Embed Size (px)
Citation preview

JOURNAL OF BACTERIOLOGY, Apr. 2005, p. 2469–2482 Vol. 187, No. 70021-9193/05/$08.00�0 doi:10.1128/JB.187.7.2469–2482.2005Copyright © 2005, American Society for Microbiology. All Rights Reserved.
Analysis of the Hypervariable Region of the Salmonella entericaGenome Associated with tRNAleuX†
Anne L. Bishop,1 Stephen Baker,2 Sara Jenks,1 Maria Fookes,2 Peadar O Gaora,1Derek Pickard,2 Muna Anjum,3 Jeremy Farrar,4 Tran T. Hien,5
Al Ivens,2 and Gordon Dougan2*The Wellcome Trust Sanger Institute, Wellcome Trust Genome Campus, Hinxton, Cambridge,2 Centre for Molecular
Microbiology and Infection, Department of Biological Sciences, Imperial College London, London,1 andDepartment of Food and Environmental Safety, Veterinary Laboratories Agency—Weybridge,
New Haw, Addlestone, Surrey,3 United Kingdom, and Oxford University ClinicalResearch Unit4 and Hospital for Tropical Diseases,5
Ho Chi Minh City, Vietnam
Received 7 April 2004/Accepted 8 December 2004
The divergence of Salmonella enterica and Escherichia coli is estimated to have occurred approximately 140million years ago. Despite this evolutionary distance, the genomes of these two species still share extensivesynteny and homology. However, there are significant differences between the two species in terms of genesputatively acquired via various horizontal transfer events. Here we report on the composition and distributionacross the Salmonella genus of a chromosomal region designated SPI-10 in Salmonella enterica serovar Typhiand located adjacent to tRNAleuX. We find that across the Salmonella genus the tRNAleuX region is a hyper-variable hot spot for horizontal gene transfer; different isolates from the same S. enterica serovar can exhibitsignificant variation in this region. Many P4 phage, plasmid, and transposable element-associated genes arefound adjacent to tRNAleuX in both Salmonella and E. coli, suggesting that these mobile genetic elements haveplayed a major role in driving the variability of this region.
Salmonella enterica and Escherichia coli are thought to havediverged from a common ancestor approximately 140 millionyears ago (37, 39). Despite this length of time, the genomes ofthese two members of the Enterobacteriaceae exhibit significanthomology and synteny (35). This conservation of the genomebackbone may be a reflection of the similar/overlapping envi-ronments occupied by E. coli and S. enterica. Dispersedthroughout the genomes of members of S. enterica and otherenteric bacteria are a number of horizontally acquired DNAsegments (38), some of which contribute to pathogenicity(pathogenicity islands, or PIs). Perhaps the best characterizedexamples of Salmonella pathogenicity islands (SPIs) are SPI-1and SPI-2, which encode type III secretion systems (24). PIsare frequently associated with mobile genetic elements, includ-ing transposons and bacteriophages, and are often found ad-jacent to tRNA genes (29, 34). Complete DNA sequencing ofthe genomes of several S. enterica and E. coli has led to theidentification of different combinations of PIs among strains.Although some of the PIs are highly conserved between dif-ferent S. enterica serovars, others are very divergent (15, 35, 40,44).
This study investigates the composition of a region adjacentto tRNAleuX, which was termed SPI-10 and is positioned at93.4 centisomes (genome coordinates 4683690 to 4716539) inthe Salmonella enterica serovar Typhi CT18 genome sequence
(40). Analysis of the tRNAleuX-associated region in silico anduse of microarrays, Southern blotting, and PCR reveal that thisis a hot spot for divergence within both Salmonella and E. coli.The many phage, plasmid, and transposable element-relatedgenes or gene fragments found within this region in both S.enterica and E. coli may be a major driving force for the ob-served hypervariability.
MATERIALS AND METHODS
Strains. The S. enterica and Salmonella bongori strains used in this study werea combination of both clinical and reference collections (12, 13). Isolates thatwere cultured for genetic analysis are detailed in Table 1. Those that wereanalyzed purely in silico are detailed in the “In silico genome analysis” sectionbelow. Bacteria were routinely cultured in Luria-Bertani broth or on Luria-Bertani agar overnight at 37°C.
In silico genome analysis. The E. coli complete genome sequences analyzedwere the nonpathogenic K-12 strain MG1655 (accession number NC_000913) (9)and pathogenic O157:H7 substrain RIMD 0509952 (28) (Sakai outbreak isolate,accession number NC_002695). The Salmonella complete genomes analyzedwere S. enterica serovar Typhi CT18 (40) (accession number NC_003198) andSalmonella enterica serovar Typhimurium LT2 (35) (accession numberNC_003197). The recently completed, but unannotated, genomes of S. bongori(strain 12419) and Salmonella enterica serovar Enteritidis (strain PT4) (www.sanger.ac.uk/Projects/Salmonella) were compared in detail with the fully se-quenced genomes. The partially sequenced genome of Salmonella enterica sero-var Paratyphi A (strain ATCC 9150) (www.genome.wustl.edu) was also analyzed,as described below.
Fully sequenced and annotated bacterial genomes were compared pairwise byusing MegaBLAST (51) and visualized by using the Artemis Comparison Tool(http://www.sanger.ac.uk/Software/ACT). Complete, but unannotated, genomesequences were compared pairwise with annotated genomes by using the MUM-mer DNA-DNA alignment tool (18). Open reading frames were predicted byGlimmer software (17), and the annotation of individual genes was refined byusing the sequence alignment tools BLASTN, BLASTX (1), and BLAST 2sequences (47) and the Conserved Domain Database (33). Artemis (http:
* Corresponding author. Mailing address: The Wellcome TrustSanger Institute, Wellcome Trust Genome Campus, Hinxton, Cam-bridge CB10 1SA, United Kingdom. Phone: 441223495381. Fax:441223494919. E-mail: [email protected].
† Supplemental material for this article may be found at http://jb.asm.org/.
2469
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
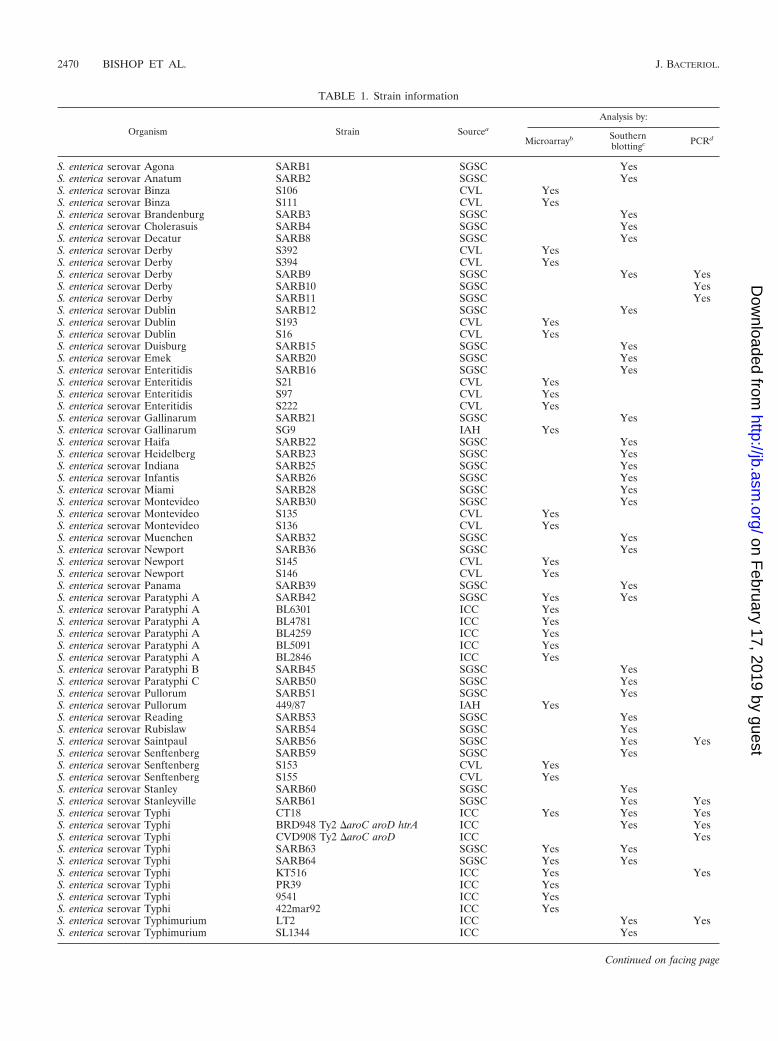
TABLE 1. Strain information
Organism Strain Sourcea
Analysis by:
Microarrayb Southernblottingc PCRd
S. enterica serovar Agona SARB1 SGSC YesS. enterica serovar Anatum SARB2 SGSC YesS. enterica serovar Binza S106 CVL YesS. enterica serovar Binza S111 CVL YesS. enterica serovar Brandenburg SARB3 SGSC YesS. enterica serovar Cholerasuis SARB4 SGSC YesS. enterica serovar Decatur SARB8 SGSC YesS. enterica serovar Derby S392 CVL YesS. enterica serovar Derby S394 CVL YesS. enterica serovar Derby SARB9 SGSC Yes YesS. enterica serovar Derby SARB10 SGSC YesS. enterica serovar Derby SARB11 SGSC YesS. enterica serovar Dublin SARB12 SGSC YesS. enterica serovar Dublin S193 CVL YesS. enterica serovar Dublin S16 CVL YesS. enterica serovar Duisburg SARB15 SGSC YesS. enterica serovar Emek SARB20 SGSC YesS. enterica serovar Enteritidis SARB16 SGSC YesS. enterica serovar Enteritidis S21 CVL YesS. enterica serovar Enteritidis S97 CVL YesS. enterica serovar Enteritidis S222 CVL YesS. enterica serovar Gallinarum SARB21 SGSC YesS. enterica serovar Gallinarum SG9 IAH YesS. enterica serovar Haifa SARB22 SGSC YesS. enterica serovar Heidelberg SARB23 SGSC YesS. enterica serovar Indiana SARB25 SGSC YesS. enterica serovar Infantis SARB26 SGSC YesS. enterica serovar Miami SARB28 SGSC YesS. enterica serovar Montevideo SARB30 SGSC YesS. enterica serovar Montevideo S135 CVL YesS. enterica serovar Montevideo S136 CVL YesS. enterica serovar Muenchen SARB32 SGSC YesS. enterica serovar Newport SARB36 SGSC YesS. enterica serovar Newport S145 CVL YesS. enterica serovar Newport S146 CVL YesS. enterica serovar Panama SARB39 SGSC YesS. enterica serovar Paratyphi A SARB42 SGSC Yes YesS. enterica serovar Paratyphi A BL6301 ICC YesS. enterica serovar Paratyphi A BL4781 ICC YesS. enterica serovar Paratyphi A BL4259 ICC YesS. enterica serovar Paratyphi A BL5091 ICC YesS. enterica serovar Paratyphi A BL2846 ICC YesS. enterica serovar Paratyphi B SARB45 SGSC YesS. enterica serovar Paratyphi C SARB50 SGSC YesS. enterica serovar Pullorum SARB51 SGSC YesS. enterica serovar Pullorum 449/87 IAH YesS. enterica serovar Reading SARB53 SGSC YesS. enterica serovar Rubislaw SARB54 SGSC YesS. enterica serovar Saintpaul SARB56 SGSC Yes YesS. enterica serovar Senftenberg SARB59 SGSC YesS. enterica serovar Senftenberg S153 CVL YesS. enterica serovar Senftenberg S155 CVL YesS. enterica serovar Stanley SARB60 SGSC YesS. enterica serovar Stanleyville SARB61 SGSC Yes YesS. enterica serovar Typhi CT18 ICC Yes Yes YesS. enterica serovar Typhi BRD948 Ty2 �aroC aroD htrA ICC Yes YesS. enterica serovar Typhi CVD908 Ty2 �aroC aroD ICC YesS. enterica serovar Typhi SARB63 SGSC Yes YesS. enterica serovar Typhi SARB64 SGSC Yes YesS. enterica serovar Typhi KT516 ICC Yes YesS. enterica serovar Typhi PR39 ICC YesS. enterica serovar Typhi 9541 ICC YesS. enterica serovar Typhi 422mar92 ICC YesS. enterica serovar Typhimurium LT2 ICC Yes YesS. enterica serovar Typhimurium SL1344 ICC Yes
Continued on facing page
2470 BISHOP ET AL. J. BACTERIOL.
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
//www.sanger.ac.uk/Software/Artemis) was used to aid in the annotation of eachtRNAleuX island.
All contigs from the incomplete Salmonella serovar Paratyphi A strain ATCC9150 genome sequence (http://genome.wustl.edu/projects/bacterial/sparatyphiA/)were compared with the fully sequenced Salmonella serovar Typhi CT18 genome(40) and ordered according to the coordinates of the best alignments on theCT18 genome. From this ordered partial genome sequence, four nonoverlappingcontigs, which cover the region depicted in Fig. 1 (STY4820-53), were thenconcatenated, compared with complete genome sequences, and visualized byusing the Artemis Comparison Tool. By comparison with Salmonella serovarTyphi CT18, it was found that tRNAleuX mapped between the first two contigsand was missing from the available Salmonella serovar Paratyphi A sequence.PCR and sequencing were carried out to confirm the presence of tRNAleuX at thestart of the island in Salmonella serovar Paratyphi A (described below). Theother two joins between contigs (within genes resembling Salmonella serovarTyphi CT18 STY4832 and STY4849) were not closed in this way.
Microarray analysis of genomic DNA. The design and construction of theSalmonella serovar Typhi CT18 microarray used in this study is described else-where (48). The array contains 4,097 screened and refined PCR products (200 to500 bases) from annotated coding sequences from the chromosome of Salmo-nella serovar Typhi CT18 (40). Genomic DNA was extracted by using the hexa-decyltrimethylammonium bromide (Sigma-Aldrich Ltd., Dorset, United King-dom) method (3). DNA from 40 S. enterica or S. bongori strains (Table 1) plusCT18 itself was sonicated (10 s, level 2, Virsonic sonicator) and labeled withFluoroLink Cy3 or Cy5 dyes by using the Bioprime kit (Invitrogen Ltd., Paisley,United Kingdom). In each case, Salmonella serovar Typhi CT18 DNA was usedas the reference strain. Labeled DNA was then purified by using an AutoseqG-50 column (Amersham Biosciences, Chalfont St. Giles, United Kingdom),denatured, and precipitated, and finally, the probes were hybridized to theSalmonella microarray slide overnight at 49°C (http://www.sanger.ac.uk/Projects/Microarrays/arraylab/methods.shtml).
After stringent washing, hybridization results were detected by using a Gene-pix 4000B scanner (Axon Instruments, Inc., Union City, Calif.) and quantifiedwith Genepix Pro software (Axon Instruments, Inc.). Data analysis was carriedout as described by Thomson et al. (48). The final readouts were mean Lowess-normalized Cy3/Cy5 ratio intensities [Ln(Cy3/Cy5)] for up to eight data points(four arrays carried out for each test strain and each gene spotted in duplicate).Data were ordered and labeled according to the Salmonella serovar Typhi CT18genome gene names (STY0001 to STY4949). The presence/conserved or ab-sence/divergent status of each gene was assigned according to its final Ln(Cy3/Cy5) ratio intensity and was given a standard GeneSpring colored representation(color scale represented in Fig. 2), as follows: absent/divergent genes, Ln(Cy3/Cy5) below 0.3, blue; genes that could not be determined present/conserved orabsent/divergent, Ln(Cy3/Cy5) between 0.3 and 0.45; present/conserved genes,Ln(Cy3/Cy5) greater than or equal to 0.45, yellow; missing data, grey.
Southern blotting. Genomic DNA (2.5 to 5 �g) was restricted with EcoRV(New England Biolabs [United Kingdom] Ltd., Hitchin, United Kingdom) over-
night at 37°C and separated on a 1% agarose gel. Using standard methodsdescribed previously (45), the gel was treated and transferred to a Hybond-N�
membrane (Amersham Biosciences), which was then UV fixed. Southern blotprobes were designed to encompass at least 600 bp of each gene, or genes, to bedetected and were based upon either Salmonella serovar Typhimurium LT2 orSalmonella serovar Typhi CT18 genome sequences. Probes were prepared byPCR with primers (see Table S1 in the supplemental material) to detect thefollowing genes: for LT2, STM4493 and STM4496-98; for CT18, aroC(STY2616) and STY4822. PCR was carried out with 10 ng of genomic templateDNA, Red Taq (Sigma-Aldrich) in the buffer provided by the manufacturer(containing 1.5 mM Mg2�), 200 �M concentrations of deoxynucleoside triphos-phates, and 10 pmol of each primer. PCR conditions were as follows: 94°C for 1min; 30 cycles of 94°C for 30 s, 54°C for 30 s, and 72°C for 2 min; and finalelongation at 72°C for 2 min. PCR products were purified by using QIA quickPCR purification kits (QIAGEN Ltd., Crawley, United Kingdom). To confirmthat the correct PCR products had been amplified, the sizes of the products werechecked and restriction patterns (restriction enzymes from New England Bio-labs) were assessed: the aroC PCR product was digested with SmaI, the STY4822PCR product was digested with KpnI, the STM4493 PCR product was digestedwith BamHI, and the STM4496-98 PCR product was digested with PstI. Randomprime labeling of probes (using fluorescein-labeled nucleic acids, random prim-ers, and Klenow polymerase), hybridizations (60°C overnight), and an enhancedchemiluminescence-based detection were carried out by using the Randomprime labeling and detection system version II (Amersham Biosciences) accord-ing to the manufacturer’s instructions.
The stringencies of the Southern hybridizations were assessed by using theCT18-drived aroC probe to blot for aroC in E. coli K-12 (83% identity within theprobe region) and Salmonella serovar Typhimurium LT2 (98% identity withinthe probe region). With 2.5 �g of DNA (the minimum amount used subse-quently), a weak, but clearly visible, aroC DNA fragment was seen with K-12DNA (data not shown). This suggests that, under the blotting conditions used,any gene with at least 83% identity to the probe should be picked up. Also, itshould be noted that the aroC probe is the shortest of the probes used in theSouthern blotting screen (see Table S1 in the supplemental material), whichshould make it the least sensitive (45).
PCR screening of tRNAleuX-associated genes in different salmonellae. PCRcomparisons of tRNAleuX-associated genes in different salmonellae were carriedout by using either the Salmonella serovar Typhimurium LT2 or Salmonellaserovar Typhi CT18 genome sequence for primer design as appropriate. For thePCRs shown in Fig. 3a, Fig. 5i and Fig. 6i to iii, amplification reactions werecarried out by using Red Taq DNA polymerase as described above for thepreparation of PCR products for Southern blot probes but with 3-min 72°Celongation steps and primers detailed in Table S1 in the supplemental material.For the PCRs shown in Fig. 5ii and Fig. 6iv, the Expand high-fidelity PCR system(Roche Diagnostics Ltd., Lewes, United Kingdom) was used according to themanufacturer’s instructions, as it contains a polymerase that is more efficientthan Red Taq for amplification of larger DNA fragments, with 8-min elongation
TABLE 1—Continued
Organism Strain Sourcea
Analysis by:
Microarrayb Southernblottingc PCRd
S. enterica serovar Typhimurium S6332 Copenhagen CVL YesS. enterica serovar Typhimurium S1055 Copenhagen CVL YesS. enterica serovar Typhimurium S56 CVL YesS. enterica serovar Typhimurium S204 CVL YesS. enterica serovar Typhimurium S23 CVL YesS. enterica serovar Typhimurium S39 CVL YesS. enterica serovar Typhimurium 4015 ICC YesS. enterica serovar Typhimurium 4311 ICC YesS. enterica serovar Typhimurium mar227 ICC YesS. enterica serovar Typhisuis SARB70 SGSC YesS. enterica serovar Wien SARB71 SGSC YesS. bongori SARC11 SGSC Yes
a SGSC, Salmonella Genetic Stock Center, Calgary, Canada; CVL, Central Veterinary Laboratories, Colindale, United Kingdom; ICC, Imperial College Collection,London, United Kingdom; IAH, Institute for Animal Health, Compton, United Kingdom.
b Results are shown in Fig. 2.c Results are shown in Table 2.d Results are shown in Fig. 3, 5, and 6.
VOL. 187, 2005 A HYPERVARIABLE REGION OF THE SALMONELLA GENOME 2471
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

steps at 68°C, an annealing temperature of 54°C, and primers that are detailed inTable S1 in the supplemental material.
A number of PCR products were cloned and sequenced as follows. To confirmthe presence of tRNAleuX between STY4820 (similar to STM4487)- andSTY4821-like genes in Salmonella serovar Paratyphi A, PCR was carried outwith Salmonella serovar Paratyphi A SARB42 template DNA, with Red Taqpolymerase (as described above) and primers STM4487_for and STY4821_rev2.Additional novel PCR products were generated from SARB61 and SARB56genomic DNA with STM4490_for and STM4492_rev primers and from SARB56with STM4492_for and STM4495_rev primers, all with Red Taq DNA polymer-ase. Amplification from SARB10 DNA with tRNA_for and STM4488_rev prim-ers was carried out by using the Expand high-fidelity PCR system (conditionsdescribed above, but 5-min 68°C elongation steps). For each of these PCRs, the
primers are detailed in Table S1 in the supplemental material. Each PCR prod-uct, from at least two separate PCRs in each case, was purified by using theQIAGEN PCR purification kit and sequenced both directly and after cloninginto pGEM-T Easy (Promega United Kingdom Ltd., Southampton, UnitedKingdom).
PCR-based detection of circularized ST46 phage. PCR for detection of circu-larized ST46 phage in genomic Salmonella serovar Typhi DNA preparations(Fig. 3b) was carried out by using Red Taq DNA polymerase as described abovebut using 3-min elongation steps. The primers used to amplify the PCR productsshown in Fig. 3b were as follows. Circular ST46 was amplified by using eitherST46_rev (ATCAATGCCCTGCACTAGCAAC, reverse-strand primer withinintegrase gene termed STY4821 in CT18) with ST46_for (CAGACCAACGGGATGTTTATGG, forward-strand primer within P4 phage � gene termed
FIG. 1. Comparisons of sequenced E. coli and Salmonella genomes reveal seven different organizations of tRNAleuX-adjacent genes. Predictedintact open reading frames and pseudogenes (marked with a cross) are shown as arrows indicating the direction of transcription. The Salmonellaserovar Typhimurium LT2 STY4854-like pseudogene is shown as a cross without an arrow because it has lost its start codon. The boundaries ofeach island are marked with thick vertical lines. Genes shaded with spots are common to two or more of the islands shown and therefore lie outsideof the tRNAleuX-adjacent islands as defined in this study. Genes of related origin or function are shaded similarly, as indicated in the key. Thecomplete fully annotated genome sequences analyzed are those of E. coli nonpathogenic K-12 (strain MG1655 accession number NC_000913) (9)and pathogenic O157:H7 substrain RIMD 0509952 (28) (accession number NC_002695), Salmonella serovar Typhi CT18 (40) (accession numberNC_003198), and Salmonella serovar Typhimurium LT2 (35) (accession number NC_003197). Gene numbers assigned by the sequencing projectsare indicated for LT2 (35) and CT18 (40). Partially sequenced or fully sequenced, but unannotated, genomes that were also compared are asfollows: S. enterica subsp. I serovars Enteritidis (strain PT4) and Paratyphi A (strain ATCC 9150) and S. bongori (stain 12419) (www.sanger.ac.uk/Projects/Salmonella and www.genome.wustl.edu). The genes and pseudogenes shown for Salmonella serovar Paratyphi A are predicted to bepresent by comparison with Salmonella serovar Typhi CT18. The incomplete Salmonella serovar Paratyphi A genome sequence contains manyadditional frameshifts and is missing regions between contigs compared with CT18, but as these features remain to be verified, they are notrepresented here.
2472 BISHOP ET AL. J. BACTERIOL.
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
STY4832 in CT18) or STY4822_rev2 (CTAGAGCACACCCTGTAATTCTTTG, reverse-strand primer within ST46-specific phage cos site insertion genetermed STY4822 in CT18) with ST46_for. Chromosomal ST46 was amplifiedusing either STY4835_rev (AATGCGGGCAGCAGTGTTTA) with ST46_for orSTM4487_for (which recognizes STY4820, listed in Table S1 in the supplementalmaterial) with ST46_rev. Control PCRs amplified either chromosomal genesoutside ST46 by using STY4835_for (TAAACACTGCTGCCCGCATT) withSTY4836a_rev (CCTGAACCTCTGCTTTGTTA) or all ST46 phage by usingSTY4822_for with STY4822_rev (listed in Table S1 in the supplemental mate-rial). Circular ST46 phage-derived PCR products amplified from BRD948genomic DNA were gel purified by using a QIAGEN gel purification kit andsequenced to confirm that they were the expected products.
Genome walking for analysis of Salmonella serovar Typhi isolate KT516tRNAleuX island. Genome walking was carried out by using the UniversalGenomeWalker kit (BD Biosciences, Palo Alto, Calif.). Genomic DNA (1.5 �g)was digested with enzyme DraI, EcoRV, or SspI (New England Biolabs), and
DNA was phenol-chloroform purified, precipitated, and resuspended in 20 �l ofdistilled H2O. Four microliters of each digested DNA solution was ligated toadaptors provided by the manufacturer, the ligase was heat inactivated, and thesolution was made up to 80 �l with Tris-EDTA buffer. PCR amplification wascarried out by using the Expand high-fidelity PCR system (conditions describedabove but with 6-min extensions that were modified for the final 20 cycles with15-s increments per cycle). First-round PCR mixtures contained 1 �l of eachligation mixture as the template and used adaptor primer 1 (AP1, GTAATACGACTCACTATAGGGC) with tRNAfor_outer (CGTTTTCCGCATACCTCTTC) or with STY4835rev_outer (CTTTGCCAGCCGGGAAATAATG) and anannealing temperature of 55°C. Second-round PCR mixtures contained 1 �l offirst-round PCR product as a template and used adaptor primer 2 (AP-2, ACTATAGGGCACGCGTGGT) with tRNAfor_inner (AAGTGGCGAAATCGGTAGAC) or with STY4835_rev (see Table S1 in the supplemental material) asappropriate and an annealing temperature of 57°C. PCR products amplified in
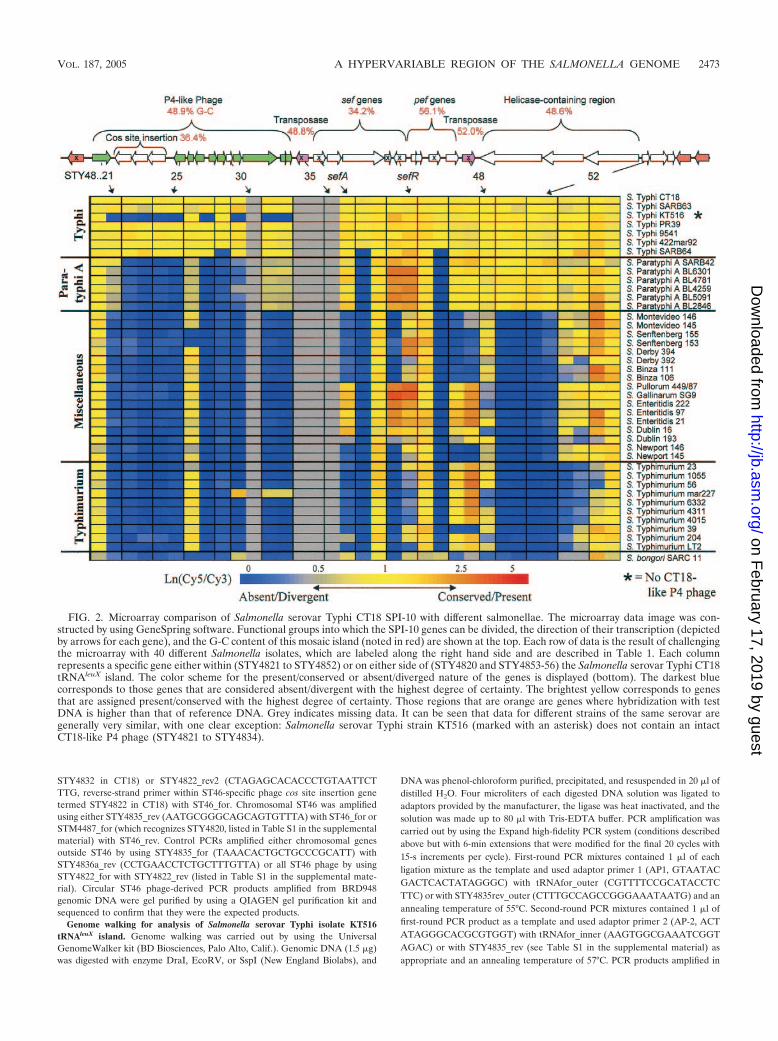
FIG. 2. Microarray comparison of Salmonella serovar Typhi CT18 SPI-10 with different salmonellae. The microarray data image was con-structed by using GeneSpring software. Functional groups into which the SPI-10 genes can be divided, the direction of their transcription (depictedby arrows for each gene), and the G-C content of this mosaic island (noted in red) are shown at the top. Each row of data is the result of challengingthe microarray with 40 different Salmonella isolates, which are labeled along the right hand side and are described in Table 1. Each columnrepresents a specific gene either within (STY4821 to STY4852) or on either side of (STY4820 and STY4853-56) the Salmonella serovar Typhi CT18tRNAleuX island. The color scheme for the present/conserved or absent/diverged nature of the genes is displayed (bottom). The darkest bluecorresponds to those genes that are considered absent/divergent with the highest degree of certainty. The brightest yellow corresponds to genesthat are assigned present/conserved with the highest degree of certainty. Those regions that are orange are genes where hybridization with testDNA is higher than that of reference DNA. Grey indicates missing data. It can be seen that data for different strains of the same serovar aregenerally very similar, with one clear exception: Salmonella serovar Typhi strain KT516 (marked with an asterisk) does not contain an intactCT18-like P4 phage (STY4821 to STY4834).
VOL. 187, 2005 A HYPERVARIABLE REGION OF THE SALMONELLA GENOME 2473
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
the second round were gel purified by using the QIAGEN gel purification kit andsequenced.
Novel DNA sequence accession numbers. The Salmonella serovar Paratyphi ASARB42 tRNAleuX region (STY4820- to STY4821-like genes) PCR product wasassigned accession number AY775127. The Salmonella enterica serovar DerbySARB10 insertion between tRNAleuX and the STM4488-like gene was assignedaccession number AY786416. The SARB56 novel region within the insertionbetween STM4490 and STM4491 was assigned accession number AY775129.The SARB61 novel region within the insertion between STM4490 and STM4491was assigned accession number AY775128. The Salmonella serovar Typhi KT516genome walking 5� sequence was assinged accession number AY795072. TheSalmonella serovar Typhi KT516 genome walking 3� sequence was assignedaccession number AY795073.
RESULTS AND DISCUSSION
Comparison of tRNAleuX-associated regions of E. coli andSalmonella genomes reveals distinct organizations. During ourefforts to annotate the Salmonella serovar Typhi CT18 ge-nome, we noted that the tRNAleuX-associated region of differ-ent S. enterica and E. coli isolates are extremely variable, oftenencoding completely unrelated DNA sequences and genes.
Thus, the tRNAleuX may be one of the most variable regions ofthe S. enterica and E. coli genomes. Consequently, we decidedto analyze this variation within S. enterica in more detail. Ini-tially, we examined the tRNAleuX-associated genes from se-quenced Salmonella and E. coli strains by using in silico meth-ods. To support and extend these studies, we utilized S.enterica-based microarrays, Southern blotting, PCR, and DNAsequencing to characterize this region from representative iso-lates of different S. enterica serovars. Our aim was to try tocapture information about the mechanisms driving this diver-sity and the type of genes acquired by this region in different S.enterica serovars. Since we analyzed a large number of differentDNA sequences, we present here the key conclusions of thisstudy but provide an in depth analysis of individual sequencesas a supplementary data set. This supplementary set can beused by others to facilitate genetic analysis of particular sero-vars and to construct broader and more representative S. en-terica-based microarrays.
The tRNAleuX-associated region of Salmonella serovar
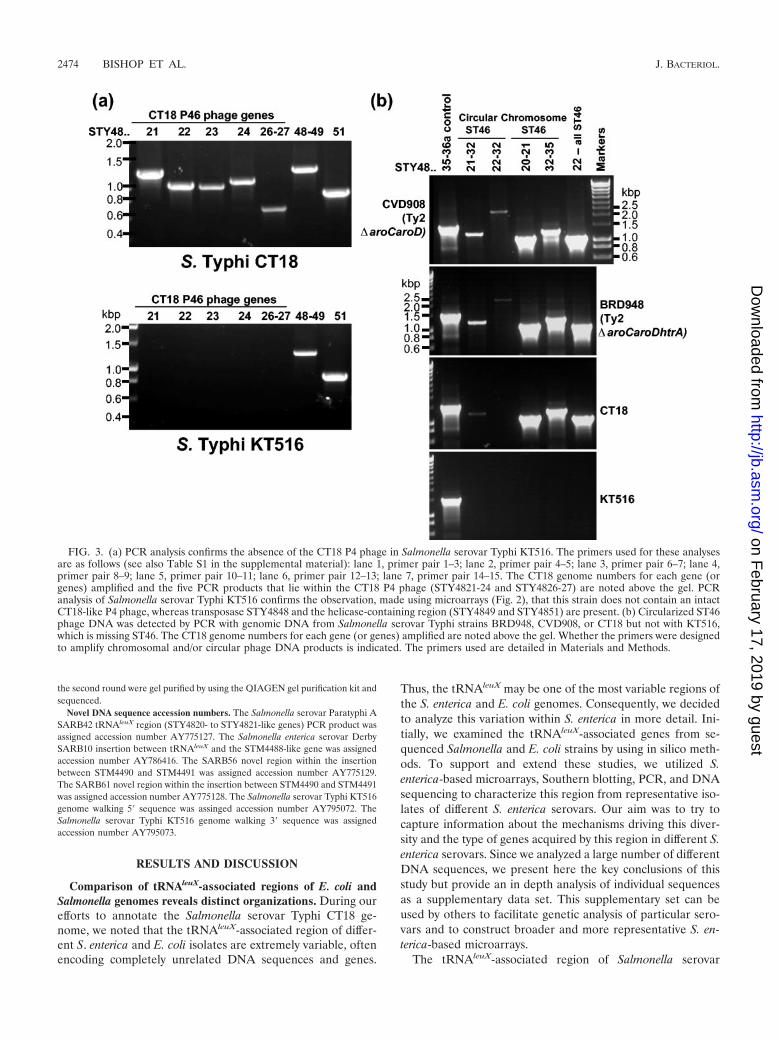
FIG. 3. (a) PCR analysis confirms the absence of the CT18 P4 phage in Salmonella serovar Typhi KT516. The primers used for these analysesare as follows (see also Table S1 in the supplemental material): lane 1, primer pair 1–3; lane 2, primer pair 4–5; lane 3, primer pair 6–7; lane 4,primer pair 8–9; lane 5, primer pair 10–11; lane 6, primer pair 12–13; lane 7, primer pair 14–15. The CT18 genome numbers for each gene (orgenes) amplified and the five PCR products that lie within the CT18 P4 phage (STY4821-24 and STY4826-27) are noted above the gel. PCRanalysis of Salmonella serovar Typhi KT516 confirms the observation, made using microarrays (Fig. 2), that this strain does not contain an intactCT18-like P4 phage, whereas transposase STY4848 and the helicase-containing region (STY4849 and STY4851) are present. (b) Circularized ST46phage DNA was detected by PCR with genomic DNA from Salmonella serovar Typhi strains BRD948, CVD908, or CT18 but not with KT516,which is missing ST46. The CT18 genome numbers for each gene (or genes) amplified are noted above the gel. Whether the primers were designedto amplify chromosomal and/or circular phage DNA products is indicated. The primers used are detailed in Materials and Methods.
2474 BISHOP ET AL. J. BACTERIOL.
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

Typhi CT18 is known as SPI-10 and encodes a P4-like phage,the sef/pef fimbrial islet, IS element remnants, and genes ofunknown function. Comparisons of the Salmonella serovarTyphi SPI-10 sequence with other sequenced enteric bacteriarevealed remarkable heterogeneity (this study and previouspartial sequence data) (21). For example, the gene comple-ments at this locus in Salmonella serovar Typhi and Salmonellaserovar Typhimurium are entirely different (Fig. 1). ThetRNAleuX-associated islands ranged in size from over 44 kbp inE. coli O157:H7 Sakai to just over 11.6 kbp in S. bongori 12418.The predicted characteristics of some of the genes adjacent totRNAleuX are depicted in Fig. 1 and are described in moredetail in the supplemental material (see Tables S2 to S7 in thesupplemental material). General themes of the tRNAleuX is-lands are the appearance of genes related to P4 phages andtransposable elements and plasmids, suggesting that mobileelements may drive the hypervariability in this region (Fig. 1).In some cases, the mosaic structures of these islands are alsoreflected in variations in G-C content. For example, the regionencompassing the sef fimbrial genes has a particularly low G-Ccontent (34.14% compared to 52.17% in Salmonella serovarEnteritidis PT4, as previously noted [21], and 34.2% in Salmo-nella serovar Typhi CT18 compared with 52.09% overall in thegenome) (Fig. 2). A region containing three nonphage genes,STY4822-24, inserted into the Salmonella serovar Typhi CT18P4 phage also has a low G-C content (36.4%) (Fig. 2).
The GntII (subsidiary) system for gluconate metabolism andL-idonic acid catabolism (5) lies immediately 5� of tRNAleuX inE. coli K-12. A series of small deletions and rearrangementscan be detected in different salmonellae, but the region ofmajor divergence begins 3� of tRNAleuX. Uropathogenic E. colistrain 563 harbors a distinct PI at this locus, the ends of whichare clearly defined by repeats of 18 bp (20). In contrast, repeatsdo not appear to mark the ends of the islands shown in Fig. 1.Orthologs of two genes, yeeN (encoding a conserved hypothet-ical protein) and yjhP (encoding a putative methyltransferase),are most commonly present at the 3� end of the tRNAleuX
islands. The S. bongori 12419 island ends with two adjacentgenes, ORF_5468 and ORF_5469, which are together 84%identical to the full Salmonella serovar Typhi CT18 yjhP-likegene STY4856. In E. coli K-12, the sgc operon, which codes fora potential phosphoenolpyruvate sugar phosphotransferasesystem, is present upstream of yjhP. This can be considered tobe the end of the K-12-specific island, as Salmonella serovarTyphi CT18 also contains orthologs of the sgc system at adifferent position in the genome (STY1447 to STY1450 andSTY1452). Delineation of the borders of the tRNAleuX islandswas supported for a number of different Salmonella by mi-croarray analysis, which yielded a positive hybridization forSTY4820, just prior to tRNAleuX, for all strains tested (Fig. 2).The 3� limit of the Salmonella serovar Typhi CT18 serovar-specific PI was less distinct (Fig. 2), but the in silico assignmentof STY5852 as the last Salmonella serovar Typhi-specificSPI-10 gene is supported by the microarray data. STY4852 isconserved in all Salmonella serovar Paratyphi A and Salmo-nella serovar Typhi strains tested but is absent from all otherSalmonella strains, whereas STY4853 is detected in many Sal-monella strains, with the marked exception of all 10 Salmonellaserovar Typhimurium strains tested (Fig. 2). STY4854 is sim-ilar in distribution to ST4853, but two Salmonella serovar Ty-
phimurium isolates (39 and 204) show positive STY4854 hy-bridizations, the majority of Salmonella isolates harborSTY4855, with exceptions being Salmonella serovar Typhi-murium isolates 23, 1055, and 56, and all Salmonella isolatestested positive for STY4856 (Fig. 2).
P4 family phages as drivers of diversity in S. enterica. AP4-related prophage, termed ST46 (48), lies adjacent totRNAleuX in Salmonella serovar Typhi CT18. P4-like phagesare known to target a 20-bp sequence within tRNAleuX, result-ing in duplication of the attachment site sequence at either endof the prophage insertion (42). Remnants of one such dupli-cation flank the P4 phage of Salmonella serovar Typhi: 19 bp ofthe 20-bp tRNAleuX attP P4 phage attachment site are dupli-cated within the predicted P4 phage-related gene STY4834 atthe extreme 3� end of the prophage element. Salmonella sero-var Paratyphi A isolate ATCC 9150 harbors a similar P4-likeprophage at the same position (Fig. 1). The only major differ-ence between the Salmonella serovar Typhi and Salmonellaserovar Paratyphi A P4 phages are the genes that are insertedinto the phage cos site, which is a common spot for integrationof nonphage cargo DNA (48). Salmonella serovar Typhi ST46contains an insertion of three genes with homology to serine/threonine protein kinases (STY4822-23) and a PP2C family-like serine/threonine protein phosphatase (STY4824) (48).The Salmonella serovar Paratyphi A P4-like phage encodes acos site insertion of three genes that code for a restrictionmodification system (36). The P4 phages of Salmonella serovarTyphi and Salmonella serovar Paratyphi A both contain anunusual feature, Salmonella serovar Typhi CT18 geneSTY4830 (96% identical over all 228 bp in Salmonella serovarParatyphi A) replaces the normal P4 phage genes ε and orf151.These data suggest that Salmonella serovar Paratyphi A andSalmonella serovar Typhi may have acquired their residentphage from a common ancestor whose P4 phage subsequentlypicked up different cos site insertions.
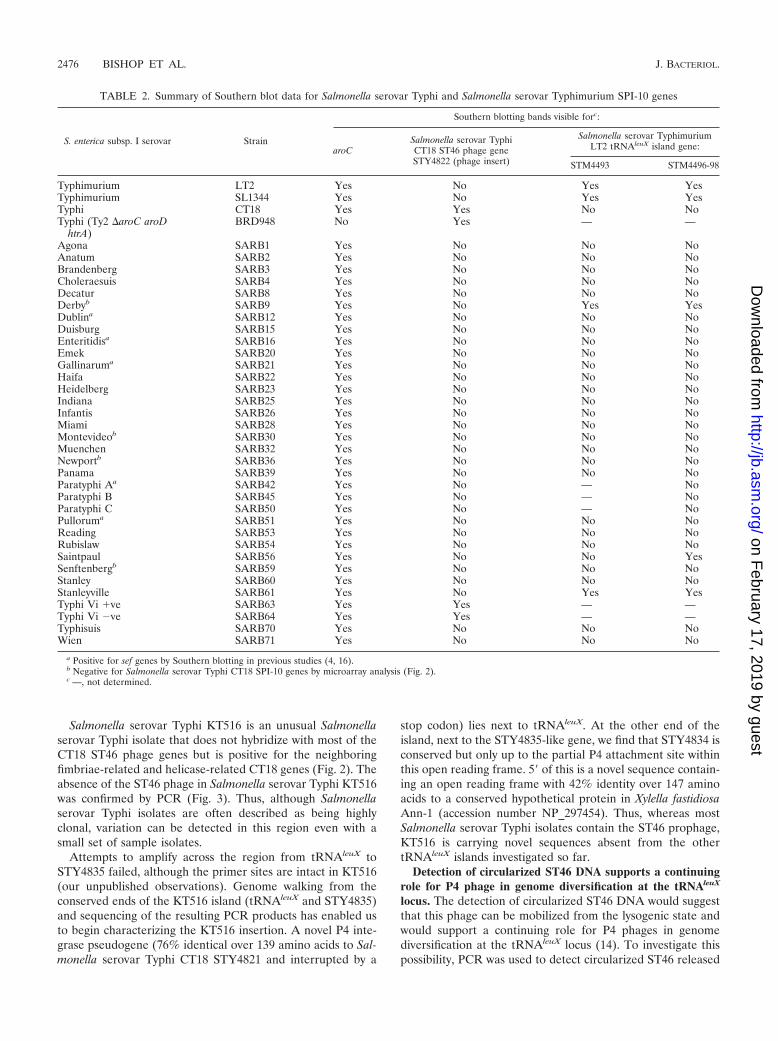
Salmonella serovar Typhi CT18 microarray data confirmedthe presence of a CT18-like P4 phage, STY4821-34, in all butone of the Salmonella serovar Typhi isolates tested and arelated phage in all Salmonella serovar Paratyphi A strains. Allother S. enterica serovars tested did not contain a closely re-lated P4 phage, although hybridization with one phage gene,STY4826, a homologue of the P4 � subunit, is common to allbut Salmonella enterica serovar Senftenberg and S. bongori(Fig. 2). The Salmonella serovar Typhi-specific nature ofSTY4822-24 was confirmed by the lack of any hybridizationwith STY4822-24 by non-serovar Typhi Salmonella isolates(microarray results are shown in Fig. 2; Southern blotting re-sults with STY4822 probe are shown in Table 2 and Fig. 4).
In addition to the full P4 prophages described above, anumber of predicted phage integrases, or fragments of inte-grases, are also associated with the tRNAleuX region (Fig. 1).For example, Salmonella serovar Typhi and Salmonella serovarParatyphi A contain identical 159-bp integrase remnants be-tween the end of the P4 phage (STY4834) and the neighboringtransposase pseudogene (STY4835). The Salmonella serovarTyphimurium LT2 tRNAleuX island begins with STM4488,which is a highly truncated P4-related integrase (33% identicalto P4 int over just 89 amino acids). S. bongori contains an evenmore truncated integrase gene that is very similar (94% iden-tical) to STM4488 over its 211-bp length.
VOL. 187, 2005 A HYPERVARIABLE REGION OF THE SALMONELLA GENOME 2475
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
Salmonella serovar Typhi KT516 is an unusual Salmonellaserovar Typhi isolate that does not hybridize with most of theCT18 ST46 phage genes but is positive for the neighboringfimbriae-related and helicase-related CT18 genes (Fig. 2). Theabsence of the ST46 phage in Salmonella serovar Typhi KT516was confirmed by PCR (Fig. 3). Thus, although Salmonellaserovar Typhi isolates are often described as being highlyclonal, variation can be detected in this region even with asmall set of sample isolates.
Attempts to amplify across the region from tRNAleuX toSTY4835 failed, although the primer sites are intact in KT516(our unpublished observations). Genome walking from theconserved ends of the KT516 island (tRNAleuX and STY4835)and sequencing of the resulting PCR products has enabled usto begin characterizing the KT516 insertion. A novel P4 inte-grase pseudogene (76% identical over 139 amino acids to Sal-monella serovar Typhi CT18 STY4821 and interrupted by a
stop codon) lies next to tRNAleuX. At the other end of theisland, next to the STY4835-like gene, we find that STY4834 isconserved but only up to the partial P4 attachment site withinthis open reading frame. 5� of this is a novel sequence contain-ing an open reading frame with 42% identity over 147 aminoacids to a conserved hypothetical protein in Xylella fastidiosaAnn-1 (accession number NP_297454). Thus, whereas mostSalmonella serovar Typhi isolates contain the ST46 prophage,KT516 is carrying novel sequences absent from the othertRNAleuX islands investigated so far.
Detection of circularized ST46 DNA supports a continuingrole for P4 phage in genome diversification at the tRNAleuX
locus. The detection of circularized ST46 DNA would suggestthat this phage can be mobilized from the lysogenic state andwould support a continuing role for P4 phages in genomediversification at the tRNAleuX locus (14). To investigate thispossibility, PCR was used to detect circularized ST46 released
TABLE 2. Summary of Southern blot data for Salmonella serovar Typhi and Salmonella serovar Typhimurium SPI-10 genes
S. enterica subsp. I serovar Strain
Southern blotting bands visible forc:
aroCSalmonella serovar TyphiCT18 ST46 phage geneSTY4822 (phage insert)
Salmonella serovar TyphimuriumLT2 tRNAleuX island gene:
STM4493 STM4496-98
Typhimurium LT2 Yes No Yes YesTyphimurium SL1344 Yes No Yes YesTyphi CT18 Yes Yes No NoTyphi (Ty2 �aroC aroD
htrA)BRD948 No Yes — —
Agona SARB1 Yes No No NoAnatum SARB2 Yes No No NoBrandenberg SARB3 Yes No No NoCholeraesuis SARB4 Yes No No NoDecatur SARB8 Yes No No NoDerbyb SARB9 Yes No Yes YesDublina SARB12 Yes No No NoDuisburg SARB15 Yes No No NoEnteritidisa SARB16 Yes No No NoEmek SARB20 Yes No No NoGallinaruma SARB21 Yes No No NoHaifa SARB22 Yes No No NoHeidelberg SARB23 Yes No No NoIndiana SARB25 Yes No No NoInfantis SARB26 Yes No No NoMiami SARB28 Yes No No NoMontevideob SARB30 Yes No No NoMuenchen SARB32 Yes No No NoNewportb SARB36 Yes No No NoPanama SARB39 Yes No No NoParatyphi Aa SARB42 Yes No — NoParatyphi B SARB45 Yes No — NoParatyphi C SARB50 Yes No — NoPulloruma SARB51 Yes No No NoReading SARB53 Yes No No NoRubislaw SARB54 Yes No No NoSaintpaul SARB56 Yes No No YesSenftenbergb SARB59 Yes No No NoStanley SARB60 Yes No No NoStanleyville SARB61 Yes No Yes YesTyphi Vi �ve SARB63 Yes Yes — —Typhi Vi �ve SARB64 Yes Yes — —Typhisuis SARB70 Yes No No NoWien SARB71 Yes No No No
a Positive for sef genes by Southern blotting in previous studies (4, 16).b Negative for Salmonella serovar Typhi CT18 SPI-10 genes by microarray analysis (Fig. 2).c —, not determined.
2476 BISHOP ET AL. J. BACTERIOL.
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
by Salmonella serovar Typhi BRD948. Putative phage particleswere purified from bacterial lysates with chloroform extraction,and these were found to contain detectable circularized phageDNA (data not shown). The phage extracts were not able tocause lysis of target bacteria (E. coli, Salmonella serovar En-teritidis, and Salmonella serovar Typhimurium strains weretested) (data not shown). Salmonella serovar Typhi BRD948genomic DNA (extracted from bacteria grown to stationaryphase) was also tested for the presence of circular ST46 phageDNA by PCR and was found to be clearly positive (Fig. 3b).
Genomic DNA from Salmonella serovar Typhi CVD908(Ty2 �aroC aroD), without the htrA mutation harbored byBRD948 that could potentially affect phage release (htrA isinvolved in the stress response) (31), was also clearly positivefor circular ST46 (Fig. 3b). Importantly, Salmonella serovarTyphi KT516 was negative for both circular and chromosomalST46 PCR products, consistent with the absence of ST46 inthis Salmonella serovar Typhi strain (Fig. 3b). Circular ST46was also detectable by using Salmonella serovar Typhi CT18genomic DNA as the template but at a much lower level thanwith the Ty2-derivative strains. We are currently testing thehypothesis that efficient circularization of ST46 is facilitated byan additional P4 lysogenic phage (ST2_27) known to bepresent within Ty2 and absent from CT18 (19, 48).
Evidence for genes from other mobile elements in the tRNAleuX-associated region. (i) Plasmid-related genes and IS elements.SPI-10 of Salmonella serovar Typhi CT18 harbors a truncatedpef (plasmid-encoded fimbrial) operon and the full sef (Salmo-nella serovar Enteritidis fimbriae) operon, forming a pathoge-nicity islet (16). This Salmonella serovar Typhi islet is associ-ated with six pseudogenes, which are also pseudogenes inSalmonella serovar Typhi strain Ty2 and Salmonella serovarParatyphi A (Fig. 1) (19, 21, 49), that are likely to inactivate thefimbrial operons. The sef/pef islet is flanked by IS elementremnants, which may have facilitated its insertion into thechromosome (16) (Fig. 1). Homologues of the CT18 sef genesSTY4836a/sefA-STY4841/sefR were clearly present in all sixSalmonella serovar Typhi isolates tested on microarrays (Fig.2). The majority of these genes, all but sefB, are also detectedin Salmonella serovar Paratyphi A, Salmonella enterica serovarPullorum, Salmonella enterica serovar Gallinarum, Salmonellaserovar Enteritidis, and Salmonella enterica serovar Dublinstrain 16. This is consistent with our in silico analysis (Fig. 1)and with previous literature describing the sef genes by South-ern blotting for sefC and sefD (4) or sefA (16) (Table 2).Hybridization with sefC was observed for all serovars tested,and in some cases, hybridization with sefR was also observed(Fig. 2), suggesting that similar fimbrial genes are present in
FIG. 4. Examples of Southern blots showing that STY4822 is Salmonella serovar Typhi specific and that the Salmonella serovar TyphimuriumSPI-10 genes tested (STM4493 and STM4496-98) are only shared with three other serovars: Salmonella serovar Derby (not shown here),Salmonella serovar Saintpaul, and Salmonella serovar Stanleyville. Data were only included in the final analysis (summarized in Table 2) where aclear aroC control band was visible (with the exception of Salmonella serovar Typhi BRD948, which is an aroC deletion mutant). Strain names andserovars are noted above each panel, and the probes used are indicated at the side. (a) Four Salmonella serovar Typhi strains tested containSTY4822, whereas strains of Salmonella serovar Typhimurium, Salmonella serovar Paratyphi A, Salmonella serovar Paratyphi B, and Salmonellaserovar Paratyphi C do not. (b) STY4822 is present in the control Salmonella serovar Typhi CT18, but it is not detected in 11 SARB collectionstrains tested here. Salmonella serovar Typhimurium LT2 tRNAleuX-adjacent genes, on the other hand, are shared with Salmonella serovarStanleyville (STM4493 and STM4496-98) and Salmonella serovar Saintpaul (STM4496-98), and are shown in lanes that are boxed.
VOL. 187, 2005 A HYPERVARIABLE REGION OF THE SALMONELLA GENOME 2477
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

serovars where the full sef operon is absent. Of the serovarstested, only Salmonella serovar Dublin and Salmonella serovarEnteritidis have been reported to actually elaborate sef fim-briae (16).
The origins of the sef operon are not known, while the pefgenes present in the chromosomes of some salmonellae arethought to be of plasmid origin, as full pef fimbrial operons arefound on the F-type virulence plasmids of Salmonella serovarTyphimurium (pSLT), Salmonella enterica serovar Cholerae-suis (pKDSC50), and Salmonella serovar Enteritidis (pS72)(26). S. bongori SARC11 is not strongly positive with microar-ray analysis for any of the CT18 plasmid-derived pef genes,consistent with in silico analysis of its tRNAleuX-associatedisland and the lack of any reported pef-containing plasmid. Ofthe other salmonellae tested, Salmonella enterica serovar Mon-tevideo, Salmonella serovar Senftenberg, Salmonella serovarDerby, Salmonella enterica serovar Binza, and Salmonella en-terica serovar Newport give positive hybridizations with justone of the four CT18 pef genes, STY4846 (Fig. 2), suggestingthat the chromosomal pef genes are not present in these sero-vars. The presence of integrated plasmid-related genes in se-rovars such as Salmonella serovar Typhi, Salmonella serovarParatyphi A, and Salmonella serovar Enteritidis strongly sug-gests that these serotypes had a common ancestor that ac-quired these genes from a plasmid. Indeed, the Salmonellaserovar Enteritidis tRNAleuX region encodes three more puta-tive plasmid-related genes, which lie between IS630 and IS1230elements at the start of the island (see Table S6 in the supple-mental material). IS elements, or their remnants, are presentat many points in the tRNAleuX islands of different Salmonellaand E. coli strains (Fig. 1) (9) and may have contributed todiversity at tRNAleuX by mediating exchanges between theSalmonella chromosome and horizontally transferred DNAsuch as plasmids.
(ii) Genes found in conjugative elements are present in theSalmonella serovar Typhimurium tRNAleuX island. The tRNAleuX
island of Salmonella serovar Typhimurium is distinct in DNAsequence and gene content from SPI-10 of Salmonella serovarTyphi. The island in Salmonella serovar Typhimurium LT2 is20,842 kbp in length and includes coding sequences designatedSTM4488 to STM4498 (Fig. 1). The genes in the Salmonellaserovar Typhimurium island do not have a clear functionalrelationship, although a possible link to DNA repair has beensuggested (43). This hypothesis is based on the presence of apredicted helicase (STM4489), a putative ATPase involved inDNA repair (STM4496), a predicted type II restriction enzymemethylase subunit (STM4495), and a predicted Mrr restrictionendonuclease (STM4490). In addition to these genes,STM4494 is predicted to be an ATPase component of anABC-type sugar/spermidine/putrescine transport system andSTM4491 is a predicted Lon protease. STM4498, STM4497,STM4492, and STM4493 are genes of unknown function.
We have observed a striking homology and synteny between6 of the 10 predicted open reading frames in Salmonella sero-var Typhimurium SPI-10 (STM4491, STM4492, STM4495,STM4496, STM4497, and STM4498) and genes, or fragmentsof genes, found within conjugative elements harbored by otherenteric bacteria (see Table S8 in the supplemental material),with these being an uncharacterized E. coli plasmid p1658/97(accession number AF550679) and two well-characterized
IncJ-related conjugative integrating genomic elements: Vibriocholerae SXT transposon-like element (accession numberAY055428) (7) and Providencia rettgeri conjugative genomicisland R391 (the similarity of R391 genes to those of LT2 hasbeen noted previously [10], accession number AY090559). Todate, no functional significance has been attributed to theseparticular genes within SXT or R391 (6). Genes STM4493 andSTM4494 are not found in SXT or R391 and may have beenacquired by Salmonella serovar Typhimurium separately.
DNA fragments encoding Salmonella serovar TyphimuriumLT2 genes STM4496-STM4498, within the integrating conju-gative element-related region, and STM4493, which we pro-pose to have been acquired through a separate event, wereused to probe Southern blots containing DNA from represen-tatives of different Salmonella enterica subsp. I serovars. Themajority of Salmonella strains tested were negative for all ofthe Salmonella serovar Typhimurium SPI-10 genes. However,STM4496-98 was detected in a small number of serovars (Sal-monella serovar Derby SARB9, Salmonella enterica serovarSaintpaul SARB56 and Salmonella enterica serovar StanleyvilleSARB61), and STM4493 was detected just in Salmonella se-rovar Derby SARB9 and Salmonella serovar StanleyvilleSARB61 (Fig. 4b and Table 2).
The presence of STM4496-98, but not STM4493, in Salmo-nella serovar Saintpaul SARB56 backs up the hypothesis thatSTM4493-94 may have been acquired by Salmonella serovarTyphimurium separately from the surrounding genes. To de-termine whether both STM4493 and STM4494 were missingfrom Salmonella serovar Saintpaul SARB56, we carried outPCR across from STM4492- to STM4495-like genes. A muchsmaller DNA fragment was amplified from SARB56 than fromSalmonella serovar Stanleyville SARB61, which was positivefor STM4493 by Southern blotting (Table 2), or the Salmonellaserovar Typhimurium LT2 control (Fig. 5i). Sequencing of thePCR product confirmed that STM4495 lies right next toSTM4492 in Salmonella serovar Saintpaul SARB56 (depictedschematically in Fig.5iii) and that neither STM4493 norSTM4494 are present. This gene arrangement in Salmonellaserovar Saintpaul SARB56 is, therefore, similar to that foundin the integrating conjugative genomic islands. These datastrongly suggest that integration of genes found within conju-gative elements into the Salmonella serovar TyphimuriumtRNAleuX island occurred prior to insertion of STM4493 andSTM4494.
Further variation within Salmonella serovar Derby, Salmo-nella serovar Saintpaul and Salmonella serovar StanleyvilletRNAleuX islands. Salmonella serovar Derby, Salmonella sero-var Saintpaul and Salmonella serovar Stanleyville were positivefor LT2 genes by Southern blotting (Table 2). To detect anyadditional variation within the tRNAleuX islands in these sero-vars a detailed comparison with the Salmonella serovar Typhi-murium LT2 tRNAleuX-adjacent island was carried out usingPCR.
(i) A novel insertion in Salmonella serovar Saintpaul andSalmonella serovar Stanleyville tRNAleuX islands. Havingfound variations at the 3� end of the Salmonella serovar Saint-paul SARB56 tRNAleuX island compared to LT2 (describedabove), the gene complement at the 5� tRNAleuX-adjacent endof the Salmonella serovar Saintpaul SARB56 and Salmonellaserovar Stanleyville SARB61 islands was investigated. PCR
2478 BISHOP ET AL. J. BACTERIOL.
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from
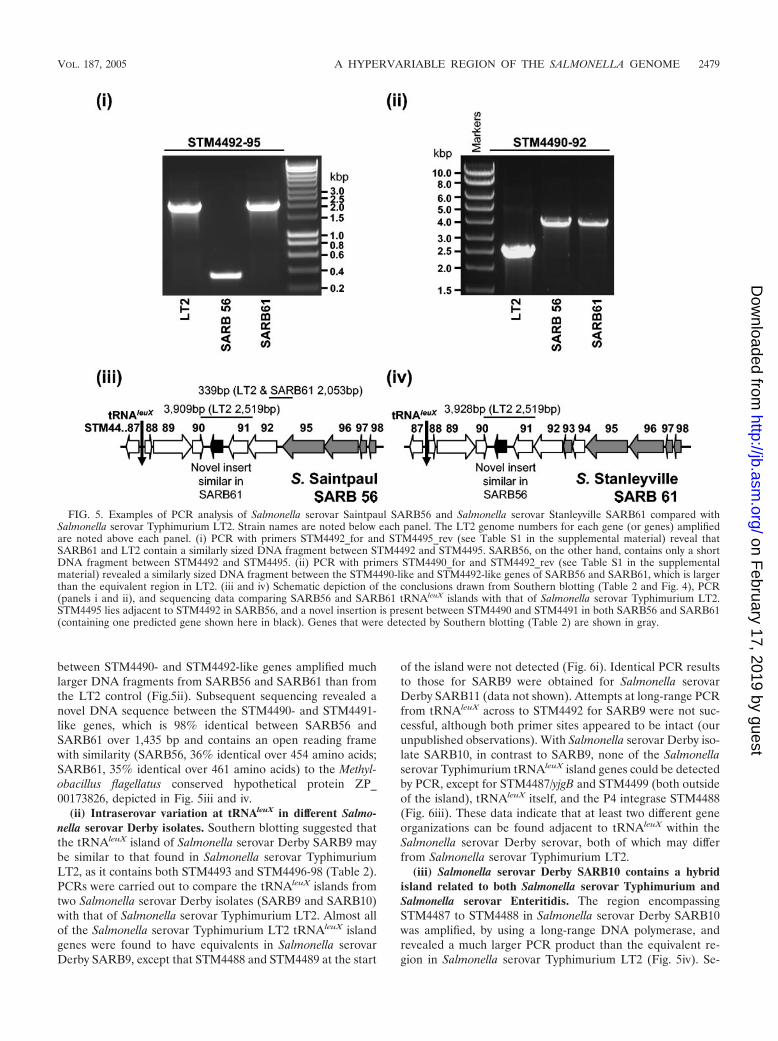
between STM4490- and STM4492-like genes amplified muchlarger DNA fragments from SARB56 and SARB61 than fromthe LT2 control (Fig.5ii). Subsequent sequencing revealed anovel DNA sequence between the STM4490- and STM4491-like genes, which is 98% identical between SARB56 andSARB61 over 1,435 bp and contains an open reading framewith similarity (SARB56, 36% identical over 454 amino acids;SARB61, 35% identical over 461 amino acids) to the Methyl-obacillus flagellatus conserved hypothetical protein ZP_00173826, depicted in Fig. 5iii and iv.
(ii) Intraserovar variation at tRNAleuX in different Salmo-nella serovar Derby isolates. Southern blotting suggested thatthe tRNAleuX island of Salmonella serovar Derby SARB9 maybe similar to that found in Salmonella serovar TyphimuriumLT2, as it contains both STM4493 and STM4496-98 (Table 2).PCRs were carried out to compare the tRNAleuX islands fromtwo Salmonella serovar Derby isolates (SARB9 and SARB10)with that of Salmonella serovar Typhimurium LT2. Almost allof the Salmonella serovar Typhimurium LT2 tRNAleuX islandgenes were found to have equivalents in Salmonella serovarDerby SARB9, except that STM4488 and STM4489 at the start
of the island were not detected (Fig. 6i). Identical PCR resultsto those for SARB9 were obtained for Salmonella serovarDerby SARB11 (data not shown). Attempts at long-range PCRfrom tRNAleuX across to STM4492 for SARB9 were not suc-cessful, although both primer sites appeared to be intact (ourunpublished observations). With Salmonella serovar Derby iso-late SARB10, in contrast to SARB9, none of the Salmonellaserovar Typhimurium tRNAleuX island genes could be detectedby PCR, except for STM4487/yjgB and STM4499 (both outsideof the island), tRNAleuX itself, and the P4 integrase STM4488(Fig. 6iii). These data indicate that at least two different geneorganizations can be found adjacent to tRNAleuX within theSalmonella serovar Derby serovar, both of which may differfrom Salmonella serovar Typhimurium LT2.
(iii) Salmonella serovar Derby SARB10 contains a hybridisland related to both Salmonella serovar Typhimurium andSalmonella serovar Enteritidis. The region encompassingSTM4487 to STM4488 in Salmonella serovar Derby SARB10was amplified, by using a long-range DNA polymerase, andrevealed a much larger PCR product than the equivalent re-gion in Salmonella serovar Typhimurium LT2 (Fig. 5iv). Se-
FIG. 5. Examples of PCR analysis of Salmonella serovar Saintpaul SARB56 and Salmonella serovar Stanleyville SARB61 compared withSalmonella serovar Typhimurium LT2. Strain names are noted below each panel. The LT2 genome numbers for each gene (or genes) amplifiedare noted above each panel. (i) PCR with primers STM4492_for and STM4495_rev (see Table S1 in the supplemental material) reveal thatSARB61 and LT2 contain a similarly sized DNA fragment between STM4492 and STM4495. SARB56, on the other hand, contains only a shortDNA fragment between STM4492 and STM4495. (ii) PCR with primers STM4490_for and STM4492_rev (see Table S1 in the supplementalmaterial) revealed a similarly sized DNA fragment between the STM4490-like and STM4492-like genes of SARB56 and SARB61, which is largerthan the equivalent region in LT2. (iii and iv) Schematic depiction of the conclusions drawn from Southern blotting (Table 2 and Fig. 4), PCR(panels i and ii), and sequencing data comparing SARB56 and SARB61 tRNAleuX islands with that of Salmonella serovar Typhimurium LT2.STM4495 lies adjacent to STM4492 in SARB56, and a novel insertion is present between STM4490 and STM4491 in both SARB56 and SARB61(containing one predicted gene shown here in black). Genes that were detected by Southern blotting (Table 2) are shown in gray.
VOL. 187, 2005 A HYPERVARIABLE REGION OF THE SALMONELLA GENOME 2479
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

quencing of the SARB10 insertion, which we narrowed downto a region between tRNAleuX and an STM4488-like gene,revealed that it is very similar to the equivalent section of theSalmonella serovar Enteritidis PT4 tRNAleuX island (comparethe Salmonella serovar Enteritidis depicted in Fig. 1 with Sal-monella serovar Derby SARB10 in Fig. 6vi). Both Salmonellaserovar Derby SARB10 and Salmonella serovar Enteritidiscontain a truncated P4-like integrase gene, an IS630-relatedtransposase, and three predicted plasmid-related genes.SARB10 diverges from Salmonella serovar Enteritidis at the
end of a partial tRNAleuX P4 phage attachment site duplication(16 bp of the 20-bp attP attachment site are conserved). TheSARB10 sequence continues with a pseudogene resemblingSalmonella serovar Typhimurium LT2 STM4488 (95% identi-cal over 164 bp but with a frameshift), while Salmonella serovarEnteritidis has instead acquired the IS element-flanked sef/pefislet at this locus. Attempts to PCR amplify the region fromSTM4488 to STM4499, to detect any genes inserted 3� ofSTM4488, were not successful, although the primer sites ap-pear to be present (our unpublished observations).
FIG. 6. Examples of PCR comparison of tRNAleuX island genes from LT2 with Salmonella serovar Derby strains SARB9 and SARB10. The LT2genome numbers for each gene (or genes) amplified are noted above each panel and numbers for genes that were not detected by PCR or whereinsertions were found to be present compared with LT2 are underlined. The primers used for the PCR analyses shown in panels (i to iii) are asfollows (see also Table S1 in the supplemental material): lane 1, primer pair 16–17; lane 2, primer pair 16–19; lane 3, primer pair 20–22; lane 4,primer pair 21–25; lane 5, primer pair 24–26; lane 6, primer pair 24–27; lane 7, primer pair 24–28; lane 8, primer pair 29–30; lane 9, primer pair29–31. (i) Salmonella serovar Derby SARB9 contains genes similar to LT2 genes STM4490 to STM4499, but STM4488- and STM4489-like geneswere not detected. (ii) Salmonella serovar Typhimurium LT2 controls for PCRs shown in panels i and iii. (iii) None of the LT2 tRNAleuX islandgenes (STM4488 to STM4498) were initially detected in Salmonella serovar Derby SARB10 by PCR. STM4499 (data not shown) and STM4487,which lie on either side of the island, were both present. (iv) PCR with a long-range DNA polymerase with primers STM4487_for andSTM4488_rev (see Table S1 in the supplemental material) revealed a larger DNA fragment between STM4487 and STM4488 in SARB10 than inthe equivalent region in LT2. (v and vi) Schematic depiction of the gene arrangements in SARB9 and SARB10 supported by PCR data, such asthat shown in panels i to iv, Southern blotting (Table 2 and Fig. 4), and sequencing of the SARB10 insertion between tRNAleuX and STM4488(amplified with primers tRNA_for and STM4488_rev). The sequences that lie between tRNAleuX and STM4490 in SARB9 and between STM4488and STM4499 in SARB10 remain unknown (shown as question marks). Genes detected by Southern blotting (Table 2) are shown in grey.
2480 BISHOP ET AL. J. BACTERIOL.
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

Conclusions. Most of the SPIs identified in Salmonella se-rovar Typhi CT18 are largely conserved between different S.enterica subsp. I serovars. In SPI-1 to SPI-5, the only majorvariation from Salmonella serovar Typhimurium is withinSPI-3 where, across a relatively short region (3 kbp), someinsertions or deletions have been detected (2). ComparingSalmonella serovar Typhi CT18 with Salmonella serovar Ty-phimurium LT2, there is a high level of conservation withinSPI-1 to SPI-6, except for a large insertion of the tcf fimbrialoperon in SPI-6 of Salmonella serovar Typhi (23). SPI-8 andSPI-9 (40) have not yet been thoroughly characterized. Ourdata suggest that SPI-10 can be placed alongside SPI-7, pre-dicted to be a conjugative transposon (41), as an island inSalmonella serovar Typhi CT18 that is highly variable betweendifferent serovars within S. enterica subsp. I.
In some cases, within a single serovar we find significantvariation at the tRNAleuX locus from one isolate to another.Even with relatively small numbers of strains, we observed thisintraserovar variation both within serovars that (by multilocusenzyme electrophoresis typing) are considered relativelyclonal, Salmonella serovar Typhi (46) (Fig. 3), or divergent,Salmonella serovar Derby (8) (Fig. 6). In a microarray-basedstudy by Boyd et al., the tRNAleuX locus was also picked out asone of a small number of regions of major chromosomal vari-ation between Salmonella serovar Typhi isolates (11). Boyd etal. identified three isolates lacking the ST46 phage, one ofwhich was isolated in Indonesia (In15 isolated in 1994), as wasthe ST46-negative strain analyzed here (KT516 isolated in1986) (30), while the other two were from different continents(3125, Chile; CDC1707, Liberia) (11). This suggests that theloss of ST46 by Salmonella serovar Typhi isolates has occurredindependently in at least three locations worldwide. Differ-ences at the tRNAleuX locus, at least for some serovars, mayeven prove prevalent enough to be of use in epidemiologicalstudies.
The tRNAleuX island of Salmonella serovar Typhi has beentermed SPI-10 (40), even though to our knowledge none of thegenes at this locus have been linked to pathogenesis. Thisisland in Salmonella serovar Typhi has instead undergone sub-stantial loss of gene function, through the acquisition of pseu-dogene mutations, which may contribute to host restrictionrather than pathogenesis. The Salmonella serovar EnteritidistRNAleuX locus has more convincing features of a Salmonellapathogenicity island, as the intact sef operon has been impli-cated in in vivo pathogenesis with mice (22). There remain alarge number of serovars with uncharacterized tRNAleuX loci(Table 2), as they have not been sequenced and contain neitherthe Salmonella serovar Typhi or Salmonella serovar Typhi-murium genes for which we screened nor the previously char-acterized sef/pef islet (4, 16). It is likely that many more novelgene sets are present in these isolates. It remains to be deter-mined whether these are of importance to the different hostrestrictions and adaptations observed within S. entericasubsp. I.
The tRNAleuX islands have mosaic structures. For example,within the hybrid island of Salmonella serovar Derby SARB10,we find a partial integrase very similar to Salmonella serovarTyphimurium STM4488 alongside plasmid-related genes andIS elements also seen in Salmonella serovar Enteritidis (Fig.
6iv); the sef/pef islet is present with or without a full P4 phagelysogen in different islands (Fig. 1).
tRNA loci are often the targets for insertion of horizontallytransferred DNA (25, 32), and several of the Salmonella patho-genicity islands are located at tRNA genes (7 of the 10 islandsidentified in Salmonella serovar Typhi are located adjacent toa tRNA) (27, 40, 50). Our in silico and molecular analysis ofthe tRNAleuX region has revealed such remarkable variationthat we believe tRNAleuX in S. enterica to be a locus that isunusually prone to insertion and excision events.
The presence of P4-like phage, many IS elements, and inte-grase remnants is striking and suggests that this is a commonspot for the insertion of transposable elements. The detectionof circularized Salmonella serovar Typhi ST46 DNA in strainsgrowing in culture and identification of an Salmonella serovarTyphi strain missing ST46 (Fig. 3) underlines the potential ofP4 phages to drive diversity at the tRNAleuX locus. The pres-ence in some serovars of a number of plasmid-derived genes inclose association with IS elements is indicative of an IS-medi-ated acquisition of plasmid DNA. Thus, tRNAleuX may providea chromosomal locus through which S. enterica can sampledifferent gene sets, a property that could significantly influencethe evolution of S. enterica as a species.
ACKNOWLEDGMENTS
This work was supported by The Wellcome Trust and the BBSRC.We thank Julian Parkhill and Nicholas Thomson for assistance with
in silico genome analysis and advice relating to phage biology.
REFERENCES
1. Altschul, S. F., W. Gish, W. Miller, E. W. Myers, and D. J. Lipman. 1990.Basic local alignment search tool. J. Mol. Biol. 215:403–410.
2. Amavisit, P., D. Lightfoot, G. F. Browning, and P. F. Markham. 2003.Variation between pathogenic serovars within Salmonella pathogenicity is-lands. J. Bacteriol. 185:3624–3635.
3. Ausubel, F. M., R. Brent, R. E. Kingston, D. D. Moore, J. G. Seidman, andJ. A. Smith. 1995. Preparation of genomic DNA from bacteria, p. 2.11–2.22.In K. Struhl (ed.), Short protocols in molecular biology, vol. 3. John Wileyand Sons, Inc., New York, N.Y.
4. Baumler, A. J., A. J. Gilde, R. M. Tsolis, A. W. van der Velden, B. M. Ahmer,and F. Heffron. 1997. Contribution of horizontal gene transfer and deletionevents to development of distinctive patterns of fimbrial operons duringevolution of Salmonella serotypes. J. Bacteriol. 179:317–322.
5. Bausch, C., N. Peekhaus, C. Utz, T. Blais, E. Murray, T. Lowary, and T.Conway. 1998. Sequence analysis of the GntII (subsidiary) system for glu-conate metabolism reveals a novel pathway for L-idonic acid catabolism inEscherichia coli. J. Bacteriol. 180:3704–3710.
6. Beaber, J. W., V. Burrus, B. Hochhut, and M. K. Waldor. 2002. Comparisonof SXT and R391, two conjugative integrating elements: definition of agenetic backbone for the mobilization of resistance determinants. Cell. Mol.Life Sci. 59:2065–2070.
7. Beaber, J. W., B. Hochhut, and M. K. Waldor. 2002. Genomic and functionalanalyses of SXT, an integrating antibiotic resistance gene transfer elementderived from Vibrio cholerae. J. Bacteriol. 184:4259–4269.
8. Beltran, P., J. M. Musser, R. Helmuth, J. J. Farmer III, W. M. Frerichs, I. K.Wachsmuth, K. Ferris, A. C. McWhorter, J. G. Wells, A. Cravioto, et al.1988. Toward a population genetic analysis of Salmonella: genetic diversityand relationships among strains of serotypes S. choleraesuis, S. derby, S.dublin, S. enteritidis, S. heidelberg, S. infantis, S. newport, and S. typhi-murium. Proc. Natl. Acad. Sci. USA 85:7753–7757.
9. Blattner, F. R., G. Plunkett III, C. A. Bloch, N. T. Perna, V. Burland, M.Riley, J. Collado-Vides, J. D. Glasner, C. K. Rode, G. F. Mayhew, J. Gregor,N. W. Davis, H. A. Kirkpatrick, M. A. Goeden, D. J. Rose, B. Mau, and Y.Shao. 1997. The complete genome sequence of Escherichia coli K-12. Sci-ence 277:1453–1474.
10. Boltner, D., C. MacMahon, J. T. Pembroke, P. Strike, and A. M. Osborn.2002. R391: a conjugative integrating mosaic comprised of phage, plasmid,and transposon elements. J. Bacteriol. 184:5158–5169.
11. Boyd, E. F., S. Porwollik, F. Blackmer, and M. McClelland. 2003. Differencesin gene content among Salmonella enterica serovar Typhi isolates. J. Clin.Microbiol. 41:3823–3828.
VOL. 187, 2005 A HYPERVARIABLE REGION OF THE SALMONELLA GENOME 2481
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from

12. Boyd, E. F., F. S. Wang, P. Beltran, S. A. Plock, K. Nelson, and R. K.Selander. 1993. Salmonella reference collection B (SARB): strains of 37serovars of subspecies I. J. Gen. Microbiol. 139(Pt 6):1125–1132.
13. Boyd, E. F., F. S. Wang, T. S. Whittam, and R. K. Selander. 1996. Moleculargenetic relationships of the salmonellae. Appl. Environ. Microbiol. 62:804–808.
14. Briani, F., G. Deho, F. Forti, and D. Ghisotti. 2001. The plasmid status ofsatellite bacteriophage P4. Plasmid 45:1–17.
15. Chan, K., S. Baker, C. C. Kim, C. S. Detweiler, G. Dougan, and S. Falkow.2003. Genomic comparison of Salmonella enterica serovars and Salmonellabongori by use of an S. enterica serovar Typhimurium DNA microarray. J.Bacteriol. 185:553–563.
16. Collighan, R. J., and M. J. Woodward. 2001. The SEF14 fimbrial antigen ofSalmonella enterica serovar Enteritidis is encoded within a pathogenicityislet. Vet. Microbiol. 80:235–245.
17. Delcher, A. L., D. Harmon, S. Kasif, O. White, and S. L. Salzberg. 1999.Improved microbial gene identification with GLIMMER. Nucleic Acids Res.27:4636–4641.
18. Delcher, A. L., S. Kasif, R. D. Fleischmann, J. Peterson, O. White, and S. L.Salzberg. 1999. Alignment of whole genomes. Nucleic Acids Res. 27:2369–2376.
19. Deng, W., S. R. Liou, G. Plunkett III, G. F. Mayhew, D. J. Rose, V. Burland,V. Kodoyianni, D. C. Schwartz, and F. R. Blattner. 2003. Comparativegenomics of Salmonella enterica serovar Typhi strains Ty2 and CT18. J.Bacteriol. 185:2330–2337.
20. Dobrindt, U., G. Blum-Oehler, G. Nagy, G. Schneider, A. Johann, G.Gottschalk, and J. Hacker. 2002. Genetic structure and distribution of fourpathogenicity islands (PAI I536 to PAI IV536) of uropathogenic Escherichiacoli strain 536. Infect. Immun. 70:6365–6372.
21. Edwards, R. A., B. C. Matlock, B. J. Heffernan, and S. R. Maloy. 2001.Genomic analysis and growth-phase-dependent regulation of the SEF14fimbriae of Salmonella enterica serovar Enteritidis. Microbiology 147:2705–2715.
22. Edwards, R. A., D. M. Schifferli, and S. R. Maloy. 2000. A role for Salmo-nella fimbriae in intraperitoneal infections. Proc. Natl. Acad. Sci. USA 97:1258–1262.
23. Folkesson, A., A. Advani, S. Sukupolvi, J. D. Pfeifer, S. Normark, and S.Lofdahl. 1999. Multiple insertions of fimbrial operons correlate with theevolution of Salmonella serovars responsible for human disease. Mol. Mi-crobiol. 33:612–622.
24. Galan, J. E. 2001. Salmonella interactions with host cells: type III secretionat work. Annu. Rev. Cell Dev. Biol. 17:53–86.
25. Hacker, J., and J. B. Kaper. 2000. Pathogenicity islands and the evolution ofmicrobes. Annu. Rev. Microbiol. 54:641–679.
26. Haneda, T., N. Okada, N. Nakazawa, T. Kawakami, and H. Danbara. 2001.Complete DNA sequence and comparative analysis of the 50-kilobase viru-lence plasmid of Salmonella enterica serovar Choleraesuis. Infect. Immun.69:2612–2620.
27. Hansen-Wester, I., and M. Hensel. 2002. Genome-based identification ofchromosomal regions specific for Salmonella spp. Infect. Immun. 70:2351–2360.
28. Hayashi, T., K. Makino, M. Ohnishi, K. Kurokawa, K. Ishii, K. Yokoyama,C. G. Han, E. Ohtsubo, K. Nakayama, T. Murata, M. Tanaka, T. Tobe, T.Iida, H. Takami, T. Honda, C. Sasakawa, N. Ogasawara, T. Yasunaga, S.Kuhara, T. Shiba, M. Hattori, and H. Shinagawa. 2001. Complete genomesequence of enterohemorrhagic Escherichia coli O157:H7 and genomic com-parison with a laboratory strain K-12. DNA Res. 8:11–22.
29. Hou, Y. M. 1999. Transfer RNAs and pathogenicity islands. Trends Biochem.Sci. 24:295–298.
30. Kidgell, C., U. Reichard, J. Wain, B. Linz, M. Torpdahl, G. Dougan, and M.Achtman. 2002. Salmonella typhi, the causative agent of typhoid fever, isapproximately 50,000 years old. Infect. Genet. Evol. 2:39–45.
31. Lowe, D. C., T. C. Savidge, D. Pickard, L. Eckmann, M. F. Kagnoff, G.Dougan, and S. N. Chatfield. 1999. Characterization of candidate live oralSalmonella typhi vaccine strains harboring defined mutations in aroA, aroC,and htrA. Infect. Immun. 67:700–707.
32. Mantri, Y., and K. P. Williams. 2004. Islander: a database of integrativeislands in prokaryotic genomes, the associated integrases and their DNA sitespecificities. Nucleic Acids Res. 32:D55–D58.
33. Marchler-Bauer, A., J. B. Anderson, C. DeWeese-Scott, N. D. Fedorova, L. Y.
Geer, S. He, D. I. Hurwitz, J. D. Jackson, A. R. Jacobs, C. J. Lanczycki, C. A.Liebert, C. Liu, T. Madej, G. H. Marchler, R. Mazumder, A. N. Nikolskaya,A. R. Panchenko, B. S. Rao, B. A. Shoemaker, V. Simonyan, J. S. Song, P. A.Thiessen, S. Vasudevan, Y. Wang, R. A. Yamashita, J. J. Yin, and S. H.Bryant. 2003. CDD: a curated Entrez database of conserved domain align-ments. Nucleic Acids Res. 31:383–387.
34. Marcus, S. L., J. H. Brumell, C. G. Pfeifer, and B. B. Finlay. 2000. Salmo-nella pathogenicity islands: big virulence in small packages. Microbes Infect.2:145–156.
35. McClelland, M., K. E. Sanderson, J. Spieth, S. W. Clifton, P. Latreille, L.Courtney, S. Porwollik, J. Ali, M. Dante, F. Du, S. Hou, D. Layman, S.Leonard, C. Nguyen, K. Scott, A. Holmes, N. Grewal, E. Mulvaney, E. Ryan,H. Sun, L. Florea, W. Miller, T. Stoneking, M. Nhan, R. Waterston, andR. K. Wilson. 2001. Complete genome sequence of Salmonella entericaserovar Typhimurium LT2. Nature 413:852–856.
36. Naderer, M., J. R. Brust, D. Knowle, and R. M. Blumenthal. 2002. Mobilityof a restriction-modification system revealed by its genetic contexts in threehosts. J. Bacteriol. 184:2411–2419.
37. Ochman, H., S. Elwyn, and N. A. Moran. 1999. Calibrating bacterial evolu-tion. Proc. Natl. Acad. Sci. USA 96:12638–12643.
38. Ochman, H., J. G. Lawrence, and E. A. Groisman. 2000. Lateral genetransfer and the nature of bacterial innovation. Nature 405:299–304.
39. Ochman, H., and A. C. Wilson. 1987. Evolution in bacteria: evidence for auniversal substitution rate in cellular genomes. J. Mol. Evol. 26:74–86.
40. Parkhill, J., G. Dougan, K. D. James, N. R. Thomson, D. Pickard, J. Wain,C. Churcher, K. L. Mungall, S. D. Bentley, M. T. Holden, M. Sebaihia, S.Baker, D. Basham, K. Brooks, T. Chillingworth, P. Connerton, A. Cronin, P.Davis, R. M. Davies, L. Dowd, N. White, J. Farrar, T. Feltwell, N. Hamlin,A. Haque, T. T. Hien, S. Holroyd, K. Jagels, A. Krogh, T. S. Larsen, S.Leather, S. Moule, P. O’Gaora, C. Parry, M. Quail, K. Rutherford, M.Simmonds, J. Skelton, K. Stevens, S. Whitehead, and B. G. Barrell. 2001.Complete genome sequence of a multiple drug resistant Salmonella entericaserovar Typhi CT18. Nature 413:848–852.
41. Pickard, D., J. Wain, S. Baker, A. Line, S. Chohan, M. Fookes, A. Barron,P. O. Gaora, J. A. Chabalgoity, N. Thanky, C. Scholes, N. Thomson, M.Quail, J. Parkhill, and G. Dougan. 2003. Composition, acquisition, anddistribution of the Vi exopolysaccharide-encoding Salmonella enterica patho-genicity island SPI-7. J. Bacteriol. 185:5055–5065.
42. Pierson, L. S., III, and M. L. Kahn. 1987. Integration of satellite bacterio-phage P4 in Escherichia coli. DNA sequences of the phage and host regionsinvolved in site-specific recombination. J. Mol. Biol. 196:487–496.
43. Porwollik, S., and M. McClelland. 2003. Lateral gene transfer in Salmonella.Microbes Infect. 5:977–989.
44. Porwollik, S., R. M. Wong, and M. McClelland. 2002. Evolutionary genomicsof Salmonella: gene acquisitions revealed by microarray analysis. Proc. Natl.Acad. Sci. USA 99:8956–8961.
45. Sambrook, J., E. F. Fritsch, and T. Maniatis. 1989. Molecular cloning: alaboratory manual, 2nd ed. Cold Spring Harbor Laboratory Press, ColdSpring Harbor, N.Y.
46. Selander, R. K., P. Beltran, N. H. Smith, R. Helmuth, F. A. Rubin, D. J.Kopecko, K. Ferris, B. D. Tall, A. Cravioto, and J. M. Musser. 1990. Evo-lutionary genetic relationships of clones of Salmonella serovars that causehuman typhoid and other enteric fevers. Infect. Immun. 58:2262–2275.
47. Tatusova, T. A., and T. L. Madden. 1999. BLAST 2 sequences, a new tool forcomparing protein and nucleotide sequences. FEMS Microbiol. Lett. 174:247–250.
48. Thomson, N., S. Baker, D. Pickard, J. Wain, D. House, M. Fookes, A. Ivens,N. Hamlin, Z. Bhutta, M. Anjum, M. Woodward, S. Falkow, K. Chan, J.Parkhill, and G. Dougan. 2004. Prophage-like elements in the genome ofSalmonella enterica serovar Typhi. Contribution to diversity in S. entericaserovars. J. Mol. Biol. 339:279–300.
49. Townsend, S. M., N. E. Kramer, R. Edwards, S. Baker, N. Hamlin, M.Simmonds, K. Stevens, S. Maloy, J. Parkhill, G. Dougan, and A. J. Baumler.2001. Salmonella enterica serovar Typhi possesses a unique repertoire offimbrial gene sequences. Infect. Immun. 69:2894–2901.
50. Wain, J., D. House, D. Pickard, G. Dougan, and G. Frankel. 2001. Acquisi-tion of virulence-associated factors by the enteric pathogens Escherichia coliand Salmonella enterica. Philos. Trans. R. Soc. Lond. B 356:1027–1034.
51. Zhang, Z., S. Schwartz, L. Wagner, and W. Miller. 2000. A greedy algorithmfor aligning DNA sequences. J. Comput. Biol. 7:203–214.
2482 BISHOP ET AL. J. BACTERIOL.
on February 17, 2019 by guest
http://jb.asm.org/
Dow
nloaded from





























