Embed Size (px)
Citation preview

Annu. Rev. Phys. Chern. 1993. 44: 115-44 Copyright © 1993 by Annual Reviews Inc. All rights reserved
ANDE]�SON LOCALIZATION AND THE E�(CEPTIONS
Philip Philli,7Js*
Department of Chemistry, Massachusetts Institute of Technology, Cambridge, Massachusetts 02 1 39
KEY WORDS: disorder, randomness
INTRODUCTION
A problem that has always been central to solid-state physics is the insulator-metal transition. The question one is attempting to answer here is why do some materials conduct and others do not. Although this problem is simple to state, it wasn't until the advent of quantum mechanics in the 1 920s that a reasonably satisfactory qualitative answer to this question was put forth" Currently, two standard models appear to capture the physics and chemistry of the insulator-metal transition: one by Mott (I), the other by Anderson (2). Mott's mechanism emphasizes the role of electron-electron interactions, whereas the Anderson transition obtains entirely from static disorder. This review focuses solely on recent advances (3-1 84) on the static disordered model pioneered by Anderson in the context of the insulator-metal transition. In particular, this review covers primarily the advances since the extensive 1 985 review on disordered systems by Lee & Ramakrishnan (3).
Anderson's original model grew out of a series of electron spin resonance experiments by Feher et al (4) on donor impurities (P, As, etc.) in Si. In the concentration regime below the formation of an impurity band, Feher et al noticed that the electron spin on each 31p nucleus retained its characteristic frequency on a time scale ranging from seconds to minutes (4). A simple golden rule calculation based on the exchange interaction between 31 P spilns predicted, however, that the expected lifetime should be
* Current address: Department of Physics, University of Illinois, Urbana, Illinois 61801.
1 1 5 0066-426XJ93Jl lOl-0l1 5$05.00
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

116 PHILLIPS
on the order of 0.001 s (2). The persistence of localized spin packets in the Feher et al (4) experiments indicated an absence of spin diffusion among the impurity 31 P spins. This observation of localized spin packets in doped semiconductors led to the formulation of the much-studied (2-186) Anderson model for the role of disorder on electron transport. Anderson (2) considered originally a tight-binding model
1. n n,m
for the impurity band in which one orbital and a single site energy en are assigned at random to the lattice sites. A constant nearest-neighbor matrix element V mediates transport between nearest-neighbor sites. In Equation 1, a'; (an) creates (annihilates) an electron on site n. This model can describe the onset of localized electronic states, as can be seen immediately by considering two simple limits. Let us assume that the site energies are chosen from a uniform distribution of width W. When W = 0, an ordered system obtains, becausc all the sites have the samc energy. The resultant eigenstates are delocalized Bloch states that remain un scattered over the size of the sample. Transport in this regime is ballistic. However, in the limit that V = 0, none of the sites are connected, and transport ceases. That is, the resultant eigenstates are localized. Hence, the model described by Equation 1 can describe a localization/delocalization transition that is governed entirely by the ratio VI W. On physical grounds, it is tempting to argue that, in all cases, gradually increasing the ratio VI W will lead to a smooth interpolation between the limit of extreme localization (V = 0) and the ballistic regime (V» W). For example, in the limit of weak disorder, the mean-free path of an electron scales as I � (2VIW)2. This sets the length scale over which the electron is localized. For sufficiently small W(W« 2V), I � O (L) where L is the size of the sample. Consequently, the electron states are extended. As W increases, 1« L, and the electron is localized. We know now from the scaling theory of localization (5), as well as from the early work of Mott & Twose (6) and Borland (7), that this heuristic argument is not always applicable. For d � 2, it is now well-established [except in some d = I models (8-15) recently developed] that an infinitesimal amount of disorder precludes the existence of extended states (5). That is, the smooth interpolation between the ballistic and localized regimes does not obtain for d = 1 , 2. In three dimensions, extended states fail to form when WI V exceeds some critical value, (WI V)c' When WIV < (WIV)n extended and localized states coexist in the energy band, but are separated at an energy now known as the mobility edge.
Because of the lack of a rigorous proof of Anderson localization, much of the recent theoretical work has focused on large-scale numerical simu-
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZATION 1 17
lations ( 1 5-57) of disordered systems, the development of new analytical methods, and the study of simplified disorder models that can be solved exactly in certain limits (58-1 70). In addition, there have been successful attempts to include electron interactions in the scaling theory of localization ( 1 7 1 -173). This review focuses on a few of these advances. We begin this review by summarizing the standard analytical approaches to the localization problem (5, 59, 60, 72). Next, we present new analytical techniques, as well as applications of the standard approaches to such problems as topological disorder (61 , 62, 1 1 8, 1 30). We then focus on numerical approaches to localization (IS-57). Counter-examples to oneparameter scaling ( 1 5, 25, 28, 35) and the multi fractal character of qitical wavefunctions ( 1 6, 22) in three-dimensional systems are highlighted. Finally, we cover the absence of localization in correlated disordered systems, particularly the random dimer model (8-1 4, 1 88, 1 89).
ANAL YT1[CAL METHODS
Green Functions
The least a theory of Anderson localization should provide is all accurate method for distinguishing between localized and extended states. The earliest attempt (58) to provide such a minimal theory can be attributed to Abou-Cha.cra, Anderson, and Thouless (AAT). To introduce the AAT method, we need to define the Green function at a particular energy, G(E), for the site-disordered Hamiltonian. As a resolvent operator, the Green function, G(E) = (E- H) - I , contains all information about the exact eigenstates of Equation 1 . From the eigenvalue equation for H,
Ean = Gnan + L v"mam m
we write the matrix elements of the single-particle Green function as
(E-en)Gnm(E) = bnm+ L v",G'rn(E) n# I
2.
3.
where v,,/ is zero unless n and I are nearest-neighbors, in which case v,,/ = V. The form of Equation 3 is particularly illuminating. Because the tightbinding states form the atomic basis, the mth eigenstate of the Green function can be written quite generally as a superposition
4. n
of these statl;s. The coefficients Crnn determine the amplitude that the mth eigenstate overlaps site n. Without the second term in Equation 3, the
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.
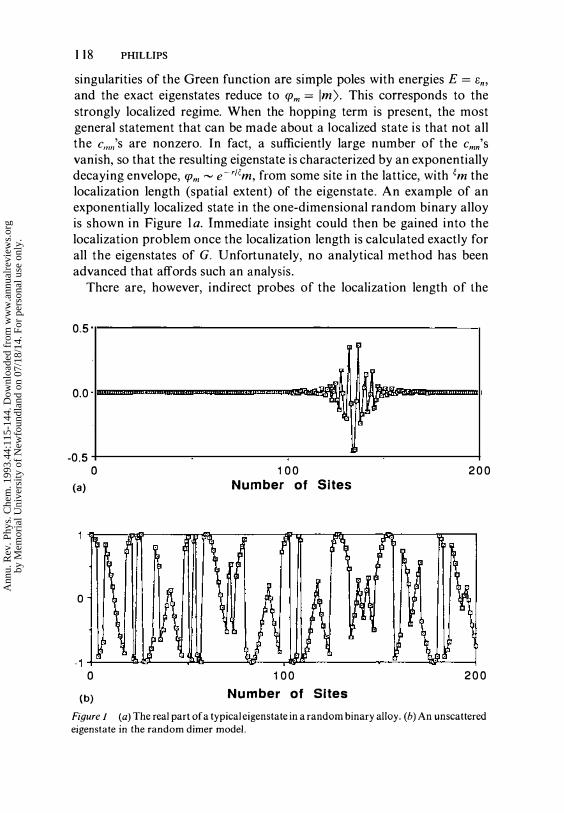
1 1 8 PHILLIPS
singularities of the Green function are simple poles with energies E = en, and the exact eigenstates reduce to ({Jm = 1m). This corresponds to the strongly localized regime. When the hopping term is present, the most general statement that can be made about a localized state is that not all the C,,".'s are nonzero. In fact, a sufficiently large number of the cmn's
vanish, so that the resulting eigenstate is characterized by an exponentially decaying envelope, ({Jm � e-r/l;m, from some site in the lattice, with �m the localization length (spatial extent) of the eigenstate. An example of an exponentially localized state in the one-dimensional random binary alloy is shown in Figure l ao Immediate insight could then be gained into the localization problem once the localization length is calculated exactly for all the eigenstates of G. Unfortunately, no analytical method has been advanced that affords such an analysis.
There are, however, indirect probes of the localization length of the
0.5
0.0
-0.5 o
(a)
o
... .!l!i' . . �-" .�. Ib"" jlJ .
�" � �
100 Number of Sites
200
_1+=--��L---��----�����--------��--��--� o
(b) 100
Number of Sites 200
Figure 1 (a) The real part of a typical eigenstate in a random binary aHoy. (b) An unscattered eigenstate in the random dimer model.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZATION 1 19
eigenstates, such as the AAT analysis of the self-energy (58). A straightforward iteration of Equation 3 leads to a perturbative expansion for the Green function in powers of the hopping term V. Because of the restriction on the indices in the second term in Equation 3, each term in the perturbation expansion corresponds to a self-avoiding walk in which no site is visited twice. If we define the diagonal elements of the Green function such that
5.
the self-avoiding perturbative terms will be contained in the site self energy, Sn(E). If as E approaches the real axis Sn(E) is purely real, then the singularities of Gnn(E) are once again simple poles, and the eigenstates are localized states. Extended states form only when Gnn(E) is complex, or equivalently when 1m Sn(E) is nonvanishing, as E approaches the Re E axis (58). That is, the inverse of 1m Sn(E) determines the lifetime of the state at energy E. However, as Anderson (2) emphasized, the more precise quantity that is relevant to the localization problem is the probability distribution of the site self-energies. The signature of the absence of extended states, then, is the vanishing of the probability distribution of the site self-en��rgy for all energies along the Re E axis. The amount of disorder required to make this state of affairs obtain defines the critical value (Wj V)c-
The primary hurdle in the Green function analysis of the localization problem is the computation of the site self-energy. The self-energy requires the enumeration of all self-avoiding walks that return to a given site. Hence, on lattices lacking closed loops, such as Cayley trees, the site self-energy can be calculated exactly (58). AAT have performed such an analysis on a Cayley tree of connectivity K and obtained
(Wj2V)c = 4Kln (Wj2V)c 6.
as the upper limit criterion for the value of the critical disorder beyond which no state is extended. This criterion was derived by neglecting the real part of the self-energy. Attempts have certainly been made to extend the AAT analysis to hypercubic lattices (57, 60). In such approaches, the self-energy is calculated through self-avoiding walks of M steps (57, 60). Economou & Cohen (60) have argued that a stability analysis of the Mth root of the term of order M in the self-energy can be used to distinguish between localized and extended states. If in the limit that M -+ 00 the Mth root of the OeM) term in the self-energy exceeds unity, then the perturbative expansion for Sn(E) diverges, and the electronic statc at energy E is exh:nded. Although this criterion is not as precise as the AA T criterion, it does appear to provide reliable results for the square lattice, as shown by Soukoulis & Economou (57). Other applications of the AAT
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 20 PHILLIPS
self-energy analysis can be found in a series of papers by Logan & Wolynes (6 1 , 62) on topological disorder. These authors have extended the AAT work to include the effects of short-range, liquid-like correlations in spatially inhomogeneous materials. From their analysis, they could obtain mobility edges, as well as the role of band structure effects (61, 62). Their analysis is in agreement with the results of Guinea and Verges ( 130). Winn & Logan (63) have expanded on the topological disorder analysis (6 1 ) and analyzed a multi band, topologically disordered model in the context of the metal-insulator transition in expanded fluid mercury. They conclude that states at the Fermi level are localized at fluid densities at which the s-p band gap closes (63). In a similar vein, Schafer & Wegner (64) have also analyzed multi band models. They have shown that disordered multiband models can be understood in the context of the nonlinear sigma-model (64, 98, liS).
In addition, several attempts (6S-68) to include the effects of inelastic phonon scattering on the localization transition have also utilized the work of AA T. A key motivating factor for this work is the experimental observation by Mooij (17S) that the temperature coefficient
din peT)
IX = dT 7.
of the resitivity [peT)] changes sign in a wide range of materials. This effect is now known as the Mooij correlation 07S). In the band transport regime, IX is positive because phonons decrease the electron mean-free path; for hopping transport lJ. is negative. Charge carrier motion is thermally assisted in the hopping regime. Girvin & Johnson (66) have stressed that Mooij correlations arise from a breakdown of the adiabatic or weak e- -phonon coupling limit. That is, in the hopping regime it is not possible to separate the timescale for charge carrier motion from the timescale for the lattice vibrations because the electron is self-trapped. Hence, standard weakcoupling theories that treat the e--phonon coupling as a linear perturbation are inapplicable. Although Girvin & Johnson (66) did treat the e--phonon coupling as a linear perturbation, they described the hopping-to-band transition by careful resummation. A more extensive treatment of the Mooij correlation can be found in the work of Zhang & Phillips (67). In this work, the weak-coupling approximation was avoided by transforming the bare electronic states to the self-trapped polaron states. The self-energy was then calculated with the renormalized polaron states. At T = 0, Zhang & Phillips showed that coupling to a frozen-in lattice distortion stabilized localized states, thereby increasing the value of VjW needed to delocalize the self-trapped electronic states (67). These
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZATION 121
results corroborated the earlier phenomenological work of Cohen et al (68). Subsequent calculations of the conductivity of the self-trapped states revealed a hopping-to-band transition (or equivalently the Mooij correlation) as the temperature was decreased (67). Because these results were obtained in strong-phonon coupling regime, a hopping-to-band transition was observed for all values of the disorder, unlike the weak-coupling results of Girvin & Johnson (66) in which hopping and band motion were not predicted to exist at the same value of the disorder. Zhang & Phillips' (67) analysis is then applicable to Ti02 and exfoliated graphites in which hopping and band motion coexist. Logan & Wolynes (65) have also analyzed the role of phonon scattering by shifting thc eigencnergy to E �
E + i1J. Thouless & Kirkpatrick (69) have discussed how this kind of energy shift leads to dephasing on a timescale of T = h/21J. In the limit of 17 = 0, the pure Anderson problem is recovered, whereas as 1J � CD all phase coherence is lost and hopping transport obtains. Hence, this approach can describe a hopping-to-band transition.
Scaling Theory
Although the Green function techniques described above can lead to reasonably accurate predictions of the mobility edge in disordered systems, much of the physics of the localization transition is suppressed in this view. For example, the dimensional dependence of the localization transition is not easily d{:termined from the Green function approach. That the dimensionality might play a strong role in the localization transition can easily be deduced from the fact that a single defect in an otherwise ordered lattice produces a bound state for d ::::;: 2, regardless of the strength of the defect. For d> 2, the defect strength must exceed a critical value for a bound state to form. Although the single defect results cannot be applied straightforwardly to the case of an infinite system containing a finite fraction of disordered sites, the strong dimensional dependence does suggest that the localization transition should somehow reflect this behavior. The simplest account that delineates the dimensional dependence of the localization transition is the one-parameter scaling theory of Abrahams et al (5). The key assumption in this account is that for each dimension, a single parameter, g, completely characterizes the localization transition. For a more complicated Hamiltonian containing random V's along with energy disorder, one-parameter scaling is not as immediately obvious as it is for the simple model in Equation I with a single matrix element Vand diagonal disorder of width W.
The scaling theory can be developed as follows (5, 70, 7 1 ): Consider increasing the size of a unit cell of a lattice by a factor p. The new lattice constant is LI ,= ap, where a is the original lattice constant, and the
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 22 PHILLIPS
new unit cell contains pd old lattice sites. By our initial assumption, the localization properties in the new unit cell are determined entirely by geL I) ' After m transformations, the new linear dimension is Lm = apm. In such a sample, g(L
m) characterizes the localization transition. A further conse
quence of the scaling assumption is that all of the g(L;)'s must be related as a result of their universal length dependence. This implies that the eigenstates in a disordered block of length Lz can be determined by those from a block of length L,; moreover, they can (by the scaling assumption) be constructed through knowledge of a single parameter. Thouless (70) has argued that when linear combinations of the eigenstates for the sample of length L \ are taken to construct those of the sample of length Lz, the required crucial ratio is I1E/(j W. Here, I1E is a measure of the overlap integral between the states, and (j W determines the spacing between the energy levels in the sample of edge length L \. The spread of the eigenenergies I1E can be computed directly by measuring the energy shift of the eigenstates when periodic or antiperiodic boundary conditions are imposed on the L'i sample. For a state that is localized, I1E is insensitive to the scale change to L2• On the other hand, I1E is large for an extended state, because such a state will extend over the new region of length L2• Hence, the ratio I1E/(j W is a single parameter that can be used to determine the localization properties in a sample of length L. Let us now determine the length dependence of this quantity. The level spacing (j W = (pLd)- \ where p is the density of states in a sample of length L. Consequently, geL) = pLdl1E. Licciardello & Thouless (7 1 ) argued that EI1 = h/T:, where T: is the time to diffuse from the center to the edge of the sample. That is, T: = (L/2)2/D, and I1E = 2haj(e2pL 2), where we have used the Einstein relationship, (J = 2e2pD, between the dc conductivity (0) and the diffusion constant (D) . If we now combine these relationships, we obtain
8 .
as the length dependence of our single parameter for the localization problem. In Equation 8, G = (JL d- 2 is the dc conductance; consequently 9 is simply the dimensionless conductance. Note that the form given here for the conductance is simply Ohm's law for a metal. The crucial assumption, then, in one-parameter scaling theory is that the dimensionless conductance 9 relates the eigenstates in a sample of edge length 2L to those in an original sample of linear dimension L (5).
Let us now define (5)
f3 =
d In [geL)] d ln L
9.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.
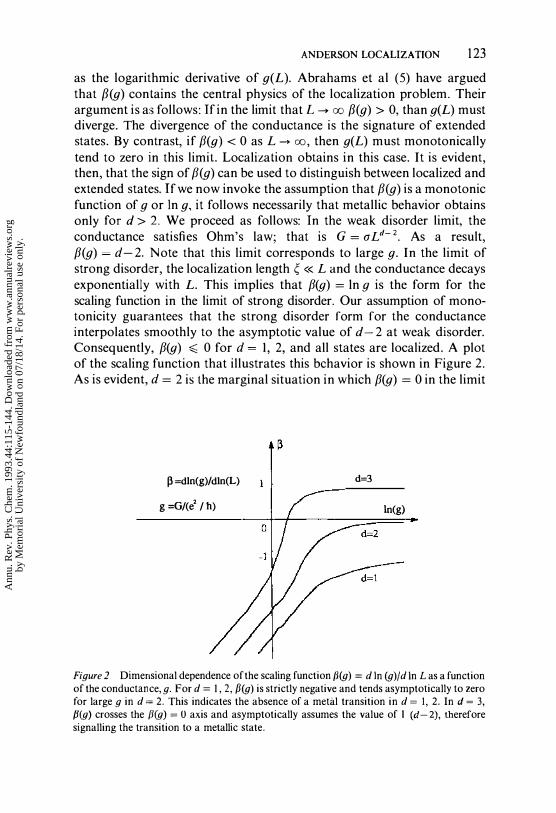
ANDERSON LOCALIZATION 123 as the logarithmic derivative of g(L). Abrahams et al (5) have argued that f3(g) contains the central physics of the localization problem. Their argument is as follows: If in the limit that L -> 00 f3(g) > 0, than g(L) must diverge. The divergence of the conductance is the signature of extended states. By contrast, if [3(g) < 0 as L -> 00, then g(L) must monotonically tend to zero in this limit. Localization obtains in this case. It is evident, then, that the sign of [3 (g) can be used to distinguish between localized and extended states. If we now invoke the assumption that [3(g) is a monotonic function of 9 or In g, it follows necessarily that metallic behavior obtains only for d> 2. We proceed as follows: In the weak disorder limit, the conductance satisfies Ohm's law; that is G = aLd-2. As a result, [3(g) = d- 2. Note that this limit corresponds to large g. In the limit of strong disorder, the localization length � « L and the conductance decays exponentially with L. This implies that [3(g) = In 9 is the form for the scaling function in the limit of strong disorder. Our assumption of monotonicity guarantees that the strong disorder form for the conductance interpolates smoothly to the asymptotic value of d- 2 at weak disorder. Consequently, [3(g) � 0 for d = I, 2, and all states are localized. A plot of the scaling function that illustrates this behavior is shown in Figure 2. As is evident, d = 2 is the marginal situation in which [3(g) = 0 in the limit
� =dln(g)/dln(L)
g =G/(e' /h)
d=3
In(g)
Figure 2 Dimensional dependence of the scaling function f3(g) = din (g)/d In L as a function of the conductance, g. For d = 1,2, f3(g) is strictly negative and tends asymptotically to zero for large g in d ,= 2. This indicates the absence of a metal transition in d = I, 2. In d = 3, P(g) crosses the f3(g) = 0 axis and asymptotically assumes the value of 1 (d-2), therefore signalling the transition to a metallic state.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 24 PHILLIPS
of large conductance. Tn d = 3, P(g) crosses the In 9 axis and asymptotically approaches unity, which indicates an onset of metallic behavior.
Although the scaling analysis of the localization problem is intrinsically phenomenological, it has received much support from more rigorous theories (72-76, 82, 83), as well as from numerical simulations ( 15-57) and experiments ( 1 76-1 84). As mentioned in the previous section, the Green function calculations of Sokoulis & Economou (57) have verified an absence of extended states in a d = 2 disordered square lattice. More extensive numerical studies are discussed in the following section. Further support for the scaling theory of localization can be found in the work of Vollhardt & Wolfle (72), who have shown that the dimensional predictions of scaling theory can be reproduced by a self-consistent diagrammatic expansion of the density response function for a system of independent particles moving in a random potential. A further key result of this work is that much of the physics of the localization problem can be obtained by analyzing the maximally crossed diagrams (72, 73, 1 25). Such diagrams contain the contribution from coherent backscattering. Other, more recent analytical work that has verified the scaling predictions is the random matrix theory approach of Muttalib (74). Based on earlier work by Tmry (76) and others (75), Muttalib has shown how the most probable value of the conductance can be calculated from the distribution of eigenvalues for the transfer matrices. One-parameter scaling theory follows immediately (74), once it is assumed that the distribution of eigenvalues of the transfer matrices is described by a single parameter. Although this assumption does not appear to hold for strong disorder, it is supported by numerical simulations in the presence of weak disorder (74). Only the weak disorder limit is of interest here, because it is in this regime that the one-parameter scaling function predicts an absence of a metal transition for d = I , 2.
Density Matrices
The dynamics of an electron on a statically disordered lattice can be described equivalently by the Liouville equation of motion
dp dt
= -i[H, p] 1 0.
for the density matrix. Because the density is a physical observable, the solution to Equation 10 will contain all information needed to understand the localization transition. For example, the site-diagonal elements of the density matrix reveal the electron population on the lattice sites. An early attempt to formulate a theory of localization based on the density matrix can be found in a series of papers by Loring and Mukamel (77-79) in which
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZA nON 125 they explore a self-consistent dephasing theory of quantum localization, based upon an effective-medium type treatment of the Liouville equation for the single particle density matrix. Their approach is surprisingly simple to implement and, at first sight, appears to recover correctly the main features of one-parameter scaling theory. The specific Liouville equation of motion that Loring and Mukamel (77-79) investigate,
dp dt
= -i[H,p]-Ap, I I .
contains the stochastic site-otT-diagonal dephasing term [AP]mn = r(1-Jmn)Pmn. The use of a dephasing term, such as in Equation II, has a long history in the study of translationaJly invariant systems, where it has provided a simple phenomenological way to introduce scattering into the coherent motion that results from the single-particle Hamiltonian, H. For a disordered system, however, Parris & Phillips (80) have shown that Equation I I is of limited utility. Consider the time derivative of the norm of the density matrix (d/dt) lip II = (d/dt)(Tr p2) = -2 Tr [(Ap)p] . Because the operator A is purely off-diagonal, the left-hand side of this equation reduces to the sum of the absolute squares of the off-diagonal elements of the density matrix in the site representation. Thus, at any time during which the density matrix has nonzero site off-diagonal elements, II p II will monotonically decrease (80). Because II p II is positive definite, it cannot decrease forever. Consequently, the system irreversibly relaxes until II p II reaches its minimum value of zero, at which point Pnm = 0 (80). The vanishing of the off-diagonal elements at long times has a profound effect on the spatial population. This can be seen most clearly from the equation of motion for the otT-diagonal elements, which depends on the diagonal elements through Hnm(Pnn -Pmm). The vanishing of Pnm at long times requires that Hnm(Pnn -Pmm) = 0, as well. Thus, any two sites directly connected by a nonzero matrix element are equally populated at t = O. Hence, a localized distribution is incompatible with a dephasing process of the kind dellineated in Equation 11. This state of affairs should come as no surprise, as Equation I I is often referred to as the high-temperature approximation (80).
There have been successful attempts, however, to investigate Anderson localization by using density matrices. For example, Dunlap et al (8 1) studied the localization problem by analyzing the generalized master equation
Pnn = f' I[w"m(t-t')Pmm(t')- Wnm(t-t')Pnn{t')]dt' Jo m 12.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 26 PHILLIPS
for the time evolution of the diagonal elements of the density matrix. They used a nearest-neighbor approximation for the memory function Wnn±l(t-t') = 2V2cos (en-en±I)(t-t'). Equation 1 2 can be derived in general from the Liouville equation of motion by using projection operators ( 1 85). The nearest-neighbor memory function approach predicts an Anderson transitions for WjV = 13 and WjV = 20 for K = 2 and K = 3 Cayley trees, respectively (8\). These results are in good agreement with the exact values of WjV = 1 7 and WjV = 29, respectively (58). Further work along these lines is needed to see how accurately this method predicts localization transitions for hypercubic lattices. Other successful density matrix methods that circumvented the effective Liouvillian approximation of Loring and Mukamel can be found in the work of Thomas & Weller (82). These authors have advanced a mode-coupling theory from which they compute the diffusion constant for an excitation moving in a site-disordered lattice (82). They accurately locate the Anderson transition for a cubic lattice and predict the dephasing time. This work, which is closely related to the self-consistent approach of Gotze (83), appears to be a promising analytical tool for studying the localization transition.
NUMERICAL METHODS
Finite-Size Scaling
Because the Hamiltonian is known exactly for the localization problem, numerical studies offer the ideal testing ground for the assumptions and predictions of the analytical methods discussed in the previous section. Key issues that numerical studies can answer include the following: the critical disorder at the localization transition; the validity of one-parameter scaling; the precise values of the critical exponents; the time dependence of the conductivity; and the spatial extent of eigenstates at the mobility edge. We focus here on recent advances and extensions of old numerical methods that can address these problems. Special attention is given to the numerical studies designed to test the validity of one-parameter scaling.
The earliest attempt to implement the scaling hypothesis of Abrahams et al (5) can be found in the work of MacKinnon & Kramer (29). The starting point for their analysis is an infinite disordered strip of some width M. Let L,(M) represent the localization length for an infinite strip of width M. The behavior of d> I dimensional systems can be obtained in the M - w limit. For any amount of disorder, recursive Green function (29), as well as recursive transfer matrix methods (30, 3 1 ), can be used to calculate accurate results for Lc(M). When a state is exponentially localized, the ratio LJM)jM is a decreasing function of M (29). In fact, Lc(M) will approach some finite value when M> LrCM). On the other
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZATION 1 27
hand, wh�m a state is extended, the ratio Lc(M)j M is an increasing function of M. The intermediate or critical case corresponds to Lc(M)jM, which exhibits constant behavior as M increases. Nonetheless, the two extremes are clearly distinguished by plotting Lc(M)jM versus M. The change in the slope of Lc(M)j M beyond some value of the disorder signifies a transition from a metal to an insulator. The quantity Lc(M)j M, however, is energy as well as disorder dependent. A more transparent way of presenting the numerical data from this approach is to scale Lc(M)jM, evaluated at a particular energy and over a range of disorder values, by a widthindependent localization length 2 that is strictly determined by the disorder. Correct implementation of this procedure, known as finite-size scaling, results in a single universal curve for all the data at a particular energy; that is, Lc(M, E)jM = f[2(E)jM] , where f is a universal function (29). This procedure is certainly correct, so long as one-parameter scaling theory is valid. MacKinnon & Kramer (29) report that this procedure produces a value for the critical disorder of We = 1 6.5 ± 0.25 for the Anderson transition on a simple cubic lattice. A uniform distribution was used to describe the static diagonal disorder. No such transition was observed for the square lattice (29) or the d = 2 triangular lattice (42), in contrast to the analytical work of Srivastava (43). More recently, Schreiber (28) has used this method to evaluate We for different distributions of the disorder. He has shown that differences with the simple cubic lattice result do arise when Gaussian and triangular distributions are used (28). The critical value of We is 20.5 ± 0.5 and 20.95 ± 0. 1 for the triangular and Gaussian distributions, respectively (28). The latter value is in agreement with earlier work of Soukoulis et al (35). On its own, this discrepancy does not imply that one-parameter scaling is not valid because, as Schreiber (28) correctly points out, it is unclear that equating the second moments guarantees that the various distributions are equal. It is essential, then, to look at the critical exponents.
The key exponent that finite-size scaling is amenable to calculating is the correlation length exponent. In the vicinity of the critical region, the correlation length diverges as e � [WjV - (Wj V)c] -v. The relationship between the exponent v and the scaling function can be made as follows: At (Wj V)n the p-function vanishes. Let us call the value of the conductance at which {l(g) = 0, gc. In the vicinity of the fixed point gn peg) has a slope (3), which we take to be Ijv, such that
peg) = (g-gJ =
[;g.
vgc v 1 3 .
This relationship i s true unless bg « l . I f we now integrate Equation l3 [coupled with the definition of peg) through Equation 8] from a length
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 28 PHILLIPS
scale comparable to the mean-free path out to the Ohmic regime at which point P(g) � I, we find that geL) = (JL where (J = (Agcjl)(Jg)' with A a proportionality constant of unit order (3). If we write the conductivity in the suggestive form (J = gR, we readily identify � = IjA (bg)- ' as the diverging correlation length with exponent v. Consequently, the conductivity and correlation length exponents are identical. In finite-size scaling, Lc(M, E)jM plays the role of the single parameter (g) that characterizes the localization transition. Hence, the argument leading to Equation 1 3 can be repeated with the parameter Lc(M, E)jM, and the correlation length can be identified accordingly. Following this procedure, MacKinnon & Kramer (29) calculated a value v = 1 .50 ± 0.05 for the Anderson transition on a cubic lattice with diagonal site-energy disorder chosen from a uniform distribution. Schreiber (28) has repeated this calculation with a slightly more accurate algorithm for three different distributions. The results are as follows: uniform distribution, v = 1 .55 ± 0. 1 5; triangular distribution, v = 1 .55 ± 0.15; and Gaussian distribution, v = 1 . 1 5 ± 0. 1 5. The first two values are well within the statistical errors of the MacKinnon & Kramer (29) results; however, the value for the Gaussian distribution is not. A difference of this magnitude certainly
suggests that the details of the insulator-metal transition depend on the details of the disorder. As a result, more than a single parameter is needed to characterize the conductance on length scales longer than the meanfree path, I. This discrepancy has been highlighted by recent subsequent calculations by Kramer et al (25), who report a value of v = 0.9 ± 0.3 for the correlation length exponent in the presence of Gaussian disorder. At this point, however, it is unclear how strongly the nonuniversality argument should be made, given the sources of error that naturally creep into numerical determinations of exponents. However, we address this point later.
As presented above, the finite-size scaling methods cannot be readily applied to localization problems on topologically disordered systems in which no underlying lattice structure exists. Such a situation arises naturally in doped semiconductors (I 76). To solve this problem, Skinner and
coworkers ( 1 5-20) have advanced the concept of a quantum connectivity. Classically, two sites are connected if a bond exists between them. The quantum analogue is not as straightforward to state, because the site probabilities obey a more complicated equation of motion (see Equation 12). Skinner and coworkers ( 1 8) have proposed
� Pij ij = (P_p_)'/2
JJ "
as the quantum analogue of the classical connectivity in which
1 4.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZA nON 1 29
1 5.
The physical motivation ( 1 8) for this form for the quantum connectivity is twofold: First, Equation 1 4 is proportional to the time-averaged probability that an electron at time t = 0 on site i is at site j at t = 00. Second, the classical connectivity results when the master equation is used to calculate Pij• Skinner and coworkers ( 15-20) then used standard percolation arguments to relate the connectivity to the correlation length and the finit{:-size scaling hypothesis to formulate phenomenological renormalization-group equations for the exponents (as discussed above). For the standard d = 3 Anderson site-diagonal disorder problem (with a uniform distribution), they obtain a critical disorder of We = 1 5.95 ± 0.25 and a correlation length exponent of v = 1 .83 ± 0.06 ( 1 8). Their value for the critical disorder is in reasonable agreement with the MacKinnon/ Kramer value of 1 6.5 ± 0.5; however, their value of the exponent is some
what higher than the values cited previously (v = 1 .55 ± 0. 1 5) for the uniform distribution of disorder ( 1 8). They also used the inverse participation ratio to evaluate v for a Gaussian distribution. A value of v = 0.99:1::0.04 was obtained ( 1 5, 1 6). The small error bars on this value would suggest that, indeed, the Gaussian disorder problem possesses a correlation length exponent � I as earlier calculations of Kramer et al (25) indicated. This calculation also supports the earlier result of Schreiber (28) that the uniform and Gaussian disorder problems are in different universality classes. More importantly, a correlation length exponent � I is consistent with experimental measurements on such alloys as Ge '_xAux and compensated doped semiconductors, which also find that the conductivity exponent s = v � I ( 1 76). Wegner (84) originally concluded from an e-expansion that v = I exactly. However, he has shown (85) in a more careful calculation that v = e- '- (9/4K(3)e2+ 0(e3) which is strictly negative for G = I (d = 3). It is unclear how seriously this calculation should be taken, because Chayes et al (86) have proven that, in a wide class of static disordered models, v � 2/d. On the other hand, Castellani et al ( 1 73) have shown that a myriad of exponents obtain in an interacting electron model
depending on the strength of the spin-flip or spin-orbit scattering. They find that s � 1 when strong spin-orbit interactions are present. If, however, there is a minimum metallic conductivity, the spin-orbit scattering rate is enhanced in the vicinity of the metal-insulator transition, and s � 1 /2 ( 1 73), as is observed in uncompensated Si : P ( 1 76). Hence, the lower bound of Chayes et al (86) can be tested by measuring experimentally the magnitude of the spin-orbit interaction in the vicinity of the metal-insulator transition. If such interactions are weak in a given sample, and a minimum
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 30 PHILLIPS
metallic conductivity is absent, then an exponent of v '" I would indicate that the metal transition is of the Anderson type.
Matrix Diagonalization and Numerical Integration
Probably the simplest of all numerical approaches to the localization problem is that of diagonaJizing the secular matrix corresponding to the Hamiltonian in Equation I. Early attempts along these lines, for example the work of Yoshino & Okazaki (41), have been limited by the memory capabilities of available computers. Recently, Schreiber & Grussbach (22) partially circumvented this problem by using the Lanczos algorithm coupled with memory storage tricks and found that matrices on the order of 1 00,000 x 1 00,000 can be diagonalized efficiently. Consequently, detailed information about thc spatial cxtent of the eigenstates can easily be obtained once the generalized inverse participation ratio
\L ICnmI2kJ(E-En)/ P k I (E) = �n,_m --;--___ -----. __
(� J(E-En)) 1 6.
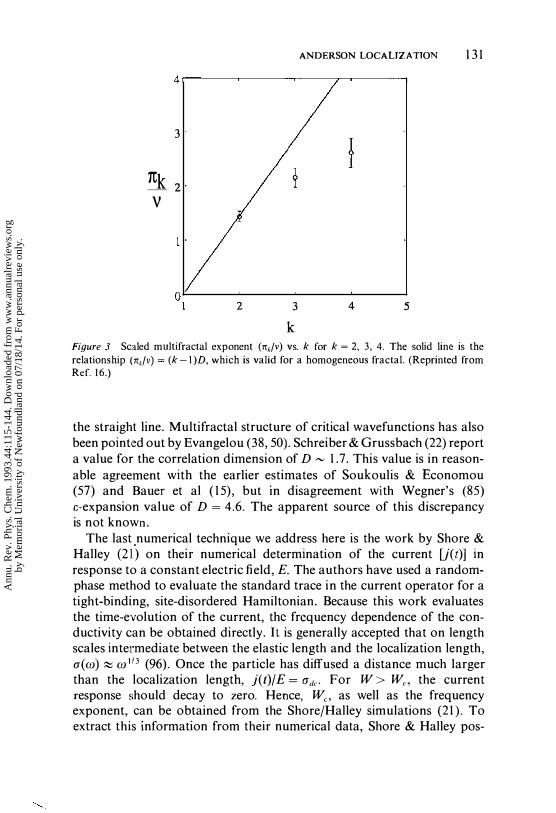
is calculated. The average in Equation 1 6 is over the random distribution for the disorder, and the cnm's are the amplitudes defined in Equation 4. Pl(E) is the inverse participation ratio that represents the average number of sites over which an eigenstate extends. For a de localized state, P zCE) '" N, where N is the number of sites along an edge in the lattice. A state at the mobility edge is critical in that it is neither extended nor localized. It has been suggested that, in general, P2(E) � LD where L = N 1/2 (38-40). If D < d, then the wave function at this energy is properly characterized by a fractal dimension, D. Physically then, fractal structure of eigenstates can be thought of as arising from spatial fluctuations in the site amplitudes of a critical state or from a localized state on length scales up to the localization or elastic length. Higher moments of the generalized participation ratio can be used to define a hierarchy of fractal exponents nk ( 1 5, 22). For a homogeneous fractal, (nk/v) = D (k- l) . Hence, deviations from this relationship indicate thc presencc of m ulti fractal structure to the electronic eigenstates. In the muItifractal language, D is actually referred to as the correlation dimension, as opposed to the fractal dimension . Schreiber & Grussbach (22) have used direct diagonalization to compute the hierarchy of fractal exponents for the localization problem in d = 3. Their work corroborates earlier results by Chang et al ( 1 5 ), which are shown in Figure 3, that critical eigenstates in the d = 3 localization problem do indeed have a multifractal structure, as evidenced by the deviations from
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.
ANDERSON LOCALJZA TTON 1 3 1
4r------r------.----;-.-----.
3
1tk 2 V
2 3 k
4 5
Figure 3 Scaled multifractal exponent (rrdv) vs. k for k = 2, 3, 4. The solid line is the relationship (rrk/v) = (k-I)D, which is valid for a homogeneous fractal. (Reprinted from Ref. 16.)
the straight line. Multifractal structure of critical wavefunctions has also been pointed out by Evangelou (38, 50). Schreiber & Grussbach (22) report a value for the correlation dimension of D '" 1 .7. This value is in reasonable agreement with the earlier estimates of Soukoulis & Economou (57) and Bauer et al ( 1 5), but in disagreement with Wegner's (85) c-expansion value of D = 4.6. The apparent source of this discrepancy is not known.
The last.numerical technique we address here is the work by Shore & Halley (21) on their numerical determination of the current [jet)] in response to a constant electric field, E. The authors have used a randomphase method to evaluate the standard trace in the current operator for a tight-binding, site-disordered Hamiltonian. Because this work evaluates the time-evolution of the current, the frcquency dependence of the conductivity can be obtained directly. It is generally accepted that on length scales intermediate between the elastic length and the localization length, O"(w) � wl13 (96). Once the particle has diffused a distance much larger than the localization length, J(t)/ E = O"dC' For W> We> the current response should decay to zero. Hence, We> as well as the frequency exponent, can be obtained from the Shore/Halley simulations (21 ). To extract this information from their numerical data, Shore & Halley pos-
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 32 PHILLIPS
tulated thatjtl/z = F(tl W - WeIVZ). This scaling function has the property that jet) --+ Ig as t --+ 00. A frequency response of ill i/3 corresponds to z = 3 (2 1 ). Because z, W,., and v are all fitting parameters, a range of exponents were produced. However, an excellent fit to their data was obtained for We = 16.5, Z = 3.0, and v = 1.5 (2 1 ). These values are in agreement with earlier results cited above ( 1 8, 28, 29) on Anderson localization with a uniform distribution, which is the relevant distribution here. Attempts were made to fit their data so that an exponent of v = 1.0 was produced (2 1 ). As remarked earlier. this is apparently the incorrect value for a uniform distribution (28, 29). The correct frequency response for a Gaussian distribution, which appears the only distribution to date that yields an exponent of v = 1 .0, thus remains an open question. Further work along these lines is certainly needed.
EXCEPTIONS TO LOCALIZATION
General Considerations
In the models discussed thus far, the site energies or matrix elements were strictly statistically independent entities assigned from a single random
distribution. In any real physical system, however, the site energies and matrix elements are correlated. Until recently, there have been strikingly few studies (108, 1 68-170) on the role of statistical correlations on the localization transition, partly because of the suspicion that correlations naturally present in real systems cannot be determined with any sort of certainty. Hence, the physical significance of proposed models would not be clear. For example, consider the eigenvalue equation
1 7.
for the site amplitudes in a nearest-neighbor, tight-binding Hamiltonian. In the event that - Cn = Vn,n+ I + Vn,n_ b the eigenvalue equation reduces to
1 8.
which is identical in form to the master equation with random nearestneighbor hopping rates (8). This equation is well-known to have a diffusion pole at E = 0; that is, the k = 0 state is extended (8, 187). Except in the context of structural disorder, the correlation leading to Equation 1 8 is at best artificial at the electronic level. Flores (186) has explored additional algebraic correlations between the site energies and matrix elements and found that an infinitesimal fraction of the electronic states possessed zero reflection coefficients and, hence, were extended in one dimension. This
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZATION 1 33
result is indeed surprising in light of the scaling arguments (5), which prohibit extended states in one dimension.
Although these models are suggestive, it remains an open question what minimal requirement must be satisfied for one-dimensional systems to possess extended states. This question has recently been answered by a series of disordered models proposed by Phillips and coworkers (8-14). The original model proposed by Dunlap et al (8, 9) was based on a general mapping between the site energies and matrix elements, so that IN (N the length of the sample) of the electronic states remained extended. The origin of the extended states in this model remained obscure until the physics of the general mapping was shown to be captured by a much simpler and physically realizable model, the random dimer model ( 10). In the RDM" the site energies are chosen from a bi-valued distribution and at least one of the energies is assigned to pairs of sites along a linear chain ( 1 0). That a nearest-neighbor correlation in the site energies is sufficient to product; a set of extended states in one dimension is the truly surprising and novel feature of this model. This model is different in kind from those discussed above in that no artificial correlations need be imposed between the site energies and the matrix elements for extended states to exist. In the next sections, we analyze this model, present generalizations, and discuss briefly physical applications.
Random Dimer Model
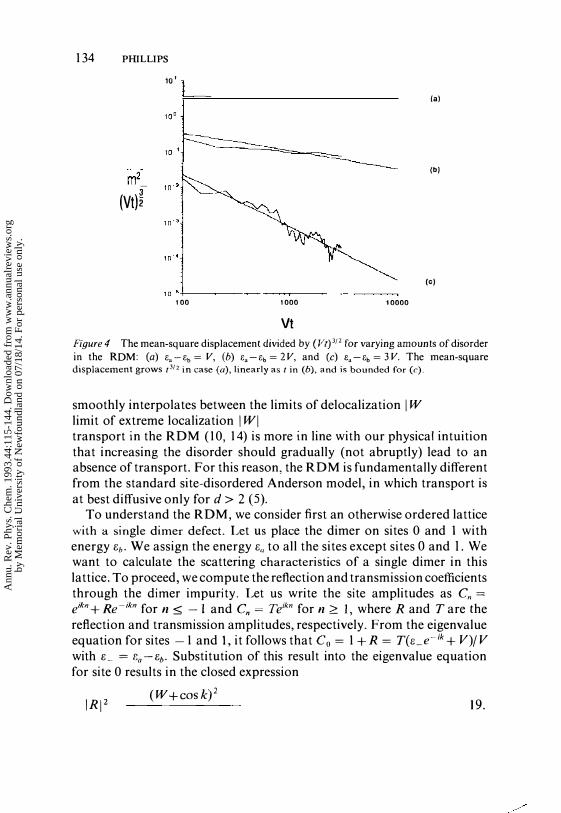
To put the: RDM in context, let us consider for the moment the random binary alloy. In the tight-binding model of a random binary alloy, site energies t:a and t:b are assigned at random to the lattice sites with probability p and l-p, respectively. All states in this model are localized. A typical wavefunction in the random binary alloy is shown in Figure lao The RDM can be obtained from the binary alloy by replacing all clusters containing an odd number of f.b'S by clusters containing an odd number of f.a's. We: referred to any such lattice in which at least one of the site energies is assigned at random to pairs of lattice sites as the RDM. A typical dimer in the RDM is shown in Figure 5a. We showed that -IN of the electronic states extend over the entire sample in the RDM provided that -I :S W:s I, with W = ( t:a-t:b)/2V( l0, 1 4). A delocalized eigenstate in the RDM is shown in Figure l b. An additional curiosity of the RDM is that the mean-square displacement of an initially localized particle grows superdiffusively as 13/2, provided that -1 < W < 1 . Diffusion obtains only when the disorder is increased, such that W = ± 1 ( 1 0, 1 4). In all other cases, the particle remains localized at long times. Numerical simulations of the mean-square displacement, which illustrate this behavior, are shown in Figure 4. It is evident, then, that increasing the disorder in the RDM
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.
1 34 PHILLIPS
10'
L 3 (Vt) 2
10·5+-_�_���-r--��_��., 100 1000 10000 Vt
(a)
(b)
(0)
Figure 4 The mean-square displacement divided by (Vt)3!2 for varying amounts of disorder in the RDM: (a) Ea-Eb= V, (b) Ea-Eb=2V, and (c) Ea-Eb= 3V. The mean-square displacement grows /3/2 in case (a), linearly as / in (b), and is bounded for (c).
smoothly interpolates between the limits of delocalization I W I < I to the limit of extreme localization I WI > 1. Unlike the Anderson model, then, transport in the RDM ( 1 0, 1 4) is more in line with our physical intuition that increasing the disorder should gradually (not abruptly) lead to an absence of transport. For this reason, the RDM is fundamentally different from the standard site-disordered Anderson model, in which transport is at best diffusive only for d > 2 (5).
To understand the RDM, we consider first an otherwise ordered lattice with a single dimer defect. Let us place the dimer on sites 0 and I with energy eb' We assign the energy en to all the sites except sites ° and 1. We want to calculate the scattering characteristics of a single dimer in this lattice. To proceed, we compute the reflection and transmission coefficients through the dimer impurity. Let us write the site amplitudes as Cn = eikn + Re -ikn for n :::;; - 1 and Cn = Teikn for n � I, where Rand T are the reflection and transmission amplitudes, respectively. From the eigenvalue equation for sites - 1 and 1, it follows that Co = 1 +R = T(Le-ik+ V)/V with L = ea-eb' Substitution of this result into the eigenvalue equation for site 0 results in the closed expression
2 (W +COSk)2 IRI =
(W+cos k)2+sin2 k 19.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZATION 1 35
for the reflection probability. We see then that the reflection coefficient vanishes when W = - cos k, which will occur for some value of k as long as - 1 s W s I . The location in the parent ordered band of the perfectly transmitted electronic state corresponds to the wavevector ko = cos- I W. At ko, there is no difference between the ordered and disordered bands; the density of states coincide at this point (8). The vanishing of the reflection coefficient through a single dimer at a particularly energy can be understood as a resonance effect. That is, the dimers are acting as resonance cavities. At a particular energy (ko = cos- 1 W), the reflection from the second site in the dimer is 1 800 out of phase from the reflection from the first. At this energy, unit transmission obtains.
Of cours1e, no transport would obtain if only a single electronic state remained unscattered. To determine the total number of states that extend over the entire sample, we expand R around ko. To lowest order we find that in the vicinity of kn, I RI2 '" (Ak) 2 where Ak = k-ko. The time between scattering events , is inversely proportional to the reflection probability. As a result, thc mean-free path A = <velocity>, '" 1 / (Ak)2 in thc vicinity of ko. Let Ak = AN/2nN. Upon equating the mean-free path to the length of the system (N), we find that the total number (AN) of states whose mean-free path is equal to the system size scales as AN = )N. Because the mean-free path '" localization length in one dimension, we find that the total number of states whose localization lengths diverge is )N. The single dimer argument presented here has been shown to hold for a random system by Bovier ( 1 88), as well as by Wu et al ( 1 4). Bovier's ( 1 88) argument is particularly elegant and establishes rigorously that the Lyapunov exponent (inverse localization length) vanishes quadratic�lly in the vicinity of the resonance energy, ko = cos- 1 W. Consequently, III the RDM V N of the electronic states remain extended over the total length of the sample.
Such states ultimately contribute to the transport properties. Because the mean-free path of the extended states in the RDM is at least the system size, such states move through the crystal ballistically with a constant group velocity [v(k)], except when they are located at the bottom or top of the band where the velocity vanishes. Because all the other electronic states are localized, the diffusion constant is determined simply by integrating v(k).�(k) over the width of k-states that participate in the transport. The upper limit of the integration is then proportional to the total fraction of unscattered states, or l /)N and Jo(k) '" N. When the velocity is a nonzero constant, we obtain that the D '" )N. Because the states that contribute to transport traverse the length of the system with a constant velocity, t and N can be interchanged or D '" t 1/2. Consequently, the meansquare displacement grows as t3!2. At the bottom or top of the band where
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 36 PHILLIPS
the group velocity vanishes, v(k) '" k and D ", 0( 1 ). Figure 4 illustrates simulations of this behavior. \,..
The functional form of the resonant state in the RDM can be constructed as follows: The transfer matrix connecting nearest-neighbor amplitudes along a linear chain with only site energy disorder is given by
(Cn+ ') = [E�Gn - 1 ]( Cn ) = Tn ( Cn ) .
Cn Cn_ , Cn_ , I 0
20.
In the RDM, two such transfer matrices exist, Ta and Tb• The 1 - 1 element in Ta and Tb is [(E- Ga)/ V] and [(E- Gb)/V], respectively. Let's assume the Gb'S are assigned in pairs. Transport across an arbitrary segment of a region of the RDM can be represented by . . . T�l T�12T�>T�I.y�s r?;'6 . . . , where the l;'s are random variables. The resonant condition W = - cos k is equivalent to E = Gb, because E is the energy of the ordered band. At this energy, the transfer matrix across a b-defect reduces to
- IJ o . 2 1 .
The square of this matrix (which corresponds to the transfer matrix across a b-dimer) is the negative of the unit matrix. At resonance then, the bdimers can be simply erased at the expense of a sign change. Consequently, the phase shift through a dimer defect is Q = - 2k + n ( 10, 1 4). The un scattered state corresponds to a Bloch state in which the dimer sites have been effectively decimated at the cost of a sign change in the wavefunction. For example, if dimers are placed at sites 0 - 1 and 2 - 3, the unscattered state is ' " e- 2ik, e- ik, 1 , _ e- ik, - 1, e-ik, 1 , eik, e2ik, . . . An example of such an unscattered state is plotted in Figure lb .
The only feature of the RDM that appears to be in dispute in the literature is the existence of superdiffusive transport. Gangopadhyay & Sen ( 1 89) have studied the RDM and have concluded that indeed IN of the electronic states possess localization lengths longer than the system size. They speculate, however, that at long times, the diffusion constant will ultimately vanish in this model. Their argument is based on an observation and a speculation: The fraction of extended states vanishes in the thermodynamic limit. And the extended states in the dimer model are critical states that ultimately decay algebraically ( 1 89). The first statement is indeed true; however, the second is not. The resonant state located at E = Gb, is not critical. It is a Bloch state, as calculated above and illustrated in Figure l b. Over no length scale does this state decay. fo of the states in the vicinity of this state support this resonance, as calculated by Bovier (1 88).
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZATION 1 37
For any finite-size sample, these states possess mean-free paths as least as long as the sample. Only the state at E = eb possesses a mean-free path independent of the sample size. We are guaranteed, then, that for any length sample, at least fo ( 14, 1 88) of the electronic states will have mean-free paths of O (N). In the limit N -t 00 , the fraction of extended states certainly tends to zero as I /JN; however, the length of these states diverges as N. Hence, it is not a foregone conclusion that either the divergence or the zero will win in the thermodynamic limit. The diffusion constant calculations, as well as the simulations, indicate that the divergence wins, thereby producing superdiffusive transport. Stated correctly, (see Ref. 1 89), because the extended states in this model possess mean-free paths longer than the system size, there is no real length scale up to which a simulation should be run to determine definitively whether a particle is localized. They do speculate that over a very long sample, the effects of localization should be observed. The longest simulations we have run correspond to 1 0,000 VI. No deviation from the trends reported here was observed. In fact, no simulation to date has produced results that deviate from these trends. Over this timescale, a particle explores distances on the order of � 3000 lattice sites. In addition, a typical solid, VI � 1 0,000, corresponds to times longer than the inelastic scattering time. Hence, in a real material, the motion predicted here will be distinct from hopping transport in which the mean-free path is typically on the order of a lattice constant. It is ccrtainly a curious feature of the RDM that so few states do so much.
Generalizations: Symmetry Condition The essence of the RDM is that at an energy in the band, the defect transfer matrices reduce to the negative of the unit matrix ( 1 4). The local dimer correlation creates this macroscopic quantum effect. It is straightforward to show that this result holds only if the site energies of the dimer are equal; that is, the dimer is symmetric. A plane of symmetry is a sufficient condition for a defect to possess a resonant state. The general statement that can be made is as follows: The standard tendency of disorder to localize electronic states is suppressed at certain energies in the band whenever the defects contain a plane of symmetry ( 14). This is the minimal requirement for extended states to exist in one-dimension. The RDM is just the simplest model in which the defects possess a plane of symmetry. Any random n-mer will suffice, as shown in Figure 5b. In addition, the single defect model shown in Figure Sc is the off-diagonal dual of the RDM ( I I) . This state of affairs is relevant whenever dilute amounts of an impurity are: added to an otherwise ordered lattice. In the dilute limit, impurities occur infrequently on neighboring sites. We refer to a lattice in
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 38 PHILLIPS
v V V V ---0 0 • •
V o 0- ( a )
�a
V V V V --0 • •
eb '------ n s i tes
-of--_V -{Of--_V -(0 V
• • 0- ( b )
• V V OJ---o- ( C )
Figure 5 (a) Dimer defect in the RDM , (b) n-site defect possessing the dimer symmetry, (c) defect in the repulsive binary alloy.
which impurities are separated by at least a single lattice site as a repulsive binary alloy ( 1 1 ). We have previously shown that the condition for a resonant state in this model is given by (E -Sb)/(2 = E - Sa or equivalently, Vlsa - sb l :0;; 2 1 V2 - )721 with K = V; V ( 1 1 , 1 4). As a consequence, in the un scattered state, only the amplitude at the defect site changes. We have also shown that in the unscattered state, the site amplitude changes by K = vI P. Aside from applying to the dilute limit of the binary alloy, the repulsive binary alloy also applies to physical systems, such as Fibonacci lattices formed out of two kinds of materials ( 190). Let a = GaAs and b = AlAs. A Fibonacci sequence of a and b will be of the form a, ab, aba, abaab, . . . where the nth entry is a product of the (n - Ost and (n - 2)nd. The b's appear singly; hence, a Fibonacci lattice is a subset of the repulsive binary alloy. Such semiconductor heterostructures have been fabricated experimentally because of the general interest in quasiperiodic order in crystals ( 1 90). The numerous theoretical treatments (see, for example, Ref. 1 9 1 ) of Fibonacci lattices have not predicted the transmission resonances
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ANDERSON LOCALIZATION 1 39
we have discovered in the repulsive binary alloy, because they have arranged either the matrix elements or the site energies in a Fibonacci sequence. As is evident from the repulsive binary alloy, the site energies in a Fibonacci lattice follow the Fibonacci rule, but the matrix elements do not. These correlations have also been pointed out by Kumar ( 192) and elsewhere ( 193-1 95) in the context of Kronig-Penney models for Fibonacci lattices. Experiments by Smith et al ( 196) on propagation of sound waves on a silicon Fibonacci substrate exhibited the resonances predicted by the repulsive binary alloy ( I I ) analysis of a Fibonacci lattice.
Two further generalizations are of interest here. The first is relevant to the origin of metallic states in single strands of the conducting polymer polyaniline ( 197-199). The predominant defects ( 1 98) present in the doped state of this polymer are bipolarons, as illustrated in Figure 6. Extensive numerical simulations on single strands of the polymer containing randomly placed bipolarons indicate the presence of extended states (200). More prec:ise calculations have revealed that these extended states have unit transmission coefficients when the Fermi level corresponds to a dopant concentration of 50% (201 ). Finite-size effects can then be ruled out as the origin of the metallic states along a linear chain containing a disordered distribution of bipolarons. From Figure 6, it is immediately evident that a bipolaron in polyaniline contains a plane of symmetry and, hence, is functionally equivalent to a dimer in the RDM ( 1 2, 1 3). As a result, a disordered distribution of bipolarons is a subset of the disorder inherent
t '2 t 2 n
v Vo �Df------(O Eo Eo Eo
n,3
n,1
n ,4
t o n,5
t 2 \ '2 n+1 (a)
t o n ,6
� � Vo � • OI---iOI---Q- (b)
Eo Eo Eo Figure 6 (a) Bipolaron quinoid defect in polyaniline and (b) its equivalence to a dimer. The circles on the ring represent carbon atoms, whereas those along the chain are nitrogen atoms.
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 40 PHILLIPS
in the RDM. Wu and Phillips ( 1 2, 1 3) have shown that the reflection coefficient through a protonated or unprotonated bipolaron vanishes precisely at the energy at which the numerical simulations found a set of extended states to exist. Consequently, the RDM can explain the occurrence of metallic states in the single strands of the conducting polyaniline ( 1 2, 1 3). The applicability of the RDM is not entirely limited to polyaniline. The disorder inherent in any bipolaron lattice is quite generally described by the ROM ( 1 3). Numerical calculations by Lavarda et al (202) reveal that the location of extended states in single strands of polyacetylene, as well as other conducting polymers in the polyaniline family, such as polyparaphenelyne, poly thiophene, and polypyrrole, are accurately described by the RDM .
The final generalization of the RDM is as follows: Consider a disordered linear chain of arbitrary length. Let us call this system L 1 . L 1 is an insulator. However, if two copies of L 1 containing identical sequences of disorder are linked end-to-end, such a system will contain a plane of symmetry (N. Goldenfeld 1 992, personal communication). Hence, although L 1 is an insulator, the composite system possesses a plane of symmetry and, as a result, must have a set of extended states, as predicted
by the ROM. This statement is true for any distribution of the disorder in L l ' An immediate experimental realization of this state of affairs is a twinned disordered crystal. According to the RDM, such a system should act as a narrow pass-band filter. Such a composite system constitutes the boldest generalization of a dimer and could provide the ideal testing ground for the predictions of the suppression of localization by symmetrical defects.
ACKNOWLEDGMENTS
I thank Patrick Lee, Paul Parris, Dave Dunlap, Jim Skinner, and Michael Schreiber for their characteristically level-headed remarks throughout the writing of this paper. I am also grateful to Qiming Li for assistance with the figures and a careful reading of the manuscript. I also thank Kalyan Kundu for critical analyses of the ROM and for making available prior to publication his preprints in support of this model. This work was funded by the National Science Foundation, the American Chemical Society Petroleum Research Fund, and the Center for Materials Research at MIT.
Literature Cited
1 . Mott, N. F. 1 949. Proc. Phys. Soc. London Sect. A 62: 4 16-22
2. Anderson, P. W. 1 958. Phys. Rev. 109: 1492�1 505; Anderson, P. W. 1978. Science 20 1 : 307� 1 6
3. Lee, P. A . , Ramakrishnan, T. V. 1 985. Rev. Afod. Phys. 57: 287�337
4. Feher, G., Fletcher, R. c., Gere, E. A. 1955. Phys. Rev. 1 00: 1 784-86
5. Abrahams, E., Anderson, P. W., Lic-
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

ciardello, D. C., Ramakrishnan, T. V. 1 979. Phys. Rev. Lett. 42: 673-76
6. Mott, N. F., Twose, W. D. 1 96 1 . Adv. Phys. 10: 107-63
7. Borland, R. E. 1 963. Proc. R. Soc. London Ser. A 274: 529-45
8. Dunlap, D. H. , Kundu, K., Phillips, P. 1 989. Phys. Rev. 40: 1 0999-1 1 006
9. Dunlap, D. H. , Phillips, P. 1 990. J. Chern. Phys. 92: 6093-97
10. Dunlap, D. H. , Wu, H.-L., Phillips, P. 1 990. Phys. Rev. Lett. 65: 88-91
I I . Wu, H.-L., Phillips, P. 1 990. J. Chern . Phys. 93: 7369-73
12 . Wu, H .-L., Phillips, P. 199 1 . Phys. Rev. Lett. 66: 1366-69
13 . Phillips, P., Wu, H.-L. 1 99 1 . Science 252: 1 805·- 12
14. Wu, H.-L., Goff, W. E., Phillips, P. 1992. Phys. Rev. B 45: 1 623-2H
1 5 . Chang, T. M. , Bauer, J. D., Skinner, J. L. 1990. J. Chern. Phys. 93: 8973-82
16. Bauer, J ., Chang, T. M., Skinner, J . L. 1 990. Phys. Rev. B 42: 8 1 2 1 -24
17 . Root, L. J. , Skinner, J. L. 1988. J. Chern. Phvs. 89: 3279-84
1 8 . Root, L. t, Bauer, J. D., Skinner, J . L. 1 988. Phys. Rev. B 37: 55 18-21
19. Bauer, J. D., Logovinsky, V., Skinner, J. L. 1 988. J. Phys. C 2 1 : L993-1 000
20. Bauer, 1. D., Logovinsky, V., Skinner, J . L. 1 989. J. Chern. Phys. 90: 2703-7
2 1 . Shore, H . 8., Halley, J . W. 1 990. Phys. Rev. Lett. 66: 205-8
22. Schreiber, M. , Grussbach, H. 1 99 1 . Phys. Rev. B 67: 607- 10
23 . Schreiber, M., MacKinnon, A., Kramer, B. 1 989. Phys. Scr. T25: 67-7 1
24. Bulka, B . , Kramer, 8., Schreiber, M . 1 987. Z. Phys. B 66: 2 1 -30
25. Kramer, EL, Broderix, K., MacKinnon, A., Schreiber, M. 1990. Physica A 1 67 : 1 63 74
26. Bulka, B. R., MacKinnon, A., Kramer, B. 1 985. Z. Phys. B 60: 1 3- 1 7
27. Bulka, B. .. Kramer, B., Schweitzer, L . 1 985. J. Non-Cryst. Solids 7 7 -78: 29 32
28. Schreiber, M. 1 99 1 . See Ref. 203, pp. 385-95
29. MacKinnon, A., Kramer, B. 1 983. Phys. Rev. Lett. 57: 2999-3003
30. Pichard, J. L., Sarma, G. 1 98 1 . J. Phys. C 1 4: L l 27-32, L61 7-25
3 1 . Economou, E. N., Soukoulis, C. M. , Cohen, M. H., Zdetsis, A. D. 1 985. Phys. Rev .. 3 1 : 6 1 72-83
32. Li, Q. M., Soukoulis, C. M., Grest, G. S. , Economou, E. N. 1989. Phys. Rev. B 40: 2825-30
33. Economou, E. N., Cohen, M. H . , Sou-
ANDERSON LOCALIZATION 1 4 1
koulis, c . M . 1988. Phys. Rev. B 37: 4399-4407
34. Soukoulis, C. M. , Economou, E. N., Grest, G. S. 1 987. Phys. Rev. B 36: 8649-55
35. Soukoulis, C. M., Economou, E. N., Zdetsis, A. D. 1986. Phys. Rev. 8 34: 2253-57
36. Soukoulis, C. M ., Grest, G. S. , Economou, E. N., Cohen, M. H. 1 989. Ph),s. Rev. Lett. 62: 575-78
37. Zhang, Z. Q., Chu, Q. J., Xue, W. G., Sheng, P. 1 990. Phys. Rev. B 42: 46 1 3-30
38. Evangelou, S. N. 1 99 1 . See Ref. 203, pp. 357-8 1
39. Aoki, H . 1 986. Ph),s. Rev. B 33: 73 10-1 3
40. DeVries, P., deRaedt, H. , Lagendijk, A. 1989. Ph),s. Rev. Lett. 62: 25 1 5-1 8
4 1 . Yoshino, S., Okazaki, M . 1 977. J. Phys. Soc. Jpn. 43: 4 1 5-23
42. Li, Q., Phillips, P. 1 992. J. Phys . Condens. Matter 4: 5647-50
43. Srivastava, V. 19R9. J. Phys. Condens. Matter 1 : 43 1 1-22
44. Zhang, Z. Q., Chu, Q. J ., Xue, W. G., Sheng, P. 1 990. Ph),s. Rev. B 42: 46 1 3-30
45. Wischmann, B. , M ullerhartmann, E. 1 990. Z. Phys. B 79: 9 1-99
46. Gibbons, M. K., Madden, P. A., Logan, D. E. 1988. Ph),s. Rev. B 38: 7292-7302
47. He, F. L. 1 988. Solid State Cornrnun. 67: 509-1 3
48. Hansel, D., Luciani, J. F. 1 987. J. Ph),s. A 20: 1 03 1 -38
49. Grabert, H . , Ingold, G. L., Schramm, P. 1 987. Phys. Rev. Lett. 58: 1 285-88
50. Evangelou, S. N. 1 9R6. J. Ph),s. C 1 9: 429 1-4302
5 1 . Rodriques, D. E., Pastawski, H. M. , Weisz, J . F. 1 986. Phys. Rev. B 33: 7738-42
52. Kasner, M. , Weller, W. 1 986. Phys. Status Solidi B 1 34: 73 1-39
53. Delyon, F., Souillard, B., Levy, Y. 1 985. Cornrnun. Math. Phys. 100: 463-70
54. Delyon, F., Souillard, B., Levy, Y. 1 985. J. Stat. Phys. 4 1 : 375-88
55. Johnston, R., Kramer, B., Schweitzer, L., Masek, J. 1 987. J. Non-Cryst. Solids 97-8: 241-44
56. Sheng, P., Zhang, Z. Q. 1 99 1 . J. Phys. 3: 4257-67
57. Soukoulis, C. M., Economou, E. N. 1 984. Phys. Rev. Lett. 52: 565-68
58. Abou-Chacra, R., Anderson, P. W., Thouless, D. J. 1 973. J. Phys. C 6: 1 734-5 1
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 42 PHILLIPS
59. K1after, J . , Joernwe, J. 1 979. J. Chern. Phys. 7 1 : 1961-66
60. Economou, E. N., Cohen, M. H. 1 970. Phys. Rev. Lett. 24: 1445-48
6 1 . Logan, D. E., Wolynes, P. G. 1986. J. Chern. Phys. 85: 937-48
62. Logan, D. E., Wolynes, P. G. 1 985. Phys. Rev. B 3 1 : 2437-50
63. Winn, M. D., Logan, D. E. 1990 . ]. Chem. Phys. 93: 6756-66
64. Schafer, L., Wegner, F. 1 980. Z. Phys. B 38: 1 1 3-26
65. Logan, D. E., Wolynes, P. G. 1987. Phys. Rev. B 36: 4 135-47
66. Girvin, S. M. , Jonson, M . 1980. Phys. Rev. B 22: 3583-97
67. Zhang, Q., Phillips, P. 1 987. J. Chern. Phys. 87: 2370-76
68. Cohen, M. H. , Economou, E. N., SOIlkou1is, C. M. 1983. Phys. Rev. Lett. 5 1 : 1 202-5
69. Thouless, D. J . , Kirkpatrick, S. 1 98 1 . J. Phys. C 14: 235-45
70, Thou1ess, D, J. 1 977, Phys. Rev. Lett. 39: 1 1 67-70
7 1 . Licciardello, D. c., Thou1ess, D. J. 1 975. J. Phys. C 8: 4 1 57-70
72. Vollhardt, D. , Wii1fie, P. 1 980. Phys. Rev. Lett. 45: 482-85
73. Kroha, J., Kopp, T., Wii1fie, P. 1 990. Phys. Rev. B 4 1 : 888-91
74. Mutta1ib, K. A. 1 990. Phys. Rev. Lett. 65: 745--47
75. Mutta1ib, K. A., Richard, 1.-L., Stone, A. D. 1987. Phys. Rev. Lett. 59: 2475-78
76. Imry, y, 1986. Europhys, Lett. I : 249-52
77. Loring, R. F., Franchi, D. S., Mukame1, S. 1 988. Phys. Rev. B 37: 1 874--83
78. Mukamel, S. , Loring, R. F. , Franchi, D. S. 1 988. J. Chem. Phys. 1 28: 99-1 23
79. Loring, R. F., Mukame1, S. 1 986. J. Chern. Phys. 85: 1 950--65
80. Parris, P. Eo, Phillips, P. 1 988. J. Chern, Phys. 88: 3561-63
8 1 . Dunlap, D. H., Kundu, K., Phillips, P. 1989. J. Phys. Condens. Matter I : 7883-7900
82 . Thomas, P., Weller, A. 1 987. J. NonCryst. Solids 97-98: 245--48
83. Gotze, W. 1 979. J. Phys. C 12 : 1 279-96
.
84, Wegner, F. 1 987. 'Nucl. Phys. B 280: 2 10--24
85. Wegner, F. 1989. Nuc/. Phys. B 3 16: 663-78
86. Chayes, J. T" Chayes, L., Fisher, D. S., Spencer, T. 1 986. Phys. Rev. Lett. 57: 2999-3003
87. Sornette, D., Souillard, B. 1990. Europhys. Lett. \ 3 : 7-1 2
88. Evensky; D. A., Sca1etter, R . T., Wolynes, P. G. 1990. J. Phys. Chern. 94: 1 149-54
89. Chalker, J. T., Siak, S. 1 990. J. Phys. Condens. Matter 2: 267 1-86
90. Hakami, S. 1990. Physica A 1 67: 1 49-62
9 1 . Kroha, J. 1 990. Physica A 1 67: 23 1-52 92. Oppermann, R. 1 990. Physica A 1 67:
301 - 1 2 9 3 . Weaver, R. L . 1990. Wave Motion 12 :
1 29--42 94. Bovier, A. 1 990. J. Stat. Phys. 59: 745-
79 95. Logan, D. E., Wolynes, P. G. 1990. J.
Chern. Phys. 93: 4994--50 1 2 96. Abrahams, E. , Lee, P . A . 1986. Phys.
Rev. B 33: 683-89 97. Hu, G. Y., O'Connell, R. F. 1 990. J.
Phys. 2: 5335--44 98. Horbach, M ., Schon, G. 1 990. Physica
B 1 6S: 3 1 S-16 99. Campanino, M., Klein, A. 1 990. Com
rnun. Math. Phys. 1 30: 441-56 1 00. Sornette, D. 1 989. J. Stat. Phys. S6:
669-80 1 0 1 . Jiang, Q., Tao, R. B. 1 989. Phys. Lett.
A 140: 443--46 102. Vondreifus, H. , Klein, A. 1 989.
Cornrnun. Math. Phys. 1 24: 285-99 103. Heinrichs, J. 1989. Europhys. Lett. 9:
581-84 104. Kravtsov, V. E., Yudson, ' V. I . ,
Agranovich, V. M . 1 989. Solid State Commun. 72: 1 10S-9
lOS. Bovier, A. 1 989 . J. Stat. Phys. S6: 645-68
106. Kolley, E., Kolley, W. 1 988. J. Phys.C 2 1 : 6099-6 109
107. Huckestein, B., Kramer, B. 1 989. Solid State Commun. 7 1 : 445--48
1 08. Zhang, Z. Q., Chu, Q. J. 1988. Phys. Lett. A 1 3 I : 5 1 7-23
109. Bealmonod, M. T., Forgacs, G. 1 988. Phys. Rev. B 37: 6646-57
1 10. Tanaka, K., Yamanaka, S., Yamabe, T., Oiji, M. 1 988. Synth. Met. 22: 247-56
I I I . Bovier, A. , Campanino, M. , Perez, J . F . , Klein, A . 1988. Commun. Math. Phys. 1 14: 439-6 1
1 12 . Bovier, A. , Klein, A . 1988. J. Stat. Phys. S I : SOI-17
1 1 3 . Markos, P. 1 988. Z. Phys. B 73: 1 7-2 1 1 14. Dorokhov, O. N. 1 988. Phys. Rev. B
37: 526--41 1 1 5 . Kravtsov, V. E., Lerner, I. V., Yudson,
V. I. 1 988. Zh. Eksp. Tear. Fiz. 94: 255-63
1 1 6. Pruiskcn, A. M. M. 1 988. Nuc!. Phys. B 295 : 653-57
1 1 7. Klein, A. , Speis, A. 1988. Ann. Phys. 183 : 352-98
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 18 . Gunnarsson, 0., Jepsen, 0. 1988. Phys. Rev. B 38: 3568�71
1 1 9. Cannons, R., Klein, A., Martinelli, F. 1987. Commun. Math. Phys. 108: 41� 66
1 20. Doucot, B., Rammal, R. 1987. Europhys. Lett. 3: 969�74
12 1. Zheng, Z. B. 1987. 1. Phys. C 20: 4627� 38
122. Condall, C. A., Kirkpatrick, T. R. 1987. Phys. Rev. B 36: 6782�93
1 23. Igarahi, J. 1987. Phys. Rev. B 35: 5 15 1� 63
124. Cohen, S. M ., Condat, C. A., Kirkpatrick, T. R., Machta, J. 1987. Phys. Rev. Lett. 58: 785�88
125. Gold, A. 1987. Solid State Commun. 64: 823�26
1 26. Crisanti, A., Paladin, 0., Vulpiani, A. 1987. Phys. Rev. B 34: 7 164-66
1 27. Martinelli, F., Scoppola, E. 1987 . Rev. Nuovo Cimento 10: 1�90
128. Delyon , F., Simon, B . , Souillard, B. 1 987. Commun. Math. Phys. 1 09: 1 57� 65
129. Derrida, B., Mecheri, K., Richard, J. L. 1 987. J. Phys. 48: 733-40
1 30. Guinea, F., Verges, J . A. 1987. Phys. Rev. B 35: 979-86
13 1 . Verges, J. A., Brey, L., Louis, E., Tejedor, C. 1987. Phys. Rev. B 35: 5270-72
1 32. Geller, V. A., Margulis, V. A. 1987. Theor. Math. Phys. 70: 1 33-40
133. Grabert, H. , Ingold, G. L., Schramm, P. 1987. Phys. Rev. Lett. 58: 1 285-88
1 34. Chalker, J. T. 1987. J. Phys. C 20: L493-98
135 . Heinrichs, J. 1987. Phys. Rev. B 36: 2867�69
1 36. Klein, A., Martinelli, F., Perez, J. F. 1986. Commun. Math. Phys. 1 06: 623-33
137. Fukuyama, H. 1986. J. Magn. Magn. Mat. 54�57: 1437-4 1
1 38. Zimbauer, M. R. 1 986. Nuc!. Phys. B 265: 375-408
139. Fisch, R. 1986. Phys. Rev. B 34: 1662--{)7 140. Kirkpatrick, T. R. 1986. Phys. Rev. B
33: 780-88 14 1 . Zekri, N., Brezini, A. 1 986. Phys.
Status Solidi B 133: 463-68 142. Ono, Y., Ohtsuki, T. 1986. Surf Sci.
1 70: 7 14-18 143. Castellani, C., Dicastro, C., Peliti, L.
1 986. 1. Phys. A 1 9: 1 099- 1 1 03 144. Berlitz, D., Kirkpatrick, T. R. 1986.
Phys. Rev. B 33: 7332-35 145. Brezini, A., Dahmani, L., Sebbani, M .
1986. Phys. Status Solidi B 1 37: 667� 73
146. Kohmoto, M. 1986. Phys. Rev. B 34: 5043-47
ANDERSON LOCALIZATION 143
147. Ping, S., Papanicolaou, G., White, B. , Zhang, Z. Q. 1 986. Phys. Rev. B 34: 4757�6 1
148. Chakravarty, S., Schmid, A. 1986. Phys. Rep. Rev. Sect. Phys. Lett. 140: 195�236
149. Dcpollicr, c., Kergomard, J . , Laloe, F. 1986. Ann. Phys. 1 1: 458�92
150. Frohlich, 1. , Spencer, T., Scoppola, E., Martinelli, F. 1985. Commun. Math. Phys. 1 0 1 : 2 1-46
l S I . Bulaevskii, L. N., Sadovskii, M. V. 1985. J. Low Temp. Phys. 59: 89�1 1 3
152. Hikami, S . 1985. J. Phys. Lett. 46: L7 19�22
1 53 . McMillan, W. L. 1985. Phys. Rev. B 3 1: 344-45
154. Canisius, 1., Vanhemmen, 1. L., Nieuwenhuizen, T. M . 1985. Physica A 1 3 1 : 1 3 1�56
1 55. Houghton, A., Wen, H. 1 985. J. Phys. C 18: L507�1 O
1 56. Kaveh, M . 1985. Philos. Mag. B 5 1: 453-64
1 57. Stone, A. D., Lee, P. A., Azbel, M. Y. 1985. Phys. Rev. B 3 1: 1707�14
158. Vanalbada, M. P., Lagendijk, A. 1985. Phys. Rev. Lett. 55: 2692�95
1 59. Neal, H. L. 1985. Phys. Rev. B 32: 5002�8
160. Simon, B., Taylor, M. , Wolff, T. 1985. Phys. Rev. Lett. 54: 1 589-92
1 6 1 . Kotani, S., Simon, B. 1987. Commun. Math. Phys. 1 1 2: 1 03�19
162. Brezini, A., Behilil, F., Sebbani, M . 1986. Phys. Status Solidi B 138: K 1 37� 42
163. Enz, C. P., Flores, J. C. 1988. Helv. Phys. Acta 61: 1079�86
1 64. Yoo, K. M., Takiguchi, Y., Alfano, R. R. 1 989. App/. Opt. 28: 2343-49
1 65. Ziazwa, Y., Shudo, A. , Kurumi, K., Goda, M., Yamada, H. 1991. Progr. Theor. Phys. 86: 1�6
166. Richard, J. L., Sanguer, M. , Slevin, K. , Debray, P. 1990. Phys. Rev. Lett. 65: 1 8 1 2�15
167. Condat, C. A. , Kirkpatrick, T . R. 1985. Phys. Rev. B 32: 495�98
168. Economou, E. N., Cohen, M. H., Soukoulis, C. M. 1988. Phys. Rev. B 37: 4399-4407
169. Kasner, M. , Weller, W. 1986. Phys. Status Solidi B 1 34: 73 1�39
1 70. Johnston, R., Kramer, B. 1 986. Z. Phys. B 63: 273�8 1
1 7 1 . Suzumura, Y., Fukuyama, H . 1984. J. Phys. Soc. Jpn. 53: 3918-28
172. Giamarchi, T., Schulz, H. J. 1 988. Phys. Rev. B 37: 325-40
1 73. Castellani, c., Kotliar, G., Lee, P. A. 1987. Phys. Rev. Lett. 59: 323�26
1 74. Smith, D. T., McCall, K. R., Lorenson,
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.

1 44 PHILLIPS
C. P., Guyer, R. A., Hallock, R. 8. 1988. Phys. Rev. Leu. 6 1 : 1 286-89
175. Mooij, J. H. 1973. Phys. Status Solidi A 1 7: 521-30
1 76. Milligan, R. F., Thomas, G. A. 1 985. Annu. Rev. Phys. Chern. 36: 1 39-58
1 77. Polishchuk, I. Y., Burin, A. L., Maksimov, L. A. 1 990. JETP Lett. 5 1 : 730-34
1 78. Iwabuchi, S., Nagaoka, Y. 1 989. J. Phys. Soc. Jpn. 58: 1 325-33
1 79. Adams, P. W., Paalanen, M. A. 1 988. Phys. Rev. Lett. 61: 45 1-54
1 80. Blumel, R., Fishman, S., Smilansky, U. 1986. J. Chern. Phys. 84: 2604 - 14
18 1 . He, S. , Maynard, J . D. 1986. Phys. Rev. Lett. 57: 3 1 7 1-74
1 82. Arya, K. , Birman, J. L., Su, Z. B. 1986. Phys. Rev. Lett. 57: 2725-28
1 83 . Pi raux, L., Bayot, V., Issi, J. P., Mareche, J. F., McRae, E., Michenaud, J. P. 1986. Solid State Commun . 59: 7 1 1-15
1 84. Dassarma, S . , Kobayashi, A. , Prange, R. E. 1 986. Phys. Rev. Lett. 56: 1280-83
1 85 . Zwanzig, R. 1964. Physics 30: 1 109-23 1 86. Flores, 1. C. 1989. J. Phys. Condens.
Matter I : 847 1-79 1 87. Alexander, S., Bernasconi, J., Schnei
der, W. R., Orbach, R. 1 98 1 . Rev. Mod. Phys. 53: 1 75-98
1 88 . Bovier, A. 1 992. J. Phvs. A 25: 1021-29
1 89. Gangopadhyay, S., Sen, A. K. 1 992. J. Phys. Conden,\'. Matter 4: 9939-54
1 90. Clarke, R., Moustakas, T., Bajema, K., Grier, D. , dos Passos, W., Merlin, R. 1988. Superlat tices Microstruct. 4: 371-74
1 9 1 . Kohmoto, M., Sutherland, B. , Tang, C. 1 987. Phys. Rev. B 35: 1020-33
192. Kumar, V. 1 99 1 . J. Phys. Condens. Matter 2: 1 349-53
193. Ananthakrishna, G. 1990. J. Phys. Condens. Maller 2: 1 343-48
1 94. Kollar, J., Suto, A. 1986. Phys. Lett. A 1 1 7: 203-9
1 95. Larnellc, F., Etienne, B. 1988. Phys. Rev. B 37: 48 1 6-19
196. Smith, D. T. , Lorenson, C. P., Hallock, R. B. 1989. Phys. Rev. B 40: 6634-47
197. Zuo, F., Angelopuolos, M. , MacDiarmid, A. G., Epstein, A. J. 1 987. Phys. Rev. B 36: 3475-78
198. Mizoguchi, K., Nechtschein, M., Travers, J.-P. 1 99 1 . Synth. Met. 41-43: 1 1 3-1 6
199. Monkman, A . P., Adams, P . 199 1 . Synth. Met. 40: 87-96
200. Galvao, D. S. , dos Santos, D. A., Laks, 8., de Melo, C. P., Caldas, M. J. 1 989. Phys. Rev. Lett. 63: 786-89
201 . Schulz, P. A., Galvao, D. S., Caldas, M. J. 1 99 1 . Phys. Rev. 8 44: 6073-77
202. Lavarda, F. c., Galvao, D. S., Laks, B. 1992. Phys. Rev. 8 45: 3 1 07-1 0
203. Gans, W . , Blumen, A . , Amann, A . , eds. 1 99 1 . Large-Scale Molecular Systems. New York: Plenum
Ann
u. R
ev. P
hys.
Che
m. 1
993.
44:1
15-1
44. D
ownl
oade
d fr
om w
ww
.ann
ualr
evie
ws.
org
by M
emor
ial U
nive
rsity
of
New
foun
dlan
d on
07/
18/1
4. F
or p
erso
nal u
se o
nly.





























