Embed Size (px)
Citation preview

Analysis of SCA1, DRPLA, MJD, SCA2, and SCA6CAG Repeats in 48 Portuguese Ataxia Families
I. Silveira,1,2 P. Coutinho,3 P. Maciel,1,2 C. Gaspar,1,2 S. Hayes,1 A. Dias,1,2 J. Guimaraes,4L. Loureiro,5 J. Sequeiros,2,6* and G.A. Rouleau1
1Centre for Research in Neurosciences, McGill University and The Montreal General Hospital Research Institute,Montreal, Quebec, Canada
2UnIGENe, IBMC, Universidade do Porto, Portugal3Servico de Neurologia, Hospital de Santo Antonio, Porto, Portugal4Servico de Neurologia, Hospital Egas Moniz, Lisboa, Portugal5Servico de Neurologia, Hospital de Sao Pedro, Vila Real, Portugal6Laboratorio de Genetica Medica, ICBAS, Universidade do Porto, Portugal
The spinocerebellar ataxias (SCAs) areclinically and genetically a heterogeneousgroup of neurodegenerative disorders. Todate, eight different loci causing SCA havebeen identified: SCA1, SCA2, Machado-Joseph disease (MJD)/SCA3, SCA4, SCA5,SCA6, SCA7, and dentatorubropallidol-uysian atrophy (DRPLA). Expansion of aCAG repeat in the disease genes has beenfound in five of these disorders. To estimatethe relative frequencies of the SCA1,DRPLA, MJD, SCA2, and SCA6 mutationsamong Portuguese ataxia patients, we col-lected DNA samples from 48 ataxia familiesand performed polymerase chain reaction(PCR) amplification of the CAG repeat mu-tations on chromosomes 6p, 12p, 14q, 12q,and 19p, respectively. Fifty-five individualsbelonging to 34 dominant families (74%) hadan expanded CAG repeat at the MJD gene.In five individuals from two kindreds with adominant pattern of inheritance (4%), an ex-panded CAG repeat at the SCA2 gene wasfound. In MJD patients, the normal allelesize ranged from 13 to 41, whereas the mu-tant alleles contained 65 to 80 repeats. Forthe SCA2 patients, normal alleles had 22 or23, while expanded alleles had between 36and 47 CAG units. We did not find the SCA1,DRPLA, or SCA6 mutations in our group offamilies. The MJD mutation remains the
most common cause of SCA in Portugal,while a small number of cases are caused bymutations at the SCA2 gene, and 22% aredue to still unidentified genes. Am. J. Med.Genet. (Neuropsychiatr. Genet.) 81:134–138,1998. © 1998 Wiley-Liss, Inc.
KEY WORDS: spinocerebellar ataxia; den-tatorubropallidoluysian at-rophy and Machado-Josephdisease
INTRODUCTIONThe autosomal dominant spinocerebellar ataxias
(SCAs) are a heterogeneous group of late onset neuro-degenerative disorders with variable clinical pheno-type, including ataxia, dysarthria, dysmetria, and in-tention tremor, resulting from the involvement of thecerebellum and its afferent and efferent pathways. Al-though the symptoms and signs of degeneration in thecerebellum, spinal cord, and brainstem are predomi-nant, changes in the basal ganglia, optic nerves, retina,and peripheral nerve may also be present. Dementiaoccurs occasionally, as well. To date, eight different lociresponsible for SCA have been mapped: the SCA1 locuson chromosome 6p [Yakura et al., 1974; Zoghbi et al.,1988], SCA2 on chromosome 12q [Gispert et al., 1993;Lopes-Cendes et al., 1994], the Machado-Joseph dis-ease (MJD)/SCA3 locus on 14q [Takiyama et al., 1993;Sequeiros et al., 1994; Twist et al., 1995], SCA4 on 16q[Gardner et al., 1994], SCA5 on the centromeric regionof chromosome 11 [Ranum et al., 1994b], SCA6 on 19p[Zhuchenko et al., 1997], SCA7 on 3p [Benomar et al.,1995; Gouw et al., 1995], and the dentatorubropallidol-uysian atrophy (DRPLA) locus on 12p [Koide et al.,1994; Nagafuchi et al., 1994]. Expansion of a CAG re-peat causes at least five of these disorders: SCA1 [Orret al., 1993], DRPLA [Koide et al., 1994; Nagafuchi etal., 1994], MJD [Kawaguchi et al., 1994], SCA2 [Pulstet al., 1996; Imbert et al., 1996; Sanpei et al., 1996],
Contract grant sponsor: FRSQ-ACAF; Contract grant sponsor:NIH; Contract grant number: NS 31687; Contract grant sponsor:Network of Centres of Excellence; Contract grant sponsor:JNICT.
*Correspondence to: Dr. J. Sequeiros, Laboratorio de GeneticaMedica, ICBAS, Largo do Prof. Abel Salazar, 2 4000 Porto, Por-tugal.
Received 26 August 1997; Revised 29 July 1997
American Journal of Medical Genetics (Neuropsychiatric Genetics) 81:134–138 (1998)
© 1998 Wiley-Liss, Inc.

and SCA6 [Zhuchenko et al., 1997]. Detection of thesetrinucleotide repeat mutations recognizes single indi-viduals for these five types of SCAs, allowing their ac-curate diagnosis. The extent of the trinucleotide CAGexpansion also explains some of the variation in age atonset and disease severity. We have screened a largegroup of Portuguese ataxia patients for the presence ofthese mutations to estimate the relative frequency ofthe SCAs in this population.
SUBJECTS AND METHODSPatients and Families
We have identified and collected 65 affected, one sus-pected, and 25 at-risk individuals from 46 Portuguesefamilies (22 previously reported, and 24 newly identi-fied) with dominant cerebellar ataxia; 18 families werefrom the islands of the Azores and 28 from mainlandPortugal. Age at onset ranged from 13 to 60 years. Pa-tients were ascertained between 1990 and 1995, in fiveAzorean and four continental field trips. Family historywas exhaustively studied and no relationship amongthem was found. In addition, two patients from oneataxia family with apparently recessive inheritance, aswell as one isolated case, were also included in thestudy.
All patients presented a progressive form of cerebel-lar ataxia often, but not always, associated with pyra-midal signs (such as hyperreflexia, Babinski’s sign,ankle clonus, and spasticity), extrapyramidal signs(dystonia or parkinsonian features), ocular findings(ophtalmoplegia, nystagmus), and dementia (two fami-lies).
Methods
Genomic DNA was isolated from peripheral bloodleukocytes following standard techniques [Sambrook etal., 1989]. Lymphoblastoid cell lines were establishedby transformation with the Epstein-Barr virus [Ander-son and Gusella, 1984].
The published primer sequences were used for poly-merase chain reaction (PCR) analysis: Rep1 and Rep2for SCA1 [Orr et al., 1993], B37 CAG repeat primersequences [Li et al., 1993] for DRPLA, MJD52 andMJD25 for MJD [Kawaguchi et al., 1994], SCA2-A andSCA2-B for SCA2 [Pulst et al., 1996], and S-5-F1 andS-5-R1 for SCA6 [Zhuchenko et al., 1997] mutations.PCR was carried out as described previously [Silveiraet al., 1996; Pulst et al., 1996; Zhuchenko et al., 1997].
To accurately assess the size of SCA1 alleles as wellas the presence of CAT interruptions, DNA sequencingwas performed. DNA was amplified using primersRep1 [Orr et al., 1993] and GCT-435 [Chung et al.,1993] in a 50-ml PCR reaction with the same conditionsas those used in PCR analysis. The PCR products wererun on a 15% nondenaturing polyacrylamide gel, andbands cut out. DNA was removed from the gel usingMaxam buffer. Sequencing reactions were performedusing 7 ml of DNA and a Sequenase Version 2.0 DNAsequencing Kit (USB, Canada). Primers Rep2 [Orr etal., 1993] and GCT-214 [Chung et al., 1993] were usedfor CAG and GCT strand sequencing, respectively.
According to previous reports, normal SCA1 alleles
have a size range of 16–36, while mutant SCA1 alleleshave 42–81 CAG repeats [Ranum et al., 1994a]. For theDRPLA locus, normal allele size ranged from 8 to 25,while mutant alleles have 54–68 CAGs [Koide et al.,1994]. In normal chromosomes, the MJD gene contains12–40, while expansions range from 62 to 84 CAGs[Maciel et al., 1995; Ranum et al., 1995]. Normal SCA2alleles size ranges from 15–29 CAGs, whereas mutatedalleles range from 35–59 CAG units [Pulst et al., 1996;Sanpei et al., 1996; Imbert et al., 1996]. At the SCA6gene, normal chromosomes have alleles containing 4–16, while expanded alleles have 21–27 CAGs[Zhuchenko et al., 1997].
Linear regression analysis was used to determinethe correlation between CAG repeat number and theage at onset of disease.
RESULTS
Among the 48 families examined for the presence ofCAG expansions at the SCA1 locus, one isolated pa-tient showed an allele in the intermediate size range(37–41 repeats) [Ranum et al., 1994a], with 37 CAGunits. The DNA from this patient was sequenced inorder to characterize this allele. Sequence analysisshowed an interrupted repeat configuration of(CAG)16CATCAGCAT(CAG)18. In spite of its overalllonger size, this was consistent with a normal allele,since the CAG repeat had two CAT interruptions[Chung et al., 1993]. Reports of normal alleles of 39[Genis et al., 1995] and 38 [Ranum et al., 1995] CAGshave been published. No mutant alleles at the SCA1locus were found among these families; the range fornormal alleles was 24–37 CAGs, which is consistentwith previous reports [Ranum et al., 1994a; Genis etal., 1995]. No expanded alleles at the DRPLA locuswere also found; normal alleles had between 7 and 23CAG repeats. No expanded SCA6 alleles were foundamong this group of families; the normal allele sizeranged from 7 to 13 CAG units.
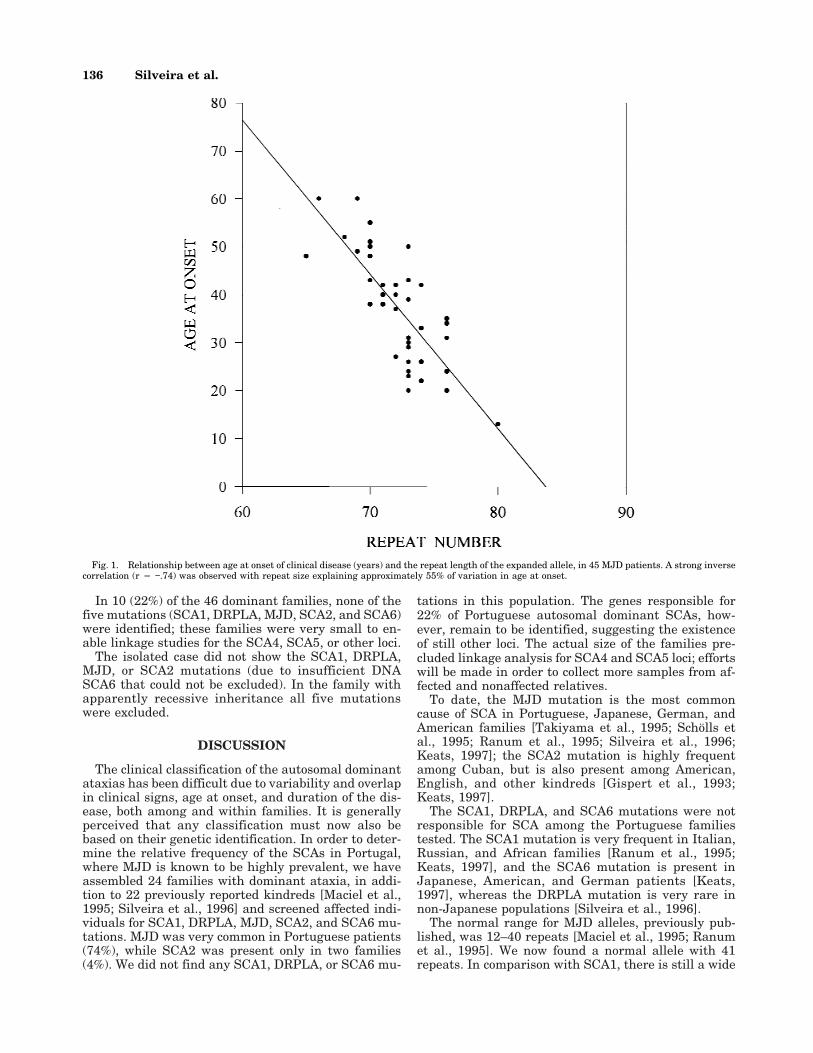
In contrast to the other mutations, we found a highnumber of expanded CAG repeats at the MJD locus: 48affected individuals from 34 dominant families (74%);16 of these families were from the Azores, and 18 frommainland Portugal. Each affected individual had a nor-mal allele ranging from 13 to 41 repeats (thus increas-ing the normal range previously reported), and an ex-panded allele with 65–80 CAGs. Instability was seen in4 of 6 transmissions, where the (CAG)n changed from−2 to +3 CAG units. Figure 1 shows the correlation ofthe age-at-onset of disease and the number of CAGrepeats, in 45 MJD patients. There was a linear corre-lation between the number of CAGs and the age atonset of symptoms with r 4 −.74 (P < .001), indicatingthat only 55% of that variation may be explained by theexpanded repeat-size.
Four individuals (three affected and one suspected)from two families had a CAG expansion at the SCA2locus; normal alleles contained 22 or 23, while the ex-panded allele size ranged from 36 to 47 CAGs. In afather-to-daughter transmission the CAG repeat de-creased two CAG units, while in a father-to-son trans-mission it has increased seven CAGs.
Analysis of SCA1, DRPLA, MJD, SCA2, and SCA6 CAG Repeats 135
In 10 (22%) of the 46 dominant families, none of thefive mutations (SCA1, DRPLA, MJD, SCA2, and SCA6)were identified; these families were very small to en-able linkage studies for the SCA4, SCA5, or other loci.
The isolated case did not show the SCA1, DRPLA,MJD, or SCA2 mutations (due to insufficient DNASCA6 that could not be excluded). In the family withapparently recessive inheritance all five mutationswere excluded.
DISCUSSION
The clinical classification of the autosomal dominantataxias has been difficult due to variability and overlapin clinical signs, age at onset, and duration of the dis-ease, both among and within families. It is generallyperceived that any classification must now also bebased on their genetic identification. In order to deter-mine the relative frequency of the SCAs in Portugal,where MJD is known to be highly prevalent, we haveassembled 24 families with dominant ataxia, in addi-tion to 22 previously reported kindreds [Maciel et al.,1995; Silveira et al., 1996] and screened affected indi-viduals for SCA1, DRPLA, MJD, SCA2, and SCA6 mu-tations. MJD was very common in Portuguese patients(74%), while SCA2 was present only in two families(4%). We did not find any SCA1, DRPLA, or SCA6 mu-
tations in this population. The genes responsible for22% of Portuguese autosomal dominant SCAs, how-ever, remain to be identified, suggesting the existenceof still other loci. The actual size of the families pre-cluded linkage analysis for SCA4 and SCA5 loci; effortswill be made in order to collect more samples from af-fected and nonaffected relatives.
To date, the MJD mutation is the most commoncause of SCA in Portuguese, Japanese, German, andAmerican families [Takiyama et al., 1995; Scholls etal., 1995; Ranum et al., 1995; Silveira et al., 1996;Keats, 1997]; the SCA2 mutation is highly frequentamong Cuban, but is also present among American,English, and other kindreds [Gispert et al., 1993;Keats, 1997].
The SCA1, DRPLA, and SCA6 mutations were notresponsible for SCA among the Portuguese familiestested. The SCA1 mutation is very frequent in Italian,Russian, and African families [Ranum et al., 1995;Keats, 1997], and the SCA6 mutation is present inJapanese, American, and German patients [Keats,1997], whereas the DRPLA mutation is very rare innon-Japanese populations [Silveira et al., 1996].
The normal range for MJD alleles, previously pub-lished, was 12–40 repeats [Maciel et al., 1995; Ranumet al., 1995]. We now found a normal allele with 41repeats. In comparison with SCA1, there is still a wide
Fig. 1. Relationship between age at onset of clinical disease (years) and the repeat length of the expanded allele, in 45 MJD patients. A strong inversecorrelation (r 4 −.74) was observed with repeat size explaining approximately 55% of variation in age at onset.
136 Silveira et al.

gap between the ranges of affected and unaffected al-lele size for MJD. In SCA1, both the size of the CAGrepeat tract and the presence of a CAT interruptiondistinguish a normal from a mutant allele [Chung etal., 1993]; in DRPLA, MJD, and SCA6, an expansion ofthe affected alleles seems to be the only major deter-minant [Kawaguchi et al., 1994].
Our results showed a strong inverse correlation (r 4−.74) in MJD between the size of the (CAG)n and theage at onset (with repeat size explaining approximately50% of the variation in age at onset), similar to previ-ous reports [Maciel et al., 1995; Ranum et al., 1995]. Acomparable correlation has been shown in other neu-rodegenerative diseases caused by trinucleotide re-peats [Snell et al., 1993; Orr et al., 1993; Koide et al.,1994; Nagafuchi et al., 1994]. The repeat size, however,cannot be used to predict accurately the age at onset;for example, in the group of MJD patients studied, twosubjects had repeat size of 70 CAGs, but their ages atonset were 38 and 55 years.
Instability in transmission and somatic mosaicismare also characteristic features of disorders caused bytrinucleotide repeat expansions [Duyao et al., 1993;Snell et al., 1993; Telenius et al., 1994; Ueno et al.,1995]. Among our MJD families, we had six parent-to-child transmissions, with a mean increase of 0.67CAGs, a meiotic instability identical to that reported byMaciel et al. [1995]. As in previous reports [Cancel etal., 1995], the MJD normal alleles appear always as asingle major band, while the MJD expanded allelesshow numerous bands, indicating the presence of so-matic mosaicism.
There are many common features among SCA1,DRPLA, MJD, SCA2, SCA6, spinal and bulbar muscu-lar atrophy (SBMA), and Huntington disease (HD). Allare neurodegenerative diseases caused by the expan-sion of a CAG repeat coding for polyglutamine tractswithin the disease causing protein [Orr et al., 1993;Koide et al., 1994; Kawaguchi et al., 1994; Pulst et al.,1996; Zhuchenko et al., 1997; La Spada et al., 1991;The Huntington’s Disease Collaborative ResearchGroup, 1993]. In SCA1, DRPLA, and HD, the sizeranges for normal and expanded alleles are almostidentical [Orr et al., 1993; The Huntington’s DiseaseCollaborative Research Group, 1993; Koide et al., 1994;Nagafuchi et al., 1994]; paternal transmissions are as-sociated with larger expansions (with a consequent an-ticipation of age at onset) [Chung et al., 1993; Nagafu-chi et al., 1994; Snell et al., 1993]. In contrast, in MJDthere is a large gap between normal and expanded al-leles and paternal effect is on instability as a whole(larger contractions in repeat size) [Maciel et al., 1995;Cancel et al., 1995]. In our group of MJD patients, thelargest anticipation observed (18 years) was in a fa-ther-to-son transmission resulting from an increase ofjust three CAG units; also, the only juvenile case (13years) received the mutant allele from the father (nottyped).
Three of the 10 dominant ataxia families excludedfor the five loci had been classified as MJD. Furtherclinical studies, based on this molecular identification,will be needed to clarify if there are relevant pheno-
typic differences between them and the MJD com-proved families.
ACKNOWLEDGMENTS
We thank Dr. H.Y. Zogbi and Dr. S. Tsuji for provid-ing positive controls for the SCA1 and DRPLA muta-tions, respectively; the patients and their families fortheir cooperation. This work was supported by: thejoint Program FRSQ-ACAF (Fonds de la Recherche enSante du Quebec and Association Canadienne del’Ataxie de Friedreich), the NIH (grant NS 31687), theNetwork of Centres of Excellence (Canadian GeneticDisease Network), and the Financiamento Plurianualde Unidades de Investigacao from JNICT (Junta Na-cional de Investigacao Cientifica e Tecnologica), Portu-gal. I.S., C.G. and P.M. are recipients of a scholarshipfrom the Praxis Programme, JNICT, Portugal.
REFERENCES
Anderson MA, Gusella JF (1984): Use of cyclosporin A in establishingEpstein-Barr virus-transformed human lymphoblastoid cell lines. InVitro 20:856–858.
Benomar A, Krols L, Stevanin G, Cancel G, LeGuern E, David G, OuhabiH, Martin JJ, Durr A, Zaim A, Ravise N, Busque C, Penet C,Regemorter NV, Weissenbach J, Yahyaoui M, Chkili T, Agid Y, Broeck-hoven CV, Brice A (1995): The gene for autosomal dominant cerebellarataxia with pigmentary macular dystrophy maps to chromosome 3p12-p21.1. Nature Genet 10:84–88.
Cancel G, Abbas N, Stevanin G, Durr A, Chneiweiss H, Neri C, DuyckaertsC, Penet C, Cann HM, Agid Y, Brice A (1995): Marked phenotypicheterogeneity associated with expansion of a CAG repeat sequence atthe spinocerebellar ataxia 3/Machado-Joseph disease locus. Am J HumGenet 57:809–816.
Chung M, Ranum LPW, Duvick LA, Servadio A, Zoghbi HY, Orr HT (1993):Evidence for a mechanism predisposing to intergenerational CAG re-peat instability in spinocerebellar ataxia type 1. Nature Genet 5:254–258.
Duyao M, Ambrose C, Myers R, Novelletto A, Persichetti F, Frontali M,Folstein S, Ross C, Franz M, Abbott M, Gray J, Conneally P, Young A,Penney J, Hollingsworth Z, Shoulson I, Lazzarini A, Falek A, Koro-shetz W, Sax D, Bird E, Vonsattel J, Bonilla E, Alvir J, Bickham CondeJ, Cha J-H, Dure L, Gomez F, Ramos M, Sanchez-Ramos J, SnodgrassS, Young M, Wexler N, Moscowitz C, Penchaszadeh G, MacFarlane H,Anderson M, Jenkins B, Srinidhi J, Barnes G, Gusella J, MacDonald M(1993): Trinucleotide repeat length instability and age of onset in Hun-tington’s disease. Nature Genet 4:387–392.
Gardner K, Alderson K, Galster B, Kaplan C, Leppert M, Ptacek L (1994):Autosomal dominant spinocerebellar ataxia: Clinical description of adistinct hereditary ataxia and genetic localization to chromosome 16(SCA4) in a Utah kindred. Neurology (suppl 2) 44:A361.
Genis D, Matilla T, Volpini V, Rosell J, Davalos A, Ferrer I, Molins A,Estivill X (1995): Clinical, neuropathologic, and genetic studies of alarge spinocerebellar ataxia type 1 (SCA1) kindred: (CAG)n expansionand early premonitory signs and symptoms. Neurology 45:24–30.
Gispert S, Twells R, Orozco G, Brice A, Weber J, Heredero L, Scheufler K,Riley B, Allotey R, Nothers C, Hillermann R, Lunkes A, Khati C, Ste-vanin G, Hernandez A, Magarino C, Klockgether T, Durr A, ChneiweissH, Enczmann J, Farrall M, Beckmann J, Mullan M, Wernet P, Agid Y,Freund H-J, Williamson R, Auburger G, Chamberlain S (1993): Chro-mosomal assignment of the second locus for autosomal dominant cer-ebellar ataxia (SCA2) to chromosome 12q23-24.1. Nature Genet 4:295–299.
Gouw LG, Kaplan CD, Haines JH, Digre KB, Rutledge SL, Matilla A,Leppert M, Zoghbi HY, Ptacek LJ (1995): Retinal degeneration char-acterizes a spinocerebellar ataxia mapping to chromosome 3p. NatureGenet 10:89–93.
Imbert G, Saudau F, Yvert G, Devys D, Trottier Y, Garnier J-M, Weber C,Mandel J-L, Cancel G, Abbas N, Durr A, Didierjean O, Stevanin G,Agid Y, Brice A (1996): Cloning of the gene for spinocerebellar ataxia 2reveals a locus with high sensitivity to expanded CAG/glutamine re-peats. Nature Genet 14:285–291.
Analysis of SCA1, DRPLA, MJD, SCA2, and SCA6 CAG Repeats 137

Kawaguchi Y, Okamoto T, Taniwaki M, Aizawa M, Inoue M, Katayama S,Kawakami H, Nakamura S, Nishimura M, Akiguchi I, Kimura J, Na-rumiya S, Kakizuka A (1994): CAG expansions in a novel gene forMachado-Joseph disease at chromosome 14q32.1. Nature Genet 8:221–228.
Keats B (1997): Population genetics of the inherited ataxias. Annual Mon-treal Neurological Meeting, May 29–June 1.
Koide R, Ikeuchi T, Onodera O, Tanaka H, Igarashi S, Endo K, TakahashiH, Kondo R, Ishikawa A, Hayashi T, Saito M, Tomoda A, Miike T, NaitoH, Ikuta F, Tsuji S (1994): Unstable expansion of CAG repeat in he-reditary dentatorubralpallidoluysian atrophy (DRPLA). Nature Genet6:9–13.
La Spada AR, Wilson EM, Lubahn DB, Harding AE, Fischbeck KH (1991):Androgen receptor gene mutations in X-linked spinal and bulbar mus-cular atrophy. Nature 352:77–79.
Li SH, McInnis MG, Margolis RL, Antonarakis SE, Ross CA (1993): Noveltriplet repeat containing genes in human brain: Cloning, expression,and length polymorphisms. Genomics 16:572–579.
Lopes-Cendes I, Andermann E, Attig E, Cendes F, Bosch S, Wagner M,Gerstenbrand F, Andermann F, Rouleau GA (1994): Confirmation ofthe SCA-2 locus as an alternative locus for dominantly inherited spi-nocerebellar ataxias and refinement of the candidate region. Am JHum Genet 54:774–781.
Maciel P, Gaspar C, DeStefano AL, Silveira I, Coutinho P, Radvany J,Dawson DM, Sudarsky L, Guimaraes J, Loureiro JE, Nezarati MM,Corwin LI, Lopes-Cendes I, Rooke K, Rosenberg R, MacLeod P, FarrerLA, Sequeiros J, Rouleau GA (1995): Correlation between CAG repeatlength and clinical features in Machado-Joseph disease. Am J HumGenet 57:54–61.
Nagafuchi S, Yanagisawa H, Sato K, Shirayama T, Ohsaki E, Bundo M,Takeda T, Tadokoro K, Kondo I, Murayama N, Tanaka Y, KikushimaH, Umino K, Kurosawa H, Furukawa T, Nihei K, Inoue T, Sano A,Komure O, Takahashi M, Yoshizawa T, Kanazawa I, Yamada M (1994):Dentatorubral and pallidoluysian atrophy expansion of an unstableCAG trinucleotide on chromosome 12p. Nature Genet 6:14–18.
Orr HT, Chung M, Banfi S, Kwiatkowski TJ, Jr., Servadio A, Beaudet AL,McCall AE, Duvick LA, Ranum LPW, Zoghbi HY (1993): Expansion ofan unstable trinucleotide CAG repeat in spinocerebellar ataxia type 1.Nature Genet 4:221–226.
Pulst S-M, Nechiporuk A, Nechiporuk T, Gispert S, Chen X-N, Lopes-Cendes I, Pearlman S, Starkman S, Orozco-Diaz G, Lunkes A, DeJongP, Rouleau GA, Auburger G, Korenberg JR, Figueroa C, Sahba S(1996): Moderate expansion of normally biallelic trinucleotide repeat inspinocerebellar ataxia type 2. Nature Genet 14:269–275.
Ranum LPW, Chung M, Banfi S, Bryer A, Schut LJ, Ramesar R, DuvickLA, McCall A, Subramony SH, Goldfarb L, Gomez C, Sandkuijl LA, OrrHT, Zoghbi HY (1994a): Molecular and clinical correlations in spino-cerebellar ataxia type 1: Evidence for familial effects on the age atonset. Am J Hum Genet 55:244–252.
Ranum LPW, Schut LJ, Lundgren JK, Orr HT, Livingston DM (1994b):Spinocerebellar ataxia type 5 in a family descended from the grand-parents of President Lincoln maps to chromosome 11. Nature Genet8:280–284.
Ranum LPW, Lundgren JK, Schut LJ, Ahrens MJ, Perlman S, Aita J, BirdTD, Gomez C, Orr HT (1995): Spinocerebellar ataxia type 1 andMachado-Joseph disease: Incidence of CAG expansions among adult-onset ataxia patients from 311 families with dominant, recessive, orsporadic ataxia. Am J Hum Genet 57:603–608.
Sambrook J, Fritsch EF, Maniatis T (1989): “Molecular Cloning: A Labo-ratory Manual.” Cold Spring Harbor, NY: Cold Spring Harbor Labora-tory Press.
Sanpei K, Takano H, Igarashi S, Sato T, Oyake M, Sasaki H, Wakisaka A,Tashiro K, Ishida Y, Ikeuchi T, Koide R, Saito M, Sato A, Tanaka T,
Hanyu S, Takiyama Y, Nishizawa M, Shimizu N, Nomura Y, SegawaM, Iwabuchi K, Eguchi I, Tanaka H, Takahashi H, Tsuji S (1996):Identification of the spinocerebellar ataxia type 2 gene using a directidentification of repeat expansion and cloning technique, DIRECT(1996). Nature Genet 14:277–283.
Schols L, Vieira-Saecker AMM, Schols S, Przuntek H, Epplen JT, Riess O(1995): Trinucleotide expansion within the MJD1 gene presents clini-cally as spinocerebellar ataxia and occurs most frequently in GermanSCA patients. Hum Mol Genet 4:1001–1005.
Sequeiros J, Silveira I, Maciel P, Coutinho P, Manaia A, Gaspar C, BurletP, Loureiro L, Guimaraes J, Tanaka H, Takiyama Y, Sakamoto H,Nishizawa M, Nomura Y, Segawa M, Tsuji S, Melki J, Munnich A(1994): Genetic linkage studies of Machado-Joseph disease with chro-mosome 14q STRPs in 16 Portuguese-Azorean kindreds. Genomics21:645–648.
Silveira I, Lopes-Cendes I, Kish S, Maciel P, Gaspar C, Coutinho P, BotezMI, Teive H, Arruda W, Steiner CE, Pinto-Junior W, Maciel JA, Jain S,Sack G, Andermann E, Sudarsky L, Rosenberg R, MacLeod P, ChitayatD, Babul R, Sequeiros J, Rouleau GA (1996): Frequency of spinocer-ebellar ataxia type 1, dentatorubropallidoluysian atrophy, andMachado-Joseph disease mutations in a large group of spinocerebellarataxia patients. Neurology 46:214–218.
Snell RG, MacMillan JC, Cheadle JP, Fenton I, Lazarou LP, Davies P,MacDonald ME, Gusella JF, Harper PS, Shaw DJ (1993): Relationshipbetween trinucleotide repeat expansion and phenotypic variation inHuntington’s disease. Nature Genet 4:393–397.
Takiyama Y, Nishizawa M, Tanaka H, Kawashima S, Sakamoto H, KarubeY, Shimazaki H, Soutome M, Endo K, Ohta S, Kagawa Y, Kanazawa I,Mizuno Y, Yoshida M, Yuasa T, Horikawa Y, Oyanagi K, Nagai H,Kondo T, Inuzuka T, Onodera O, Tsuji S (1993): The gene for Machado-Joseph disease maps to human chromosome 14q. Nature Genet 4:300–304.
Takiyama Y, Igarashi S, Rogaeva EA, Endo K, Rogaev EI, Tanaka H,Sherrington R, Sanpei K, Liang Y, Saito M, Tsuda T, Takano H, IkedaM, Lin C, Chi H, Kennedy JL, Lang AE, Wherret JR, Segawa M, No-mura Y, Yuasa T, Weissenbach J, Yoshida M, Nishizawa M, Kidd KK,Tsuji S, St George-Hyslop PH (1995): Evidence for inter-generationalinstability in the CAG repeat in the MJD1 gene and for conservedhaplotypes at flanking markers amongst Japanese and Caucasian sub-jects with Machado-Joseph disease. Hum Molec Genet 4:1137–1146.
Telenius H, Kremer B, Goldberg YP, Theilmann J, Andrew SE, Zeisler J,Adam S, Greenberg C, Ives EJ, Clarke LA, Hayden MR (1994): Somaticand gonadal mosaicism of the Huntington disease gene CAG repeat inbrain and sperm. Nature Genet 6:409–414.
The Huntington’s Disease Collaborative Research Group (1993): A novelgene containing a trinucleotide repeat that is expanded and unstableon Huntington’s disease chromosomes. Cell 72:971–983.
Twist EC, Casaubon LK, Ruttledge MH, Rao VS, Macleod PM, Radvany J,Zhao Z, Rosenberg RN, Farrer LA, Rouleau GA (1995): Machado Jo-seph disease maps to the same region of chromosome 14 as the spino-cerebellar ataxia type 3 locus. J Med Genet 32:25–31.
Ueno S, Kondoh K, Kotani Y, Komure O, Kuno S, Kawai J, Hazama F, SanoA (1995): Somatic mosaicism of CAG repeat in dentatorubral-pallidoluysian atrophy (DRPLA). Hum Mol Genet 4:663–666.
Yakura H, Wakisaka A, Fujimoto S, Itakura K (1974): Hereditary ataxiaand HL-A genotypes. N Engl J Med 291:154–155.
Zoghbi HY, Pollack MS, Lyons LA, Ferrell RE, Daiger SP, Beaudet AL(1988): Spinocerebellar ataxia: Variable age of onset and linkage tohuman leukocyte antigen in a large kindred. Ann Neurol 23:580–584.
Zhuchenko O, Bailey J, Bonnen P, Ashizawa T, Stockton DW, Amos C,Dobyns WB, Subramony SH, Zoghbi HY, Lee CC (1997): Autosomaldominant cerebellar ataxia (SCA6) associated with small polygluta-mine expansions in the a1A-voltage-dependent calcium channel. Na-ture Genet 15:62–69.
138 Silveira et al.





























