Embed Size (px)
Citation preview

Dédicaces
UNIVERSITE MOHAMMED V- SOUISSI
FACULTE DE MEDECINE ET DE PHARMACIE -RABAT-
ANNEE: 200 THESE N°:
LLaa ddrrééppaannooccyyttoossee ddaannss uunn sseerrvviiccee ddee mmééddeecciinnee iinntteerrnnee
EEttuuddee ddee 3333 ccaass cchheezz ll’’aadduullttee
THESE
Présentée et soutenue publiquement le :………………………..
PAR
Mlle. GNAGO NINA Prisca
Née le 12 Février 1983 à Abidjan
Pour l'Obtention du Doctorat en
Médecine
MOTS CLES Drépanocytose – Complications - Ostéonécrose – Lithiase - Ostéite .
JURY
Mr. M. AOUNI PRESIDENT &
Professeur de Médecine Interne RAPPORTEUR
Mr. M. ADNAOUI
Professeur de Médecine Interne
Mr. J. CHAARI
Professeur de Médecine Interne
Mr. H. HARMOUCHE
Professeur de Médecine Interne
JUGES

Dédicaces
DEDICACES

Dédicaces
GLOIRE A
CELUI PAR QUI et POUR QUI
TOUTES
CHOSES ONT ETE FAITES

Dédicaces
A mon très cher père
A ma très chère mère
Nulle dédicace n’est susceptible de vous exprimer mon profond amour et mon immense
gratitude.
Que Dieu vous garde et vous prodigue santé et prospérité.
A mon tendre et bien aimé Jean-Christophe
Fidèle compagnon de route. Que Dieu nous garde et nous donne d’aller jusqu’au bout.
JE t’aime
A Massaba, Yannick et Manou
Que vous trouviez ici le témoignage d’une fraternité sans égale.
A toute la famille Kouadio,
Pour son amour et son hospitalité, Spécialement à Maman pour ses encouragements ;
Soyez bénis
A la famille Anyawu,
Thanks to Mammy Dolapo

Dédicaces
A la famille Dogbré,,
A la famille Vors,
A la famille Yoro,
Vos prières, vos encouragements, votre présence ont été pour moi une source de motivation.
Soyez honorés en ce jour par ce travail
A mes amis :
Guy blaise et Malory, Arlette et Doudou, Rodrigue et Cécile
Ange , Maguy, Rose, Josiane, Aimé, Mohammed, Bertin,
A Mes Compagnons :
Fidèle,Marèse,Ephrem,Ludjer,Rodrigue,Crepin,Régine,Tedy,Sandra,Kounta,Dao,
A toute la grande famille de l’AC pour votre soutient
A tous mes amis
A tous ceux qui me sont chers
Je dédie ce travail
Nina.

Remerciements
Remerciements

Remerciements
A notre Maître Président et Rapporteur de thèse : Monsieur le Professeur M. AOUNI
Professeur de médecine interne
Nous vous remercions pour la confiance que vous nous avez accordée en
acceptant de nous confier ce travail et de le diriger.
Votre dynamisme et votre rigueur sont un exemple.
Nous sommes fiers de compter parmi vos élèves.
Veuillez trouver ici l’expression de notre sincère considération.

Remerciements
A notre Maître et Juge de thèse : Monsieur le Professeur M. ADNAOUI
Professeur de médecine interne
Nous vous remercions de nous faire l’honneur de juger ce travail.
Veuillez trouver ici l’expression de notre respect et notre profonde
admiration pour vos qualités scientifiques et humaines.

Remerciements
A notre Maître et Juge de thèse : Monsieur le Professeur J. CHAARI
Professeur de médecine interne
Nous avons été touchés par la bienveillance et la cordialité de votre accueil.
Nous sommes très sensibles à l’honneur que vous nous faites en acceptant
de juger notre travail.
C’est pour nous l’occasion de vous témoigner estime et respect.

Remerciements
A notre Maître et Juge de thèse Monsieur le Professeur H. HARMOUCHE
Professeur de médecine interne
Nous vous remercions de l’attention que vous avez bien voulu porter à ma
thèse.
Nous vous prions d’accepter nos remerciements.

Remerciements
A Monsieur le Docteur SERRAJ Khalid
Merci pour tous les efforts que vous avez consenti pour ma réalisation du
travail
Recevez ainsi l’expression de ma profonde gratitude.

Listes des figures
LISTE DES FIGURES
Figure 1: Bases génétiques de la drépanocytose 6
Figure 2 : Origine des différents haplotypes 7
Figure 3 : Globules rouges en faucille au cours de la drépanocytose 11
Figure 4 : Electrophorèse de l’hémoglobine à pH alcalin d’un patient SS 31
Figure 5 : Focalisation isoélectrique 33
Figure 6 : Analyse de l’hémoglobine d’un patient homozygote SS par CLHP 33
Figure 7 : arbre de décision diagnostic et examens à réaliser 35
Figure 8: Répartition selon la nature des crises 52
Figure 9: Répartition selon le nombre de transfusion 57
Figure 10 : Répartition selon le statut vaccinal 57
Figure 11 : Répartition selon la durée du suivi 58
Figure 12 : Répartition géographique de la drépanocytose dans le monde 65
Figure 13 : Distribution comparative du paludisme et de l’hémoglobinose S 66

Listes des tableaux
LISTE DES TABLEAUX
Tableau I : volume des échanges transfusionnels dans la drépanocytose 39
Tableau II : Caractéristiques générales des patients 50
Tableau III : Répartition selon la présence de cas familiaux 51
Tableau IV : Répartition selon le type de complications ischémiques 53
Tableau V: Répartition selon les complications liée à l’anémie 53
Tableau VI: Répartition selon les complications infectieuses 54
Tableau VII : Répartition selon le taux d’hémoglobine 55
Tableau VIII : Répartition selon le taux de plaquettes 55
Tableau IX : Répartition selon le phénotype hémoglobinique 56
Tableau X: facteurs influençant les complications ischémiques 59
Tableau XI : facteurs influençant les complications infectieuses 60
Tableau XII: Complications évolutives et comparaison avec d’autres séries 68
Tableau XIII: Comparaison des complications infectieuses avec une série Africaine 71
Tableau XIV : Hémogramme chez le patient homozygote comparaison avec
d’autres séries 72
Tableau XV: Répartition des phénotypes et comparaison avec d’autres 72
Tableau XVI : Comparaison de statut vaccinal à d’autres séries 73

Listes des abréviations
LISTE DES ABREVIATIONS
Hb : Hémoglobine
Hb F : Hémoglobine fœtale
CCMH : Concentration corpusculaire moyenne en hémoglobine
VCAM : Vascular cell adhesion molecule
STA : Syndrome thoracique aigu
CVO : Crises vaso-occlusives
AVC : Accident vasculaire cérébral
IEF : Isoélectrofocalisation
CLHP : Chromatographie liquide haute performance
ADN : Acide désoxyribonucléase
OMS : Organisation mondiale de la santé
AINS : Anti-inflammatoires non stéroïdiens

Listes des abréviations
INTRODUCTION ............................................................................................................. 1
PARTIE I : GENERALITES SUR LA DREPANOCYTOSE
HISTORIQUE ................................................................................................................... 3
Chapitre I : Aspects Génétiques ...................................................................................... 5
Chapitre II : Physiopathologie ......................................................................................... 9
I. La Polymérisation ............................................................................................................ 10
II. La Déshydratation ........................................................................................................... 12
III. L’Adhésion Endothéliale ............................................................................................... 12
IV. L’Hémolyse ................................................................................................................... 13
V. Le Déficit Immun ........................................................................................................... 14
Chapitre III : Aspects Cliniques ...................................................................................... 15
I. L’Enfant Drépanocytaire Hétérozygote ........................................................................... 16
II. L’Enfant Drépanocytaire Homozygote .......................................................................... 16
III. L’Adulte Drépanocytaire Homozygote ......................................................................... 17
III.1 Les Complications Ostéoarticulaires ...................................................................... 18
III.1.1 L’Infarctus Osseux .......................................................................................... 18
III.1.2 Les Ostéonécroses ........................................................................................... 18
III.1.2.1 De la tête fémorale .................................................................................. 19
III.1.2.2 De la tête humérale ................................................................................. 19
III.1.3 Les Ostéomyélites ........................................................................................... 19
III.1.4 L’Hyperplasie Médullaire ............................................................................... 20
III.1.5 Les Manifestations Articulaires ...................................................................... 20
III.2 Les Complications Rénales .................................................................................... 21
III.2.1 L’Hématurie .................................................................................................... 21
III.2.2 La Nécrose Papillaire ...................................................................................... 21
III.2.3 L’Insuffisance Rénale Sans Syndrome Néphrotique ...................................... 22
III.2.4 L’Insuffisance Rénale avec Syndrome Néphrotique ...................................... 22
III.2.5 Le Carcinome Médullaire Rénal ..................................................................... 22

Listes des abréviations
III.3 Le Syndrome Thoracique Aigu (STA) ................................................................... 23
III.4 Le Priapisme ........................................................................................................... 24
III.5 Les Complications Oculaires .................................................................................. 24
III.6 Les Complications Bilio - Digestives ..................................................................... 25
III.7 Les Complications Neurologiques ......................................................................... 26
III.8 Les Complications Dermatologiques ..................................................................... 26
III.9 Les Complications Cardiaques ............................................................................... 26
Chapitre IV : Aspects Paracliniques ................................................................................ 28
I. L’Hémogramme ............................................................................................................... 29
I.1 En dehors des Crises ................................................................................................. 29
I.2 Pendant la Crise ......................................................................................................... 30
II. La Confirmation de la Drépanocytose ............................................................................ 30
II.1 Les Méthodes Standards .......................................................................................... 30
II.1.1 L’Electrophorèse de l’Hémoglobine ..................................................................... 31
II.1.2 Le Test de Falciformation ..................................................................................... 31
II.2 Les Nouvelles Méthodes .......................................................................................... 32
II.2.1 L’Isoélectrofocalisation ........................................................................................ 32
II.2.2 La Chromatographie Liquide Haute Performance CLHP ..................................... 33
III. Diagnostics Néonatal et Prénatal ................................................................................... 34
Chapitre V : Traitement ................................................................................................... 36
I. Les Traitements Palliatifs ................................................................................................ 37
I.1 Les Antalgiques ......................................................................................................... 37
I.2 L’Hyperhydratation Parentérale ................................................................................ 38
I.3 La Transfusion .......................................................................................................... 38
I.4 La Prévention des Infections ..................................................................................... 41
I.4.1 L’Antibioprophylaxie ........................................................................................ 41
I.4.2 La Vaccination ................................................................................................... 41
I.4.3 Prophylaxie Anti-Palustre ................................................................................. 42
I.4.4 Prévention des Parasitoses Intestinales ............................................................. 42
I.4.5 Supplémentation Orale en Folates et en Fer ...................................................... 42
II. Les Traitements de Fond ................................................................................................ 43
II.1 L’Hydroxyurée ......................................................................................................... 43
II.2 La Transplantation Médullaire ................................................................................. 43

Listes des abréviations
PARTIE II: ETUDE PRATIQUE
MATERIEL ET METHODES ......................................................................................... 46
RESULTATS ..................................................................................................................... 49
DISCUSSION .................................................................................................................... 61
CONCLUSION .................................................................................................................. 76
RESUMES .......................................................................................................................... 78
REFERERENCES BIBLIOGRAPHIQUES .................................................................. 82

Introduction
1
INTRODUCTION
La drépanocytose, encore appelée hémoglobinose S ou sicklémie de
l’anglais « sikcle cell anemia », est une anémie hémolytique corpusculaire due à
une anomalie qualitative de l’hémoglobine. Elle tire son appellation de l’aspect
des globules rouges observés au microscope chez certains patients [1].
Il s’agit de la pathologie génétique héréditaire la plus répandue dans le
monde puisqu’elle touche plus de 50 millions personnes. La répartition
géographique de la drépanocytose se limite à l’Afrique subsaharienne, le moyen
orient, le sud est asiatique, quelques pays d’Amérique et à la méditerranée dont
le Maroc où la prévalence est estimée entre 1% à 2% [2].
Les bases physiopathologiques de la drépanocytose étant connues depuis
presque une cinquantaine d’année, l’accentuation des recherches se tourne
actuellement de plus en plus vers l’amélioration de la prise en charge
diagnostique et thérapeutique des patients. Les manifestations cliniques de la
drépanocytose sont liées aux propriétés anormales conférées aux globules
rouges drépanocytaires par l’hémoglobine mutante et peuvent être regroupées en
trois catégories : l’anémie hémolytique chronique, les phénomènes vaso-
occlusifs et l’extrême susceptibilité aux infections. Ces manifestations sont
responsables d’une morbi-mortalité importante qui rend compte de l’importance
d’une prise en charge aussi bien précoce qu’adaptée.
Dans ce travail, nous nous proposons d’une part, de faire une revue
exhaustive de la littérature afin de faire le point sur les données actuelles
physiopathologiques, diagnostiques et thérapeutiques de la drépanocytose, et
d’autre part de préciser les particularités de cette pathologie à l’âge adulte à
travers une étude réalisée sur une série de patients colligés dans le service de
médecine interne du CHU IBN SINA de Rabat.

Partie I : généralité sure la drépanocytose
2

Historique
3
HISTORIQUE

Historique
4
Historique
La drépanocytose a été décrite pour la première fois en 1910 par James
Herrick qui avait remarqué chez un jeune étudiant noir jamaïcain âgé de 20 ans,
la présence d’hématies inhabituelles en forme de faucille ou feuille d'acanthe au
frottis sanguin périphérique. En 1929, Hahn et Gillespie ont montré
expérimentalement que cette falciformation des globules rouges ne se produisait
que lors de la désoxygénation et qu’elle était réversible au moment de la
réoxygénation. [3]
Pauling, Itano, Singer et Wells apportent un progrès majeur en 1949 par la
mise en évidence à l’électrophorèse d’une migration anormale l’hémoglobine
des globules rouges drépanocytaires mais ce n’est qu’en 1959 que Igram avait
pu identifier la substitution d’un acide aminé au niveau de la chaine β de la
globine comme étant le mécanisme principal de la maladie. [1]
Dans les années soixante, les avancées réalisées dans le domaine de la
génétique se sont soldées par la mise en évidence et la localisation des gènes
codant pour l’hémoglobine au niveau du chromosome 11. [1]

Aspects Génétiques
5
CHAPITRE I:
ASPECTS GENETIQUES

Aspects Génétiques
6
La drépanocytose est une maladie due à une mutation ponctuelle du gène
codant pour la chaîne β de l’hémoglobine situé sur le chromosome 11. Il s'agit
d’une substitution de l’adénine par la thymine (CAG >>> GTG) ayant comme
conséquence le remplacement de l’acide glutamique hydrophile par la valine qui
est un acide aminé hydrophobe [figure1]. On obtient ainsi l’hémoglobine S dont
la solubilité est diminuée par rapport à l’hémoglobine A normale [4]
.
Figure 1: Bases génétiques de la drépanocytose [5]
Aspects Génétiques
7
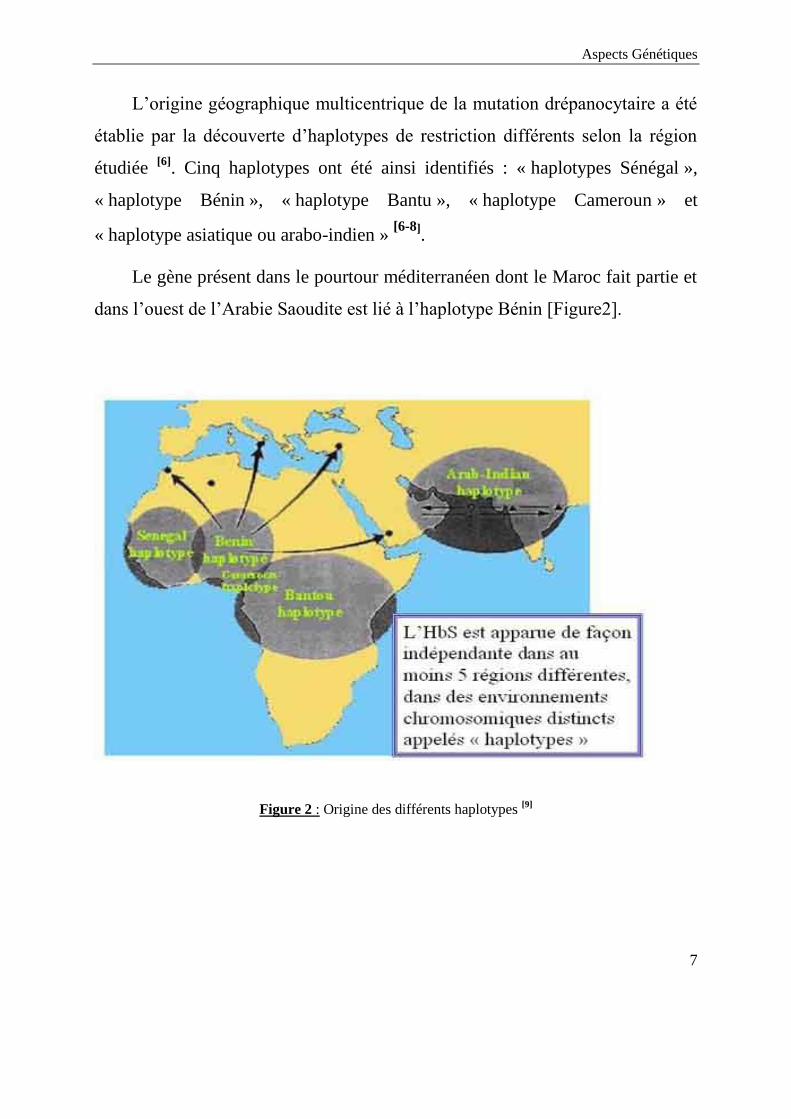
L’origine géographique multicentrique de la mutation drépanocytaire a été
établie par la découverte d’haplotypes de restriction différents selon la région
étudiée [6]
. Cinq haplotypes ont été ainsi identifiés : « haplotypes Sénégal »,
« haplotype Bénin », « haplotype Bantu », « haplotype Cameroun » et
« haplotype asiatique ou arabo-indien » [6-8]
.
Le gène présent dans le pourtour méditerranéen dont le Maroc fait partie et
dans l’ouest de l’Arabie Saoudite est lié à l’haplotype Bénin [Figure2].
Figure 2 : Origine des différents haplotypes [9]

Aspects Génétiques
8
La transmission de la drépanocytose est autosomique récessive.
Schématiquement, les sujets hétérozygotes sont AS et cliniquement sains ou
pauci symptomatiques, alors que les homozygotes SS sont très symptomatiques
et présentent généralement le tableau typique. D'autres anomalies aussi bien
qualitatives que quantitatives de l’hémoglobine peuvent s'associer à la
drépanocytose dont les principales sont l’hémoglobinose C et la β thalassémie
(doubles hétérozygotes). Ces formes composites peuvent être asymptomatiques
ou à l’inverse très bruyantes [10]
.

Physiopathologie
9
CHAPITRE II :
PHYSIOPATHOLOGIE

Physiopathologie
10
I. La Polymérisation
L’hémoglobine S a la propriété de polymériser sous l’influence de
différents facteurs : acidose, hypoxie, fièvre et déshydratation. La
polymérisation s’explique par le remplacement de l’acide glutamique acide
aminé hydrophile par la valine en position 6 qui est un résidu hydrophobe. Les
globines étant entourées par un film d’eau, la présence d’un site hydrophobe
crée ainsi un point de « collage » entre deux molécules d’hémoglobines
voisines. Celui-ci s’établit d’une part entre la leucine en position 88 et la
phénylalanine en position 85 d’une chaîne α et d’autre part la valine située en
position 6 de la chaîne β de l’hémoglobine voisine d’où la création d’une
structure cristalline en fibres. Dans les globules rouges SS, la polymérisation est
d’autant plus importante que :
la concentration corpusculaire moyenne en hémoglobine [CCMH] est
élevée,
-la saturation en oxygène est basse,
le taux d’ (HbF) hémoglobine fœtale est bas,
la température est anormale,
-le pH est acide,
La CCMH a la plus forte influence sur la polymérisation de la désoxy-
Hémoglobine S [11]
. L’Hb F inhibe également fortement la polymérisation de
l’hémoglobine S, c’est la raison pour laquelle la drépanocytose ne s’exprime pas
cliniquement avant l’âge de trois mois.
Physiopathologie
11
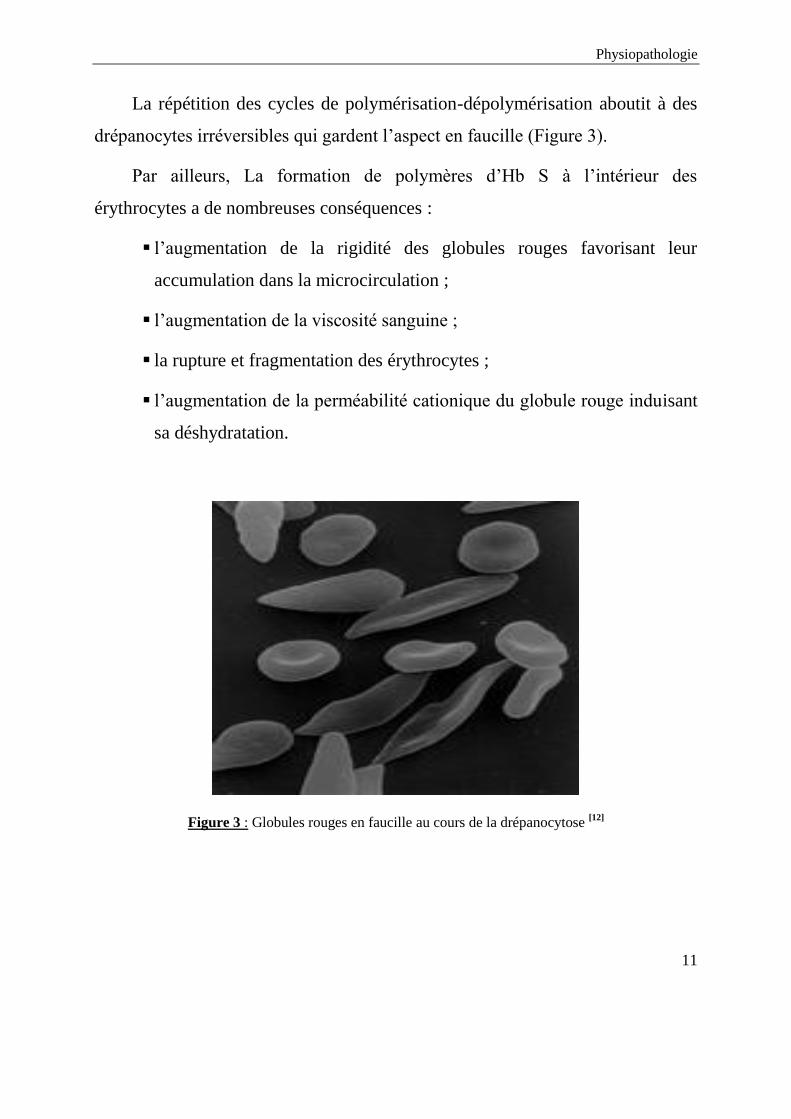
La répétition des cycles de polymérisation-dépolymérisation aboutit à des
drépanocytes irréversibles qui gardent l’aspect en faucille (Figure 3).
Par ailleurs, La formation de polymères d’Hb S à l’intérieur des
érythrocytes a de nombreuses conséquences :
l’augmentation de la rigidité des globules rouges favorisant leur
accumulation dans la microcirculation ;
l’augmentation de la viscosité sanguine ;
la rupture et fragmentation des érythrocytes ;
l’augmentation de la perméabilité cationique du globule rouge induisant
sa déshydratation.
Figure 3 : Globules rouges en faucille au cours de la drépanocytose [12]

Physiopathologie
12
II. La Déshydratation
Elle dépend de trois systèmes de transports ioniques :
Les canaux K+ dépendant du calcium: Lors des phases de
désoxygénation, l’augmentation de la perméabilité membranaire induite
par la polymérisation, favorise l’entrée de Calcium extracellulaire en
échange du potassium qui est rejeté hors de la cellule. La nécessité de
maintenir un équilibre osmotique et hydrique transmembranaire conduit
à une perte d’eau et de chlore dans le milieu extracellulaire [13]
.
L’activation du co-transport K+/Cl- par l’acidose favorisant une
déshydratation rapide des hématies.
La pompe Na+/K+ qui, dans le but de corriger les perturbations de
l’homéostasie cationique, excrète trois ions sodium pour deux ions
potassium ingérés.
C’est en modifiant les processus de déshydratation et de perméabilité
induits par la polymérisation que les cellules contenant de grandes quantités
d’Hb F réduisent ou préviennent la falciformation.
III. L’Adhésion Endothéliale
Les résultats de l’étude ex-vivo des flux montrent que la vaso-obstruction
est initiée par l’adhérence des réticulocytes, plus jeunes et encore déformables,
aux endothéliums vasculaires, puis amplifiée par le piégeage de drépanocytes
irréversibles ayant une faible déformabilité. Ce serait le facteur essentiel du
ralentissement de la vitesse sanguine dans la microcirculation laissant le temps
à la desoxy-Hb S de polymériser.

Physiopathologie
13
L’adhérence aux cellules endothéliales se fait directement via les intégrines
α4 β1 réticulocytaires, les molécules (vascular cell adhesion molecule) VCAM-1
des cellules endothéliales et indirectement via un certain nombre de ligands dont
un facteur von willebrand de poids moléculaire inhabituellement élevé, les
immunoglobulines, la fibronectine, le fibrinogène et la thrombospondine. Ce
dernier étant le plus actif. Le nombre de cellules endothéliales activées, c'est-à-
dire marquée par la présence à leur surface de ces molécules d’adhérence est
plus élevé chez le sujet drépanocytaire homozygote que chez le sujet sain ou
hétérozygote. Il se majore au cours des crises vaso-occlusives soulignant
davantage le rôle de l’endothélium [1]
.
IV. L’Hémolyse
L’hémolyse des cellules drépanocytaires est à la fois extra et
intravasculaire. L’hémolyse extravasculaire est due aux conséquences de
l’instabilité de l’Hb S et des falciformations récurrentes qui entraînent des
lésions oxydatives des membranes globulaires. L’Hb S dénaturé par l’oxydation
se lie à la portion cytoplasmique de la protéine 3 favorisant la fixation de Ig G et
du complément sur cette dernière ; le complexe ainsi formé est reconnu par le
macrophage et détruit. Aussi les drépanocytes irréversibles très rigides sont
piégés dans le secteur extravasculaire ce qui explique leur courte durée de vie.
L’hémolyse intravasculaire quant à elle s’explique par l’exocytose, induite par la
falciformation, de vésicules riches en protéines membranaires rendant les
globules rouges sensibles à la lyse, et par la fragilité mécanique qui accélère
l’hémolyse durant l’exercice.

Physiopathologie
14
V. Le Déficit Immun
La sensibilité des enfants atteints de drépanocytose aux infections et
notamment par Streptococcus pneumoniae est due à une altération de la fonction
splénique et de la diminution de l’activité opsonisante du sérum [14]
.

Aspects Cliniques
15
CHAPITRE III :
ASPECTS CLINIQUES

Aspects Cliniques
16
La sévérité du tableau clinique est fonction du profil phénotypique. Les
manifestations sont plus intenses chez les sujets homozygotes SS et certains
composites SC et Sβ0 que chez les sujets AS. L’expression clinque débute après
l’âge de trois mois car la présence d’hémoglobine fœtale à des taux élevés chez
le nouveau –né empêche la polymérisation.
Nous aborderons très brièvement les aspects cliniques chez l’enfant et
nous nous attarderons ensuite sur l’adulte dont il est question dans notre étude.
I. L’Enfant Drépanocytaire Hétérozygote
Il est pauci voire asymptomatique dans la majorité des cas. Cependant, au
décours de situations d’hypoxie sévère, des manifestations vaso-occlusives sont
possibles.
II. L’Enfant Drépanocytaire Homozygote
L’histoire clinique d’un enfant drépanocytaire est rythmée par trois types
de situations à savoir :
les phases stationnaires
les complications aiguës faites de crises douloureuses, d’infections et
d’aggravation multifactorielle et dont la fréquence diminue avec l’âge
sans pour autant disparaître à l’âge adulte,
l’installation progressive des complications chroniques dues aux
accidents vaso-occlusifs répétés.
L’anémie hémolytique chronique est une manifestation constante et
précoce. La triade pâleur, ictère, splénomégalie est de règle avant l’âge de 5 ans
chez l’homozygote. Les micro- infarctus répétés de la rate entraînent une

Aspects Cliniques
17
atrophie et la disparition de la splénomégalie .A long terme, l’anémie est
responsable d’un retard staturo-pondéral et d’un retard pubertaire de 4 à 5 ans.
Le syndrome mains- pieds est la crise vaso-occlusive caractéristique de l’enfant
[15]. L’aggravation de l’anémie chronique peut se faire par une séquestration
splénique ou encore par une érytroblastopénie [16 ,17]
.
III. L’Adulte Drépanocytaire Homozygote
A ce stade, la maladie se manifeste toujours par les crises douloureuses
vaso-occlusives qui ponctuent la vie des patients et sur lesquelles vont se greffer
les différentes complications hémolytiques, infectieuses et ischémiques qui
influencent la qualité de vie, le pronostic fonctionnel et le pronostic vital des
patients. Les complications aiguës de la drépanocytose sont généralement liées
aux trois mécanismes précédents à la fois, alors que les complications
chroniques sont surtout la conséquence lésionnelle tissulaire de l’ischémie
répétée. Les crises vaso-occlusives (CVO) sont le plus souvent osseuses ou
ostéoarticulaires, moins souvent thoraciques et rarement abdominales. Mais
c’est aussi la période où Elles sont variables de par leur type, leur intensité, leur
durée, leur localisation, leur fréquence et dominent la symptomatologie.
La douleur aigue est bien souvent le premier symptôme de la maladie et le
motif de consultation le plus fréquent. Elle peut intéresser n’importe quelle
partie du corps.
Certes, il existe une relation entre la sévérité des épisodes d’obstruction
vasculaire et le génotype ; cependant au sein des différents génotypes, des
variabilités interindividuelles et intra-individuelles apparaissent au fil des temps.
Ces épisodes douloureux sont bien des douleurs aigues. Cependant, les

Aspects Cliniques
18
répétitions leur confèrent un caractère chronique aux répercussions
psychologiques non négligeables [18]
. Plusieurs complications marquent
l’évolution de la maladie et influence son pronostic.
Dans ce qui suit, nous aborderons les différentes complications de la
drépanocytose en fonction de l’organe atteint.
III.1 Les Complications Ostéoarticulaires
III.1.1 L’Infarctus Osseux
L’infarctus osseux est toujours associé à des infarctus médullaires. La
symptomatologie initiale est celle d’une crise vaso-occlusive. Les infarctus du
squelette axial (bassin, rachis, côtes) sont possibles, plus fréquemment
rencontrées chez l’adulte. Le Sternum, le bassin et mandibules sont plus
rarement touchés. La durée de la crise peut aller de dix minutes à plusieurs
semaines mais la persistance des crises au-delà de deux semaines est rare lors
des crises non compliquées [19]
. Une majoration importante de l’anémie doit
faire suspecter la survenue d’une complication hémorragique ou d’une
complication plus spécifique telle qu’une nécrose ostéomédullaire. Les
radiographies sont initialement normales, ne montrant qu’un épaississement des
parties molles ou des signes en rapport avec l’hyperplasie médullaire. Puis,
apparaissent des zones d’ostéosclérose en plage associées à des débris calcifiés.
III.1.2 Les Ostéonécroses
Les ostéonécroses aseptiques épiphysaires de la drépanocytose atteignent
principalement la tête fémorale, les condyles fémoraux, la tête humérale, les os
du tarse voire même les vertèbres [20]
.

Aspects Cliniques
19
III.1.2.1 De la tête fémorale
Elle représente la localisation la plus fréquente. La radiographie est
souvent normale au stade de simple douleur de la hanche. Le diagnostic se fait
classiquement par les radiographies, la scintigraphie et surtout l’IRM qui est à
l’heure actuelle le moyen le plus performant pour assurer le diagnostic et évaluer
la taille de la nécrose épiphysaire. La radiographie montre une densification de
la tête fémorale comme signe le plus précoce, et recherche une dissection sous
chondrale ainsi que la classique coquille d’œuf ou perte de la sphéricité de la
tête fémorale.
III.1.2.2 De la tête humérale [21 ,22]
Souvent moins bruyante, elle doit être systématiquement recherchée. Elle
est bilatérale dans plus de 80 % des cas et pratiquement toujours associée à une
nécrose de hanche [23]
. Les séquelles radiologiques, présentes chez plus de la
moitié des drépanocytaires, sont à type de caput magna ou caput plana, avec
parfois hypoplasie totale ou partielle de la glène. Habituellement bien tolérée,
elle peut devenir gênante en cas d’arthrose secondaire, ce qui est rencontré dans
environ 10 % des cas. Elle peut également être responsable de dysmorphie de
l’épaule, exposant au risque de conflit antérieur à l’origine d’épanchements
articulaires.
III.1.3 Les Ostéomyélites
Tous les os peuvent être atteints mais surtout les os longs. Les localisations
plurifocales ne sont pas rares, souvent symétriques. Le diagnostic est évoqué
devant une douleur intense, des signes inflammatoires locaux avec tuméfaction
des tissus mous adjacents, une fièvre élevée, une altération de l’état général.

Aspects Cliniques
20
L’évolution vers une ostéomyélite chronique conditionne le pronostic
fonctionnel de ces patients.
III.1.4 L’Hyperplasie Médullaire
Elle est liée à la régénération secondaire à l’hémolyse chronique. En zone
tropicale, sont observées déformations du faciès et du crâne (faciès mongoloïde,
acrocéphalie) et retards staturaux importants. Les anomalies radiologiques du
squelette associent hypertransparence des os des membres et trabéculation
accentuée, amincissement des corticales diaphysaires et élargissement des
métaphyses des métatarsiens et métacarpiens. L’épaississement des os de la
voûte du crâne est moins marqué. L’ostéoporose est parfois importante sur les os
du rachis, avec aspect de « vertèbre en H » quand il s’y associe une dépression
centrale. Des modifications de la statique vertébrale peuvent se voir au cours de
la croissance à type d’accentuation de la cyphose dorsale ou d’hyperlordose
lombaire [24]
.
III.1.5 Les Manifestations Articulaires
L’arthrite aigue infectieuse est moins fréquente que l’ostéomyélite.
Néanmoins elle doit être évoquée en premier devant toute atteinte
articulaire avec un épanchement. La ponction articulaire permet de
mettre en évidence le germe.
Elles sont habituellement de mauvais pronostic : la gravité est due à la
possibilité de séquelles secondaires aux déformations et au possible passage à la
chronicité.

Aspects Cliniques
21
l’arthrite goutteuse est liée à une hyper production d’acide urique et à
une diminution de son excrétion en rapport avec l’atteinte rénale. Le
diagnostic est évoqué devant des crises articulaires aigues qui ne
s’accompagnent pas de signes de déglobulisation et confirmé par la mise
en évidence de cristaux d’urate dans le liquide articulaire [13]
.
III.2 Les Complications Rénales
Chez le jeune patient drépanocytaire SS, le flux sanguin et plasmatique
rénal, ainsi que le débit de filtration glomérulaire sont augmentés de plus de
50% pouvant entraîné des dysfonctions tubulaires rénales [25 ,26]
.
III.2.1 L’Hématurie
L’hématurie asymptomatique est un des symptômes les plus fréquents de la
maladie, que le patient soit homozygote ou non, et ce à n’importe quel âge.
L’hématurie microscopique peut être chronique, ponctuée par des épisodes
d’hématurie macroscopique. Elle résulte d’infarctus microthrombotiques,
favorisant la falciformation dans les vasa recta adjacents. L’importance de
l’hématurie peut, dans certains cas, nécessiter une transfusion, voire entraîner un
caillotage avec obstruction de l’arbre urinaire et tableau de colique néphrétique.
Ailleurs, une pyurie accompagne l’hématurie, devant faire discuter le diagnostic
de pyélonéphrite aiguë [27 ,28]
.
III.2.2 La Nécrose Papillaire
Elle peut survenir chez les patients homozygotes ou hétérozygotes. Le plus
souvent, elle est diagnostiquée rétrospectivement, lors d’un examen d’imagerie,
par urographie intraveineuse, échographie, scanner ou imagerie par résonance
magnétique. Au plan clinique, l’hématurie macroscopique douloureuse est la

Aspects Cliniques
22
présentation la plus commune. Parfois, elle peut occasionner une infection, voire
une septicémie, un tableau de colique néphrétique lorsque la nécrose papillaire
crée un obstacle urétéral, ou/et une insuffisance rénale aiguë. Ailleurs,
l’association pyurie, hématurie, lombalgies fait évoquer un épisode de
pyélonéphrite aiguë ; l’analyse du sédiment urinaire et l’examen bactériologique
des urines redressent le diagnostic.
III.2.3 L’Insuffisance Rénale Sans Syndrome Néphrotique
La rhabdomyolyse non traumatique liée aux crises vaso-occlusives sévères
en est l’un des principaux mécanismes [29]
. Cette complication est plus fréquente
lors des atteintes systémiques, en particulier thoraciques, hépatiques.
L’insuffisance rénale contribue à la mortalité chez les patients drépanocytaires
âgés. Une hypertension artérielle inhabituelle est classique chez ces patients.
III.2.4 L’Insuffisance Rénale avec Syndrome Néphrotique
Les lésions initiales commencent par une hypertrophie glomérulaire avec
développement progressif d’une glomérulosclérose focale prédominant au pôle
vasculaire [30-32]
. Une fibrose interstitielle et une atrophie tubulaire sont souvent
associées à l’existence de la glomérulosclérose. D’autres types de
glomérulonéphrites sont possibles à type de glomérulonéphrites
poststreptococciques, des glomérulonéphrites extramembraneuses, des
glomérulonéphrites membranoprolifératives [33, 34]
.
III.2.5 Le Carcinome Médullaire Rénal
Le carcinome médullaire rénal fait partie des complications évolutives
possibles. Son incidence dans la population drépanocytaire est de 1,74/1 000
patients par an. Le taux de mortalité est de 1,04 cas par année/patients [35,36]
.

Aspects Cliniques
23
L’hématurie macroscopique et les douleurs lombaires sont les symptômes les
plus fréquents, à l’inverse de l’amaigrissement et de la présence d’une masse
tumorale palpable. L’association d’une drépanocytose du sujet jeune et d’un
carcinome rénal a fait suggérer le rôle d’un facteur génétique [37]
. Il faut le
rechercher chez les patients drépanocytaires au stade le plus précoce. Une
échographie rénale devrait ainsi, faire partie du bilan systématique annuel de
surveillance.
III.3 Le Syndrome Thoracique Aigu (STA)
C’est la principale cause de décès et la deuxième cause d’hospitalisation
des patients drépanocytaires. Le STA correspond à des phénomènes de vaso-
occlusions siégeant dans la microcirculation pulmonaire auxquels participent la
réactivité vasculaire et l’adhésion anormale des globules rouges drépanocytaires
à l’endothélium vasculaire. Les étiologies en cause sont l’infection, l’œdème
pulmonaire, embolie graisseuse, infarctus pulmonaire [38,39]
.Toute complication
pulmonaire aiguë associant des signes fonctionnels et physiques respiratoires et
des signes radiologiques chez un drépanocytaire est un STA. En pratique, il
correspond à l’existence d’un nouvel infiltrat radiologique pulmonaire au moins
segmentaire (syndrome alvéolaire), en dehors d’une atélectasie, associé à des
signes respiratoires (tachypnée, wheezing, toux, hémoptysie…) ou des douleurs
thoraciques survenant parfois dans un contexte fébrile [40]
. A long terme, le STA
favorise la survenue de la maladie respiratoire chronique. Le STA s’accompagne
le plus souvent d’une crise vaso-occlusive qui le précède dans 50% des cas, ce
qui nécessite une surveillance régulière des patients hospitalisés pour CVO [39]
.

Aspects Cliniques
24
L’atteinte radiologique est plutôt multilobaire, associée à une atteinte
pleurale alors qu’elle est plutôt lobaire supérieur chez l’enfant. L’évolution peut
conduire à une insuffisance respiratoire aigue en moins de 48h. Par ailleurs, des
complications extra respiratoires peuvent survenir au cours du STA notamment
neurologiques à type d’accident vasculaire cérébral.
III.4 Le Priapisme
Les priapismes intermittents : ils sont habituellement brefs (10 à 3Omn). La
fonction pénienne reste le plus souvent normale. Cependant, ils peuvent
entraîner des lésions ischémiques des corps caverneux. L’augmentation de leur
fréquence et de leur durée précède souvent l’apparition d’un priapisme prolongé.
Le priapisme prolongé : d’une durée supérieure à trois heures pouvant
atteindre plusieurs jours et entraîner dans ce cas une impuissance quasi
constante surtout après la puberté [41]
. Apres 24 heures de priapisme non traité,
des lésions apparaissent avec évolution vers la fibrose et impuissance définitive.
III.5 Les Complications Oculaires
La rétinopathie drépanocytaire débute à la périphérie de la rétine, tout
d’abord par une simple tortuosité des veines évoluant vers une occlusion
vasculaire périphérique (capillaires et artérioles pré capillaires). Cette occlusion
est responsable d’une ischémie rétinienne visible au fond d’œil sous forme de
pâleur rétinienne en plage isolée à l’ora serrata. Puis, se mettent en place des
anastomoses atérioveineuses en bordure du territoire ischémique. Il s’en suit une
néo vascularisation. L’hémorragie intravitréenne survient lors de la rupture des
néovaisseaux. Le décollement rétinien est le dernier stade par traction vitréenne
La conjonctive peut être siège de dilatations vasculaires sans connexion avec le

Aspects Cliniques
25
réseau vasculaire au biomicroscope [42]
. Cette atteinte doit être recherchée au
niveau de la conjonctive inférieure.
D’autres atteintes notamment palpébrales et iriennes sont possibles.
III.6 Les Complications Bilio - Digestives
La Lithiase biliaire : c’est la principale complication abdominale de la
drépanocytose. Elle est secondaire à l’hémolyse chronique et atteint
préférentiellement les drépanocytaires homozygotes.
L’atteinte hépatique : l’hépatomégalie sans anomalies biologiques
associées est constatée chez la moitié des patients drépanocytaires. Les
CVO hépatiques sont parfois difficiles à distinguer d’une cholécystite
aiguë. La guérison est obtenue en règle en une à trois semaines, bien
qu’il y ait d’authentiques évolutions vers l’insuffisance. Il faut rappeler
que les complications hépatiques peuvent aussi être celles de la
transfusion sanguine : hépatites B, C et surcharge en fer.
L’atteinte du tube digestif : la survenue d’un iléus paralytique lors
d’une CVO de l’intestin grêle est la conséquence d’une ischémie et/ou
de lésions de reperfusion. Les infarctus sont rares en raison de la
richesse de la vascularisation du grêle. La douleur est parfois
épigastrique, éventuellement en rapport avec un ulcère gastrique ou
duodénal.
Le pancréas : Les pancréatites aiguës sont rares.

Aspects Cliniques
26
III.7 Les Complications Neurologiques
La vasculopathie cérébrale et les accidents vasculaires cérébraux (AVC)
constituent les principales complications neurologiques de la drépanocytose. Ils
engagent à court terme le pronostic vital et à long terme le pronostic fonctionnel,
requièrant ainsi un traitement d’urgence. Les AVC sont de trois types :
l’infarctus, l’hémorragie et l’embolie graisseuse. La mortalité globale est
inférieure à 10% mais les séquelles motrices, mentales et comitiales sont
fréquentes.
Il existe des infarctus silencieux repérer par la pratique de plus en plus
fréquente de l’IRM. On retrouve chez ces sujets une proportion notable de
vitesse anormale du flux cérébral mesuré avec le doppler transcranien .Les
récidives sont très fréquentes (46 à 90 %) et maximales dans les deux à trois ans,
surtout lorsque le premier AVC est survenu avant 20 ans [43, 44]
.
III.8 Les Complications Dermatologiques
Les ulcères de jambe sont les plus fréquents. Le mode de survenue peut
être spontané ou plutôt annoncé par des prodromes à types de : douleur ou de
dysesthésies. Il est parfois favorisé par un traumatisme minime, un grattage, une
piqûre d’insecte ou par des injections intraveineuses locales. L’infection
secondaire de l’ulcère est quasi constante, le plus souvent à Staphylococcus
aureus et Pseudomonas aeruginosa, plus rarement à germes anaérobies.
III.9 Les Complications Cardiaques
La myocardiopathie est symptomatique en cas d’insuffisance cardiaque,
d’angor ou de trouble du rythme. La douleur angineuse, les palpitations, les
signes d’insuffisance cardiaque sont à rechercher à l’examen clinique. Des

Aspects Cliniques
27
souffles systoliques éjectionnels ou d’insuffisance mitrale fonctionnelle, une
hyperpulsatilité artérielle, une cardiomégalie radiologique et des signes
électriques d’hypertrophie ventriculaire gauche peuvent être constatés chez le
drépanocytaire.

Aspects Paracliniques
28
CHAPITRE IV :
ASPECTS PARACLINIQUES

Aspects Paracliniques
29
I. L’Hémogramme
Chez le sujet hétérozygote AS, l’hémogramme est normal et il n’existe pas
de drépanocytes sur la lame de frottis sanguin [45]
. Ces derniers peuvent être
obtenus par la réalisation des tests de falciformation qui seront détaillés plus
loin.
Chez le sujet homozygote SS, il est important de faire la différence entre
l’hémogramme en période de crise et l’hémogarmme de base en intercritique.
I.1 En dehors des Crises
L’anémie est constante, d’intensité variable entre 6 et 10 g/dl ; elle est
normocytaire très régénérative. Une microcytose oriente vers une carence
martiale ou vers une association à une thalassémie. L’aspect du frottis montre de
multiples anomalies morphologiques érythrocytaires telles l’anisocytose avec
des cellules hypochromes et des cellules cibles, une poikilocytose, un nombre
variables d’érythroblastes et un petit nombre de corps de howell-jolly témoins
de l’hyposplénisme. Le nombre de drépanocytes circulants est variable d’un
patient à l’autre ; en moyenne 5 à10% des hématies du frottis. Une
hyperleucocytose à polynucléaires neutrophiles est habituelle en dehors des
infections. Le nombre des plaquettes, quant à lui, est normal jusqu’à ce que le
phénomène d’autosplénectomie fonctionnelle ou organique se soit manifesté.
Dans ce cas, les plaquettes peuvent être augmentées de façon sensible [45]
.

Aspects Paracliniques
30
I.2 Pendant la Crise
Il se produit une chute du taux d’hémoglobine pouvant atteindre 4 ou5 g /dl
contemporaine d’une réticulocytose basse précédant une phase d’hyper
réticulose qui persiste jusqu’à ce que le taux habituel de l’hémoglobine soit
atteint.
Chez l’hétérozygote composite SC, le taux d’hémoglobine est plus élevé
avec des leucocytes et des plaquettes moins élevés en comparaison au patient
homozygote. L’étude sur la lame ne retrouve pas de drépanocytes [46]
.
II. La Confirmation de la Drépanocytose
Le diagnostic de la drépanocytose est biologique. Il repose sur
l’identification de l’Hb S et le dosage des fractions de l’hémoglobine. Les
méthodes de diagnostic qui s’offrent au biologiste se basent soit sur la différence
de charge, soit sur la diminution de la solubilité pour révéler l’hémoglobine S.
II.1 Les Méthodes Standards
La mise en évidence de l’hémoglobine S doit se faire par deux méthodes au
minimum vu la multiplicité des anomalies de l’hémoglobine. Elle se fait par
l’électrophorèse à pH alcalin confirmée soit par l’électrophorèse à pH acide ou
par le test de solubilité.

Aspects Paracliniques
31
II.1.1 L’Electrophorèse de l’Hémoglobine
Elle est réalisée sur acétate de cellulose à pH =8,5 0,1 ou sur ph acide à
ph =6. Selon qu’il s’agisse d’une électrophorèse sur PH acide ou alcalin.
L’électrophorèse à ph alcalin est une méthode qui permet une bonne séparation
des Hb S , Hb A , et Hb C en fonction de leur charge. Lors de la technique, l’Hb
S migre moins rapidement vers l’anode que l’Hb A et l’Hb C qui, elles, migrent
plus lentement que l’Hb S. Cet examen est généralement utilisé en première
intention [47]
. (Figure 4).
Figure 4 : Electrophorèse de l’hémoglobine à pH alcalin chez un patient SS[48]
II.1.2 Le Test de Falciformation
C’est un test non spécifique dont la positivité témoigne seulement de la
présence d’hématies falciformes dans le sang du sujet ; d’autres hémoglobinoses
que l’Hb S peuvent donner un test de falciformation positif comme
l’hémoglobine I [49 ]
et l’hémoglobine bart’s [50]
à forte concentration.
Le test est basé sur le fait qu’à l’état désoxygène, l’Hb S se cristallise et le
globule rouge prend la forme de faucille. Le métabisulfate à 2%, agent

Aspects Paracliniques
32
réducteur, provoque la falciformation. Les hématies falciformes sont alors
visibles au microscope optique.
II.2 Les Nouvelles Méthodes
II.2.1 L’Isoélectrofocalisation
Les limites de l’électrophorèse à pH alcalin chez le nouveau -né ont
conduit à l’utilisation de (IEF) l’isoélectrofocalisation. Il s’agit d’une technique
très résolutive qui constitue à l'heure actuelle la technique de référence dans le
diagnostic néonatal des hémoglobinopathies. Les différentes hémoglobines sont
séparées en fonction de leur point isoélectrique (pHi).
L'isoélectrofocalisation diffère de l'électrophorèse classique par le fait que
la migration, sous l'effet du champ électrique, ne s'effectue plus dans un tampon
de pH fixe mais dans un gradient de pH (gradient de pH formé grâce à
l'utilisation de molécules amphotères). L'électrofocalisation permet de séparer
des protéines dont les pHi sont différents de 0,005 unité pH. C’est une méthode
plus sensible qui permet de traiter des séries importantes d’échantillons [51]
(voir
Figure 5 à la page suivante).
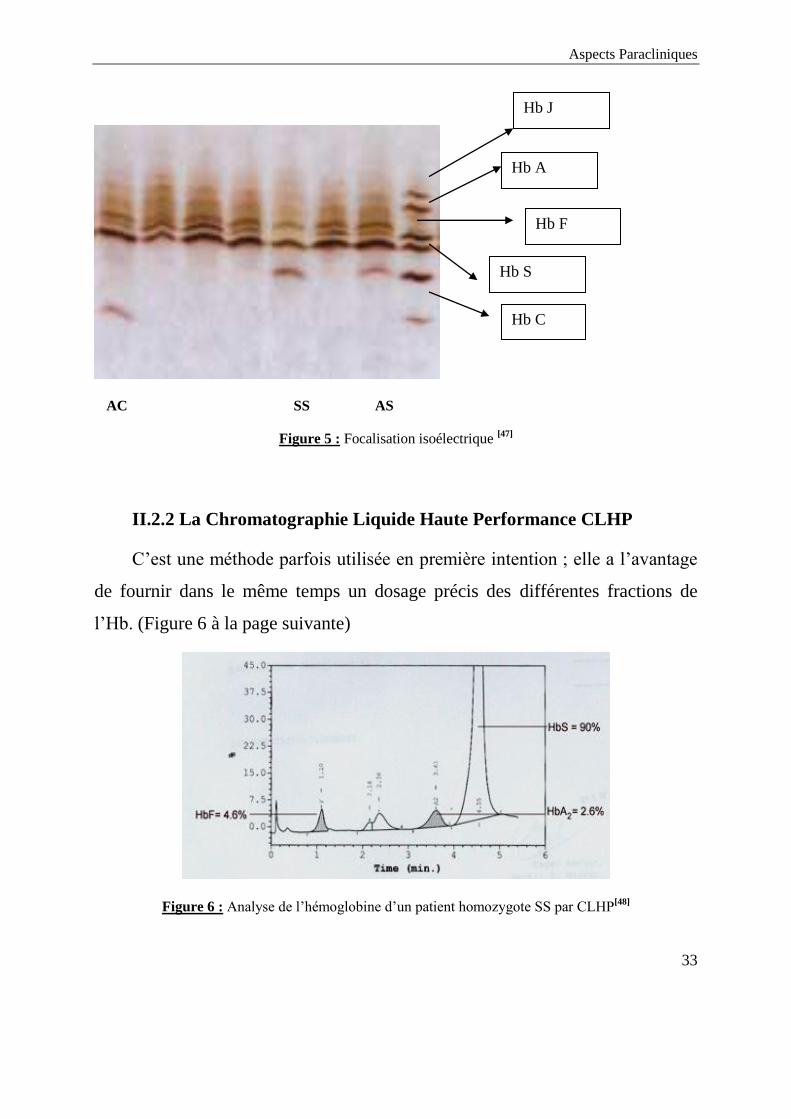
Aspects Paracliniques
33
AC SS AS
Figure 5 : Focalisation isoélectrique [47]
II.2.2 La Chromatographie Liquide Haute Performance CLHP
C’est une méthode parfois utilisée en première intention ; elle a l’avantage
de fournir dans le même temps un dosage précis des différentes fractions de
l’Hb. (Figure 6 à la page suivante)
Figure 6 : Analyse de l’hémoglobine d’un patient homozygote SS par CLHP[48]
Hb C
Hb F
Hb S
Hb A
Hb J

Aspects Paracliniques
34
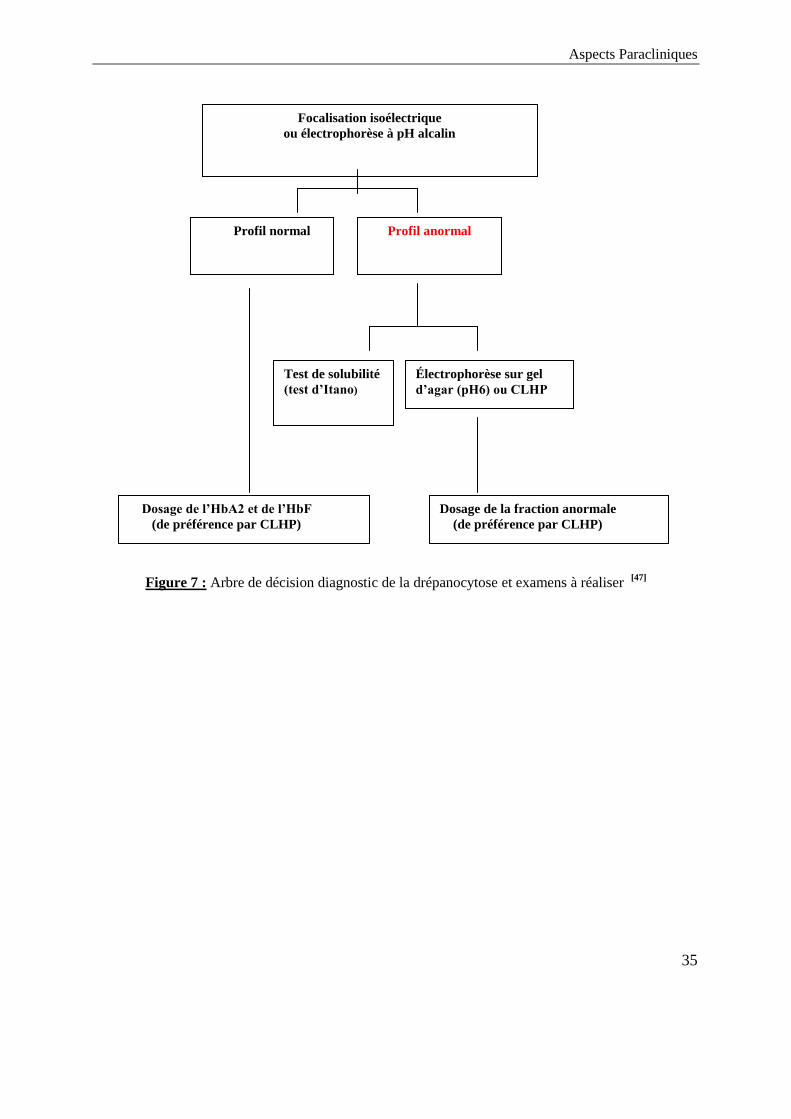
La figure 7 résume l’arbre décisionnel concernant le diagnostic positif de la
drépanocytose [47]
.
III. Diagnostics Néonatal et Prénatal
La technique utilisée est l’isoélectrofocalisation en première intention ; la
confirmation se fera par l’électrophorèse à pH acide soit par la chromatographie
liquide haute pression selon les laboratoires. Ce diagnostic se fait dans les
familles à risque et permet un suivi précoce réduisant ainsi les complications
chez le jeune enfant. Il est nécessaire de prime abord d’évaluer les
connaissances et de restaurer l’image de la maladie auprès de la famille.
Le diagnostic prénatal repose sur l’analyse de (ADN) l’acide
désoxyribonucléase fœtale obtenue à partir de trophoblastes prélevés entre huit
et douze semaines d’aménorrhée ou de cellules amniotiques prélevées entre la
quinzième et la vingtième semaine d’aménorrhée [52]
.
Aspects Paracliniques
35
Figure 7 : Arbre de décision diagnostic de la drépanocytose et examens à réaliser [47]
Focalisation isoélectrique
ou électrophorèse à pH alcalin
Profil normal Profil anormal
Électrophorèse sur gel
d’agar (pH6) ou CLHP
Test de solubilité
(test d’Itano)
Dosage de l’HbA2 et de l’HbF
(de préférence par CLHP)
Dosage de la fraction anormale
(de préférence par CLHP)

Traitement
36
CHAPITRE V :
TRAITEMENT

Traitement
37
Les différents outils thérapeutiques dont nous disposons à ce jour ont une
approche palliative dans la grande majorité des cas. Des progrès se font toujours
dans la recherche pour obtenir une curabilité de la pathologie utilisable pour tous
les patients. Les indications thérapeutiques dépendent de la sévérité des crises.
I. Les Traitements Palliatifs
I.1 Les Antalgiques
Le traitement antalgique ne peut se concevoir sans une évaluation correcte
de la douleur. Il existe plusieurs méthodes d’évaluation de la douleur comme
l’échelle visuelle analogique, l’échelle numérique ou encore l’échelle verbale
simple. La distinction entre douleur chronique et douleur aigue est importante
lors de cette évaluation. La crise vaso-occlusive est une douleur par excès de
nociception dont la prise en charge fait appel aux agents antalgiques classés dans
l’échelle de l’OMS organisation mondiale de la santé repartie en trois paliers.
Les anti-inflammatoires non stéroïdiens (AINS) et le paracétamol :
palier I
Le palier I est approprié en cas de douleur légère à modérées de la
drépanocytose. Ces molécules peuvent être associées à des opioïdes en cas de
douleur sévère. Les AINS présentent un grand nombre d’effets secondaires et
d’interférences médicamenteuses. Leur maniement est moins simple de ce fait.
Par ailleurs, ils peuvent interférer avec l’évolution d’une infection sous jacente
en masquant les signes [53]
.

Traitement
38
Les antalgiques morphiniques faibles : palier II
Une grande place est donnée aux agents associant paracétamol à des
morphiniques. L’indication est faite dans les douleurs modérées et dans celles
qui résistent au palier I.
La morphine : palier III
C’est le médicament de référence dans les douleurs sévères. Compte tenu
des effets indésirables de la morphine tels que l’hypotension, les
bronchospasmes, les œdèmes laryngés et la détresse respiratoire, il est
nécessaire que les services hospitaliers disposent de moyens de réanimation
conséquents à proximité.
I.2 L’Hyperhydratation Parentérale
C’est un geste essentiel. La réhydratation orale toute seule est insuffisante
même soigneusement surveillée. C’est bien à la réhydratation veineuse qu’il faut
avoir recours systématiquement. Elle joue un important rôle dans le contrôle de
la crise vaso-occlusive. Elle se fait par le sérum glucosé isotonique à raison de
150ml/ kg/24h en faisant attention aux éventuels déséquilibres électrolytiques.
I.3 La Transfusion
Elle constitue l’élément majeur du traitement du patient drépanocytaire. Il
existe trois modalités différentes dans la drépanocytose : la transfusion sanguine
simple, l’échange transfusionnel et la transfusion sanguine au long cours.

Traitement
39
La transfusion simple : L’objectif est de ramener un taux d’Hb abaissé
à sa valeur habituelle. 3ml /kg de culot globulaire ou 6ml/kg de sang
total permettent d’obtenir un gain de 1g/dl d’hémoglobine.
L’échange transfusionnel [54]
: L’objectif de l’échange transfusionnel
est de remplacer les hématies drépanocytaires par des hématies
contenant de l’HbA. Il associe une saignée et une transfusion. Cet
échange doit se faire en règle générale à hématocrite constant. Les
techniques manuelles supposent deux voies d’abord veineuse, l’une pour
la soustraction (saignée) l’autre pour les apports (transfusion). On
procède en trois temps :
o Une saignée de 10 à 15 ml/kg associée à une perfusion concomitante
du même volume de soluté isotonique par la seconde voie d’abord ;
o Une transfusion réglée au même débit que la saignée jusqu’à
obtention du volume à dépléter ;
o La poursuite de la transfusion jusqu’à obtention du volume que l’on
veut apporter. (tableau I à la page suivante).
Tableau I : Volume des échanges transfusionnels dans la drépanocytose [54]
Taux d’Hb souhaité Volume de sang à soustraire Volume de concentré
érythrocytaire à transfuser
<25 %
<40 %
60 ml /kg
40 ml/kg
45 ml/kg
30 ml/kg

Traitement
40
Les échanges transfusionnels au long cours sont proposés chez certains
drépanocytaires après un AVC, lors d’une détérioration viscérale sévère rénale,
respiratoire ou cardiaque [55]
.
En pratique, les transfusions sont indiquées dans les situations où il y a :
Une anémie profonde cliniquement mal tolérée (le chiffre de
l'hémoglobine doit toujours être interprété en fonction du chiffre observé
à l'état basal)
Une crise vaso-occlusive qui se prolonge (> 8 jours) malgré un
traitement symptomatique bien conduit ou la présence d’effets
secondaires limitants de la morphine lors d'une crise hyperalgique
Un syndrome thoracique grave (hypoxémie profonde, images
thoraciques bilatérales et extensives) ou ne répondant pas au traitement
symptomatique après 48 à 72 heures d'évolution (extension des images
radiographiques, persistance de la fièvre et des douleurs thoraciques,
majoration de la dyspnée et de l'hypoxie)
Un priapisme aigu avec plus de 3 heures d'évolution et absence
d'efficacité des injections intracaverneuses d'étiléfrine
Un accident vasculaire cérébral ischémique ou hémorragique aigu
(l'intérêt de la poursuite des transfusions au long cours au cours des
accidents hémorragiques est en revanche discuté).
Une infection sévère intercurrente
Toute complication grave intercurrente pouvant mettre en jeu le
pronostic vital ou fonctionnel

Traitement
41
Une intervention chirurgicale majeure
Des manifestations vaso-occlusives pendant la grossesse (avis
spécialisé).
Il est important de rappeler que la thérapeutique transfusionnelle expose
aux risques d’alloimmunisation, de transmission virale et de surcharge en fer qui
s’ajoutent chez le drépanocytaire au risque d’hyperviscosité, d’où l’intérêt d’en
mesurer soigneusement l’indication et de veiller sur les modalités
transfusionnelles [56]
.
I.4 La Prévention des Infections
Les patients drépanocytaires, sujets à l’asplenie fonctionnelle, sont exposés
à des risques infectieux. La prophylaxie devient alors une nécessité.
I.4.1 L’Antibioprophylaxie
L’antibioprophylaxie par la pénicilline orale est prescrite chez l’enfant de
moins de cinq ans à raison de 50000 UI/kg/j en deux ou trois prises. Les
infections étant moins fréquentes chez l’adulte, la prescription de pénicilline au
long cours n’est pas indispensable. Une exception est présentée pour les sujets
infectés par le virus d’immunodéficience acquise qui doivent bénéficier d’une
prophylaxie continue [57]
.
I.4.2 La Vaccination
En plus des vaccins usuels retrouvés dans le calendrier légal, certains
vaccins sont particulièrement recommandés chez les patients drépanocytaires. Il
s’agit des vaccins anti-pneumococciques et anti-haemophilus B, vaccin contre
l’hépatite B [58]
.

Traitement
42
I.4.3 Prophylaxie Anti-Palustre
En milieu tropical, d’autres mesures anti-infectieuses doivent être prises,
notamment contre le paludisme. La survenue d’un accès palustre chez le
drépanocytaire constitue un facteur déclenchant des crises vaso-occlusives et
risque d’aggraver l’hyperhémolyse.
I.4.4 Prévention des Parasitoses Intestinales
La prophylaxie des parasitoses intestinales se justifie par l’extrême
fréquence de ces pathologies chez les enfants en milieu tropical. Ce contexte
épidémiologique amène à préconiser une cure d’antihelminthique tous les trois
mois chez les enfants de moins de cinq ans et tous les six mois pour les autres
[59].
I.4.5 Supplémentation Orale en Folates et en Fer
Les folates présentent une activité régénératrice de la moelle. Leur
prescription est d’autant plus utile que l’alimentation ne couvre pas dans
certaines régions suffisamment les besoins. Les folates sont prescrits à la dose
de 5mg /j ou de 15/mois [59]
. La supplémentation en fer se fait à raison de
3à5mg /kg /j de fer métal pendant deux ou 3 mois chez tout enfant présentant
une baisse du taux d’Hb et du taux de réticulocytes associée à une microcytose
et une hypochromie récente [60]
.

Traitement
43
II. Les Traitements de Fond
II.1 L’Hydroxyurée
D’un point de vue physiopathologique, l’hydroxyurée est le premier
traitement ayant potentiellement un impact sur le cours évolutif de la
drépanocytose en diminuant la fréquence des crises douloureuses chez la plupart
des patients et en allongeant leur espérance de vie. L’hydroxyurée bloque
préférentiellement le développement des cellules érythroides mures. Il en résulte
un recrutement des précurseurs érythrocytaires jeunes avec une production
supérieure d’Hb F [61 ,62]
.Une plus grande concentration d’Hb F réduit la
polymérisation et le nombre d’hématies falciformes. Elle agit aussi en
améliorant la déformabilité érythrocytaire et en diminuant l’adhésion des
hématies à l’endothélium vasculaire. Par ailleurs, elle augmente la production
objective de monoxyde d’azote [63]
.
II.2 La Transplantation Médullaire
La transplantation médullaire a un intérêt curatif dans la drépanocytose.
Elle constitue le seul traitement potentiellement curateur de la drépanocytose
mais a été jusqu'à présent limitée aux patients ayant un donneur HLA –identique
familial et ses risques et résultats au long cours doivent être comparé de façon
prospective à ceux obtenus par les autres thérapeutiques . Il s’agit de remplacer
les cellules hématopoïétiques du malade de façon définitive par celles du
donneur de phénotype AA.
L’expérience est encore récente et le recul peu important mais, à 6 ans, la
survie globale est de 94% et la survie sans événements de 84 %. Près de 10 %
des drépanocytaires greffés ont un rejet de la greffe ou une récidive de la

Traitement
44
drépanocytose. Les traitements utilisés pour la préparation, les
immunosuppresseurs et les antimitotiques, posent le problème de l’infertilité et
de l’oncogénicité qu’ils induisent. Ces risques incitent à la prudence concernant
les indications de la greffe [64,65]
.

Résumés
45

Matériel & Méthodes
46
MATERIEL & METHODES

Matériel & Méthodes
47
Il s’agit d’une étude rétrospective descriptive et analytique portant sur
l’ensemble des patients drépanocytaires suivi dans le service de Médecine A de
l’hôpital Ibn Sina de Rabat sur une période de onze ans allant du 1er janvier 1996
au 31 décembre 2006.
Critères d’inclusion et d’exclusion :
Nous avons inclus dans le présent travail tous les patients étant suivis au
service de médecine interne en 1996 et tous les patients ayant commencé le
suivi à partir de 1996. Il s’agissait soit de patients chez qui le diagnostic a été
porté de novo, soit de patients suivis antérieurement dans d’autres structures
pendant l’enfance.
Nous n’avons retenu que les patients avec un diagnostic certain de la
drépanocytose sur les bases de l’hémogramme, l’électrophorèse de
l’hémoglobine et de l’enquête familiale pour certains.
Les dossiers médicaux ont été notre outil de base. Des fiches
d’exploitations pré-établies ont été élaborées afin de permettre une étude
descriptive homogène des paramètres épidémiologiques, cliniques, biologiques,
thérapeutiques et de la prise en charge des patients.
Les informations recueillies à partir du dossier médical de chaque patient a
permis d’étudier les caractères suivants :
o Les aspects épidémiologiques : le sexe, l’âge du diagnostic, l’âge au
début du suivi en Médecine A, la notion de consanguinité, le lieu
d’habitation, les cas familiaux.

Matériel & Méthodes
48
o Les aspects cliniques : le nombre d’hospitalisations, la splénomégalie,
la nature des crises, l’état général, la présence ou non de complications
évolutives infectieuses, ischémiques et liée à l’anémie chronique.
o Les aspects biologiques : le phénotype, les taux de base de
l’hémoglobine, des plaquettes et des globules blancs.
o Les données de la prise en charge : le nombre de transfusion, la
régularité du suivi médical, le statut vaccinal, la durée du suivi et la
présence ou non de traitements par l’ hydroxyurée et l’acide folique.
Une étude analytique a été aussi faite pour rechercher une éventuelle
corrélation entre le sexe, l’âge médian du diagnostic, le phénotype, le taux
moyen de plaquettes, le taux moyen d’hémoglobine, d’une part, et la survenue
des complications infectieuses et ischémique d’autre part.
Méthodologie statistique :
Toutes les données ont été traitées grâce au logiciel SPSS. Le test khi-2 a
été utilisé pour les comparaisons des variables qualitatives et le test non
paramétrique de Mann- Whitney pour les comparaisons des variables
quantitatives. Les résultats étaient considérés comme étant significatifs quand le
P était inférieur à 0,05.

Résultats
49
RESULTATS

Résultats
50
Entre 1996 et 2006, nous avons identifié 33 patients répondant aux critères
d’inclusion suscités, dont les principales caractéristiques sont résumées dans le
tableau II.
Tableau II : Caractéristiques générales des patients
Nombre de patients 33
Homme /femme (sex. ratio) 1,36
Age du diagnostic (médiane) 7ans
Consanguinité parentale 24,2 %
Origine marocaine 93,9%
Splénomégalie 24,3%
Crises Vaso-occlusives 53,4 %
Crises Hémolytiques 33,3 %
Crises Mixtes 13,3 %
Taux moyen d’hémoglobine 9 ,02g /dl
Taux moyen de plaquettes 464707 ,14 /mm3
Phénotype SS 42,4%
Complications liée à l’anémie 30 ,3%
Complications ischémiques : 45,4%
Complications infectieuses 39 ,4%
ASPECTS EPIDEMIOLOGIQUES
Il y avait 19 hommes (58% des patients) soit une sex-ratio de 1, 36. L’âge
médian du diagnostic était de 7 ans avec le quartile à 4ans et demi. Les extrêmes
d’âge étaient 6 mois et 30 ans. L’âge moyen au début du suivi en médecin A
était 22,9 ans avec une prédominance de la tranche d’âge de 21 à 25 ans
(42,4%). Il y avait une notion de consanguinité parentale chez 8 patients
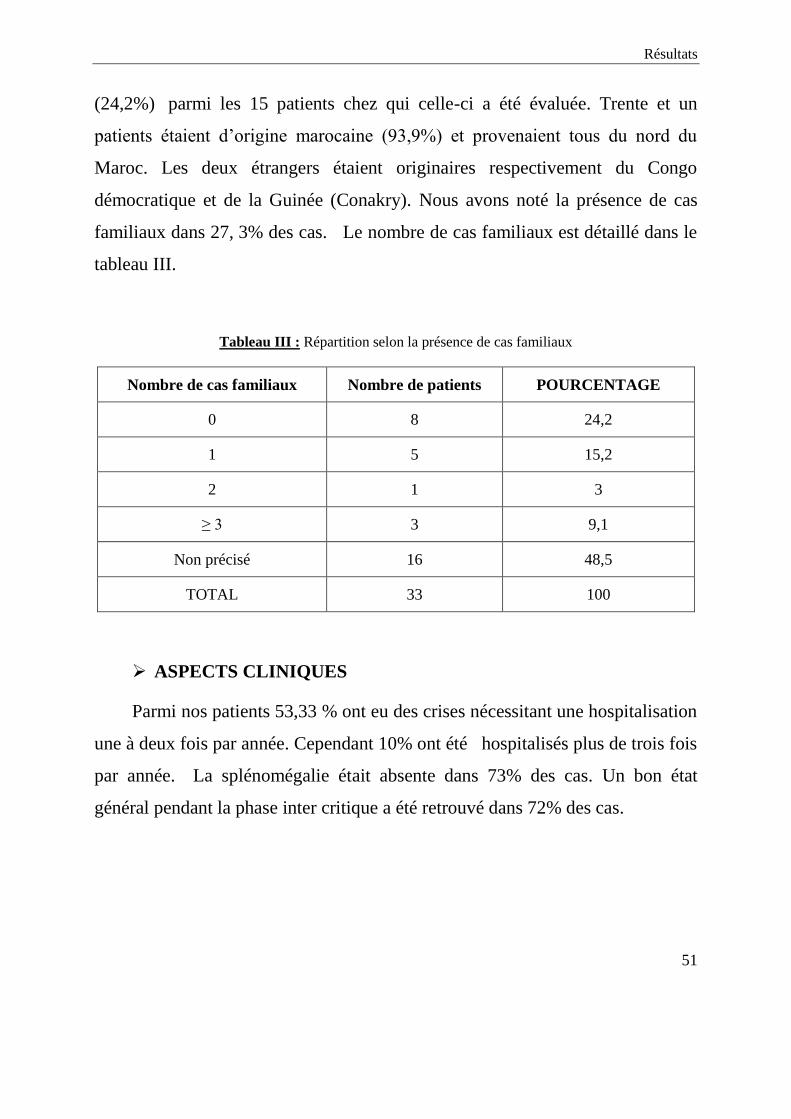
Résultats
51
(24,2%) parmi les 15 patients chez qui celle-ci a été évaluée. Trente et un
patients étaient d’origine marocaine (93,9%) et provenaient tous du nord du
Maroc. Les deux étrangers étaient originaires respectivement du Congo
démocratique et de la Guinée (Conakry). Nous avons noté la présence de cas
familiaux dans 27, 3% des cas. Le nombre de cas familiaux est détaillé dans le
tableau III.
Tableau III : Répartition selon la présence de cas familiaux
Nombre de cas familiaux Nombre de patients POURCENTAGE
0 8 24,2
1 5 15,2
2 1 3
≥ 3 3 9,1
Non précisé 16 48,5
TOTAL 33 100
ASPECTS CLINIQUES
Parmi nos patients 53,33 % ont eu des crises nécessitant une hospitalisation
une à deux fois par année. Cependant 10% ont été hospitalisés plus de trois fois
par année. La splénomégalie était absente dans 73% des cas. Un bon état
général pendant la phase inter critique a été retrouvé dans 72% des cas.
Résultats
52
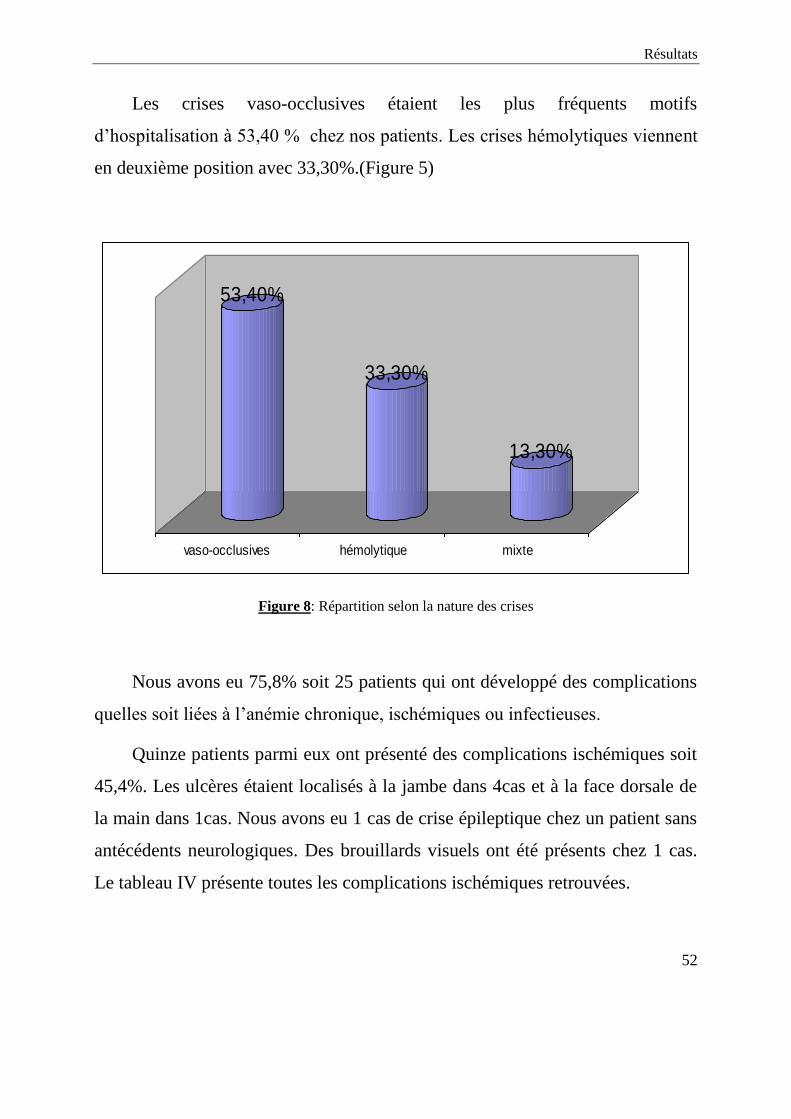
Les crises vaso-occlusives étaient les plus fréquents motifs
d’hospitalisation à 53,40 % chez nos patients. Les crises hémolytiques viennent
en deuxième position avec 33,30%.(Figure 5)
53,40%
33,30%
13,30%
vaso-occlusives hémolytique mixte
Figure 8: Répartition selon la nature des crises
Nous avons eu 75,8% soit 25 patients qui ont développé des complications
quelles soit liées à l’anémie chronique, ischémiques ou infectieuses.
Quinze patients parmi eux ont présenté des complications ischémiques soit
45,4%. Les ulcères étaient localisés à la jambe dans 4cas et à la face dorsale de
la main dans 1cas. Nous avons eu 1 cas de crise épileptique chez un patient sans
antécédents neurologiques. Des brouillards visuels ont été présents chez 1 cas.
Le tableau IV présente toutes les complications ischémiques retrouvées.

Résultats
53
Tableau IV : Répartition selon le type de complications ischémiques
- Ulcère de jambe : 4 cas
- Autre localisation d’ulcère : 1 cas
- Infarctus osseux : 1 cas
- Nécrose aseptique des deux têtes fémorales : 3 cas
- Infarctus splénique : 5 cas
- Priapisme : 1 cas
- Insuffisance rénale fonctionnelle : 1 cas
- Ophtalmologique : 1 cas
- Neurologique : 1cas
Dix patients dans notre série ont développé des complications liées à
l’anémie chronique soit 30,3%. Les lithiases vésiculaires étaient les plus
fréquentes (8 cas). La dilatation ventriculaire gauche (4 cas) et la tachycardie (1
cas).
Le dernier cas était une dépression des vertèbres dorsales et lombaires.
Tableau V: Répartition selon les complications liée à l’anémie
- lithiase vésiculaire : 8cas
- cœur anémique : 5cas
- dépression vertébrale : 1cas

Résultats
54
Nous avons eu 13 patients qui ont développé des complications infectieuses
soit 39,4 %. Les infections urinaires basses ont été retrouvées dans 3 cas et 1 cas
de pyélonéphrite. Les infections à salmonella étaient 1 cas de fièvre typhoïde et
1 cas d’abcès de la partie antérieur du tibia se compliquant en ostéomyélite
chronique. Les ostéites étaient localisées dans 2 cas au niveau du tibia et 1 cas
au niveau du péroné. Les cas de complications gastriques étaient une duodénite.
(voir TableauVI ).
Tableau VI: Répartition selon les complications infectieuses
- infection urinaire : 4 cas
- ostéite : 3 cas
- péricardite : 1 cas
- gastrique : 2 cas
- hépatite : 1 cas
- infection à salmonella : 2 cas
ASPECTS BIOLOGIQUES
o L’Hémogramme :
Le taux d’hémoglobine en phase inter critique variait entre 6,5 g/dl (2 cas)
et 13,6 g /dl (1 cas) avec un taux moyen de 9 ,1g/dl. Dix-sept patients avaient
des taux inférieurs ou égaux à 10 g/dl soit 74 % sur les 23 patients pris en
compte (tableau VII à la page suivante).
On note une hyperleucocytose chez 14 patients soit 63,64% avec des
extrêmes allant de 5633 leucocytes /mm3 à 14925 leucocytes /mm3. Le taux
moyen des leucocytes était 10735 ,08 /mm3.
Résultats
55
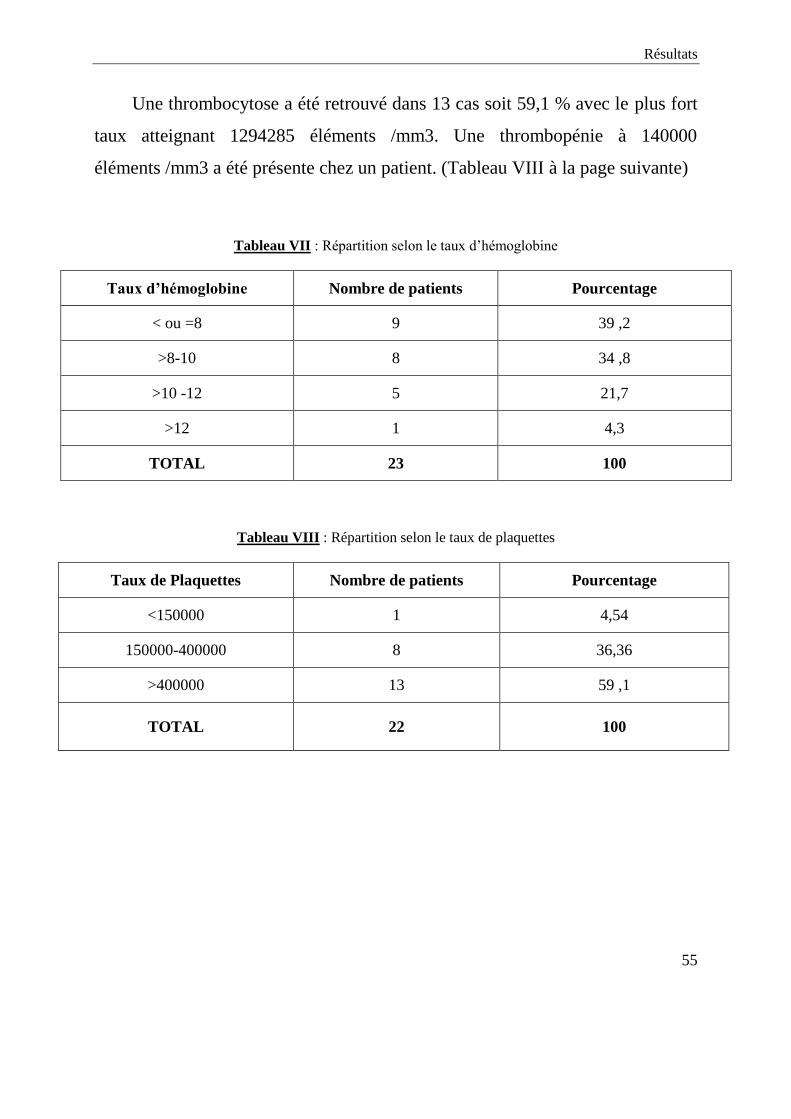
Une thrombocytose a été retrouvé dans 13 cas soit 59,1 % avec le plus fort
taux atteignant 1294285 éléments /mm3. Une thrombopénie à 140000
éléments /mm3 a été présente chez un patient. (Tableau VIII à la page suivante)
Tableau VII : Répartition selon le taux d’hémoglobine
Taux d’hémoglobine Nombre de patients Pourcentage
< ou =8 9 39 ,2
>8-10 8 34 ,8
>10 -12 5 21,7
>12 1 4,3
TOTAL 23 100
Tableau VIII : Répartition selon le taux de plaquettes
Taux de Plaquettes Nombre de patients Pourcentage
<150000 1 4,54
150000-400000 8 36,36
>400000 13 59 ,1
TOTAL 22 100

Résultats
56
o L’étude de l’hémoglobine
Nous avons observé une prédominance du phénotype SS à hauteur de
42,4% parmi les patients dont phénotype a été précisé. (Tableau IX)
Tableau IX : Répartition selon le phénotype hémoglobinique
Phénotype Nombre de patients Pourcentage
SS 14 42 ,4
SC 5 15,1
AS 2 6,1
Non Précisé 12 36,4
TOTAL 33 100
LA PRISE EN CHARGE THERAPEUTIQUE
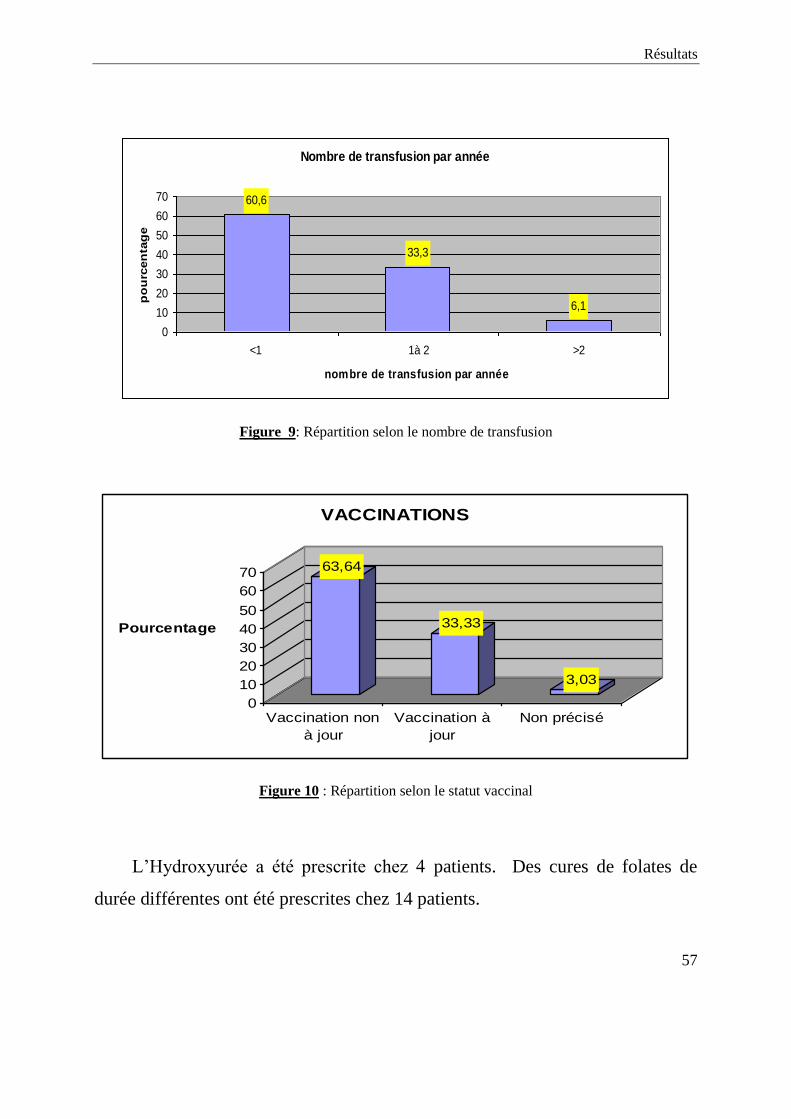
Le nombre de transfusion reçu dans le service de Médecine A variait de 0
(13 cas) à 14 fois (1cas). 2 patients ont eu plus de deux transfusions par année
soit 6,1%. (Figure 9).
La vaccination n’est pas à jour chez 21 patients soit 63,64 % ; Le statut
vaccinal n’a pas été précisé chez 1 patient. (Figure 10 à la page suivante).
Résultats
57
Nombre de transfusion par année
60,6
33,3
6,1
0
10
20
30
40
50
60
70
<1 1à 2 >2
nombre de transfusion par année
po
urc
en
tag
e
Figure 9: Répartition selon le nombre de transfusion
63,64
33,33
3,03
0
10
20
30
40
50
60
70
Pourcentage
Vaccination non
à jour
Vaccination à
jour
Non précisé
VACCINATIONS
Figure 10 : Répartition selon le statut vaccinal
L’Hydroxyurée a été prescrite chez 4 patients. Des cures de folates de
durée différentes ont été prescrites chez 14 patients.
Résultats
58
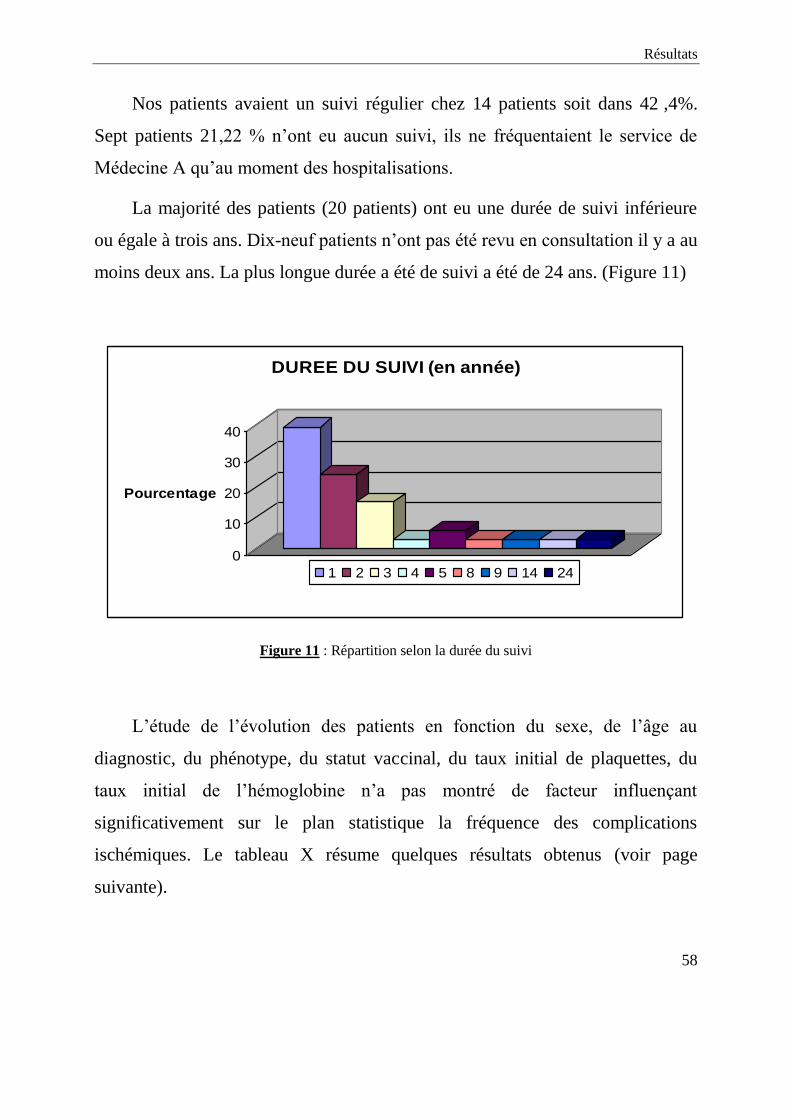
Nos patients avaient un suivi régulier chez 14 patients soit dans 42 ,4%.
Sept patients 21,22 % n’ont eu aucun suivi, ils ne fréquentaient le service de
Médecine A qu’au moment des hospitalisations.
La majorité des patients (20 patients) ont eu une durée de suivi inférieure
ou égale à trois ans. Dix-neuf patients n’ont pas été revu en consultation il y a au
moins deux ans. La plus longue durée a été de suivi a été de 24 ans. (Figure 11)
0
10
20
30
40
Pourcentage
DUREE DU SUIVI (en année)
1 2 3 4 5 8 9 14 24
Figure 11 : Répartition selon la durée du suivi
L’étude de l’évolution des patients en fonction du sexe, de l’âge au
diagnostic, du phénotype, du statut vaccinal, du taux initial de plaquettes, du
taux initial de l’hémoglobine n’a pas montré de facteur influençant
significativement sur le plan statistique la fréquence des complications
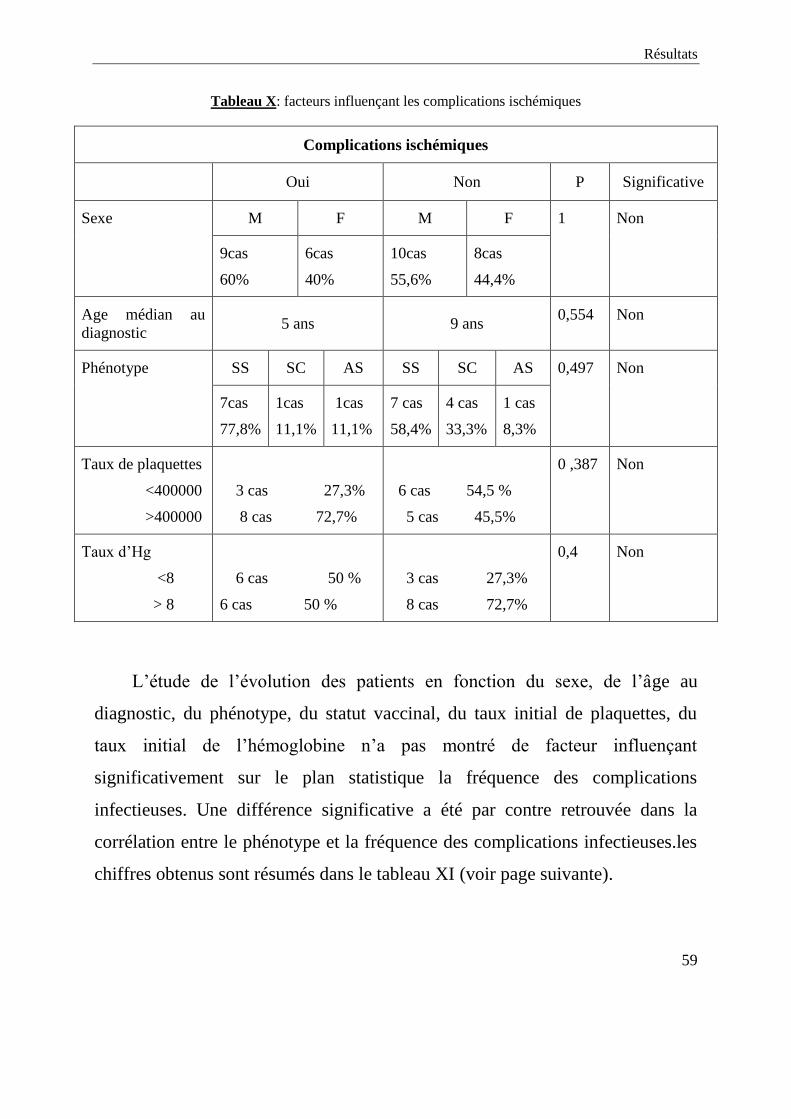
ischémiques. Le tableau X résume quelques résultats obtenus (voir page
suivante).
Résultats
59
Tableau X: facteurs influençant les complications ischémiques
Complications ischémiques
Oui Non P Significative
Sexe M F M F 1 Non
9cas
60%
6cas
40%
10cas
55,6%
8cas
44,4%
Age médian au
diagnostic 5 ans 9 ans
0,554 Non
Phénotype SS SC AS SS SC AS 0,497 Non
7cas
77,8%
1cas
11,1%
1cas
11,1%
7 cas
58,4%
4 cas
33,3%
1 cas
8,3%
Taux de plaquettes
<400000
>400000
3 cas 27,3%
8 cas 72,7%
6 cas 54,5 %
5 cas 45,5%
0 ,387 Non
Taux d’Hg
<8
> 8
6 cas 50 %
6 cas 50 %
3 cas 27,3%
8 cas 72,7%
0,4 Non
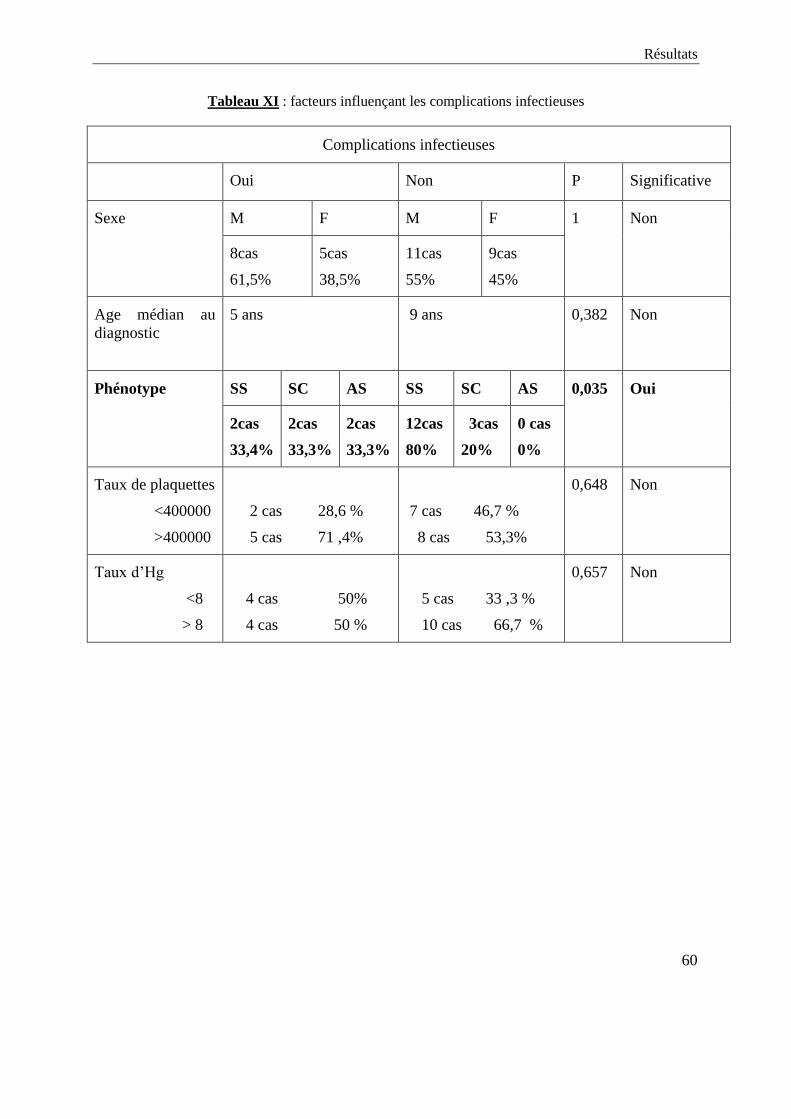
L’étude de l’évolution des patients en fonction du sexe, de l’âge au
diagnostic, du phénotype, du statut vaccinal, du taux initial de plaquettes, du
taux initial de l’hémoglobine n’a pas montré de facteur influençant
significativement sur le plan statistique la fréquence des complications
infectieuses. Une différence significative a été par contre retrouvée dans la
corrélation entre le phénotype et la fréquence des complications infectieuses.les
chiffres obtenus sont résumés dans le tableau XI (voir page suivante).
Résultats
60
Tableau XI : facteurs influençant les complications infectieuses
Complications infectieuses
Oui Non P Significative
Sexe M F M F 1 Non
8cas
61,5%
5cas
38,5%
11cas
55%
9cas
45%
Age médian au
diagnostic
5 ans 9 ans 0,382 Non
Phénotype SS SC AS SS SC AS 0,035 Oui
2cas
33,4%
2cas
33,3%
2cas
33,3%
12cas
80%
3cas
20%
0 cas
0%
Taux de plaquettes
<400000
>400000
2 cas 28,6 %
5 cas 71 ,4%
7 cas 46,7 %
8 cas 53,3%
0,648 Non
Taux d’Hg
<8
> 8
4 cas 50%
4 cas 50 %
5 cas 33 ,3 %
10 cas 66,7 %
0,657 Non

Discussion
61
DISCUSSION

Discussion
62
Le nombre de 33 patients que compte notre série est certes nettement plus
réduit par rapport aux effectifs des autres séries de la littérature notamment
celles de l’Afrique subsaharienne. Ceci s’expliquerait en partie par les chiffres
de prévalence de la drépanocytose dans le bassin méditerranéen dont le Maroc
(1 à 2%) et qui restent très inférieurs aux autres régions du monde concernées
par cette maladie [2]
. Ce taux de 2% n’est tout de même pas du tout négligeable
et il est donc à prendre en compte d’autant plus que plus de 90% de nos patients
sont autochtones. Par ailleurs, il est important à noter que l’effectif de notre
étude ne reflète pas ces chiffres de prévalence précités et pourrait donc
témoigner plus d’un sous diagnostic que d’une réelle rareté de la pathologie dans
notre pays.
D’un autre côté, la drépanocytose est certes une maladie génétique qui, par
définition, se déclarerait plus volontiers à l’enfance qu’à l’âge adulte. Il est
important donc de mettre l’accent sur la fréquence des formes hétérozygotes qui
peuvent rester longtemps asymptomatiques et ne se révéler que par des
complications chez un adulte chez qui le diagnostic de drépanocytose n’est pas
forcément évoqué comme première hypothèse diagnostique.
Nous en citons notamment les manifestations extra-hématologiques qui ne
sont que des séquelles du processus hémolytique et/ou vaso-occlusif comme la
lithiase biliaire, la goutte, les atteintes tubulaires et les crises comitiales [66]
.
Dans notre série, le cas d’un patient hétérozygote AS ayant développé des
complications tant infectieuses qu’hémolytiques sévères illustre bien ce constat.

Discussion
63
Une sensibilisation des praticiens à reconnaître les signes cliniques même
mineurs et les circonstances déclenchantes des crises est de ce fait plus
qu’indispensable. Cette démarche permettrait de corriger le sous diagnostic et de
prévenir le développement de complications chez l’adulte.
L’origine génétique autosomique de la maladie laisse supposer qu’il
n’existe pas de prédominance d’un sexe par rapport à un autre. Cependant, on
note constamment dans les différentes séries de la littérature une prédominance
d’un sexe par rapport à l’autre. Dans notre série, il y avait une prédominance
masculine avec un pourcentage de 58 % et une sex-ratio de 1,36. Nos résultats
rejoignent ceux de certaines séries africaines comme celle de TUO et al. [67]
qui
avaient trouvé une prédominance masculine et une sex-ratio de 1,23. En
revanche, BERTRAND et al. [68]
, dans leur étude portant sur 111cas, ont
rapporté une prédominance féminine avec une sex-ratio de 0,7.
Notons enfin que des différences importantes sont retrouvées dans des
séries provenant du même pays. L’origine géographique et ethnique des patients
ne peut pas être évoquée comme explication à ces différences.
L’âge médian au diagnostic qui a été de 7 ans dans notre série se rapproche
des résultats d’une autre étude marocaine publiée par LAHLOU [69]
qui avaient
trouvé un âge supérieur à 6 ans dans 61,5 % des cas. La littérature nous montre
que la maladie drépanocytaire se répartit en deux groupes selon l’âge du début
des manifestations : un groupe à début précoce se manifestant entre 6 et 18 mois
[70] et un à révélation tardive après l’âge de 4-5ans dont les cas ne sont pas
exceptionnelles comme ça a été le cas chez une grande partie de nos patients
(57,1% début après 5 ans dans notre série ) [71]
. Le diagnostic tardif peut

Discussion
64
s’expliquer par le fait que la maladie est souvent décelée à la suite d’un accident
aigu chez des patients avec des signes d’anémie déjà présents et souvent
longtemps bien tolérée. Nous avons eu trois patients diagnostiqués à un âge se
situant entre 26 et 30 ans. L’enquête familiale prend donc tout son intérêt dans
ce contexte dans la mesure elle permet de diagnostiquer la drépanocytose au
stade infraclinique.
La consanguinité accroît le risque d’expression de la drépanocytose. Elle
est de l’ordre de 24,2 % dans notre série. Ce chiffre se rapproche du taux de
26,1% retrouvé dans une étude Nigérienne de DAN et al [72]
. Chez plus de la
moitié de nos patients (54,4%), la notion de consanguinité n’avait pas été
précisée, ce qui laisse supposer une certaine sous estimation des mariages
consanguins dans notre série. En 2007, le taux de mariages consanguins a été
estimé à 22 ,79% selon une étude anthropogénique marocaine [73]
.
Les régions de forte prévalence de la drépanocytose sont plutôt retrouvées
en Afrique subsaharienne avec des taux moyen de 10% à 30 % [11]
. Aux États-
Unis, la fréquence du trait drépanocytaire chez les Afro-américains se situe entre
7 et 9% [11]
. Dans des parties de l’est de l’Arabie saoudite et de l’Inde, la
fréquence de l’hémoglobine S atteint les 20 %. Une vue globale de la répartition
mondiale est représentée dans la Figure 12 à la page suivante. La répartition
géographique de nos patients est bien en corrélation avec la répartition générale
de la drépanocytose puisque nos deux patients étrangers provenaient de
l’Afrique subsaharienne (la guinée et le Congo démocratique).

Discussion
65
Figure 12 : Répartition géographique de la drépanocytose dans le monde [74]
La répartition dans le monde de la drépanocytose se superpose facilement
à celle du paludisme à Plasmodium falciparum comme la montre la figure
10.Des études statistiques sur la question ont permis les conclusions suivantes :
- Les sujets homozygotes SS résistent au paludisme mais meurent de leur
hémoglobinopathie.
- Les sujets hétérozygotes quant à eux sont résistants au paludisme et
présente une hémoglobinopathie asymptomatique ; cet avantage est
essentiellement dû aux rapports entre le parasite et l’hématie.

Discussion
66
La réplication du parasite est gênée par la falciformation et par la
destruction prématurée des globules rouges infectés. Ainsi la pression de
sélection due au paludisme a rendu le gène plus fréquent surtout dans les zones à
forte transmission palustre [75]
.
Figure 13 : Distribution comparative du paludisme et de l’hémoglobinose S [76]
Dans notre série, 53,3% des patients ont été hospitalisés à raison de 1 à 2
fois par année et 10% avaient plus de trois hospitalisations par année. Ces
résultats sont proches de ceux de Le Turdu-Chicot et al, dans son étude en
Guadeloupe, qui avaient 1,38 hospitalisations par année, de Brozovic et al en

Discussion
67
Grande Bretagne (trois hospitalisations par patient), de Pollack et al aux États-
Unis (deux hospitalisations par patient) et de Sergeant et al en Jamaïque avec
1,55 hospitalisations par patient et par an [77 - 80]
.
La nature des crises a été dominée par les crises vaso-occlusive à 53,4%.
Dans son étude de 108 cas de patients âgés de plus de 20 ans à Dakar, Diop et al
ont fait le même constat avec un taux de 50 % [81]
. Bakary, quant à lui, avait
trouvé des taux de 54,2 % de crises vaso-occlusives, 13,39 % de crises
hémolytiques et 32,4 % de crises mixtes [82]
. Tous ces chiffres concordent avec
nos résultats. Par ailleurs, cette fréquence de la crise vaso-occlusive représente
la cause majeure derrière l’altération de la qualité de vie voire le pronostic vital
des patients. La grande fréquence des crises vaso-occlusives est de plus en plus
établie comme indice de sévérité de la maladie. Dans une étude multicentrique
américaine, Platt et al. [83]
ont observé que les décès par infection sont rares chez
l’adulte, contrairement à l’enfant. La majorité des patients adultes décédaient en
effet dans un contexte de crise vaso-occlusive. Godeau et al ont observé des
résultats similaires dans une étude multicentrique européenne ayant porté sur 61
décès et où 45% des décès étaient dans un contexte de crise vaso-occlusive [84]
.
Devant des crises répétées, il est indispensable de rechercher un facteur
déclenchant qui devra être corrigé : traiter une acidose satellite d’une atteinte
rénale, éliminer un foyer infectieux profond (dents, sinus, urines, vésicule…),
rechercher des épisodes nocturnes de désaturation en oxygène en rapport avec
une apnée du sommeil, insister de nouveau sur les règles hygiénodiététiques
(éviter les variations thermiques brutales, l’exposition au froid ou au contraire la
déshydratation…). Il peut être envisagé la mise en route d’un « traitement de
fond » qui vise à prévenir la survenue d’accidents vaso-occlusifs graves.
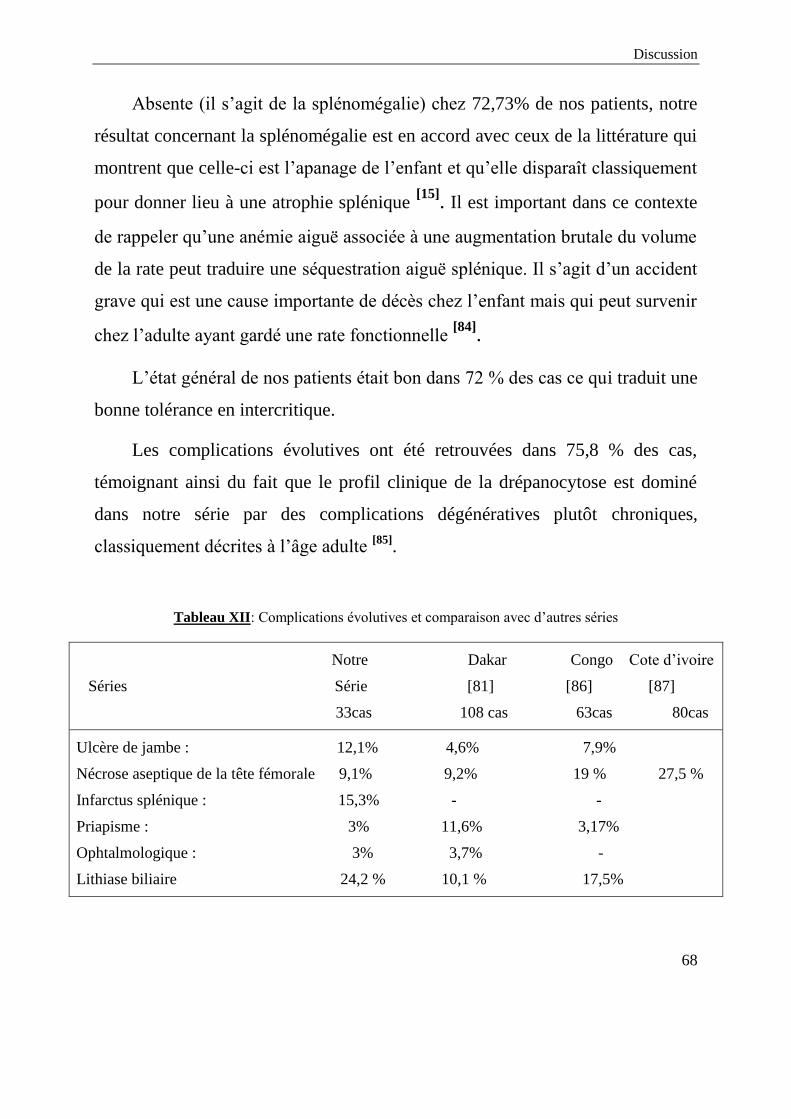
Discussion
68
Absente (il s’agit de la splénomégalie) chez 72,73% de nos patients, notre
résultat concernant la splénomégalie est en accord avec ceux de la littérature qui
montrent que celle-ci est l’apanage de l’enfant et qu’elle disparaît classiquement
pour donner lieu à une atrophie splénique [15]
. Il est important dans ce contexte
de rappeler qu’une anémie aiguë associée à une augmentation brutale du volume
de la rate peut traduire une séquestration aiguë splénique. Il s’agit d’un accident
grave qui est une cause importante de décès chez l’enfant mais qui peut survenir
chez l’adulte ayant gardé une rate fonctionnelle [84]
.
L’état général de nos patients était bon dans 72 % des cas ce qui traduit une
bonne tolérance en intercritique.
Les complications évolutives ont été retrouvées dans 75,8 % des cas,
témoignant ainsi du fait que le profil clinique de la drépanocytose est dominé
dans notre série par des complications dégénératives plutôt chroniques,
classiquement décrites à l’âge adulte [85]
.
Tableau XII: Complications évolutives et comparaison avec d’autres séries
Notre Dakar Congo Cote d’ivoire
Séries Série [81] [86] [87]
33cas 108 cas 63cas 80cas
Ulcère de jambe : 12,1% 4,6% 7,9%
Nécrose aseptique de la tête fémorale 9,1% 9,2% 19 % 27,5 %
Infarctus splénique : 15,3% - -
Priapisme : 3% 11,6% 3,17%
Ophtalmologique : 3% 3,7% -
Lithiase biliaire 24,2 % 10,1 % 17,5%

Discussion
69
Les complications ischémiques ont représenté 45,4% de toutes les
complications sont détaillées en comparaison aux études Sénégalaise et
Congolaise et Ivoirienne dans le tableau XII. L’infarctus splénique a occupé une
grande place (15,3%). L’ulcère de jambe dans notre série (12,1%) est plus
fréquent par rapport au chiffre de séries Sénégalaise (4,6 %) et Congolaise
(7,9%). L’étude Ivoirienne avait rapporté des complications ischémiques dans
68,75% des cas dont une ostéonécroses de la tête fémorale dans 27,5% et une
rétinopathie dans 11,25% des cas. La spécificité de cette étude sur les doubles
hétérozygotes SC pourrait être mise en cause ici [87]
.
Les phénotypes de nos 3 patients (9,1%) ayant développé l’ostéonécrose
n’étaient pas de phénotype SC mais plutôt SS dans deux cas et non précisé pour
le troisième. Dans la littérature, il est clairement établi que l’ostéonécrose est
deux fois plus fréquente chez les patients SC comme l’ont si bien montré
plusieurs études dont celles de Homawoo et al, Mijiyawa et al et Tobossi et al.
Cette fréquente était par ailleurs indépendante des autres facteurs de sévérité
potentielle [88 -90].
Nos trois patients avaient des âges respectifs très différents
(18, 22, et 43 ans) au moment du diagnostic.
Ces chiffres nous interpellent sur la nécessité de rechercher
systématiquement cette complication aux symptômes très peu spécifiques et
donc source fréquente de retard diagnostique comme une simple douleur fessière
ou de hanche. Il faut noter que la bilatéralité a été présente chez tous les patients
témoignant de la sévérité de l’atteinte. L’atteinte oculaire noté seulement dans
un seul cas chez nous pourrait être sous estimée vus les résultats des séries sus-
cités. Un examen ophtalmologique pourrait bien aussi avoir sa place dans le

Discussion
70
cadre du suivi d’un drépanocytaire en raison de la symptomatologie pas
forcement patente.
La lithiase biliaire a prédominé dans les complications liées à l’anémie en
touchant 8 patients soit 24,2% avec une fréquence plus élevée que dans la
littérature .Chez les huit patients, la lithiase a été diagnostiquée après l’âge de
16 ans dans le service de médecine interne et permettait de renseigner
indirectement sur la longue durée du processus hémolytique chronique. L’autre
complication assez fréquemment retrouvée en rapport avec l’anémie chronique
était la dilatation du ventricule gauche retrouvée chez 4 patients.
Les complications infectieuses retrouvées dans 39,4 % des cas étaient
dominées par les infections osseuses comme ce fut le cas dans la série Ivoirienne
(tableau XIII). La salmonellose était le germe en cause dans ce cas. Dans leur
étude S.Diop et al ont retrouvés que les patients SS faisaient plus d’infections
bactérienne [91]
comme le témoigne les résultats significatifs p=0,035 obtenus
dans l’étude analytique. La greffe osseuse est favorisée par les modifications de
la structure osseuse consécutives aux épisodes de nécrose préexistants. Le
Staphylocoque doré et les germes à Gram négatif sont également souvent en
cause [92, 93]
. Un cas d’hépatite virale a été retrouvé ce qui nous interpelle sur la
prévention lors des techniques transfusionnelles. Un patient est passé à la
chronicité dans l’ostéomyélite qu’il a faite ce qui est une évolution possible de
l’ostéomyélite. Le risque élevé d’infections nosocomiales des patients, du fait de
leurs fréquentes hospitalisations pour CVO et le recours habituel à l’hydratation
par voie veineuse, favorisent les métastases septiques ostéoarticulaires qui
peuvent survenir avec retard en particulier l’ostéomyélite. Ainsi une surveillance

Discussion
71
prolongée est justifiée en cas de bactériémie pour ne pas méconnaitre une
localisation septique.
Tableau XIII: Comparaison des complications infectieuses avec une série Africaine
Série Notre série Cote d’ivoire
[87]
Infections osseuses 9,1 % 30%
Salmonelloses 6,1 % 3,7%
Infections urinaire 12,1% 1,25%
Sur le plan biologique, le taux d’hémoglobine de base a oscillé entre
6,5g/dl et 13g/dl, avec une moyenne à 9,02 g/dl. Dans deux études Sénégalaise
et burkinabée, les taux moyens respectifs étaient de 8,3g/dl et de 7,8 g/dl [81, 93].
Il faut noter que ces deux études ne s’intéressaient qu’aux homozygotes, ce qui
concorde donc avec nos données concernant nos patients homozygotes dont le
taux moyen d’hémoglobine était de 8,5 g/dl. Une hyperplaquettose a été notée
fréquemment dans notre série contrairement aux séries de la littérature reflétant
très probablement l’état d’asplénie fonctionnelle classiquement rencontrée chez
les patients adultes (TableauXIV à la page suivante).
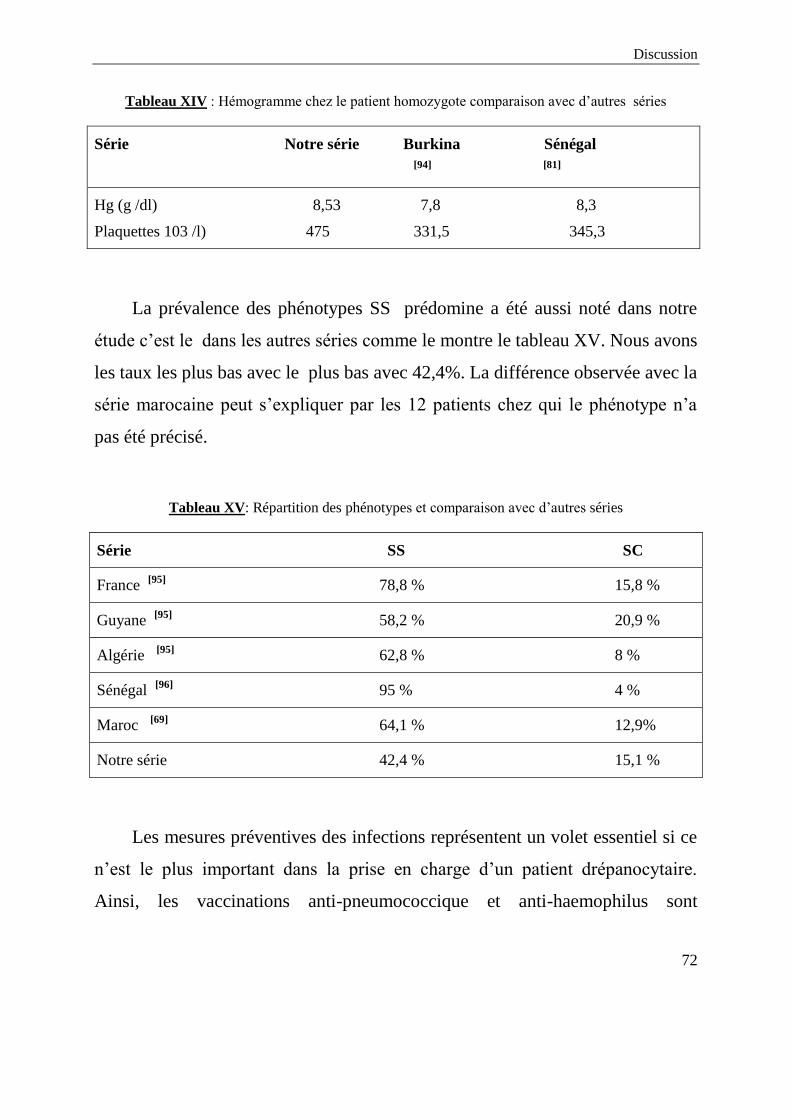
Discussion
72
Tableau XIV : Hémogramme chez le patient homozygote comparaison avec d’autres séries
Série Notre série Burkina Sénégal
[94] [81]
Hg (g /dl) 8,53 7,8 8,3
Plaquettes 103 /l) 475 331,5 345,3
La prévalence des phénotypes SS prédomine a été aussi noté dans notre
étude c’est le dans les autres séries comme le montre le tableau XV. Nous avons
les taux les plus bas avec le plus bas avec 42,4%. La différence observée avec la
série marocaine peut s’expliquer par les 12 patients chez qui le phénotype n’a
pas été précisé.
Tableau XV: Répartition des phénotypes et comparaison avec d’autres séries
Série SS SC
France [95]
78,8 % 15,8 %
Guyane [95]
58,2 % 20,9 %
Algérie [95]
62,8 % 8 %
Sénégal [96]
95 % 4 %
Maroc [69]
64,1 % 12,9%
Notre série 42,4 % 15,1 %
Les mesures préventives des infections représentent un volet essentiel si ce
n’est le plus important dans la prise en charge d’un patient drépanocytaire.
Ainsi, les vaccinations anti-pneumococcique et anti-haemophilus sont
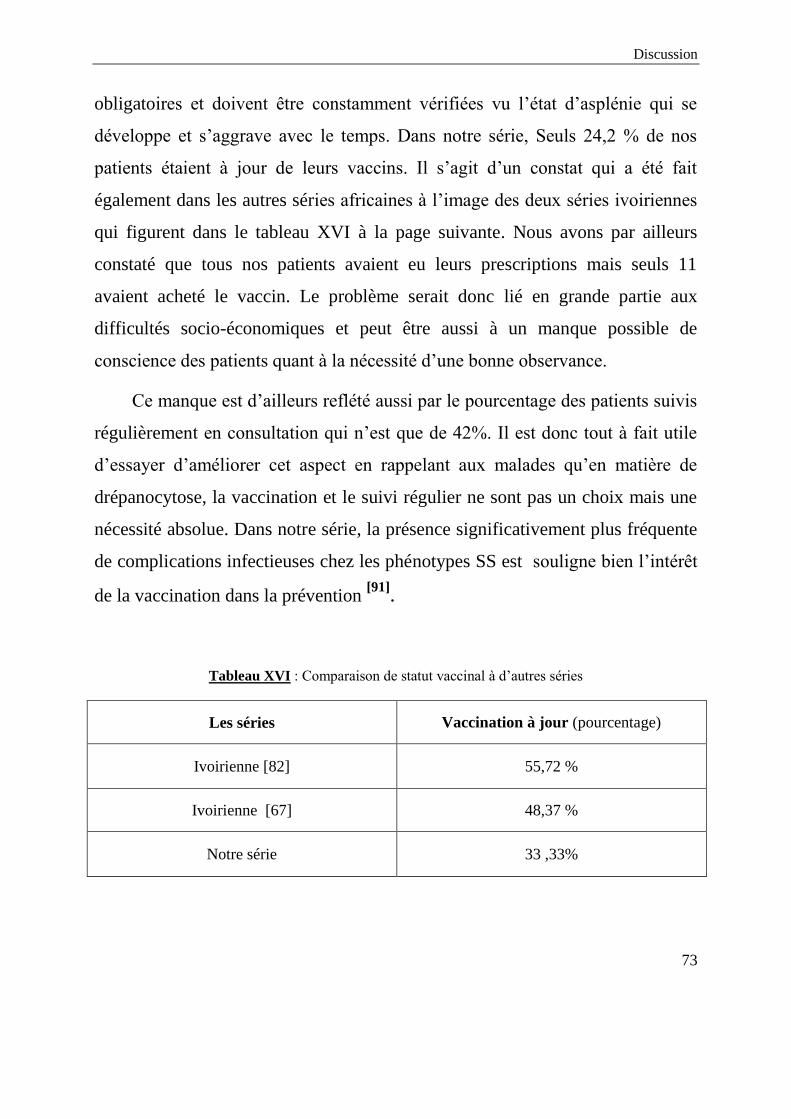
Discussion
73
obligatoires et doivent être constamment vérifiées vu l’état d’asplénie qui se
développe et s’aggrave avec le temps. Dans notre série, Seuls 24,2 % de nos
patients étaient à jour de leurs vaccins. Il s’agit d’un constat qui a été fait
également dans les autres séries africaines à l’image des deux séries ivoiriennes
qui figurent dans le tableau XVI à la page suivante. Nous avons par ailleurs
constaté que tous nos patients avaient eu leurs prescriptions mais seuls 11
avaient acheté le vaccin. Le problème serait donc lié en grande partie aux
difficultés socio-économiques et peut être aussi à un manque possible de
conscience des patients quant à la nécessité d’une bonne observance.
Ce manque est d’ailleurs reflété aussi par le pourcentage des patients suivis
régulièrement en consultation qui n’est que de 42%. Il est donc tout à fait utile
d’essayer d’améliorer cet aspect en rappelant aux malades qu’en matière de
drépanocytose, la vaccination et le suivi régulier ne sont pas un choix mais une
nécessité absolue. Dans notre série, la présence significativement plus fréquente
de complications infectieuses chez les phénotypes SS est souligne bien l’intérêt
de la vaccination dans la prévention [91]
.
Tableau XVI : Comparaison de statut vaccinal à d’autres séries
Les séries Vaccination à jour (pourcentage)
Ivoirienne [82] 55,72 %
Ivoirienne [67] 48,37 %
Notre série 33 ,33%

Discussion
74
La fréquence et la sévérité intolérables des crises étaient à l’origine de la
mise en route d’un traitement de fond par l’hydroxyurée chez 4 patients. Il s’agit
de la seule molécule ayant prouvé son efficacité à diminuer la fréquence des
crises douloureuses, des syndromes thoraciques aigus et des besoins
transfusionnels chez les patients drépanocytaires atteints d’une forme sévère. Il
existe toutefois une grande variabilité de la réponse au traitement, les enfants
répondant en règle mieux que les adultes. La tolérance à court et à moyen terme
est bonne. Une incertitude subsiste sur la fertilité ultérieure des garçons traités
jeunes et longtemps. L’hydroxyurée vient d’obtenir une autorisation de mise sur
le marché en Europe dans la drépanocytose sévère [97]
.
L’étude «Multicenter Study of Hydroxyurea (MSH)» [98]
a montré à travers
une comparaison versus placebo l’efficacité de l’hydroxyurée chez les patients
drépanocytaires adultes souffrant d’une forme sévère (au moins trois crises
douloureuses par an). Dans ce travail, le groupe des patients recevant
l’hydroxyurée a eu significativement moins de crises douloureuses (2,5 versus
4,5 crises par an, p < 0,001), moins de syndrome thoracique aigus (25 versus 51,
p < 0,001) et moins de besoins transfusionnels sur la durée de l’étude
(48 versus 73, p = 0,001).
D’autres approches dans le domaine de la génothérapie sont en étude avec
pour axes :
l’activation de gènes de compensation comme l’activation des gènes γ
de l’HbF

Discussion
75
la correction d’un gène muté par ciblage, réparation ou recombinaison
entre une séquence correctrice d’acide aminé et la région altérée d’un
gène, en l’occurrence celle du 6éme cordon du gène β de la globine
l’addition d’un gène de globine « anti drépanocytaire » dans le
patrimoine génétique des cellules souches hématopoïétiques, dont
l’expression doit survenir uniquement dans les érythroblastes, à un
niveau élevé.
Les thérapies par le monoxyde d’azote, inhibiteurs de l’adhésion des
globules rouges à l’endothélium sont aussi en cours d’essai [99]
. L’objectif
principal, celui de la guérison génétique après ou, si possible, avant l’apparition
des symptômes est maintenant nettement visible, au bout d’un tunnel long d’un
demi- siècle de progrès dans la compréhension de la maladie, la prise en charge
des patients et des thérapeutiques non spécifiques.

Conclusion
76
CONCLUSION

Conclusion
77
Notre étude nous permet de tirer les conclusions suivantes :
La répartition géographique de nos patients (93% autochtones) nous
montre bien que drépanocytose est une affection qui concerne le patient
marocain.
L’adulte drépanocytaire est sujet à des complications tant
infectieuses, ischémiques que des complications en rapport avec
l’anémie chronique.
L’ostéonécrose de la tête fémorale rend compte de l’importance de sa
recherche systématique chez tout patient au delà de 15ans par un bon
interrogatoire lors des consultations et une sensibilisation des patients
sur les examens radiologiques réguliers.
La lithiase biliaire constituait 24% des complications dans notre
série et apparaît aussi comme un élément devant faire l’objet de
dépistage régulier dans le suivi d’une drépanocytose.
L’étude analytique nous a confirmé la différence significative
attendue concernant l’influence du phénotype sur la fréquence des
complications infectieuses.
Enfin, le dernier point essentiel est sans doute la nécessité d’améliorer
le suivi et la sensibilisation des patients quant à l’importance capitale
de la vaccination régulière.

Résumés
78
RESUMES

Bibliographie-webographie
79
RESUME
La drépanocytose est la maladie génétique la plus répandue dans le monde.
Il s’agit d’une hémoglobinopathie qualitative due à la substitution du 6ème acide
aminé de la chaine β l’acide glutamique par la valine. Le Maroc est concerné
avec une prévalence de 1à 2%.
Notre travail a eu pour objectif, après une revue de la littérature, de mettre
en évidence les caractéristiques de l’adulte drépanocytaire par une étude
rétrospective descriptive et analytique chez 33 patients colligés au service de
médecine interne du CHU Ibn Sina.
Les phénotypes obtenus étaient SS dans 42,4 %, SC dans 15,1% et AS
dans 6,1% des cas. Les crises vaso-occlusives étaient le motif le plus fréquent
des hospitalisations (53 ,4%). Les complications chroniques ayant marqué
l’évolution chez nos patients étaient par ordre de fréquence décroissante la
lithiase biliaire, l’ulcère de jambe et l’ostéonécrose. Seuls un tiers des patients
avaient une vaccination à jour. Le phénotype SS était corrélé à une fréquence
plus élevée des complications infectieuses.
Ces différents résultats rendent compte de la gravité de la maladie et à
fortiori de la nécessité d’une prise en charge adéquate portant sur deux volets
essentiels à savoir, la gestion préventive , curative ,rapide et efficace des crises
et la mise en place d’un traitement de fond par hydroxyurée chez tout patient
avec une atteinte sévère incompatible avec une qualité de vie acceptable.

Bibliographie-webographie
80
ABSTRACT
The sickle cell disease is the genetic disease the most wide-spread in the
world. It is a qualitative hemoglobinopathy due to the replacement of the 6th
amino acid of the ß chain, the glutamic acid by the valine. It concerned Morocco
with a prevalence of 1à 2 %.
Our work had for objective, after a revision of the literature, to bring to
light the characteristics of the adult patients affected with sickle cell anemia by a
descriptive, retrospective and analytical study of about 33 patients brought
together from the service of internal medicine of the teaching hospital Ibn
Sina(CHU).
The phenotypes obtained were SS in 42, 4 %, SC in 15, 1 % and AS in 6, 1
% of the cases. The acute vaso-occlusive crisis was the most frequent motive for
the hospitalizations (53, 4%). The chronic complications which marked the
evolution of our patients were, in descending order of frequency the biliary
calculi, the ulcer of leg and the osteonecrosis .Only one- third of the patients
had a vaccination up to date. The phenotype SS was correlated with a higher
frequency of the infectious complications.
These various results report the gravity of the disease and deem it
necessary of an adequate care concerning two essential points namely, the fast
and effective preventive and curative management of the vaso-occlusive crisis
and the implementation of a thorough treatment by hydroxyurea to every patient
with a severe form of the disease and to whom an acceptable quality of life is
impossible.

Bibliographie-webographie
81
%
SS%SC%AS%
%
SS

Bibliographie-webographie
82
BIBLIOGRAPHIE - WEBOGRAPHIE

Bibliographie-webographie
83
[1] Bennett, Plum, Gill, Kokko, Mandell, Ockner, Smith .
Drépanocytose et hémoglobinopathies associées : Traité de médecine
interne Edition1997 ; p. 882
[2] Rapport de Organisation Mondiale de la Santé.
Cinquante-neuvième assemblée mondiale de la santé A59/9 point 11.4
de l’ordre du jour provisoire 24 avril 2006
[3] Marc Gentilini, Bernard Dufflo.
Anémie Tropicale. Médecine tropicale, Edition Flammarion
Médecine-sciences 1986 ; p.460
[4] XXXVIII congrès Français de Médecine, Beyrouth 1971.
Les anémies Hémolytiques. Edition Masson & Cie ;p 169
[5] www.chups.jussieu.fr
[6] Chebloune Y, Pagnier J, Trabuchet G, Faure C, Verdier G, Labie
D et al.
Structural analysis of the 5’flanking region of the [beta]-globingenein
African sickle cell anemia patients: further evidence for three origins
of the sickle cell mutation in Africa. Proc Natl Acad Sci USA 1988;
85: 4431-4435

Bibliographie-webographie
84
[7] Lapoumeroulie C, Dunda O, Ducrocq R, Trabuchet G, Mony-
Lobe M , Bodo JM et al.
A novel sickle cell mutation of yet another origin in Africa: the
Cameroon type.Hum Genet 1992; 89: 333-337
[8] Labie D, Srinivas R, Dunda O, Dode C, Lapoumeroulie C, Devi V
et al.
Haplotypes in tribal Indians bearing the sickle gene: evidence for the
unicentric origin of the beta S mutation and the unicentric origin of the
tribal populations of India.Hum Biol 1989 ; 61 : 479-49
[9] http://www.acguadeloupe.fr/Cati971/PEDAGO/SVT/pedagogique/
Ladrepanocytose.htm
[10] Hptt://globin.cse.psu.edu
[11] C Arnal, R. Girot.
Drépanocytose chez l’adulte.Encyclopédie Médicochirurgicale
hématologie 13-006-D-16
[12] Http:/upload.wikimedia.org/wikipedia/commons/9/92/sicklecell.jpg
[13] Gardos G.
The function of calcium in the perméability of human erytrocytes;
Biochim biophys acta 1958; 30:653-654

Bibliographie-webographie
85
[14] Larcher V.F, Wyke R.J, Davis L R, Stroud C.E, Williams R .
Defective yeast opsonisation and functional deficiency of complement
in sickle cell disease. Arch .Dis. Child.1982 , 57,343-346
[15] R. Girot, M. de Montalembert.
La drépanocytose chez l’enfant. Encyclopédie médicochirurgicale ;
2006 Elsevier SAS. . 4-080-A-20
[16] Emond AM ,Collins R , Darvills D , Higgs DR , Maude GH ,
Serjent GR .
Acute splenic sequestration in homozygous sickle cell disease: natural
history and Management. J Pediatr 1985. 107 : 201-6
[17] Thomas C , Lemerle S , Bernaudin F , Feingold J, Guilloud-
bataille M ,Reinert P.
Réseau de recherche clinique sur la drépanocytose .
Drépanocytose : étude de la mortalité pédiatrique en Ile –de – France
de 1985 à 1992. ;Archiv pédiatrique 1986 ; 3 : 445-51.
[18] Ballas SK .
Treatment of pain in adult with sickle cell disease.
Am J Hematolo 1990 ; 34 : 49-54

Bibliographie-webographie
86
[19] Georges JB, De Ceulaer K.
Actualités des manifestations rhumatologiques des hémo-
globinopathies.
Revue de Rhumatologie et Maladies Ostéoarticulaires 2003; 70:157-
61
[20] Saborio P, Scheinman JI. Sickle cell nephropathy.
J AmSoc Nephrol 1999 ; 10 : 187-192
[21] David HG, Bridgman SA, Davies SC, Hine AL, Emery RJ.
The shoulder in sickle-cell disease. J Bone Joint Surg Br 1993;75:538-
45.
[22] Hernigou P, Allain J, Bachir D, Galacteros F.
Abnormalities of the adult shoulder due to sickle cell osteonecroses
during childhood.
Rev Rhum Engl Ed 1998;65:27-32.
[23] Hernigou P, BernaudinF , Reinert P , Kuentz M , Vernant JP .
Bone marrow transplantation in sikcle cell disease; effect on
osteonecrosis.
J Bone Joint Surg ; 79-A , 1726-30
[24] Ph. Collet.
Manifestations ostéoarticulaires des anémies. Encyclopédie
médicochirurgicale.14-027-A-10

Bibliographie-webographie
87
[25] Saborio P, Scheinman JI. Sickle cell nephropathy.
J Am Soc Nephrol 1999 ; 10 : 187-192
[26] Sklar AH, Campbell H, Caruana RJ, Lightfoot BO, Gaier JG,
Milner P.
A population study of renal function in sickle cell anemia
Int J Artif Organs 1990 ; 13 : 231-236
[27] Scheinman JI.
Sickle cell nephropathy. In : Greenberg A ed. rimer on kidney
diseases. San Diego :
Academic Press, 1998 : 309-313
[28] Ataga KI, Orringer EP.
Renal abnormalities in sickle cell disease.
Am J Hematol 2000 ; 63 : 205-211
[29] Sklar AH, Perez JC, Harp RJ, Caruana RJ.
Acute renal failure
in sickle cell anemia. Int J Artif Organs 1990 ; 13 : 347-351
[30] Remy P. GalacterosF, Reins et hémoglobinopathies.
Encyclopedie medicochirurgicale . Nephropathie
Urologie. Paris :Elsevier,2001 : 18-053L-10.

Bibliographie-webographie
88
[31] Bhathena DB, Sondheimer JH.
The glomerulopathy of homozygous sickle hemoglobin (SS) disease:
morphology and pathogenesis.
J AmSoc Nephrol 1991; 1: 1241-1252
[32] Verani RR, Conley SB.
Sickle cell glomerulopathy with focal
segmentalglomerulosclerosis.
ChildNephrolUrol1991; 11: 206-208
[33] Roy S,MurphyWM,Pitcock JA, Rimer RL. Sickle-cell disease
and poststreptococcal acute glomerulonephritis.
Am J Clin Pathol 1976; 66: 986-990
[34] Iskandar SS, Morgann RG, Browning MC, Lorentz WB.
Membranoproliferative glomerulonephritis associated
with sickle cell disease in two siblings. Clin Nephrol 1991; 35: 47-51
[35] 35 DawkinsFW,KimKS, Squires RS, Chisholm R , Kark JÁ,
Perlin E
Cancer incidence rate and mortality rate in sickle cell
disease patients atHowardUniversity hospital:1986-1995.
Am J Hematol 1997; 55 : 18-192

Bibliographie-webographie
89
[36] Miller BA, Reis LA, Hankey BF, Kosary CS, Harras A, Devessa
SS et al.
Cancer statistics review: 1973-1990. National Cancer Institute,
National Institute of Health, 1993
[37] Avery RA, Harris JE, Davis CJ, Borgaonkar DS, Byrd JC, Weiss
RB.
Renal medullary carcinoma: clinical and therapeutic aspects of a newly
described tumor. Cancer 1996; 78: 128-132
[38] Platt OS, Brambilla DJ, Rosse WF, et al.
Mortality in sickcle cell disease .Life expectancy and risk factors for
early death.
N Engl J Med 1994; 330: 1639-44
[39] Vichinsky E, Neumayr L , Earles AN, Williams R, Lennette ET,
Dean D et al.
For the National Acute chest Syndrome Study Groupe.
Causes and outcomes of the acute chest syndrome in sickle cell
disease.
N Engl J Med 2000; 342: 1855-65
[40] Vichinsky EP, Williams R, Das M, Earles AN, Lewis N, Adler A et
al.
Pulmonary fat embolism: A distinct cause of severe acute chest
syndrome in sickle cell anemia. Blood 1994 ; 83 : 3107-3112

Bibliographie-webographie
90
[41] Melman A, Serels S . Priapism. Int J Impot Res 2000; S 133-9
[42] Condon PL , Serjeant GR.
Ocular finding in homozygous sickle cell anemia in Jamaica .Am J
Ophtalmologie 1972; 73: 533-43
[43] Balkaran B ,cha G , Morris JS Thomas PW, Serjeant BE, Serjeant
GR.
Stroke in a cohort of patients with sickcle cell disease.
J Pediatr 1992 ; 120: 350- 6
[44] Ohene-Frempong K, Weiner SJ, Sleeper LA, Miller ST,
Embury S, Moohr JW et al. Cerebrovascular accidents in sickle cell
disease: rates and risk factors. Blood 1998 ; 91 : 288-294
[45] Robert Girot, Pierre Begué, Fréderic Galacteros
La drépanocytose ; diagnostic biologique des syndromes
drépanocytaires
Edition 2003 ; P 13-17
[46] Pierre Bégué.
La maladie drépanocytaire.Edition 1984 ;
[47] Henri Wajcman.
Diagnostic et dépistage de la drépanocytose
Revue du praticien 2004 ; 54 :1543-7

Bibliographie-webographie
91
[48] Ah image bank (2001) ; doi ;10.1182/ashimage bank-2001-100248
[49] Altwater J , Schwartz I.R. , Erslev A. J. , Montgomery T. D. et
Tocantins L. M.
Sickcling of erythrocytes in a patient with thalassemia-hemoglobin I
disease.New England Journal of medicine 1960, 263
[50] Lie –Injo Hemoglobin bart’s and the sickling phenomena.
Nature (Lond.)1961; 191 :13-14
[51] Basset P, Beuzard Y, Garel MC, Rosa J .
The isoelectric focusing of human hemoglobins and it’s applications to
screenig to the Caracterization of seventy variants, and to study of
modified fractions of normal Hemoglobins. Blood 1978; 51:971-82
[52] R. Girot, M. de Montalembert Encyclopedie Medicochirurgicale; la
drépanocytose chez l’enfant ¶ 4-080-A-20
[53] Serrie A, Langlade A.
Les analgésiques périphériques: utilisations en pratique quotidienne.
In: Serrie A, Thurel C, eds, La douleur en pratique quotidienne :
diagnostic et traitements. Paris : Arnette ,1994 :65-80
[54] D. Bachir,M. Bonnet Gajdos ,F. Galacteros.
La transfusion dans la drépanocytose.
La presse médical 1990 , volume 19 (35) : 1627-1631

Bibliographie-webographie
92
[55] Ohene- Frempong K,
Indications for red cells transfusion in sickle cell disease.
Semin Hematol 2001;38: 5-13
[56] A. Habibi, B. Godeau, F. Galacteros.
Drépanocytose et réanimation
Elsevier Masson. Réanimation 16 (2007) 310–317
[57] Robert Girot, Pierre Begué, Fréderic Galacteros.
La drépanocytose ; l’infection chez l’adulte .Edition 2003 ; P 121
[58] P. Begue, Castello-Herbreteau.
La drépanocytose : de l’enfant à l’adolescent, prise en charge en 2001.
Bull Soc Pathol exot 2001, volume 94 (2) :85-89
[59] A.D G badoe, N.Kampatibe, B. Bakonde, J. K. Assimadi, K. Kessie
Attitude thérapeutiques chez lz drepanocytaire en phase critique et
intercritique au Togo . Medecine d’Afrique noire 1998,volume 45
(3) :154-160
[60] Diagne I , N.D.R Diagne –Gueye , H.Signate –Sy ,B. Camara Ph.
Lopes –Shall , A. Diack-Mbaye et al.
Prise en charge de la drépanocytose chez l’enfant en Afrique :
expérience de la cohorte de L’hôpital d’enfants Albert Robert de
Dakar. Médecine tropical 2003 ; 63 : 53-520

Bibliographie-webographie
93
[61] Chara+che S,Terri ML, More RD , et al.,
And the investigators of the Multicnter Study of Hydroxyurea in sickle
Cell Anemia. Effect of hydroxyurea on the frequency of painful crises
in sickle cell anemia.
N Engl J Med 1995; 322: 1317-22
[62] Streinberg MH , Barton F , Castro O ,Ramirez G, Bellvue R ,
Terrin M ,
Multicenter Study of Hydroxyurea in sickle Cell Anemia.
Hydroxyurea is associated with reduced mortality in adult with sickle
cell anemia .Blood 2000 ; 96: 485a
[63] Cokies VP, Smith RD, Belestin-Cokies BB, Njoroge JM, Miller JL,
Gladwin MT et al Hydroxyurea induces fetal haemoglobin by the
nitric oxide dependant activation of the soluble activation of the
guanylate cyclise. J Clin Invest 2003, volume 111(2):231-239
[64] Bernaudin F.
Résultats et indications actuelles de l’allogreffe de moelle dans la
drépanocytose. Pathol Biol 1999 ;47 : 59-64
[65] Walters MC, Storb R, Patience M, Leisenring W, Taylor
T,Sanders JE et al.
Impact of bone marrow transplantation for symptomatic sickle cell
disease: an interim report. Multicenter investigation of bone marrow
transplantation for sickle cell disease. Blood 2000 ; 95 :1918-1924

Bibliographie-webographie
94
[66] www.med.univ-
angers.fr/discipline/lab_hema/PATHOL2007/ERYTHR/11drepano
[67] Tuo M.
Prévalence des complications ischémiques au cours de la
drépanocytose. Thèse de médecine. Faculté de médecine d’Abidjan :
2003 ; n°3522.
[68] Bertrand E, Chauvet J, Lebras M, Remanbot J, Odi AM, Beda B
et al.
Les signes cardiaques dans la drépanocytose de l’adulte à propos de
111 cas homozygotes ou hétérozygotes. Cardiol Trop 1975 ; 1 : 63-70
[69] Lahlou H.
La drépanocytose au Maroc à travers l’activité du laboratoire
d’hématologie du CHU Ibn Sina. Thèse en médecine. Faculté de
médecine et de pharmacie de Rabat. 2003 ; n°14
[70] Pierre Bégué .
La maladie drépanocytaire .Sandoz édition 1984
[71] Reinert Ph, Doppelt E, Bernaudin F, Lemerle S, Zelensky A.
Drépanocytose chez l’enfant. Sem Hop Paris ,1991 ; 67, 24 ; 1105-
1110

Bibliographie-webographie
95
[72] Dan Nouhou B.
Diagnostic et suivi des malades drépanocytaires à l’hôpital national de
Niamey. Thèse de médecine. Faculté de médecine et de pharmacie de
Rabat : 2005 ; 33
[73] Tabili J, et al.
Etude de la consanguinité dans la population marocaine. Impact sur le
profil de la santé. 2007. Antropo, 15, 1-11. www.didac.ehu.es/antropo
[74] Naoum Salamé .
Donnée moléculaire de la recherche à l’enseignement
WWW.cndp.fr/archivage/valid/10856-2367-2491.pdf
[75] Drépanocytose .Hématologie édition Flammarion science-
médecine 1976 p 841
[76] http://www.rofsed.fr/media/image2__047278200_1446_24042006.j
pg
[77] C. Le Turdu-Chicot, L. Foucan, M. Etienne-Julanl, Y. Leborgne-
Samuel, R. Fanhan, C.
Berchel Analyse des hospitalisations chez les patients drépanocytaires
adultes en Guadeloupe .Rev Méd Interne 2000 : 21 : 24-9
[78] Brozovic M, Davies SC, Brownell AI.
Acute admissions of patients with sickle cell disease who live in
Britain. Br Med J 1987 ; 294 : 1206-8.

Bibliographie-webographie
96
[79] Pollack CV, Jorden RC. Kolb JC.
Usefulness of empiric chest radiography and urinalysis testing in
adults with acute sickle cell pain crisis. Ann Emerg Med 1991; 20 : 12
10-4.
[80] Serjeant GR. Ceulaer CDE. Lethbridpe R. Morris J, Singhal A,
ThomasPW.
The painful crisis of homozygous sickle cell disease: clinical features.
Br Med J 1994; 87: 586-91.
[81] S. Diop, S.O. Mokono, M. Ndiaye, A.O. Touré Fall, D. Thiam, L.
Diakhaté.
La drépanocytose homozygote après l’âge de 20 ans: suivi d’une
cohorte de 108 patients au CHU de Dakar .Rev Méd Interne 2003 ;
24 :711-715
[82] K.Bakary
Profil hématologique de la drépanocytose homozygote en Cote
d’Ivoire. Expérience du service d’hématologie clinique du CHU de
Yopougon.
Thèse de médecine. Faculté de médecine et de pharmacie d’Abidjan.
2006 n°4305.
[83] Platt O, Branbilla DJ, Rosse WF, Milner PF, Castro O, Steinberg
MH, et al.
Mortality in sickle cell disease. Life expectancy and risk factors for
early death. N Engl J Med 1994 ; 330 : 1639-44.

Bibliographie-webographie
97
[84] B. Godeau, V. Noël, A. Habibi, A. Schaeffer, D. Bachir, F.
Galactéros
La drépanocytose chez l’adulte : quelles urgences pour l’interniste ?
Rev Méd Interne 2001 ; 22 : 440-51
[85] Serjeant .G.R
Natural History and determinant of clinical severity of sickle cell
disease Curr.Opin .Hematol, 1995, 2,2, 103-108
[86] Elira Dokekias Alexis et Nzingoula S ;
Profil clinique du sujet drépanocytaire après l’âge de 30 ans. Tunisie
médicale 2003 ; 81 ;2 :101-108
[87] A. Véronique.
Profil de la drépanocytose double hétérozygote SC. Expérience du
service d’hématologie clinique du CHU de Yopougon à propos de 80
cas.
Thèse de médecine. Faculté de médecine et de pharmacie d’Abidjan.
2005 n°4071.
[88] K. Homawoo, K. Bissang, B. Songne, A. Ayite.
Drépanocytose et ostéonécrose de la tête fémorale .considérations
thérapeutiques à propos de 38cas. Médécine d’Afrique Noire : 1991;38

Bibliographie-webographie
98
[89] I .M .Muyawa .
La nécrose aseptique de la tête fémorale d l’adulte au CHU de Lomé.
Etude prospective à propos de 20 observations. Thèse de médecine.
Lomé
[90] Tobossi C .
A propos de 16 cas d’ostéonécrose de la tete fémorale chez les
drépanocytaires observés au Centre National Hospitalier et
Universitaire de cotonou. Thèse de médecine. Faculté de médecine de
Montpellier ; 1979
[91] S. Diop , G. Koffi , E. N’Dahtz , O. Allangba , M. A. Aka Adjo , I.
Sanogo et A. Sangaré.
Profil infectieux chez le drépanocytaire. « clinique » 1997
[92] Anand AJ, Glatt AE.
Salmonella osteomyelitis and arthritis in sicklecell disease. Semin
Arthritis Rheum 1994;24:211-21.
[93] Chambers JB, Forsythe DA, Bertand SL, Iwinski HJ, Steflik DE.
Retrospective review of osteoarticular infections in a pediatric sickle
cell age group. J Pediatr Orthop 2000;20:682-5.
[94] E.Nacoulma, J. Sakande, E. Kadanfo, E.Kpowbié, I.P. Guissou
Profil hématologique et biochimique des drépanocytaires SS et SC en
phase stationnaire au centre Hospitalier National Yalgado Ouedraogo
de Ouagadougou. Mali médical 2006 ,XXI , n°1

Bibliographie-webographie
99
[95] De montalembert M, Guillou bataille M, Feingold J , Girot.
Epidemiological and clinical study of sickle cell disease in France,
Guyane and Algeria . Eur. J. Hematol. 1993 : 51; 136-140
[96] Diagne I et al.
Les syndromes drépanocytaires majeurs en pédiatrie à Dakar. Arch.
Pediatr. 2000. 7. 16-24
[97] De montalembert M,
Traitement des patients drépanocytaires par hydroxyurée: efficacité et
tolérance, Transfusion Clinique et Biologique (2008),
doi:10.1016/j.tracli.2008.03.010
[98] Charache S, Terrin ML, Moore RD, Dover GJ, Barton FB, Eckert
SV, et al.
Effect of hydroxyurea on the frequency of painful crises in sickle cell
anemia.
N Engl J Med 1995; 332:1317–22
[99] Robert Girot, Pierre Begué, Fréderic Galacteros
La drépanocytose ; Vers de nouvelle thérapeutiques spécifiques
Edition 2003 ; P 31-40

AAuu mmoommeenntt dd''êêttrree aaddmmiiss àà ddeevveenniirr mmeemmbbrree ddee llaa pprrooffeessssiioonn mmééddiiccaallee,, jjee
mm''eennggaaggee ssoolleennnneelllleemmeenntt àà ccoonnssaaccrreerr mmaa vviiee aauu sseerrvviiccee ddee ll''hhuummaanniittéé..
JJee ttrraaiitteerraaii mmeess mmaaîîttrreess aavveecc llee rreessppeecctt eett llaa rreeccoonnnnaaiissssaannccee qquuii lleeuurr
ssoonntt dduuss..
JJee pprraattiiqquueerraaii mmaa pprrooffeessssiioonn aavveecc ccoonnsscciieennccee eett ddiiggnniittéé.. LLaa ssaannttéé ddee mmeess
mmaallaaddeess sseerraa mmoonn pprreemmiieerr bbuutt..
JJee nnee ttrraahhiirraaii ppaass lleess sseeccrreettss qquuii mmee sseerroonntt ccoonnffiiééss..
JJee mmaaiinnttiieennddrraaii ppaarr ttoouuss lleess mmooyyeennss eenn mmoonn ppoouuvvooiirr ll''hhoonnnneeuurr eett lleess
nnoobblleess ttrraaddiittiioonnss ddee llaa pprrooffeessssiioonn mmééddiiccaallee..
LLeess mmééddeecciinnss sseerroonntt mmeess ffrrèèrreess..
AAuuccuunnee ccoonnssiiddéérraattiioonn ddee rreelliiggiioonn,, ddee nnaattiioonnaalliittéé,, ddee rraaccee,, aauuccuunnee
ccoonnssiiddéérraattiioonn ppoolliittiiqquuee eett ssoocciiaallee nnee ss''iinntteerrppoosseerraa eennttrree mmoonn ddeevvooiirr eett
mmoonn ppaattiieenntt..
JJee mmaaiinnttiieennddrraaii llee rreessppeecctt ddee llaa vviiee hhuummaaiinnee ddééss llaa ccoonncceeppttiioonn..
MMêêmmee ssoouuss llaa mmeennaaccee,, jjee nn''uusseerraaii ppaass ddee mmeess ccoonnnnaaiissssaanncceess mmééddiiccaalleess
dd''uunnee ffaaççoonn ccoonnttrraaiirree aauuxx llooiiss ddee ll''hhuummaanniittéé..
JJee mm''yy eennggaaggee lliibbrreemmeenntt eett ssuurr mmoonn hhoonnnneeuurr..
Serment d'Hippocrate


-
2008 145
–––





























