Embed Size (px)
Citation preview

0002-9 270/93/8803-O436$()3,00/0Tiih AMIRICAN Joi HNAI. OF- GASTROENTEROI-(X;Y
Copyright c, 1993 by Am, Coll, of GasirocnterologyVol-8K. No, 3, 1993
PriniL-d in U,S,A,
Angioedema Presenting As Chronic Gastrointestinal Symptoms
Stephen L. Eck, M.D., Ph.D.. James H. Morse, M.D., David A. Janssen, M.D..Stephen G. Kmerson, M.D., Ph.D., and David M. Markovitz, M.D.
Department of Internal Medicine. Univcrsily ot Miclii.i;an Medical Cenlcr. Ann Arbor. Michigan
Gastrointestinal complaints may be the presentingfeature of patients with aequired or hereditary angioe-dema. We descrihe two patients with episodie nausea,abdominal pain, and cramping secondary to Cl inhibitordeficiency. In one patient, an acquired deficiency aroseas a paraneoplastic syndrome with abdominal com-plaints preceding the diagnosis of an occult lymphoma.Ihe second patient presented at age 61 with ahdominalcomplaints secondary to a hereditary deficiency of Clinhibitor. The patients' symptoms were due to gastroin-testinal angioedema, resulting from episodic unregu-lated complement activation. The biochemical mecha-nism of tbis unusual syndrome and its diagnostic im-portance are discussed. A CI inhibitor deficiency sbouldbe considered in patients witb unexplained abdominalsymptoms suggestive of intestinal pseudo-obstruction.
INTRODUCTION
Intestinal pseudo-obstruction may occur as a primaryidiopathic disorder, or secondarily as a result of anunderlying illness, including neoplastic, rheumatologic.endocrine, neurologic, and other diseases {I). Intestinalpseudo-obstruction often presents as a recurrent illnesswith symptoms ofobstruction of the intestines (crampyabdominal pain and distention. nausea, and vomiting)without evidence of mechanical obstruction. We de-scribe two patients with a Cl complement inhibitordeficiency causing angioedema who had recurrent epi-sodes of intestinal pseudo-obstruction as their primarymanifestation of this disorder. In one case, an under-lying lymphoma was identified as the cause of theangioedema. and in the second, case a hereditar\' defi-ciency of Cl inhibitor caused the angioedema.
CASE REPORTS
Paiienl 1
A previously healthy 28-yr-old man presented onSeptember 21. 1978, with a fever to 39.5''C. chills, andprofound fatigue, and was found to have cervicallymphadcnopathy and a palpably enlarged liver and
Received . IS. 2: ticccptcd Nov. 9, 1992.
spleen. A heterophile test was negative, as was a throateuiture for streptococcal pharyngitis. Over the followingyear, his spleen remained palpably enlarged, and radio-nuclide scanning documented splenomegaly. This wasattributed to the residua of mononucleosis. In June1980, the patient developed recurrent episodes of ab-dominal bloating, nausea, and post-prandial erampyabdominal pain. These symptoms persisted intermit-tently over the next 10 yr. The patient underwentextensive evaluations ineluding two esophagogastro-duodenoscopies (EGD). abdominal ultrasound, tworadionuclide scans of the liver and spleen, three eolon-oscopies. an ERCP. proctoscopy. and multiple stoolevaluations for occult blood, ova, and parasites. Thesetests were unrevealing. except for the persistent findingof splenomegaly. At different times the patient wasdiagnosed as having irritable bowel syndrome, inflam-matory bowel disease (based on the finding of lymphoidnodular hyperplasia of the sigmoid colon on one colon-oscopy). and a somatization disorder. In June 1990, thepatient developed pain and swelling of the left ankle.which resolved after easting for a presumed stress frac-ture. Similar swelling appeared transiently in the rightankle a few weeks later. In July 1990. he developedtransient arthralgias and non-pitting edema of the rightwrist and. over the next year, he continued to havemigratory arthralgias with concomitant, transient, non-pitting edema involving the wrists and ankles. His CBCwas normal on multiple occasions. He had no furtherepisodes of fever since 1978. and he denied chills, nightsweats, or significant weight changes over the preceding13 yr.
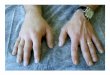
A CT scan of the abdomen, performed in July 1991,showed a moderately enlarged spleen, increased in sizefrom Eebruary 1989. and new nodular densities in thesplenic hilum suggestive of adenopathy (Fig. 1). He wasthen referred to our medical center. Physical examina-tion revealed a palpably enlarged, non-tender spleen,and non-pitting edema of the wrist, hand, ankle, andscrotum. Non-blanching, pigmented, macuiar skin le-sions with "petechial" spots on the lower legs, clinicallydiagnosed as Schamberg's pigmented purpura. werebiopsied and revealed atypical lymphoeytes surround-ing and infiltrating dermal vessels with localized edema
436

Mareh 1993 Cl INHIBITOR DHKICIENCY & GASTROINTESTINAL SYMPTOMS 437
FIG, 1. Computed tomographic scan of the abdomen (patientshowing enlarged spleen (*) and perisplenic adcnopalhy /arrow).subsequently confirmed at laparotomy.
Fi(.i. 2. Photomicrographs of hematoxylin and cosin-stainedsplenic tissue (patient 1) showing multiple foci of lymphoma (arrow)
and microhcmonhage. The patient was found to havedecreased Cl inhibitor (1.9 mg/dl. normal 8-24 mg/dl). Clq (5 mg/dl, normal 7-15) and C4 {8 mg/dl.normal 10-46 mg/dl) levels with normal C3 levels.confirming the diagnosis of angioedema. His CBC.ANA. ENA. and rheumatoid factors al! were normal.Serum prolein electrophoresis revealed an IgM mono-clonal gammopathy (IgM 746 mg/dl. normal 21-393).with normal IgG and IgA levels. In addition, his angio-tensin-converting enzyme was elevated (67.6 U/ml.normal 20-50 U/ml).
Splenectomy (l500-g spleen) and bone marrow bi-opsy revealed extensive involvement of these tissueswith a low grade follicular lymphoma (Fig. 2). Splenicartery and splenic hilar lymph nodes also were involved:however, multiple liver biopsies were negative for lym-phoma. In the postoperative period, the patient's symp-toms of fatigue, arthralgias. nausea, bloating, and ab-dominal pain resolved, as did his peripheral and scrotaledema, and they have remained absent for more than
1 yr. His serum complement levels have remained low,but his Cl inhibitor and atigiotensin-converting en-zyme levels returned to normal (8 mg/dl units and 43.7U/ml. respectively) 2 wk after splenectomy. Fifteenmonths after splenectomy. the patient was found tohave a large cell lymphoma (Richter's syndrome) withpulmonary involvement., but without symptoms of an-gioedema. He subsequently began systemic chemother-apy.
Patient 2A 61-yr-old man was admitted to the hospital with
complaints of abdominal cramping and distention.nausea, vomiting, and anorexia. He had been in goodhealth until 6 months prior to admission, when hedeveloped abdominal eramps. occurring in four self-limited episodes, each of successively greater intensityand characterized by a crescendo pattern. Colonoscopicand abdominal ultrasound examinations were per-formed and were unremarkable. In the three days priorto admission, his abdominal cramping became verysevere, with assoeiated nausea, vomiting, anorexia, andabdominal bloating. He noted that his daily bowelmovements were "loose" and that he had significantflatus. Melena. hematochezia. and hematemesis wereabsent. The patient reported a history of migratoryedema for several months, with episodic swelling of hisextremities and scrotum.
Physical examination was remarkable for orthostaticblood pressure changes and a soft, non-tender abdomenthat was tympanitic to pereussion. with high-pitchedtinkles on auscultation. No peripheral or scrotal edemawas present. Serum chemistries and a complete bloodcount were normal. Stool evaluation for occult bloodwas negative. An abdominal roentgenogram revealedmultiple dilated loops of small bowel with regular mu-cosal thickening. The patient was treated with bowelrest, nasogastric suction, and intravenous fluids. Anupper gastrointestinal series performed the day afteradmission revealed localized "coin stacking" in thejejunum, compatible with angioedema (Fig. 3). By thesecond hospital day. the patient's condition had im-proved and his diet was gradually reinstituted. Serumcomplement studies obtained shortly after admissionrevealed decreased levels of C4 (7.9 mg/dl; 11.5-50mg/dl normal). CH50 (20 kU/ml: 22-84 kU/ml nor-mal), and CI inhibitor (6.8 mg/dl; 8-24 mg/dl normal),while the serum Clq and C3 levels were normal. Sub-sequent small bowel follow-through and enteroclysisstudies were normal. The patient was discharged on thelOth hospital day. His symptoms returned approxi-mately 1 month later, with small bowel dilation and airfluid levels noted on abdominal roentgenogram studies.He was begun on danazol for the treatment of the

438 ECK el al. Vol.S,S.No.3. 1993
ci. 3. Abdominal film with barium contrast showing locali<redmucosal thickening of the small bowel ["coin-stacking" (arrow)]during an acute episode of abdominal pain (patient 2),
angioedema, and has had no further episodes of gas-trointestinal distress in over 2 yr of follow-up.
DISCUSSION
Angioedema is often unrecognized, although boththe acquired and the hereditary forms are well described(2). Acquired angioedema most often appears as aparaneoplastic syndrome associated with a variety oflymphoproliferative (2-6) and other neoplastic (7-9)and autoimmune disorders (10, II). It is most com-monly associated with lymphomas. multiple myeloma.Waldenstrom's maeroglobulinemia. and ehronic lym-phocytic leukemia. This condition results from anti-idiotype antibodies directed against a monoclonal par-aprotein (type I) (2). The resultant immune complexesbind the Cl eomplex (composed ofClq. Clr. and Cls)through the Clq subunit. leading to sequential activa-tion of Clr and Cls. Cls. when activated, is able tocleave C2 and C4. This loads to the depletion of C2,C4, and Cl inhibitor and the release of vasoactivepeptides. Normal serum levels of Cl inhibitor preventthe conversion of inactive Cl to its active form and thesubsequent activation of the complement system. Theabsenee of the inhibitor, whether acquired or hereditary(a dominant trait with irregular penetranee). results inthe continued activity of Clr and Cls. leading to eon-tinued complement activation and depletion of C2 andC4 (12). A portion of the C2 molecule is believed toaccount for the increased vascular permeability (13)which results in tissue edema in diverse loeations. Bothacquired and hereditary angioedema can have a varietyof manifestations, including malaise, ailhralgias.crampy abdominal pain, diarrhea, laryngeal edema,and non-pitting edema of the extremities, scrotum, andface. The episodic submucosal and subcutaneousedema which results during attacks of angioedema wasdescribed by William Osier in 1888 (14). He noted thatgastrointestinal symptoms were almost invariably pres-ent in patients with hereditary angioedema, a feature
that has not been fully appreciated in subsequent re-ports of acquired angioedema. Patients with acquiredangioedema may have symptoms for many years priorto the diagnosis of an underlying malignancy (3), orsymptoms may appear once therapy for the malignancyhas been initiated (15). In rare instances, angioedemaresults from an acquired anti-Cl inhibitor antibody(type II) (16).
In the first case, it is highly likely that the oceurrenceof the symptoms of angioedema coincided with thedevelopment of the lymphoma. The patient's gastroin-testinal complaints had been documented for 11 yr.and the joint swelling had been present intermittentlyfor 11/? yr. Splenomegaly had been intermittently notedover a i3-yr period by physieal examination, radio-nuelide scanning, and computed tomography. Hissymptoms of angioedema were manifested as peripheraland scrotal edema, Schamberg's pigmented purpura(17) of the lower extremities (secondary to repeatedepisodes of edema), nausea, abdominal bloating, andabdominal pain. His abdominal symptoms predatedthe other manifestations by approximately 10 yr. prob-ably resulting from recurrent episodes of localized sub-mucosal edema which occurred as a result of comple-ment activation. In a retrospective analysis, the inei-dence of acquired angioedema was reported to be lessthan one per thousand cases o\' lymphoproliferativedisorders (2).
The second patient was strongly suspected of havingthe hereditary form of angioedema because he had anormal serum protein electrophcresis and a normalClq level (wV/c infra). Furthermore, no predisposingdisorder has been identified in more than 2 yr of follow-up. The absence o fa family history of angioedema isnot inconsistent with this conclusion, given its variablepenetranee (25% of individuals with hereditary angioe-dema have no family history of the disease).
The rarity of the identification of angioedema maybe due. in part, to lack of recognition of its multiplemanifestations. The diagnosis of angioedema should beconsidered in patients whose symptoms can be attrib-uted to episodes of localized edema, including edemaof the small intestine. Several laboratory tests may behelpful in establishing this diagnosis. Serum levels ofC4, C2. and Cl inhibitor are typically decreased,whereas C3 levels are nortnal. Serum protein electro-phoresis reveals an M component in most reportedcases of acquired angioedema associated with lympho-proiiferative and autoimmune disorders, Clq levels canditTerentiate the acquired form (low levels), from theinherited form (normal levels) (6), In addition, thefamily history may be helpful in establishing the pres-enee of the inherited form; however, a negative familyhistory does not exclude it. The regular thickening ofsmall bowel folds seen on abdominal films ("coin stack-

March 1993 Cl INHIBITOR DEFICIENCY & GASTROINTESTINAL SYMPTOMS 439
ing") tends to be more localized in angioedema than ingeneral intestinal edema due to other causes (18). Theradiographic findings are entirely reversible and areseen only during an acute episode. Both the small andlarge intestine may be involved, including jejunum,ileum. duodenum, stomach, and colon in descendingfrequency of involvement.
Clinieians should consider angioedema as an under-lying process in eases of unexplained chronie abdomi-nal pain. The multiple, transient, and often subtlemanifestations of angioedema may make diagnosis dif-ficult if the clinician is not appropriately suspicious.Gastrointestinal symptoms may predate the develop-ment of cutaneous or respiratory symptoms by manyyears, or may be the only lifelong manifestation inpatients with the hereditary form of angioedema (19).Indeed, the first patient was seen by several physiciansover a period of many years before this diagnosis wasentertained. Once the diagnosis of angioedema is estab-lished, an oecult lymphoproliferative disease should beseriously considered. The clinical significance of ac-quired angioedema. as it relates to the prognosis of theunderlying disorder, is unknown. It may. however,provide a clue to an occult malignancy and thus leadto an earlier diagnosis and treatment of the underlyingprocess and the symptoms arising from angioedema.
Reprint requests and correspondence: Stephen L, lick,. M,D.. TheInstitute for Human Cienc Therapy. The Wistar Institute. Room 204.36th and Spruee Streets. Philadelphia. PA 19104-4268.
REFERENCES
1, Faulk DI-, Anuras S. Christensen J. Chronie intestinal pseudo-obstruction, Gastroenterology 1978;74:922-31.
2, Geha RS, Quinti I. Austen KF. et al. Acquired Cl-inhihitordeficiency associated with antiidiotypic antibody to monoclonalimmunoglobuhns, N EnglJ Med 19S5:.'ll2:534-40,
3, Gottlieb M, Campbell K. Pelzmann K. Hu C-H, Long-standing
angioedema with Cl esterase inhibitor defieieney associated withoccuh Ivmphoma, West J Med 1983:1 38:258-60,
4. Schreiber AD. Zweiman B. Atkins P. et, al, Aequired angioedemawith lyniphoproliTerative disorder: Assoeiation ot Cl inhibitordeficiency with cellular abnormality. Blood 1976:48:567-80.
5. Hauptmann G. L.ang J-M. Notih M-L. et a!. Acquired Cl-inhibitor deficiency in lymphoproliferative diseases with serumimmunoglobulin abnormalities, Blut 1976:32:195-206,
6. Gelfand JA. Boss GR. Conley CL. et al. Aequired Cl esteraseinhibitor deficiency and angioedema: A review. Medicine1979:58:321-8.
7. Nilsen A. Matre R. Aequired angioedema and hypoeomplemen-temia in a patient with myelofibrosis. Aeta Med Seand1980:207:123-5.
8. Cohen SH. Koethe SM. Kozen F. et al. Aequired angioedemaassoeiated with rectal carcinoma and its response to danazoltherapy. Acquired angioedema treated with dana/ol, J AllergyClin Immunol 1978:62:217-21,
9. Ross TF. Coleman DL. Naughton JL. Angioedema and small-cell carcinoma of the lung, thorax 1982:37:950-1,
10, Wallington TB, Acquired Cl esterase deficiency with cold agglu-tinin disease assoeiated with monoelonal IgM antihodies, Br JDermatol 1978:99(suppl 16):39-40.
11, Costan/i JJ. Coltman CA. Donaldson VH, Aetivation of com-plement by a monoclonal cr>oglobulin associated with coldUrticaria, J Lab Clin Med 1969:74:902-10,
12, Rosse WF. Clinical immunohematology: Basie eoneepts andclinical applications. 1st ed. Boston: Blackwell Seientifie Publi-cations, 1990:72-7,
13, Donaldson VH. RatnofTOD. Diasda Silva W. et al. Permeability-increasing activity in hereditary angioncurotic edema plasma, II,Mechanism of formation and partial characterization, J ClinInvest 1969:48:642-53.
14, Osier W, Hereditary angioneurotic edema. Am J Med Sci1888:95:362-7.
15, Beretta KR. Spath PJ. Pedra//ini A. et al, Angioixiem durcherworbenen Komplement-Cl-lnhibitor-Mangel bei einer Patien-tin mit Non-Hodgkin-Lymphom und autoimmun-hamolytischerAnamie, Sehwei/ Med Woehensehr 1991:121:943-7,
16, Spath PJ. Wuthrich B. Matter L. et al, Aequired angioedema andanti-Cl-inhibitor autoantibody. Arch Intern Med1989:149:1213-4,
17, Moschella SL. Hurley HJ. eds. Dermatology, vol 1. Philadelphia:W,B, Saunders. 1985:1071-3,
18, Pearson KD. Buchignani JS. Shimkin PM. et al. Hereditary-angioedema of the gastrointestmal traet, AJR 1972:116:256-61
19, Weinstoek LB. Kothain T. Sharma RN. et al. Recurrent abdom-inal pain as the sole manifestation of hereditan,' angioedema inmultiple family members. Gastroenterology 1987:93:11 16-8,