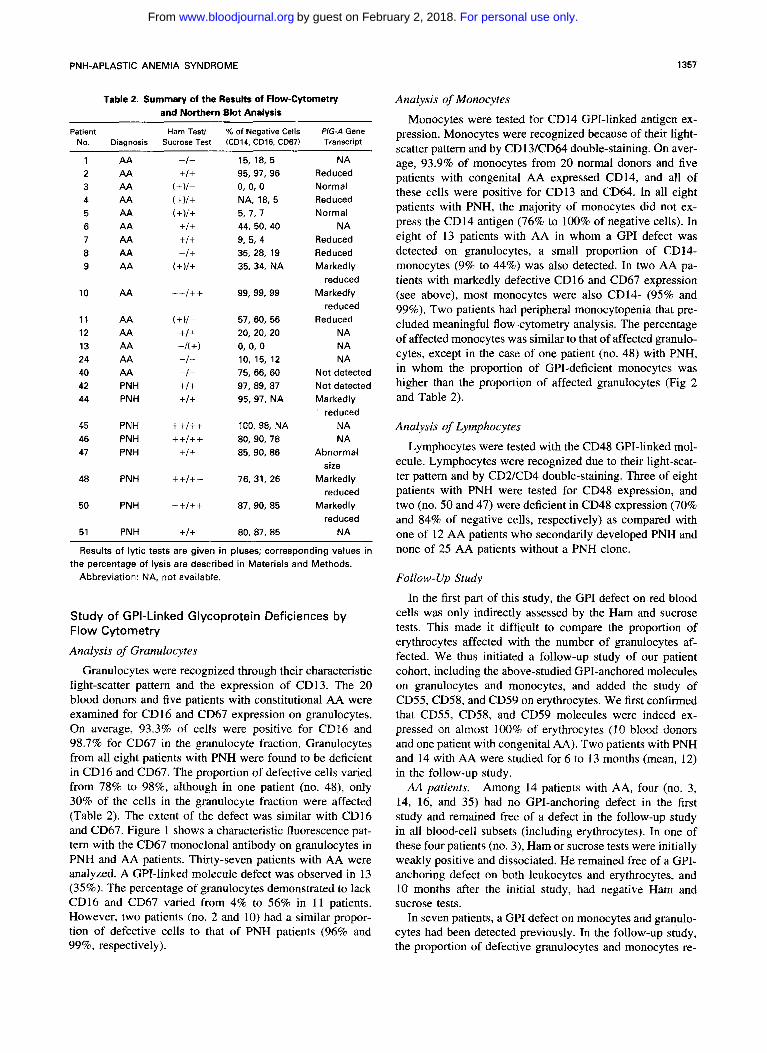
Table 2. Summary of the Results of Flow-Cytometry and Northern Blot Analysis
Patient Ham Test/ % of Negative Cells PIG-A Gene No. Diagnosis Sucrose Test (CD14, CD16. CD67) Transcript
1 2 3 4 5 6 7 8 9
10
11 12 13 24 40 42 44
45 46 47
48
50
51
AA AA AA AA AA AA AA AA AA
AA
AA AA AA AA AA PNH PNH
PNH PNH PNH
PN H
PNH
PNH
-1- +l+
(+)l- (+)/+ (+V+ +I+ +l+ -l+
(+)l+
++l+ +
(+)l- +I+ -l(+) -1- -1- +l+ +I+
++l+ + ++l++
+l+
f +l+ i
t tlt f
+l+
15, 18, 5 95, 91, 96 0, 0. 0 NA, 18, 5 5.7,7 44, 50, 40 9. 5. 4 35, 28, 19 35, 34, NA
99, 99, 99
57, 60, 56 20, 20, 20 0, 0, 0 10, 15, 12 75, 66, 60 91, 89, 87 95, 91, NA
100.98, NA 80, 90, 78 85, 90, 86
76, 31, 26
87, 90, 85
80, 87, 85
NA Reduced Normal Reduced Normal
NA Reduced Reduced Markedly
reduced Markedly
Reduced NA NA NA
Not detected Not detected Markedly ' reduced
reduced
NA NA
Abnormal size
Markedly reduced
Markedly reduced
NA
Results of lytic tests are given in pluses; corresponding values in
Abbreviation: NA, not available. the percentage of lysis are described in Materials and Methods.
Study of GPI-Linked Glycoprotein Deficiences by Flow Cytometry
Analysis of Granulocytes
Granulocytes were recognized through their characteristic light-scatter pattern and the expression of CD13. The 20 blood donors and five patients with constitutional AA were examined for CD16 and CD67 expression on granulocytes. On average, 93.3% of cells were positive for CD16 and 98.7% for CD67 in the granulocyte fraction. Granulocytes from all eight patients with PNH were found to be deficient in CD16 and CD67. The proportion of defective cells varied from 78% to 98%, although in one patient (no. 48), only 30% of the cells in the granulocyte fraction were affected (Table 2). The extent of the defect was similar with CD16 and CD67. Figure 1 shows a characteristic fluorescence pat- tern with the CD67 monoclonal antibody on granulocytes in PNH and AA patients. Thirty-seven patients with AA were analyzed. A GPI-linked molecule defect was observed in 13 (35%). The percentage of granulocytes demonstrated to lack CD16 and CD67 varied from 4% to 56% in I1 patients. However, two patients (no. 2 and 10) had a similar propor- tion of defective cells to that of PNH patients (96% and 99%, respectively).
1357
Analysis of Monocytes
Monocytes were tested for CD14 GPI-linked antigen ex- pression. Monocytes were recognized because of their light- scatter pattern and by CD13/CD64 double-staining. On aver- age, 93.9% of monocytes from 20 normal donors and five patients with congenital AA expressed CD14, and all of these cells were positive for CD13 and CD64. In all eight patients with PNH, the majority of monocytes did not ex- press the CD14 antigen (76% to 100% of negative cells). In eight of 13 patients with AA in whom a GP1 defect was detected on granulocytes, a small proportion of CD14- monocytes (9% to 44%) was also detected. In two AA pa- tients with markedly defective CD16 and CD67 expression (see above), most monocytes were also CD14- (95% and 99%). Two patients had peripheral monocytopenia that pre- cluded meaningful flow-cytometry analysis. The percentage of affected monocytes was similar to that of affected granulo- cytes, except in the case of one patient (no. 48) with PNH, in whom the proportion of GPI-deficient monocytes was higher than the proportion of affected granulocytes (Fig 2 and Table 2).
Analysis of Lymphocytes
Lymphocytes were tested with the CD48 GPI-linked mol- ecule. Lymphocytes were recognized due to their light-scat- ter pattern and by CD2KD4 double-staining. Three of eight patients with PNH were tested for CD48 expression, and two (no. 50 and 47) were deficient in CD48 expression (70% and 84% of negative cells, respectively) as compared with one of 12 AA patients who secondarily developed PNH and none of 25 AA patients without a PNH clone.
Fotlow-Up Study
In the first part of this study, the GP1 defect on red blood cells was only indirectly assessed by the Ham and sucrose tests. This made it difficult to compare the proportion of erythrocytes affected with the number of granulocytes af- fected. We thus initiated a follow-up study of our patient cohort, including the above-studied GPI-anchored molecules on granulocytes and monocytes, and added the study of CD55, CD58, and CD59 on erythrocytes. We first confirmed that CD55, CD58, and CD59 molecules were indeed ex- pressed on almost 100% of erythrocytes (10 blood donors and one patient with congenital AA). Two patients with PNH and 14 with AA were studied for 6 to 13 months (mean, 12) in the follow-up study. AA patients. Among 14 patients with AA, four (no. 3,
14, 16, and 35) had no GPI-anchoring defect in the first study and remained free of a defect in the follow-up study in all blood-cell subsets (including erythrocytes). In one of these four patients (no. 3), Ham or sucrose tests were initially weakly positive and dissociated. He remained free of a GPI- anchoring defect on both leukocytes and erythrocytes, and 10 months after the initial study, had negative Ham and sucrose tests.
In seven patients, a GP1 defect on monocytes and granulo- cytes had been detected previously. In the follow-up study, the proportion of defective granulocytes and monocytes re-
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom

1358 GRISCELLI-BENNACEUR ET AL
A B m .I :~ " e:
P
e. rl -4- .
H I 2: 8
2: B p v-
+ *. 3 Fig 1. Double-staining flow-cytometry analysis .
on granulocytes using CD67 and CD13 monoclonal antibodies. (A) Defect in a patient with PNH (87%
7 CD67 defect). (B) Defect in a patient with AA (20% 1111111 I 1111111 I I r . l l l l r , , , , , m I I
4 " .l l l9 i a a l a m .I l l a cm-r-rzTc coG1-rzTc "' *'" CD67 defect).
mained roughly the same in six patients. A GPI-anchoring defect was detected on erythrocytes in five patients with positive Ham and sucrose tests. In one patient (no. l), who had a GPI-anchoring defect on leukocytes, no defect was observed on erythrocytes (Ham and sucrose tests were also negative) (Table 3). The seventh patient (no. 40) underwent allogeneic BMT. Seven and 9 months after transplant, flow- cytometry analysis demonstrated disappearance of the PNH clone.
In three patients (no. 22, 26, and 49) without a GPI-an- choring defect after the initial study, a defect was detected in the follow-up study. This defect was limited to leukocytes with persistent negative Ham and sucrose tests in patient no. 22. It was found in all cell subsets with Ham and sucrose tests, becoming positive in patient no. 26, and on erythro- cytes only (isolated reduced expression of the CD59 mole- cule), with persistent negative Ham and sucrose tests in pa- tient no. 49.
PNH patients. For the two patients with PNH, the GPI- defect on leukocytes was of the same order of magnitude as in the initial study. The defect on erythrocytes reached 35% and 75%, on average, for patients no. 44 and 5 1 , respectively.
Ham and Sucrose Tests: Correlation With Flow-Cytometry Analysis
blood-product transfusion. In all but five patients, a good correlation was observed between the positivity of Ham and sucrose tests and a GPI-linked molecule defect on granulo- cytes and monocytes (Table 2). In two patients with AA (no. 3 and 13) in whom the Ham or sucrose test was weakly positive and dissociated (Table 2), no PNH defect could be detected on granulocytes and monocytes by immunofluores- cence. One of these patients (no. 3) was recently reevaluated as part of the follow-up study (see previous section). In contrast, defective GP1 molecule expression was found on leukocytes in three patients with AA (no. I , 24, and 40) whose Ham and sucrose tests were negative. One patient (no. 1 ) was reevaluated in the follow-up study. Twelve months after the first study, we confirmed a GPI-anchoring defect on granulocytes and monocytes, while erythrocytes were not affected (see previous section).
Cytogenetic Studies
Cytogenetic analyses were performed in 15 cases ( 1 2 pa- tients with AA and three with PNH). Monosomy 7 was diagnosed in an AA patient who had no GP1 defect. One patient with PNH had a constitutional abnormality [Robert- sonian translocation t( 13; 14)].
Coagulation Tests
Ham and sucrose tests and flow-cytometry analysis were Proteins C and S, thrombomodulin, and antithrombin 111 performed on the same day in the absence of any recent levels were within the normal range in the group of patients
A B 0 .I
4
m m rl 0 m
0 0 *
0 m 4
U W L
B !? B
A: :f
Fig 2. Double-staining flow-cytometry analysis on monocytes using CD14 and CD13 monoclonal an- tibodies. (A) Defect in a patient with PNH (97% CD14 defect). (B) Defect in a patient with AA 120% CD14 defect). .l l
CDl4-FITC 18 CD14-PITC
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom

PNH-APLASTIC ANEMlA SYNDROME 1359
Table 3. Flow-Cytometry Analysis: Summary of the FoIIow-UP Study
% of Negative Cells Interval First Patient Ham Test/ (CD14/CD16-CD67/ StudyIFollow-Up
No. Diagnosis Sucrose Test CD55-CD58-CD591 Study (mol
1 A A -/- 16/6-6/0-0-0 12 4 A A -/( +) 18/5-5/0-0-34* 11 6 A A +I++ 40139-39121-48-47 11 8 A A (+M+) 35/51-55/11-11-37 11
10 AA + +/++ 93/92-92/33-58-88 12 12 AA +I+ NA/33-32122-18-80 12 22 AA -l- NA/37/30/0-0-0 10 26 AA (+){+ 35/12-1218-16-49 9 49 AA -/- 010-0/0-0~33* 12 44 PNH +/+ 90195-94134-35-34 13 51 PNH +/+ 90/88-88P5-73-68 6
Results of lytic tests are given in pluses; corresponding values in
* Protein reduced, rather than absent on abnormal cells. the percentage of lysis are described in Materials and Methods.
with PNH and in patients with AA (with or without PNH) (Table 4).
Study of PIG-A Gene Expression by Northern Blot Analysis
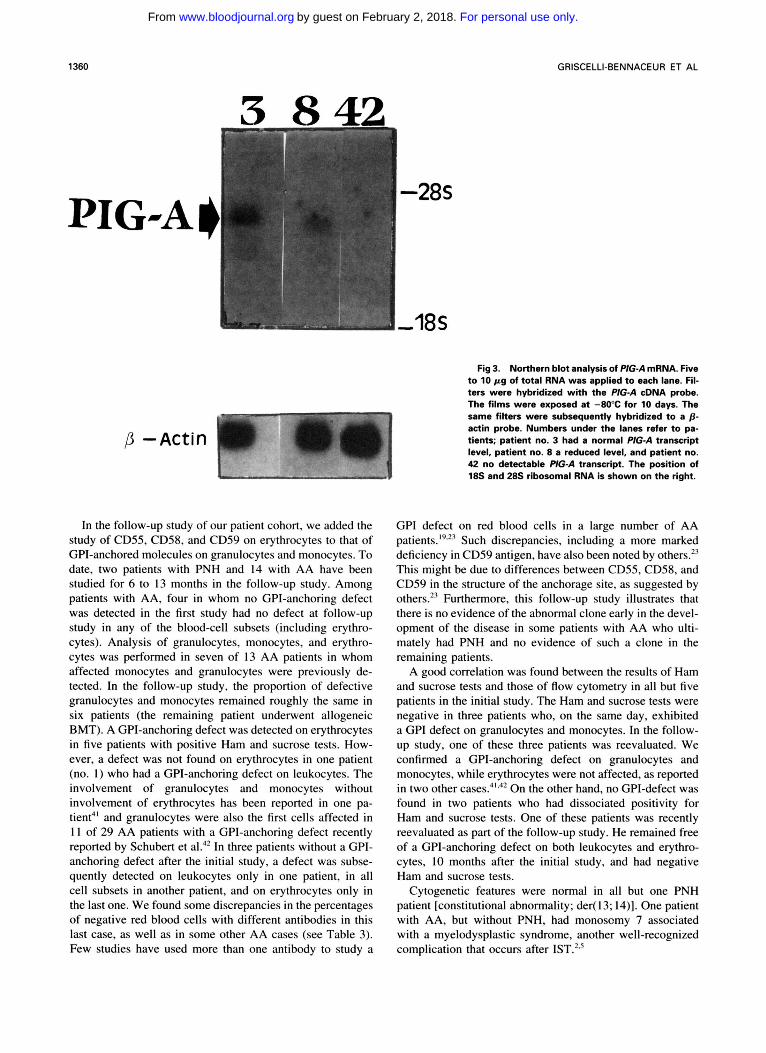
Sufficient RNA could be recovered from frozen PBMNC in only five of eight patients with PNH. In four of these patients, the P G A transcript was barely visible in two and undetectable in two (Table 2 and Figs 3 and 4). An abnor- mally small transcript was detected in the remaining patient (no. 47). The PIG-A transcript was analyzed in nine of 12 patients with AA in whom a GP1 defect was detected by flow cytometry. A normal-sized transcript was observed in all nine (Table 2 and Figs 3 and 4). The level of PIG-A gene expression was quantified by the PIG-A/@-actin gene ratio using densitometry, and was apparently reduced, since in both healthy blood donors and AA patients, the ratio was 1.93 arbitrary units (range, 1.10 to 2.42) and was 0.46 arbi- trary units (range, 0.25 to 0.8) in eight patients with AA in whom a GP1 defect was detected by flow cytometry.
DISCUSSION
AA and PNH are clinically related syndromes. During the course of PNH, pancytopenia is common and about one third of patients will die of bone marrow failure. Similarly, a fairly high proportion of patients with pancytopenia and marrow hypocellularity have either a positive sucrose hemolysis test or Ham test during the evolution of the di~ease.~' The overlap with AA is particularly striking among younger patients with
Table 4. Coagul
Patient Group
Protein C Activity (%)
Protein C Protein S Antigen (%l Total (%l
PNH, who much less frequently develop hemoglobinuria and more often have moderate to severe marrow failure, often leading to an initial diagnosis of AA:' In addition to their concurrent or sequential appearance in individual patients, PNH and AA share several pathophysiologic features. He- matopoietic progenitor numbers are markedly decreased in patients with AA and PNH, even when the marrow appears ~ e l l u l a r . ' ~ * ~ ~ However, the question as to whether these two disorders are two forms of the same disease is unres~lved. '~*l~ To search for a pathogenetic link between these two syn- dromes, we compared patients with PNH with those with AA who secondarily developed PNH after IST, using the results of Ham and sucrose tests at diagnosis as the classic diagnostic criteria.
All patients with PNH had a mixture of the classic clinical manifestations (hemolytic anemia, thrombosis, and AA), while only six of 12 AA patients who developed PNH had clinical symptoms (essentially hemolytic anemia). The other six patients had a purely laboratory-based diagnosis. The Basel group2 has also reported that four of their 13 patients who developed PNH after IST were diagnosed by laboratory tests while they were free of clinical manifestations.
During the past few years, flow-cytometry analysis has proved to be a powerful tool for the diagnosis of the GPI- linked protein defect in PNH. As in this report, the various cell lineages studied include granulocytes (using CD161 CD67), monocytes (using CD14), erythrocytes (using CD551 CD58/CD59), and lymphocytes (using CD48).'7-27 Although the nature of the primary disease (PNH or AA) is generally omitted from such reports, it seems that patients with PNH exhibit a pronounced GPI-linked protein d e f e ~ t . ~ ~ - ~ ~ . * ~ . * ' We found that 87.5% of monocytes and 84% of granulocytes had a GP1 defect in patients with PNH. In contrast, in 10 of 12 AA patients who secondarily developed PNH, the defect was less pronounced, affecting a mean of 27% and 20% of monocytes and granulocytes, respectively. In the remaining two patients, the GPI-linked protein defect reached the same level as that found in PNH patients (>90%). Such a quantita- tive difference in the extent of the defect in these two forms of the disease has also been noted by others.'8.20.26 The involvement of lymphocytes in the GPI-anchoring defect has been the subject of conflicting reports,18*20 with lymphocytes being affected in 11 of 12 patients in the study by Schubert et a]," compared with only two of 10 patients in the report by Van der Schoot et al.'' In our study, two of three patients with PNH and one of 12 patients who secondarily developed PNH exhibited a CD48 defect on lymphocytes. Further stud- ies are clearly warranted to determine whether both lympho- cytes and myeloid involvement (thus suggesting a stem-cell level defect) are more frequent in PNH.
lation Test Results
Protein S Free Form Antithrombin 111
(96) Thrombomodulin
(%) (ng/mL)
PNH 119.1 L 43.0 99.3 f 20.6 84.7 2 14.2 92.9 t 17 106 f 13.8 AA Controls 82.6 rt 17.2 83.4 It 13.5 88.9 2 10.8 89.9 f 22.8 87.2 2 18.2 35.9 f 24.5
29.3 % 71.7 88.5 rt 14.3 79.1 2 6.8 78.6 f 5.7 83.8 f 7.6 NA 46.8 f 26
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom
1360 GRISCELLI-BENNACEUR ET AL
PI G
Fig 3. Northern blot analysis of P/G-A mRNA. Five to 10 p g of total RNA was applied to each lane. RI- ters were hybridized with the PIG-A cDNA probe. The films were exposed at -80°C for 10 days. The same filters were subsequently hybridized to a p - actin probe. Numbers under the lanes refer to pa- tients; patient no. 3 had a normal PIG-A transcript level, patient no. 8 a reduced level, and patient no. 42 no detectable PlGA transcript. The position of 18s and 28s ribosomal RNA is shown on the right.
In the follow-up study of our patient cohort, we added the study of CD55, CD58, and CD59 on erythrocytes to that of GPI-anchored molecules on granulocytes and monocytes. To date, two patients with PNH and 14 with AA have been studied for 6 to 13 months in the follow-up study. Among patients with AA, four in whom no GPI-anchoring defect was detected in the first study had no defect at follow-up study in any of the blood-cell subsets (including erythro- cytes). Analysis of granulocytes, monocytes, and erythro- cytes was performed in seven of 13 AA patients in whom affected monocytes and granulocytes were previously de- tected. In the follow-up study, the proportion of defective granulocytes and monocytes remained roughly the same in six patients (the remaining patient underwent allogeneic BMT). A GPI-anchoring defect was detected on erythrocytes in five patients with positive Ham and sucrose tests. How- ever, a defect was not found on erythrocytes in one patient (no. 1) who had a GPI-anchoring defect on leukocytes. The involvement of granulocytes and monocytes without involvement of erythrocytes has been reported in one pa- tient4' and granulocytes were also the first cells affected in 11 of 29 AA patients with a GPI-anchoring defect recently reported by Schubert et a1.4' In three patients without a GPI- anchoring defect after the initial study, a defect was subse- quently detected on leukocytes only in one patient, in all cell subsets in another patient, and on erythrocytes only in the last one. We found some discrepancies in the percentages of negative red blood cells with different antibodies in this last case, as well as in some other AA cases (see Table 3). Few studies have used more than one antibody to study a
GP1 defect on red blood cells in a large number of AA patient^.'^.^' Such discrepancies, including a more marked deficiency in CD59 antigen, have also been noted by others2' This might be due to differences between CD55, CD58, and CD59 in the structure of the anchorage site, as suggested by others2? Furthermore, this follow-up study illustrates that there is no evidence of the abnormal clone early in the devel- opment of the disease in some patients with AA who ulti- mately had PNH and no evidence of such a clone in the remaining patients.
A good correlation was found between the results of Ham and sucrose tests and those of flow cytometry in all but five patients in the initial study. The Ham and sucrose tests were negative in three patients who, on the same day, exhibited a GP1 defect on granulocytes and monocytes. In the follow- up study, one of these three patients was reevaluated. We confirmed a GPI-anchoring defect on granulocytes and monocytes, while erythrocytes were not affected, as reported in two other cases."'.42 On the other hand, no GPI-defect was found in two patients who had dissociated positivity for Ham and sucrose tests. One of these patients was recently reevaluated as part of the follow-up study. He remained free of a GPI-anchoring defect on both leukocytes and erythro- cytes, 10 months after the initial study, and had negative Ham and sucrose tests.
Cytogenetic features were normal in all but one PNH patient [constitutional abnormality; der( 13; 14)]. One patient with AA, but without PNH, had monoscmy 7 associated with a myelodysplastic syndrome, another well-recognized complication that occurs after IST.'.'
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom

PNH-APLASTIC ANEMIA SYNDROME 1361
47 9 11 16 7 2 -28s
Fig 4. Northern blot analysis of PIG-A mRNA. Techniques used were as described in the text and legend of Fig 3. A nor- mal (patient no. 161, reduced (no. 11, 7, and 21 and markedly re- duced (no. 9) amount of PIG-A transcript was detected. An ab- normally small transcript was detected in patient no. 47.
The pathophysiologic factors involved in the occurrence of thrombosis in patients with PNH are not yet fully eluci- dated. A recent study showed that the lack of the receptor for urokinase-type plasminogen activator observed on PNH leukocytes may be related to the increased tendency toward thrombosis? A protein Uprotein S/thrombomodulin system impairment and antithrombin I11 deficiency are considered the main causes of thrombophilia. To test this hypothesis, we determined protein S , protein C, antithrombin 111, and thrombomodulin plasma levels in patients with PNH and AA. Proteins C and S , thrombomodulin, and antithrombin I11 levels were within the normal range in PNH patients and in those with AA (with or without PNH). Another hypothesis for the increased risk of thrombosis in PNH patients is through platelet activation. In this regard, it is interesting to note that it was recently demonstrated that PNH platelets with undetectable levels of CD59 antigen show increased sensitivity to activation by C5b-9 proteins compared with normal controls, resulting in markedly increased production of procoagulant vesicles." This may contribute, at least in part, to the tendency toward thrombosis in PNH patients.
PIG-A gene expression studied by Northern blot analysis showed that among five patients with PNH, from whom sufficient RNA could be recovered from PBMNC for analy- sis, the PIG-A transcript was barely visible in two, undetect- able in two, and abnormal in length in the fifth. In these five patients, a GPI-anchoring defect affected the majority of monocytes. Lymphocytes were studied in two of these five patients, and in these two patients, 70% and 84% of the lymphocytes had a GPI-anchoring defect. Thus, the mRNA extracted from PBMNC originated mostly from affected cells (at least in the two patients in whom both monocyte and lymphocyte populations could be studied). The fact that
A
the PIG-A transcript was barely visible in two cases and undetectable in two further cases may reflect to a consider- able extent, instability of the affected transcripts.3".32 Although Ware et a14" found a normal level of PIG-A transcript in four patients, Takeda et aI3' found barely detectable PIG-A gene expression in GPI-deficient lym- phoblastoid cell lines. Our fifth patient (no. 47) had an abnormally small transcript, similar to that described in a patient by Takeda et aL3" Recently, Bessler et a132 de- scribed three different PIG-A mRNA species in four cell lines deficient in GPI-linked proteins. The two smaller mRNA species (2.8 and 3.3 kb), which were not detected in our analyses, are probably present in a small amount and could probably only be detected in the poly(A)-en- riched RNA fraction." The exact molecular alterations (ie, point mutations or deletions) responsible for the reduction in the amount of full-length mRNA or for its abnormal length remain to be determined. Using reverse-transcribed amplification of the PIG-A transcripts and subsequent cloning in plasmids, the Osaka group further extended their sequencing studies in granulocytes isolated from five patients with PNH4' and, recently, Ware et a14" also dem- onstrated point mutations within the coding regions of the PIG-A gene in four patients with PNH.
In conclusion, the only significant difference we were able to establish in this study, between PNH arising in patients with previous AA and patients with de novo PNH, was the size of the PNH clone. Whether the PNH clone already exists in some AA patients at diagnosis but is undetectable by the methods used in this study remains to be determined by more sensitive techniques. Whether a PIG-A gene alteration is also the underlying genetic defect in otherwise typical AA also seems to warrant attention.
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom

1362 GRISCELLI-BENNACEUR ET AL
ACKNOWLEDGMENT
We thank Dr J. Takeda for the generous gift of the P G A cDNA probe; Drs D. Blanchard and F. Mami-Chouaib for the generous gifts of the CD55-59 and CD48 monoclonal antibodies, respectively; 0. Romagnt, V. Ctzard, and M.Y. Lisch for their help in blood sample collection; C. Lafon for excellent technical assistance in the Northern blot analysis; C. Glossek and C. Guyonnet for help in typing the manuscript; Y. Bretagne for data on Ham and sucrose tests; and L. Saint-Ange for careful review of English grammar and usage.
REFERENCES I . Nissen C: The pathophysiology of aplastic anemia. Semin
Hematol 28:313, 1991 2. Tichelli A, Gratwohl A, Wursch A, Nissen C, Speck B: Late
haematological complications in severe aplastic anaemia. Br J Haematol 69:413, 1989
3. De Planque MM, Kluin-Nelemans HC, Van Krieken HJM, Kluin P, Brand A, Beverstock GC, Willemze R, Van Rood JJ: Evolu- tion of acquired severe aplastic anaemia to myelodysplasia and sub- sequent leukemia in adults. Br J Haematol 70:55, 1988
4. DePlanque MM, Bacigalupo A, Wiirsch A, Hows JM, Devergie A, Frickhofen N, Brand A, Nissen C: Long-term follow-up of severe aplastic anaemia patients treated with anti-thymocyte globulin. Br J Haematol 73: 12 1, 1989
5 . Socit G, Henry-Amar M, Bacigalupo A, Hows J, Tichelli A, Ljungman P, McCann S , Frickhofen N, Van’t Veer-Korthof E, Gluckman E: Malignant tumors occurring after treatment of aplastic anemia. N Engl J Med 329:1152, 1993
6. Rosse WF: Paroxysmal nocturnal hemoglobinuria: The bio- chemical defects and the clinical syndrome. Blood 3:192, 1989
7. Rosse WF: Phosphatidylinositol-linked proteins and paroxys- mal nocturnal hemoglobinuria. Blood 75:1595, 1990
8. Rotoli B, Luzzatto L: Paroxysmal nocturnal hemoglobinuria. Semin Hematol 26:201, 1989
9. Josten KM, Tooze JA, Borthwick-Clarke C, Gordon-Smith EC, Rutherford TR: Acquired aplastic anemia and paroxysmal nocturnal hemoglobinuria: Studies on clonality. Blood 78:3162, 1991
IO. Young NS: The problem of clonality in aplastic anemia: Dr Dameshek‘s Riddle, restated. Blood 79:1385, 1992
1 1. Ham TH: Studies on destruction of red blood cells. 1. Chronic hemolytic anemia with paroxysmal nocturnal hemoglobinuria: inves- tigation of mechanism of hemolysis with observation on 5 cases. Arch Intern Med 64:1271, 1939
12. Hartman RC: The sugar-water test for paroxysmal nocturnal hemoglobinuria. N Engl J Med 275:155, 1966
13. Birgens HS, Hancke S , Rosenklint A, Hansen NE: Ultrasonic demonstration of clinical and subclinical hepatic venous thrombosis in paroxysmal nocturnal haemoglobinuria. Br J Haematol 64:585, 1986
14. Marsh JCW, Geary CG: Is aplastic anaemia a pre-leukaemic disorder? Br J Haematol 77:447, 1991
15. Nissen C, Gratwohl A, Speck B, Wursch A, Moser Y, Weis J: Acquired aplastic anaemia: A PNH-like disease? Br J Haematol 62:355, 1986
16. Rotoli B, Bessler M, Alfinito F, Del Vecchio L: Membrane proteins in paroxysmal nocturnal haemoglobinuria. Blood 7:75, 1993
17. Low M, Saltiel A: Structural and functional roles of glycosyl- phosphatidylinositol in membranes. Science 239:268, 1988
18. Schubert J, Uciechowski P, Delany P, Tischler HJ, Kolanus W, Schmidt R: The GPI-anchoring defect in NK lymphocytes of PNH patients. Blood 76:1181, 1990
19. Schubert .I, Alvarado M, Uciechowski P, Zielinska-Skowro- nek M, Freund M, Vogt H, Schmidt E: Diagnosis of paroxysmal
nocturnal hemoglobinuria using immunophenotyping of peripheral blood cells. Br J Haematol 79:487, 1991
20. Van der Schoot E, Huizinga T, Van’t Veer-Korthof E, Wij- mans R, Pinkster J, Van dem Borne A: Deficiency of glycosyl- phosphatidylinositol-linked membrane glycoproteins of leukocytes in paroxysmal nocturnal hemoglobinuria: Description of a new diag- nosis cytofluorometric assay. Blood 76:1853, 1990
21. Okuda K, Kanamaru A, Ueda E, Kitani T, Nagai K: Mem- brane expression of decay-accelerating factor on neutrophils from normal individuals and patients with paroxysmal nocturnal hemoglo- binuria. Blood 75: I 186, 1990
22. Mahoney JF, Urakaze M, Hall S , De Gasperi R, Chang HM, Sugiyama E, Warren CD, Borowitz M, Nicholson-Weller A, Rosse WF, Yeh ETH: Defective glycosylphosphatidylinositol anchor syn- thesis in paroxysmal nocturnal hemoglobinuria granulocytes. Blood 79: 1400, 1992
23. Hillmen P, Hows J , Luzzatto L: Two distinct patterns of glycosylphosphatidylinositol (GPI) linked protein deficiency in the red cells of patients with paroxysmal nocturnal haemoglobinuria. Br J Haematol 80:399, 1992
24. Nicholson-Weller A, Douglas M, Spicer D, Austen F: Defi- ciency of the complement regulatory protein, “decay-accelerating factor”, on membranes of granulocytes, monocytes, and platelets in paroxysmal nocturnal haemoglobinuria. N Engl J Med 312:1091, 1985
25. Bessler M, Fehr J: FcgRIII receptors (FcRIII) on granulo- cytes: A specific and sensitive diagnostic test for paroxysmal noctur- nal hemoglobinuria (PNH). Eur J Haematol 47:179, 1991
26. Bessler M, Hillmen P, Luzzatto L: Clonal origin of abnormal granulocytes in paroxysmal nocturnal hemoglobinuria. Blood 80:844, 1992
27. Nagakura S , Nakakuma H, Horikawa K, Hidaka M, Kagimoto T, Kawakita M, Tomita M, Takatsuki K: Expression of decay-accel- erating factor and CD59 in lymphocyte subsets of healthy individuals and paroxysmal nocturnal hemoglobinuria patients. Am J Hematol 43:14, 1993
28. Hyman R: Somatic genetic analysis of the expression of cell surface molecules. Trends Genet 4:5, 1988
29. Takahashi M, Takeda J, Hirose S , Hyman R, Inoue N, Miyata T, Ueda E, Kitani T, Medof M, Kinoshita T: Deficient biosynthesis of N-acetylglucosaminyl-phosphatidylinositol, the first intermediate of glycosyl phosphatidylinositol anchor biosynthesis in cell lines established from patients with paroxysmal nocturnal hemoglobin- uria. J Exp Med 177517, 1993
30. Takeda J, Miyata T, Kawagoe K, Lida Y, Endo Y, Fujita T, Takahashi M, Kitani T, Kinoshita T: Deficiency of the GP1 anchor caused by a somatic mutation of the PIG-A gene in paroxysmal nocturnal hemoglobinuria. Cell 73:703, 1993
31. Miyata T, Takeda J, Lida Y, Yamada N, Inoue N. Takahashi M, Maeda K, Kitani T, Kinoshita T: The cloning of PIG-A, a compo- nent in the early step of GPI-anchor biosynthesis. Science 259: 1318, 1993
32. Bessler M, Mason P, Hillmen P, Miyata T, Yamada N, Takeda J, Luzzatto L, Kinoshita T: Paroxysmal nocturnal hemoglobinuria is caused by somatic mutations in the PIG-A gene. EMBO J 13: 110, 1994
33. Kamitani T, Chang HM, Rollins C, Waneck GL, Yeh ETH: Correction of the class H defect in glycosyl phosphatidylinositol anchor biosynthesis in Ltk- cells by a human cDNA clone. J Biol Chem 268:20733, 1993
34. Camitta B, Thomas ED, Nathan DG, Santos G, Gordon-Smith EC, Gale RP, Rappeport JM, Storb R: Severe aplastic anemia: A prospective study on the effect of early marrow transplantation on acute mortality. Blood 48:63, 1976
35. Mitelman F (ed): Guidelines for Cancer Cytogenetics: Sup-
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom

PNH-APLASTIC ANEMIA SYNDROME 1363
plement to an International System for Human Cytogenetic Nomen- clature. Basel, Switzerland, Karger, 1991
36. Sandberg AA (ed): The Chromosome in Human Cancer and Leukemia. New York, NY, Elsevier, 1990
37. Amiral J, Adalbert B, Adam M Application of enzyme immu- noassays to coagulation testing. Clin Chem 301512, 1984
38. Fared J: New Methods in Hemostatic Testing, in Fareed J, Messmore HL, Fenton I1 JW, Brinkhouse KM (eds): Perspectives in Hemostasis. New York, NY, Pergamon, 1981, p 310
39. Young NS, Alter BP: Bone marrow failure secondary to ge- netic injury: radiation; myelodysplasia and related syndrome, in Young NS, Alter BP (eds): Aplastic Anemia Acquired and Inherited. Philadelphia, PA, Saunders, 1993, p 46
40. Ware RE, Hall SE, Rosse WF: Paroxysmal nocturnal hemoglo- binuria in childhood and adolescence. N Engl J Med 325:9!91, 1991
41. Fujioka S, Yamada T: Decay accelerating factor and CD59 expression in peripheral blood cells in aplastic anaemia and report of a case of paroxysmal nocturnal haemoglobinuria secondary to aplastic anaemia. Br J Haematol 83:660, 1993
42. Schubert J, Vogt HG, Zielinska M, Freund M, Kaltwasser JP, Hoelzer D, Schmidt RE: Development of the glycosylphosphatidyl- inositol-anchoring defect characteristic for paroxysmal nocturnal he- moglobinuria in patients with aplastic anemia. Blood 83:2323, 1994
43. Ploug M, Plesner T, Rome E, Ellis V, Hoyer-Hansen G, Hanse N, Dan0 K: The receptor for urokinase-type plasminogen activator is deficient on peripheral blood leukocytes in patients with paroxysmal nocturnal hemoglobinuria. Blood 79: 1447, 1992 44. Wiedmer T, Hall SE, Ortel TL, Kane WH, Rosse WF, Sims
PJ: Complement-induced vesiculation and exposure of membrane prothrombinase sites in platelets of paroxysmal nocturnal hemoglo- binuria. Blood 82:1192, 1993
45. Miyata T, Yamada N, Iida Y, Nishimura J, Takeda J, Kitani T, Kinoshita T: Abnormalities of PIG-A transcripts in granulocytes from patients with paroxysmal nocturnal hemoglobinuria. N Engl J Med 330:249, 1994
46. Ware R E , Rosse WF, Howard TA: Mutations within the PIG- A gene in patients with paroxysmal nocturnal hemoglobinuria. Blood 83:2418, 1994
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom

1995 85: 1354-1363
Carosella, F Sigaux and G SocieA Griscelli-Bennaceur, E Gluckman, ML Scrobohaci, P Jonveaux, T Vu, A Bazarbachi, ED for a pathogenetic linkAplastic anemia and paroxysmal nocturnal hemoglobinuria: search
http://www.bloodjournal.org/content/85/5/1354.full.htmlUpdated information and services can be found at:
Articles on similar topics can be found in the following Blood collections
http://www.bloodjournal.org/site/misc/rights.xhtml#repub_requestsInformation about reproducing this article in parts or in its entirety may be found online at:
http://www.bloodjournal.org/site/misc/rights.xhtml#reprintsInformation about ordering reprints may be found online at:
http://www.bloodjournal.org/site/subscriptions/index.xhtmlInformation about subscriptions and ASH membership may be found online at:
Copyright 2011 by The American Society of Hematology; all rights reserved.Society of Hematology, 2021 L St, NW, Suite 900, Washington DC 20036.Blood (print ISSN 0006-4971, online ISSN 1528-0020), is published weekly by the American
For personal use only.on February 2, 2018. by guest www.bloodjournal.orgFrom





























