Embed Size (px)
Citation preview

ANTHROPOLOGICAL GENETIC ANALYSIS OF HUMAN DISEASE FROM EVOLUTIONARY, POPULATION, AND CLINICAL PERSPECTIVES
By
REBECCA R. GRAY
A DISSERTATION PRESENTED TO THE GRADUATE SCHOOL OF THE UNIVERSITY OF FLORIDA IN PARTIAL FULFILLMENT
OF THE REQUIREMENTS FOR THE DEGREE OF DOCTOR OF PHILOSOPHY
UNIVERSITY OF FLORIDA
2008
1

© 2008 Rebecca R. Gray
2

ACKNOWLEDGMENTS
Foremost, I thank my Ph.D. advisor Dr. Connie Mulligan for her mentorship and affording
me the opportunity to conduct this research. I thank the members of my Ph.D. committee;
specifically I thank Dr. Maureen Goodenow for her training and expertise; I thank Dr. John
Krigbaum and Dr. Marta Wayne for their early guidance; and I thank Dr. David Reed for his
helpful analytical advice. I thank Dr. Marco Salemi for his instruction and involving me in
additional projects. I thank my collaborators on the Treponema project at the University of
Washington, including Dr. Sheila Lukehart and Dr. Arturo Centurion. I thank my collaborators at
the National Institutes of Health including Dr. Jordi Clarimon, Dr. Andrew Singleton, Dr. David
Goldstein, and Dr. Mary-Anne Enoch. I thank Dr. Grace Aldrovandi at the Children’s Hospital in
Los Angeles who collaborated on the breastmilk project. I thank my undergraduate advisor Dr.
Carole Counihan for my initial and continued enthusiasm for anthropological research. I
appreciate the suggestions and feedback on these projects from the members of Drs. Mulligan
and Goodenow’s lab. I acknowledge the undergraduate students who assisted me on these
projects, including Danielle Muchnick and Lindsay Williams. I profess deep gratitude to the
Native American individuals and the Zambian women who participated in the studies which are
part of this dissertation. Finally, I thank my parents for the intellectual foundation they provided,
and my husband for his encouragement and profound patience.
3

TABLE OF CONTENTS page
ACKNOWLEDGMENTS ...............................................................................................................3
LIST OF TABLES...........................................................................................................................7
LIST OF FIGURES .........................................................................................................................8
ABSTRACT...................................................................................................................................10
CHAPTER
1 INTRODUCTION ..................................................................................................................12
2 MOLECULAR EVOLUTION OF THE TPRC, D, I, K, G, AND J GENES IN THE PATHOGENIC GENUS Treponema .....................................................................................25
Introduction.............................................................................................................................25 Materials and Methods ...........................................................................................................29
Treponemal Strains and tpr Sequencing..........................................................................29 Evolutionary Analysis of Sequences ...............................................................................30 Phylogenetic Analyses.....................................................................................................30 Detection of Recombination............................................................................................31
Results.....................................................................................................................................32 Phylogenetic Analyses.....................................................................................................32
Phylogenetic analyses of subfamily I.......................................................................33 Phylogenetic analyses of subfamily II......................................................................36 Phylogenetic analyses of subfamily III ....................................................................37
Statistical Tests for Recombination.................................................................................38 Analysis of Nucleotide Diversity and Composition........................................................40
Discussion...............................................................................................................................41
3 LINKAGE DISEQUILIBRIUM AND ASSOCIATION ANALYSIS OF ALPHA SYNUCLEIN (SNCA) AND ALCOHOL AND DRUG DEPENDENCE IN TWO AMERICAN INDIAN POPULATIONS ...............................................................................59
Introduction.............................................................................................................................59 Materials and Methods ...........................................................................................................61
Sampling Strategy ...........................................................................................................61 Testing Instruments, Interviews, and Psychiatric Diagnoses ..........................................62 Genotyping ......................................................................................................................63 Statistical Analysis ..........................................................................................................63
Results.....................................................................................................................................65 Discussion...............................................................................................................................68
4

4 LACK OF ASSOCIATION BETWEEN ADH/ALDH MARKERS AND SUBSTANCE USE DISORDER IN NATIVE AMERICAN POPULATION ..............................................75
Introduction.............................................................................................................................75 Materials and Methods ...........................................................................................................78
Samples............................................................................................................................78 Testing Instruments, Interviews, and Psychiatric Diagnoses ..........................................78 Genotyping ......................................................................................................................79 Statistical Analysis ..........................................................................................................79
Results.....................................................................................................................................80 Discussion...............................................................................................................................82
5 DYNAMIC AND DISTINCT EVOLUTION OF HIV-1 IN BREASTMILK OVER TWO YEARS POST-PARTUM ............................................................................................89
Introduction.............................................................................................................................89 Background.............................................................................................................................91
Human Immunodeficiency Virus Type 1 Infection.........................................................91 Stages of Breastmilk Production .....................................................................................93 Cellular Composition of Breastmilk................................................................................95 Compartmentalization of Breastmilk Virus.....................................................................96 Risk of Transmission via Breast-feeding ........................................................................98 Our Study.......................................................................................................................102
Materials and Methods .........................................................................................................104 Subject ...........................................................................................................................104 Viral Isolation, Amplification, and Sequencing ............................................................104 Sequence Analysis and Recombination.........................................................................105 Phylogenetic Analyses...................................................................................................106
Branch Selection Analysis .....................................................................................108 Compartmentalization ............................................................................................108
Results...................................................................................................................................109 Subtype Analysis ...........................................................................................................109 Sequence Analysis.........................................................................................................109
Variable regions 1 and 2 sequence analysis ...........................................................109 Variable region 3 loop analysis ..............................................................................111
Recombination Analysis................................................................................................112 Phylogenetic Analyses...................................................................................................113
Bayesian tip-date phylogeny ..................................................................................113 Rooting the phylogeny ...........................................................................................116 Branch selection analysis .......................................................................................117 Inclusion of breastmilk month 1 sequences ...........................................................118
Migration Analysis ........................................................................................................119 Discussion.............................................................................................................................119
6 CONCLUSION.....................................................................................................................139
APPENDIX
5

A LIST OF QUESTIONS FOR SUBSTANCE ABUSE CATEGORIZATION .....................145
LIST OF REFERENCES.............................................................................................................147
BIOGRAPHICAL SKETCH .......................................................................................................172
6

LIST OF TABLES
page Table 2-1. Treponema isolates used in this study ..........................................................................50
Table 2-2. T. pallidum primers used in this study..........................................................................51
Table 2-3. Polymorphism at the tprC and tprD loci among pathogenic treponemes. ...................52
Table 2-4. Recombinant regions identified by RDP2....................................................................53
Table 2-6. Average GC content at combined 1st + 2nd (GC1+2) and 3rd codon (GC3) positions .............................................................................................................................54
Table 3-1. Demographic and phenotypic characterstics of southwest (SW) and plains populations.........................................................................................................................70
Table 4-1. Loci, primers, cycling conditions and restriction enzymes for 12 loci studied. ...........85
Table 4-2. Phenotypic characteristics of the dataset. ....................................................................86
Table 4-3. Haplotype frequencies and p-value for comparisons of cases vs. controls. ................86
Table 4-4. Chi-squared and regression p-values for genotype and allele associations for each marker........................................................................................................................87
Table 5-1. Number of sequences generated for each tissue.........................................................125
Table 5-2. Sequence characteristics of V1 and V2. .....................................................................125
Table 5-3. Combination of V1 and V2 haplotypes. .....................................................................126
Table 5-4. Hudson test for population structure. .........................................................................126
Table 5-5. Number of putative recombinant clones.....................................................................127
Table 5-6. Marginal likelihoods for models used in the Bayesian analysis.................................127
7

LIST OF FIGURES
Figure page Figure 1-1. Model for studying human diseases............................................................................24
Figure 2-1. Unrooted ML phylogenies of multiple tpr genes........................................................55
Figure 2-2. ML phylogenies for tprD, C, and I. ............................................................................56
Figure 2-3. ML phylogeny of tprG and J.......................................................................................57
Figure 2-4. ML phylogeny of tprK. ...............................................................................................58
Figure 3-1. Relative positions of single nucleotide polymorphisms assessed in α-synuclein (SNCA) gene.......................................................................................................................72
Figure 3-2. Single-marker analyses representing p values for each marker on a logarithmic scale....................................................................................................................................73
Figure 3-3. Allelic distribution of the NACP-REP1 microsatellite repeats...................................74
Figure 4-1. Linkage disequilbrium of markers assessed in the ADH gene family. .......................88
Figure 5-1. Sampling times and tissues. ......................................................................................128
Figure 5-2. Neighbor-joining phylogeny of all subtypes in group M plus this patient. ..............128
Figure 5-3. Haplotype analysis of V1. .........................................................................................129
Figure 5-4. Haplotype analysis of V3. .........................................................................................130
Figure 5-5. Recombination alignments........................................................................................131
Figure 5-6. Network of breast milk sequences from week 1. ......................................................131
Figure 5-7. Bayesian consensus phylogeny for C2V5 (A). .........................................................132
Figure 5-8. Bayesian consensus phylogeny for C2V5 (B). .........................................................133
Figure 5-9. Best-rooted maximum likelihood phylogeny............................................................134
Figure 5-10. Bayesian consensus phylogeny for the C2V5 (A) dataset with branches under significant selection. ........................................................................................................135
Figure 5-11. Bayesian consensus phylogeny with breast milk month 1 sequences.....................136
8

Figure. 5-12. Migration analysis for two tissues..........................................................................137
Figure 5-13. Migration analysis for three tissues.........................................................................138
9

Abstract of Dissertation Presented to the Graduate School of the University of Florida in Partial Fulfillment of the Requirements for the Degree of Doctor of Philosophy
ANTHROPOLOGICAL GENETIC ANALYSIS OF HUMAN DISEASE FROM EVOLUTIONARY, POPULATION, AND CLINICAL PERSPECTIVES
By
Rebecca R. Gray
May 2008
Chair: Connie Mulligan Major: Anthropology
In this dissertation, I used genetic data from both humans and pathogens to explore the
evolution and etiology of three diseases from temporally distinct perspectives. I employed an
evolutionary framework to address the the origin of syphilis, a population perspective to
determine genetic components contributing to alcoholism in Native Americans, and a clinical
perspective to study factors relating to the transmission of HIV-1 via breastfeeding. In the first
study, I used sequence data from six genes from three subspecies of Treponema pallidum, the
spirochetes that cause venereal syphilis, yaws, and endemic syphilis in humans, as well as two
other Treponema species, to determine their evolutionary origin and relationships using
phylogenetic and population genetic analyses. My data discriminate between key components of
several of the leading theories of treponemal evolution, and provide new loci that are distinct
among the treponemes and can be used for diagnosis. Second, I genotyped ~1000 Native
American individuals for markers in the alpha-synuclein gene (SNCA) and used sequence data
from ~400 Native Americans from the alcohol dehydrogenase gene (ADH) and the aldehyde
dehydrogenase gene (ALDH) to test for an association with substance abuse in these populations.
I used both dichotomous and continuous measures of addition and several statistical tests to
determine association. Despite the high power of the study, no significant association was
10

detected. This may be the result of the past evolutionary history of Native Americans, who
experienced a severe genetic bottleneck during the migration from Asia and may have lost
variants that have been previously associated with substance abuse. I concluded that a focus on
environmental causes and solutions may be most appropriate in these populations. Finally, I
sequenced and analyzed the env gene of the human immunodeficiency virus type-1 (HIV-1) in
breast milk and blood plasma from an HIV-1 positive woman who transmitted the virus to her
infant via breastfeeding. This was the first longitudinal study of HIV-1 in breast milk, and major
findings included the distinctiveness of the virus in milk during the first month post-partum, the
compartmentalization of the virus over time, and the dynamic evolutionary pattern of the virus in
the milk. These results provided information about the biological mechanism responsible for
differential transmission risks associated with various modes of breastfeeding.
Genetic anthropologists are equipped with the analytical tools to study the biological
mechanisms of diseases and to incorporate information about the relevant underlying population
structure and evolutionary history. This unique perspective allows genetic anthropologists to
provide comprehensive clinical and policy recommendations based on genetic data. Finally, the
multi-disciplinary approach employed by anthropologists can be valuable in ensuring that
resulting applications of the data are culturally appropriate and provide maximum health benefits
to communities in need.
11

CHAPTER 1
INTRODUCTION
Disease has been a major component of the human experience for the past 10,000 years,
and likely long before that time as well (Cockburn 1971; Omran 1971; Armelagos and Dewey
1975; Cohen and Armelagos 1984; Barrett et al. 1998). Human health and diseases are studied in
a wide range of fields, including anthropology, biology and medicine. Anthropologists in
particular pride themselves on the holistic nature of their discipline, which not only incorporates
incredibly diverse research, but also encourages interdisciplinary communicastion and
interpretation. Dialogue between subfields within anthropology and across disciplines such as
medicine and public health allows for a more comprehensive and cross-cultural perspective on
the nature of disease. Anthropological genetics is a subfield of biological anthropology and
applies which uses genetic data and evolutionary concepts to address anthropological questions,
including the nature of human and non-human primate relationships (Krings et al. 1997; Krings
et al. 1999; Krings et al. 2000; Relethford 2001; Lalueza-Fox et al. 2005; Caramelli et al. 2006;
Plagnol and Wall 2006; Krause et al. 2007), the routes and timing of human migrations around
the world (i.e. Cann, Stoneking, and Wilson 1987; Kolman, Sambuughin, and Bermingham
1996; Quintana-Murci et al. 1999; Macaulay et al. 2005; Ramachandran et al. 2005), and the
demographic forces that have shaped human history (i.e. Harpending et al. 1993; Harpending
1994; Sherry et al. 1994; Harpending et al. 1998; Harpending and Rogers 2000). The impact of
disease on human genetic diversity is often addressed as well, in part because alleles affecting
and affected by diseases are often population specific and provide information about the
questions posed above. In addition, many anthropologists are interested in determining the
relative contribution of genetics to the etiology and severity of human disease.
12

In this dissertation, I use genetic data from both humans and pathogens to explore the
evolution and etiology of one complex disease and two infectious diseases from temporally
distinct perspectives. I have adapted a model that incorporates three major perspectives
(evolutionary, population, and clinical) from which the anthropological study of the genetics of
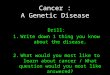
human disease can be approached (Figure 1-1). Although the original model was applied to
infectious disease (Quintana-Murci et al. 2007), I have broadened the model to include complex
disease as well. This modification provides a temporal framework for considering genetic
diversity and disease, which enables a more holistic treatment of the evolutionary, demographic,
and cultural forces that are operating on, and in concert with, genetic variability. I used an
evolutionary perspective to address the origin of syphilis, a population perspective to determine
genetic components contributing to alcoholism in Native Americans, and a clinical perspective to
study factors relating to the transmission of HIV via breastfeeding.
The evolutionary perspective typically incorporates the greatest genetic variation, because
the questions addressed in this framework are often rooted in the distant past. For example,
constant exposure to pathogens has shaped the human genome (Nielsen et al. 2007), either
through negative selection (Schwartz et al. 1995; Diaz et al. 2000; Hugot et al. 2001) or
balancing selection (Schroeder, Gaughan, and Swift 1995; Allen et al. 1997; Verrelli et al. 2002).
An intriguing example is the chemokine receptor CCR5 locus, at which homozygosity for the
delta32 mutation confers resistance to the human immunodeficiency virus type 1 (HIV-1)
infection (Samson et al. 1996). Under the assumption of neutrality, the high frequency (up to
10%) of the variant in European populations would suggest that the allele arose over 100ka
(Stephens et al. 1998b; Galvani and Novembre 2005). However, using linkage disequilibrium
and geographic structure analyses, the origin of the allele has been estimated ca.1,000 years ago,
13

which suggests that strong selection has driven the allele to its current high frequency (Libert et
al. 1998; Stephens et al. 1998b; Lucotte 2001). Clearly HIV-1, which only entered the human
population less than one hundred years ago (Sharp et al. 2001), could not have been the selective
force. However, smallpox, which is phenotypically similar to HIV-1 and has caused high rates of
mortality episodically over the past millennium, may have been the selective force (Galvani and
Slatkin 2003; Galvani and Novembre 2005). For comparison, the nucleotide diversity at the
CCR5 gene was compared between humans and chimpanzees, which are subject to a simian
immunodeficiency virus (SIVcpz) similar to HIV-1 but much less pathogenic. An excess of rare
variants was found in the chimpanzee gene, suggesting that the locus was influenced by a
selective sweep (Wooding et al. 2005). CCR5 was much more diverse in humans and
characterized by an excess of common variants, suggesting balancing selection (Wooding et al.
2005). These studies of the CCR5 gene demonstrate the global nature of the evolutionary
perspective, which considers comprehensive human genetic variation and the impact on human
and nonhuman primate genomes from past pathogen experiences. In chapter two, I used genetic
information from pathogens themselves to address evolutionary questions of anthropological
interest, such as where and when in human history veneareal syphilis evolved. This project
elucidates an important evolutionary mechanism of emerging pathogens, gene conversion, which
may have a significant impact on our approach to treatment and vaccination.
The second stage of the model is the population perspective, which considers the genetic,
demographic and cultural influences that shape the distribution of diseases between and within
geographic and ethnic groups. Disease may differentially affect populations either because
particular disease-causing variants may exist at higher frequencies in populations due to
demographic history, or because environmental forces within a population may exacerbate the
14

effect of variants predisposing a disease (Schork 1997). One well studied example is the extreme
difference in diabetes prevalence between different ethnic groups (Fujimoto 1996) which can
range from over 50% in the Native American Pima (Knowler et al. 1990) and in Pacific Islanders
(Amos, McCarty, and Zimmet 1997; McCarthy and Zimmet 2001) to a fraction of that
prevalence in European populations with the same risk factors (West 1974; Young et al. 2000).
The “thrifty-gene” hypothesis proposed that repeated exposure to famine in certain hunter-
gatherers led to the selection of genes which promote storage of fat; however, in modern times,
an over-abundance of food has led to the high prevalence of diabetes and metabolic disorders
(Neel 1962; Neel 1999), which is especially exacerbated in non-Western populations that have
possibly had less time to adapt to changing conditions (Neel 1982). Although this hypothesis
helped to explain Native American rates of diabetes (Johnson and McNutt 1964; Doeblin, Evans,
and Ingall 1969; Wise 1976) and continues to be discussed (Benyshek and Watson 2006;
Paradies, Montoya, and Fullerton 2007), numerous objections have been raised, including the
ethnographic validity of the premise that hunter-gatherers experience more famines than
agriculturalists (Dirks 1993; Benyshek and Watson 2006), the importance of the fetal
environmental component (Hales and Barker 1992; Barker et al. 1993; Hales and Barker 2001;
Lindsay and Bennett 2001; Ordovas, Pittas, and Greenberg 2003), and the reductionist approach
to human variability encompassed by a typological (race-based) approach (Fee 2006; Paradies,
Montoya, and Fullerton 2007), among other points. However, the longevity of the hypothesis
demonstrates both the attraction of an anthropological theory that incorporates evolution and
culture, as well as the complications inherent in the etiology of complex diseases. In chapters
three and four, I investigated the potential association between genetic data markers and
substance abuse in Native American population using the population perspective. The past
15

genetic history of this population may have contributed to the non-significance of the genetic
data, and I ultimately concluded that a focus on environmental causes and solutions might be
most appropriate in these populations.
Finally, the clinical perspective focuses on individuals involved in experimental and
intervention studies usually run by medical practitioners. This perspective typically measures
either the response to an intervention or the frequency with which healthy individuals succumb
to a particular disease over time. Studies from the clinical perspective often do not explicitly
account for genetic and cultural diversity, which risks misinterpretation of results due to the
underlying population genetic stratification and/or cultural influences that may impact the
outcome of such trials. For example, the majority of drug trial studies for HIV-1 have been
conducted in the developed world (Perrin, Kaiser, and Yerly 2003). Host genetics, such as the
human leukocyte antigen genes (HLA) are certainly involved in HIV-1 infection (Moore et al.
2002; O'Brien and Nelson 2004; Fellay et al. 2007; Brass et al. 2008), which are differentially
distributed among geographic groups (Cavalli-Sforza, Menozzi, and Piazza 1994; Monsalve,
Helgason, and Devine 1999; Blanco-Gelaz et al. 2001; Cao et al. 2004; Prugnolle et al. 2005). If
aspects of host genetics affect the efficacy of drug therapy or vaccines, then only incorporating a
subset of the total human genetic variation in these trials can lead researchers to misinterpret the
value of their discoveries, since treatments may not have the same efficacy in every human
group. Furthermore, the need for such drugs is much greater in developing countries than in the
West, and ignoring the particular genetic and cultural aspects of these populations hinders the
development of effective treatments. Another potential concern with clinical studies is the
generalized use of race, which is often used as a quick proxy by the medical community to
represent perceived differences in genetic ancestry and cultural lifestyle, when in fact the factors
16

underlying a person’s genetic ancestry and choices are much more complex (Duster 2007;
Hoover 2007). For example, in a controversial decision by the FDA, approval was granted for
the drug to be marketed towards African-Americans (Carmody and Anderson 2007; Yancy et al.
2007). Some have argued that the identification of the efficacy in African-Americans but not
Causasians was prospective and questionable (Bibbins-Domingo and Fernandez 2007; Duster
2007), although others suggest that acknowledging the interplay between human genomic
variation and pharmacogenomics may improve drug development and global health care (Seguin
et al. 2008). Lastly, the ethics of clinical studies can be questionable when indigenous
populations are used as study subjects who may not have the expertise to fully give their
voluntary informed consent, and who may receive no benefit from their participation. The
expertise of anthropologists is sorely needed in the clinical realm to advise, plan and interpret
studies and data that make use of clinical trials so that maximum benefit for the eventual
recipients of the intervention can be achieved. In chapter five, I use a clinical perspective to
investigate potential molecular mechanisms involved in transmission of HIV-1 via breastfeeding.
I believe that current recommendations about breastfeeding by HIV-1 positive women in the
developing world should both account for the difficulties inherent in the practice and its
cessation for women and the infants, as well as ensure that all aspects of the guidelines are
scientifically sound.
In this dissertation, I chose to study three diseases affecting humans corresponding to the
three perspectives outlined above. I used genetic variation from both the pathogen itself and from
humans to address anthropological, evolutionary, public health, and medical questions. I used an
evolutionary framework to address the the origin of syphilis, a population perspective to
determine genetic components contributing to alcoholism in Native Americans, and a clinical
17

perspective to study factors relating to the transmission of HIV-1 via breastfeeding. Thus, my
results have broad relevance not only to a range of anthropological questions, such as the origin
of venereal syphilis, but can also be translated into clinical significance and inform health
policies.
In chapter two, I examined the evolution of three human treponemes: Treponema pallidum
subsp. pallidum, which is the etiological agent of venereal syphilis, T.p. subsp. pertenue, which
causes yaws, and T.p. subsp. endemicum, which causes endemic syphilis. Previous knowledge of
these diseases has come primarily from archaeological and historical evidence; however it is
difficult to discern the three diseases in the archeological record because the bone pathologies
caused by the three diseases are similar, and a major diagnostic criterion is therefore the
frequency and distribution of treponemal pathology among skeletons at burial sites and the
anatomical distribution of lesions (reviewed in (Powell and Cook 2005). Even the diagnosis of
contemporary samples is difficult because the clinical manifestations are similar and there is a
dearth of distinct molecular markers defining the three diseases (Centurion-Lara et al. 2006).
Several prominent hypotheses have been advanced describing the evolution of the treponemes
(Baker and Armelagos 1988; Powell and Cook 2005). Rothschild (2003) proposed that yaws (T.
p. subsp. pertenue) was the most ancestral of the three T. pallidum subspecies and was present at
least as far back as the origin of modern humans in Africa, and the other two subspecies each
derived from yaws, with T. p. subsp. pallidum evolving in the New World no more than ~2000
years ago (Rothschild 2003). A New World origin of T. p. subsp. pallidum is central to the
original Columbian hypothesis that suggested venereal syphilis was brought to Europe by
Columbus’ crews returning from the New World (Crosby 1969). An alternative Columbian
hypothesis was advanced by Baker and Armelagos (1988) that suggested venereal syphilis
18

evolved very rapidly during Columbus’ return voyage from the New World from a non-venereal
treponeme and was subsequently introduced to Europe (molecular data supporting this view were
published recently, (Harper et al. 2008) as well as a critical review (Mulligan, Norris, and
Lukehart 2008) and both attracted astonishingly widespread interest among the general public).
In contrast, the Pre-Columbian hypothesis suggests that treponemal diseases, including venereal
syphilis, existed in the Old World prior to Columbus’ voyages but were diagnosed incorrectly.
For example, pinta was the original form present throughout the world during the Pleistocene,
followed by the evolution of yaws (12,000 years ago), then endemic syphilis (9,000 years ago)
and, finally, venereal syphilis (5,000 years ago) (Hackett 1963). Lastly, the Unitarian hypothesis
suggests that venereal syphilis, endemic syphilis, yaws, and pinta are not in fact distinct diseases,
but rather are environmentally determined manifestations of the same disease (Hudson 1965).
My goal was to use molecular genetic data sampled from contemporary strains of each of the
three main treponemes (no molecular data exist for T. carateum that causes pinta), as well as two
outgroup species, to determine the support for any of these hypotheses (Chapter 2, Gray et al.
2006). This was the first phylogenetic study of the treponemes, and it provided valuable
information on the possible evolutionary scenarios of these pathogens. Furthermore, I was able
to establish particular alleles that are specific to each of the three subspecies that could be used in
future clinical investigations to aid in diagnosis. Finally, I provided new data that suggests
treponemal genome evolution has been driven by recombination, specifically gene conversion,
much more often than was previously known or predicted.
In the second study (chapters three and four), I used a population perspective to study
alcoholism in Native Americans. This group experiences alcohol related deaths at more than five
times the rate of the general United States population (IHS 2006) and are twice as likely to die of
19

chronic liver disease than Caucasians (CDC 2006a). The possible cultural reasons for this
disparity include high rates of poverty and unemployment, lack of access to health care, and
overall poor health (IHS 2006). However, high rates of alcoholism in Native Americans are
surprising in light of the research that shows ancestral Asian populations have a low level of
alcoholism, most likely mediated by a very high frequency of two alleles at the two main genes
involved in alcohol metabolism (alcohol dehydrogenase gene [ADH] and aldehyde
dehydrogenase [ALDH]). These alleles slow the body’s metabolism of alcohol resulting in toxic
accumulation of acetaldehyde that produces an intensely uncomfortable sensation, i.e. flushing
response, that ultimately protects against alcoholism through the behavioral response of
consuming less alcohol (Chao et al. 1994; Thomasson et al. 1994; Chen et al. 1996; Nakamura et
al. 1996; Tanaka et al. 1996; Shen et al. 1997; Osier et al. 1999). Because Native Americans are
genetically descended from a north-central Asian source population within the last 20,000 years
(Meltzer 1993; Merriwether, Rothhammer, and Ferrell 1995; Kolman, Sambuughin, and
Bermingham 1996), it might be expected that they would have inherited these protective genes.
However, these protective alleles were found to be absent in a Southwest population, although a
significant association was found between other alleles at the ADH locus and the behavior of
binge drinking (Mulligan et al. 2003). In addition, a genome–wide association study performed
with the same Native American population found a strong association signal with alcoholism on
chromosome four near the ADH gene (Long et al. 1998). In order to further investigate the
possible genetic basis of alcoholism in Native Americans, I examined 12 single nucleotide
polymorphisms (SNPs) at both the ADH and ALDH genes of ~400 individuals from a Plains
population for association with multiple dichotomous and continue measures of alcohol and drug
abuse (Chapter 4). Despite the numerous phenotypes and the extensive genetic dataset, no
20

significant associations were detected. I also analyzed genotype data from the alpha-synuclein
(SNCA) gene, also located on chromosome four near ADH and therefore another attractive
candidate gene for alcoholism (Chapter 3, Clarimon et al. 2007). α-synuclein is involved in
dopaminergic neurotransmission, and the overexpression of the protein has been implicated in
the etiology of Parkinson’s disease (Polymeropoulos et al. 1997; Kruger et al. 1998) and
Alzheimer’s disease (Ueda et al. 1993), possibly because of neurodegeneration of dopamine
neurons due to toxic build-up of the protein (Mash et al. 2003). More recently, α-synuclein has
also been associated with alcoholism (Liang et al. 2003; Bonsch et al. 2005a; Bonsch et al.
2005b; Bonsch et al. 2005c) and drug addiction (Mash et al. 2003; Kobayashi et al. 2004).
Specifically, increased mRNA and protein are elevated in alcohol-preferring individuals in
humans, rats, and macaque monkeys (Liang et al. 2003; Spence et al. 2005; Walker and Grant
2006) and are associated with alcohol craving in humans (Bonsch et al. 2005a; Bonsch et al.
2005c). I genotyped and analyzed 15 SNPs at the SNCA locus in ~1000 individuals from a Plains
and a Southwest population and again found no significant association between any SNP and
alcohol or drug abuse or dependence. Since genetic variability and promoter polymorphisms
upstream of SNCA may mediate the increase in mRNA and protein expression (Bonsch et al.
2005b) my results suggest that study of upstream polymorphisms may represent a productive
avenue for future research. However, the results of these two studies suggest that the
environment may be a more influential component in substance abuse among Native Americans,
and therefore further resources should be devoted to address the underlying economic and social
problems in these populations.
In the final study (chapter five), I used molecular data to investigate recent observations
that, contrary to previous wide-held opinion, breastfeeding by HIV-1 positive women in
21

resource-poor areas is more beneficial to the long-term health of their children than complete or
partial replacement feeding (feeding of any substance other than breastmilk). This study used a
clinical perspective, as I investigated the evolution of HIV-1 in the breastmilk and blood over
time from a woman who participated in a clinical trial on breastfeeding-mediated transmission of
HIV. This study addressed many anthropological issues. Worldwide, an estimated 420,000
children were infected with HIV-1 in 2007, the vast majority through mother-to-child-
transmission (MTCT) (WHO 2007). Breast-feeding accounts for one-third to one-half of all
MTCT events during the first 24 months of life (Dabis et al. 1999; Iliff et al. 2005). In the US,
women are counseled by the CDC to replace breastfeeding with formula if infected with HIV-1
(CDC 2007), and the World Health Organization (WHO) previously recommended that HIV-1
positive women in all countries avoid all breastfeeding (WHO 2003). However, formula-feeding
is impractical for women in resource poor regions of the world where they do not have consistent
access to clean water, formula, and health care, and breast feeding may be the only practical
option. Cultural pressures also make women reluctant to eschew breast feeding as this can be
seen as a tacit admission of HIV-1 status. However, recent observational studies have suggested
that exclusive breast feeding, as opposed to the simultaneous feeding of milk and other foods,
may significantly reduce the risk of transmission of HIV-1 (Coutsoudis 2000; Coutsoudis et al.
2001; Coutsoudis et al. 2002; Iliff et al. 2005; Kuhn et al. 2007). The WHO subsequently
changed its recommendations to women in developing countries to encourage exclusive
breastfeeding up to six months followed by abrupt weaning (WHO 2006). However, the
biological mechanisms underlying the reduction of risk through exclusive breastfeeding have not
been clearly elucidated. Also, the benefits of abruptly weaning at six months are not at all clear,
while the practice is difficult and painful for the mother who would typically wean over a period
22

of months. I amplified and sequenced the env gene from viral populations present in the breast
milk and plasma over a two-year period from an HIV-1 positive woman participating in a
clinical trial in Zambia. I used phylogenetic and sequence-based analyses to examine the
evolution of the virus over time and within tissues. I concluded that the breastmilk virus was
genotypically distinct from the plasma virus during the early stages of breastfeeding, and the
virus in both tissues was subject to changing evolutionary dynamics and selective pressures over
time. The benefit of an anthropological genetic perspective that I bring to this study is the ability
to use evolutionary analyses to investigate the molecular basis of modulated risk of MTCT, with
the goal of advocating a scientifically sound and culturally sensitive breastfeeding management
plan to women while eliminating unnecessarily onerous measures.
In sum, this dissertation demonstrates how genetic anthropology can be used to address
both anthropological and clinical concerns from three temporally and philosophically distinct
perspectives. My studies incorporate pathogen genetics in addition to human genetics, which can
broaden our evolutionary understanding of the interaction between humans and pathogens. My
dissertation demonstrates the value of using an anthropological perspective in arenas often
dominated by medical practitioners. My unique advantage as a genetic anthropologist is that I
can apply analytical tools of evolutionary genetics to study the biological mechanisms of
diseases, while maintaining a multi-discinplinary approach that considers the cultural, historical,
and demographic factors that influence etiology. In addition, my training as an anthropologist
allows me to interpret the clinical results from these studies in a culturally appropriate context
for the maximum benefit of the participants.
23

Time
Div
ersi
ty
Time
Div
ersi
ty
Figure 1-1. Model for studying human diseases.
24

CHAPTER 2
MOLECULAR EVOLUTION OF THE TPRC, D, I, K, G, AND J GENES IN THE PATHOGENIC GENUS Treponema1
Introduction
The evolution of bacterial genomes has been heavily influenced by processes such as
horizontal gene transfer and homologous recombination, both of which can accelerate adaptation
through the generation of new alleles (Feavers et al. 1992; Baldo et al. 2006). Horizontal (or
lateral) gene transfer occurs through the uptake of genetic material from another genome, i.e. an
inter-genomic event, and includes transformation, conjugation, and transduction (Ochman,
Lawrence, and Groisman 2000). Homologous recombination, which is typically an intra-
genomic event, also occurs with high frequency in bacterial genomes (Smith, Dowson, and
Spratt 1991; Feil et al. 2001; Feil and Spratt 2001). Several outcomes may arise from a
recombination event, including translocations, deletions, duplications, inversions, and gene
conversions (Hughes 2000). Gene conversions are intra-genomic events that are the result of a
non-reciprocal transfer of genetic information from a donor locus to a recipient locus, either
through the permanent transfer of genetic material to the recipient locus or through the temporary
use of the donor sequence as a template for DNA synthesis on the recipient strand (Santoyo and
Romero 2005).
Gene conversion is especially important in the evolution of gene families (Slightom,
Blechl, and Smithies 1980; Drouin et al. 1999; Lathe and Bork 2001; Noonan et al. 2004). Gene
families are comprised of paralogous genes, which are defined as two or more genes within the
same genome that are so similar in DNA sequence they are assumed to have originated from one
1 Gray, R. R., C. J. Mulligan, B. J. Molini, E. S. Sun, L. Giacani, C. Godornes, A. Kitchen, S. A. Lukehart, and A. Centurion-Lara. 2006. Molecular evolution of the tprC, D, I, K, G, and J genes in the pathogenic genus Treponema. Mol Biol Evol 23:2220-2233.
25

ancestral gene (King and Stansfield 1997). The initial event creating the gene family was thus
likely to be one or more duplication events. The high sequence homology between paralogous
genes that signals a past duplication event also sets the stage for potential future homologous
recombination events (Schimenti 1994; Posada, Crandall, and Holmes 2002). Orthologous genes,
on the other hand, share sequence homology and are assumed to be descendant from a common
ancestral gene, but are present in different species (King and Stansfield 1997; Gogarten and
Olendzenski 1999). In this case, the genes most likely evolved through speciation rather than
duplication. Recombination can significantly impact inferred phylogenetic relationships (Feil et
al. 1999; Holmes, Urwin, and Maiden 1999; Feil and Spratt 2001; Worobey 2001). In the case of
gene families, gene conversion can cause paralogous genes to cluster more closely than
orthologous genes, thus confusing the order of evolution of the organisms (Drouin et al. 1999).
There are two seemingly opposite outcomes of gene conversion, concerted evolution and
increased sequence diversity, which may be distinctive of different stages of multi-gene
evolution (Santoyo and Romero 2005). After a gene family has been generated by ancient
duplication events, paralogous and orthologous comparisons should exhibit the same degree of
divergence. If the paralogous comparisons are more similar, then the genes in a multi-gene
family are evolving in a non-independent manner leading to homogenization of the genes, or
concerted evolution (Ohta 1992; Howell-Adams and Seifert 2000; Liao 2000; Lathe and Bork
2001). This may be beneficial in the case where a weakly advantageous point mutation arises in
one gene, and its effect is multiplied when the entire gene sequence is converted to other loci
(Dover 2002). This is consistent with the proposal that purifying selection may operate on genes
that have undergone duplication on the assumption that a duplicated gene must have an initial
benefit for the organism and, thus, its sequence must be conserved (Lynch and Conery 2000;
26

Kondrashov et al. 2002). As the sequences accumulate neutral diversity, though, the process of
gene conversion becomes less efficient. After time, only small “islands” of homology exist and a
site-specific system of shorter regions of gene conversion may take over, the outcome of which
is increased sequence variation (Zhang et al. 1992; Zhang et al. 1997; Zhang and Norris 1998;
Santoyo and Romero 2005; Taguchi et al. 2005). This is consistent with Ohno (1970), who
suggested that duplicated genes are under less selective pressure and may accumulate more
mutations leading to loss of the paralog or creation of a new function (Kimura and King 1979;
Walsh 1995; Wagner 1998; Lynch and Force 2000). Thus, concerted evolution and increased
sequence diversity may indicate earlier and later stages, respectively, in the evolution of gene
families (Santoyo and Romero 2005).
In this study, we examine genes in the tpr (Treponema pallidum repeat) gene family in
members of the genus Treponema (Spirochete family of bacteria) to investigate the evolution of
the gene family and, possibly, evolution of the treponemes themselves. The tpr gene family
consists of 12 paralogous genes that comprise 2% of the T. pallidum genome and have probably
evolved through gene duplication and gene conversion. These genes are related to the major
outer sheath protein (Msp) in Treponema denticola (TDE0405); however it appears that T.
denticola did not experience a history of gene duplication and gene conversion at this locus since
T. denticola possesses only one tpr-like gene (Seshadri et al. 2004). The tpr gene family in T.
pallidum is believed to encode potential virulence factors and is divided into three families:
Subfamily I (tprC, D, I, and F), Subfamily II (tprE, G, and J), and Subfamily III (tprA, B, H, K,
and L). The gene products from Subfamilies I and II have conserved amino and carboxyl
terminal sequences with unique central regions, while Subfamily III has scattered conserved and
unique or variable regions (Centurion-Lara et al. 1999). Gene conversion has previously been
27

reported in tprK (Centurion-Lara et al. 2000a; Centurion-Lara et al. 2004). Seven variable
regions within tprK were proposed to have been created by gene conversion using sequences
from the flanking regions of tprD as donors (Centurion-Lara et al. 2004). The degree of diversity
in these variable regions appears to increase in the presence of adaptive immune pressure,
suggesting that a function of these gene conversions may be to create antigenic diversity
(Centurion-Lara et al. 2004).
The pathogenic treponemes include three Treponema pallidum subspecies, T. carateum, T.
paraluiscuniculi (rabbit syphilis), and the unclassified Fribourg-Blanc (simian) isolate. The three
T. pallidum subspecies include pallidum, which is the causative agent of human venereal syphilis
and pertenue and endemicum, which cause yaws and bejel, respectively. T. carateum is the
etiological agent of pinta, although no isolates of this organism are known to exist. None of the
pathogenic treponemes mentioned above can be propagated in vitro. The complete T. p. subsp.
pallidum genome (from the Nichols strain) was sequenced in 1998 and is considered the
reference strain (Fraser et al. 1998). T. denticola, considered a non-pathogenic treponeme,
probably had an ancient divergence with T. pallidum based on the large difference in GC content
between T. pallidum and T. denticola (52.8% and 37.9%, respectively) and in genome length
(1.14 Mb and 2.84 Mb, respectively) (Seshadri et al. 2004) and thus the T. denticola sequence
was not considered in this study. Although lateral gene transfer has been identified as a probable
evolutionary force in the genome of T. denticola, no evidence exists for lateral gene transfer in T.
pallidum (Seshadri et al. 2004).
In this project, we examined eight strains of T. pallidum subsp. pallidum, and two strains
each of T. pallidum subsp. pertenue and T. pallidum subsp. endemicum, representing all known
propagated human strains (two additional T. pallidum subsp. pertenue strains have recently been
28

obtained and are under study) as well as two non-human strains, T. paraluiscuniculi and the
simian isolate. Six tpr genes, representing all three subfamilies, were sequenced: tprC, D, G, J, I,
and K. In order to investigate the evolution of these tpr genes, we utilized phylogenetic methods,
general measures of nucleotide diversity, and specific methods to detect recombination events.
Materials and Methods
Treponemal Strains and tpr Sequencing
All treponemal isolates used in this study were propagated in New Zealand White rabbits
(Lukehart et al. 1980) with the approval of the University of Washington Institutional Animal
Care and Use Committee. The Fribourg-Blanc strain was isolated from the popliteal lymph node
of a baboon from a yaws-endemic area (Fribourg-Blanc, Mollaret, and Niel 1966); a single report
describes an experimental infection of humans with this strain (Smith et al. 1971). Strain
designations and origins of the isolates are indicated in Table 2-1. Organisms were extracted by
mincing infected testicular tissue in 0.9% saline and were quantitated by darkfield microscopy.
Treponemal suspensions were mixed with an equal volume of 2x DNA lysis buffer (20mM Tris,
pH 8; 0.2 M EDTA, pH 8; 1.0% sodium dodecyl sulfate). DNA from treponemes was extracted
as previously described (LaFond et al. 2003).
Full-length open reading frames (ORFs) of 1791-2268 bp (Table 2-2) from each strain
were amplified, cloned, and sequenced as previously described (Giacani et al. 2004; Sun et al.
2004). The ORFs were amplified from T. pallidum strains by PCR using primers (Table 2-2)
located in the flanking regions of the genes, cloned into the TOPO II vector (Invitrogen,
Carlsbad, CA) and sequenced in both directions by the primer walking approach as previously
described (Centurion-Lara et al. 2000b); the amplicons at tprG and J from MexicoA were
obtained using primers internal to the start and stop codons and contained no flanking sequence.
A minimum of two clones were sequenced for each amplicon and ambiguities were resolved by
29

sequencing a third clone from an independent PCR, except for the Gauthier tprG, I, and J ORFs,
for which a single clone for each ORF was sequenced in both directions. For most sequences,
five clones were analyzed. The T. paraluiscuniculi sequences were described previously
(Giacani et al. 2004). GenBank accession numbers for the sequences are: tprC - NC_000919,
AY536645-6, AY550204, AY542157, AY590560, AY550206, AY542153-5, AY685236,
DQ886671-73; tprD - AF217537-41, AF187952, AY685237, AE000520, AY533515,
AY542156; tprI - AY533508-14, NC_000919, DQ886678-82; tprG/tprJ - NC_000919,
AF073527, AY685239-40, DQ886674-77; TprK - NC_000919, AY685248-50, DQ886683-700.
Evolutionary Analysis of Sequences
Six loci were considered in this analysis: tprC, D, G, I, J and K. Sequences were aligned
using ClustalX (Thompson et al. 1997) as well as manually using BioEdit to ensure proper amino
acid alignment (http://www.mbio.ncsu.edu/BioEdit/bioedit.html). Frameshift mutations in T.
paraluiscuniculi (bp 439 in tprC and tprD, bp 653 in tprG1 and tprG2) and T. p. subsp. pallidum
Sea81-4 (bp 1860 at tprG ) were removed from the alignment, as this would have created a
misalignment of the amino acids for the rest of the sequence. Levels of nucleotide diversity
within and between human treponemal subspecies (π and Dxy, respectively) were calculated
using DNAsp v. 4.10.4 (Rozas et al. 2003). GC content, using all available tpr sequences from
human treponemes (see Table 2-1), was calculated using PAML (Yang 1997). An AMOVA
(Analysis of Molecular Variance) was performed for tprC, I, and K using Arlequin version 3.0
(Excoffier, Laval, and Schneider 2005).
Phylogenetic Analyses
Maximum likelihood (ML) methods were used to infer the phylogenetic relationships
among the tested loci. First, the most appropriate substitution model for each locus was
determined using MODELTEST 3.06 (Posada and Crandall 1998). The following models were
30

selected for each locus: tprC (without T. paraluiscuniculi) - HKY+I+Γ , which allows for
different base frequencies and a separate transition and transversion rate (HKY model;
(Hasegawa, Kishino, and Yano 1985) as well as a proportion of invariant sites (I) and a gamma
distribution of mutation rates (Γ); tprD - HKY+Γ; tprG/J – GTR+Γ, which is a general time
reversible model that allows six different mutation rate categories (GTR) as well as a gamma
distribution of mutation rates (Γ) (with Nichols J) and HKY+Γ (without Nichols J); tprI – HKY;
tprK - HKY+Γ; tprC, D and I – GTR + Γ. The HKY + Γ model was used for the phylogeny
including all twelve Nichols tpr genes to reduce computational time due to the complexity of the
dataset. A maximum likelihood phylogeny was inferred using PAUP* 4.0b10 (Swofford 2002)
and the indicated substitution model. Full heuristic searching with the simple addition of
sequences and tree-bisection-reconnection (TBR) branch-swapping algorithms were used to
traverse the tree-space. Bootstrap analysis (1000 maximum likelihood replicates) was performed
using PAUP* 4.0b10 to determine the relative support for internal nodes. Third positions were
excluded in a separate analysis in order to determine if these positions had been subject to
mutational saturation.
Detection of Recombination
The RDP2 package (Martin, Williamson, and Posada 2005) was used to detect
recombination. This program implements several non-parametric methods to identify
recombinant and parental sequences and to estimate breakpoint positions that identify the limits
of the recombinant DNA in the sequences (Martin, Williamson, and Posada 2005). We used four
methods implemented in the RDP2 program: the RDP method, which is a phylogenetic method
that uses discordant branching patterns to infer recombination; the Maximum Chi-squared
(MaxChi) method (Smith 1992; Posada and Crandall 2001), which uses a sliding-window
31

approach along pairwise comparisons to identify discrepancies; the Chimera method (Smith
1992; Posada and Crandall 2001), which is similar to MaxChi but uses triplets of sequences
instead of pairs; and GENECONV, which compares fragments of sequence pairs (Padidam,
Sawyer, and Fauquet 1999). Non-default settings that were used consisted of a window size of
100, linear sequences, maximum p-value of 0.01 or 0.001 and a Bonferroni correction. All events
were listed. For the RDP method, internal and external references sequences were used, the
window size was set to 10, and 0-100 sequence identity was used. For both the MaxChi and the
Chimera methods, the number of variable sites was set to 30 with 1000 permutations and a max
p-value of 0.05. For the GENECONV method, the program was set to scan sequence triplets. In
all cases, the same alignment files from the phylogenetic analyses were used for the
recombination analyses.
Results
We examined six genes of the tpr gene family (tprC, D, G, J, I and K) in three human
treponemal subspecies (T. pallidum subsp. pallidum, endemicum and pertenue) and in two non-
human treponemes (T. paraluiscuniculi and the simian isolate) (Table 2-1). We were interested
in the relationship of the genes and alleles to one another as well as evidence for recombination.
Because of the well documented evidence for gene conversion in gene families and because no
evidence exists for lateral gene transfer in T. pallidum, we were specifically interested in
identifying intra-genomic recombination events, i.e. gene conversion. In order to investigate the
evolution of these tpr genes, we utilized 1) phylogenetic methods, 2) specific methods to detect
recombination events, and 3) general measures of nucleotide diversity and composition.
Phylogenetic Analyses
In order to obtain an overall view of the genetic diversity at all of the studied loci, a
maximum likelihood (ML) tree was created using an alignment of 2708 nucleotides from all
32

twelve available tpr gene sequences for T. p. subsp. pallidum Nichols strain (obtained from
GenBank) (fig. 1a). Sequences from Subfamily I (tprC, D, I, F) and Subfamily II (tprE, J, G)
cluster in two separate clades that are each clearly separated from the rest of the phylogeny. In
contrast, Subfamily III (tprA, B, H, L, K) sequences do not cluster with each other or any other
sequences and are distributed with varying branch lengths between the Subfamily I and II clades.
These results are consistent with previous studies in which Subfamily III membership was less
clearly defined than the other subfamilies (Centurion-Lara et al. 2000b).
Phylogenetic analyses of subfamily I
In order to focus on Subfamily I diversity, a ML phylogeny was generated for all available
DNA sequences for all Subfamily I loci: tprC, D, and I (Figure 2-1). All of the tprI sequences
clade together, while the tprC and D sequences are interspersed with each other such that there
are no major monophyletic tprC or D clades. There are three instances in which paralogous
sequences cluster more closely than their orthologous counterparts, all of which involve tprC and
D: 1) The tprC and D sequences for four of the T. pallidum subsp. pallidum strains; 2) the tprC
and D sequences from pertenue Gauthier (along with SamoaD tprC); 3) the tprC and D
sequences for T. paraluiscuniculi. The eight pallidum sequences are identical, while the pertenue
Gauthier and T. paraluiscuniculi tprC and D sequences differ by a maximum of one and three
point mutations, respectively, highlighting the paralogous relationship of tprC and D in these
strains.
Individual ML trees were also created for each of the Subfamily I loci examined in this
study: tprC, D, and I. In the tprD phylogeny, two distinct clades are evident (Figure 2-2). One
clade is comprised of four identical T. p. subsp. pallidum sequences and T. paraluiscuniculi, all
of which carry the D2 allele (using terminology of Centurion-Lara et al. 2000b; sequences that
differ by a few base pairs but have the same defining motif are considered the same allele). The
33

second clade is comprised of the other four identical T. p. subsp. pallidum sequences that carry
the D allele and the T. p. subsp. pertenue Gauthier strain, which carries the D3 allele that is 95%
homologous to the D allele (Centurion-Lara et al. 2000b). Although sequence data are
unavailable, PCR analysis suggests that T. p. subsp. endemicum and non-Gauthier strains of T. p.
subsp. pertenue would cluster in the D2 clade (Centurion-Lara et al. 2000b). The D and D2
alleles differ from each other by a 330bp central region at bp 855-1180 and three smaller variable
regions at bp1275-1306, 1425-1503 and 1569-1626 (relative to the Nichols strain sequence). A
contiguous expanse encompassing the four variable regions (bp 855-1626) was removed and the
new alignment was used to generate a ML tree in which the eight T. p. subsp. pallidum
sequences comprise a monophyletic clade (data not shown).
An initial tprC phylogeny included all strains (not shown). This phylogeny contained a
very long branch leading to T. paraluiscuniculi which increased the scale by an order of
magnitude (data not shown). The long T. paraluiscuniculi branch, along with the paralogous
grouping of T. paraluiscuniculi tprC and D sequences in Figure 2-1, suggested a gene conversion
event in T. paraluiscuniculi that replaced the ancestral sequence at tprC with tprD. Table 2-3
summarizes the proposed gene conversion events between tprC and D). The T. paraluiscuniculi
sequence was removed and an alternative phylogeny was generated (Figure 2-2). In the new
phylogeny, the three human subspecies cluster separately with strong bootstrap support (94-
100%). Simian is contained in the T. p. subsp. pertenue clade although it is distinct from the two
T. p. subsp. pertenue sequences. The T. p. subsp. pallidum sequences form three well-supported
clusters within a monophyletic clade. Four of the T. p. subsp. pallidum sequences are identical to
each other as well as to the tprD sequences in these same strains and are considered to carry the
D allele at both loci (see examples of paralogous sequences clustering above). The tprC alleles in
34

the other four T. p. subsp. pallidum strains show high sequence homology with the D allele and
are labeled D-like alleles (Centurion-Lara et al. 2004) (Table 2-3). The fact that there is higher
similarity between the paralogous tprC and D sequences in the strains carrying the D allele (they
are identical) than between their respective homologs suggests that a gene conversion event has
occurred between tprC and tprDin the D allele strains. In this case, tprC appears to be the likely
donor since there is detectable homology among all of the subspecies at this locus, whereas the D
and D2 alleles differ by a long central variable region. Furthermore, the tprC and tprD sequences
in T. p. subsp. pertenue Gauthier strain are identical suggesting a third gene conversion event,
again with the tprC locus serving as the donor due to the detectable homology among the tprC
homologs (Table 2-3).
The tprI phylogeny includes the same isolates as the tprC phylogeny with the exception of
T. paraluiscuniculi, which does not have a tprI locus (Figure 2-2). The phylogeny for tprI shows
a relatively long branch between T. p. subsp. pallidum and the other treponemes, similar in
length (0.016 substitutions/site) to the corresponding branch in the tprC phylogeny (0.014
substitutions/site). There is 100% bootstrap support for the two monophyletic clades consisting
of T. p. subsp. pallidum (all eight pallidum sequences are identical) and T. p. subsp. endemicum,
respectively, moderate support for clustering of simian with T. p. subsp. pertenue SamoaD
(85%), and little support for a T. p. subsp. pertenue + simian clade (62%), although the simian
sequence clearly does not belong with the other two clades. The tprI phylogeny confirms the
close relationship between the unclassified Fribourg-Blanc simian isolate and T. p. subsp.
pertenue that was also evident in the tprC phylogeny. Phylogenetic clustering of these sequences
suggests that there is no strong species boundary, a conclusion that is supported by the fact that
the simian treponeme is reported to infect humans (Smith et al. 1971).
35

Phylogenetic analyses of subfamily II
The tpr Subfamily II consists of tprE, G, and J. Previous studies have shown that the T. p.
subsp. pallidum Nichols tprG and J sequences are highly homologous at the 5’ and 3’ ends while
the central regions show extreme divergence (Giacani, Hevner, and Centurion-Lara 2005),
specifically at two variable regions (V1 = sites 976-1510, with a small internal region of
homology at sites 1168-1295, and the much smaller V2 = sites 1879-1947) that are unlikely to
have evolved through point mutation.. Different V1 and V2 sequences are classified as “G” and
“J” motifs, which are used to define the G, J, and G/J alleles present at tprG and J loci. The G
allele is defined as a “G motif” at V1 and V2, the J allele is defined as a “J motif” at V1 and V2,
and the G/J allele is defined as a “G motif” at V1 and a “J motif” at V2. At the tprG locus,
analysis of our alignment shows that two of the three pallidum strains analyzed at this locus
(Nichols and Sea81-4) carry the G allele, while the other pallidum strain (MexicoA) and the
pertenue strain (Gauthier) carry the G/J allele. At tprJ, Nichols and MexicoA carry the J allele,
while Sea81-4 and pertenue Gauthier carry the G/J allele (data not shown). PCR analysis
indicates that the other five pallidum strains discussed in this study also carry the J allele at tprJ,
although sequence data do not exist for these strains. PCR analysis also indicates that the other T.
p. subsp. pertenue strain (SamoaD) as well as a T. p. subsp. endemicum strain (IraqB) carry the
G/J allele (Centurion-Lara, unpublished). In T. paraluiscuniculi, the positions corresponding to
tprE and J contain two almost identical G/J allele sequences that are designated the G2 and G1
alleles, respectively (Giacani et al. 2004). In T. paraluiscuniculi G1 and G2 alleles, the second
half of V1 is somewhat different than V1 in the human G/J allele, although it is still much more
similar to the G/J allele than to the J allele. Furthermore, in T. paraluiscuniculi, tprG has
recombined with tprI (Subfamily I) to form a single allele termed the “G/I” hybrid at the tprG
locus (Giacani et al. 2004).
36

For our phylogenetic analysis, tprG and J sequences were grouped together because of the
high amount of gene conversion at and between these loci (Figure 2-3). In this phylogeny, a long
branch with 100% bootstrap support leads to the Nichols and MexicoA tprJ sequences, while the
MexicoA tprG, Sea81-4 tprJ, and Gauthier tprG and J form a polytomy (no bootstrap support).
The tprG sequences from Nichols and Sea81-4 form a highly supported clade (99%) and are
clearly closer to the rest of the sequences than Nichols and MexicoA tprJ. Because the J allele is
only found in T. p. subsp. pallidum, while the G/J allele is found in T. p. subsp. pallidum,
pertenue, and endemicum and T. paraluiscuniculi, the latter is most likely ancestral. The “J
motif” at V2 may be the result of a gene conversion or lateral gene transfer, although no
sequence homology was found in a search of the public database. The “G” motif at V2 in the G
allele also occurs in Nichols tprE (data not shown) and may represent a small gene conversion
event from tprE to tprG that replaced only V2 of the ancestral G/J allele in Nichols and Sea81-4
(although more tprE sequence data are needed to be certain). The Gauthier tprG and J sequences
differ by only 2 bp and may also represent a paralogous clustering reflective of a gene
conversion event, although the polytomy makes it difficult to be certain. The T. paraluiscuniculi
clade is also highly supported (100%), which represents a paralogous clustering of closely
related G/J alleles at tprE and J.
Phylogenetic analyses of subfamily III
The tprK phylogeny includes multiple clones from all represented strains because the locus
is highly variable and accumulates mutations within a single infection (Figure 2-4). Seven
variable regions have been identified in tprK that are likely the result of gene conversion events,
with the probable donor sites located in the 3’ and 5’ flanking regions of tprD (Centurion-Lara et
al. 2004). These variable regions were removed from our analysis in order to focus on the non-
recombinant history of the locus (variable regions were slightly modified to capture additional
37

flanking sites, i.e. bp 132-180, 596-671, 749-834, 866-920, 963-1059, 1141-1215, and 1291-
1390). T. paraluiscuniculi appears to be an appropriate outgroup for tprK as the scale is on the
same order of magnitude as tprC and I. Strong bootstrap support is shown for the T.
paraluiscuniculi (100%) and T. p. subsp. endemicum (97%) clades, as well as for a combined T.
p. subsp. pallidum + T. p. subsp. pertenue clade (96%) in which these two subspecies are
unresolved relative to each other. However, the fact that variable regions in tprK appear to
accumulate more variation in response to selective pressure (Centurion-Lara et al. 2004) and
clones from single individuals show single nucleotide polymorphisms (SNPs) even after removal
of variable regions suggests that tprK may evolve differently than the other tpr loci.
Statistical Tests for Recombination
Four tests (RDP, MaxChi, Chimera and GENECONV) in the RDP2 package were used to
investigate recombination events in the Subfamily I, II and III genes (Table 2-4). We use these
methods to identify significant recombination and the location of recombinant breakpoints, but
we do not infer donor and recipient alleles because there is likely inter-locus recombination also
occurring that will be undetected because these methods focus on a single locus at a time. In all
cases, the same alignment files from the phylogenetic analyses were used for the recombination
analyses. Our primary interest was to investigate support for the putative regions of gene
conversion identified in the phylogenetic analyses. Relatively strong overlap was shown in the
results from all four methods, and, in general, MaxChi found the most recombination events,
which was previously shown to be the most powerful test in the RDP2 package (Posada and
Crandall 2001).
In tprD, one region of recombination was identified in all of the T. p. subsp. pallidum D2
allele sequences, pertenue Gauthier, and T. paraluiscuniculi (see Table 2-4 for exact location of
recombinant regions). These results are consistent with a recombination breakpoint present at
38

site 855, which marks the beginning of the central variable region that differentiates the D and
D2 alleles. In tprC, two regions of recombination in the T. p. subsp. pallidum D allele sequences
were identified at bp 137-889 and 1459-1728. This result is consistent with the 100% clustering
of these sequences within the T. p. subsp. pallidum clade (Figure 2-2). Multiple recombination
events were identified in MexicoA and Sea81-3 that are consistent with a branch leading to a
monophyletic clade containing MexicoA and Sea81-3 in the tprC phylogeny (Figure 2-2). In tprI
only one recombination event was identified in pertenue SamoaD although the sequence has
only two unique single nucleotide polymorphisms in this region and point mutation seems a
more likely evolutionary mechanism than recombination in this case.
In tprG and J, more than 40 recombination events were identified when the significance
level was set to p=0.01. This result was impossible to interpret precisely so the analysis was
performed again with more stringent settings of p=0.001 and the requirement that more than one
method was necessary to identify a recombination event. Five sequences showed no evidence of
recombination under these conditions: pallidum Sea81-4 tprJ (G/J allele), pertenue Gauthier
tprG (G/J allele), pertenue Gauthier tprJ (G/J allele), pallidum Nichols tprJ (J allele), and
pallidum MexicoA tprJ (J allele). However, all four methods identified recombination at the
region containing V2 in tprG sequences for both pallidum G alleles (Sea81-4 and Nichols) as
well as the pallidum G/J allele (MexicoA). There are four polymorphisms between V1 and V2
that are shared between the pallidum G alleles and MexicoA G/J, although they are not found in
any of the other G/J alleles from other subspecies, which may contribute to this result.
In tprK, with the extended variable regions excluded, no recombination events were found
by any of the methods. These results agree with the phylogenetic analyses, which also do not
indicate any recombination outside of the variable regions (although Giacani and colleagues
39

(2004) did identify a putative region of recombination at tprK in CuniculiA between V5 and V6
that was not detected in any of our analyses).
The results of the tests for recombination were consistent with the phylogenetic analyses in
the overall detection of a high level of recombination across the studied loci. The recombination
tests also identified new regions of recombination, particularly at tprC. Overall, far more
recombination was indicated at tprG and J than for any other locus studied here and this result is
consistent with our phylogenetic analyses that revealed multiple instances of paralogous
clustering and the presence of multiple divergent alleles at tprG and J.
Analysis of Nucleotide Diversity and Composition
Additional measures, such as nucleotide diversity and GC content, can be used to
investigate recombination events, with the acknowledgement that other phenomena also affect
these measures (Baldo et al. 2006). The amount of within-subspecies genetic diversity is low for
all three subspecies at loci tprC, I, and K (π = 0-.0076) (Table 2-5). At tprD and J, however, the
diversity within T. p. subsp. pallidum is very high (π = 0.101 and 0.0958, respectively),
reflecting the intra-subspecies gene conversion events discussed above. The amount of diversity
at tprG within T. p. subsp. pallidum is intermediate (π = 0.0154), and specifically lower than
tprJ, reflecting a smaller putative gene conversion event, i.e. the V2 region.
The pattern of genetic diversity between subspecies of T. pallidum differs among loci,
especially for T. p. subsp. pallidum. The Dxy nucleotide diversity between T. p. subsp. pertenue
and T. p. subsp. endemicum is fairly consistent within tprC, I, and K (no tprD sequence data
currently exist for T. p. subsp. endemicum). The Dxy distance between T. p. subsp. pallidum and
the other two subspecies is approximately doubled relative to the distance between T. p. subsp.
pertenue and T. p. subsp. endemicum at tprC and I, consistent with the long branches leading to
40

T. p. subsp. pallidum in these phylogenies. However, at tprK, the distance between T. p. subsp.
pallidum and T. p. subsp. pertenue is much smaller than the distance between T. p. subsp.
endemicum and the others (0.0028 vs 0.011 and 0.013), consistent with the clustering of T. p.
subsp. pallidum and T. p. subsp. pertenue in the tprK phylogeny. At tprG, the distance between
T. p. subsp. pallidum and T. p. subsp. pertenue is intermediate (Dxy = 0.0130), while at tprJ the
distance between T. p. subsp. pallidum and T. p. subsp. pertenue is only slightly lower than
between these same two subspecies at tprD (Dxy = 0.0962). Again, this is in agreement with the
proposed gene conversion or horizontal gene transfer event that created the highly divergent J
allele in most T .p. subsp. pallidum strains.
Previous studies have suggested that gene conversion events lead to increased GC content
at third codon positions (Eyre-Walker 1993; Galtier et al. 2001; Galtier 2003; Noonan et al.
2004). Although the molecular mechanism is unknown, it may be due to a GC bias in mismatch
repair, which is required to resolve conversion events (Galtier et al. 2001; Noonan et al. 2004).
Third positions reflect this bias more strongly because they are under less selective constraint
since base changes at this position are less likely to result in a change in amino acid. At each of
the six tpr loci studied here, GC content was increased at the third position (GC3) relative to the
first and second positions combined (GC1+2) (table 6) although not as dramatically as reported
in other systems (Galtier et al. 2001; Noonan et al. 2004). This analysis supports our general
finding of multiple gene conversion events at the studied loci.
Discussion
Intra-genomic homologous recombination appears to have been a major force in the
evolution of the tpr gene family in the pathogenic Treponema species. After the gene duplication
events that created the gene family, our phylogenetic analyses of tprC, D, I, G, J, and K suggest
that the high levels of homology among the loci have supported multiple gene conversion events
41

both within and between these tpr genes. Although lateral gene transfer can theoretically produce
the genetic signatures we describe, this mechanism has not been reported in T. pallidum (lateral
gene transfer has been identified as a probable evolutionary force in the genome of T. denticola
due to the signature presence of phage-mediated integration events and restriction-modification
systems that may serve as a barrier against lateral gene transfer, but neither of these signatures is
present in T. pallidum (Seshadri et al. 2004) and gene conversion appears more likely,
particularly at tprC, D, G and K where donor regions can be identified within the same genome.
No donor sequence was identified for the V1 region of the J allele at tprJ and, thus, horizontal
gene transfer cannot be definitively ruled out.
In Subfamily I, we propose three gene conversion events between loci tprC and tprD; 1) a
tprC-to-tprD conversion that introduced the D allele into tprD in the D pallidum strains, 2) a
tprC-to-tprD conversion in T. p. subsp. pertenue Gauthier strain that introduced the D3 allele
into tprD, and 3) a tprD-to-tprC conversion in T. paraluiscuniculi that introduced the D2 allele
into tprC (table 3). At this point, there are insufficient data to determine the order of the three
proposed gene conversion events. However, it is clear that the tprC-to-tprD conversions (#1 and
2) represent two distinct events since the pertenue Gauthier sequences differ by only two bp and
the pallidum sequences are identical, but the pertenue Gauthier and pallidum sequences differ
from each other by 55 bp. Furthermore, our results suggest that the tprC locus is likely to be
older than the tprD locus because there is more variation among the pallidum D-like alleles at
tprC compared to the pallidum D2 sequences at tprD that are identical (fig. 2a and b). At tprD,
the D2 allele is most likely the ancestral allele since it is found in multiple subspecies (i.e.
pallidum, pertenue, endemicum) as well as in T. paraluiscuniculi, while the D allele is only
42

found in a subset of pallidum strains. Non-D2 alleles in the four pallidum strains and pertenue
Gauthier are likely the result of two subsequent gene conversion events, as described above.
At tprC, a single gene conversion event (originating from tprD) is posited in T.
paraluiscuniculi based on the phylogenetic analyses. The RDP2 recombination analysis
identified several small, additional recombinant regions in all of the T. p. subsp. pallidum
sequences, which is consistent with the higher diversity observed in the T. p. subsp. pallidum
tprC sequences (Figure 2-2, Table 2-4). Close inspection of the tprC alignment (including
pallidum and non-pallidum strains) revealed the presence of a high number of non-synonymous
mutations in the pallidum strains that were grouped in clusters rather than scattered throughout
the alignment. The transition/transversion ratio was decreased in these clusters and initially
revealed a significant signal for positive selection at tprC in T. p. subsp. pallidum (data not
shown). However, when we examined an alignment of tprC, D, and I together, we found that the
majority of the transversions and non-synonymous mutations were unique to T. p. subsp.
pallidum at tprC. The presence of clustered mutations, with a high frequency of transversions,
argues against accumulated point mutations and instead suggests that multiple smaller, ‘site-
specific’ gene conversion events may have occurred at tprC in T. p. subsp. pallidum. This may
be similar to the presence of multiple, variable regions in tprK, although the tprC regions do not
appear to undergo rapid sequence variation as occurs in tprK. These putative recombination
events at tprC would have to have occurred prior to the major tprC-to-tprD gene conversion that
replaced the D2 allele with the D allele at tprD since the D alleles at tprC and D are identical
(table 3). In proteins with antigenic relevance, recombination produces variation that may have
an adaptive purpose. However, it is not understood whether these proteins are more likely to
undergo recombination or whether high variability simply increases the power of detection of
43

these events (Baldo et al. 2006). In the case of tprC, which may be a cell-surface protein as
predicted by PSORT analysis (data not shown), it appears that the majority of tprC variation may
be a result of gene conversion events supporting an adaptive explanation.
At the tprI locus, variation among the treponemes was scattered and did not highlight a
specific region that might have undergone gene conversion as described above for other loci.
However, the fact that all eight of the T. p. subsp. pallidum tprI sequences were identical is
intriguing, considering this was not the case at any other locus in our study. There are several
possible explanations for the 100% sequence homology including a significantly lower (point)
mutation rate at tprI in T. p. subsp. pallidum, a more recent divergence of the T. p. subsp.
pallidum tprI sequences, functional constraint at tprI in T. p. subsp. pallidum, or a gene
conversion event that occurred prior to evolution of the T. p. subsp. pallidum tprI sequences (if a
sequence longer than that in our dataset were replaced, the recombination event would go
undetected by our recombination analysis that looked for the endpoints of recombination events).
Both a lower mutation rate and more recent evolution of T. p. subsp. pallidum tprI seem unlikely
because the lengths of branches leading to T. p. subsp. pallidum at tprI and tprC are comparable
(0.016 and 0.014 substitutions/site, respectively). Using a tprI phylogeny, a test for selection on
the branch leading to the eight T. p. subsp. pallidum sequences indicated that the non-
synonymous/synonymous rate ratio was not significantly different from 1.0 (data not shown).
Thus, functional constraint does not appear to explain the lack of mutations at this locus in the
pallidum subspecies. No specific gene conversion events were identified at tprI in our analyses
(although GC3 content was highest at tprI). The most likely explanation may be that the rate of
point mutations is generally low at all tpr genes and the pallidum tprI sequences have escaped
44

(by chance) the frequent gene conversion events that are mainly responsible for the diversity
seen at tprC and D in T. p. subsp. pallidum.
In Subfamily II, the various alleles that occur at tprG and J (and at tprE and J in T.
paraluiscuniculi), i.e. the G, J, and G/J alleles, are strongly suggestive of multiple gene
conversion events although the directionality of these events is difficult to determine due to the
complexity of the DNA sequences at these loci. Because the G/J allele occurs in multiple
subspecies and at multiple loci (Figure 2-3), it appears to be the ancestral sequence. The Nichols
and Mexico tprJ sequences have a divergent central region that is suggestive of a gene
conversion event (or horizontal gene transfer) that replaced the G/J allele (most likely only the
V1 region was replaced with a “J motif” V1). Unlike the scenario proposed above for gene
conversions at tprC and D, no donor region is immediately apparent for the gene conversion that
created the “J motif” V1 (BLAST searches did not identify any homology between the “J motif”
at the VI variable region and any other treponemal or non-treponemal sequences). Interestingly,
the clustering of the pallidum strains is not consistent between loci. At tprJ, only Sea81-4 has
apparently escaped the gene conversion which created the divergent J allele. At tprG, however,
Nichols and Sea81-4 appear to have shared a gene conversion event creating the “G motif” at V2
to the exclusion of MexicoA. This is in contrast with tprD, where the ancestral D allele was
replaced by the D2 allele in Nichols, but not in MexicoA or Sea81-4. A consistent history of the
evolution of the subspecies pallidum strains therefore cannot be ascertained from these data.
Previous studies have demonstrated a high frequency of gene conversion events at the tprK
locus in T. p. subsp. pallidum (Centurion-Lara et al. 2004). Seven variable regions have been
identified at tprK that are likely the result of multiple gene conversion events, with the probable
donor sites located in the 3’ and 5’ flanking regions of tprD. A multi-site/multi-step
45

recombination process has been described for the accumulation of diversity within the variable
regions. This diversity was shown to accumulate more dramatically in the presence of adaptive
immune pressure suggesting a mechanism to generate antigenic diversity in T. pallidum
(Centurion-Lara et al. 2004). It is probable that the gene conversion operating at tprK is different
than that affecting the tprC and D loci. Gene conversion at the tprK locus generates diversity and
each event seems to affect a relatively small region (each variable region is 48-99 bases). In
contrast, gene conversions at the tprD loci appears to result in concerted evolution and affect a
larger portion of the gene since the T. p. subsp. pallidum D alleles are identical at both tprC and
D. These seemingly contradictory outcomes of concerted evolution and increased diversity, both
mediated by gene conversion, may be explained by differing stages of evolution in a multi-gene
family (Santoyo and Romero 2005). In the first stage after initial gene duplication to create the
multi-gene family, homogenization or concerted evolution is likely the dominant force because
the high sequence homology drives gene conversion at a faster rate than point mutation occurs.
As point mutations accumulate over time, homologous recombination is no longer as effective
and the rate of point mutations may surpass that of gene conversion. At this stage of evolution of
a multi-gene family, smaller scale ‘site-specific’ gene conversion may become more significant,
thus allowing concerted evolution to occur in small regions while antigenic variation is created
throughout the gene (Santoyo and Romero 2005). This explanation may indicate a younger
history for the tprD sequences (i.e. concerted evolution stage) relative to tprK, while the tprK
(and possibly tprC) sequences are experiencing site-specific gene conversion events, possibly
leading to increased antigenic variation.
Several scenarios have been proposed for the evolution of the human treponemal species
(Baker and Armelagos 1988; Powell and Cook 2005). A New World vs. Old World origin of
46

venereal syphilis has been long debated (For a recent review, see Powell and Cook 2005). The
Columbian hypothesis originally suggested that venereal syphilis (T. p. subsp. pallidum)
originated in the New World and was brought to Europe by Columbus’ crews returning from the
New World (Crosby 1969). This was based on the paucity of skeletal and historical evidence for
treponemal disease in the Old World prior to the early 1500s. For example, Rothschild (2003)
proposed that yaws (T. p. subsp. pertenue) was the most ancestral of the three T. pallidum
subspecies and was present at least as far back as the origin of modern humans in Africa, and the
other two subspecies each derived from yaws, with T. p. subsp. pallidum evolving in the New
World no more than ~2000 years ago (Rothschild 2003). Baker and Armelagos (1988) have
proposed an alternative Columbian hypothesis which suggests that venereal syphilis evolved in
Europe from a New World non-venereal treponeme that was introduced to Europe by Columbus’
crews. This hypothesis is based on the lack of specific evidence for venereal syphilis in the New
World, despite the overwhelming evidence of treponemal disease. The Pre-Columbian
hypothesis suggests that treponemal diseases, including venereal syphilis, existed in the Old
World prior to Columbus’ voyages but were diagnosed incorrectly. One scenario suggests that
pinta was the original form present throughout the world during the Pleistocene, followed by the
evolution of yaws (12,000 years ago), then endemic syphilis (9,000 years ago) and, finally,
venereal syphilis (5,000 years ago) (Hackett 1963). Finally, a Unitarian hypothesis, based on
skeletal morphology data, has been advanced by Hudson (1965), who suggests that venereal
syphilis, endemic syphilis, yaws, and pinta are not in fact distinct diseases, but rather are
environmentally determined manifestations of the same disease. More recently, Armelagos and
colleagues (2005) have reviewed the molecular literature on human treponemes and they suggest
a lack of molecular distinction between these subspecies.
47

Our molecular data suggest that the three subspecies are legitimately classified as distinct
entities (Figure 2-2). The phylogenies for tprC and tprI demonstrate high bootstrap support for
the separation of the three subspecies into separate clades, with relatively long branches leading
to endemicum and pallidum. In tprD, pertenue is artificially closer to some pallidum sequences
because of the gene conversion event that separates pallidum D and D2 alleles. In the tprK
phylogeny, there is no bootstrap support to separate pallidum and pertenue, but the tprK
phylogeny is difficult to interpret because this locus has an exceedingly high mutation rate as
demonstrated by the fact that multiple tprK sequences exist in a single individual (even when the
variable regions are removed). Furthermore, AMOVA results reveal a significant amount of
among-subspecies variation (70-95%, p=0.000) when analyzing tprC, I and K from all three
subspecies further supporting the genetic distinctiveness of the subspecies (data not shown). It is
clear that recombination has played a significant role in the evolution of the tpr genes, and
possibly in the evolution of the treponemes. Therefore, studies that look only at SNPs, as
reviewed by Armelagos et al. (2005), will miss this high level of recombination and it may
appear there are few subspecies-specific variants because recombination has frequently
scrambled alleles within a subspecies, e.g. the D and D2 alleles at tprD. Moreover, the fact that
these recombination events are unique to a subspecies argues strongly in favor of the genetic
distinctiveness of the three subspecies.
Ascertaining the distinctiveness of the subspecies is prerequisite to resolving their
evolutionary history. In general, our analyses do not appear to support a dramatically older origin
of yaws relative to venereal syphilis contra Rothschild (2003), i.e. we do not see greater levels of
variation or longer tree branches for T. p. subsp. pertenue relative to T. p. subsp. pallidum (Table
2-4, Figures 2-2 and 2-4). Furthermore, our results do not clearly support current hypotheses that
48

49
consider venereal syphilis to be the most recently evolved of the treponemal syndromes. For
instance, multiple gene conversion events have occurred within subsets of T. p. subsp. pallidum
strains at tprC, D, G, and J (Figures 2-2 and 2-3) arguing for an older evolution of the entire
subspecies of pallidum in order to allow sufficient time for these events to have occurred. The
long branch in the tprI phylogeny leading to T. p. subsp. pallidum reflects a large number of
point mutations, which are assumed to evolve in a clock-like manner, suggesting that more
evolution has occurred on the branch to pallidum than on the branches to the other two
subspecies. Our results are generally consistent with a relatively coincident evolution of the three
human treponemal subspecies as proposed by Hackett (1965) but contra Rothschild (2003) who
proposed dramatically different timeframes for evolution of yaws and venereal syphilis.
Moreover, the T. p. subsp. pallidum sequences appear to carry too much variation to support the
modified Columbian hypothesis of evolution of venereal syphilis within the past 500 years
(Baker and Armelagos 1988). This is further supported by the fact that at least one T. p. subsp.
pallidum strain (Nichols) was collected in the early 1900s and is identical at the loci examined to
several other strains collected later in the 20th century, suggesting that the mutation rate is not
high enough to have created variants within this time frame. Additional samples, e.g. more
representatives of T. p. subsp. endemicum and T. p. subsp. pertenue, and analysis of more loci
will be needed to definitively answer questions concerning the origin and evolution of the
treponemes. Moreover, the high levels of recombination revealed in our study suggest that the
analysis of contiguous sequence data, as opposed to analysis of scattered SNPs, will be necessary
to identify possible recombination events prior to reconstruction of the evolutionary history of
the treponemes.

50
Table 2-1. Treponema isolates used in this study Species/ Subspecies
Strain Geographical source
Year isolated
tprC tprD tprG tprI tprJ tprK Reference
T. p. subsp. pallidum
Nicholsa Washington, D.C.
1912 +e + + + + + (Nichols and Hough 1913)
Sea81-4b Seattle 1980 + + + + + + (Lukehart et al. 1988) Chicagoc Chicago 1951 + + -f + - - (Turner and Hollander 1957) Bal73-1c Baltimore 1968 + + - + - - (Hardy et al. 1970) Bal3c Baltimore Unknown + + - + - - Bal7c
Sea81-3bBaltimore Seattle
1976 1981
+ +
+ +
- -
+ +
- -
- -
(Tramont 1976) (Lukehart et al. 1988)
MexicoAc Mexico 1953 + + + + + - (Turner and Hollander 1957) T. p. subsp. pertenue
Gauthierd Brazzaville 1960 + + + + + + (Gastinel et al. 1963)
SamoaDc Western Samoa
1953 + - - + - + (Turner and Hollander 1957)
T. p. subsp. endemicum
BosniaAc Bosnia 1950 + - - + - + (Turner and Hollander 1957)
IraqBc Iraq 1951 + - - + - + (Turner and Hollander 1957) T. paraluiscuniculi
CuniculiAc Baltimore Unknown + + + - + +
Simian species Fribourg-Blancc
Guinea 1966 + - - + - - (Fribourg-Blanc, Mollaret, and Niel 1966)
a Originally provided by James N. Miller, University of California, Los Angeles, CA b Strain isolated in Seattle by Sheila A. Lukehart, University of Washington, Seattle, WA c Strains provided by Paul Hardy and Ellen Nell, Johns Hopkins University, Baltimore, MD d Provided by Peter Perine, Centers for Diseases Control and Prevention, Atlanta, GA e ‘+’ indicates sequence data that were generated in the current study f ‘-‘ indicates sequence data were not generated in the current study

Table 2-2. T. pallidum primers used in this study. Locusa Strains
sequenced Sense primer Antisense Primer Length of
ORFs analyzed (bp)
tprC
BosniaA, Simian, SamoaD, CuniculiA
5’-gggggtgaggtagaagtgaga 5’-ccctcatccgagacaaaat 1797
Nichols, Bal7, Bal73-1, Chicago, Sea81-3, Sea81-4, MexicoA, Bal3, IraqB, Gauthier
5’-ttagaggaggcgtcagaacg 5’-ggtccgtgagcaggaagtaa 1797
tprD
Nichols, Bal7, Bal73-1, Chicago, Sea81-3, Sea81-4, MexicoA, Bal3, Gauthier
5’- cgcgtaccgctttgcagttca 5’- catggcattggtgagaaagacg 1791
CuniculiA 5’-aagaggttcaggaagcaacg 5’-acttcgtaggagcagcagga 1791
tprI BosniaA, IraqB, Simian, SamoaD
5’-cgtcaccctctcctggtagt 5’-atccctcgcctgtaaactga 1830
Nichols, Bal7, Bal73-1, Chicago, Sea81-3, MexicoA, Bal3, Sea81-4
5’-tgggagcttgtatgcagatg 5’-gggaaccctctcccttcc 1830
Gauthier 5’-agggtgagggggctactaga 5’-atccctcgcctgtaaactga 1830
tprG Gauthier, Sea81-4 5’-ccctgcgtttcccatctg 5’-gtactaccttcccccggtct 2268
MexicoA 5'-caggttttgccgttaagc 5'-aatcaagggagaataccgtc 1812 CuniculiA 5’-cgcgtacccacttctctctc 5’-gtactaccttcccccggtct 2268
tprJ MexicoA 5'-caggttttgccgttaagc 5'-aatcaagggagaataccgtc 1812
Gauthier 5’-agggtgagggggctactaga 5’-atccctcgcctgtaaactga 2268
Sea81-4 5’-cgagtgaggctcatcaagaa 5’-agtaagccctgcccaagaac 2268 CuniculiA 5’-aagtttgctttcagat 5’-gtactaccttcccccggtct 2268
tprK
BosniaA, IraqB, Simian, SamoaD, Gauthier
5’-agtaatggttttcggcatcg 5’-ccatacatccctaccaaatca 1470
Sea81-4, CuniculiA 5’-tcccccagttgcagcactat 5’-tcgcggtagtcaacaatacca 1470
51

a PCR Cycling conditions: tprC (Other than IraqB or CuniculiA): Denaturation at 94°C for 3 minutes, then 40 cycles of 94°C for 1 minute, 60°C for 2 minutes, and 72°C for 1 minute, with a final elongation step of 72°C for 10 minutes. tprC (IraqB): 40 cycles of 94°C for 1 minute, 58°C for 1 minute, and 72°C for 2 minutes, with a final elongation step of 72°C for 2 minutes. tprC, D ,G, J, K (CuniculiA): Denaturation at 95°C for 2 minutes, then 35 cycles of 95°C for 1 minute, 60°C for 1 minutes, and 72°C for 2 minute, with a final elongation step of 72°C for 3 minutes. tprD: (Other than CuniculiA)94 °C denaturation for 3 minutes, then 30 cycles of 94°C for 1 minute, 60°C for 2 minutes, and 72 °C for 1 minute, with a final extension step of 72 °C for 10 minutes. tprI: Denaturation at 94°C for 3 minutes, then 30 (Nichols, Bal7, Bal73-1, Chicago, Sea81-3, , MexicoA, Bal3, Sea81-4) or 40 cycles (BosniaA, IraqB, Simian, SamoaD) of 94°C for 1 minute, 60°C for 2 minutes, and 72°C for 1 minute, with a final elongation step of 72°C for 10 minutes. tprI, J (Gauthier): Denaturation at 94°C for 1 minute, then 40 cycles of 98°C for 10 seconds, 63°C for 5 minutes, with a final elongation step of 72°C for 10 minutes (used LA Kit, Takara Bio Inc. Shiga, Japan). tprG (Gauthier): Denaturation at 94°C for 1 minute, then 45 cycles of 98°C for 10 seconds, 62°C for 5 minutes, with a final elongation step of 72°C for 10 minutes (used LA Kit, Tokara Bio Inc. Shiga, Japan). tprG (Sea81-4): Denaturation at 94°C for 1 minute, then 35 cycles of 98°C for 20 seconds, 68°C for 5 minutes, with a final elongation step of 72°C for 10 minutes (used LA Kit, Tokara Bio Inc. Shiga, Japan). tprG, J (MexicoA): Denaturation at 94°C for 3 minutes, then 40 cycles of 94°C for 1 minute, 63°C for 2 minutes, and 68°C for 1 minute, with a final elongation step of 68°C for 7 minutes. Note that these primers amplify a partial ORF. tprK: (All except CuniculiA) Denaturation at 94°C for 3 minutes, then 40-45 cycles of 94°C for 1 minute, 60°C for 2 minutes, and 72°C for 1 minute, with a final elongation step of 72°C for 10 minutes. Table 2-3. Polymorphism at the tprC and tprD loci among pathogenic treponemes.
Species/Subspecies Strains tprD allele Directionality of proposed gene
conversion events
tprC allele
T. p. subsp. pallidum Nichols Chicago Bal73-1
Bal7
D D D D
← ← ← ←
D D D D
Sea81-3 Sea81-4
Bal3 MexicoA
D2 D2 D2 D2
No conversion No conversion No conversion No conversion
D-like D-like D-like D-like
T. p. subsp. endemicum Iraq Bosnia
D2 ND
No conversion NDa
D3-like ND
T. p. subsp. pertenue Gauthier D3 ← D3 SamoaD D2 No conversion D3-like
a ND = No sequence data
52

Table 2-4. Recombinant regions identified by RDP2. Subspecies Strain tprC tprD tprI tprK tprG/J T. p. subsp. pallidum
MexicoA ~800-1600a,c,d,e
~1450-1750a,c,d,1-854e
NCf NC 1669-2149
(G/J)b,c
Sea81-3 ~1450-1750a,c,d,
1-1500c1-854e
NC ND NDg
Sea81-4 NC 1-854e
NC NC 1669-2149
(G)b,c,d,e
Bal3 NC 1-854e
NC ND ND
Nichols 137-889c
1459-1728c,dNC NC ND 1556-2176
(G)b,c,d,e
Chicago 137-889c
1459-1728c,dNC NC ND ND
Bal73-1 137-889c
1459-1728c,dNC NC ND ND
Bal7 137-889c
1459-1728c,dNC NC ND ND
T. p. subsp. pertenue
Gauthier NC 1-854e
1701-1768c,d,e
1627-1796d
NC NC NC
SamoaD NC 143-621c NC ND T. p. subsp. endemicum
BosniaA 1459-endc,d ND NC NC ND
IraqB 714-1471c,d ND NC NC ND Simian NC ND NC ND ND a Multiple regions were identified by all four programs; the consensus is reported here. b RDP program identified this region as recombinant. c MaxChi program identified this region as recombinant. d Chimera program identified this region as recombinant. e GENECONV program identified this region as recombinant. f NC = no conversion identified. g ND = no DNA sequence data.
53

Table 2-5. Levels of nucleotide diversity within and between subspecies. Within
subspecies (π)
Between subspecies (Dxy)
Within subspecies (π)
Between subspecies (Dxy)
Locus pallidum pertenue Locus pallidum pertenue Locus tprD 0.10105 NCa tprD 0.10105 NCa tprD tprC 0.00659 0.00111 tprC 0.00659 0.00111 tprC tprI 0.0 0.00765 tprI 0.0 0.00765 tprI tprK 0.00066 0.00443 tprK 0.00066 0.00443 tprK tprG 0.01541 NC tprG 0.01541 NC tprG tprJ 0.09584 NC tprJ 0.09584 NC tprJ a NC = not calculated because only one DNA sequence was available. b ND = no DNA sequence data Table 2-6. Average GC content at combined 1st + 2nd (GC1+2) and 3rd codon (GC3) positions Locusa GC1+2 GC3 tprD 0.524 0.634 tprC 0.537 0.625 tprI 0.535 0.654 tprK 0.481 0.569 tprG+J 0.523 0.599 a All available human sequences were used for each locus; see Table 1 for list of sequences used.
54

Figure 2-1. Unrooted ML phylogenies of multiple tpr genes.
(a) Unrooted ML phylogeny of twelve Nichols tpr nucleotide sequences based on an alignment of 2708 bp. (b) ML phylogeny of tprC, D, and I nucleotide sequences based on an alignment of 1797 bp using mid-point rooting. Subspecies designations are indicated by vertical lines. Grey boxes indicate clades in which paralogous sequences group together. The specific tpr locus designation is appended to each strain name. Bootstrap values based on (a) 250 and (b) 1000 replications are shown next to branches.
55

Figure 2-2. ML phylogenies for tprD, C, and I.
Phylogenies are based on nucleotide sequences of (a) tprD; (b) tprC; (c) tprI. Human subspecies are circled and labeled. Bootstrap values based on 1000 replications are shown next to branches.
56

Figure 2-3. ML phylogeny of tprG and J.
The specific tpr locus designation is appended to each strain name. Subspecies are circled and labeled. For T. paraluiscuniculi, G1 signifies the G/J allele at tprJ and G2 signifies the G/J at tprE. Bootstrap values based on 1000 replications are shown next to branches.
57

Figure 2-4. ML phylogeny of tprK.
Rooted using CuniculiA strains. Subspecies designations are indicated by vertical lines and labeled. Bootstrap values based on 1000 replications are shownnext to branches. .
58

CHAPTER 3 LINKAGE DISEQUILIBRIUM AND ASSOCIATION ANALYSIS OF ALPHA SYNUCLEIN
(SNCA) AND ALCOHOL AND DRUG DEPENDENCE IN TWO AMERICAN INDIAN POPULATIONS2
Introduction
Alpha synuclein is involved in dopaminergic neurotransmission and has been implicated in
a number of neurological disorders. An association between α-synuclein and neurodegenerative
disorders is well established. Overexpression of α-synuclein has been implicated in the etiology
of Parkinson’s disease (Polymeropoulos et al. 1997; Kruger et al. 1998) and Alzheimer’s disease
(Ueda et al. 1993), possibly because of neurodegeneration of dopamine neurons due to toxic
build-up of α-synuclein (Mash et al. 2003).
More recently, α-synuclein has been associated with neuropsychiatric disorders, such as
alcoholism (Liang et al. 2003; Bonsch et al. 2005a; Bonsch et al. 2005b; Bonsch et al. 2005c)
and drug addiction (Mash et al. 2003; Kobayashi et al. 2004). Alpha synuclein is located at a
quantitative trait locus for a(Bonsch et al. 2005a; Bonsch et al. 2005c) Alcohol preference in
humans and levels of its mRNA and protein are elevated in alcohol-preferring individuals in rats
and macaque monkeys (Liang et al. 2003; Spence et al. 2005; Walker and Grant 2006). The
complex microsatellite repeat, NACP-REP1, which is located ~10kb upstream of the α-synuclein
gene (SNCA) has been associated with alcohol dependence in humans; specifically, longer alleles
were correlated with elevated levels of α-synuclein and were more frequent in alcohol dependent
patients (Bonsch et al. 2005b). Increased methylation of the SNCA promoter was also detected in
alcoholic patients (Bonsch et al. 2005c), and elevated SNCA mRNA and protein levels have also
been associated with craving in alcoholic patients (Bonsch et al. 2005a; Bonsch et al. 2005c). 2 Clarimon, J., R. R. Gray, L. N. Williams, M. A. Enoch, R. W. Robin, B. Albaugh, A. Singleton, D. Goldman, and C. J. Mulligan. 2007. Linkage disequilibrium and association analysis of alpha-synuclein and alcohol and drug dependence in two American Indian populations. Alcohol Clin Exp Res 31:546-554.
59

With respect to drug abuse, three out of four SNPs assayed in intron 1 of SNCA were found to be
significantly associated with methamphetamine psychosis/dependence (Kobayashi et al. 2004).
An overexpression of SNCA was also observed in the dopamine neurons of cocaine abusers
although α-synuclein may not directly increase the risk of drug abuse and it was speculated that
SNCA overexpression may be a protective response to changes in dopamine turnover resulting
from cocaine abuse (Mash et al. 2003). In general, overexpression of α-synuclein is thought to
interfere with dopaminergic neurotransmission, which has been proposed as a main mechanism
for withdrawal and craving (Self et al. 1995), two important factors for the development,
maintenance and relapse of addictive disorders.
The gene for α-synuclein has been mapped to chromosome position 4q21.3-22 (Chen et
al. 1995; Shibasaki et al. 1995; Spillantini, Divane, and Goedert 1995). Independent genome-
wide linkage studies have provided modest evidence that a locus in this region contributes to
alcohol dependence in one of the American Indian populations analyzed in the current study
(Southwest population; (Long et al. 1998; Mulligan et al. 2003) as well as in Euro-Americans
(Reich et al. 1998). Follow-up studies on the Euro-American population demonstrated strong
linkage to a phenotype defined by the maximum number of drinks consumed on one occasion
(Saccone et al. 2000). A recent study of Mission Indians reported no linkage in this region to a
diagnosis of alcohol dependence, but detected modest support for linkage to a more narrowly
defined phenotype of drinking severity (Ehlers et al. 2004a). This region has also been associated
with drug abuse through a genome-wide single-nucleotide polymorphism (SNP) genome scan
(Uhl et al. 2001).
In our study, we assayed 15 SNPs in the α-synuclein gene and one upstream
microsatellite repeat (NACP-REP1) in participants belonging to Southwest (n=514) and Plains
60

(n=420) American Indian populations. Patterns of linkage disequilibrium (LD) across the
assayed SNPs were similar in both tested populations and were consistent with the LD patterns
of the SCNA region in Caucasians, Africans and Asians, as reflected in the HapMap database
(www.hapmap.org). The assayed alleles and constructed haplotypes were tested for association
with alcohol dependence or alcohol use disorders (pooled diagnoses of alcohol dependence and
alcohol abuse) and drug dependence or drug use disorders (pooled diagnoses of drug dependence
and drug abuse), which are disorders that reach lifetime prevalences as high as 64% in the study
populations. Individual alleles and constructed haplotypes were also tested against two symptom
count phenotypes (all 18 questions and the eight questions that are diagnostic for alcohol
dependence).
Materials and Methods
Sampling Strategy
Blood samples and clinical data were collected from 514 adult members of a SW American
Indian tribe and from 420 adult members of a Plains American Indian tribe. Participants were
initially chosen at random from the tribal registry, and family members of ascertained alcoholics
were subsequently included. Descriptive data on both populations are presented in Table 3-1
(only a subset of these individuals was typed for a sufficient number of SNPs to be included in
the haplotype analysis, see Table 3-2). All participants were >21 years and required to have a
minimum of 25% ancestry to be included on the register. Williams et al. (1992) found a high
correspondence between overall levels of stated ancestry and ancestry estimated from genetic
markers and they found evidence for <5% non-American Indian admixture in the SW population.
Belfer et al. (2006) report low admixture with non-American Indian tribes in the Plains
population. Informed consent was obtained under a human subjects research protocol approved
61

by the respective Tribal Councils of each population group and the Institutional Review Board
(IRB) of the National Institute on Alcohol Abuse and Alcoholism.
Testing Instruments, Interviews, and Psychiatric Diagnoses
Focus groups comprised of tribal staff and community members reviewed testing
instruments and questionnaires for potential cultural biases and general suitability to the
population. Research diagnoses for lifetime alcohol dependence and abuse and lifetime drug
dependence and abuse were based on: 1) semi-structured psychiatric interviews using the
Schedule for Affective Disorders and Schizophrenia - Lifetime Version (SADS-L) with probes
added to enable diagnoses using both Research Diagnostic Criteria and Diagnostic and Statistical
Manual of Mental Disorders, Third Edition-Revised (DSM-III-R, American Psychiatric
Association, 1987) criteria (Robin et al. 1998), 2) medical, educational, court, and other records,
and 3) corroborative information from family members. The SADS-L was administered to all
subjects by a psychologist experienced with psychiatric assessment in this tribe and other
American Indian populations. DSM-III-R diagnoses of alcohol dependence were made from the
SADS-L by following operationally defined criteria and using the instructions of Spitzer et al.
(1989) (an additional criterion of heavy drinking for one year or more was added for a diagnosis
of alcohol dependence in the Plains population; (Belfer et al. 2006). Diagnoses were made from
the SADS-L interview data independently by two raters: a clinical social worker and a clinical
psychologist. Diagnostic differences were resolved in a consensus conference that included a
senior psychiatrist experienced in diagnosis in American Indian people. Sampling strategy,
interview procedure, and diagnosis protocol are summarized from Belfer et al. (2006), Long et
al. (1998), and Robin et al. (1998).
62

Genotyping
Genotype data from the HapMap project (www.hapmap.org) were used to generate an
optimal set of six tagging SNPs through the use of TagIT software. However, because the
HapMap SNP frequencies were calculated using Euro-American populations, we added nine
more SNPs in order to provide full coverage of genetic variation within this locus in American
Indian populations (Figure 3-1). Thirteen of the SNPs were assayed using Taqman® Assays-by-
DesignSM SNP Genotyping Services (Applied Biosystems). Thermal cycling and end-point PCR
analysis was performed on an ABI PRISM® 7900HT Sequence Detection System. Primer and
probe sequences are available upon request. Two SNPs, rs920624 and rs3775423, were assayed
as restriction fragment length polymorphisms by digesting amplification products with restriction
enzymes PsiI or MseI and SspI (New England Biolabs, Beverly, MA), respectively, and
electrophoresing digests on 2% agarose gels. Primers for rs920624 were 5’-
ACTACTTCTCTGTTGGATTGC-3’ and 5’-AAGATTCTTCACCTCTGTGTG-3’ and for
rs3775423 were 5’-GTATCCAATGCCCAAAGG-3’ and 5’-TGCCTCAGAAAGAACAGATG-
3’. The NACP-REP1 dinucleotide repeat polymorphism was amplified as previously described
(Farrer et al., 2001). PCR products were run on an ABI PRISM® 3100 (Applied Biosystems)
automated sequencer. GeneScanTM analysis software (version 3.7) and Genotyper® (version 3.7)
were used to assess fragment sizes.
Statistical Analysis
Previous studies have demonstrated that the degree of genetic relationship (kinship) in both
the Plains and SW samples is low and close to the average for the source populations, thus
permitting analyses that assume independence of individual samples (Robin et al. 1998; Belfer et
al. 2006). Hardy-Weinberg equilibrium was assessed by Fisher’s exact test, implemented in
63

Arlequin ver.2.000 program (Schneider, Roessli, and Excoffier 2000). PHASE ver.2.1 (Stephens
and Donnelly 2003) uses a Bayesian approach to reconstruct haplotypes from unphased
population genotypic data and takes into account recombination events and decay of LD with
distance between markers, thus leading to a more accurate inference of the real haplotype. One
thousand permutations were performed for each comparison. Haploview (Barrett et al. 2005) was
used to visualize LD relationships between SNCA SNPs as well as to ascertain the tagSNPs that
resulted from the LD analyses. LD blocks were constructed following the D’ method by Gabriel
et al. (2002) also implemented in Haploview.
A total of four clinical phenotypes were tested: alcohol dependence, alcohol use disorder
(diagnoses of alcohol dependence and alcohol abuse were pooled), drug dependence, and drug
use disorder (diagnoses of drug dependence and drug abuse were pooled). Differences in allele
and genotype distributions were analyzed using the chi square test and two-tailed P values are
presented. Data were analyzed using SPSSTM ver.11.0 for Windows (SPSS, Chicago, IL).
Haplotype frequency comparisons between cases and controls were performed with PHASE
ver.2.1 (Stephens and Donnelly 2003). In order to correct for multiple comparisons, global P
values were calculated using the COCAPHASE module of the UNPHASED statistical package
(Dudbridge 2003). Permutation correction was performed using 10,000 permutations. Symptom
counts were also used as two additional phenotypes, which were calculated as 1) the total
number of affirmative responses to the SADS-L interview questions (Endicott and Spitzer 1978)
and 2) the total number of affirmative responses to eight questions used in the diagnosis of
alcohol dependence. ANOVA tests were used to analyze the variance of the two symptom count
phenotypes among the four most common haplotypes and among the six SNPs used to define
haplotypes using the R program (R Foundation for Statistical Computing, Vienna, Austria). The
64

CLUMP program (Sham and Curtis 1995) was used to analyze contingency tables resulting from
NACP-REP1 alleles. Significance was assessed using Monte Carlo approach by performing a
total of 10,000 simulations.
A power calculation, based on haplotypes, indicated that our study had 83-100% power
to detect an association at an odds ratio of 2.0 using pooled haplotype frequencies, 91% power
based on the most frequent haplotype in the SW population and 87% power based on the most
frequent haplotype in the Plains population (p = 0.05).
Results
Based on 14 common SNPs located within the SNCA gene, similar LD patterns were
detected in both American Indian populations that are also consistent with the LD structures of
European Caucasian, Yoruban (African) and Chinese and Japanese Asian populations in the
HapMap database (Figure 3-1; SNP rs920624 and the NACP-REP1 polymorphism were not
included in the comparative LD analysis because there are no associated frequency data in the
HapMap database). Based on a strict criterion of continuous LD >90%, one small and one large
LD block were defined in the SW population and three small LD blocks were identified in the
Plains population. A less stringent criterion based on the presence of a recombination region
between SNPs 4 and 5 suggests two large LD blocks (SNPs 2-4 and 5-14) present in both
populations. Haplotype tests can be more powerful than using only one SNP to define a
haplotype block (Schaid 2004), so SNPs 3, 4, 8, 9, 12, and 13 (as depicted in Figure 3-1) were
chosen to define haplotypes in our analysis. The high level of LD suggests that we have assayed
the full spectrum of SNCA variation present in our study populations, i.e. effectively all SNPs
currently identified at SNCA (433 SNPs, http://www.ncbi.nlm.nih.gov/SNP/) were assayed.
Phenotypes were investigated with respect to individual SNPs as well as to the haplotype blocks
defined above.
65

Frequencies of all analyzed SNPs were in Hardy-Weinberg equilibrium for cases and
controls. We did not find any allele or genotype frequency differences between cases and
controls for alcohol dependence, alcohol use disorders (pooled diagnoses of alcohol dependence
and alcohol abuse), or drug use disorders (pooled diagnoses of drug dependence and drug abuse)
when total populations were tested (p values for allele frequency comparisons are presented in
Figure 3-2). Drug dependence was significantly associated with SNPs rs2583978, rs356186,
rs356198 and rs3775423 in the SW population (Figure 3-2). We tested whether this association
was gender-dependent. When we focused on males, the association with drug dependence
persisted for SNPs rs356186 and rs3775423 and SNPs rs3775439 and rs356165 also were
significantly associated (Figure 3-2). However, when we adjusted for multiple testing in order to
control for the family-wise type I error (FWER) by means of a permutation test with 10,000
replicates, the global P-value was 0.113 for the entire population and 0.112 when only males
where counted. None of the SNP associations were significant in females (Figure 3-2).
Stratification of the Plains population by gender revealed a significant association between
rs356163 and alcohol dependence and alcohol use disorders and between rs2572324 and alcohol
use disorders in males (Figure 3-2). However, the global COCAPHASE P value in Plains males
was not significant (p > 0.1 for both alcohol dependence and alcohol use disorders).
Association of the four addiction phenotypes was also tested against haplotypes
constructed across the entire gene using SNPs 3, 4, 8, 9, 12, and 13 as described above. Several
SNPs within the SNCA gene may be contributing to an addiction phenotype in the form of a
“super-allele” (Schaid 2004); therefore we combined the six markers into a haplotype to increase
the power of detecting an association. Four major haplotypes were detected in the SW and Plains
populations (>5 % frequency in all phenotype categories) (Table 3-2). No difference in haplotype
66

frequencies between cases and controls were detected in either population. A power calculation
using pooled haplotype frequency data from both populations indicated 83-100% power to detect
an association at an odds ratio of 2.0 (p = 0.05).
Categorical diagnoses of substance abuse may not adequately describe a disease that is
both heterogeneous and occurs on a continuum, and symptom counts may be used as a
quantitative variable to provide more information (Helzer et al. 2006). Thus, the total number of
affirmative responses on the SADS-L interview (n=18) and the eight diagnostic questions for
alcohol dependence were used as additional phenotypes. No significant association between the
symptom count and the four most common haplotypes was detected using an ANOVA in either
the SW or the Plains populations. When each of the six SNPs used to define the haplotypes was
tested individually against the symptom count only SNP rs356198 was marginally significant
(p=0.044) in the SW population; however, that significance disappeared after correction for
multiple testing. No comparisons with individual SNPs were significant in the Plains population.
Since recent reports have described an association between the NACP-REP1 repeat
polymorphism and the risk of alcohol dependence (Bonsch et al. 2005b), we tested this variant in
our populations. Distribution of the NACP-REP1 alleles in both populations is shown in Figure
3-3. In contrast to other studied populations, both American Indian populations exhibited
reduced variation at the REP1 locus with a very high frequency of the 267 bp repeat allele (80-
85%). The 267 bp allele was associated with virtually all haplotypes in both populations
suggesting that no linkage disequilibrium exists between the SNCA SNPs and the REP1 locus,
thus the REP1 locus was analyzed independently of the SNCA SNPs and haplotypes. No
association was found between the REP1 locus and any of the four addiction phenotypes in the
67

two populations (Figure 3-3). The same lack of association was found when each gender was
analyzed independently (data not shown).
Discussion
Several recent studies have suggested that α-synuclein may play a role in the development
and maintenance of certain addictive disorders (Liang et al. 2003; Mash et al. 2003; Kobayashi et
al. 2004; Bonsch et al. 2005b). We assayed 15 SNPs in the α-synuclein gene, SNCA, and one
upstream microsatellite repeat (NACP-REP1) in two American Indian populations with a high
lifetime prevalence of alcohol and drug dependence and abuse (Table 3-1). The assayed alleles
and constructed haplotypes were tested for association with one of four clinical phenotypes,
including alcohol dependence and alcohol use disorders (pooled diagnoses of alcohol
dependence plus abuse) and drug dependence and drug use disorders (pooled diagnoses of drug
dependence plus abuse), as well as two symptom count phenotypes (total number of questions
and eight questions diagnostic for alcohol dependence).
Single allele tests revealed significant associations between four SNPs and drug
dependence in the SW population. Two of those SNPs plus another two SNPs were found to be
associated with drug dependence in SW males only. In the Plains population, a significant
association was detected only in males with two SNPs and alcohol use disorders and one SNP
and alcohol dependence. An association with alcoholism in males is consistent with an
overrepresentation of alcohol dependence in Native American males (66-70%) compared to
females (30-53%) (Kinzie et al. 1992; Kunitz et al. 1999; Ehlers et al. 2004b; Gilder, Wall, and
Ehlers 2004). However, none of the global p-values, calculated to adjust for multiple testing,
reached the level of significance. Haplotype analyses did not reveal any association between
SNCA and substance abuse or dependence. Furthermore, when corrected for multiple
68

comparisons, no significant associations were detected when symptom counts were tested against
haplotypes or individual SNPs.
The polymorphic REP1 complex dinucleotide repeat is located ~10kb upstream of the
SNCA transcriptional start site and is polymorphic for alleles of length 265-273 (Chiba-Falek,
Touchman, and Nussbaum 2003). Longer REP1 alleles have been associated with increased
expression of the SNCA gene and loss of the REP1 repeat in vitro resulted in a 4-fold reduction
in expression of the SNCA gene (Touchman et al. 2001; Chiba-Falek, Touchman, and Nussbaum
2003). Several studies have correlated elevated levels of SNCA mRNA and protein with various
phenotypes related to alcohol dependence (Liang et al. 2003; Bonsch et al. 2004; Walker and
Grant 2006). In contrast, our results do not support an association between NACP-REP1 and
alcohol dependence/use disorders or drug dependence/use disorders in the American Indians
populations analyzed here. Lack of association could be explained by the low allelic variability
present at this locus in both populations and, in particular, lack of the longer alleles implicated in
the previous study (alleles 271 and 273, Bönsch et al. 2005a).
To our knowledge, this is the most exhaustive analysis performed to date of genetic
variation at the SNCA locus and possible association with risk of alcohol and drug addiction. The
study is strengthened by analysis of two different populations. Although several SNPs initially
returned significant p values, none of the results remained significant after correction for
multiple testing. It is possible that the low genetic variation in these American Indians may mask
a significant association, or actually contribute to SNCA being a non-factor in addiction
vulnerability in these particular populations. It is likely that a search for genes involved in
addiction disorders will be complicated by population-specific genetic effects, as well as varying
effects of social and environmental factors. Additionally, cannabis is the most frequently abused
69

drug in our populations in contrast to previous studies that found a correlation between SNCA
and cocaine or methamphetamine addiction. Alcohol craving was not examined in this study, and
thus its association with the SNCA gene in these populations cannot be ruled out. Nonetheless,
our results in two American Indian populations do not support a role for a genetic variant in the
SNCA gene that contributes to alcohol or drug addiction. These results do not preclude a role for
this gene, particularly in other populations exhibiting more diversity at NACP-REP1. Altered
methylation patterns in the SNCA promoter have already been associated with alcoholism and
may contribute to differential expression levels of this gene (Bonsch et al. 2005c). Thus, future
research may focus on additional variants in the promoter region of SNCA that could cause the
changes in mRNA and protein levels observed in previous studies.
Table 3-1. Demographic and phenotypic characterstics of southwest (SW) and plains populations.
SW population n=514a Plain population n=420a
Age ± SD (range) 36.5 ± 13.6 (21–90) 42 ± 14.1 (18–87) Gender (% males) 42.8 43.6
Alcohol dependence 316 239 Alcohol use disorder 348 248
Drug dependence 44 (47% cannabis, 49% amphetamines, 9% cocaine)
49 (69% cannabis, 36% amphetamines, 15% cocaine)
Drug use disorder 185 (65% cannabis, 28% amphetamines, 34% cocaine)
96 (75% cannabis, 40% amphetamines, 17% cocaine)
Controlsb 135 159 aPhenotype counts do not equal population sample sizes because individuals had multiple diagnoses. For example, under drug dependence, 42 SW and 46 Plains individuals also had alcohol use disorders and, under drug use disorder, 170 SW and 88 Plains individuals also had alcohol use disorders. Furthermore, percentages of abused drugs do not sum to 100 because several individuals had multiple drug dependence. bControls are defined as those individuals with no diagnosis of alcohol dependence, alcohol use disorder, drug dependence, or drug use disorder.
70

Table 3-2. Frequency of Major Haplotypes in Cases and Controls in Southwest and Plains Populations. Population Southwest Plains Phenotype AD AUD DD DUD Controla AD AUD DD DUD Controla
Nb 312 344 43 184 130 224 233 47 91 147 221212c 0.412 0.411 0.337 0.416 0.4 0.281 0.281 0.328 0.258 0.265 222121c 0.24 0.23 0.302 0.217 0.208 0.337 0.339 0.34 0.324 0.313 111212c 0.164 0.166 0.186 0.152 0.204 0.152 0.15 0.192 0.154 0.119 111221c 0.08 0.076 0.081 0.092 0.077 0.132 0.129 0.075 0.126 0.16 % total haplotypes
0.896 0.883 0.896 0.877 0.889 0.902 0.899 0.935 0.862 0.857
aControls are defined as those individuals with no diagnosis of alcohol dependence, alcohol use disorder, drug dependence, or drug use disorder. bNumber of individuals in each category differs slightly from Table 1 because only individuals typed for a majority of the 6 single-nucleotide polymorphism (SNPs) were included. cHaplotypes were constructed from the following SNPs (in order): rs2737020, rs1812923, rs356164, rs356198, rs3775423, rs10033209. AD = alcohol dependence, AUD = alcohol use disorder, DD = drug dependence, DUD = drug use disorder.
71

rs25
8398
5
rs25
8397
8
rs37
7543
9
rs35
6163
rs25
7232
4
rs35
6168
rs10
0332
09
rs27
3702
0rs
1812
923
rs35
6186
rs35
6164
rs35
6198
rs35
6165
109,8 Kb
5’ 3’
rs37
7542
3
rs25
8398
5
rs25
8397
8
rs37
7543
9
rs35
6163
rs25
7232
4
rs35
6168
rs10
0332
09
rs27
3702
0rs
1812
923
rs35
6186
rs35
6164
rs35
6198
rs35
6165
109,8 Kb
5’ 3’
rs37
7542
3
Figure 3-1. Relative positions of single nucleotide polymorphisms assessed in α-synuclein
(SNCA) gene.
Single-nucleotide polymorphisms (SNP) rs920624 is not included because there are no associated frequency data in the HapMap database. The gene structure of SNCA is shown (top), with vertical bars indicating exons. Linkage disequilibrium (LD) structure is presented for the CEU population allele frequencies from the HapMap project (top), the Southwest population (middle), and the Plains population (bottom). Numbers within the diamonds are D' values for the respective SNP pairs. Solid red diamonds represent absolute LD (D'=1), blue diamonds represent strong LD with low level of significance. Numbers in gray within white diamonds represent a high probability or evidence of historical recombination. Haplotype blocks, as determined with the use of Haploview software, are depicted.
72

Southwest
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Alc-dep Alc-abu Drug-dep Drug-abu
Southwest males
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Southwest females
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
luesPlains
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Alc-dep Alc-abu Drug-dep Drug-abu
Plains males
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Plains females
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Alc-dep Alc-use-dis Drug-dep Drug-use-dis Alc-dep Alc-use-dis Drug-dep Drug-use-dis
Southwest
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Alc-dep Alc-abu Drug-dep Drug-abu
Southwest males
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Southwest females
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
luesPlains
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Alc-dep Alc-abu Drug-dep Drug-abu
Plains males
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Plains females
0
0.5
1
1.5
2
2.5
3
rs258
3985
rs258
3978
rs920
624
rs273
7020
rs181
2923
rs377
5439
rs356
186
rs356
163
rs356
164
rs356
198
rs257
2324
rs356
168
rs377
5423
rs100
3320
9
rs356
165
-log
p-va
lues
Alc-dep Alc-use-dis Drug-dep Drug-use-dis Alc-dep Alc-use-dis Drug-dep Drug-use-dis
Figure 3-2. Single-marker analyses representing p values for each marker on a logarithmic scale.
A dotted line indicating a p value of 0.05 is represented in each graph, so values above correspond to significant associations. Graphs on the left refer to the Southwest population whereas graphs on the right refer to the Plains population. The first row represents the entire data set for each population, while the second row refers to males only and the third row refers to females only.
73

Figure 3-3. Allelic distribution of the NACP-REP1 microsatellite repeats.
Shown for the Southwest and Plains populations. Allele size is depicted on the X-axis while allele frequency is depicted on the Y-axis.
74

CHAPTER 4 LACK OF ASSOCIATION BETWEEN ADH/ALDH MARKERS AND SUBSTANCE USE
DISORDER IN NATIVE AMERICAN POPULATION
Introduction
Native Americans had the highest rate of alcohol related deaths of all ethnic groups in the
United States in 2001 (age-adjusted death rate=42.1/100,000) which was more than five times
higher than the alcohol-related death rate for the general US population (6.9/100,000) (Health,
United States, 2006, http://info.ihs.gov/Files/DisparitiesFacts-Jan2006.pdf ). Native Americans
were also 2.5 times as likely to die of chronic liver disease or cirrhosis than Caucasians
(22.7/100,000 vs. 9.2/100,000) (Facts on Indian Health Disparities,
http://www.cdc.gov/nchs/data/hus/hus06.pdf#031). In addition, this group also had the highest
frequency of current drinkers who reported drinking more than four drinks in one day in the past
year compared to other ethnic groups (40.9% vs. 32.7% for Caucasians, 24.3% for African-
Americans, and 20.8% for Asians) (Facts on Indian Health Disparities,
http://www.cdc.gov/nchs/data/hus/hus06.pdf#031). It is unclear to what extent this is a result of
genetic determinants or cultural influences such as poverty and lack of health care on
reservations. Many studies have investigated the role of genes in substance abuse, and multiple
studies have provided evidence for a locus contributing to alcohol dependence on chromosome 4
(Long et al. 1998; Reich et al. 1998; Williams et al. 1999; Zinn-Justin and Abel 1999; Mulligan
et al. 2003; Ehlers et al. 2004a). Several candidate genes proximally located on that chromosome
include alpha synuclein (Bonsch et al. 2004; Bonsch et al. 2005a; Bonsch et al. 2005b; Bonsch et
al. 2005c; Clarimon et al. 2007) the GABA receptors (Long et al. 1998) and the alcohol
dehydrogenase gene family (ADH) (Chen et al. 1996; Osier et al. 1999; Mulligan et al. 2003).
The seven ADH genes encode enzymes that convert alcohol to acetaldehyde, which is then
75

processed into acetate by enzymes encoded by the ALDH genes. The ADH genes are located in a
cluster (ADH7-ADH1C-ADH1B-ADH1CA-ADH6-ADH4-ADH5) on chromosome 4 (Osier et al.
2002b). Certain variants within the ADH and ALDH genes have been found to protect against
alcoholism; in particular, the ADH1B*47His allele (and possibly the ADH1B*369Cys allele)
results in increased blood levels of acetaldehyde leading to an unpleasant flushing response that
is proposed to have a protective effect against alcoholism (Thomasson et al. 1991; Goedde et al.
1992; Thomasson et al. 1993; Nakamura et al. 1996). However, the ADH1B*47His allele is only
present at polymorphic frequencies in Asian and Jewish populations (Chao et al. 1994;
Thomasson et al. 1994; Chen et al. 1996; Nakamura et al. 1996; Tanaka et al. 1996; Shen et al.
1997; Neumark et al. 1998; Osier et al. 1999; Ehlers et al. 2004a), although it has been found at
low levels in European, African, and Middle Eastern populations as well (Borras et al. 2000;
Ehlers et al. 2001; Osier et al. 2002b). A protective effect has also been found for the
ADH1C*349Ile allele in Asians, Native Americans and Europeans (Chao et al. 1994; Chen et al.
1996; Shen et al. 1997; Borras et al. 2000; Konishi et al. 2003; Mulligan et al. 2003), although
this may be a result of linkage disequilibrium with ADH1B*47His in Asian populations that
carry this allele (Chen et al. 1999; Osier et al. 1999). An additional variant in the mitochondrial
ALDH2 gene, ALDH2-2, blocks the conversion of actetaldehyde to acetate resulting in an
accumulation of acetaldehyde and a more pronounced flushing response than the ADH1B*47His
allele (Harada, Agarwal, and Goedde 1982; Thomasson et al. 1991; Goedde et al. 1992;
Thomasson et al. 1993; Novoradovsky et al. 1995; Peterson et al. 1999). The allele has been
found to be protective in Asian populations (Iwahashi et al. 1995; Chen et al. 1996; Chen et al.
1999; Hara, Terasaki, and Okubo 2000; McCarthy et al. 2000). In addition, the combination of
76

the ADH1B*47His and the ALDH2-2*487Glu alleles have been found to drastically increase the
risk of alcoholism in Koreans (Kim et al. 2008).
Although ADH1B*47His and ALDH2-2 are absent in most Native American populations,
several lines of evidence suggest that genetic variants in or near the ADH genes contribute to
alcohol dependence in Native Americans. First, an autosomal genome scan in a Native American
population identified this region of chromosome 4 as a possible alcoholism risk locus (Long et
al. 1998). Subsequent studies have identified additional polymorphisms within the ADH genes
that are present in Native Americans (Wall et al. 1997; Ehlers et al. 1998; Osier et al. 2002a).
Two ADH1C variants (including ADH1C*349Ile) and a neighboring microsatellite marker were
associated with alcohol dependence in a subset of individuals from a Southwest Native American
population (Mulligan et al. 2003). In order to determine if these alleles contribute to risk of
substance abuse in a Native American Plains population, we assayed nine single nucleotide
polymorphisms (SNPs) across the ADH1A, ADH1B, and ADH1C genes and three markers in the
ALDH gene in 359 members of a Plains Native American population. Two different diagnoses
for alcohol use were investigated (alcohol dependence and abuse) as well as two continuous
measures of alcohol use based on survey responses. The survey consisted of 18 questions
concerning the impact of alcohol use on daily life (see Appendix A). The number of positive
responses to both the full 18 questions and a subset of eight diagnostic questions was used as the
outcome variable in a regression analysis. We also included two measures of drug use (drug
dependence and abuse) as a recent study found an association between drug dependence and
ADH variants independent of alcohol behavior (Luo et al. 2007).
77

Materials and Methods
Samples
The sampling methodology was previously described in Mulligan et al. (2003) and is
briefly summarized here. Blood samples and clinical data were initially collected from 420 adult
members of a Plains Native American population. Ultimately, a subset of 359 individuals that
had full diagnostic information was selected for genotyping. Informed consent was obtained
under a human subjects research protocol approved by the Plains Tribal Council and the
Institutional Review Board (IRB) of the National Institute on Alcohol Abuse and Alcoholism.
Testing Instruments, Interviews, and Psychiatric Diagnoses
Focus groups comprised of tribal staff and community members reviewed testing
instruments and questionnaires for potential cultural biases and general suitability to the
population. Research diagnoses for lifetime alcohol dependence and abuse and lifetime drug
dependence and abuse were based on: (1) semi-structured psychiatric interviews using the
Schedule for Affective Disorders and Schizophrenia—Lifetime Version (SADS-L) with
additional information to enable diagnoses using both the Research Diagnostic Criteria and
Diagnostic and Statistical Manual of Mental Disorders, Third Edition, Revised (DSM-III-R,
American Psychiatric Association, 1987), (2) medical, educational, court, and other records, and
(3) corroborative information from family members. The SADS-L was administered to all
subjects by a psychologist experienced with psychiatric assessment in this tribe and other
American Indian populations. Sampling strategy, interview procedure, and diagnosis protocol
are summarized from (Long et al. 1998; Robin et al. 1998; Mulligan et al. 2003; Belfer et al.
2006).
78

Genotyping
Three hundred and fifty-nine individuals were typed for five ADH1A, three ADH1B, one
ADH1C, and three ALDH polymorphisms. Loci, primers, thermocycling conditions, and
restriction enzymes are listed in Table 1. PCR amplicons were analyzed by electrophoresis on
2% agarose or 4% Metaphor (FMC BioProducts, Rockland, Me.) gels. Marker ADH1Ain8 BccI
was analyzed on a Beckman Coulter CEQ 8000 using the Beckman SNP genotyping kit
according to the manufacturer’s instructions (Beckman Inc, Fullerton CA).
Statistical Analysis
Haploview (Barrett et al. 2005) was used to visualize linkage disequilibrium (LD)
relationships between the assayed ADH markers, and linkage disequilibrium blocks were
constructed following the D' method by (Gabriel et al. 2002). PHASE ver. 2.1 (Stephens and
Donnelly 2003) was used to reconstruct haplotypes from unphased population genotypic data.
The program R (R Foundation for Statistical Computing, Vienna, Austria) was used to
implement tests for Hardy-Weinberg equilibrium.
A total of four clinical phenotypes were tested: alcohol dependence, alcohol use disorder
(diagnoses of alcohol dependence and alcohol abuse were pooled), drug dependence, and drug
use disorder (diagnoses of drug dependence and drug abuse were pooled). The phenotypic
information is summarized in Table 4-2 (categories were not mutually exclusive). Six continuous
variable measures of substance use were also tested for association with the genetic data. Two
measures of symptom count data were tested, which were calculated as (1) the total number of
affirmative responses to the SADS-L interview questions (Endicott and Spitzer 1978) and (2) the
total number of affirmative responses to eight questions used in the diagnosis of alcohol
dependence (see Appendix 1 for questions). The smaller subset of questions captured the most
severe aspects of alcohol dependence and was tested separately to determine association with
79

genetic markers among the most afflicted individuals. The other four phenotypes included
maximum number of drinks ever consumed in one day, maximum drinks ever consumed in one
month, age at which regular drinking began, and age at which heavy drinking began.
Genotype and allele frequencies for each marker were compared for each of the four
diagnostic groups with the frequencies in the control group. The chi-square test was used for all
comparisons as implemented in the program R. For each analysis, males and females were tested
separately, then pooled together. Genotype frequencies for each marker were tested for
association with the six continuous phenotypes. These data were analyzed in SAS v.9.1 (SAS
Institute Inc, Cary NC) using a regression model that incorporated sex and age as well as
genotype information. A subset of individuals (n=325) were used in the regression analysis due
to missing phenotype information for some individuals.
In addition, frequencies for the haplotypes identified through the statistical analysis
described above were compared for all cases (individuals with any of the four diagnoses) and
controls, defined as individuals without any of the four diagnoses. Haplotype association tests
incorporate information from multiple sites and possibly are more powerful than single-marker
tests (Shaid et al 2004).
Results
Three hundred and fifty seven individuals were genotyped, of which 212 were females and
142 were males. Of the nine ADH markers, four (ADH1BArg47His, ADH1B RsaI,
ADH1BArg369Cys, and ADH1Ain8 BccI ) were not polymorphic in this population and were
thus not informative for our analyses. All markers were in HW equilibrium (data not shown).
Among the five polymorphic ADH markers, 98% linkage disequilibrium (LD) was determined
by Haploview analysis (Figure 4-1). Because of the very high level of LD, only three major
80

haplotypes were present in the population and represented 93% of the diversity. The frequencies
of these haplotypes were not significantly different between cases and controls (Table 4-3).
The genotype, allelle and haplotype frequencies of each of the seven polymorphic markers
were tested for association with the four discrete phenotypes (alcohol dependence, alcohol use
disorder, drug dependence, and drug use disorder) and the control individuals (Table 4-4). A
marginally significant association (p=0.0421) was found between drug dependence and genotype
at marker ADH1C EcoRI (Table 4-4). However, the allele comparison at this locus was not
significant, and after Bonferroni correction for multiple testing, the p-value for the genotype
association was no longer significant (for each phenotype, adjusted alpha = 0.05/ number of
comparisons = 0.0035). When males and females were tested separately for association with the
four discrete diagnoses and the genotype data, males had lower p-values in general but none
were significant after correction for multiple testing (data not shown).
A regression analysis was also performed using six continuous phenotypic categories
(total number of affirmative answers on the symptom count questionnaire, number of affirmative
responses for a reduced set of eight questions on the same questionnaire, maximum number of
drinks ever consumed in one day, maximum drinks ever consumed in one month, age at which
regular drinking began, and age at which heavy drinking began) and the genotype for each of the
seven markers along with sex and age as additional predictor variables (Table 4-4). For both
symptom count variables, only sex was consistently significant after correction for multiple
testing (alpha=0.002). For both maximum number of drinks variables, again only sex was
consistently significant. For the categories related to age/years of drinking, age was significantly
associated, and sex was only occasionally significantly associated with these variables. The
genotype data were not significantly associated with any of the continuous phenotype variables.
81

Discussion
The ADH and ALDH genes have been extensively studied in many ethnically diverse
populations, and the strongest associations with substance abuse have been found in Asian
populations that carry the ADH1B*47His and ALDH2-2 alleles known to protect against
alcoholism through toxic accumulation of acetaldehyde (Chao et al. 1994; Thomasson et al.
1994; Chen et al. 1996; Nakamura et al. 1996; Tanaka et al. 1996; Shen et al. 1997; Osier et al.
1999). A previous study found significant association between alcohol dependence and two
ADH1C markers (ADH1CHaeIII and ADHCIle349Val) , between binge drinking and three
markers (ADH1C EcoRI, ADH1C HaeIII, and ADHC Ile349Val) and between flushing and one
marker (ALDH2-In6A) in a subset of individuals in a Southwest Native American tribe (Mulligan
et al. 2003). Thus, we sought to replicate the study in a different Native American population in
order to strengthen support for ADH as a risk locus. We used a continuous measure of
alcoholism and information about behavior in a regression model in addition to traditional
dichotomous diagnoses, since considering substance abuse as a continuum might enable us to
detect associations that would be masked by broad diagnoses which include individuals who
abuse substances for social or cultural reasons.
Three hundred and fifty-nine individuals were assayed for nine markers from the ADH1A,
ADH1B, and ADH1C genes and three from the ALDH gene. As previously reported (Mulligan et
al. 2003), neither of the protective alleles (ADH1B*47His and ALDH2-2) were detected in this
population. In contrast to the previous study (Mulligan et al. 2003), neither of the significantly
associated markers in the Southwest population were found to be significantly associated with
any of the tested phenotypes in this Plains population. Age and/or sex were significant in most of
the regression analyses, suggesting that the demographic information is strongly correlated with
substance abuse.
82

It is somewhat surprising that this study did not uncover any significant association
between the genetic data and substance use disorder phenotypes, despite the multiple ways in
which the genetic data were analyzed and several innovative measures of abuse. However, the
previous study investigating the association between the ADH/ALDH genes and alcohol use in a
Southwest population found only nominally significance and only in a subsets of the study
sample (Mulligan et al. 2003) In addition, a study use a whole-genome scan only found
significance for the ADH locus on chromosome 4 using two-point linkage analysis, but not
multipoint linkage analysis which takes into account all marker information from one
chromosome (Long et al. 1998). While these results were initially suggestive of ADH/ALDH
having genetic determinants linked to alcohol abuse and were the impetus for conducting a
similar study in a different Native American population, the results from this study suggest that
genetic variants in the ADH and ALDH genes may not be major determinants of substance abuse
disorders in Native Americans. There are several possible reasons for this result. First, during the
migration from Asia to the New World, a population bottleneck severely reduced the genetic
variability present in the ancestral population (Kolman et al. 1995; Kolman and Bermingham
1997; Ramachandran et al. 2005), which also resulted in higher LD at the ADH genes in Native
Americans than in Chinese or Africans (Mulligan et al. 2003). Variants conferring protection/risk
at this locus may therefore have been lost in all or some Native American populations. The fact
that modest association was found in the Southwest population is consistent with a recent study
that found higher diversity in western populations compared with eastern populations in South
American Native Americans, suggesting that more ancestral variants might have been retained in
western populations (Wang et al. 2007) This pattern could result from an initial coastal migration
83

84
from Asia, followed by expansion into the interior regions, as has been recently suggested
(Anderson and Gillam 2000; Dixon 2001; Fix 2002; Surovell 2003).
Another explanation is that different cultural factors may be influencing substance abuse in
the Plains population and may play a larger role than genetic factors. This is supported by the
demographic component consistently being the only significantly associated factor. The genetic
component of alcoholism in this case may be swamped by the high number of individuals who
exhibit substance abuse phenotypes, because many individuals in the affected category may
abuse substances for cultural or social reasons but do not have a genetic pre-disposition, despite
the fact that we attempted to use continuous measures of abuse to capture degrees of severity.
Native Americans experience much poorer health in general than the rest of the population in the
U.S. Their infant death rate is almost double that of Caucasians, they have a 40% higher
prevalence of AIDS, and are more than twice as likely to be diagnosed with diabetes. These
statistics may be associated with poorer access to health care as well: 30% of Native Americans
had no health coverage in 2005, and 25% of this group lives at the poverty level (Office of
Minority Health, 2006). Therefore, it is likely that environmental factors contribute heavily to the
high prevalence of alcoholism in this Native American population, and suggest that resources
could productively be devoted to addressing the poverty and poor health care in this ethnic
group.

85
Table 4-1. Loci, primers, cycling conditions and restriction enzymes for 12 loci studied. Marker Primer Name Primer Sequence Thermocycling Conditions Restriction
Enzymes ADH1CEcoRI A3EX2DW
A3EcoUP2 5’-TTGCACCTCCTAAGGCTC-3’ 5’-TCTAATGCAAATTGATTGTGAAC-3’
94 °C (15 s), 51 °C (15 s), 72 °C (75 s); 40 cycles
EcoRI
ADH1CHaeIII A3EX5FOR2 A3EX5REV1
5’-TGAGTTTGCACATTAGTTATGG-3’ 5’-TGCTCTCAGTTCTTTCTGGG-3’
94 °C (40 s), 56 °C (30 s), 72 °C (60 s); 35 cycles
HaeIII
ADH1CArg271Gln A3EX6FXNFOR1 A3EX6FXNREV3
5’-TTGTTTATCTGTGATTTTTTTTGT-3’ 5’-CGTTACTGTAGAATACAAAGC-3’
94 °C (15 s), 54 °C (15 s), 72 °C (60 s); 35 cycles
ADHCIle349Val A3FXNFOR1 A3FXNREV3
5’-TTGTTTATCTGTGATTTTTTTTGT-3’ 5’-CGTTACTGTAGAATACAAAGC-3’
94 °C (15 s), 51 °C (15 s), 72 °C (75 s); 40 cycles
SspI
ADH1CPro351Thr ADH1CSNPFOR1 A3FXNREV3
5’-GTTTTCACTGGATGCACTAATAAC-3’ 5’-CGTTACTGTAGAATACAAAGC-3’
94 °C (30 s), 51 °C (30 s), 72 °C (75 s); 40 cycles
ADH1BArg47His A2FXNFOR A2FXNREV
5’-ATTCTAAATTGTTTAATTCAAGAAG-3’ 5’-ACTAACACAGAATTACTGGAC-3’
95 °C (30 s), 56 °C (30 s), 72 °C (60 s); 35 cycles
MslI
ADH1BRsaI A2IN3DW3 A2IN3UP2
5’-ATATTTATTTTACCCTAAACTTATG-3’ 5’-GAGCTAAAACATACTTTGGATAG-3’
94 °C (30 s), 60 °C (30 s), 72 °C (30 s); 35 cycles
RsaI
ADH1BArg369Cys HE39 HE40
5’-TGGACTTCACAA CAAGCATGT-3’ 5’-TTGATAACATCTCTGAAGAGCTGA-3’
95°C (15 s), 58°C (15 s), 72°C (60 s); 35 cycles
AluNI
ADH1Ain8BccI A1BccIDW A1IN8UPI A1BccITUP
5’-ATTGTCTAGCAGAAAATGAAAAG-3’ 5’-AGTTTCTTTCCCTCCTCAAGAATG-3’ 5’-TTTTTTTTTTTTCTAATTTTTCTCATCCTTCCA-3’
94 °C (15 s), 54 °C (15 s), 72 °C (60 s); 35 cycles
NA
ALDH2.5' 5 .for 5 .rev
5’-GCAGTGCCGTCTGCCCCATCCATGT-3’ 5’-GGCCCGAGCCAGGGCGACCCTGAGCT-3’
94 °C (30 s), 60–62 °C (30 s), 72 °C (30 s); 40 cycles
SacI
ALDH2.In6A In6A.For In6A.Rev
5’-AAATATTGCTCTAGGCCAGGC-3’ 5’-TGGGAATTCTAAATGGGACGG-3’
94 °C for 10 cycles/89 °C for 30 cycles(30 s), 55 °C (30 s), 72 °C (30 s); 40 cycles
HaeIII
ALDH2.Def L12 R12
5’-TTTGGTGGCTAGAAGATGTC-3’ 5’-CACACTCACAGTTTTCTCTT-3’
94 °C (30 s), 57 °C (30 s), 72 °C (30 s); 40 cycles
MboII

Table 4-2. Phenotypic characteristics of the dataset. Phenotype Females Males Total Alcohol dependencea 101 105 206 Alcohol Abusea 106 111 217 Drug Dependencea 15 32 47 Drug Abusea 42 51 92 All Casesb 113 112 225 Controlb 99 30 129 Average Total Symptom Count (18)
5.5 9.6 7.1
Average Reduced Symptom Count (8)
3.3 5.2 4.1
aIndividuals can be part of multiple phenotypes, and therefore the totals do not sum to the toal number of individuals (359). bAll cases and controls sum to 359. Table 4-3. Haplotype frequencies and p-value for comparisons of cases vs. controls. Haplotype Frequency All
individuals Frequency Cases Frequency
Controls p-value
11212 0.51 0.49 0.55 0.173 12122 0.34 0.35 0.33 0.490 22121 0.08 0.08 0.05 0.138 For haplotype designations, the order of the markers is as follows: ADH1CEcoRI, ADH1CHaeIII, ADH1CArg271Gln, ADHCIle47Val, ADH1CPro351Thr. “1” refers to the presence of a restriction site, and “2” refers to the absence of a restriction site.
86

Table 4-4. Chi-squared and regression p-values for genotype and allele associations for each marker.
Marker P-value ADH1C EcoRI
ADH1C HaeIII
ADH1C Arg271Gln
ADHC Ile349Val
ADH1C Pro351Thr
ALDH 2.5'
ALDH 2.In6A
Frequency Major Allele 0.9 0.54 0.53 0.54 0.92 0.65 0.94 Dichotomous Diagnosesa
genotype 0.2725 0.1366 0.0914 0.1682 0.2062 0.2662 0.2775 Alcohol Dependence allele 0.19 0.168 0.204 0.195 0.091 0.146 0.184
genotype 0.2642 0.1005 0.0746 0.1377 0.2062 0.3864 0.2624 Alcohol Abuse allele 0.171 0.125 0.176 0.166 0.09 0.233 0.177
genotype 0.0421 0.1136 0.05997 0.0954 0.2436 0.2352 0.7812 Drug Dependence allele 0.588 0.565 0.44 0.523 0.365 0.107 0.567
genotype 0.1525 0.2407 0.2702 0.2966 0.1319 0.341 0.3318 Drug Abuse allele 0.121 0.188 0.417 0.327 0.257 0.174 0.142 Continuous Diagnosesb
genotype 0.0989 0.1485 0.136 0.1873 0.0265 0.2377 0.3569 age 0.9461 0.8225 0.7142 0.7464 0.9693 0.9464 0.9952 Symptom
Count (18) sex <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 genotype 0.0868 0.3751 0.267 0.3551 0.0304 0.1151 0.5301 age 0.396 0.3553 0.2432 0.257 0.4223 0.352 0.4559 Symptom
Count (8) sex <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 genotype 0.5815 0.7387 0.9867 0.7225 0.6649 0.835 0.5663 age 0.354 0.3983 0.4531 0.4677 0.2355 0.3546 0.1893 Max
Drinks/month sex 0.001 0.0009 0.0028 0.0017 0.0006 0.0013 0.0006 genotype 0.5815 0.6398 0.6768 0.6014 0.0839 0.7642 0.3218 age 0.354 0.1074 0.1322 0.0736 0.1048 0.117 0.0591 Max
Drinks/Day sex 0.001 <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 genotype <0.0001 0.5548 0.515 0.6154 0.0709 0.62 0.9289 age <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 <0.0001 Years of Heavy
Drinking sex 0.0215 0.0448 0.0208 0.0235 0.0434 0.0414 0.049 genotype 0.0403 0.2558 0.1969 0.2071 0.0482 0.9888 0.7319 age 0.0004 0.0004 0.0007 0.0006 0.0003 0.001 0.0008 Start Heavy
Drinking sex 0.0542 0.0819 0.0625 0.0823 0.0631 0.082 0.0709 genotype 0.1465 0.7951 0.7094 0.7449 0.2044 0.9803 0.1766 age 0.0056 0.006 0.0288 0.0308 0.0055 0.01 0.0059 Start regular
drinking sex 0.0006 0.0017 0.0019 0.0018 0.0009 0.0019 0.0009 aAll analyses were performed using the chi-squared test. bThese analyses were performed using a regression model with each marker separately, and age and sex included in the model. Values in bold indicate significance after correction for multiple testing.
87

Figure 4-1. Linkage disequilbrium of markers assessed in the ADH gene family.
Linkage disequilibrium (LD) structure is presented for the Plains population. Numbers above the red blocks indicate polymorphic assayed markers in ADH1A, B, and C: 1=ADH1CEcoRI, 2=ADH1CHaeIII, 3=ADH1CArg271Gln, 4=ADHCIle47Val, 5=ADH1CPro351Thr. Numbers within the diamonds are D' values for the respective marker pairs. Solid red diamonds represent absolute LD (D'=1). One haplotype blocks, as determined with the use of Haploview software, was determined.
88

CHAPTER 5 DYNAMIC AND DISTINCT EVOLUTION OF HIV-1 IN BREASTMILK OVER TWO
YEARS POST-PARTUM
Introduction
As of 2007, 33.2 million people were living with the human immuno-deficiency virus type
1 (HIV-1) worldwide. 22.5 million of those people (68%) live in sub-Saharan Africa. World-
wide, an estimated 420,000 children were infected in 2007, the vast majority through mother-to-
child-transmission (MTCT) of HIV-1 (WHO 2007). Breast-feeding accounts for one-third to
one-half of all MTCT events over 24 months (Dabis et al. 1999; Iliff et al. 2005). In the US,
women are counseled by the CDC to replace breastfeeding with formula if infected with HIV-1
(CDC 2007), which (along with anti-retroviral drugs and cesarean-sections) resulted in only
~300 infants becoming infected perinatally in 2000 (CDC 2006b). Because of the seemingly
obvious reduction of transmission with formula feeding, as demonstrated in studies in developed
countries, the World Health Organization recommended that “when replacement feeding is
acceptable, feasible, affordable, sustainable, and safe, avoidance of all breastfeeding by HIV-
infected mothers is recommended” (WHO 2003). However, formula-feeding is impractical for
women in resource poor regions of the world where they do not have consistent access to clean
water, formula, and health care, and breast feeding may be the only practical option. Cultural
pressures also make women reluctant to eschew breastfeeding as this can be seen as a tacit
admission of HIV-1 status. The result was an ineffectual policy because women were essentially
left with no practical guidelines for reducing the risk of transmission. This situation highlights
the critical need for an anthropological perspective in international health policy, which is often
based on ideal conditions and Western epidemiology.
A shift in the traditional thinking about breastfeeding was influenced by the supposition
that breastfeeding by infected mothers actually reduced infant mortality (Ross and Labbok
89

2004). Recent observational studies have suggested that exclusive breastfeeding, as opposed to
the simultaneous feeding of milk and other foods, may significantly reduce the risk of
transmission of HIV-1 (Coutsoudis 2000; Coutsoudis et al. 2001; Coutsoudis et al. 2002; Iliff et
al. 2005; Kuhn et al. 2007). The WHO subsequently changed its recommendations to women in
developing countries to state that “breastfeeding is preferable to artificial feeding in the first six
months of life, regardless of the mother’s HIV status, as replacement feeding poses a greater risk
of death to the infant than breastfeeding from an HIV-infected mother in first months. HIV-
infected mothers are advised to wean their infants early to avoid prolonged exposure of the infant
and to avoid combining breastfeeding with replacement feeding, which appears to heighten the
risk of transmission” (WHO 2006). While this revision addresses some of the cultural barriers
presented by the former recommendation, there are still problematic aspects. The rapid weaning
advocated by the WHO is often painful for the mother and stressful for the infant, often leading
to a relapse in breastfeeding after the introduction of other foods. Furthermore, in many African
countries the typical duration of breastfeeding lasts well after the infant’s first year. Therefore,
the recommendation to wean early still presents the mother with the challenges of finding and
affording formula, clean water, and social stigmas. Furthermore, the biological mechanisms
underlying the reduction of risk through exclusive breastfeeding have not been clearly
elucidated, and the optimal duration of breastfeeding is still debated. Several studies suggest that
the risk of transmission actually declines over time, and that exclusive breastfeeding is only
necessary for the first four months (Kuhn et al. 2007). If this can be bourne out, women would be
able to avoid the painful abrupt weaning process and the problems associated with complete
replacement feeding without undue risk of transmission, and would be a more culturally
appropriate recommendation. The benefit of the anthropological genetic perspective which I
90

bring to this study is the ability to use evolutionary analyses to investigate the molecular basis of
modulated risk of MTCT, with the goal in mind of advocating a scientifically sound and
culturally sensitive breastfeeding management plan to women while eliminating unnecessarily
onerous measures.
Background
Human Immunodeficiency Virus Type 1 Infection
There are two major types of HIV: HIV-1 and HIV-2. HIV-1 is categorized into three main
subtypes, M, N, and O, which derive from separate zoonotic events in which the simian immuno-
deficiency virus (SIV) was transmitted from Pan troglodytes troglodytes (chimpanzee) (Gao et
al. 1999). Group O viruses have only been found in people living or having contact with central
Africa (mainly Cameroon and some neighbouring countries) (Gurtler et al. 1994; Loussert-Ajaka
et al. 1995), and group N viruses have only been reported from Cameroon (Simon et al. 1998).
Within group M, eleven major subtypes of HIV-1 (A-D, F-H, J-K) and at least 16 published
circulating recombinant forms (CRFs) make up the majority of infections worldwide (Leitner et
al. 2005). Subtypes are specific to geographic regions reflecting the initial routes and modes of
transmission. In the United States, Western Europe, and Australia, subtype B is found almost
exclusively, while in sub-Saharan Africa and India, subtype C is most prevalent. Subtypes can be
up to 25% different in nucleotide diversity (Perrin, Kaiser, and Yerly 2003).
The HIV-1 genome is ~10,000 bp in size and codes for the gag, pol, and env genes
characteristic of all retroviruses (Steffy and Wong-Staal 1991). Env encodes for the
glycoproteins (gp)120 and gp41, which are located on the surface of the lipid membrane
surrounding the viral particle. Gp120 binds to the host cell-surface molecule CD4, which is
expressed primarily on the surface of T-lymphocytes and macrophrages, i.e. leukocytes critical
to the immune response to infection (Maddon et al. 1986). HIV-1 also requires a co-receptor to
91

enter the host cell, principally either the CCR5 or CXCR4 chemokine receptors, which are
differentially expressed on subsets of macrophages and lymphocytes (Broder and Collman
1997). CCR5 is mainly expressed on macrophages and antigen-primed memory T-cells, while
CXCR4 is expressed on unactivated naïve T-cells and some macrophages. Different HIV viruses
differ in their ability to use each co-receptor, and are categorized as an R5 virus (uses the CCR5
co-receptor), X4 virus (uses the CXCR4 co-receptor), or R5X4 (uses both) (Berger et al. 1998;
Garzino-Demo et al. 2000). Typically, only R5 viruses are found early in infection, while X4
viruses emerge later in the majority of infected individuals (Bjorndal et al. 1997; Connor et al.
1997), and appear to evolve from earlier R5 viruses (Salemi et al. 2007).
After attachment to CD4 and the co-receptor, the viral lipid envelope fuses with the target
cell lipid membrane, which allows the viral core to enter the cell. The core is comprised of the
double stranded RNA, proteins, and enzymes. After entry, the viral RNA is reverse-transcribed
into double-stranded DNA in the cytoplasm and transported to the nucleus as a pre-integration
complex. The viral integrase enzyme integrates the viral DNA through linkage between the long
terminal repeats at each end of the viral DNA and in the host DNA. The viral DNA is then
transcribed into viral RNA, which either remains intact or else is spliced and transported to the
cytoplasm for translation into regulatory and polyproteins. These proteins along with the full-
length RNA transcripts are packaged into viral particles that bud from the surface of the infected
cells and enveloped by the host cell membrane. The viral particle must undergo a final
maturation step in which the gag polyproteins are cleaved by viral protease (Goodenow et al.
2003).
Env gp120 is comprised of five conserved regions (C1-C5) and five variable regions (V1-
V5). The determinants for co-receptor use are localized to the V3 loop of gp120 (Carrillo and
92

Ratner 1996; Hung, Vander Heyden, and Ratner 1999). In subtype B viruses, the net charge of
the amino acids in the V3 loop in gp120 is predictive of co-receptor usage (Briggs et al. 2000).
However, in other subtypes, the correlation may not be as strong. Env can differ by up to 5%
within an individual (Lamers et al. 1993), and can differ up to 30% among subtypes. N-linked
glycosylation sites are prevalent in gp120, and are involved in the structure and folding of the
protein. The glycosylation sites can also form a glycan shield to block neutralizing antibody
response (Wei et al. 2003). The V1 and V2 regions display considerable diversity in terms of
number of glycosylation sites, length, and amino acid variation over the course of infection in
one individual (Hughes, Bell, and Simmonds 1997a; Klevytska et al. 2002; Kitrinos et al. 2003;
Nabatov et al. 2004; Ritola et al. 2004; Sagar et al. 2006). Furthermore, functional (Pastore et al.
2006) and phylogenetic (Salemi et al. 2007) studies suggest that X4 sequences evolve according
to a consistent program of development that is recapitulated in individual patients and that
requires compensatory mutations in V1V2 to occur prior to emergence of high charge V3
domains. Thus, analyzing the diversity in gp120 provides both phylogenetic signal as well as
identification of potentially relevant functional changes over time.
Stages of Breastmilk Production
The initial HIV-1 infection in the breastmilk is poorly understood. Breastmilk is a complex
composition of cells, proteins, water, ions, fats, vitamins and minerals. Secretory alveolar
epithelial cells in the mammary gland surround multiple lumina, which are storage chambers for
the milk. Lactogenesis I refers to the onset of secretion of milk components to form colostrum
(i.e. the early milk), which occurs at 16-24 weeks of pregancy (Arthur, Smith, and Hartmann
1989). During this phase, the epithelial cells of the alveoli differentiate into secretory cells,
which then begin to synthesize lactose, casein, and milk fat triglycerides, and are secreted into
the lumen. The alveolar epithelial cells also extract water, vitamins, and minerals from the blood
93

capillaries surrounding the milk duct. During this stage, the epithelial cells are not tightly joined,
which allows elements from the blood such as immune cells and plasma protein to directly enter
the lumen via the paracellular pathway (Neville and Neifert 1983), as well as HIV-1 infected
cells or viral particles from the plasma. Colostrum contains higher concentrations of sodium,
chlorine, and proteins than mature milk (Georgeson and Filteau 2000), as well as a higher
proportion of leukocytes (white blood cells) (Goldman 1993), and is optimized for the infant’s
requirements in the first days after birth. All of the milk components remain in the lumina until
the suckling infant stimulates activation of the hormone oxytocin, which in turn causes the
contraction if the alveolar cells and the flow of the milk through the duct system (Fuchs 1991).
Lactogenesis II is defined as the initiation of large amounts of milk which is triggered by a
decrease in progesterone a few days post-partum (Neville et al. 1991). Biochemical changes
include increase in lactose and glucose and a decrease in sodium and protein resulting from the
closure of tight junctions between the epithelial cells. This closure is reversed during weaning
when the milk volume falls to <400 ml/day which corresponds to <2 feedings per day and
corresponds with significant increases in sodium, chloride, and protein and a decrease in lactose
(Neville et al. 1991). Inflammation of the breast (mastitis) also causes the opening of the
paracellular pathway, and increased sodium and albumin levels are associated with mastitis
(Shuster, Kehrli, and Baumrucker 1995; Semba et al. 1999b; Becquart et al. 2000; Rollins et al.
2001). The increased HIV-1 loads associated with both weaning (Thea et al. 2006) and mastitis
have been suggested to result from these “leaky ducts” in which cell associated (viral DNA) and
cell-free (viral RNA) virus can more efficient transfer from the plasma to the milk (Kuhn et al.
2007).
94

Cellular Composition of Breastmilk
The relative composition of cells in the breastmilk has not been consistently reported, due
to the changing composition of the milk over time, the storage conditions of the expressed milk,
and the use of different measurement techniques (Kourtis et al. 2003). Early studies suggested
that macrophages predominate both in the colostrum (Crago et al. 1979) and in mature
breastmilk (Ho, Wong, and Lawton 1979; Pitt 1979). However, later studies suggest that
polymorphonuclear cells (neutrophils) comprise 80% of all cell types, followed by macrophages
(15%) and lymphocytes (5-10%), most of which are T-lymphocytes (Goldman, Chheda, and
Garofalo 1998). The leukocyte concentration is highest during early lactation and decreases 5-10
fold by the end of the first week post-partum (Goldman 1993; Georgeson and Filteau 2000).
Since macrophages and T-lymphocytes are the primary cells infected by HIV-1, the high
frequency of these cells in the breast milk represent a large target cell population.
The phenotype and functional characteristics of milk T cells are different than peripheral
blood T cells (Bertotto et al. 1990b). Almost all of the breastmilk T-cells are memory cells, as
evidenced by high expression of activation markers such as HLA-DR, CD25, and CD45RO
(Bertotto et al. 1990b; Wirt et al. 1992; Rivas, el-Mohandes, and Katona 1994; Kourtis et al.
2003). A large percentage of T cells express mucosal homing markers such as CD49f, alpha4-
beta7 integrin, and CD103+, suggesting that the T cells found in milk migrate from other tissues
in the body where they were originally activated (Bertotto et al. 1990b; Kourtis et al. 2003). A
possible source for the T-cells is the gut associated lymphoid tissue (GALT) (Manning and
Parmely 1980; Kourtis et al. 2003), which is supported by the high frequency of cells carrying
the gamma/delta T-cell receptor in both the GALT (Ullrich et al. 1990) and in the breast milk,
but not the plasma (Bertotto et al. 1990a). The GALT contains the majority of the T-cells in the
human body, and during initial HIV infection experiences almost a complete depletion of T-cells
95

due to high levels of infection (Douek 2007b; Douek 2007a). If the milk is being populated by
GALT-derived T-cells, many of the cells are likely infected with HIV-1 and are the source of
infection.
Macrophages in the breastmilk also have distinct characteristics from those in the blood.
For example, they spontaneously produce granulocyte monocyte colony stimulating factor (GM-
CSF) and can differentiate into CD1+ dendritic cells in the presence of interluken-4 (IL4) alone,
in contrast with monocytes in the peripheral blood mononuclear cells (PBMC), which require
GM-CSF and IL4 (Ichikawa et al. 2003). IL-4 stimulated breastmilk macrophages express DC-
SIGN, a dendritic cell-specific receptor for HIV-1. During mastitis, IL-4 is locally produced,
which may then up-regulate DC-SIGN expression, which could lead to increased HIV-1
infection of macrophages (Ichikawa et al. 2003). This is an alternative explanation for the high
levels of viral load in the breastmilk associated with mastitis, rather than the leaky duct
hypothesis.
Epithelial cells from the mammary gland itself were reported present in mature milk at low
levels (Xanthou 1997) although another study found that almost 80% of all cells were epithelial
(Petitjean et al. 2007). Mammary epithelial cells can become productively infected with HIV-1
(Toniolo et al. 1995), and therefore the infection in the breastmilk could originate from either
infected epithelial cells shedding into the lumina, or else cell-free viral particles produced by the
epithelial cells pass into the milk and infect T-cells and macrophages already there.
Compartmentalization of Breastmilk Virus
As discussed above, the origin of the virus in breastmilk is unclear, and the molecular
genetic evolution of the virus in breastmilk over time has not been extensively investigated. One
important question that remains to be answered is whether the virus in the breastmilk
compartmentalizes after the initial infection, i.e, forms a separate population initially due to
96

restricted gene flow with other tissues (Nickle et al. 2003), and accelerated by the high in vivo
mutation rate (Saag et al. 1988) and differential selective pressures (Haase et al. 1996; Pilcher et
al. 2001). This is an important question because viruses in different compartments may exhibit
differential pathogenesis (Donaldson et al. 1994), response to drug therapy (Si-Mohamed et al.
2000; Venturi et al. 2000; Smit et al. 2004) and potentially different transmission rates.
Compartmentalization has already been established for the genital tract in men and women (Zhu
et al. 1996; Poss et al. 1998; Ping et al. 2000; De Pasquale et al. 2003; Kemal et al. 2003;
Philpott et al. 2005; Pillai et al. 2005; Sullivan et al. 2005), which as the most common route of
HIV transmission worldwide (Royce et al. 1997) has tremendous implications for our
understanding of the transmission and initial seeding of infection. If particular characteristics of
the virus infecting the genital tract can be identified, such as tropism, co-receptor usage, viral
epitopes, or structural characteristics, then vaccines and drug intervention could potentially be
developed to target these attributes. Because the virus both within and between individuals is so
variable, interventions will be more efficient if particular subsets of viruses can be targeted and
neutralized, and eradicating the early virus before it can infect other tissues is critical.
Distinct viral populations have also been identified in the central nervous system due to the
blood-brain barrier, which is important because many anti-retroviral drugs are unable to
penetrate the brain, and the proliferation of the virus there can lead to AIDS-associated dementia
(Korber et al. 1994; Hughes, Bell, and Simmonds 1997b; Gatanaga et al. 1999; Morris et al.
1999; Shapshak et al. 1999; Staprans et al. 1999; Venturi et al. 2000; Smit et al. 2001; Wang et
al. 2001; Ohagen et al. 2003; Langford et al. 2004; Petito 2004; Smit et al. 2004; Thompson et al.
2004; Abbate et al. 2005; Burkala et al. 2005; Ritola et al. 2005; Salemi et al. 2005; Strain et al.
2005; Pillai et al. 2006). Lymphoid, spleen, and lung tissues are also subject to
97

comparmentalization (Wong et al. 1997; Salemi et al. 2007). The few studies that have
investigated compartmentalization in the breastmilk have found conflicting results. One study
found compartmentalization between breastmilk virus DNA and RNA with respect to plasma and
PBMC virus (Becquart et al. 2002). A second study found that viral variants were similar
between plasma and milk (Henderson et al. 2004). However, both studies only considered the
virus from a single timepoint in each patient. In addition, the median time since delivery in the
first study was 3 months versus 12 months in the second study, which may explain the
apparently contradictory results if compartmentalization is not static, but changes over time.
Risk of Transmission via Breast-feeding
Increased transmission has been associated with the mother’s health status and the method
of feeding. In particular, an increased risk of transmission is correlated with a high viral load in
the mothers plasma (John et al. 2001; Fawzi et al. 2002a; Rousseau et al. 2003) and breastmilk
(Van de Perre et al. 1993; Lewis et al. 1998; Semba et al. 1999a; Pillay et al. 2000; Rousseau et
al. 2003; Rousseau et al. 2004). Breastmilk RNA concentrations are typically 2-3 log lower than
the plasma viral load, though the two are highly correlated (Rousseau et al. 2004). RNA viral
loads were found to be significantly higher in the colostrum than in the mature milk produced 14
days after birth (2.59 log10 copies/ml vs. 2.19 log10 copies/ml) but did not significantly change
from 14 days to 15 months (Rousseau et al. 2004). Although this may explain earlier studies
suggesting that most transmission events occur during the first 6 weeks after delivery (Miotti et
al 1999), it can be difficult to determine if an infant was infected via breastfeeding or intra-
partum early in life.
Both cell-free (RNA) and cell-associated (DNA) viral load independently increases the
risk, although the two measures are highly correlated (Richardson et al. 2003; Koulinska et al.
2006). A ten-fold increase in breastmilk RNA is associated with a two-fold increase in risk of
98

transmission, while a ten-fold increase in infected cells (DNA) is associated with a three-fold
increase in transmission (Rousseau et al. 2003; Rousseau et al. 2004). RNA was detected in one
study in 57% of all breastmilk samples from HIV-1+ women, and in 74% of all samples from
women who transmitted the virus (Koulinska et al. 2006). Cell associated (DNA) virus was
detected in 74% of all breast milk samples from HIV+ women and in 87% of the samples from
women who transmitted via breastfeeding (Koulinska et al. 2006). Interestingly, RNA virus was
significantly associated with late transmission of HIV-1 (>9 months post-partum) while DNA
virus had no time-dependent association.
Other factors increasing the risk of transmission via breastmilk include a low CD4 count
(Leroy et al. 2003; Iliff et al. 2005), mastitis (Van de Peere et al. 1992), and potentially sub-
clinical mastitis (the symptoms are not evident), which are all associated with high viral loads
(Semba et al. 1999b; Willumsen et al. 2000). If the mother was infected after birth, the risk of
MTCT may be increased 6-fold due to the high viral loads associated with primary infection
(Embree et al. 2000). Maternal nutrition status may also be associated with transmission; vitamin
A supplementation was associated with an increased risk, while multivitamin use was associated
with a lower risk (Fawzi et al. 2002b). A possible explanation for these results is the recent
observation that env gp120 binds to an activated form of the integrin alpha4-beta7 on CD4 T
cells which facilitates infection of neighboring cells. Retinoic acid, which is derived from
vitamin A, activates alpha4-beta7 and promotes binding to gp120. Interestingly, the function of
alpha4-beta7 is to act as a homing receptor for T-cells migrating to the GALT (Arthos et al.
2008). Therefore, a possible hypothesis is that increased vitamin A supplementation by the
mother may be passed to the infant through the breast milk, which activates alpha4-beta7 in the
99

gut of the infant, which in turn promotes binding of gp120 to CD4+ cells in the gut, leading to
increased infection.
The mode of breastfeeding is also associated with differential risk and is the basis for
modification of the WHO guidelines to HIV+ mothers concerning breastfeeding. Recent
observational studies have found that exclusive breastfeeding appears to lower the risk of
transmission over mixed feeding, which is defined as providing the infant with any food other
than water. A study in Durban, South Africa demonstrated an equal rate of infant HIV-1
infection by three months among mothers who had never breastfed and whose infants were
negative at birth (13.2%) and mothers who had exclusively breastfed up to 3 months (8.3%),
compared to a significantly higher infection probability for infants who were mixed fed (19.9%)
(Coutsoudis et al. 1999; Coutsoudis et al. 2001). In a follow-up study of the same cohort, the
cumulative number of HIV-infections (including inter-uterine and intra-partum) was 19.4% for
both the non-breastfeeding and the exclusively breastfeeding mothers, compared to 26.1% for
the mixed-feeding mothers by six months (Coutsoudis et al. 2001). In another study in South
Africa, the cumulative probability of infection at six months for exclusively breastfed infants was
15% compared with 7% for non-breast fed infants and 27% for mixed-fed infants (Coovadia et
al. 2007). In a study of >2000 mothers in Zimbabwe, infants who were HIV negative at six
weeks and who were exclusively breast fed were significantly less likely to be infected at six
months (1.31%) than mixed fed infants (4.4%) (Iliff et al. 2005), while in a study in Zambia, 4%
of exclusively breastfed infants who were negative at six weeks were infected by 4 months,
compared to 10% of mixed fed infants (Kuhn et al. 2007). The mechanism for the reduced risk
via exclusive breastfeeding has not been demonstrated. One possibility is that damage to the
infant’s gut mucosa from early introduction of food compromises the intestinal mucosal barrier
100

or intestinal immune activation from early introduction of foreign antigens or pathogens
facilitates transmission (Smith and Kuhn 2000). Mixed feeding could be associated with
suboptimal breastfeeding, subclinical mastistis (in which no swelling of the mammary gland is
visible during inflammation), or poor health in general which leads to higher viral loads
(Willumsen et al. 2000), so the association between mixed feeding and transmission is complex
(Chisenga et al. 2005). However, the study in Zambia controlled for viral load and maternal
health (as measured by CD4 cells) and still found a significant association (Kuhn et al. 2007).
The increased risk in transmission upon cessation of exclusive breastfeeding may also be related
to the increase in mammary epithelial permeability upon weaning (Kourtis et al. 2003). This
hypothesis is supported by both the finding that the mean concentration of plasma albumin was
higher in the milk of transmitting mothers (Becquart et al. 1999) as well as the increase in
breastmilk viral load following weaning (Thea et al. 2006).
In addition, the duration of the breastfeeding has been suggested influence the risk of
transmission, although the association is not clear. Over a two year period, the risk of
transmitting the virus through breastfeeding was estimated at 16% (37% for mothers who did
breastfeed, vs. 21% for mothers who did not) (Nduati et al. 2000). A study in Zimbabwe found
that more than 2/3 of all post-natal MTCT occurred after six months (Iliff et al. 2005) while a
study in Kenya found that the majority of transmission events occurred before six months
(Nduati et al. 2000). Other studies have found an approximately constant risk over time
(Coutsoudis et al. 2004) or a reduced risk over time (Kuhn et al. 2007). The contradictory results
are difficult to interpret, although it is difficult to compare across studies because of the different
interpretation of intra-partum vs. early breastfeeding transmission and the non-distinction of
exclusive vs. non-exclusive breastfeeding. In a study in Zambia, the risk of HIV-1 transmission
101

during non-exclusive breastfeeding was shown to be greater during the first four months (~2.4%
per month) compared with 1% from 5-12 months, and 0.5% from 12-24 months, suggesting that
the greatest benefit for exclusive breastfeeding is only until month 4. The risk of continuing to
breastfeed non-exclusively was compensated by the mortality associated with not breastfeeding,
and no difference in HIV-1 free survival was detected between mothers who stopped breast-
feeding at four months versus mothers who continued up to 16 months (Kuhn et al. 2007;
Sinkala et al. 2007). Therefore, while the cumulative probability of HIV-1 transmission may
increase over time, the more important measure of HIV-1 free survival may be increased with
continued breastfeeding.
Our Study
The current WHO recommendations concerning breastfeeding by HIV+ mothers are based
on several observational studies that provide strong evidence for the attenuation of risk of MTCT
by exclusive breastfeeding. However, although the WHO advises women to abruptly wean at 6
months, recent data suggest a decreased risk of MTCT over time that is outweighed by the
benefits of continued breastfeeding (Kuhn et al 2007). Furthermore, while exclusive
breastfeeding is critical during the first several months post-partum, after 4 months the benefits
are less evident (Kuhn et al 2007). Clearly, a better understanding of the molecular mechanisms
underlying the observations is needed in order to provide HIV-1 positive women with the
optimal recommendations.
HIV-1 rapidly evolves within a patient in response to constantly changing host pressures.
Within several months of infection a new viral population may emerge, often with functionally
significant differences. New viral populations may have an altered entry, replication, or immune
evasion phenotype, which could impact the overall pathogenicity or transmissibility of the virus.
Therefore, investigating the viral evolution over several years can provide important information
102

about both the characteristics of the virus as well as the dynamic host environment. It is likely
that features of the virus at particular junctures during the course of breastfeeding may impact its
ability to initiate a new infection in the infant. Understanding the evolutionary dynamics of the
milk virus may elucidate the underlying cause for the attenuated risk of MTCT associated with
exclusive breastfeeding as well as the reduced risk of transmission over time.
This study is the first to investigate the longitudinal evolution of HIV-1 in breastmilk with
a focus on elucidating important factors underlying transmission to the infant. I have posed
several questions designed to address aspect of the viral evolution that may be important in
transmission. First, I wanted to determine the means by which virus enters the breastmilk.
Specifically, I investigated if breastmilk is infected by virus from the plasma during the initial
stages of lactogenesis, or if the early breastmilk virus is distinct from the virodeme (HIV-1
subpopulations) in the peripheral blood system suggesting an alternative infection. Second, I
wanted to learn more about the characteristics of the virus when it is in breastmilk. Specifically, I
investigated if virus in the breastmilk evolves into a separate population distinct from the
peripheral virus population, indicating compartmentalization. Related to that question, I also
investigated to what degree the virus migrates between tissues. Third, I wanted to learn more
about what role natural selection might play in the evolution of the virus. Specifically, I explored
what selective pressures are present during the evolution of the breastmilk virus, and whether
they differed between the plasma and the milk. Finally, I wanted to know if the characteristics of
the virus in breastmilk change over time. Specifically, I considered whether particular
evolutionary dynamics between four and six months could be associated with either the
documented cessation of exclusive breastfeeding or the transmission of the virus to the infant. To
address these questions, I analyzed the population of viruses, or virodeme, present in the breast
103

milk (left and right) and plasma RNA over a two-year period (timepoints at birth, 1, 4, 12, 18,
and 24 months) from an HIV-1 positive woman who was documented to have transmitted the
virus to her infant via breastfeeding at four months post-partum.
Materials and Methods
Subject
The HIV-1 positive female subject in this study was enrolled in the Zambia Exclusive
Breast Feeding Study (ZEBS). The goal of the ZEBS was to determine whether exclusive
breastfeeding was associated with a lower risk of transmission (Thea et al. 2004; Kuhn et al.
2007). All women were encouraged to breastfeed exclusively until month 4, at which time
women were randomized into two groups: the control group, in which women were encouraged
to exclusively breastfeed until six months and then gradually introduce complementary foods,
and the intervention group, in which women were encouraged to rapidly wean their infants after
four months (Thea et al. 2004; Kuhn et al. 2007). This patient was part of the control group and
breastfed until 18 months post-partum. She reported exclusively breastfeeding until four months,
and reported mixed feeding at six months, which was defined as providing the infant with any
non-maternal milk substance including water and cow’s milk. Her infant had the first positive
PCR for HIV at 4 months, and negative PCRs at 1 week, and 1, 2, and 3 months, strongly
suggesting that breastmilk was the vehicle of transmission. Breast milk and blood were serially
sampled from the mother at 1 week, 1 month, 4 months, 12 months, 18 months, and 24 months
(see Figure 5-1).
Viral Isolation, Amplification, and Sequencing
RNA was isolated from breastmilk and plasma samples using a Qaigen RNA Easy kit
(Qiagen, Valencia, CA, USA) and 10 ul of RNA extract was used as template in a One-Step RT-
PCR kit (Invitrogen, Carlsbad, CA, USA) according to manufacturer’s instructions. To amplify
104

the V1V5 region of the env gp120 gene, I used first round primers pol-env (5’-
GAGCAGAAGACAGTGGCAATGA-3’) and 192H (5’-CCATAGTGCTTCCTGCTGCT-3’).
Cycling conditions were as follows: 50ºC for 30’, 94ºC for 2’, followed by 35 cycles of 94ºC for
15s, 58ºC for 30s, 72ºC for 2’, and a final extension step of 72ºC for 10’. A second nested PCR
was performed using the GoTaq PCR Supermix (Promega, Madison, WI, USA) according to
manufacturer’s instructions using 5ul of the first round amplification as template and primers D1
(5’-CACAGTCTATTATGGGGTACCTGTGTGGAA-3’) and 194G (5’-
CTTCTCCAATTGTCCCTCATA-3’) with cycling conditions as follows: an initial denaturing
step of 95ºC for 5’, 35 cycles of 94ºC for 1’, 58ºC for 1’, and 72º for 2’, followed by a final
extension step of 72ºC for 10’. In some cases, a third round PCR was necessary using 5ul of the
second round as template, primers Env5 (5’-
GGGGATCCGGTAGAACAGATGCATGAGGAT-3’) and 194G, and the same cycling
conditions as described for the second round PCR. Additional sequences were generated for the
plasma 1 week, 12 month, and 24 month samples using 1ul of RNA template to determine
whether larger input volumes of RNA were contributing to PCR-generated recombination. PCR
products were ligated into TopoTA vector (Invitrogen, Carlsbad, CA, USA) according to the
manufacturer’s instructions, transformed into top10F’ cells for 30’ on ice, followed by a 30s
heatshock at 42 ºC, and incubation overnight at 30 ºC. In sum, approximately 30 clones from
each sample were sequenced in both directions by the Genome Sequence Service Laboratory at
the University of Florida yielding 284 independent sequences.
Sequence Analysis and Recombination
The V1V5 sequences were manually aligned and checked for accuracy using BioEdit v7.0
(Hall, 1999) and Mega v4.0 (Tamura et al. 2007). V1 and V2 haplotypes were assigned based on
sequence similarity. Length and number of glycosylation sites were calculated manually.
105

The entire V1V5 region as well as shorter regions of the gp120 gene were evaluated for
recombinant sequences based on an algorithm that I helped developed that uses the pairwise
homoplasy index (PHI) test in conjunction with phylogenetic networks (Salemi MM, Gray RR,
Goodenow MM, unpublished). The PHI test statistic is a modified sum of incompatibility scores
calculated for pairs of informative sites in an alignment. The PHI statistic is then assessed with a
normal approximation of a permutation test, and has been shown to be a powerful method with
simulation studies (Bruen, Philippe, and Bryant 2006). In our method, viral populations were
tested for significant population structure using the K*s test (Hudson, Slatkin, and Maddison
1992; Achaz et al. 2004) and divided into smaller datasets by time point if significant population
structure was detected. A neighbor-net phylogenetic network was inferred for each dataset,
which allows the presence of phylogenetic uncertainty, and sequences were progressively
removed until the PHI test statistic was no longer significant (p>0.05). The smaller datasets were
again combined, and the procedure repeated to detect inter-time point recombinants. All putative
recombinants were removed from the dataset for further analysis. Pairwise calculations were
performed for the non-recombinant alignment using Mega v4.0 with the Kimura 2-Parameter
model with pairwise deletions and uniform rates.
Phylogenetic Analyses
Phylogenies were estimated for non-recombinant datasets using a Bayesian analysis with
the BEAST software package 1.4 (Drummond et al. 2005; Drummond and Rambaut 2007). The
evolutionary rate was initially estimated using a model assuming a strict clock, the SRD6 model
of nucleotide substitution (HKY + the 1st and 2nd codon positions were considered separately
from the 3rd , hereafter referred to as two partitions (Shapiro, Rambaut, and Drummond 2006)),
and a constant population size with sampling time information included in the model
(Drummond et al. 2006). The rate obtained from this model was then used as a prior in further
106

analyses that assumed a relaxed clock with log-normally distributed rates with three models of
nucleotide substitution: the SRD6 model, the gamma time-reversible (GTR) model with two
partitions, and the GTR model with three partitions (all three codon positions considered
separately). The Markov Chain Monte Carlo analysis was run for 100,000,000 generations with
sampling every 10,000th generation. The results were visualized in Tracer v.1.3, and convergence
of the Markov chain was assessed by calculating the effective sampling size (ESS) for each
parameter (Drummond et al. 2006). All ESS values were >500 indicating sufficient sampling.
The time to the most recent common ancestor (TMRCA) was estimated for each analysis. The
marginal likelihoods of each model were compared using the Bayes Factor method, in which a
difference of greater than 20 log units between the marginal likelihoods of any two models is
considered significant evidence for the alternative model (the more complex model). The correct
root of the phylogeny was analyzed by constraining all sequences except particular groups from
the first timepoint as the ingroup using the SRD6 model with the relaxed clock and log-normal
distribution of rates. This was performed for five sets of outgroup sequences, and the marginal
likelihoods were compared using the Bayes Factor test. A 50% consensus tree based on the
posterior distribution of trees with a 50% burnin was calculated using Mr. Bayes and
manipulated in FigTree v.1.0.
A maximum likelihood (ML) phylogeny was inferred using PAUP v. 4.0 (Swofford 2002).
The best-fitting nucleotide substitution model was tested with a hierarchical likelihood ratio test
using a neighbor-joining tree with Jukes and Cantor corrected distances. Statistical support for
internal branches in the tree was obtained by bootstrapping (1,000 replicates). All rootings of the
best ML phylogeny were generated in MacClade (Maddison and Maddison 1989). The most
likely rooting was determined using baseml in the PAML package (Yang 1997) with the clock
107

hypothesis and sampling information included with the sequences. The best rooted tree was then
re-estimated without the clock hypothesis in PAML.
To determine the subtype of this patient, one sequence from each tissue (plasma, left and
right breast milk) from week 1 was aligned to representative sequences from each major subtype
in macrogroup M obtained from the Los Alamos database
(http://www.hiv.lanl.gov/content/sequence/HIV/SUBTYPE_REF/align.html). A neighbor-
joining tree was inferred using PAUP v. 4.0 with the HKY model of substitution, estimated
transition/transversion ratio, an estimated gamma distribution of rates and an estimated
proportion of invariable sites. Statistical support for internal branches in the tree was obtained by
bootstrapping (1,000 replicates).
Branch Selection Analysis
A branch selection analysis was performed using HYPHY (Pond, Frost, and Muse 2005).
The synonymous/non-synonymous rate ratio (dN/dS) was estimated for each branch leading to a
major clade, as well as the 95% confidence interval (CI). All branches for which the CI did not
include 1.0 were determined to significantly deviate from neutral evolution.
Compartmentalization
Compartmentalization between tissues was investigated using a modified version of the
Slatkin-Maddison test (Slatkin and Maddison 1989) implemented in MacClade (Maddison and
Maddison 1989) using the State Changes and Stasis option. The number of unambiguous
instances in which an ancestral state changed from one tissue to the other, as well as all instances
in which no change occurred, were calculated for each phylogeny sampled from the posterior
distribution in BEAST with a 25% burnin and 10,000 random splitting and joining trees. The
Kolmogorov-Smirnov nonparametric test was used to compare the distribution of each category
(breastmilk to breastmilk, breastmilk to plasma, plasma to plasma, and plasma to breastmilk) for
108

the estimated and random trees. The nonparametric Wilcoxon Rank Sum test was used to
compare the median value for each category between the estimated and random phylogenies.
Results
Subtype Analysis
The neighbor-joining tree using representative sequences from each major subgroup in
macro-group M is shown in Figure 5-2. A high bootstrap value (100) supports the placement of
this patient into subtype C.
Sequence Analysis
An initial dataset of 184 sequences was obtained and used in the majority of analyses
(Table 5-1). Additional sequences were generated for three plasma samples (1W, 12M, 24M) to
rule out the possibility of PCR-generated recombination. These sequences were not included in
any of the phylogenetic analyses. Additional sequences were also generated for milk samples
from month 1. These sequences were included in a subset of phylogenetic analyses as noted
below.
Variable regions 1 and 2 sequence analysis
All generated sequences (n=282) were included in the sequence analysis. Because the
length variation in the V1V2 region may confound phylogenetic analyses, this variation was
analyzed separately. All generated sequences (n=282) were included. For V1, six different
haplotypes were assigned (A-F) based on amino acid (aa) motif, length, and number of
glycosylation sites. Representative sequences for each haplotype are shown in Figure 5-3. The
length of V1 ranged from 29aa-51aa, and the number of gylcosylation sites ranged from 1-5.
Haplotypes D, B, and C were most prevalent (in that order), and share similar aa motifs but differ
in length. For V2, five haplotypes (A-E) were assigned. Representative sequences for each
haplotype are shown in Figure 5-4. The length of V2 ranged from 40aa-51aa, and the number of
109

gylcosylation sites ranged from 1-3. Haplotype B was most prevalent, with the remaining
sequences somewhat evenly divided between the other haplotypes. The amino terminus was
fairly well conserved among sequences, as was the leu-asp-iso motif that mediates binding of
activated integrin a4b7, which mediates migration of CD4 T-cells to the gut (Arthos et al. 2008).
In V1, the plasma shows a trend of increasing diversity over time (number of haplotypes),
with the haplotypes present at the earliest timepoints being retained (Table 5-2). The longer
haplotypes C and D are present at the first timepoint, while the shorter haplotype B emerges at
month 12 at a low frequency. Haplotype B increases to almost 50% by month 24, which suggests
that a trend of shortening V1 length and loss of glycosylation sites over time. However, because
haplotype B is present in the breastmilk at the first time point (BMR1W), it is unclear whether
the presence of haplotype B in the plasma by 12 months is this is the result of a migration from
the breastmilk to the plasma, or whether the viral population in the plasma evolved into
haplotype B independently (Table 5-2). Haplotype A, which is the most divergent from
haplotypes B, C, and D, remains at a low frequency at all timepoints in the plasma. Haplotype E,
which has a unique aa motif, emerges only at month 24. There is less diversity in the breastmilk
sequences at most timepoints relative to the plasma. At week 1, the left breast has only a subset
of haplotypes found in the right breast. Haplotype B is unique to the breastmilk, and while two of
the haplotypes (A and C) are present in both the plasma and the breastmilk, haplotype A is found
at very low frequencies in the plasma. By month 1, a new haplotype emerges in the breastmilk
(F), which is not found at any other timepoint or tissue, but is found in both breasts. In fact, the
viral population in the left breast seems to have experienced a complete population replacement
from haplotype A in week 1 to haplotypes B and F at month 1. In contrast, the viral population in
the right breast displays more continuity from week 1 to month 1. This pattern suggests either
110

limited gene flow from the right to the left breast, or else the left breast is infected with only a
subset of the viral population infecting the right breast. Haplotype D is present in the right breast
(but not the left) at month 1, which at week 1 was in the plasma but not the breastmilk. At month
4, haplotype D is the most frequent in the both the breastmilk and the plasma, and by the last two
timepoints only haplotype D is found in the breastmilk. Interestingly, this is the opposite trend of
the plasma, in which diversity increased over time. This could suggest that at month 1, the right
breast began to experience some gene flow from the plasma, though the left breast remained
compartmentalized. By month 4, there was complete geneflow between both breasts and the
plasma. By the last timepoints, the gene flow from the plasma subsided (Table 5-2).
In V2, a similar trend of increasing diversity over time in the plasma is apparent (Table 5-
2). Again, the haplotypes present at month 24 includes all haplotypes seen at previous timepoints
with the exception of haplotype E that was present at month 4. Neither the length nor the number
of glycosylation sites appear increase or decrease over time, although the diversity again
increases over time. In the breastmilk, again the haplotypes in the left breast are a subset of those
found in the right at both week 1 and month 1, and again a population turnover is apparent in the
left breast from week 1 to month 1. At month 4, the same haplotypes are found in the breastmilk
and the plasma, and again by 12 and 18 months only one haplotype is found in the breastmilk.
There is no clear association between particular haplotypes in V1 and V2. This may be due
to recombination between the two regions or to convergent evolution, as the difference between
some of the haplotypes is only a few amino acids. Overall, there is a clear trend in the plasma of
increasing combinations of haplotypes, while the opposite trend in true in the milk.
Variable region 3 loop analysis
All sequences had V3 loop charges of <5, which in subtype B is predictive of CCR5 co-
receptor usage (data not shown). However, the association between charge and co-receptor usage
111

is not as defined in subtype C. Further functional analyses are being performed to determine
actual tropism and co-receptor usage.
Recombination Analysis
The presence of population structure was tested for each group of sequences from
different tissues and timepoints. All inter-tissue and inter-timepoint comparisons demonstrated
significant population structure (p>0.001) except for the three tissues sampled at month four,
which showed no population structure (Table 5-4). These sequences were therefore considered as
one group for the recombination test.
The V1-V5 alignment was initially tested for the presence of putative recombinants using
the PHI/phylogenetic method described above with a dataset of 184 sequences. However, a non-
significant inter-timepoint dataset could not be obtained using more than half of the sequences.
Therefore, the alignment was shortened into four new alignments: V1-V3, V1-V2, C2-V3, and
C2-V3 (see Figure 5-5). For the V1-V3 dataset, again a non-recombinant dataset could not be
obtained. The full V1-V2 dataset initially suggested the presence of recombinants (p=8.43 x 10-
7), and 27 sequences could be removed so that no recombination was detected (Table 5-5). For
the C2-V3 dataset, no recombinants were detected. For the C2V5 dataset, initially the dataset
was again unable to be resolved. However, the removal of ~10 amino acids at the carboxyl end
of V3 as well as all of V4 resulted in a non-recombinant dataset. These results suggested that a
hotspot for recombination is located at the amino terminus the V2 region or the carboxyl
terminus of the C2 region of the gp120 gene.
Two alternate datasets could be produced for the C2V5 alignment, depending on which week
1 breastmilk sequences were removed. The network for these sequences is shown in Figure 5-6
(when all sequences were included, p=4.06 x 10-4, indicating a significantly recombinant
dataset). Four distinct groups are evident: Group 1 contains all of the sequences from the left
112

breast, while the sequences from the right breast form groups 2-4. Two of these groups could be
removed separately to result in a non-recombinant dataset: Group 3 plus two additional non-
Group 3 sequences (p= 0.076) or Group 1 (p=0.376). Because choosing the first alternative
resulted in less putative recombinants overall (31 vs. 34), and to avoid removing all of the
representatives from the left breast, the first alternative (C2V5 [A]) was considered the better of
the two options. However, to confirm some of the phylogenetic results, the second alternative
(C2V5 [B]) was also used. Other than the breastmilk week 1 sequences and the three additional
recombinants, the other sixteen recombinants were identical between C2V5 (A) and (B).
In order to confirm that the multitude of recombinant sequences was not due to PCR-
generated recombination, additional plasma samples from week 1, month 12, and month 24 were
re-amplified using 1/10 the input RNA, as increased template has been suggested to cause in
vitro recombination. The new sequences were added to the C2V5 alignment, and the dataset was
re-tested for recombinants. Similar variants were recovered for all three timepoints, and again the
majority of the plasma recombinants were found in the month 24 sample, suggesting that the
recombinant sequences are actually present in vivo.
The breastmilk sequences sampled from month 1 were added to the C2V5 (A) alignment. No
additional recombinants were detected among these sequences.
Phylogenetic Analyses
Bayesian tip-date phylogeny
The C2V5 (A) and C2V5 (B) alignments were used to infer a posterior distribution of trees
using the program BEAST under three models of nucleotide substitution (SRD6, GTR + 2
partitions, and GTR + 3 partitions) with a relaxed molecular clock, and the SDR6 model
assuming a a strict molecular clock. For the C2V5 (A) dataset, the Bayes Factor of the GTR + 2
partition model was >20 log higher than either the SRD6 model or the strict clock model,
113

suggesting a significantly better fit to the data, while the GTR + 3 model was <20 log higher than
the GTR +2 model, suggesting unnecessary parameterization (Table 6). For the C2V5 (B)
dataset, both GTR models resulted in very low ESS values after 100,000,000 generations of the
MCMC chain, and thus these models were not considered. The Bayes Factor for the SRD6
model with the relaxed clock was >20 log higher than the strict clock (data not shown), again
suggesting that the strict clock could be rejected for both datasets.
The posterior distribution of trees generated under the best model (as identified above) was
used to generate a 50% consensus phylogeny for C2V5 (A) (Figure 5-7) and C2V5 (B) (Figure
5-8). Both phylogenies demonstrated similar topologies with strong temporal evolution. In
phylogeny (A), breastmilk week 1 samples formed three groups (1, 2, 3) that clustered in
separate clades at the base of the tree. Groups 1 and 2 were well supported (>95%), while group
3 (from the right breast) was moderately supported (72%). Groups 2 and 3 showed moderate
support for clustering together (79%). In phylogeny (B), breastmilk week 1 Groups 2 and 4 also
form well supported clades (95% and 100%, respectively). Group 3 is again moderately
supported (73%), but in this case does not cluster with Group 2 and is part of the larger clade of
the remaining sequences. In both phylogenies, the week 1 plasma sequences do not cluster with
the breastmilk sequences and with one exception are part of the larger clade of remaining
sequences. These results indicate that the breastmilk and plasma viruses, as well as the virus in
both breasts, are distinct one week after birth. Furthermore, the virus in the right breast is also
more diverse than the left, as shown in the V1V2 analysis.
The majority of the month 4 sequences form a moderately well-supported paraphyletic
clade (90% in [A] and 84% in [B]), and a well-supported clade containing a subset of month 4
plasma, right and left breastmilk sequences is present within the larger clade (99% [A] and 94%
114

[B]), consistent with a lack of population structure among these sequences. Thus, at month 4, it
appears that no biological barrier is restricting gene flow between the tissues. In general, this
large population of month four viruses does not give rise to later viral populations, with the
exception of a few month 12 plasma sequences. Furthermore, this group of month 4 sequences
appears to evolve from the earlier plasma virus rather than from the breastmilk virus.
The rest of the later timepoint sequences form two major clades that cluster together with
moderate support (89% [A] and 83% [B]). In the first major clade, the majority of the plasma
sequences from 12 months form a well-supported clade (92% [A] and 82% [B]) which gives rise
to the clade of 24 month sequences (93% [A] and 80% [B]). In the second major clade the few
remaining month four sequences from all three tissues cluster with all of the month 12 breastmilk
sequences as well as three plasma month 12 sequences with very high support (98% [A] and
81% [B]). In (A), the month 12 breastmilk sequences cluster with the month 18 breastmilk
sequences as well as a few plasma month 24 sequences with moderate support (81%).
Alternatively, in (B), the month 18 breast milk sequence cluster with the majority of the plasma
sequences from months 12 and 24. This suggests that either the virus in the breastmilk re-
compartmentalizes after month 12 and evolves independently from the plasma virus (A), or that
the breast milk was re-infected by the virus in the plasma at month 18 (B). For the breastmilk
month 18 sequences, only a truncated region amplified, probably due to low viral load. This may
explain the phylogenetic uncertainty as to their true location. Irrespective of the true position of
the month 18 sequences, at month 12, there seems to be limited migration of the virus between
the breastmilk and the plasma. Possibilities for the apparent lack of gene flow include a
biological barrier such as a renewed tight junction between epithelial cells or limited production
of breastmilk.
115

Rooting the phylogeny
Because the origin of the virus in both tissues is of great interest, obtaining the correct root
for the phylogeny is essential before drawing conclusions. Because the model chosen for the
Bayesian analyses assumed a relaxed molecular clock, the most likely position of the root is
estimated along with possible topologies. To formally test the position of the root, five
alternative consensus Bayesian phylogenies were estimated for the C2V5 (A) dataset (Table 5-
6). Both the original analysis with no constrained outgroup and the analysis with the breastmilk
group 1 sequences as the outgroup had the highest marginal likelihoods, though the marginal
likelihoods for all analyses were very similar to each other and no significant difference could be
ascertained. In order to further investigate the correct rooting, a maximum likelihood (ML)
phylogeny was estimated with an assumption of no molecular clock, and the most likely root was
estimated from the entire set of all possible roots (Figure 5-9). The topology of the correctly
rooted ML tree was similar to the Bayesian consensus phylogeny, although most branches were
not well-supported and the phylogeny is less resolved; specifically the temporal structure of the
virus evolution is much less clear. The root was placed at the breastmilk week 1 sequences, with
a bootstrap value of 100. The group 2 breastmilk sequences are still well supported (87%). The
group 3 breastmilk sequences were not supported as a clade in the ML rooted phylogeny,
however, and appear to give rise to the plasma sequences. The breastmilk month 12 sequences
clade together and appear to give rise to the month 18 breastmilk sequences, with only a few
plasma sequences are present in this clade. The plasma month 24 sequences also clade together
(along with one week 1 plasma sequence), although not with the month 12 plasma sequences as
in the original Bayesian phylogeny. However, none of these clades are well supported (<50
bootstrap value).
116

In general, the ML rooted phylogeny supports the conclusions drawn from the Bayesian
phylogeny, in particular the distinct groups in the first timepoint breastmilk sequences. The
Bayesian outgroup analysis as well as the ML analysis suggests that the breastmilk sequences did
not arise from the plasma and that they share a common ancestor well before birth. The TMRCA
was estimated for each of Bayesian outgroup analyses, and the range was between 1053-1171
days before present (=24 months). This corresponds to about one year before birth, before the
pregnancy and well before the initiation of breastmilk. This supports a model in which the
breastmilk infection is initiated locally, or else is seeded by infected cells which traffic from
other tissues in the body than the plasma. By month 4, the breastmilk virus appears to be
replaced by a virus that shares an origin with the plasma virus, while by month 12, the breastmilk
virus is again compartmentalized. In general, both the Bayesian and maximum likelihood
methods and both alternative alignments result in similar topologies, indicating high
phylogenetic signal and robust results.
Branch selection analysis
The dN/dS ratio was tested for each of the branches leading to major clades on the best-
model Bayesian consensus tree. For several of the branches, the ratio was significantly different
from 1, indicating a deviation from neutrality (data not shown). The branch leading to the right
breastmilk sequences at the first timepoint was under negative selection, as was the branch
leading to the majority of the month four plasma and breastmilk sequences (Figure 5-10). Three
branches towards the later part of the phylogeny were under significant positive selection: the
branch leading to all of the late breastmilk sequences, the branch leading to the month 18 milk
sequences plus some month 24 plasma sequences, and the branch leading to the majority of the
plasma month 24 sequences. These results suggest changing selective pressures in both the
plasma and breastmilk later in infection.
117

Inclusion of breastmilk month 1 sequences
Because the characteristics of the week 1 and the month 4 breastmilk viruses appear very
different, i.e. a shift from compartmentalization to mixing with the plasma, additional sequences
from month 1 were included in the C2V5 (A) alignment. The Bayesian analysis was re-estimated
using the SRD6 model of nucleotide substitution, a relaxed clock with a log-normal distribution
of rates and a constant population size, and the 50% consensus tree was calculated (Figure 5-11)
The left breastmilk sequences from month 1 cluster together with a moderate probability (82%),
suggesting a common origin, while they cluster with the left breastmilk week 1 group 1
sequences with low probability (57%). The initial right breastmilk week 1 virus does not appear
to evolve into the month 1 virus as these sequences do not cluster together. Rather, the right
breastmilk virus appears to have been replaced by a different, but similar, viral population. This
supports a model in which infected cells traffic to the breastmilk from other tissues, and
continuously seeds a new infection. Only a subset of infected cells may migrate from the tissue
to the milk, which would explain the observed pattern of variation. If the locally infected
mammary cells were infected, a stronger evolutionary relationship would be expected between
the week 1 and month 1 virus. There is still no support for the plasma having seeded the month 1
virus population.
Another piece of information to be learned from the phylogeny is the relative branch
lengths. Typically, longer internal branch lengths relative to the terminal branches is indicative
of a constant population size. Conversely, longer terminal branches relative to the internal branch
lengths can be indicative of exponential growth (Grenfell et al. 2004). In general, the internal
branch lengths leading to the month 1 clades are much longer than those in the plasma clades,
which is indicative of a constant population size. This supports a model in which the infection in
the breastmilk is a slowly evolving infection, possibly being reseeded with virus from other
118

tissues. This could also support a model in which a limited number of cells are available for
infection in the breastmilk relative to the plasma, which restricts the potential growth of the
infection.
Migration Analysis
To test the hypothesis that the virus in the breastmilk is compartmentalized with respect
to the plasma virus, a modified version of the Slatkin-Maddison test was used which compares
both the distribution and the median number of four possible events (migration from breastmilk
to plasma or plasma to breastmilk, and constant state of breastmilk or plasma) for each tree from
the posterior distribution and a set of randomly generated trees. The distributions for all four
possible scenarios were significantly different (p<0.0001) than the random expectation. The
median for each of the four events was also significantly different (p<0.0001) than the random
expectation. The minimum, maximum, and average for each event for the actual and random
trees is shown in Figure 5-12. These results suggest that significant compartmentalization of the
breastmilk does occur over the two-year period. In order to determine if there was migration
between the left and right breasts, the analysis was repeated using nine potential events. Again,
both the distribution and the median for each of the nine events in the observed trees were
significantly greater than expected by chance (p<0.001 for all comparisons). The minimum,
maximum, and averages are shown in Figure 5-13. This suggests that each breast functions as a
separate compartment for the virus over time.
Discussion
This study represents the first longitudinal analysis of the evolution of HIV in breastmilk
and was designed to answer several questions about the evolution of the breastmilk virus over
time. The initial question was whether the virus in the breastmilk originates from the plasma
during lactogenesis, or is seeded by either a local infection of the mammary cells or infected
119

cells trafficking from another tissue. The haplotype analysis of V1V2 and the phylogenetic
analysis of C2V5 suggest that the milk virus does not derive from the plasma. V1 and V2
haplotypes in the milk are not present in the plasma at the first timepoint, and the phylogenies
clearly show well-supported clades for the milk viruses that do not include plasma virus. The
month 1 milk sequences show the same pattern, although they are not unequivocally derived
from the week 1 virus. This lends more support to the model of trafficking infected cells, which
are infected with related viruses but do not represent the full range of variation in the source
tissue. Furthermore, the majority of the mutations are located on the internal branches of the milk
clades, rather than on the external branches as for the plasma sequences. This suggests a constant
size population of virus in the milk, which could be consistent with a low migration rate of
trafficking cells. This hypothesis is also consistent with earlier observations that the T-cells and
macrophages in the milk are phenotypically different than the cells in the blood (Bertotto et al.
1990b; Ichikawa et al. 2003; Kourtis et al. 2003). Because only two tissues were included in this
study, the tissue of origin cannot be determined. However, one likely source of infection could
be the gut associated lymphoid tissue (GALT) (G. Aldrovandi, personal communication). The
GALT contains the majority of T-cells in the body, which are the primary target cells of HIV-1.
During acute infection in the initial stage of disease, HIV-1 targets this tissue which results in the
massive depletion of T-cells (Douek 2007b; Douek 2007a). The high frequency of infected cells
in this tissue is consistent with a model in which T-cells trafficking to the breast milk during the
first month post-partum initiate a new infection. Because the GALT is infected early in the
course of disease, this scenario would also explain the observation that the milk virus appears to
emerge from the root of the tree and shares an ancestor with the plasma virus >1 year pre-
partum.
120

A second question addressed by this study was whether the onset of weaning was
associated with any evolutionary changes. The mother reported exclusive breastfeeding at four
months, although by six months she reported mixed feeding. The milk and plasma virus were
part of the same population at the month 4 sample, which appeared to have evolved from the
earlier plasma virus rather than from either the milk virus or another tissue. Therefore, it seems
that the plasma virus infected the milk before the onset of weaning, which is contrary to the
initial expectation that weaning would precede panmixia. The opening of the paracellular
pathway at the onset of weaning (<2 feedings per day) is associated with increased levels of
plasma derived minerals and nutrients, and would provide an opportunity for the plasma virus to
migrate to the milk. It is interesting that the infant was infected at 4 months as well, even though
the mother was reportedly exclusively breast feeding. Unfortunately, the precise timing of the
events around four months cannot be ascertained, so an exact order of weaning and HIV
infection cannot be determined.
A third question was whether the breastmilk virus compartmentalized over time. The
earliest virus is clearly derived from a different population than the plasma virus, but at month 4
the two tissues are experiencing nearly complete migration. By month 12, the milk virus appears
again compartmentalized with respect to the plasma virus, and appears to derive from the month
4 milk virus. Overall, this does suggest compartmentalization of the virus, and the analysis for
migration indicated much greater compartmentalization than expected by chance. By month 12
and 18, the mother was most likely breastfeeding much less frequently than during the few
months after birth, and therefore milk production would have been lower and fewer opportunities
may have existed for the plasma virus to enter the milk. In addition, the initial source of infection
does not appear to have re-infected the milk, possibly again due to lowered milk production.
121

These results may also explain the discrepancies reported earlier in the literature concerning
whether the breastmilk virus compartmentalized, since those studies considered very different
timepoint. This study demonstrates the importance in choosing sampling times carefully before
making generalizations about the origin and evolution of breastmilk virus.
Finally, I examined the selective pressures that were acting on the virus over time. My
analysis indicated that an episode of negative selection had occurred during the evolution of the
right breastmilk virus at week 1, as well as the breastmilk and plasma at month 4. During the
later stages of evolution, several independent episodes of positive selection occurred on branches
leading to the month 12 and 24 viruses in both tissues. This could suggest a relaxation of host
pressure allowing the viral population to acquire new beneficial mutations. This is also consistent
with the longer terminal branch lengths in the later samples, which is indicative of exponential
growth. . In another study, I showed that the diversity and effective population size of the virus
increases as host selective pressure relaxes and the immune system begins to fail (as measured
by decline of CD4 cells) (Gray et al, in prep). No clinical information was available for the
patient in the current study so this hypothesis could not be tested. However, this is an avenue of
investigation for future studies.
The conclusions drawn from this study have implications for the management of
breastfeeding by HIV-infected mothers. First, because the milk virus is distinct from the plasma
virus until at least month 1 and because it originated in tissues with potentially different selective
pressures, the initial milk virus may have different pathogenicity and/or transmissibility than the
plasma virus. If so, this could modulate the risk of MTCT transmission during the first few
months and impact the recommendations currently given to women. Second, the population
dynamics of the virus clearly changes by month 4. Although the mother reported exclusive
122

breastfeeding, she did initiate weaning sometime within the weeks after this sample was
collected. Therefore, although it appears that migration of the virus between tissues occurred
prior to weaning, it is difficult to confidently conclude that the two are unlinked. Third, this
study conclusively demonstrates that the milk virus is compartmentalized throughout much of its
production. The again suggests that the milk virus may evolved different characteristics that
modulate transmission. Further, if the infection in the milk maintains a constant population size
as suggested by the phylogeny, and is protected from the more rapidly growing plasma virus,
fewer infected cells are available to transmit the virus. Determining exactly why the virus is or is
not compartmentalized in the milk may aid our understanding of how to lower the risk of MTCT.
Lastly, this study demonstrates that the evolution of the virus in the milk is a dynamic process. I
am currently studying two additional patients for whom samples are available over a one-year
period to determine whether the same dynamics occur in other patients as well. Understanding
the complexity of the evolutionary process is of great importance in understanding how
exclusive breastfeeding attenuates the risk of transmission on a molecular level, so that more
precise recommendations for avoiding MTCT can be made to HIV-1 infected women worldwide.
This study was conducted within an evolutionary anthropological framework on several
levels. Traditional analytical techniques used for population genetics were implemented here to
investigate the evolution of a human pathogen, although on a much shorter timescale than
traditionally considered. Because the rate of evolution in HIV-1 is many orders of magnitude
faster than in other pathogens, the population dynamics of infection, including response to
selective pressures, changes in population size, and migration between compartments, can all be
measured within one individual, rather than within and between populations of humans as is
usually the case in anthropological genetics. This difference highlights the fact that pathogen
123

dynamics are similar in a micro and a macro environment, and lessons from each level can
inform the other. In addition, women in third-world countries who do not enjoy the benefits in
developed countries cannot safely choose to formula feed their infants to minimize risk of
transmission. However, comparatively little research has been conducted on the molecular
characteristics of the virus in milk as compared to other tissues. The recent observations that
exclusive breastfeeding may attenuate the risk of transmission are extremely important in both a
clinical and an anthropological context. The management of breastfeeding practices by the
women themselves in developing countries represents an economical, culturally appropriate, and
non-invasive method to control transmission to their infants, without intervention by medical
staff, drug companies, or governments. The uniquely interdisciplinary approach of anthropology
promotes the importance in providing marginalized women with the tools to manage their
infant’s health, as well as the analytical framework to investigate and understand the molecular
mechanisms underlying the recommendations.
124

Table 5-1. Number of sequences generated for each tissue.
Tissue Timepoint Number of Clones
Part of initial dataset
PL 1W 23 Yes PL 1W 13a No PL 4M 12 Yes PL 12M 37a Yes PL 12M 22 No PL 24M 28a Yes PL 24M 22 No BML 1W 15 Yes BMR 1W 31 Yes BML 1M 28b No BMR 1M 13b No BML 4M 11 Yes BMR 4M 9 Yes BMR 12M 14 Yes BML 18M 4 Yes
aThese sequences were generated using less starting template in the PCR reaction and were not included in any of the phylogenetic analyses. bThese sequences were included in a subset of analyses. Table 5-2. Sequence characteristics of V1 and V2. V1 V2 Tissue/Timepoint Haplotypes #GlySites Length Haplotypes #GlySites LPL1W A(.03) C(.11) D(.86) 1-4 24-37 A(.06) B(.94) 2-3 4PL4M A(.08) C(.16) D(.75) 2-4 33-37 B(.84) C(.08) E(.08) 2-3 4
PL12M A(.03) B(.05) C(.31) D(.61) 2-4 25-37 A(.05) B(.46) C(.05) D(.41) E(.3) 2-3 4
PL24M A(.04) B(.48) C(.1) D(.22) E(.16) 2-5 22-44 A(.38) B(.32) C(.16) D(.12) 1-3 4BML1W A(1) 1 23 E(1) 3 5BMR1W A(.29) B(.32) C(.39) 1-3 23-36 A(.42) C(.29) E.29) 2-3 4BML1M B(.96) F(.04) 1-4 27-46 A(.25) C(.75) 1-2 4BMR1M A(.08) B(.23) D(.08) F(.54) 1-4 24-46 A(.15) B(.08) C(.77) 1-2 4BML/R4M A(.05) D(.95) 2-4 29-37 B(.8) C(.1) E(.1) 2-3 4BMR12M D(1) 4 37 D(1) 3 4BML18M D(1) 4 37 D(1) 3 4
Haplotypes were assigned based on aa motif, length, and glycosylation sites. The frequency of each haplotype is given in parentheses.
125

Table 5-3. Combination of V1 and V2 haplotypes. V1 HAP A A A B B B C C D D D D D E FV2 HAP A C E A B C A B A B C D E C CPLA1W 1 4 1 30 PLA4M 1 2 7 1 1 PLA12M 2 3 18 9 3 24 PLA24M 2 15 9 1 4 1 3 1 6 8 BML1W 15 BMR1W 9 1 9 12 BML1M 7 20 1BMR1M 2 3 1 7BML4M 1 9 1 1 BMR4M 7 1 BML12M 14 BMR18M 4
The number of clones displaying each combination of V1 and V2 haplotypes is given for each tissue/timepoint. Haplotypes were defined by length, aa motif, and number of glycosylation sites. Table 5-4. Hudson test for population structure. Comparison Group 1 Group 2 p-value intra-timepoint tissues BML 1W BMR 1W p<0.001intra-timepoint tissues BML 4M BMR 4M p=0.935intra-timepoint tissues BML1W PLA1W p<0.001intra-timepoint tissues BMLR 1W PLA1W p<0.001intra-timepoint tissues BML/R 4M PLA 4M p=0.428intra-timepoint tissues BMR 12M PLA 12M p<0.001inter-timepoint, same tissue PLA 1W PLA 4M p<0.001inter-timepoint, same tissue PLA 1W PLA 12M p<0.001inter-timepoint, same tissue PLA 1W PLA 24M p<0.001inter-timepoint, same tissue PLA 4M PLA 12M p<0.001inter-timepoint, same tissue PLA 4M PLA 24M p<0.001inter-timepoint, same tissue PLA 12M PLA 24M p<0.001inter-timepoint, same tissue BML 1W BML/R 4M p<0.001inter-timepoint, same tissue BML 1W BMR 12M p<0.001inter-timepoint, same tissue BMR 1W BML/R 4M p<0.001inter-timepoint, same tissue BMR 1W BMR 12M p<0.001inter-timepoint, same tissue BML/R 4M BMR 12M p<0.001inter-timepoint, same tissue BMR 12M BML 18M p<0.001
126

Table 5-5. Number of putative recombinant clones. V1V5 v1v3 v1v2 c2v3 C2V5 (A) C2V5 (B) C2V5 (A) c C2V5 (A)d
Initial p-value p<10
-19p<10
-19p=8.43x10-7 p=0.191 p=0.002 p=0.002 p=0.027 p=0.006
PL 1wk UNDETb UNDET 3 0 2 2 2 3 PL 1wk a UNDET UNDET * * * * * 0 PL 4m UNDET UNDET 1 0 6 6 6 6
PL 12m UNDET UNDET 0 0 4 5 4 6 PL 12m a UNDET UNDET * * * * * 0 PL 24m UNDET UNDET 10 0 0 2 0 5 PL 24m a UNDET UNDET * * * * * 9 BML 1w UNDET UNDET 0 0 2 15 2 2 BMR 1w UNDET UNDET 12 0 14 0 14 14 BMl 4m UNDET UNDET 1 0 2 2 2 2 BMR 4m UNDET UNDET 0 0 2 2 2 2
BMR 12m UNDET UNDET 0 0 0 0 0 0 BML 18m UNDET UNDET 0 0 0 0 0 0 The number of identified clones in each alignment is given for each tissue/timepoint. aThese sequences were generated using 1/10 of the amount of starting template and were only used in this analysis. bFor these alignments, a dataset that did not include recombinants could not be obtained. cThis alignment included the additional breastmilk sequences from month 1 in addition to the non-recombinant C2V5 (A) dataset. dThis alignment included the additional plasma sequences in addition to the non-recombinant C2V5 (A) dataset. Table 5-6. Marginal likelihoods for models used in the Bayesian analysis. Model Outgroup Marg. Lik. SRD6, SC none -4362.9985 SRD6, RC none -4321.3865 GTR2, RC none -4278.6483 GTR3, RC none -4270.3296 SRD6, RC BML 1W (Group 1) -4321.3865 SRD6, RC BMR 1W (Group 2) -4322.9025 SRD6, RC BMR 1W (Group 3) -4322.7647 SRD6, RC PLA 1W (4 seq.) -4323.1764 SRD6, RC PLA 1W (2 seq.) -4322.8887
The definition of each outgroup is given in the text. SRD6 = Shapiro, Rambaut, and Drummond (2006) model. GTR = general time reversible with two or three partitions of the data based on codon position. SC = strict clock assumption. RC = relaxed clock assumption.
127

128
Figure 5-1. Sampling times and tissues.
Sampling times are on the top of the graph. W=week, M=month. Tissues which were sampled from each time are on the bottom: PL=plasma, BML=left breastmilk, BMR=right breastmilk.
Three sequences from week 1 are in red. Bootstrap values based on 1,000 neighbor-joining replicates are indicated above each major branch. Branch lengths are in units of substitutions/site.
Figure 5-2. Neighbor-joining phylogeny of all subtypes in group M plus this patient.

HA
PLO
TYPE
LEN
GTH
#GLY
# C
LON
ES
A C T N L A N D T A R R I I A K S M E E E V K N C 24 1 27A C T E I R N I T G N N R T I D F S M K G E V Q N C 25 2 2A C T E I R N I T D G G N R T I D F S M R E E I K N C 26 2 2A C T E I R N I T G G G T D N N R T I D F S M K G E V K N C 29 2 1A C T E I Y N S T S G N S T D G G K D N N R N I D L S M Q G E V K N C 34 2 1B C T N L N R T F V N Y T S S M K E E I K N C 22 2 2B C T N L N R T I A N D T S G I S M Q E E I K N C 24 2 3B C T N L N R T I G N D T S D A N G M K E E I K N C 25 2 1B C T N L K N T I V N D T S G S D T S S M K E E I K N C 27 1 7B C T N L N R T I V N D T S G S D T S S M K E E I K N C 27 2 54C C T N L K N T T V N G T S G N G A R T I D S N M K G E V K N C 31 2 1C C T N L T N T T V N G T G S G N D T K R T V D S S M E G E V K N C 33 3 6C C T N L K N T T V N G T S D T S G G N N N N R T I D N S M K E E I K N C 36 3 33D C T N L K N T T V N G T V G N G T G G G R T I D S N M K G E V K N C 34 3 2D C T N L K N T T V N S T S G T S N G N D K K R T I D S S M K G E V K N C 36 2 3D C T N L K N T T V N G T S G T S G G T D N N R T I D F S M K G E V K N C 36 3 1D C T N L K N I T V N G T S G N S T S G G N E N R T I D S S M K G E V K N C 37 4 44D C T N L K N I T V N G I S G N S T G G G T A I N R T I D F S M K E E I K N C 38 3 6D C T N L K N T T V N G T S G N S T G G G N G N N R T I D L S M K G E V K N C 38 4 61D C T N L K N I T V N G T S S N S T G D G N D T N R T I D S S M T E E V K N C 38 5 2D C T N L Q N I T V N G T S S N S T S G N S T G G G T D Y N R T I D F S M K G E V K N C 43 5 3E C E E L N G T I V N D T Y S N D A T F N N N T S G G I K R N K T I V R S M E G E V K N C 44 4 8F C T K P S G T I V N D T A G N G T M N D T A G - N G T M N G G D R K I E I S M E E E V K N C 46 5 8
129
Figure 5-3. Haplotype analysis of V1.
Haplotypes were assigned based on amino acid (aa) motif, length, and number of glycosylation sites (highlighted in green). Representative sequences for each haplotype are shown. Sequences within a haplotype that differed in aa motif but shared the same length and number of glycosylation sites are not shown.

130
HA
PLO
TYPE
LEN
GTH
#GLY
# C
LON
ES
A C S F N T T T E I H D K Q Q K V H A L F Y R L D I A Q L D K D R N D Y R L I N C 40 1 1A C S F N T T T E I H D K Q Q K V H A L F Y R L D I A Q L D N N N E T Y R L I N C 40 2 20A C S F N T T T E I H D K Q Q K V H A L F Y R L D I A Q L D N D S R T Y R L I N C 40 2 25B C S F K T T T E I R D R E Q K V H A L F Y R L D I Q P L G N E T E E G G T Y R L I N C 43 1 3B C S F N T T T E I N D R K Q K V H A L F Y R L D I Q P L G N E T E E G G T Y R L I N C 43 2 9B C S F N T T T E I R D R E Q K V H A L F Y R L D I Q P L G N E T E E N S T Y R L I N C 43 3 71B C S F N T T T E V N D R K Q K V H A L F Y R L D I Q P L G N E T K E G N G T Y R L I N C 44 3 13B C S F N T T T E I N D R Q Q K V R A L F Y R L D I Q P L G N E T N K E G N G T Y R L I N C 45 3 2B C S F N T T T E I N D R K Q K V H A L F Y R L D I Q P L G N E T T E G N G T Y T Y R L I N C 46 3 4C C S F R A N T E K D R K Q N V T A L F Y R L D I A P L D K G N K T Y R L I N C 40 1 8C C S F N T T T E I H D R Q Q K V H A L F Y R L D I Q Q L D K E R N D T Y R L I N C 41 2 3C C S F N T T T E I Q D K Q Q K V H A L F Y R L D I Q Q L D K E G D D T S E T Y R L I N C 44 1 2C C S F N T T T E I Q D R Q Q K V H A L F Y R L D I Q P L D K E G N D T F E T Y T L I N C 44 2 43D C S F N T T T E I Q D R Q Q K V H A L F Y R L D I Q P L G N K T T K E G N D T F E T Y T L I N C 48 3 48E C S F N T T T E I H D R Q Q K V H A L F Y R L D I Q P L E E G N K G G N D T T E R E N G T Y T L I N C 51 3 29
Haplotypes were assigned based on amino acid (aa) motif, length, and number of glycosylation sites (highlighted in green). Representative sequences for each haplotype are shown. Sequences within a haplotype that differed in aa motif but shared the same length and number of glycosylation sites are not shown.
Figure 5-4. Haplotype analysis of V3.

V V C V3 C3 V C V5
131 157 196 296 331 385 418 460 471
V V C V3 C V C V5
131 157 196 296 331 385 418 460 471
V1VV1V
C2V5
V1V2 C2V5
Figure 5-5. Recombination alignments.
Red lines represent alignments for which a non-recombinant datatset could not be obtained. Green lines represent alignments for which non-recombinant datasets were obtained. .
Figure 5-6. Network of breast milk sequences from week 1.
Breastmilk sequences from week 1 are categorized into four groups basaed on clustering in the network. Branch lengths are in substitutions/site.
131

Figure 5-7. Bayesian consensus phylogeny for C2V5 (A).
Branches are color coded according to tissue as follows: blue = right breast, red = left breast, green = plasma. Posterior probabilities are given above each major branch. The timepoint at which sequences were sampled is indicated by a symbol (see legend). The three groups of breastmilk sequences at week 1 are labeled as categorized in Figure 5-6.
132

Figure 5-8. Bayesian consensus phylogeny for C2V5 (B).
Branches are color coded according to tissue as follows: blue = right breast, red = left breast, green = plasma. Posterior probabilities are given above each major branch. The timepoint at which sequences were sampled is indicated by a symbol (see legend). The three groups of breastmilk sequences at week 1 are labeled as categorized in Figure 5-6.
133

Figure 5-9. Best-rooted maximum likelihood phylogeny.
Branches are color coded according to tissue as follows: blue = right breast, red = left breast, green = plasma. Bootstrap values >50 based on 1,000 replicates are given above each major branch. The timepoint at which sequences were sampled is indicated by a symbol (see legend). The three groups of breastmilk sequences at week 1 are labeled as categorized in Figure 5-6.
134

Figure 5-10. Bayesian consensus phylogeny for the C2V5 (A) dataset with branches under
significant selection.
Branches are color coded according to tissue as follows: blue = right breast, red = left breast, green = plasma. The timepoint at which sequences were sampled is indicated by a symbol (see legend). The three groups of breastmilk sequences at week 1 are labeled as categorized in Figure 5-6. Monophyletic sequences from the same tissue are collapsed. Thick black branches indicate significant negative selection, and thick red branches indicate significant positive selection.
135

Figure 5-11. Bayesian consensus phylogeny with breast milk month 1 sequences.
Branches are color coded according to tissue as follows: blue = right breast, red = left breast, green = plasma. Posterior probabilities are given above each major branch. The timepoint at which sequences were sampled is indicated by a symbol (see legend). The three groups of breastmilk sequences at week 1 are labeled as categorized in Figure 5-6.
136

Figure. 5-12. Migration analysis for two tissues.
(a) The migration matrix calculated from the posterior distribution of trees estimated using the C2V5 (A) dataset. Circles are proportional to the minimum (dark blue), average (light blue) and maximum (white) number of events for each category. (b) The migration matrix calculated from 10,000 random trees.
137

Figure 5-13. Migration analysis for three tissues.
(a) The migration matrix calculated from the posterior distribution of trees estimated using the C2V5 (A) dataset. Circles are proportional to the minimum (dark blue), average (light blue) and maximum (white) number of events for each category. (b) The migration matrix calculated from 10,000 random trees.
138

CHAPTER 6 CONCLUSION
The impact of disease on the human population is of immediate and practical concern.
Emerging infectious diseases, defined as "infections that have newly appeared in a population or
have existed previously but are rapidly increasing in incidence or geographic range" (Morse
1995) have been significantly increasing over the past 50 years (Jones et al. 2008). Emergence is
largely due to increasing urbanization, wars, environmental degradation, and global inter-
connectivity (Fauci 1998; Stephens et al. 1998a; Desselberger 2000; Pollard and Dobson 2000;
Feldmann et al. 2002; Fauci, Touchette, and Folkers 2005). Tuberculosis, malaria, and HIV-1, as
well as emerging threats such as SARS, West Nile Virus, and influenza, are major threats to
international public health (Fauci 1998; Morens, Folkers, and Fauci 2004; Fauci, Touchette, and
Folkers 2005). Infectious diseases are the second leading cause of death in the pre-industrialized
world (Fauci, Touchette, and Folkers 2005) and account for >25% of all deaths worldwide
(Morse 1995). In addition, complex diseases including cancer, diabetes, and heart disease are
responsible for 87% of deaths in high income countries and 43% of deaths in low income
countries, of which 40-80% of these deaths could be avoided with lifestyle changes (WHO
2008a; WHO 2008b) though myriad studies suggest have these diseases have a genetic
component as well. By 2030, heart disease and HIV/AIDS are predicted to be the largest
components of the burden of human disease (Mathers and Loncar 2006)
Due to the increasing global impact and complicated manner of transmission and
inheritance of infectious and complex diseases, a comprehensive approach is essential to
diagnose, treat and eradicate human diseases. My dissertation demonstrates how genetic
anthropology can be used to address both anthropological and clinical concerns from three
perspectives. As anthropological geneticists, we are uniquely positioned to use the analytical
139

tools of evolutionary genetics to study the biological mechanisms of diseases, while maintaining
a holistic approach that considers the cultural, historical, and demographic factors which
influence etiology. We can also use our expertise to influence US and international policy by
advocating for culturally sensitive implementation of scientific findings. I believe that lack of
dialogue between fields, even those that use similar molecular and bioinformatics techniques,
can substantially impede progress towards shared goals of treating and eradicating human
disease. However, my study also demonstrates that a conscientious treatment of the clinical and
policy implications does not preclude simultaneously addressing more traditional biological
anthropological questions involving human evolution and migration.
Both anthropological and clinical implications were addressed for the four projects
included in this dissertation. First, with regard to the evolution of treponemal diseases, I found
that the genetic variation present within and between treponemal subspecies was largely the
result of a particular type of homologous recombination called gene conversion. Furthermore, the
largest number of gene conversion events took place in the venereal syphilis genome, which is
largely regarded as the most recently evolved of the three subspecies. These recombinant events
distort the phylogenetic and molecular relationships among the subspecies, and therefore I
conclude that relying on single nucleotide polymorphisms without considering the contextual
sequence information are insufficient to discern evolutionary relationships among the
treponemes. Second, my molecular data suggest that the three subspecies are molecularly distinct
based on high bootstrap values for the phylogenetic branches separating the subspecies, as well
as a significant amount of among-subspecies variation. However, this could be the result of
geographic structure and does not necessarily support a classification of three diseases. Third, the
molecular data do not appear to support a dramatically older origin of yaws relative to venereal
140

syphilis but instead are consistent with a relatively coincident evolution of the three human
treponemal subspecies. Moreover, the venereal syphilis sequences harbor more variation than
would be expected under the modified Columbian hypothesis of evolution of venereal syphilis
within the past 500 years (Baker and Armelagos 1988). This study was able to compare support
for the leading anthropological hypotheses for the evolution of syphilis, as well as provide new
genomic regions suitable for diagnosing between the three diseases.
In the second project, I investigated the association between genotypic data from the ADH
and ALDH alcohol metabolism genes and both dichotomous and continuous substance abuse
phenotypes in a Plains population of Native Americans. In the third project, I genotyped and
analyzed genotype data from the SNCA gene in ~1000 individuals from the same Plains
population and a second Native American population from the Southwest United States. Despite
the extensive genetic data, I found no correlation between any of the ADH, ALDH, or SNCA
markers and the substance abuse phenotypes. For the SNCA gene, this may suggest that
unassayed promoter polymorphisms are affecting the expression of the gene and risk of
substance abuse rather than variants within the gene itself, since excessive mRNA and protein
levels have been associated with alcohol use disorders. Thus, future studies should investigate
the genetic variation upstream of the SNCA for an association with alcohol use. Alternatively, the
evolutionary history of Native Americans could explain the lack of association between the
assayed genetic markers and substance abuse. Native Americans experienced a severe population
bottleneck during migration from Asia to the New World, in which much of the ancestral genetic
variability was lost (Mulligan et al. 2004; Ramachandran et al. 2005). It was known from
previous research that the ADH and ALDH alleles already identified as protective against
alcoholism in Asians were not present in the tested Native American populations although the
141

fact that the genes were implicated in the risk of alcoholism motivated my analysis of additional
ADH and ALDH alleles. My research demonstrated that the SNCA upstream polymorphism
previously associated with alcoholism was not present in the tested populations, consistent with a
severe population bottleneck during Native American evolutionary history. Thus, these results
represent a fairly comprehensive investigation of the major candidate genes and alleles for
association with alcoholism in Native Americans and lack of association with such alleles
suggests that a non-genetic approach may represent the best strategy for treatment of alcoholism
in these populations. I believe that additional resources should be dedicated to substance abuse
intervention efforts that target societal and cultural causes, such as poverty, lack of health care
and unemployment.
In the final project, I conducted the first longitudinal study of the evolution of HIV-1 in the
breast milk and blood plasma of a HIV-positive mother over a two year period. The goal of the
study was to elucidate the molecular mechanisms responsible for the observation that exclusive
breastfeeding reduces the risk of transmission over mixed feeding (Coutsoudis et al. 1999;
Coutsoudis 2000; Coutsoudis et al. 2001; Coutsoudis et al. 2002; Iliff et al. 2005; Coovadia et al.
2007), as well as to understand the general evolutionary patterns of the virus in different tissues
within a single patient over time. I determined that the virus in the breastmilk post-partum is
distinct from the virus contemporaneously circulating in the plasma, which suggests a tissue
other than the plasma is seeding the milk infection. Because the initial milk virus may have
different pathogenicity and/or transmissibility than the plasma virus, future studies should
investigate the phenotypic characteristics of the early milk virus. Intriguingly, the population
dynamics of the infection in this patient clearly changes by month 4, coinciding with the
transmission of the virus to the infant in this case. The mother also reported ceasing exclusive
142

breastfeeding shortly thereafter. However, the exact timing of these events is unclear and
therefore conclusions about causality between changes in the breastmilk virus population and
weaning with HIV transmission to the baby are uncertain. This study also demonstrates
conclusively that the milk virus is compartmentalized throughout much of its production, and
that the evolution of the virus in the milk is a dynamic process. Understanding the complexity of
the evolutionary process is of great importance in determining the relationship between feeding
practices and transmission on a molecular level, so that women can be empowered to effectively
manage their breastfeeding practices to minimize the risk of transmission. Thus, the uniquely
interdisciplinary approach of anthropology promotes the importance in providing marginalized
women with the tools to manage their infant’s health, as well as the analytical framework to
investigate and understand the molecular mechanisms underlying the recommendations.
My dissertation also has broader implications for the field of genetic anthropology. First, I
use a model that incorporates three distinct perspectives from which human disease can be
approached. Each perspective is temporally and philosophically distinct and includes different
levels of human variation. While anthropologists traditionally employ the evolutionary or
population perspective, we have the tools and the expertise to inform studies from the clinical
perspective as well. Second, I have demonstrated that incorporating studies of pathogen genetics
is valuable in understanding the interaction between humans and disease. Pathogens can be
studied from a clinical perspective, such as HIV-1 in an individual, as well as an evolutionary
perspective, such as the movement of treponemes across continents. Pathogen evolutionary
dynamics appear to be similar across temporally disparate perspectives, for example, the high
degree of gene conversion/recombination that appears to be characteristic of both T. pallidum
and HIV-1. Uncovering uniform processes recurring in the evolution of infectious diseases will
143

allow us to better understand human-pathogen interactions, and may aid in developing strategies
to eradicate infectious diseases. Lastly, because both infectious and complex diseases are
increasing concerns to global health, genetic anthropologists should widen their focus to address
clinical and policy implications implicit in their studies that may primarily address more
evolutionary questions. Genetic anthropologists are equipped with the analytical tools to study
the biological mechanisms of diseases and incorporate information about the underlying
population structure and evolutionary history. This unique perspective allows genetic
anthropologists to provide comprehensive clinical and policy recommendations based on a
comprehensive evaluation of genetic data. Finally, the multi-discinplinary approach employed by
anthropologists can be valuable in ensuring that resulting applications of the data are culturally
appropriate and provide maximum health benefits to communities in need.
144

APPENDIX A. LIST OF QUESTIONS FOR SUBSTANCE ABUSE CATEGORIZATION
1. Was there ever a time when, because of your drinking, you often missed work, had trouble on
the job, or were unable to take care of household responsibilities?
2. Did you ever lose a job because of your drinking?
3. Did you often have difficulties with your family, friends, or acquaintances because of your
drinking?
4. Was there ever a period in your life when you drank too much?
5. Has anyone in your family - or anyone else- ever objected to your drinking?
6. Was there ever a time when you often couldn't stop drinking when you wanted to?
7. Have you ever had traffic difficulties because of your drinking - like reckless driving,
accidents, or speeding?
8. Have you often had tremors that were most likely due to drinking? (i.e. when you cut down or
stopped or cut down? After not drinking for a few hours or more, did you often drink to keep
yourself from getting the shakes or becoming sick?)
9. Was there ever a time when you frequently had a drink before breakfast?
10. Were you ever divorced or separated primarily because of your drinking?
11. Have you ever gone on a bender? (Definition: drinking steadily for 3 or more days, more
than a fifth of whiskey daily/24 bottles of beer/3 bottles of wine. Must have occurred 3 or more
times.)
12. Have you ever been physically violent while drinking? (Must have occurred on at least 2
occasions.)
145

13. Have you ever been picked up by the police because of how you were acting while you were
drinking (e.g. disturbing the peace, fighting, public intoxication. Do not include traffic
difficulties.)
14. Have you ever had blackouts? (Definition: memory loss for events that occurred while
conscious during a drinking episode.)
15. Have you ever had the DT's? (Definition: confused state following stopping drinking that
includes disorientation and illusions or hallucinations.)
16. Did you ever hear voices or see things that weren't really there, soon after you stopped
drinking (Hallucinations - must have occurred on at least two separate occasions)
17. Have you ever had a seizure or fit (non-epileptic) after you stopped drinking?
18. Did a doctor ever tell you that you had developed a physical complication of alcoholism, like
gastritis, pancreatitis, cirrhosis, or neuritis? (Include good evidence of Korsakoff's Syndrome -
chronic brain syndrome with anterograde amnesia as the predominant feature.
146

LIST OF REFERENCES
Abbate, I., G. Cappiello, R. Longo, A. Ursitti, A. Spano, S. Calcaterra, F. Dianzani, A. Antinori,
and M. R. Capobianchi. 2005. Cell membrane proteins and quasispecies compartmentalization of CSF and plasma HIV-1 from aids patients with neurological disorders. Infect Genet Evol 5:247-253.
Achaz, G., S. Palmer, M. Kearney, F. Maldarelli, J. W. Mellors, J. M. Coffin, and J. Wakeley. 2004. A robust measure of HIV-1 population turnover within chronically infected individuals. Mol Biol Evol 21:1902-1912.
Allen, S. J., A. O'Donnell, N. D. Alexander, M. P. Alpers, T. E. Peto, J. B. Clegg, and D. J. Weatherall. 1997. alpha+-Thalassemia protects children against disease caused by other infections as well as malaria. Proc Natl Acad Sci U S A 94:14736-14741.
Amos, A. F., D. J. McCarty, and P. Zimmet. 1997. The rising global burden of diabetes and its complications: estimates and projections to the year 2010. Diabet Med 14 Suppl 5:S1-85.
Anderson, D. G., and J. C. Gillam. 2000. Paleoindian colonization of the Americas: implications from an examination of physiography, demography, and artifact distribution. Am Antiquity 65:43-66.
Armelagos, G. J., and J. Dewey. 1975. Evolutionary response to human infectious disease. Bioscience 20:271-275.
Armelagos, G. J., K. N. Harper, and P. S. Ocampo. 2005. On the Trail of the Twisted Treponeme: Searching for the Origins of Syphilis. Evolutionary Anthropology: Issues, News, and Reviews 14:240-242.
Arthos, J., C. Cicala, E. Martinelli, K. Macleod, D. Van Ryk, D. Wei, Z. Xiao, T. D. Veenstra, T. P. Conrad, R. A. Lempicki, S. McLaughlin, M. Pascuccio, R. Gopaul, J. McNally, C. C. Cruz, N. Censoplano, E. Chung, K. N. Reitano, S. Kottilil, D. J. Goode, and A. S. Fauci. 2008. HIV-1 envelope protein binds to and signals through integrin alpha(4)beta(7), the gut mucosal homing receptor for peripheral T cells. Nat Immunol.
Arthur, P. G., M. Smith, and P. E. Hartmann. 1989. Milk lactose, citrate, and glucose as markers of lactogenesis in normal and diabetic women. J Pediatr Gastroenterol Nutr 9:488-496.
Baker, B. J., and G. J. Armelagos. 1988. The origin and antiquity of syphilis: paleopathological diagnosis and interpretation. Curr Anthropol 29:703-738.
Baldo, L., S. Bordenstein, J. J. Wernegreen, and J. H. Werren. 2006. Widespread recombination throughout Wolbachia genomes. Mol Biol Evol 23:437-449.
Barker, D. J., C. N. Hales, C. H. Fall, C. Osmond, K. Phipps, and P. M. Clark. 1993. Type 2 (non-insulin-dependent) diabetes mellitus, hypertension and hyperlipidaemia (syndrome X): relation to reduced fetal growth. Diabetologia 36:62-67.
Barrett, J. C., B. Fry, J. Maller, and M. J. Daly. 2005. Haploview: analysis and visualization of LD and haplotype maps. Bioinformatics 21:263-265.
Barrett, R., C. W. Kuzawa, T. McDade, and G. J. Armelagos. 1998. Emerging and re-emerging infectious diseases: The third epidemiological transition. Annual Review of Anthropology 27:247-271.
Becquart, P., N. Chomont, P. Roques, A. Ayouba, M. D. Kazatchkine, L. Belec, and H. Hocini. 2002. Compartmentalization of HIV-1 between breast milk and blood of HIV-infected mothers. Virology 300:109-117.

Becquart, P., H. Hocini, B. Garin, A. Sepou, M. D. Kazatchkine, and L. Belec. 1999. Compartmentalization of the IgG immune response to HIV-1 in breast milk. Aids 13:1323-1331.
Becquart, P., H. Hocini, M. Levy, A. Sepou, M. D. Kazatchkine, and L. Belec. 2000. Secretory anti-human immunodeficiency virus (HIV) antibodies in colostrum and breast milk are not a major determinant of the protection of early postnatal transmission of HIV. J Infect Dis 181:532-539.
Belfer, I., H. Hipp, C. McKnight, C. Evans, B. Buzas, A. Bollettino, B. Albaugh, M. Virkkunen, Q. Yuan, M. Max, D. Goldman, and M. Enoch. 2006. Association of galanin haplotypes with alcoholism and anxiety in two ethnically distinct populations. Molecular Psychiatry 11:301-311.
Benyshek, D. C., and J. T. Watson. 2006. Exploring the thrifty genotype's food-shortage assumptions: a cross-cultural comparison of ethnographic accounts of food security among foraging and agricultural societies. Am J Phys Anthropol 131:120-126.
Berger, E. A., R. W. Doms, E. M. Fenyo, B. T. Korber, D. R. Littman, J. P. Moore, Q. J. Sattentau, H. Schuitemaker, J. Sodroski, and R. A. Weiss. 1998. A new classification for HIV-1. Nature 391:240.
Bertotto, A., G. Castellucci, G. Fabietti, F. Scalise, and R. Vaccaro. 1990a. Lymphocytes bearing the T cell receptor gamma delta in human breast milk. Arch Dis Child 65:1274-1275.
Bertotto, A., R. Gerli, G. Fabietti, S. Crupi, C. Arcangeli, F. Scalise, and R. Vaccaro. 1990b. Human breast milk T lymphocytes display the phenotype and functional characteristics of memory T cells. Eur J Immunol 20:1877-1880.
Bibbins-Domingo, K., and A. Fernandez. 2007. BiDil for heart failure in black patients: implications of the U.S. Food and Drug Administration approval. Ann Intern Med 146:52-56.
Bjorndal, A., H. Deng, M. Jansson, J. R. Fiore, C. Colognesi, A. Karlsson, J. Albert, G. Scarlatti, D. R. Littman, and E. M. Fenyo. 1997. Coreceptor usage of primary human immunodeficiency virus type 1 isolates varies according to biological phenotype. J Virol 71:7478-7487.
Blanco-Gelaz, M. A., A. Lopez-Vazquez, S. Garcia-Fernandez, J. Martinez-Borra, S. Gonzalez, and C. Lopez-Larrea. 2001. Genetic variability, molecular evolution, and geographic diversity of HLA-B27. Hum Immunol 62:1042-1050.
Bonsch, D., V. Greifenberg, K. Bayerlein, T. Biermann, U. Reulbach, T. Hillemacher, J. Kornhuber, and S. Bleich. 2005a. Alpha-synuclein protein levels are increased in alcoholic patients and are linked to craving. Alcohol Clin Exp Res 29:763-765.
Bonsch, D., T. Lederer, U. Reulbach, T. Hothorn, J. Kornhuber, and S. Bleich. 2005b. Joint analysis of the NACP-REP1 marker within the alpha synuclein gene concludes association with alcohol dependence. Hum Mol Genet 14:967-971.
Bonsch, D., B. Lenz, J. Kornhuber, and S. Bleich. 2005c. DNA hypermethylation of the alpha synuclein promoter in patients with alcoholism. Neuroreport 16:167-170.
Bonsch, D., U. Reulbach, K. Bayerlein, T. Hillemacher, J. Kornhuber, and S. Bleich. 2004. Elevated alpha synuclein mRNA levels are associated with craving in patients with alcoholism. Biol Psychiatry 56:984-986.
Borras, E., C. Coutelle, A. Rosell, F. Fernandez-Muixi, M. Broch, B. Crosas, L. Hjelmqvist, A. Lorenzo, C. Gutierrez, M. Santos, M. Szczepanek, M. Heilig, P. Quattrocchi, J. Farres, F. Vidal, C. Richart, T. Mach, J. Bogdal, H. Jornvall, H. K. Seitz, P. Couzigou, and X.

Pares. 2000. Genetic polymorphism of alcohol dehydrogenase in europeans: the ADH2*2 allele decreases the risk for alcoholism and is associated with ADH3*1. Hepatology 31:984-989.
Brass, A. L., D. M. Dykxhoorn, Y. Benita, N. Yan, A. Engelman, R. J. Xavier, J. Lieberman, and S. J. Elledge. 2008. Identification of host proteins required for HIV infection through a functional genomic screen. Science 319:921-926.
Briggs, D. R., D. L. Tuttle, J. W. Sleasman, and M. M. Goodenow. 2000. Envelope V3 amino acid sequence predicts HIV-1 phenotype (co-receptor usage and tropism for macrophages). Aids 14:2937-2939.
Broder, C. C., and R. G. Collman. 1997. Chemokine receptors and HIV. J Leukoc Biol 62:20-29. Bruen, T. C., H. Philippe, and D. Bryant. 2006. A simple and robust statistical test for detecting
the presence of recombination. Genetics 172:2665-2681. Burkala, E. J., J. He, J. T. West, C. Wood, and C. K. Petito. 2005. Compartmentalization of HIV-
1 in the central nervous system: role of the choroid plexus. Aids 19:675-684. Cann, R. L., M. Stoneking, and A. C. Wilson. 1987. Mitochondrial DNA and human evolution.
Nature 325:31-36. Cao, K., A. M. Moormann, K. E. Lyke, C. Masaberg, O. P. Sumba, O. K. Doumbo, D. Koech, A.
Lancaster, M. Nelson, D. Meyer, R. Single, R. J. Hartzman, C. V. Plowe, J. Kazura, D. L. Mann, M. B. Sztein, G. Thomson, and M. A. Fernandez-Vina. 2004. Differentiation between African populations is evidenced by the diversity of alleles and haplotypes of HLA class I loci. Tissue Antigens 63:293-325.
Caramelli, D., C. Lalueza-Fox, S. Condemi, L. Longo, L. Milani, A. Manfredini, M. de Saint Pierre, F. Adoni, M. Lari, P. Giunti, S. Ricci, A. Casoli, F. Calafell, F. Mallegni, J. Bertranpetit, R. Stanyon, G. Bertorelle, and G. Barbujani. 2006. A highly divergent mtDNA sequence in a Neandertal individual from Italy. Curr Biol 16:R630-632.
Carmody, M. S., and J. R. Anderson. 2007. BiDil (isosorbide dinitrate and hydralazine): a new fixed-dose combination of two older medications for the treatment of heart failure in black patients. Cardiol Rev 15:46-53.
Carrillo, A., and L. Ratner. 1996. Cooperative effects of the human immunodeficiency virus type 1 envelope variable loops V1 and V3 in mediating infectivity for T cells. J Virol 70:1310-1316.
Cavalli-Sforza, L. L., P. Menozzi, and A. Piazza. 1994. The History and Geography of Human Genes. Princeton University Press, Princeton.
CDC. 2006a. Health, United States, 2006: with Chartbook on Trends in the Health of Americans. CDC. 2006b. Mother to Child (Perinatal) HIV Transmission and Infection. Department of Health
and Human Services. CDC. 2007. What Women Can Do. Department of Health and Human Services. Centurion-Lara, A., C. Castro, L. Barrett, C. Cameron, M. Mostowfi, W. C. Van Voorhis, and S.
A. Lukehart. 1999. Treponema pallidum major sheath protein homologue TprK is a target of opsonic antibody and the protective immune response. J Exp Med 189:647-656.
Centurion-Lara, A., C. Godornes, C. Castro, W. C. Van Voorhis, and S. A. Lukehart. 2000a. The tprK gene is heterogeneous among Treponema pallidum strains and has multiple alleles. Infect Immun 68:824-831.
Centurion-Lara, A., R. E. LaFond, K. Hevner, C. Godornes, B. J. Molini, W. C. Van Voorhis, and S. A. Lukehart. 2004. Gene conversion: a mechanism for generation of heterogeneity in the tprK gene of Treponema pallidum during infection. Mol Microbiol 52:1579-1596.

Centurion-Lara, A., B. J. Molini, C. Godornes, E. Sun, K. Hevner, W. C. Van Voorhis, and S. A. Lukehart. 2006. Molecular differentiation of Treponema pallidum subspecies. J Clin Microbiol 44:3377-3380.
Centurion-Lara, A., E. S. Sun, L. K. Barrett, C. Castro, S. A. Lukehart, and W. C. Van Voorhis. 2000b. Multiple alleles of Treponema pallidum repeat gene D in Treponema pallidum isolates. J Bacteriol 182:2332-2335.
Chao, Y. C., M. F. Wang, H. S. Tang, C. T. Hsu, and S. J. Yin. 1994. Genotyping of alcohol dehydrogenase at the ADH2 and ADH3 loci by using a polymerase chain reaction and restriction-fragment-length polymorphism in Chinese alcoholic cirrhotics and non-alcoholics. Proc Natl Sci Counc Repub China B 18:101-106.
Chen, C. C., R. B. Lu, Y. C. Chen, M. F. Wang, Y. C. Chang, T. K. Li, and S. J. Yin. 1999. Interaction between the functional polymorphisms of the alcohol-metabolism genes in protection against alcoholism. Am J Hum Genet 65:795-807.
Chen, W. J., E. W. Loh, Y. P. Hsu, C. C. Chen, J. M. Yu, and A. T. Cheng. 1996. Alcohol-metabolising genes and alcoholism among Taiwanese Han men: independent effect of ADH2, ADH3 and ALDH2. Br J Psychiatry 168:762-767.
Chen, X., H. A. de Silva, M. J. Pettenati, P. N. Rao, P. St George-Hyslop, A. D. Roses, Y. Xia, K. Horsburgh, K. Ueda, and T. Saitoh. 1995. The human NACP/alpha-synuclein gene: chromosome assignment to 4q21.3-q22 and TaqI RFLP analysis. Genomics 26:425-427.
Chiba-Falek, O., J. W. Touchman, and R. L. Nussbaum. 2003. Functional analysis of intra-allelic variation at NACP-Rep1 in the alpha-synuclein gene. Hum Genet 113:426-431.
Chisenga, M., L. Kasonka, M. Makasa, M. Sinkala, C. Chintu, C. Kaseba, F. Kasolo, A. Tomkins, S. Murray, and S. Filteau. 2005. Factors affecting the duration of exclusive breastfeeding among HIV-infected and -uninfected women in Lusaka, Zambia. J Hum Lact 21:266-275.
Clarimon, J., R. R. Gray, L. N. Williams, M. A. Enoch, R. W. Robin, B. Albaugh, A. Singleton, D. Goldman, and C. J. Mulligan. 2007. Linkage disequilibrium and association analysis of alpha-synuclein and alcohol and drug dependence in two American Indian populations. Alcohol Clin Exp Res 31:546-554.
Cockburn, J. 1971. Infectious disease in ancient populations. Current Anthropology 12:45-62. Cohen, M. N., and G. J. Armelagos. 1984. Paleopathology at the Origins of Agriculture.
Academic Press, Orlando. Connor, R. I., K. E. Sheridan, D. Ceradini, S. Choe, and N. R. Landau. 1997. Change in
coreceptor use coreceptor use correlates with disease progression in HIV-1--infected individuals. J Exp Med 185:621-628.
Coovadia, H. M., N. C. Rollins, R. M. Bland, K. Little, A. Coutsoudis, M. L. Bennish, and M. L. Newell. 2007. Mother-to-child transmission of HIV-1 infection during exclusive breastfeeding in the first 6 months of life: an intervention cohort study. Lancet 369:1107-1116.
Coutsoudis, A. 2000. Influence of infant feeding patterns on early mother-to-child transmission of HIV-1 in Durban, South Africa. Ann N Y Acad Sci 918:136-144.
Coutsoudis, A., F. Dabis, W. Fawzi, P. Gaillard, G. Haverkamp, D. R. Harris, J. B. Jackson, V. Leroy, N. Meda, P. Msellati, M. L. Newell, R. Nsuati, J. S. Read, and S. Wiktor. 2004. Late postnatal transmission of HIV-1 in breast-fed children: an individual patient data meta-analysis. J Infect Dis 189:2154-2166.

Coutsoudis, A., L. Kuhn, K. Pillay, and H. M. Coovadia. 2002. Exclusive breast-feeding and HIV transmission. Aids 16:498-499.
Coutsoudis, A., K. Pillay, L. Kuhn, E. Spooner, W. Y. Tsai, and H. M. Coovadia. 2001. Method of feeding and transmission of HIV-1 from mothers to children by 15 months of age: prospective cohort study from Durban, South Africa. Aids 15:379-387.
Coutsoudis, A., K. Pillay, E. Spooner, L. Kuhn, and H. M. Coovadia. 1999. Influence of infant-feeding patterns on early mother-to-child transmission of HIV-1 in Durban, South Africa: a prospective cohort study. South African Vitamin A Study Group. Lancet 354:471-476.
Crago, S. S., S. J. Prince, T. G. Pretlow, J. R. McGhee, and J. Mestecky. 1979. Human colostral cells. I. Separation and characterization. Clin Exp Immunol 38:585-597.
Crosby, A. 1969. The Early History of Syphilis: A Reappraisal. American Anthropologist 71:218-227.
Dabis, F., P. Msellati, N. Meda, C. Welffens-Ekra, B. You, O. Manigart, V. Leroy, A. Simonon, M. Cartoux, P. Combe, A. Ouangre, R. Ramon, O. Ky-Zerbo, C. Montcho, R. Salamon, C. Rouzioux, P. Van de Perre, and L. Mandelbrot. 1999. 6-month efficacy, tolerance, and acceptability of a short regimen of oral zidovudine to reduce vertical transmission of HIV in breastfed children in Cote d'Ivoire and Burkina Faso: a double-blind placebo-controlled multicentre trial. DITRAME Study Group. DIminution de la Transmission Mere-Enfant. Lancet 353:786-792.
De Pasquale, M. P., A. J. Leigh Brown, S. C. Uvin, J. Allega-Ingersoll, A. M. Caliendo, L. Sutton, S. Donahue, and R. T. D'Aquila. 2003. Differences in HIV-1 pol sequences from female genital tract and blood during antiretroviral therapy. J Acquir Immune Defic Syndr 34:37-44.
Desselberger, U. 2000. Emerging and re-emerging infectious diseases. J Infect 40:3-15. Diaz, G. A., B. D. Gelb, N. Risch, T. G. Nygaard, A. Frisch, I. J. Cohen, C. S. Miranda, O.
Amaral, I. Maire, L. Poenaru, C. Caillaud, M. Weizberg, P. Mistry, and R. J. Desnick. 2000. Gaucher disease: the origins of the Ashkenazi Jewish N370S and 84GG acid beta-glucosidase mutations. Am J Hum Genet 66:1821-1832.
Dirks, R. 1993. Starvation and Famine: cross-cultural codes and some hypothesis tests. Cross Cult Res 27:28-69.
Dixon, E. J. 2001. Human colonization of the Americas: timing, technology and process. Quat Sci Rev 20:277-299.
Doeblin, T. D., K. Evans, and G. B. Ingall. 1969. Diabetes and hyperglycemia in Seneca Indians. Human Hered 19:613-627.
Donaldson, Y. K., J. E. Bell, J. W. Ironside, R. P. Brettle, J. R. Robertson, A. Busuttil, and P. Simmonds. 1994. Redistribution of HIV outside the lymphoid system with onset of AIDS. Lancet 343:383-385.
Douek, D. 2007a. HIV Disease Progression. Top HIV Med 15:114-117. Douek, D. 2007b. HIV disease progression: immune activation, microbes, and a leaky gut. Top
HIV Med 15:114-117. Dover, G. 2002. Molecular Drive. Trends Genet 18:587-589. Drouin, G., F. Prat, M. Ell, and G. D. Clarke. 1999. Detecting and characterizing gene
conversions between multigene family members. Mol Biol Evol 16:1369-1390. Drummond, A. J., S. Y. Ho, M. J. Phillips, and A. Rambaut. 2006. Relaxed phylogenetics and
dating with confidence. PLoS Biol 4:e88.

Drummond, A. J., and A. Rambaut. 2007. BEAST: Bayesian evolutionary analysis by sampling trees. BMC Evol Biol 7:214.
Drummond, A. J., A. Rambaut, B. Shapiro, and O. G. Pybus. 2005. Bayesian coalescent inference of past population dynamics from molecular sequences. Mol Biol Evol 22:1185-1192.
Dudbridge, F. 2003. Pedigree disequilibrium tests for multilocus haplotypes. Genet Epidemiol 25:115-121.
Duster, T. 2007. Medicalisation of race. Lancet 369:702-704. Ehlers, C. L., C. Garcia-Andrade, T. L. Wall, D. F. Sobel, and E. Phillips. 1998. Determinants of
P3 amplitude and response to alcohol in Native American Mission Indians. Neuropsychopharmacology 18:282-292.
Ehlers, C. L., D. A. Gilder, L. Harris, and L. Carr. 2001. Association of the ADH2*3 allele with a negative family history of alcoholism in African American young adults. Alcohol Clin Exp Res 25:1773-1777.
Ehlers, C. L., D. A. Gilder, T. L. Wall, E. Phillips, H. Feiler, and K. C. Wilhelmsen. 2004a. Genomic screen for loci associated with alcohol dependence in Mission Indians. Am J Med Genet B Neuropsychiatr Genet 129:110-115.
Ehlers, C. L., T. L. Wall, M. Betancourt, and D. A. Gilder. 2004b. The clinical course of alcoholism in 243 Mission Indians. Am J Psychiatry 161:1204-1210.
Embree, J. E., S. Njenga, P. Datta, N. J. Nagelkerke, J. O. Ndinya-Achola, Z. Mohammed, S. Ramdahin, J. J. Bwayo, and F. A. Plummer. 2000. Risk factors for postnatal mother-child transmission of HIV-1. Aids 14:2535-2541.
Endicott, J., and R. L. Spitzer. 1978. A diagnostic interview: the schedule for affective disorders and schizophrenia. Arch Gen Psychiatry 35:837-844.
Excoffier, L., G. Laval, and S. Schneider. 2005. Arlequin ver. 3.0: An integrated software package for population genetics data analysis. Evolutionary Bioinformatics Online 1:47-50.
Eyre-Walker, A. 1993. Recombination and mammalian genome evolution. Proc Biol Sci 252:237-243.
Fauci, A. S. 1998. New and reemerging diseases: the importance of biomedical research. Emerg Infect Dis 4:374-378.
Fauci, A. S., N. A. Touchette, and G. K. Folkers. 2005. Emerging infectious diseases: a 10-year perspective from the National Institute of Allergy and Infectious Diseases. Emerg Infect Dis 11:519-525.
Fawzi, W., G. Msamanga, D. Spiegelman, B. Renjifo, H. Bang, S. Kapiga, J. Coley, E. Hertzmark, M. Essex, and D. Hunter. 2002a. Transmission of HIV-1 through breastfeeding among women in Dar es Salaam, Tanzania. J Acquir Immune Defic Syndr 31:331-338.
Fawzi, W. W., G. I. Msamanga, D. Hunter, B. Renjifo, G. Antelman, H. Bang, K. Manji, S. Kapiga, D. Mwakagile, M. Essex, and D. Spiegelman. 2002b. Randomized trial of vitamin supplements in relation to transmission of HIV-1 through breastfeeding and early child mortality. Aids 16:1935-1944.
Feavers, I. M., A. B. Heath, J. A. Bygraves, and M. C. Maiden. 1992. Role of horizontal genetic exchange in the antigenic variation of the class 1 outer membrane protein of Neisseria meningitidis. Mol Microbiol 6:489-495.

Fee, M. 2006. Racializing narratives: Obesity, diabetes, and the "aboriginal" thrifty genotype. Social Science and Medicine 62:2988-2997.
Feil, E. J., E. C. Holmes, D. E. Bessen, M. S. Chan, N. P. Day, M. C. Enright, R. Goldstein, D. W. Hood, A. Kalia, C. E. Moore, J. Zhou, and B. G. Spratt. 2001. Recombination within natural populations of pathogenic bacteria: short-term empirical estimates and long-term phylogenetic consequences. Proc Natl Acad Sci U S A 98:182-187.
Feil, E. J., M. C. Maiden, M. Achtman, and B. G. Spratt. 1999. The relative contributions of recombination and mutation to the divergence of clones of Neisseria meningitidis. Mol Biol Evol 16:1496-1502.
Feil, E. J., and B. G. Spratt. 2001. Recombination and the population structures of bacterial pathogens. Annu Rev Microbiol 55:561-590.
Feldmann, H., M. Czub, S. Jones, D. Dick, M. Garbutt, A. Grolla, and H. Artsob. 2002. Emerging and re-emerging infectious diseases. Med Microbiol Immunol 191:63-74.
Fellay, J., K. V. Shianna, D. Ge, S. Colombo, B. Ledergerber, M. Weale, K. Zhang, C. Gumbs, A. Castagna, A. Cossarizza, A. Cozzi-Lepri, A. De Luca, P. Easterbrook, P. Francioli, S. Mallal, J. Martinez-Picado, J. M. Miro, N. Obel, J. P. Smith, J. Wyniger, P. Descombes, S. E. Antonarakis, N. L. Letvin, A. J. McMichael, B. F. Haynes, A. Telenti, and D. B. Goldstein. 2007. A whole-genome association study of major determinants for host control of HIV-1. Science 317:944-947.
Fix, A. G. 2002. Colonization models and initial genetic diversity in the Americas. Hum Biol 74:1-10.
Fraser, C. M., S. J. Norris, G. M. Weinstock, O. White, G. G. Sutton, R. Dodson, M. Gwinn, E. K. Hickey, R. Clayton, K. A. Ketchum, E. Sodergren, J. M. Hardham, M. P. McLeod, S. Salzberg, J. Peterson, H. Khalak, D. Richardson, J. K. Howell, M. Chidambaram, T. Utterback, L. McDonald, P. Artiach, C. Bowman, M. D. Cotton, C. Fujii, S. Garland, B. Hatch, K. Horst, K. Roberts, M. Sandusky, J. Weidman, H. O. Smith, and J. C. Venter. 1998. Complete genome sequence of Treponema pallidum, the syphilis spirochete. Science 281:375-388.
Fribourg-Blanc, A., H. H. Mollaret, and G. Niel. 1966. [Serologic and microscopic confirmation of treponemosis in Guinea baboons]. Bull Soc Pathol Exot Filiales 59:54-59.
Fuchs, A. R. 1991. Physiology and endocrinology of lactation. in S. G. Gabbe, ed. Obstetrics - Normal and Problem Pregnancies. Churchill Livingstone, New York.
Fujimoto, W. Y. 1996. Overview of non-insulin-dependent diabetes mellitus (NIDDM) in different population groups. Diabet Med 13:S7-10.
Gabriel, S. B., S. F. Schaffner, H. Nguyen, J. M. Moore, J. Roy, B. Blumenstiel, J. Higgins, M. DeFelice, A. Lochner, M. Faggart, S. N. Liu-Cordero, C. Rotimi, A. Adeyemo, R. Cooper, R. Ward, E. S. Lander, M. J. Daly, and D. Altshuler. 2002. The structure of haplotype blocks in the human genome. Science 296:2225-2229.
Galtier, N. 2003. Gene conversion drives GC content evolution in mammalian histones. Trends Genet 19:65-68.
Galtier, N., G. Piganeau, D. Mouchiroud, and L. Duret. 2001. GC-content evolution in mammalian genomes: the biased gene conversion hypothesis. Genetics 159:907-911.
Galvani, A. P., and J. Novembre. 2005. The evolutionary history of the CCR5-Delta32 HIV-resistance mutation. Microbes Infect 7:302-309.

Galvani, A. P., and M. Slatkin. 2003. Evaluating plague and smallpox as historical selective pressures for the CCR5-Delta 32 HIV-resistance allele. Proc Natl Acad Sci U S A 100:15276-15279.
Gao, F., E. Bailes, D. L. Robertson, Y. Chen, C. M. Rodenburg, S. F. Michael, L. B. Cummins, L. O. Arthur, M. Peeters, G. M. Shaw, P. M. Sharp, and B. H. Hahn. 1999. Origin of HIV-1 in the chimpanzee Pan troglodytes troglodytes. Nature 397:436-441.
Garzino-Demo, A., A. L. DeVico, K. E. Conant, and R. C. Gallo. 2000. The role of chemokines in human immunodeficiency virus infection. Immunol Rev 177:79-87.
Gatanaga, H., S. Oka, S. Ida, T. Wakabayashi, T. Shioda, and A. Iwamoto. 1999. Active HIV-1 redistribution and replication in the brain with HIV encephalitis. Arch Virol 144:29-43.
Georgeson, J. C., and S. M. Filteau. 2000. Physiology, immunology, and disease transmission in human breast milk. AIDS Patient Care STDS 14:533-539.
Giacani, L., K. Hevner, and A. Centurion-Lara. 2005. Gene organization and transcriptional analysis of the tprJ, tprI, tprG, and tprF loci in Treponema pallidum strains Nichols and Sea 81-4. J Bacteriol 187:6084-6093.
Giacani, L., E. S. Sun, K. Hevner, B. J. Molini, W. C. Van Voorhis, S. A. Lukehart, and A. Centurion-Lara. 2004. Tpr homologs in Treponema paraluiscuniculi Cuniculi A strain. Infect Immun 72:6561-6576.
Gilder, D. A., T. L. Wall, and C. L. Ehlers. 2004. Comorbidity of select anxiety and affective disorders with alcohol dependence in southwest California Indians. Alcohol Clin Exp Res 28:1805-1813.
Goedde, H. W., D. P. Agarwal, G. Fritze, D. Meier-Tackmann, S. Singh, G. Beckmann, K. Bhatia, L. Z. Chen, B. Fang, R. Lisker, and et al. 1992. Distribution of ADH2 and ALDH2 genotypes in different populations. Hum Genet 88:344-346.
Gogarten, J. P., and L. Olendzenski. 1999. Orthologs, paralogs and genome comparisons. Curr Opin Genet Dev 9:630-636.
Goldman, A. S. 1993. The immune system of human milk: antimicrobial, antiinflammatory and immunomodulating properties. Pediatr Infect Dis J 12:664-671.
Goldman, A. S., S. Chheda, and R. Garofalo. 1998. Evolution of immunologic functions of the mammary gland and the postnatal development of immunity. Pediatr Res 43:155-162.
Goodenow, M. M., S. L. Rose, D. L. Tuttle, and J. W. Sleasman. 2003. HIV-1 fitness and macrophages. J Leukoc Biol 74:657-666.
Gray, R. R., C. J. Mulligan, B. J. Molini, E. S. Sun, L. Giacani, C. Godornes, A. Kitchen, S. A. Lukehart, and A. Centurion-Lara. 2006. Molecular evolution of the tprC, D, I, K, G, and J genes in the pathogenic genus Treponema. Mol Biol Evol 23:2220-2233.
Grenfell, B. T., O. G. Pybus, J. R. Gog, J. L. Wood, J. M. Daly, J. A. Mumford, and E. C. Holmes. 2004. Unifying the epidemiological and evolutionary dynamics of pathogens. Science 303:327-332.
Gurtler, L. G., P. H. Hauser, J. Eberle, A. von Brunn, S. Knapp, L. Zekeng, J. M. Tsague, and L. Kaptue. 1994. A new subtype of human immunodeficiency virus type 1 (MVP-5180) from Cameroon. J Virol 68:1581-1585.
Haase, A. T., K. Henry, M. Zupancic, G. Sedgewick, R. A. Faust, H. Melroe, W. Cavert, K. Gebhard, K. Staskus, Z. Q. Zhang, P. J. Dailey, H. H. Balfour, Jr., A. Erice, and A. S. Perelson. 1996. Quantitative image analysis of HIV-1 infection in lymphoid tissue. Science 274:985-989.

Hackett, C. J. 1963. On the origin of the human treponematoses (pinta, yaws, endemic syphilis and venereal syphilis). . Bulletin of the World Health Organization 29:7-41.
Hales, C. N., and D. J. Barker. 1992. Type 2 (non-insulin-dependent) diabetes mellitus: the thrifty phenotype hypothesis. Diabetologia 35:595-601.
Hales, C. N., and D. J. Barker. 2001. The thrifty phenotype hypothesis. Br Med Bull 60:5-20. Hara, K., O. Terasaki, and Y. Okubo. 2000. Dipole estimation of alpha EEG during alcohol
ingestion in males genotypes for ALDH2. Life Sci 67:1163-1173. Harada, S., D. P. Agarwal, and H. W. Goedde. 1982. Mechanism of alcohol sensitivity and
disulfiram-ethanol reaction. Subst Alcohol Actions Misuse 3:107-115. Harpending, H., and A. Rogers. 2000. Genetic perspectives on human origins and differentiation.
Annu Rev Genomics Hum Genet 1:361-385. Harpending, H., S. T. Sherry, A. R. Rogers, and M. Stoneking. 1993. The genetic structure of
ancient human populations. Curr Anthropol 34:483-496. Harpending, H. C. 1994. Signature of ancient population growth in a low-resolution
mitochondrial DNA mismatch distribution. Hum Biol 66:591-600. Harpending, H. C., M. A. Batzer, M. Gurven, L. B. Jorde, A. R. Rogers, and S. T. Sherry. 1998.
Genetic traces of ancient demography. Proc Natl Acad Sci U S A 95:1961-1967. Harper, K. N., P. S. Ocampo, B. M. Steiner, R. W. George, M. S. Silverman, S. Bolotin, A.
Pillay, N. J. Saunders, and G. J. Armelagos. 2008. On the origin of the treponematoses: a phylogenetic approach. PLoS Negl Trop Dis 2:e148.
Hasegawa, M., H. Kishino, and T. Yano. 1985. Dating of the human-ape splitting by a molecular clock of mitochondrial DNA. J Mol Evol 22:160-174.
Helzer, J. E., K. K. Bucholz, L. J. Bierut, D. A. Regier, M. A. Schuckit, and S. E. Guth. 2006. Should DSM-V include dimensional diagnostic criteria for alcohol use disorders? Alcohol Clin Exp Res 30:303-310.
Henderson, G. J., N. G. Hoffman, L. H. Ping, S. A. Fiscus, I. F. Hoffman, K. M. Kitrinos, T. Banda, F. E. Martinson, P. N. Kazembe, D. A. Chilongozi, M. S. Cohen, and R. Swanstrom. 2004. HIV-1 populations in blood and breast milk are similar. Virology 330:295-303.
Ho, F. C., R. L. Wong, and J. W. Lawton. 1979. Human colostral and breast milk cells. A light and electron microscopic study. Acta Paediatr Scand 68:389-396.
Holmes, E. C., R. Urwin, and M. C. Maiden. 1999. The influence of recombination on the population structure and evolution of the human pathogen Neisseria meningitidis. Mol Biol Evol 16:741-749.
Hoover, E. L. 2007. There is no scientific rationale for race-based research. J Natl Med Assoc 99:690-692.
Howell-Adams, B., and H. S. Seifert. 2000. Molecular models accounting for the gene conversion reactions mediating gonococcal pilin antigenic variation. Mol Microbiol 37:1146-1158.
Hudson, E. H. 1965. Treponematosis and man's social evolution. American Anthropologist 67:885-901.
Hudson, R. R., M. Slatkin, and W. P. Maddison. 1992. Estimation of levels of gene flow from DNA sequence data. Genetics 132:583-589.
Hughes, D. 2000. Co-evolution of the tuf genes links gene conversion with the generation of chromosomal inversions. J Mol Biol 297:355-364.

Hughes, E. S., J. E. Bell, and P. Simmonds. 1997a. Investigation of population diversity of human immunodeficiency virus type 1 in vivo by nucleotide sequencing and length polymorphism analysis of the V1/V2 hypervariable region of env. J Gen Virol 78 ( Pt 11):2871-2882.
Hughes, E. S., J. E. Bell, and P. Simmonds. 1997b. Investigation of the dynamics of the spread of human immunodeficiency virus to brain and other tissues by evolutionary analysis of sequences from the p17gag and env genes. J Virol 71:1272-1280.
Hugot, J. P., M. Chamaillard, H. Zouali, S. Lesage, J. P. Cezard, J. Belaiche, S. Almer, C. Tysk, C. A. O'Morain, M. Gassull, V. Binder, Y. Finkel, A. Cortot, R. Modigliani, P. Laurent-Puig, C. Gower-Rousseau, J. Macry, J. F. Colombel, M. Sahbatou, and G. Thomas. 2001. Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn's disease. Nature 411:599-603.
Hung, C. S., N. Vander Heyden, and L. Ratner. 1999. Analysis of the critical domain in the V3 loop of human immunodeficiency virus type 1 gp120 involved in CCR5 utilization. J Virol 73:8216-8226.
Ichikawa, M., M. Sugita, M. Takahashi, M. Satomi, T. Takeshita, T. Araki, and H. Takahashi. 2003. Breast milk macrophages spontaneously produce granulocyte-macrophage colony-stimulating factor and differentiate into dendritic cells in the presence of exogenous interleukin-4 alone. Immunology 108:189-195.
IHS. 2006. Facts on Indian Health Disparities. Indian Health Services. Iliff, P. J., E. G. Piwoz, N. V. Tavengwa, C. D. Zunguza, E. T. Marinda, K. J. Nathoo, L. H.
Moulton, B. J. Ward, and J. H. Humphrey. 2005. Early exclusive breastfeeding reduces the risk of postnatal HIV-1 transmission and increases HIV-free survival. Aids 19:699-708.
Iwahashi, K., Y. Matsuo, H. Suwaki, K. Nakamura, and Y. Ichikawa. 1995. CYP2E1 and ALDH2 genotypes and alcohol dependence in Japanese. Alcohol Clin Exp Res 19:564-566.
John, G. C., R. W. Nduati, D. A. Mbori-Ngacha, B. A. Richardson, D. Panteleeff, A. Mwatha, J. Overbaugh, J. Bwayo, J. O. Ndinya-Achola, and J. K. Kreiss. 2001. Correlates of mother-to-child human immunodeficiency virus type 1 (HIV-1) transmission: association with maternal plasma HIV-1 RNA load, genital HIV-1 DNA shedding, and breast infections. J Infect Dis 183:206-212.
Johnson, J. E., and C. W. McNutt. 1964. Diabetes mellitus in an American Indian population isolate. Tex Rep Biol Med 22:110-125.
Jones, K. E., N. G. Patel, M. A. Levy, A. Storeygard, D. Balk, J. L. Gittleman, and P. Daszak. 2008. Global trends in emerging infectious diseases. Nature 451:990-993.
Kemal, K. S., B. Foley, H. Burger, K. Anastos, H. Minkoff, C. Kitchen, S. M. Philpott, W. Gao, E. Robison, S. Holman, C. Dehner, S. Beck, W. A. Meyer, 3rd, A. Landay, A. Kovacs, J. Bremer, and B. Weiser. 2003. HIV-1 in genital tract and plasma of women: compartmentalization of viral sequences, coreceptor usage, and glycosylation. Proc Natl Acad Sci U S A 100:12972-12977.
Kim, D.-J., I.-G. Choi, B. L. Park, B.-C. Lee, B.-J. Ham, S. Yoon, J. S. Bae, H. S. Cheong, and H. D. Shin. 2008. Major genetic components underlying alcoholism in Korean population. Hum Mol Genet 17:854-858.
Kimura, M., and J. L. King. 1979. Fixation of a deleterious allele at one of two "duplicate" loci by mutation pressure and random drift. Proc Natl Acad Sci U S A 76:2858-2861.

King, R. C., and W. D. Stansfield. 1997. A Dictionary of Genetics. Oxford University Press, New York.
Kinzie, J. D., P. K. Leung, J. Boehnlein, D. Matsunaga, R. Johnson, S. Manson, J. H. Shore, J. Heinz, and M. Williams. 1992. Psychiatric epidemiology of an Indian village. A 19-year replication study. J Nerv Ment Dis 180:33-39.
Kitrinos, K. M., N. G. Hoffman, J. A. Nelson, and R. Swanstrom. 2003. Turnover of env variable region 1 and 2 genotypes in subjects with late-stage human immunodeficiency virus type 1 infection. J Virol 77:6811-6822.
Klevytska, A. M., M. R. Mracna, L. Guay, G. Becker-Pergola, M. Furtado, L. Zhang, J. B. Jackson, and S. H. Eshleman. 2002. Analysis of length variation in the V1-V2 region of env in nonsubtype B HIV type 1 from Uganda. AIDS Res Hum Retroviruses 18:791-796.
Knowler, W. C., D. J. Pettitt, M. F. Saad, and P. H. Bennett. 1990. Diabetes mellitus in the Pima Indians: incidence, risk factors and pathogenesis. Diabetes Metab Rev 6:1-27.
Kobayashi, H., S. Ide, J. Hasegawa, H. Ujike, Y. Sekine, N. Ozaki, T. Inada, M. Harano, T. Komiyama, M. Yamada, M. Iyo, H. W. Shen, K. Ikeda, and I. Sora. 2004. Study of association between alpha-synuclein gene polymorphism and methamphetamine psychosis/dependence. Ann N Y Acad Sci 1025:325-334.
Kolman, C. J., and E. Bermingham. 1997. Mitochondrial and nuclear DNA diversity in the Choco and Chibcha Amerinds of Panama. Genetics 147:1289-1302.
Kolman, C. J., E. Bermingham, R. Cooke, R. H. Ward, T. D. Arias, and F. Guionneau-Sinclair. 1995. Reduced mtDNA diversity in the Ngobe Amerinds of Panama. Genetics 140:275-283.
Kolman, C. J., N. Sambuughin, and E. Bermingham. 1996. Mitochondrial DNA analysis of Mongolian populations and implications for the origin of New World founders. Genetics 142:1321-1334.
Kondrashov, F. A., I. B. Rogozin, Y. I. Wolf, and E. V. Koonin. 2002. Selection in the evolution of gene duplications. Genome Biol 3:RESEARCH0008.
Konishi, T., M. Calvillo, A. S. Leng, J. Feng, T. Lee, H. Lee, J. L. Smith, S. H. Sial, N. Berman, S. French, V. Eysselein, K. M. Lin, and Y. J. Wan. 2003. The ADH3*2 and CYP2E1 c2 alleles increase the risk of alcoholism in Mexican American men. Exp Mol Pathol 74:183-189.
Korber, B. T., K. J. Kunstman, B. K. Patterson, M. Furtado, M. M. McEvilly, R. Levy, and S. M. Wolinsky. 1994. Genetic differences between blood- and brain-derived viral sequences from human immunodeficiency virus type 1-infected patients: evidence of conserved elements in the V3 region of the envelope protein of brain-derived sequences. J Virol 68:7467-7481.
Koulinska, I. N., E. Villamor, B. Chaplin, G. Msamanga, W. Fawzi, B. Renjifo, and M. Essex. 2006. Transmission of cell-free and cell-associated HIV-1 through breast-feeding. J Acquir Immune Defic Syndr 41:93-99.
Kourtis, A. P., S. Butera, C. Ibegbu, L. Beled, and A. Duerr. 2003. Breast milk and HIV-1: vector of transmission or vehicle of protection? Lancet Infect Dis 3:786-793.
Krause, J., C. Lalueza-Fox, L. Orlando, W. Enard, R. E. Green, H. A. Burbano, J. J. Hublin, C. Hanni, J. Fortea, M. de la Rasilla, J. Bertranpetit, A. Rosas, and S. Paabo. 2007. The derived FOXP2 variant of modern humans was shared with Neandertals. Curr Biol 17:1908-1912.

Krings, M., C. Capelli, F. Tschentscher, H. Geisert, S. Meyer, A. von Haeseler, K. Grossschmidt, G. Possnert, M. Paunovic, and S. Paabo. 2000. A view of Neandertal genetic diversity. Nat Genet 26:144-146.
Krings, M., H. Geisert, R. W. Schmitz, H. Krainitzki, and S. Paabo. 1999. DNA sequence of the mitochondrial hypervariable region II from the neandertal type specimen. Proc Natl Acad Sci U S A 96:5581-5585.
Krings, M., A. Stone, R. W. Schmitz, H. Krainitzki, M. Stoneking, and S. Paabo. 1997. Neandertal DNA sequences and the origin of modern humans. Cell 90:19-30.
Kruger, R., W. Kuhn, T. Muller, D. Woitalla, M. Graeber, S. Kosel, H. Przuntek, J. T. Epplen, L. Schols, and O. Riess. 1998. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson's disease. Nat Genet 18:106-108.
Kuhn, L., M. Sinkala, C. Kankasa, K. Semrau, P. Kasonde, N. Scott, M. Mwiya, C. Vwalika, J. Walter, W. Y. Tsai, G. M. Aldrovandi, and D. M. Thea. 2007. High Uptake of Exclusive Breastfeeding and Reduced Early Post-Natal HIV Transmission. PLoS ONE 2:e1363.
Kunitz, S. J., K. R. Gabriel, J. E. Levy, E. Henderson, K. Lampert, J. McCloskey, G. Quintero, S. Russell, and A. Vince. 1999. Alcohol dependence and conduct disorder among Navajo Indians. J Stud Alcohol 60:159-167.
LaFond, R. E., A. Centurion-Lara, C. Godornes, A. M. Rompalo, W. C. Van Voorhis, and S. A. Lukehart. 2003. Sequence diversity of Treponema pallidum subsp. pallidum tprK in human syphilis lesions and rabbit-propagated isolates. J Bacteriol 185:6262-6268.
Lalueza-Fox, C., M. L. Sampietro, D. Caramelli, Y. Puder, M. Lari, F. Calafell, C. Martinez-Maza, M. Bastir, J. Fortea, M. de la Rasilla, J. Bertranpetit, and A. Rosas. 2005. Neandertal evolutionary genetics: mitochondrial DNA data from the iberian peninsula. Mol Biol Evol 22:1077-1081.
Lamers, S. L., J. W. Sleasman, J. X. She, K. A. Barrie, S. M. Pomeroy, D. J. Barrett, and M. M. Goodenow. 1993. Independent variation and positive selection in env V1 and V2 domains within maternal-infant strains of human immunodeficiency virus type 1 in vivo. J Virol 67:3951-3960.
Langford, D., A. Grigorian, R. Hurford, A. Adame, R. J. Ellis, L. Hansen, and E. Masliah. 2004. Altered P-glycoprotein expression in AIDS patients with HIV encephalitis. J Neuropathol Exp Neurol 63:1038-1047.
Lathe, W. C., 3rd, and P. Bork. 2001. Evolution of tuf genes: ancient duplication, differential loss and gene conversion. FEBS Lett 502:113-116.
Leitner, T., B. Korber, M. Daniels, C. Calef, and B. Foley. 2005. HIV-1 Subtype and Circulating Recombinant Form (CRF) Reference Sequences, 2005. The Human Retroviruses and AIDS 2005 Compendium. Theoretical Biology and Biophysics, Los Alamos.
Leroy, V., J. M. Karon, A. Alioum, E. R. Ekpini, P. van de Perre, A. E. Greenberg, P. Msellati, M. Hudgens, F. Dabis, and S. Z. Wiktor. 2003. Postnatal transmission of HIV-1 after a maternal short-course zidovudine peripartum regimen in West Africa. Aids 17:1493-1501.
Lewis, P., R. Nduati, J. K. Kreiss, G. C. John, B. A. Richardson, D. Mbori-Ngacha, J. Ndinya-Achola, and J. Overbaugh. 1998. Cell-free human immunodeficiency virus type 1 in breast milk. J Infect Dis 177:34-39.
Liang, T., J. Spence, L. Liu, W. N. Strother, H. W. Chang, J. A. Ellison, L. Lumeng, T. K. Li, T. Foroud, and L. G. Carr. 2003. alpha-Synuclein maps to a quantitative trait locus for

alcohol preference and is differentially expressed in alcohol-preferring and -nonpreferring rats. Proc Natl Acad Sci U S A 100:4690-4695.
Liao, D. 2000. Gene conversion drives within genic sequences: concerted evolution of ribosomal RNA genes in bacteria and archaea. J Mol Evol 51:305-317.
Libert, F., P. Cochaux, G. Beckman, M. Samson, M. Aksenova, A. Cao, A. Czeizel, M. Claustres, C. de la Rua, M. Ferrari, C. Ferrec, G. Glover, B. Grinde, S. Guran, V. Kucinskas, J. Lavinha, B. Mercier, G. Ogur, L. Peltonen, C. Rosatelli, M. Schwartz, V. Spitsyn, L. Timar, L. Beckman, M. Parmentier, and G. Vassart. 1998. The deltaccr5 mutation conferring protection against HIV-1 in Caucasian populations has a single and recent origin in Northeastern Europe. Hum Mol Genet 7:399-406.
Lindsay, R. S., and P. H. Bennett. 2001. Type 2 diabetes, the thrifty phenotype - an overview. Br Med Bull 60:21-32.
Long, J. C., W. C. Knowler, R. L. Hanson, R. W. Robin, M. Urbanek, E. Moore, P. H. Bennett, and D. Goldman. 1998. Evidence for genetic linkage to alcohol dependence on chromosomes 4 and 11 from an autosome-wide scan in an American Indian population. Am J Med Genet 81:216-221.
Loussert-Ajaka, I., M. L. Chaix, B. Korber, F. Letourneur, E. Gomas, E. Allen, T. D. Ly, F. Brun-Vezinet, F. Simon, and S. Saragosti. 1995. Variability of human immunodeficiency virus type 1 group O strains isolated from Cameroonian patients living in France. J Virol 69:5640-5649.
Lucotte, G. 2001. Distribution of the CCR5 gene 32-basepair deletion in West Europe. A hypothesis about the possible dispersion of the mutation by the Vikings in historical times. Hum Immunol 62:933-936.
Lukehart, S. A., S. A. Baker-Zander, R. M. Lloyd, and S. Sell. 1980. Characterization of lymphocyte responsiveness in early experimental syphilis. II. Nature of cellular infiltration and Treponema pallidum distribution in testicular lesions. J Immunol 124:461-467.
Luo, X., H. R. Kranzler, L. Zuo, S. Wang, N. J. Schork, and J. Gelernter. 2007. Multiple ADH genes modulate risk for drug dependence in both African- and European-Americans. Hum Mol Genet 16:380-390.
Lynch, M., and J. S. Conery. 2000. The evolutionary fate and consequences of duplicate genes. Science 290:1151-1155.
Lynch, M., and A. Force. 2000. The probability of duplicate gene preservation by subfunctionalization. Genetics 154:459-473.
Macaulay, V., C. Hill, A. Achilli, C. Rengo, D. Clarke, W. Meehan, J. Blackburn, O. Semino, R. Scozzari, F. Cruciani, A. Taha, N. K. Shaari, J. M. Raja, P. Ismail, Z. Zainuddin, W. Goodwin, D. Bulbeck, H. J. Bandelt, S. Oppenheimer, A. Torroni, and M. Richards. 2005. Single, rapid coastal settlement of Asia revealed by analysis of complete mitochondrial genomes. Science 308:1034-1036.
Maddison, W. P., and D. R. Maddison. 1989. Interactive analysis of phylogeny and character evolution using the computer program MacClade. Folia Primatol (Basel) 53:190-202.
Maddon, P. J., A. G. Dalgleish, J. S. McDougal, P. R. Clapham, R. A. Weiss, and R. Axel. 1986. The T4 gene encodes the AIDS virus receptor and is expressed in the immune system and the brain. Cell 47:333-348.

Manning, L. S., and M. J. Parmely. 1980. Cellular determinants of mammary cell-mediated immunity in the rat. I. The migration of radioisotopically labeled T lymphocytes. J Immunol 125:2508-2514.
Martin, D. P., C. Williamson, and D. Posada. 2005. RDP2: recombination detection and analysis from sequence alignments. Bioinformatics 21:260-262.
Mash, D. C., Q. Ouyang, J. Pablo, M. Basile, S. Izenwasser, A. Lieberman, and R. J. Perrin. 2003. Cocaine abusers have an overexpression of alpha-synuclein in dopamine neurons. J Neurosci 23:2564-2571.
Mathers, C. D., and D. Loncar. 2006. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 3:e442.
McCarthy, D. J., and P. Zimmet. 2001. Pacific Island Populations. Pp. 239-245 in J. M. Ekoe, P. Zimmet, and R. WIlliams, eds. The epidemiology of diabetes mellitus. An international perspective. Wiley, Chichester.
McCarthy, D. M., T. L. Wall, S. A. Brown, and L. G. Carr. 2000. Integrating biological and behavioral factors in alcohol use risk: the role of ALDH2 status and alcohol expectancies in a sample of Asian Americans. Exp Clin Psychopharmacol 8:168-175.
Meltzer, D. J. 1993. Pleistocene peopling of the Americas. Evol Anthropol 1:157-169. Merriwether, D. A., F. Rothhammer, and R. E. Ferrell. 1995. Distribution of the four founding
lineage haplotypes in Native Americans suggests a single wave of migration for the New World. Am J Phys Anthropol 98:411-430.
Monsalve, M. V., A. Helgason, and D. V. Devine. 1999. Languages, geography and HLA haplotypes in native American and Asian populations. Proc Biol Sci 266:2209-2216.
Moore, C. B., M. John, I. R. James, F. T. Christiansen, C. S. Witt, and S. A. Mallal. 2002. Evidence of HIV-1 adaptation to HLA-restricted immune responses at a population level. Science 296:1439-1443.
Morens, D. M., G. K. Folkers, and A. S. Fauci. 2004. The challenge of emerging and re-emerging infectious diseases. Nature 430:242-249.
Morris, A., M. Marsden, K. Halcrow, E. S. Hughes, R. P. Brettle, J. E. Bell, and P. Simmonds. 1999. Mosaic structure of the human immunodeficiency virus type 1 genome infecting lymphoid cells and the brain: evidence for frequent in vivo recombination events in the evolution of regional populations. J Virol 73:8720-8731.
Morse, S. S. 1995. Factors in the emergence of infectious diseases. Emerg Infect Dis 1:7-15. Mulligan, C. J., K. Hunley, S. Cole, and J. C. Long. 2004. Population genetics, history, and
health patterns in native americans. Annu Rev Genomics Hum Genet 5:295-315. Mulligan, C. J., S. J. Norris, and S. A. Lukehart. 2008. Molecular Studies in Treponema pallidum
Evolution: Towards Clarity? PLoS Neg Trop Dis 2:e184. Mulligan, C. J., R. W. Robin, M. V. Osier, N. Sambuughin, L. G. Goldfarb, R. A. Kittles, D.
Hesselbrock, D. Goldman, and J. C. Long. 2003. Allelic variation at alcohol metabolism genes ( ADH1B, ADH1C, ALDH2) and alcohol dependence in an American Indian population. Hum Genet 113:325-336.
Nabatov, A. A., G. Pollakis, T. Linnemann, A. Kliphius, M. I. Chalaby, and W. A. Paxton. 2004. Intrapatient alterations in the human immunodeficiency virus type 1 gp120 V1V2 and V3 regions differentially modulate coreceptor usage, virus inhibition by CC/CXC chemokines, soluble CD4, and the b12 and 2G12 monoclonal antibodies. J Virol 78:524-530.

Nakamura, K., K. Iwahashi, Y. Matsuo, R. Miyatake, Y. Ichikawa, and H. Suwaki. 1996. Characteristics of Japanese alcoholics with the atypical aldehyde dehydrogenase 2*2. I. A comparison of the genotypes of ALDH2, ADH2, ADH3, and cytochrome P-4502E1 between alcoholics and nonalcoholics. Alcohol Clin Exp Res 20:52-55.
Nduati, R., G. John, D. Mbori-Ngacha, B. Richardson, J. Overbaugh, A. Mwatha, J. Ndinya-Achola, J. Bwayo, F. E. Onyango, J. Hughes, and J. Kreiss. 2000. Effect of Breastfeeding and Formula Feeding on Transmission of HIV-1: A Randomized Clinical Trial. Pp. 1167-1174.
Neel, J. V. 1962. Diabetes mellitus: a "thrifty" genotype rendered detrimental by "progress"? Am J Hum Genet 14:353-362.
Neel, J. V. 1999. The "thrifty genotype" in 1998. Nutr Rev 57:S2-9. Neel, J. V. 1982. The thrifty genotype revisited. in J. Kobberling, and J. Tattersall, eds. The
genetics of diabetes mellitus. Academic Press, New York. Neumark, Y. D., Y. Friedlander, H. R. Thomasson, and T. K. Li. 1998. Association of the
ADH2*2 allele with reduced ethanol consumption in Jewish men in Israel: a pilot study. J Stud Alcohol 59:133-139.
Neville, M. C., J. C. Allen, P. C. Archer, C. E. Casey, J. Seacat, R. P. Keller, V. Lutes, J. Rasbach, and M. Neifert. 1991. Studies in human lactation: milk volume and nutrient composition during weaning and lactogenesis. Am J Clin Nutr 54:81-92.
Neville, M. C., and M. R. Neifert. 1983. Lactation: Physiology, Nutrition, and Breastfeeding. Plenum Press, New York.
Nickle, D. C., M. A. Jensen, D. Shriner, S. J. Brodie, L. M. Frenkel, J. E. Mittler, and J. I. Mullins. 2003. Evolutionary indicators of human immunodeficiency virus type 1 reservoirs and compartments. J Virol 77:5540-5546.
Nielsen, R., I. Hellmann, M. Hubisz, C. Bustamante, and A. G. Clark. 2007. Recent and ongoing selection in the human genome. Nat Rev Genet 8:857-868.
Noonan, J. P., J. Grimwood, J. Schmutz, M. Dickson, and R. M. Myers. 2004. Gene conversion and the evolution of protocadherin gene cluster diversity. Genome Res 14:354-366.
Novoradovsky, A. G., J. Kidd, K. Kidd, and D. Goldman. 1995. Apparent monomorphism of ALDH2 in seven American Indian populations. Alcohol 12:163-167.
O'Brien, S. J., and G. W. Nelson. 2004. Human genes that limit AIDS. Nat Genet 36:565-574. Ochman, H., J. G. Lawrence, and E. A. Groisman. 2000. Lateral gene transfer and the nature of
bacterial innovation. Nature 405:299-304. Ohagen, A., A. Devitt, K. J. Kunstman, P. R. Gorry, P. P. Rose, B. Korber, J. Taylor, R. Levy, R.
L. Murphy, S. M. Wolinsky, and D. Gabuzda. 2003. Genetic and functional analysis of full-length human immunodeficiency virus type 1 env genes derived from brain and blood of patients with AIDS. J Virol 77:12336-12345.
Ohta, T. 1992. The Nearly Neutral Theory of Molecular Evolution. Annual Review of Ecology and Systematics 23:263-286.
Omran, A. R. 1971. The epidemiologic transition. A theory of the epidemiology of population change. Milbank Mem Fund Q 49:509-538.
Ordovas, J., A. Pittas, and A. S. Greenberg. 2003. Might the diabetic environment in utero lead to type 2 diabetes? Lancet 361:1839-1840.
Osier, M., A. J. Pakstis, J. R. Kidd, J. F. Lee, S. J. Yin, H. C. Ko, H. J. Edenberg, R. B. Lu, and K. K. Kidd. 1999. Linkage disequilibrium at the ADH2 and ADH3 loci and risk of alcoholism. Am J Hum Genet 64:1147-1157.

Osier, M. V., A. J. Pakstis, D. Goldman, H. J. Edenberg, J. R. Kidd, and K. K. Kidd. 2002a. A proline-threonine substitution in codon 351 of ADH1C is common in Native Americans. Alcohol Clin Exp Res 26:1759-1763.
Osier, M. V., A. J. Pakstis, H. Soodyall, D. Comas, D. Goldman, A. Odunsi, F. Okonofua, J. Parnas, L. O. Schulz, J. Bertranpetit, B. Bonne-Tamir, R. B. Lu, J. R. Kidd, and K. K. Kidd. 2002b. A global perspective on genetic variation at the ADH genes reveals unusual patterns of linkage disequilibrium and diversity. Am J Hum Genet 71:84-99.
Padidam, M., S. Sawyer, and C. M. Fauquet. 1999. Possible emergence of new geminiviruses by frequent recombination. Virology 265:218-225.
Paradies, Y. C., M. J. Montoya, and S. M. Fullerton. 2007. Racialized genetics and the study of complex diseases: the thrifty genotype revisited. Perspect Biol Med 50:203-227.
Pastore, C., R. Nedellec, A. Ramos, S. Pontow, L. Ratner, and D. E. Mosier. 2006. Human immunodeficiency virus type 1 coreceptor switching: V1/V2 gain-of-fitness mutations compensate for V3 loss-of-fitness mutations. J Virol 80:750-758.
Perrin, L., L. Kaiser, and S. Yerly. 2003. Travel and the spread of HIV-1 genetic variants. Lancet Infect Dis 3:22-27.
Peterson, L. E., J. S. Barnholtz, G. P. Page, T. M. King, M. de Andrade, and C. I. Amos. 1999. A genome-wide search for susceptibility genes linked to alcohol dependence. Genet Epidemiol 17 Suppl 1:S295-300.
Petitjean, G., P. Becquart, E. Tuaillon, Y. Al Tabaa, D. Valea, M. F. Huguet, N. Meda, P. Van de Perre, and J. P. Vendrell. 2007. Isolation and characterization of HIV-1-infected resting CD4+ T lymphocytes in breast milk. J Clin Virol 39:1-8.
Petito, C. K. 2004. Human immunodeficiency virus type 1 compartmentalization in the central nervous system. J Neurovirol 10 Suppl 1:21-24.
Philpott, S., H. Burger, C. Tsoukas, B. Foley, K. Anastos, C. Kitchen, and B. Weiser. 2005. Human immunodeficiency virus type 1 genomic RNA sequences in the female genital tract and blood: compartmentalization and intrapatient recombination. J Virol 79:353-363.
Pilcher, C. D., D. C. Shugars, S. A. Fiscus, W. C. Miller, P. Menezes, J. Giner, B. Dean, K. Robertson, C. E. Hart, J. L. Lennox, J. J. Eron, Jr., and C. B. Hicks. 2001. HIV in body fluids during primary HIV infection: implications for pathogenesis, treatment and public health. Aids 15:837-845.
Pillai, S. K., B. Good, S. K. Pond, J. K. Wong, M. C. Strain, D. D. Richman, and D. M. Smith. 2005. Semen-specific genetic characteristics of human immunodeficiency virus type 1 env. J Virol 79:1734-1742.
Pillai, S. K., S. L. Pond, Y. Liu, B. M. Good, M. C. Strain, R. J. Ellis, S. Letendre, D. M. Smith, H. F. Gunthard, I. Grant, T. D. Marcotte, J. A. McCutchan, D. D. Richman, and J. K. Wong. 2006. Genetic attributes of cerebrospinal fluid-derived HIV-1 env. Brain 129:1872-1883.
Pillay, K., A. Coutsoudis, D. York, L. Kuhn, and H. M. Coovadia. 2000. Cell-free virus in breast milk of HIV-1-seropositive women. J Acquir Immune Defic Syndr 24:330-336.
Ping, L. H., M. S. Cohen, I. Hoffman, P. Vernazza, F. Seillier-Moiseiwitsch, H. Chakraborty, P. Kazembe, D. Zimba, M. Maida, S. A. Fiscus, J. J. Eron, R. Swanstrom, and J. A. Nelson. 2000. Effects of genital tract inflammation on human immunodeficiency virus type 1 V3 populations in blood and semen. J Virol 74:8946-8952.
Pitt, J. 1979. The milk mononuclear phagocyte. Pediatrics 64:745-749.

Plagnol, V., and J. D. Wall. 2006. Possible ancestral structure in human populations. PLoS Genet 2:e105.
Pollard, A. J., and S. R. Dobson. 2000. Emerging infectious diseases in the 21st century. Curr Opin Infect Dis 13:265-275.
Polymeropoulos, M. H., C. Lavedan, E. Leroy, S. E. Ide, A. Dehejia, A. Dutra, B. Pike, H. Root, J. Rubenstein, R. Boyer, E. S. Stenroos, S. Chandrasekharappa, A. Athanassiadou, T. Papapetropoulos, W. G. Johnson, A. M. Lazzarini, R. C. Duvoisin, G. Di Iorio, L. I. Golbe, and R. L. Nussbaum. 1997. Mutation in the alpha-synuclein gene identified in families with Parkinson's disease. Science 276:2045-2047.
Pond, S. L., S. D. Frost, and S. V. Muse. 2005. HyPhy: hypothesis testing using phylogenies. Bioinformatics 21:676-679.
Posada, D., and K. A. Crandall. 2001. Evaluation of methods for detecting recombination from DNA sequences: computer simulations. Proc Natl Acad Sci U S A 98:13757-13762.
Posada, D., and K. A. Crandall. 1998. MODELTEST: testing the model of DNA substitution. Bioinformatics 14:817-818.
Posada, D., K. A. Crandall, and E. C. Holmes. 2002. Recombination in evolutionary genomics. Annu Rev Genet 36:75-97.
Poss, M., A. G. Rodrigo, J. J. Gosink, G. H. Learn, D. de Vange Panteleeff, H. L. Martin, Jr., J. Bwayo, J. K. Kreiss, and J. Overbaugh. 1998. Evolution of envelope sequences from the genital tract and peripheral blood of women infected with clade A human immunodeficiency virus type 1. J Virol 72:8240-8251.
Powell, M. L., and D. C. Cook. 2005. Treponematosis: Inquires into the Nature of Protean Disease in M. L. Powell, and D. C. Cook, eds. The Myth of Syphilis: the Natural History of Treponematosis in North America. University of Florida Press, Gainesville
Prugnolle, F., A. Manica, M. Charpentier, J. F. Guegan, V. Guernier, and F. Balloux. 2005. Pathogen-driven selection and worldwide HLA class I diversity. Curr Biol 15:1022-1027.
Quintana-Murci, L., A. Alcais, L. Abel, and J. L. Casanova. 2007. Immunology in natura: clinical, epidemiological and evolutionary genetics of infectious diseases. Nat Immunol 8:1165-1171.
Quintana-Murci, L., O. Semino, H. J. Bandelt, G. Passarino, K. McElreavey, and A. S. Santachiara-Benerecetti. 1999. Genetic evidence of an early exit of Homo sapiens sapiens from Africa through eastern Africa. Nat Genet 23:437-441.
Ramachandran, S., O. Deshpande, C. C. Roseman, N. A. Rosenberg, M. W. Feldman, and L. L. Cavalli-Sforza. 2005. Support from the relationship of genetic and geographic distance in human populations for a serial founder effect originating in Africa. Proc Natl Acad Sci U S A 102:15942-15947.
Reich, T., H. J. Edenberg, A. Goate, J. T. Williams, J. P. Rice, P. Van Eerdewegh, T. Foroud, V. Hesselbrock, M. A. Schuckit, K. Bucholz, B. Porjesz, T. K. Li, P. M. Conneally, J. I. Nurnberger, Jr., J. A. Tischfield, R. R. Crowe, C. R. Cloninger, W. Wu, S. Shears, K. Carr, C. Crose, C. Willig, and H. Begleiter. 1998. Genome-wide search for genes affecting the risk for alcohol dependence. Am J Med Genet 81:207-215.
Relethford, J. H. 2001. Absence of regional affinities of Neandertal DNA with living humans does not reject multiregional evolution. Am J Phys Anthropol 115:95-98.
Richardson, B. A., G. C. John-Stewart, J. P. Hughes, R. Nduati, D. Mbori-Ngacha, J. Overbaugh, and J. K. Kreiss. 2003. Breast-milk infectivity in human immunodeficiency virus type 1-infected mothers. J Infect Dis 187:736-740.

Ritola, K., C. D. Pilcher, S. A. Fiscus, N. G. Hoffman, J. A. Nelson, K. M. Kitrinos, C. B. Hicks, J. J. Eron, Jr., and R. Swanstrom. 2004. Multiple V1/V2 env variants are frequently present during primary infection with human immunodeficiency virus type 1. J Virol 78:11208-11218.
Ritola, K., K. Robertson, S. A. Fiscus, C. Hall, and R. Swanstrom. 2005. Increased human immunodeficiency virus type 1 (HIV-1) env compartmentalization in the presence of HIV-1-associated dementia. J Virol 79:10830-10834.
Rivas, R. A., A. A. el-Mohandes, and I. M. Katona. 1994. Mononuclear phagocytic cells in human milk: HLA-DR and Fc gamma R ligand expression. Biol Neonate 66:195-204.
Robin, R. W., J. C. Long, J. K. Rasmussen, B. Albaugh, and D. Goldman. 1998. Relationship of binge drinking to alcohol dependence, other psychiatric disorders, and behavioral problems in an American Indian tribe. Alcohol Clin Exp Res 22:518-523.
Rollins, N. C., S. M. Filteau, A. Coutsoudis, and A. M. Tomkins. 2001. Feeding mode, intestinal permeability, and neopterin excretion: a longitudinal study in infants of HIV-infected South African women. J Acquir Immune Defic Syndr 28:132-139.
Rothschild, B. 2003. Infectious processes around the dawn of civilization in C. L. Greenblatt, and M. Spigelman, eds. Emerging Pathogens: The Archaeology, Ecology, and Evolution of Infectious Disease. Oxford University Press, London.
Rousseau, C. M., R. W. Nduati, B. A. Richardson, G. C. John-Stewart, D. A. Mbori-Ngacha, J. K. Kreiss, and J. Overbaugh. 2004. Association of levels of HIV-1-infected breast milk cells and risk of mother-to-child transmission. J Infect Dis 190:1880-1888.
Rousseau, C. M., R. W. Nduati, B. A. Richardson, M. S. Steele, G. C. John-Stewart, D. A. Mbori-Ngacha, J. K. Kreiss, and J. Overbaugh. 2003. Longitudinal analysis of human immunodeficiency virus type 1 RNA in breast milk and of its relationship to infant infection and maternal disease. J Infect Dis 187:741-747.
Royce, R. A., A. Sena, W. Cates, Jr., and M. S. Cohen. 1997. Sexual transmission of HIV. N Engl J Med 336:1072-1078.
Rozas, J., J. C. Sanchez-DelBarrio, X. Messeguer, and R. Rozas. 2003. DnaSP, DNA polymorphism analyses by the coalescent and other methods. Bioinformatics 19:2496-2497.
Saag, M. S., B. H. Hahn, J. Gibbons, Y. Li, E. S. Parks, W. P. Parks, and G. M. Shaw. 1988. Extensive variation of human immunodeficiency virus type-1 in vivo. Nature 334:440-444.
Saccone, N. L., J. M. Kwon, J. Corbett, A. Goate, N. Rochberg, H. J. Edenberg, T. Foroud, T. K. Li, H. Begleiter, T. Reich, and J. P. Rice. 2000. A genome screen of maximum number of drinks as an alcoholism phenotype. Am J Med Genet 96:632-637.
Sagar, M., X. Wu, S. Lee, and J. Overbaugh. 2006. Human immunodeficiency virus type 1 V1-V2 envelope loop sequences expand and add glycosylation sites over the course of infection, and these modifications affect antibody neutralization sensitivity. J Virol 80:9586-9598.
Salemi, M., B. R. Burkhardt, R. R. Gray, G. Ghaffari, J. W. Sleasman, and M. M. Goodenow. 2007. Phylodynamics of HIV-1 in Lymphoid and Non-Lymphoid Tissues Reveals a Central Role for the Thymus in Emergence of CXCR4-Using Quasispecies. PLoS ONE 2:e950.
Salemi, M., S. L. Lamers, S. Yu, T. de Oliveira, W. M. Fitch, and M. S. McGrath. 2005. Phylodynamic analysis of human immunodeficiency virus type 1 in distinct brain

compartments provides a model for the neuropathogenesis of AIDS. J Virol 79:11343-11352.
Samson, M., F. Libert, B. J. Doranz, J. Rucker, C. Liesnard, C. M. Farber, S. Saragosti, C. Lapoumeroulie, J. Cognaux, C. Forceille, G. Muyldermans, C. Verhofstede, G. Burtonboy, M. Georges, T. Imai, S. Rana, Y. Yi, R. J. Smyth, R. G. Collman, R. W. Doms, G. Vassart, and M. Parmentier. 1996. Resistance to HIV-1 infection in caucasian individuals bearing mutant alleles of the CCR-5 chemokine receptor gene. Nature 382:722-725.
Santoyo, G., and D. Romero. 2005. Gene conversion and concerted evolution in bacterial genomes. FEMS Microbiol Rev 29:169-183.
Schaid, D. J. 2004. Evaluating associations of haplotypes with traits. Genet Epidemiol 27:348-364.
Schimenti, J. C. 1994. Gene conversion and the evolution of gene families in mammals. Soc Gen Physiol Ser 49:85-91.
Schneider, S., D. Roessli, and L. Excoffier. 2000. Arlequin: A software for population genetics data analysis. Genetics and Biometry Laboratory, Department of Anthropology, University of Geneva.
Schork, N. J. 1997. Genetics of complex disease: approaches, problems, and solutions. Am J Respir Crit Care Med 156:S103-109.
Schroeder, S. A., D. M. Gaughan, and M. Swift. 1995. Protection against bronchial asthma by CFTR delta F508 mutation: a heterozygote advantage in cystic fibrosis. Nat Med 1:703-705.
Schwartz, K., L. Carrier, P. Guicheney, and M. Komajda. 1995. Molecular basis of familial cardiomyopathies. Circulation 91:532-540.
Seguin, B., B. Hardy, P. A. Singer, and A. S. Daar. 2008. Bidil: recontextualizing the race debate. Pharmacogenomics J.
Self, D. W., A. W. McClenahan, D. Beitner-Johnson, R. Z. Terwilliger, and E. J. Nestler. 1995. Biochemical adaptations in the mesolimbic dopamine system in response to heroin self-administration. Synapse 21:312-318.
Semba, R. D., N. Kumwenda, D. R. Hoover, T. E. Taha, T. C. Quinn, L. Mtimavalye, R. J. Biggar, R. Broadhead, P. G. Miotti, L. J. Sokoll, L. van der Hoeven, and J. D. Chiphangwi. 1999a. Human immunodeficiency virus load in breast milk, mastitis, and mother-to-child transmission of human immunodeficiency virus type 1. J Infect Dis 180:93-98.
Semba, R. D., N. Kumwenda, T. E. Taha, D. R. Hoover, Y. Lan, W. Eisinger, L. Mtimavalye, R. Broadhead, P. G. Miotti, L. Van Der Hoeven, and J. D. Chiphangwi. 1999b. Mastitis and immunological factors in breast milk of lactating women in Malawi. Clin Diagn Lab Immunol 6:671-674.
Seshadri, R., G. S. Myers, H. Tettelin, J. A. Eisen, J. F. Heidelberg, R. J. Dodson, T. M. Davidsen, R. T. DeBoy, D. E. Fouts, D. H. Haft, J. Selengut, Q. Ren, L. M. Brinkac, R. Madupu, J. Kolonay, S. A. Durkin, S. C. Daugherty, J. Shetty, A. Shvartsbeyn, E. Gebregeorgis, K. Geer, G. Tsegaye, J. Malek, B. Ayodeji, S. Shatsman, M. P. McLeod, D. Smajs, J. K. Howell, S. Pal, A. Amin, P. Vashisth, T. Z. McNeill, Q. Xiang, E. Sodergren, E. Baca, G. M. Weinstock, S. J. Norris, C. M. Fraser, and I. T. Paulsen. 2004. Comparison of the genome of the oral pathogen Treponema denticola with other spirochete genomes. Proc Natl Acad Sci U S A 101:5646-5651.

Sham, P. C., and D. Curtis. 1995. Monte Carlo tests for associations between disease and alleles at highly polymorphic loci. Ann Hum Genet 59:97-105.
Shapiro, B., A. Rambaut, and A. J. Drummond. 2006. Choosing appropriate substitution models for the phylogenetic analysis of protein-coding sequences. Mol Biol Evol 23:7-9.
Shapshak, P., D. M. Segal, K. A. Crandall, R. K. Fujimura, B. T. Zhang, K. Q. Xin, K. Okuda, C. K. Petito, C. Eisdorfer, and K. Goodkin. 1999. Independent evolution of HIV type 1 in different brain regions. AIDS Res Hum Retroviruses 15:811-820.
Sharp, P. M., E. Bailes, R. R. Chaudhuri, C. M. Rodenburg, M. O. Santiago, and B. H. Hahn. 2001. The origins of acquired immune deficiency syndrome viruses: where and when? Philos Trans R Soc Lond B Biol Sci 356:867-876.
Shen, Y. C., J. H. Fan, H. J. Edenberg, T. K. Li, Y. H. Cui, Y. F. Wang, C. H. Tian, C. F. Zhou, R. L. Zhou, J. Wang, Z. L. Zhao, and G. Y. Xia. 1997. Polymorphism of ADH and ALDH genes among four ethnic groups in China and effects upon the risk for alcoholism. Alcohol Clin Exp Res 21:1272-1277.
Sherry, S. T., A. R. Rogers, H. Harpending, H. Soodyall, T. Jenkins, and M. Stoneking. 1994. Mismatch distributions of mtDNA reveal recent human population expansions. Hum Biol 66:761-775.
Shibasaki, Y., D. A. Baillie, D. St Clair, and A. J. Brookes. 1995. High-resolution mapping of SNCA encoding alpha-synuclein, the non-A beta component of Alzheimer's disease amyloid precursor, to human chromosome 4q21.3-->q22 by fluorescence in situ hybridization. Cytogenet Cell Genet 71:54-55.
Shuster, D. E., M. E. Kehrli, Jr., and C. R. Baumrucker. 1995. Relationship of inflammatory cytokines, growth hormone, and insulin-like growth factor-I to reduced performance during infectious disease. Proc Soc Exp Biol Med 210:140-149.
Si-Mohamed, A., M. D. Kazatchkine, I. Heard, C. Goujon, T. Prazuck, G. Aymard, G. Cessot, Y. H. Kuo, M. C. Bernard, B. Diquet, J. E. Malkin, L. Gutmann, and L. Belec. 2000. Selection of drug-resistant variants in the female genital tract of human immunodeficiency virus type 1-infected women receiving antiretroviral therapy. J Infect Dis 182:112-122.
Simon, F., P. Mauclere, P. Roques, I. Loussert-Ajaka, M. C. Muller-Trutwin, S. Saragosti, M. C. Georges-Courbot, F. Barre-Sinoussi, and F. Brun-Vezinet. 1998. Identification of a new human immunodeficiency virus type 1 distinct from group M and group O. Nat Med 4:1032-1037.
Sinkala, M., L. Kuhn, C. Kankasa, P. Kasonde, C. Vwalika, M. Mwiya, N. Scott, K. Semrau, G. Aldrovandi, D. M. Thea, and Z. E. B. S. Group. 2007. No Benefit of Early Cessation of Breastfeeding at 4 Months on HIV-free Survival of Infants Born to HIV-infected Mothers in Zambia: The Zambia Exclusive Breastfeeding Study Conference on Retroviruses and Opportunistic Infections, San Francisco.
Slatkin, M., and W. P. Maddison. 1989. A cladistic measure of gene flow inferred from the phylogenies of alleles. Genetics 123:603-613.
Slightom, J. L., A. E. Blechl, and O. Smithies. 1980. Human fetal G gamma- and A gamma-globin genes: complete nucleotide sequences suggest that DNA can be exchanged between these duplicated genes. Cell 21:627-638.
Smit, T. K., B. J. Brew, W. Tourtellotte, S. Morgello, B. B. Gelman, and N. K. Saksena. 2004. Independent evolution of human immunodeficiency virus (HIV) drug resistance

mutations in diverse areas of the brain in HIV-infected patients, with and without dementia, on antiretroviral treatment. J Virol 78:10133-10148.
Smit, T. K., B. Wang, T. Ng, R. Osborne, B. Brew, and N. K. Saksena. 2001. Varied tropism of HIV-1 isolates derived from different regions of adult brain cortex discriminate between patients with and without AIDS dementia complex (ADC): evidence for neurotropic HIV variants. Virology 279:509-526.
Smith, J. L., N. J. David, S. Indgin, C. W. Israel, B. M. Levine, J. Justice, Jr., J. A. McCrary, 3rd, R. Medina, P. Paez, E. Santana, M. Sarkar, N. J. Schatz, M. L. Spitzer, W. O. Spitzer, and E. K. Walter. 1971. Neuro-ophthalmological study of late yaws and pinta. II. The Caracas project. Br J Vener Dis 47:226-251.
Smith, J. M. 1992. Analyzing the mosaic structure of genes. J Mol Evol 34:126-129. Smith, J. M., C. G. Dowson, and B. G. Spratt. 1991. Localized sex in bacteria. Nature 349:29-31. Smith, M. M., and L. Kuhn. 2000. Exclusive breast-feeding: does it have the potential to reduce
breast-feeding transmission of HIV-1? Nutr Rev 58:333-340. Spence, J., T. Liang, T. Foroud, D. Lo, and L. Carr. 2005. Expression profiling and QTL
analysis: a powerful complementary strategy in drug abuse research. Addict Biol 10:47-51.
Spillantini, M. G., A. Divane, and M. Goedert. 1995. Assignment of human alpha-synuclein (SNCA) and beta-synuclein (SNCB) genes to chromosomes 4q21 and 5q35. Genomics 27:379-381.
Spitzer, R. L., J. Endicott, and E. Robins. 1989. Research Diagnostic Criteria (RDC) for a selected group of psychiatric disorders. . Department of Research Assessment and Training, New York Psychiatric Institute, New York.
Staprans, S., N. Marlowe, D. Glidden, T. Novakovic-Agopian, R. M. Grant, M. Heyes, F. Aweeka, S. Deeks, and R. W. Price. 1999. Time course of cerebrospinal fluid responses to antiretroviral therapy: evidence for variable compartmentalization of infection. Aids 13:1051-1061.
Steffy, K., and F. Wong-Staal. 1991. Genetic regulation of human immunodeficiency virus. Microbiol Rev 55:193-205.
Stephens, D. S., E. R. Moxon, J. Adams, S. Altizer, J. Antonovics, S. Aral, R. Berkelman, E. Bond, J. Bull, G. Cauthen, M. M. Farley, A. Glasgow, J. W. Glasser, H. P. Katner, S. Kelley, J. Mittler, A. J. Nahmias, S. Nichol, V. Perrot, R. W. Pinner, S. Schrag, P. Small, and P. H. Thrall. 1998a. Emerging and reemerging infectious diseases: a multidisciplinary perspective. Am J Med Sci 315:64-75.
Stephens, J. C., D. E. Reich, D. B. Goldstein, H. D. Shin, M. W. Smith, M. Carrington, C. Winkler, G. A. Huttley, R. Allikmets, L. Schriml, B. Gerrard, M. Malasky, M. D. Ramos, S. Morlot, M. Tzetis, C. Oddoux, F. S. di Giovine, G. Nasioulas, D. Chandler, M. Aseev, M. Hanson, L. Kalaydjieva, D. Glavac, P. Gasparini, E. Kanavakis, M. Claustres, M. Kambouris, H. Ostrer, G. Duff, V. Baranov, H. Sibul, A. Metspalu, D. Goldman, N. Martin, D. Duffy, J. Schmidtke, X. Estivill, S. J. O'Brien, and M. Dean. 1998b. Dating the origin of the CCR5-Delta32 AIDS-resistance allele by the coalescence of haplotypes. Am J Hum Genet 62:1507-1515.
Stephens, M., and P. Donnelly. 2003. A comparison of bayesian methods for haplotype reconstruction from population genotype data. Am J Hum Genet 73:1162-1169.
Strain, M. C., S. Letendre, S. K. Pillai, T. Russell, C. C. Ignacio, H. F. Gunthard, B. Good, D. M. Smith, S. M. Wolinsky, M. Furtado, J. Marquie-Beck, J. Durelle, I. Grant, D. D.

Richman, T. Marcotte, J. A. McCutchan, R. J. Ellis, and J. K. Wong. 2005. Genetic composition of human immunodeficiency virus type 1 in cerebrospinal fluid and blood without treatment and during failing antiretroviral therapy. J Virol 79:1772-1788.
Sullivan, S. T., U. Mandava, T. Evans-Strickfaden, J. L. Lennox, T. V. Ellerbrock, and C. E. Hart. 2005. Diversity, divergence, and evolution of cell-free human immunodeficiency virus type 1 in vaginal secretions and blood of chronically infected women: associations with immune status. J Virol 79:9799-9809.
Sun, E. S., B. J. Molini, L. K. Barrett, A. Centurion-Lara, S. A. Lukehart, and W. C. Van Voorhis. 2004. Subfamily I Treponema pallidum repeat protein family: sequence variation and immunity. Microbes Infect 6:725-737.
Surovell, T. A. 2003. Simulating coastal migration in New World colonization. Curr Anthropol 44:580-591.
Swofford, D. L. 2002. PAUP*: Phylogenetic Analysis Using Parsimony (and Other Methods). Sinauer.
Taguchi, Y., T. Koide, T. Shiroishi, and T. Yagi. 2005. Molecular evolution of cadherin-related neuronal receptor/protocadherin(alpha) (CNR/Pcdh(alpha)) gene cluster in Mus musculus subspecies. Mol Biol Evol 22:1433-1443.
Tamura, K., J. Dudley, M. Nei, and S. Kumar. 2007. MEGA4: Molecular Evolutionary Genetics Analysis (MEGA) software version 4.0. Mol Biol Evol 24:1596-1599.
Tanaka, F., Y. Shiratori, O. Yokosuka, F. Imazeki, Y. Tsukada, and M. Omata. 1996. High incidence of ADH2*1/ALDH2*1 genes among Japanese alcohol dependents and patients with alcoholic liver disease. Hepatology 23:234-239.
Thea, D. M., G. Aldrovandi, C. Kankasa, P. Kasonde, W. D. Decker, K. Semrau, M. Sinkala, and L. Kuhn. 2006. Post-weaning breast milk HIV-1 viral load, blood prolactin levels and breast milk volume. Aids 20:1539-1547.
Thea, D. M., C. Vwalika, P. Kasonde, C. Kankasa, M. Sinkala, K. Semrau, E. Shutes, C. Ayash, W. Y. Tsai, G. Aldrovandi, and L. Kuhn. 2004. Issues in the design of a clinical trial with a behavioral intervention--the Zambia exclusive breast-feeding study. Control Clin Trials 25:353-365.
Thomasson, H. R., D. W. Crabb, H. J. Edenberg, and T. K. Li. 1993. Alcohol and aldehyde dehydrogenase polymorphisms and alcoholism. Behav Genet 23:131-136.
Thomasson, H. R., D. W. Crabb, H. J. Edenberg, T. K. Li, H. G. Hwu, C. C. Chen, E. K. Yeh, and S. J. Yin. 1994. Low frequency of the ADH2*2 allele among Atayal natives of Taiwan with alcohol use disorders. Alcohol Clin Exp Res 18:640-643.
Thomasson, H. R., H. J. Edenberg, D. W. Crabb, X. L. Mai, R. E. Jerome, T. K. Li, S. P. Wang, Y. T. Lin, R. B. Lu, and S. J. Yin. 1991. Alcohol and aldehyde dehydrogenase genotypes and alcoholism in Chinese men. Am J Hum Genet 48:677-681.
Thompson, J. D., T. J. Gibson, F. Plewniak, F. Jeanmougin, and D. G. Higgins. 1997. The CLUSTAL_X windows interface: flexible strategies for multiple sequence alignment aided by quality analysis tools. Nucleic Acids Res 25:4876-4882.
Thompson, K. A., M. J. Churchill, P. R. Gorry, J. Sterjovski, R. B. Oelrichs, S. L. Wesselingh, and C. A. McLean. 2004. Astrocyte specific viral strains in HIV dementia. Ann Neurol 56:873-877.
Toniolo, A., C. Serra, P. G. Conaldi, F. Basolo, V. Falcone, and A. Dolei. 1995. Productive HIV-1 infection of normal human mammary epithelial cells. Aids 9:859-866.

Touchman, J. W., A. Dehejia, O. Chiba-Falek, D. E. Cabin, J. R. Schwartz, B. M. Orrison, M. H. Polymeropoulos, and R. L. Nussbaum. 2001. Human and mouse alpha-synuclein genes: comparative genomic sequence analysis and identification of a novel gene regulatory element. Genome Res 11:78-86.
Ueda, K., H. Fukushima, E. Masliah, Y. Xia, A. Iwai, M. Yoshimoto, D. A. Otero, J. Kondo, Y. Ihara, and T. Saitoh. 1993. Molecular cloning of cDNA encoding an unrecognized component of amyloid in Alzheimer disease. Proc Natl Acad Sci U S A 90:11282-11286.
Uhl, G. R., Q. R. Liu, D. Walther, J. Hess, and D. Naiman. 2001. Polysubstance abuse-vulnerability genes: genome scans for association, using 1,004 subjects and 1,494 single-nucleotide polymorphisms. Am J Hum Genet 69:1290-1300.
Ullrich, R., H. L. Schieferdecker, K. Ziegler, E. O. Riecken, and M. Zeitz. 1990. gamma delta T cells in the human intestine express surface markers of activation and are preferentially located in the epithelium. Cell Immunol 128:619-627.
Van de Peere, P., P. Lepage, A. Simonon, C. Desgranges, D. G. Hitimana, A. Bazubagira, C. Van Goethem, A. Kleinschmidt, F. Bex, K. Broliden, and et al. 1992. Biological markers associated with prolonged survival in African children maternally infected by the human immunodeficiency virus type 1. AIDS Res Hum Retroviruses 8:435-442.
Van de Perre, P., A. Simonon, D. G. Hitimana, F. Dabis, P. Msellati, B. Mukamabano, J. B. Butera, C. Van Goethem, E. Karita, and P. Lepage. 1993. Infective and anti-infective properties of breastmilk from HIV-1-infected women. Lancet 341:914-918.
Venturi, G., M. Catucci, L. Romano, P. Corsi, F. Leoncini, P. E. Valensin, and M. Zazzi. 2000. Antiretroviral resistance mutations in human immunodeficiency virus type 1 reverse transcriptase and protease from paired cerebrospinal fluid and plasma samples. J Infect Dis 181:740-745.
Verrelli, B. C., J. H. McDonald, G. Argyropoulos, G. Destro-Bisol, A. Froment, A. Drousiotou, G. Lefranc, A. N. Helal, J. Loiselet, and S. A. Tishkoff. 2002. Evidence for balancing selection from nucleotide sequence analyses of human G6PD. Am J Hum Genet 71:1112-1128.
Wagner, A. 1998. The fate of duplicated genes: loss or new function? Bioessays 20:785-788. Walker, S. J., and K. A. Grant. 2006. Peripheral blood alpha-synuclein mRNA levels are
elevated in cynomolgus monkeys that chronically self-administer alcohol. Alcohol 38:1-4.
Wall, T. L., C. Garcia-Andrade, H. R. Thomasson, L. G. Carr, and C. L. Ehlers. 1997. Alcohol dehydrogenase polymorphisms in Native Americans: identification of the ADH2*3 allele. Alcohol Alcohol 32:129-132.
Walsh, J. B. 1995. How often do duplicated genes evolve new functions? Genetics 139:421-428. Wang, S., C. M. Lewis, M. Jakobsson, S. Ramachandran, N. Ray, G. Bedoya, W. Rojas, M. V.
Parra, J. A. Molina, C. Gallo, G. Mazzotti, G. Poletti, K. Hill, A. M. Hurtado, D. Labuda, W. Klitz, R. Barrantes, M. C. Bortolini, F. M. Salzano, M. L. Petzl-Erler, L. T. Tsuneto, E. Llop, F. Rothhammer, L. Excoffier, M. W. Feldman, N. A. Rosenberg, and A. Ruiz-Linares. 2007. Genetic Variation and Population Structure in Native Americans. PLoS Genet 3:e185.
Wang, T. H., Y. K. Donaldson, R. P. Brettle, J. E. Bell, and P. Simmonds. 2001. Identification of shared populations of human immunodeficiency virus type 1 infecting microglia and tissue macrophages outside the central nervous system. J Virol 75:11686-11699.

Wei, X., J. M. Decker, S. Wang, H. Hui, J. C. Kappes, X. Wu, J. F. Salazar-Gonzalez, M. G. Salazar, J. M. Kilby, M. S. Saag, N. L. Komarova, M. A. Nowak, B. H. Hahn, P. D. Kwong, and G. M. Shaw. 2003. Antibody neutralization and escape by HIV-1. Nature 422:307-312.
West, K. M. 1974. Diabetes in American Indians and other native populations of the New World. Diabetes 23:841-855.
WHO. 2003. HIV and Infant Feeding. World Health Organization. WHO. 2007. Mother-to-child transmission of HIV. WHO. 2008a. Impact of chronic disease by World Bank income groups: High Income Countries.
World Health Organization. WHO. 2008b. Impact of chronic disease by World Bank income groups: Low Income Countries.
World Health Organization. WHO. 2006. Comprehensive HIV Prevention: 2006 Report on the Global AIDS Epidemic in W.
H. Organization, ed. Williams, J. T., H. Begleiter, B. Porjesz, H. J. Edenberg, T. Foroud, T. Reich, A. Goate, P. Van
Eerdewegh, L. Almasy, and J. Blangero. 1999. Joint multipoint linkage analysis of multivariate qualitative and quantitative traits. II. Alcoholism and event-related potentials. Am J Hum Genet 65:1148-1160.
Williams, R. C., W. C. Knowler, D. J. Pettitt, J. C. Long, D. A. Rokala, H. F. Polesky, R. A. Hackenberg, A. G. Steinberg, and P. H. Bennett. 1992. The magnitude and origin of European-American admixture in the Gila River Indian Community of Arizona: a union of genetics and demography. Am J Hum Genet 51:101-110.
Willumsen, J. F., S. M. Filteau, A. Coutsoudis, K. E. Uebel, M. L. Newell, and A. M. Tomkins. 2000. Subclinical mastitis as a risk factor for mother-infant HIV transmission. Adv Exp Med Biol 478:211-223.
Wirt, D. P., L. T. Adkins, K. H. Palkowetz, F. C. Schmalstieg, and A. S. Goldman. 1992. Activated and memory T lymphocytes in human milk. Cytometry 13:282-290.
Wise, P. H. 1976. Diabetes and associated variables in the South Australian Aboriginal. Aust NZ J Med 6:191-196.
Wong, J. K., C. C. Ignacio, F. Torriani, D. Havlir, N. J. Fitch, and D. D. Richman. 1997. In vivo compartmentalization of human immunodeficiency virus: evidence from the examination of pol sequences from autopsy tissues. J Virol 71:2059-2071.
Wooding, S., A. C. Stone, D. M. Dunn, S. Mummidi, L. B. Jorde, R. K. Weiss, S. Ahuja, and M. J. Bamshad. 2005. Contrasting effects of natural selection on human and chimpanzee CC chemokine receptor 5. Am J Hum Genet 76:291-301.
Worobey, M. 2001. A novel approach to detecting and measuring recombination: new insights into evolution in viruses, bacteria, and mitochondria. Mol Biol Evol 18:1425-1434.
Xanthou, M. 1997. Human milk cells. Acta Paediatr 86:1288-1290. Yancy, C. W., J. K. Ghali, V. M. Braman, M. L. Sabolinski, M. Worcel, W. T. Archambault, and
J. A. Franciosa. 2007. Evidence for the continued safety and tolerability of fixed-dose isosorbide dinitrate/hydralazine in patients with chronic heart failure (the extension to African-American Heart Failure Trial). Am J Cardiol 100:684-689.
Yang, Z. 1997. PAML: a program package for phylogenetic analysis by maximum likelihood. Comput Appl Biosci 13:555-556.
Young, T. K., J. Reading, B. Elias, and J. D. O'Neil. 2000. Type 2 diabetes mellitus in Canada's first nations: status of an epidemic in progress. Cmaj 163:561-566.

Zhang, J. R., J. M. Hardham, A. G. Barbour, and S. J. Norris. 1997. Antigenic variation in Lyme disease borreliae by promiscuous recombination of VMP-like sequence cassettes. Cell 89:275-285.
Zhang, J. R., and S. J. Norris. 1998. Genetic variation of the Borrelia burgdorferi gene vlsE involves cassette-specific, segmental gene conversion. Infect Immun 66:3698-3704.
Zhang, Q. Y., D. DeRyckere, P. Lauer, and M. Koomey. 1992. Gene conversion in Neisseria gonorrhoeae: evidence for its role in pilus antigenic variation. Proc Natl Acad Sci U S A 89:5366-5370.
Zhu, T., N. Wang, A. Carr, D. S. Nam, R. Moor-Jankowski, D. A. Cooper, and D. D. Ho. 1996. Genetic characterization of human immunodeficiency virus type 1 in blood and genital secretions: evidence for viral compartmentalization and selection during sexual transmission. J Virol 70:3098-3107.
Zinn-Justin, A., and L. Abel. 1999. Genome search for alcohol dependence using the weighted pairwise correlation linkage method: interesting findings on chromosome 4. Genet Epidemiol 17 Suppl 1:S421-426.

BIOGRAPHICAL SKETCH
I graduated from Great Valley High School in Malvern, Pennsylvania in 1997. I attended
Reed College in Portland, Oregon, from August to December 1997. I attended Millersville
University in Millersville, Pennsylvania, from January 1999 to December 2001. I graduated in
December 2001 with a Bachelor of Arts degree in anthropology. I began graduate school at the
University of Florida in August 2002 in anthropology. I received my Master of the Arts degree in
May of 2005 and Ph.D. in May 2008.