Embed Size (px)
Citation preview

ANALYTICAL VALIDATION CHALLENGES DURING THE RAPID DEVELOPMENT OF KEYTRUDA
CMC Strategy ForumIndustry Considerations for Phase-Appropriate Method ValidationsAthena Nagi
January 29, 2018

Abstract and Outline
2
Early positive clinical results drove rapid development of Keytruda, including analytical method development and establishment, product characterization, and specification setting. This presentation will provide several examples of method development challenges when early-phase transitions quickly to late-phase, method updates during commercialization and beyond, and points to consider in establishing specifications and reference standards with limited product history.
• Background• PTMs• HP-SEC• Charge Distribution / HP-IEX• Potency ELISA and EC50

KEYTRUDA® - Pembrolizumab
3
KEYTRUDA® (Pembrolizumab, MK-3475) • Potent and selective humanized mAB blocking interaction of ligands with PD-1
(Programmed cell death 1)• Produced in rDNA CHO cells• Formulated as freeze-dried or liquid formulation
Biological License Application (BLA) Approved by FDA on September 04, 2014 for the treatment of patients with unresectable or metastatic melanoma under Breakthrough (BT) status
Multiple indications now approved in US and other countries.
Various indications studied globally
POC – clinical results SeptapprovalDec fileValidationsTransfers
beginSite
readiness
2012 2013 2014

Standard Release Panel EvolutionMethod Ph 1 Ph3 / Commercial Post-approval
optimizationAppearance/Color/Clarity Compendial methods Compendial methodsIdentity ELISA, HP-IEX ELISA, HP-IEX, pep mapProtein Concentration A280 A280Potency V1 comp binding ELISA Optimized ELISA YesCharge (HP-IEX) V1 HP-IEX Optimized HP-IEX YesSize (HP-SEC) V1 HP-SEC Optimized HP-SEC YesCE-SDS (R/NR) V1 CE-SDS Optimized CE-SDSPTM Not tested New methodProcess Residuals (CHO, DNA, ProA)
Kit-based Kit-based, with transition to process-product specific as PMC
Yes
Bioburden/endotoxin Compendial methods Compendial methodsExcipients FIO Release
4
Ph1: fit for use/purpose assessment, limited robustnessPh3: full ICH assessments, comprehensive robustness, utilizing factorial designs

• Multiple DS, DP sites, different presentations, early development batches were manufactured using different processes (and at different scales) – complex supply chain that grew and evolved during method validation
• Platform methods were used for early development and early clinical manufacturing; transitioned to verified and validated methods for duration of clinical and into commercial manufacturing
• ICH validations of commercial methods were conducted during Site 1 clinical campaign: dual testing conducted for a period of time
• Testing at multiple sites (efficiency with transfer and qualification activities completed together)
Site1 LYO Site2 LYO
Drug product
DS Site 1 DS Site 2Drug substance
Analytical Comparability Study Site2 liquid
DS Site 3
Site4 liquid
DS Site 4
Site3 liquid
Extensive analytical activities across multiple sites
Method transfers occurring simultaneously
Co-validation strategies employed (intermediate precision)

Implications of post-translational modifications and product experience
• Keytruda includes common post translational modifications, present in many mAbs at similar levels
• Modifications can occur via a variety of mechanisms (at different points in the manufacturing process) – often there is no one single root cause
• PTMs in Keytruda are understood (location on structure, abundance). Typical levels for one modification ~4-6% as measured by a specific HPLC method.
• At levels up to 20% – nearly full potency of Keytruda (monitored by 3 different orthogonal potency methods)
• Batches at up to 7% have shown expected safety and efficacy in the clinic
• Batches at ~6% have been dosed in higher dosage clinical arms (at 10mg/kg vs 2 mg/kg): effective levels of 30%
6
DP levels remain within specification limit through 24 month approved shelf life. No impact to product safety or efficacy

Representative chromatogram and peak characterization
Pre-peak 1 and Pre-peak 2~ variants with modification on one heavy chain in moleculePre-peak 3~ variants with modifications on both heavy chains
• Characterization work completed during clinical manufacturing
• Method developed and validated
• Retains tested with newly developed method were included in original file
• Limited number of batches used to establish specifications – did not sample all sources of variability
• Method capability pressures near statistically-driven spec limits

HP-SEC for size variants
• Original file included a typical phosphate-based HP-SEC method; introduction of amino acid modifier to mobile phase provides more consistent and precise quantitation of HMW
• HMW values increase (as would be expected given better retention of species); Higher acceptance criterion for HMW justified
• Bridging study included retest of frozen retains for batches, stability studies8

HP-IEX Challenges
• As typical for mAbs, HP-IEX results in a complex chromatogram: multiple species in non-baseline-resolved peaks
• Method tracks 7 peaks/peak groups
• Isolation and characterization of peaks revealed:– Observed modifications are typical of other monoclonal antibodies; no safety concerns– Purified charge variants showed no decrease in potency (multiple different orthogonal
potency assays)– Range of clinical experience showed no issues with immunogenicity or change in
PK/PD• No concerns with product efficacy
• Initial file included two-sided specification limits; subsequent PAS justified use of one-sided limits based on broader method and product understanding
9

Difference in chromatograms highlight method performance issue
Ref standard from Run with 30 month samples shows greater resolution than column at previous time points
Ref standard from Run with 24 month samples with less resolution
Pre-main peak is not a distinct peak, more a faint shoulder, making consistent manual integration difficultThe area% level is only ~2% and the run-to-run variation is relatively large (min-max values reference standard from 0.7-2.1%)
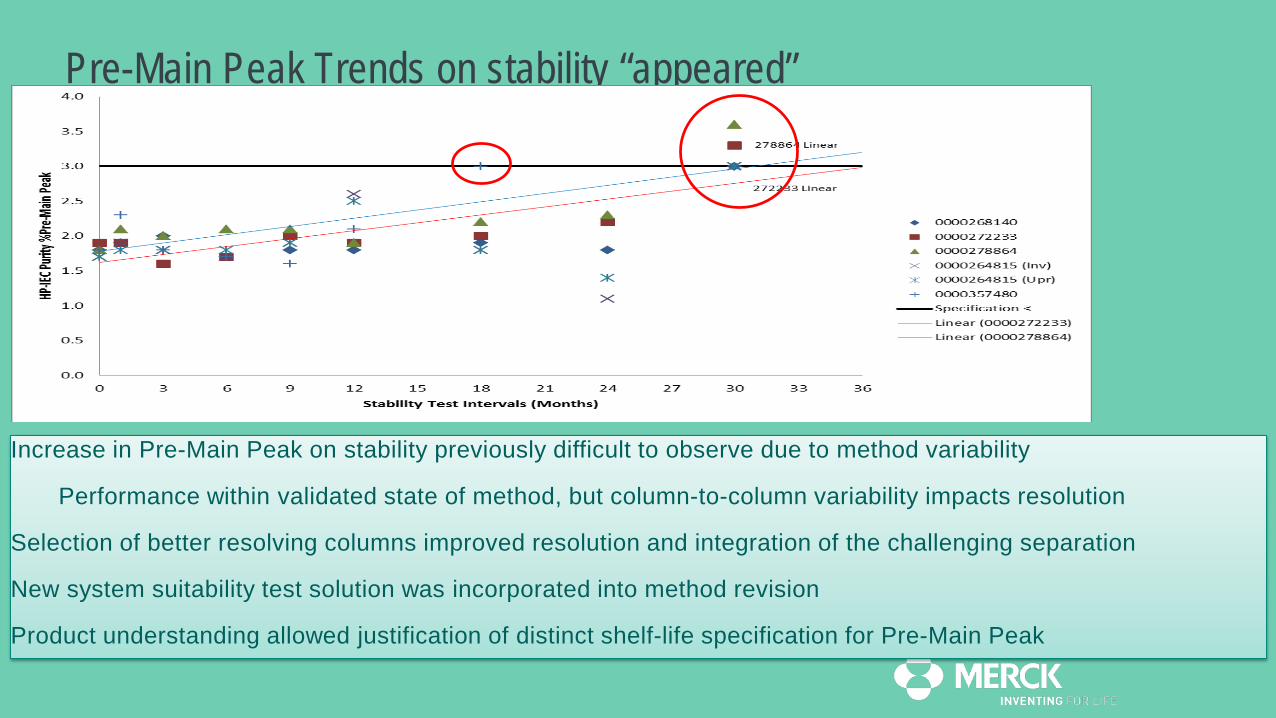
Pre-Main Peak Trends on stability “appeared”
Increase in Pre-Main Peak on stability previously difficult to observe due to method variability
Performance within validated state of method, but column-to-column variability impacts resolution
Selection of better resolving columns improved resolution and integration of the challenging separation
New system suitability test solution was incorporated into method revision
Product understanding allowed justification of distinct shelf-life specification for Pre-Main Peak

Limited data for ELISA method led to establishment of a very challenging parameter (EC50) for Reference Standard recertification
• EC50 values are one of the four parameters derived from the shape of an ELISA curve
• There is no EC50 acceptance criterion for routine batch release and stability
• To help control for potential reference standard drift, an EC50 criterion for ref std stability (requalification) testing was incorporated
• Acceptance criterion was set based on limited data from method validation (one lab, two operators)(5.0 – 8.0 ng/mL); was very difficult to meet during PRS/SRS requalification– Critical reagents exhibit lot-to-lot variability with impact on
EC50 (but not necessarily on relative potency)– Typical values at different sites over time were considerably
broader than the requalification range
Upper Asymptote (UA)
Lower Asymptote (LA)
RS Slope
Control/Sample Slope
Slope Ratio =Control/Sample Slope
RS Slope
UA Ratio =Control/Sample UA
RS UA
LA Ratio =Control/Sample LA
RS LA
Root Sum of SquaresError (RSSE)
UA/LA Ratio
EC50

Primary Root Cause of EC50 variability• Critical reagent lots used in method impact EC50 values
13
DKL714101
DKL2015031
Representative binding curves with two different ligand lots (other reagents identical between runs)Lot DKL2015031: lower RLU, shallower curve, lower EC50Lot DKL714101: higher RLU, steeper curve, higher EC50
Variability in EC50 has also been influenced by age of reagents lots (trends over time for lots shown above)
Above: EC50 of assay control(blue) and ref std (green) move together (not due to changes in stability)

PAS/Variations established new approach for Ref Std/EC50• Introduced updated Ref Std/EC50/Potency ELISA • NO EC50 requirement for reference standard
(re)qualification• Rather than relying on an absolute EC50 criterion for
reference standard stability, enhanced control strategy includes:
– Bolstered annual stability components with orthogonal methods (see right), including ELISA Assay Control potency over time (top), size exclusion (middle), and charge distribution (bottom)
– ELISA Method Performance: In addition to monitoring relative potency, will monitor reference standard EC50 with site-independent alert limit
– Critical Reagents: Established EC50 range as AC for BCR qualifications (ensuring quality reagents for the method)
ELIS
A As
say C
ontro
l Re
lPot
ency
HP-S
EC H
MW
HP-IE
X M
ain P
eak

Summary and Points to Consider
• Rapid product development applies pressure on method and process development
• Limited information about the product, process, and methods increases risk– What is true method performance and variability?– What is an appropriate release or stability specification? Can your methods handle it?
Can your process handle it?
• Platform methods now used: streamlines development and leverages previous experiences
• It is very challenging to “fix” a method that was “good enough” for commercialization– Bridging studies and what one may find– Global implementation (change control and regulatory rollout)
15





























