Embed Size (px)
Citation preview

Analysis of Cyclin-Dependent Kinase InhibitorExpression and Methylation Patterns in Human
Prostate Cancers
TuDung T. Nguyen,1 Carvell T. Nguyen,1 Felicidad A. Gonzales,1
Peter W. Nichols,2 Mimi C. Yu,3 and Peter A. Jones1*1Department of Biochemistry and Molecular Biology, USC/Norris Comprehensive Cancer
Center, University of Southern California School of Medicine, Los Angeles, California2Department of Pathology, USC/Norris Comprehensive Cancer Center, University of
Southern California School of Medicine, Los Angeles, California3Department of Preventive Medicine, USC/Norris Comprehensive Cancer Center, University
of Southern California School of Medicine, Los Angeles, California
BACKGROUND. Downregulation of genes which negatively control cell cycle progressionrepresents a possible mechanism for prostate tumorigenesis. We examined the expressionlevels of the p16, p15, p14, and retinoblastoma-susceptibility (RB) genes in primary prostatecancers and human prostate cancer cell lines, and correlated this with the DNA methylationlevels of two loci in p16.METHODS. The mRNA levels of p16, p15, and p14 were examined by reverse transcriptase-PCR (RT-PCR). DNA methylation of the p16 58 CpG island was determined by bisulfitegenomic sequencing, while methylation of exon 2 shared by the p16 and p14 genes wasmeasured by a quantitative bisulfite-based technique, methylation-sensitive single-nucleotideprimer extension (Ms-SNuPE). RB protein levels were assessed by immunohistochemicalstaining of histologic sections of normal and tumor prostate tissues, using a monoclonalantibody (mAB).RESULTS. Overexpression of p16 mRNA was found in 6/9 (67) of prostate tumors comparedto the adjacent normal prostate, whereas elevated p14 and p15 levels were only observed in2/9 (22) and 1/6 (17) of prostate cases, respectively. There was no statistically significantassociation of grade (P = 0.18) and stage (P = 1.00) of prostate cancer to the elevated p16 levelsin the tumors. The p16 58 CpG island was completely unmethylated in these tissues. Incontrast, exon 2 of p16/p14 was methylated in both the tumor and normal adjacent prostates,and was increased in 8/11 (73) of tumors relative to normal tissues. There was no associationbetween p16 overexpression and increased p16/p14 exon 2 methylation in these tumors (P =1.00). Diminished RB levels in prostate tumors that had upregulated p16 mRNA were found,although absent RB was also detected in tumors without elevated p16 levels. The expressionlevels of the two genes, RB and p16, were not correlated statistically (P = 0.16).CONCLUSIONS. Our studies show that although the levels of the cell cycle regulators p16,p15, p14, and Rb are altered in prostate cancers, there is no apparent correlation to grade,stage, or any pattern of regulation between the related genes. Exon 2 of p16/p14 is methylatedin a majority of prostate tumors compared to the unmethylated upstream 58 region, and may
*Grant sponsor: NCI; Grant number: R35 CA 49758.*Correspondence to: Peter A. Jones, Ph.D., D.Sc., USC/Norris Com-prehensive Cancer Center, 1441 Eastlake Ave., Room 8302L, MailStop #83, Los Angeles, CA 90089-9181.E-mail: [email protected] 2 September 1999; Accepted 5 January 2000
The Prostate 43:233–242 (2000)
© 2000 Wiley-Liss, Inc.

be a potential tumor marker for human prostate cancer. Prostate 43:233–242, 2000.© 2000 Wiley-Liss, Inc.
KEY WORDS: cyclin-dependent kinase inhibitor; p16; RB; DNA methylation
INTRODUCTION
Prostate cancer is one of the most common malemalignancies in the United States and is the secondmost common cause of cancer deaths in males. Previ-ous research has shown that steroids such as testos-terone, dihydrotestosterone, and vitamin D are in-volved in prostate cancer pathogenesis [1–3]. Thesehormones act through their receptors to regulate thetranscription of downstream genes controlling growthand differentiation. Transformed prostate cells havealtered cell proliferation and death rates. Berges et al.[4] demonstrated that high-grade prostatic intraepi-thelial neoplastic cells have an increased percentage ofproliferating cells compared to apoptotic cells, in con-trast to normal prostatic glandular cells, which have abalanced rate of cell proliferation and death. Prolifera-tion in localized prostatic cancer cells was also in-creased compared to normal prostate cells, althoughthe death rate was decreased, resulting in continuousgrowth of the localized prostatic cancers [4].
The cyclin-dependent kinase (CDK) inhibitor,CDKN2 or p16, is inactivated in many primary tumorsand in cell lines [5]. It encodes a protein that inhibitsthe cyclin-dependent kinase (CDK4) which phos-phorylates the product of the retinoblastoma-susceptibility gene (RB), thus inhibiting progression ofcells through the G1 phase of the cell cycle. p16 is oftenlost by deletion, mutation, or methylation events inmany tumor types [6–10], and prostate cancer celllines also show similar modes of inactivation of thep16 gene [8,11,12]. However, mutations of p16 are veryrare in primary prostate tumors [13–17], and promotermethylation is also not common [17]. In addition top16, chromosome 9p21 also contains two other nega-tive cell cycle growth regulators, p14 and p15 [18,19].p14 shares a common exon 2 with the p16 transcript,but has a different exon 1b, and is translated in analternative reading frame [20]. Loss of p14 also resultsin formation of tumors in the course of the life span ofp14 knockout mice [21]. p15, which is located centro-meric to the p16/p14 gene locus, is a negative growthregulator that is a downstream effector of transform-ing growth factor beta (TGFb)-induced cell cycle ar-rest [19]. TGFb, a multifunctional protein that regu-lates cell proliferation, cell cycle inhibition,morphogenetic movements, apoptosis, and differen-tiation [22,23], has been associated with prostate can-cer. Although TGFb is a growth inhibitor of epithelialcells in vitro, its levels are increased in vivo in prostate
cancer and benign prostatic hypertrophy (BPH) com-pared to normal prostatic epithelial cells [24–28].TGFb is regulated by steroids [22,29], which have beenshown to upregulate cyclin-dependent kinases inother studies [30,31].
The association between DNA methylation andtranscriptional downregulation has been observed formany cancers for many genes, including p16 [6,7]. In-creased DNA methylation of Glutathione S-transferasepi (GSTpi) [32,33] and Endothelin B receptor [34,35] wasassociated with inactivation of these gene products inprostate tumors, and expression of both genes couldbe reactivated in vitro by the DNA demethylatingagent 5-aza-2-deoxycytidine. We have now examinedthe expression of the cyclin-dependent kinase inhibi-tors, p16, p15, and p14 by quantitative RT-PCR, and thelevels of RB protein by immunohistochemistry inprostate cancers and adjacent normal cells to deter-mine their effects on the growth/proliferation of pros-tate tumors compared to the visibly normal adjacentprostate cells. Two CpG islands in the p16/p14 locus inprostate tumors were examined to assess if DNAmethylation played a role in transcriptional regulationand contributed to the development of prostate tu-mors.
MATERIALS AND METHODS
Samples
Eleven prostate tumors and adjacent normal speci-mens were obtained as radical prostatectomy casesfrom the USC/Norris Comprehensive Cancer Center(Los Angeles, CA). Samples were frozen in O.C.T.Compound (Miles Inc. Diagnostics Division, Elkhart,IN), and frozen sections made which were subse-quently stained in hematoxylin and eosin. All frozenspecimens were thinly sliced (#5 mm diameter). Spe-cific tumor sections with >90% tumor cells were dis-sected from the frozen block, using the microscopefrozen hematoxylin and eosin (H&E)-stained slides asa guide to determine the boundaries of the tumor inthe frozen block. The same criteria were applied toobtain corresponding pure “normal” prostate tissuefrom the same frozen tissue block. DNA and RNAwere extracted from the same tissue, using standardprotocols [7,36]. Due to the above method of tissuecollection to ensure sample purity, limiting DNA andRNA was available for the analyses. Two normal pros-tate samples were also obtained from patients who
234 Nguyen et al.

underwent total cystectomy, where the adjacent nor-mal prostate was removed. DU145, LNCap, and PC3are prostate cell lines. Sperm DNA also served as acontrol for complete unmethylation in the methylationassay.
Bisulfite Genomic Sequencing
Methylation of individual CpGs in the p16 58 UTRand exon 1 (region D in Gonzalgo et al., 1998) wasdetermined, using the protocol and primers previ-ously described [37].
Ms-SNuPE Analysis
The mean methylation level of the p16/p14 exon 2was the average of three CpG sites. Bisulfite DNA wasprepared as described previously [38,39]. Primersused to generate top strand-specific PCR product forthe p16/p14 exon 2 MS-SNuPE methylation analysiswere: sense, 58-TTG ATT ATT TTG TTT TTT TTGGTA GGT T-38, and antisense, 58-CAA ATT CTC AGATCA TCA GTC CTC ACC T-38. PCR reactions for thep16/p14 exon 2 were performed in 25 ml total volumeunder the following conditions: ∼100 ng bisulfitetreated DNA, 1 × PCR Buffer (Boehringer Mannheim),10% DMSO, 0.5 mM final concentration of eachprimer, 200 mM of each dNTP, and 1 U Taq polymer-ase (Roche Molecular Biochemical, Mannheim, Ger-many). All reactions were hot-started manually. PCRconditions for the human p16/p14 exon 2 were: 95°Cfor 3 min, 36 cycles of 95°C for 1 min, 58°C for 45 sec,and 72°C for 1 min 20 sec, and then final extension of72°C for 2 min. PCR products were gel purified with aQiaQuick Gel Extraction Kit (Qiagen, Valencia, CA),and the template was resuspended in 20–40-ml volumeof H2O. The methylation assay was performed using acombination of three primers in the Ms-SNuPE reac-tions, which monitor the methylation status of threeCpG sites in the amplified region, and are as follows:16 mer, 58-GGT GGT GGT GTT GTA T-38; 19 mer,58-AGG TTA TGA TGA TGG GTA G-38; 22 mer, 58-TAT TAG AGG TAG TAA TTA TGT T-38. Ms-SNuPEreactions were performed in a 10 ml volume. DNA(50–100 ng) was incubated in a final concentration of 1× PCR buffer, 1 mM of each Ms-SNuPE primer, and 1mCi of either [32P]dCTP or [32P]dTTP. One unit of Taqpolymerase (Roche Molecular Biochemical, Mann-heim, Germany) complexed with Taq polymerase an-tibody (Clontech, Palo Alto, CA) in a 1:1 ratio wasused in the reaction to hot-start the PCR. Ms-SNuPEreaction conditions were: 95°C for 1 min, 51°C for 2min, and 72°C for 1 min. Stop solution (4.6 ml ) wasadded to reaction mixtures, and samples were heat-denatured and then loaded onto 15% denaturing poly-acrylamide gels (7 M urea). A band in the cytosine (C)
lane from the Ms-SNuPE PCR reaction represents theamount of methylated cytosines (signal from incorpo-ration of [32P]dCTP), while a band in the thymine (T)lane was the product of [32P]dTTP incorporation andunmethylated cytosine molecules at a particular CpGsite. Radioactivity of bands was quantitated via aphosphorimager (Molecular Dynamics, Sunnyvale,CA). Ms-SNuPE analysis conditions were standard-ized with the mixtures of methylated (SssI methylase(New England Biolabs, Beverly, MA)-treated DNA) tounmethylated (untreated) DNA. Normal humansperm methylated to unmethylated mixes gave ex-pected results for each of the percent methylatedmixes (0, 25, 50, 75, and 100) in the Ms-SNuPE assayfor the p16 exon 2 region. All p16 exon 2 methylationvalues were determined from a minimum of two in-dependent experiments to obtain an averaged quanti-tative methylation value for each sample analyzed.
RT-PCR
RT-PCR was performed as described previously [7],using 5 mg of total cellular RNA to generate cDNA.One hundred nanograms of cDNA were used for eachset of primers and are as follows: p16 sense, 58-AGCCTT CGG CTG ACT GGC TGG-38; p16 antisense, 58-GCG CTG CCC ATC ATC ATG AC-38; p14 sense, 58-TGG CGC TGC TCA CCT CTG GT-38; p14 antisense,58-GCG CTG CCC ATC ATC ATG AC-38; p15 sense,58-GGC GCG CGA TCC AGG TCA TGA TGA T-38;p15 antisense, 58-CGG CTG GGG AAC CTG GCGTCA-38; GAPDH sense, 58-CAG CCG AGC CAC ATCG-38; and GAPDH antisense, 58-TGA GGC TGT TGTCAT ACT TCT C-38. PCR for GAPDH served as a con-trol that RNA was a suitable substrate for the analysis.PCR products were resolved on a nondenaturing 6%acrylamide gel, DNA was transferred to nylon mem-brane by electroblotting, and samples were probed bya 32P-labeled internal oligonucleotide probe, usingprotocols previously described [40]. Conditions for thep14 and GAPDH PCR were: 94°C for 3 min, 24 cyclesof 94°C for 1 min, 57°C for 30 sec, and 72°c for 40 sec,and then a final extension of 72°C for 1 min. Condi-tions for the p16 PCR were: 94°C for 3 min, 25 cycles of94°C for 1 min, 57°C for 30 sec, and 72°C for 40 sec,and then a final extension of 72°C for 1 min. Condi-tions for the p15 PCR were: 95°C for 3 min, 26 cycles of95°C for 1 min 40 sec, 60°C for 45 sec, and 72°C for 40sec, and then a final extension of 72°C for 2 min. Un-der the above conditions, PCR amplification for allgenes were within the linear range of the assay. Thesame 38 PCR primer was used for the p16 and p14expression analysis, and GAPDH was a control foramount of input cDNA. All PCR analyses were re-peated at least twice to determine a quantitative ex-
CDK Inhibitor and Methylation Patterns 235

pression value ratio (of CDK inhibitor to GAPDH ex-pression levels). Due the limiting amount of RNA andDNA obtained from the same tissue, since a strict cri-teria for “pure” tumor or normal was used for ourstudies, some mRNA expression analyses were notdetermined (ND or NA).
Rb Immunohistochemistry
Parrafin-embedded prostate cancers cases whichhad been fixed previously in 10% buffered formalinfrom the USC/Kenneth Norris Cancer Center (LosAngeles, CA) were sectioned with a microtometer induplicate and examined first by hematoxylin and eo-sin staining to confirm the presence of tumor cells.Serial sections were then stained for the presence ofthe RB human protein using the monoclonal antibodyRB-G3-245 (Pharmingen, San Diego, CA), which rec-ognizes an epitope between amino acids 300–380 ofhuman RB. Immunolocalization was carried out withthe avidin-biotin (ABC Detection Kit, Ventana, Tuc-son, AZ) method, as described by the manufacturer’sprotocol. Two investigators (P.W.N. and T.T.N.) re-viewed the pathological specimens by light micros-copy and evaluated the patterns. A normal prostatefrom a nonprostate cancer case served as a control forpositive RB staining. Homogeneous endothelial andmesenchymal cell nuclei staining was also present inthe tumors that failed to stained for RB, and served asan internal control.
Statistical Analysis
We used the two-sample Wilcoxon rank sum test tocompare indices of p16 overexpression by grade (7 orhigher vs. 6 or below) and stage (2 vs. 3) of disease,and by degree of nuclear RB staining in prostate tumorcells (no to weak vs. weak to moderate staining) [41].Fisher’s exact test was used to assess the associationbetween increased p16/p14 exon 2 methylation and p16overexpression in prostate tumors [41].
RESULTS
Expression of CDK Inhibitors in Prostate Cancers
The expression levels of the mRNAs for the CDKinhibitors, p16, p15, and p14 in nine primary prostatetumors were compared to the normal adjacent tissues.Figure 1 shows representative prostate cancer caseswhich show upregulated p16 expression and relativelyuniform expression levels of p15 and p14 between thetumors and normal matched prostates. The levels ofthe p16 mRNA transcripts were increased in 6/9 (67)of prostate tumors, with levels in the tumors ranging
from 3–24-fold increased compared to the normal ad-jacent prostate glands (Table I). The p15 and p14 levelsin the prostate tumors were also elevated, but lessfrequently and to a lower degree, in 1/6 (17) and 2/9(22) of the prostate cases, respectively (Table I).Samples which overexpressed p15 and p14 in the tu-mors compared to the normal prostate also had up-regulated p16 levels (Table I). Increased p16 in the tu-mors appeared selective, since the levels of p14 wereconsistently unchanged, even though the two genesshare a common exon 2 and the same 38 primer wasused for both p16 and p14 RT-PCRs. GAPDH expres-sion was uniformly observed between the paired nor-mal and tumor sample from each radical prostatec-tomy case though p16 was upregulated, suggestingthat variation in input cDNA was not the cause ofelevated p16 expression levels detected in the tumors.There was no statistically significant association ofgrade (P = 0.18) and stage (P = 1.00) of prostate cancerto the elevated p16 levels in the tumors (Table I).
Increased p16/p14 Exon 2 Methylation inProstate Tumors
The methylation status of 28 CpGs in the p16 58UTR/exon 1 in prostate tumor and adjacent normal
Fig. 1. Expression analysis of CDK inhibitors in prostate. RT-PCR was used to examine the expression of the p16, p15, and p14in matched tumor and adjacent normal tissues from radical pros-tatectomy cases. p16 expression levels were increased in the tu-mors compared to the corresponding normal prostate (768, 857,and 870), while the levels of p15 and p14 remained relativelyunchanged. N, normal prostate; T, tumor prostate.
236 Nguyen et al.

DNAs was examined by bisulfite genomic sequencing;however, no methylation was observed as expected,since all the tissues expressed p16 (Table I), and meth-ylation of this region is associated with gene inactiva-tion [37]. The methylation status of p16/p14 exon 2, adownstream CpG island, was next determined forcomparison. The p16/p14 exon 2 was unmethylated inhuman sperm DNA, but extensively methylated in theprostate cancer cell line DNAs from DU145 andLNCaP (Fig. 2). Normal prostate DNA was methylat-ed to a low degree in p16/p14 exon 2, and the adjacentprostate tumor in 8/11 (73) of the prostate casesshowed increased methylation levels (Figs. 2, 3). Exon2 methylation levels were increased in some DNAsobtained from apparently normal cells in prostate can-cer patients, compared to the lower methylation levelsin two normal prostate glands from individuals with-out prostate cancer (Fig. 3). Seven of the eight tumorswith increased p16/p14 exon 2 methylation had beenassessed for p16 mRNA level; 5/7 had p16 overexpres-sion compared to 1/2 in tumors without increasedp16/p14 exon 2 methylation. There was no associationbetween p16 overexpression and increased p16/p14exon 2 methylation in these tumors (P = 1.00). Meth-ylation of the exon 2 region shared by both p16 andp14 does not block expression of the p16 gene.
RB Protein Is Absent in Majority of ProstateTumors But Is Not Correlated With Upregulated
p16 mRNA Levels
The status of RB protein in the prostate cancer caseswas determined by immunohistochemistry and corre-lated to the levels of p16 mRNA measured earlier. Anormal prostate obtained from a patient without pros-tate cancer had 100% of the cells with nuclear staining
for RB, and 0% cytoplasmic staining (results notshown). The normal prostate cells from the cancer pa-tients showed abundant RB nuclear staining in a ma-jority of the cells, while the corresponding tumor foreach case had weak and sparse RB nuclear staining(Fig. 4). Most of the cases showed RB expression in thenormal cells except for case 870, and weak or absentRB expression in the corresponding tumor cells (TableII). Two cases, 735 and 491, had different patterns ofstaining (A and B), since two different pathologicalgrades of tumor were present in each case, demon-strating the heterogeneous nature of prostate cancers.Only cases 768 and 1406 showed weak to moderatenuclear RB protein expression in the tumor, and alsohad upregulated p16 mRNAs in the tumor. We classi-fied tumors from patients 768, 857, and 1406 as pos-sessing higher nuclear RB protein expression than theremaining 6 patients (478, 507, 549, 735, 870, and 917)(Table II). There was no statistically significant differ-ence in p16 mRNA expression levels between the twogroups of tumors stratified by RB protein expressionlevel (P = 0.16). Abnormal cytoplasmic RB localizationwas only observed in two tumor cases (917 and 784).These data suggest that abrogation of the Rb pathwayin prostate cancer is achieved by downregulation ofthe RB protein (or mutation) rather than by p16changes.
DISCUSSION
Although prostate cancer is the most common malemalignancy in the United States, its etiology and mo-lecular biology are still largely undefined. Family his-tory of prostate cancer is an important risk factor aswell as endocrine control, which contributes to theevolution of the tumor [2,42,43]. These endocrine in-
TABLE I. Expression of CDK Inhibitors inProstate Cancer*
Patient Grade Stage p14 p16 p15
478 4 pT2bN0 0.5 1.0 0.6507 7 pT3aN0 0.7 1.2 1.0549 6 pT2bN0 2.3 3.2 ND735 8 pT3aN1 2.9 16.0 5.5768 6 pT3aN0 1.3 24.0 ND857 7 pT2bN0 0.9 12.0 0.6870 5 pT3aN0 0.8 7.0 ND917 7 pT3bN0 0.6 0.3 0.4
1406 9 pT3aN0 0.9 3.3 0.7
*Grade, Gleason sum; Stage, pathologic stage by the TNM sys-tem; ND, not determined due to limiting RNA. Numerical val-ues are relative expression ratio of tumor to corresponding nor-mal compared to GAPDH.
Fig. 2. Quantitative methylation analysis of p16/p14 exon 2 inthe prostate, using Ms-SNuPE. Sperm served as a negative controlfor methylation, while DU145 and LNCaP were positive controls.Four matched tumor and normal prostates from patients showedp16/p14 exon 2 methylation, with increased levels of methylationin the tumor compared to the normal tissue. N, normal prostate;Tu, prostate tumor; C, methylated cytosine at a CpG site; T,unmethylated cytosine at the same CpG site.
CDK Inhibitor and Methylation Patterns 237
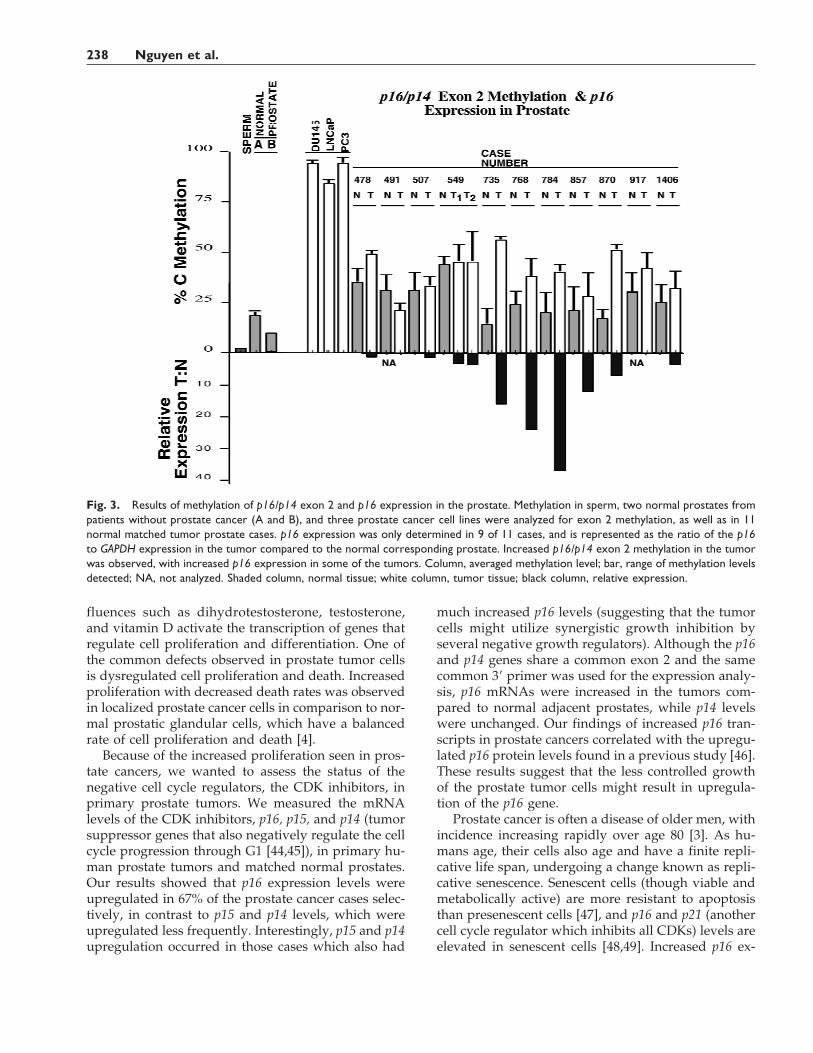
fluences such as dihydrotestosterone, testosterone,and vitamin D activate the transcription of genes thatregulate cell proliferation and differentiation. One ofthe common defects observed in prostate tumor cellsis dysregulated cell proliferation and death. Increasedproliferation with decreased death rates was observedin localized prostate cancer cells in comparison to nor-mal prostatic glandular cells, which have a balancedrate of cell proliferation and death [4].
Because of the increased proliferation seen in pros-tate cancers, we wanted to assess the status of thenegative cell cycle regulators, the CDK inhibitors, inprimary prostate tumors. We measured the mRNAlevels of the CDK inhibitors, p16, p15, and p14 (tumorsuppressor genes that also negatively regulate the cellcycle progression through G1 [44,45]), in primary hu-man prostate tumors and matched normal prostates.Our results showed that p16 expression levels wereupregulated in 67% of the prostate cancer cases selec-tively, in contrast to p15 and p14 levels, which wereupregulated less frequently. Interestingly, p15 and p14upregulation occurred in those cases which also had
much increased p16 levels (suggesting that the tumorcells might utilize synergistic growth inhibition byseveral negative growth regulators). Although the p16and p14 genes share a common exon 2 and the samecommon 38 primer was used for the expression analy-sis, p16 mRNAs were increased in the tumors com-pared to normal adjacent prostates, while p14 levelswere unchanged. Our findings of increased p16 tran-scripts in prostate cancers correlated with the upregu-lated p16 protein levels found in a previous study [46].These results suggest that the less controlled growthof the prostate tumor cells might result in upregula-tion of the p16 gene.
Prostate cancer is often a disease of older men, withincidence increasing rapidly over age 80 [3]. As hu-mans age, their cells also age and have a finite repli-cative life span, undergoing a change known as repli-cative senescence. Senescent cells (though viable andmetabolically active) are more resistant to apoptosisthan presenescent cells [47], and p16 and p21 (anothercell cycle regulator which inhibits all CDKs) levels areelevated in senescent cells [48,49]. Increased p16 ex-
Fig. 3. Results of methylation of p16/p14 exon 2 and p16 expression in the prostate. Methylation in sperm, two normal prostates frompatients without prostate cancer (A and B), and three prostate cancer cell lines were analyzed for exon 2 methylation, as well as in 11normal matched tumor prostate cases. p16 expression was only determined in 9 of 11 cases, and is represented as the ratio of the p16to GAPDH expression in the tumor compared to the normal corresponding prostate. Increased p16/p14 exon 2 methylation in the tumorwas observed, with increased p16 expression in some of the tumors. Column, averaged methylation level; bar, range of methylation levelsdetected; NA, not analyzed. Shaded column, normal tissue; white column, tumor tissue; black column, relative expression.
238 Nguyen et al.

pression could be due to accumulation of p16 in pros-tate tumor cells undergoing senescence [49,50], sincethe death rates of prostate tumor cells are also de-creased [4].
In addition to finding increased p16 levels in theprostate tumors, several studies have found that TGFbis increased in primary prostate tumor cells comparedto the normal prostate glands [24–28]. However, al-though TGFb has been shown to induce p15 expres-sion up to 30-fold [19], we did not detect frequent p15increases as seen for the p16 gene. Thus, although p15mediates the cell cycle arrest of TGFb, prostate cancercells do not demonstrate a consistent feedback loopbetween TGFb and p15 levels.
Prostate cancers have few reported p16 mutationevents [13–17], so that growth control via a nonmu-tated p16 protein is possible. We also previouslyscreened for p16 exon 2 mutations in prostate cancers,but did not detect any base changes in our prostatetumor specimens (unpublished results). p16 increasesseem paradoxical, since the prostate cancer cells haveincreased proliferation rates. However, other groups
have observed increased levels of p16 in primary tu-mors (including lung, rhabdomyosarcoma, and uro-thelium) and cell lines [49,51–54]. One common fea-ture of p16 upregulation in these tumors is that thesecancer cells also have Rb alterations. This could be thecase in prostate cancers, since Rb inactivations fre-quently arise in prostate tumors by mutation (16%),promoter deletion, and loss of heterozygosity (30–60%) [16,55–58]. RB immunohistochemistry showedthat the RB protein was absent in the our sample pros-tate tumors, which agrees with previous studies[16,57]. However, RB loss was not associated with el-evation of the p16 mRNA in all the cases as seen inother tumors, and the lack of consistent correlationbetween the RB and p16 levels in the prostate cellscould be due the inherent heterogeneity of the pros-tate tumors and does not necessarily mean that thetwo genes are not inversely correlated in the prostatetumors. Future analysis using more prostate speci-mens will determine the relationship between thesetwo cell cycle regulators more clearly.
Since transcription of p16 is influenced by the levelsof promoter methylation [6,7] as well as by the statusof the RB protein [49], we determined the methylationlevels of the p16 58 CpG island (including exon 1) bygenomic sequencing, and found the 58 CpG island ofp16 unmethylated in both the normal and tumor pros-tate tisssues (in 28 CpG sites); this agrees with theprevious results of Gu et al., who determined that p1658 CpG region methylation was not common in earlystaged prostate cancer by Southern blot analysis [17].However, we found extensive methylation in thedownstream p16/p14 exon 2 (also a CpG island) in theprostate cell lines and in both normal and tumor pros-tate tissues, whereas sperm were unmethylated. Thelevels of methylation in p16/p14 exon 2 were increasedin the tumor compared to the corresponding normaltissue, and the normal tissues from prostate cancerpatients also showed increased DNA methylation lev-els compared to normal prostate from patients with-out prostate cancer. Although the function of down-stream p16/p14 exon 2 methylation in prostate canceris currently unknown, this could be used as a potentialmarker for clinical diagnosis.
Increased p16/p14 exon 2 methylation also corre-lates with the incidence of upregulated p16 transcriptsin the prostate tumor, although the association wasnot statistically significant. There are several possiblehypotheses for this phenomenon, one of which is thattranscription of the p16 transcript (as well as p14)could induce de novo downstream exon 2 methyl-ation. We observed a similar situation in chronic my-elogenous leukemia, where transcription of Bcr-ablwas associated with de novomethylation of the trans-located, downstream c-abl CpG island [59]. Previous
Fig. 4. Immunohistochemical detection of retinoblastoma (RB)in prostate tissue. A: Benign prostatic tissue (×25). Note the pres-ence of strong nuclear RB staining in a majority of prostatic glan-dular epithelium, and lack of cytoplasmic RB staining. B: Tumorprostatic tissue (from the same case as in A; ×25). Note theabsence or lack of nuclear and cytoplasmic RB staining in thetumor cells from this case, while the stromal cells show strongnuclear staining, demonstrating the successful detection of RB inthis specimen.
CDK Inhibitor and Methylation Patterns 239

studies have shown that prostate cancer shows fre-quent hypermethylation of the 58 regulatory CpG is-land regions (i.e., promoters) of genes that control thenormal cellular environment of prostatic epithelialcells [32,33,35,60,61], and is associated with transcrip-tional inactivation. Our finding of de novo p16/p14exon 2 methylation correlating with increased tran-scription of p16 (and p14) in the prostate tumors sug-gests a more complex function for DNA methyltrans-ferase in the development and progression of prostatecancers.
In summary, we detected remarkable increases ofp16 transcripts in prostate tumor compared to normaladjacent prostate. Increased cellular proliferation ischaracteristic of the prostate tumors and BPH, so it iscounterintuitive to observe increased negative growthregulator, p16, levels in these tumors. However, theupregulation of p16 is specific, since p14 and p15 tran-scripts were not correspondingly increased, except ina few cases where p16 levels were greatly increased,suggesting a feedback mechanism to counteract in-creased cell proliferation in prostate cancer cells. Ac-cumulation of p16 could also arise as a result of non-functional Rb, which we found was true in our samplecases. However, since RB inactivation was not alwayscorrelated with upregulated p16 mRNA levels, p16 up-regulation may also be a consequence of prostate can-cer cell senescence (resulting in resistance to apopto-sis). Mutations and 58 CpG island methylation of p16
are rare in prostate cancer, and we have confirmedthese findings in our studies. However, extensive p16/p14 exon 2 methylation was frequently observed, withthe prostate tumors more methylated in this regioncompared to the matched normal prostate. These find-ings suggest that p16/p14 exon 2 methylation has con-sequences to the pathogenesis of prostate tumor, al-though further studies are needed to clarify what theycould be. The role of DNA methylation in cancer iscomplicated by our observation that increased tran-scription of p16 was observed with de novo down-stream exon 2 methylation of the p16/p14 region inprostate cancer, in contrast to the often observed cor-relation between 58 CpG island hypermethylation andtranscriptional inactivation of genes in many neo-plasms.
REFERENCES
1. Kallio PJ, Palvimo JJ, Janne OA. Genetic regulation of androgenaction. Prostate [Suppl] 1996;6:45–51.
2. Wilding G. Endocrine control of prostate cancer. Cancer Surv1995;23:43–62.
3. Feldman D, Skowronski RJ, Peehl DM. Vitamin D and prostatecancer. Adv Exp Med Biol 1995;375:53–63.
4. Berges RR, Vukanovic J, Epstein JI, CarMichel M, Cisek L,Johnson DE, Veltri RW, Walsh PC, Isaacs JT. Implication of cellkinetic changes during the progression of human prostatic can-cer. Clin Cancer Res 1995;1:473–480.
5. Foulkes WD, Flanders TY, Pollock PM, Hayward NK. TheCDKN2A (p16) gene and human cancer. Mol Med 1997;3:5–20.
TABLE II. Relative p16 mRNA Levels vs. RB Protein Expression inProstate Cancers*
Patient no. p16
% of cells staining for Rb
Nuclear Cytoplasmic
Tumor Nontumor Tumor Nontumor
478 1 <5 ++ 75 + <10 + 10 +507 1.2 <5 ++ 50–75 + 0 0549 3.2 0 20–90 +/++ 0 0735 16 A: <5 ++
B: <5 ++A: <5 ++B: 75 ++
A: 0B: 0
0
768 24 5 +++70–90 +
50–75 ++ 0 0
857 12 5 ++/+++ 100 ++ 0 0870 7 5 ++ 5 ++ 0 NA917 0.3 <5 +++ 100 +++ 70 + 0
1406 3.3 80–90 +/++ 50 +++ 0 0491 NA A: <5 +
B: 95 +++NA A: 0
B: 0NA
784 NA 10 +++<5 ++
95 + 070–90 ++
0
*Intensity of staining; +, weak; ++, moderate; +++, strong; NA, not available.Tumor and normal adjacent prostate cells’ RB staining were examined for eachprostate cancer case.
240 Nguyen et al.

6. Merlo A, Herman JG, Mao L, Lee DJ, Gabrielson E, Burger PC,Baylin SB, Sidransky D. 58 CpG island methylation is associatedwith transcriptional silencing of the tumour suppressor p16/CDKN2/MTS1 in human cancers. Nat Med 1995;1:686–692.
7. Gonzalez-Zulueta M, Bender CM, Yang AS, Nguyen TD, BeartRW, Van Tornout JM, Jones PA. Methylation of the 58CpG is-land of the p16/CDKN2 tumor suppressor gene in normal andtransformed human tissues correlates with gene silencing. Can-cer Res 1995;55:4531–4535.
8. Cairns P, Polascik TJ, Eby Y, Tokino K, Califani J, Merlo A, MaoL, Herath J, Jenkins R, Westra W, Rutter JL, Buckler A, Gabri-elson E, Tockman M, Cho KR, Hedrick L, Bova GS, Isaacs W,Koch W, Schwab D, Sidransky D. Frequency of homozygousdeletion at p16/CDKN2 in primary human tumors. Nat Genet1995;11:210–212.
9. Quesnel B, Preudhomme C, Fenaux P. p16ink4a gene and he-matological malignancies. Leuk Lymphoma 1996;22:11–24.
10. Hirama T, Koeffler HP. Role of the cyclin-dependent kinaseinhibitors in the development of cancer. Blood 1995;86:841–854.
11. Herman JG, Merlo A, Mao L, Lapidus RG, Issa JP, Davidson NE,Sidransky D, Baylin SB. Inactivation of the CDKN2/p16/MTS1gene is frequently associated with aberrant DNA methylation inall common human cancers. Cancer Res 1995;55:4525–4530.
12. Liu A, Neuhausen S, McClure M, Frye C, Weaver-Feldhaus J,Gruis NA, Eddington K, Allalunis-Turner MJ, Skolnick MH,Fujimura FK, Kamb A. CDKN2 (MTS1) tumor suppressor genemutations in human tumor cell lines. Oncogene 1995;10:1061–1067.
13. Komiya A, Suzuki H, Aida S, Yatani R, Shimazaki J. Mutationalanalysis of CDKN2 (CDK4I/MTS1) gene in tissues and cell linesof human prostate cancer. Jpn J Cancer Res 1995;86:622–625.
14. Chen W, Weghorst CM, Sobourin CLK, Wang Y, Wang D,Bostwick DG, Stoner GD. Absence of p16/MTS1 gene mutationsin human prostate cancer. Carcinogenesis 1996;17:2603–2607.
15. Tamimi Y, Bringuier PP, Smit F, van Bokhoven A, DebruyneFMJ, Schalken JA. p16 mutations/deletions are not frequentevents in prostate cancer. Br J Cancer 1996;74:120–122.
16. Konishi N, Hiasa Y, Tsuzuki T, Matsuda H, Tao M, NakamuraM, Naito H, Kitahori Y, Shiraishi T, Yatani R, Shimazaki J, LinJ-C. Detection of RB, p16/CDKN2 and p15INK4B gene alter-ations with immunohistochemical studies in human prostatecarcinomas. Int J Oncol 1996;8:107–112.
17. Gu K, Mes-Masson A-M, Gauthier J, Saad F. Analysis of the p16tumor suppressor gene in early-stage prostate cancer. Mol Car-cinog 1998;21:164–170.
18. Quelle DE, Zindy F, Ashmun RA, Sherr CJ. Alternative readingframes of the INK4a tumor suppressor gene encode two unre-lated proteins capable of inducing cell cycle arrest. Cell 1995;83:993–1000.
19. Hannon GJ, Beach D. p15INK4B is a potential effector of TGF-beta-induced cell cycle arrest. Nature 1994;371:257–261.
20. Haber DA. Splicing into senescence: the curious case of p16 andp19ARF. Cell 1997;91:555–558.
21. Kamijo T, Zindy F, Roussel MF, Quelle DE, Downing JR, Ash-mun RA, Grosveld G, Sherr CJ. Tumor suppression at themouse INK4a locus mediated by the alternative reading frameproduct p19ARF. Cell 1997;91:649–659.
22. Knabbe C, Klein H, Zugmaier G, Voigt KD. Hormonal regula-tion of transforming growth factor beta-2 expression in humanprostate cancer. J Steroid Biochem Mol Biol 1993;47:137–142.
23. Hoodless PA, Wrana JL. Mechanism and function of signalingby the TGF beta superfamily. Curr Top Microbiol Immunol1998;228:235–272.
24. Perry KT, Anthony CT, Case T, Steiner MS. Transforming
growth factor beta as a clinical biomarker for prostate cancer.Urology 1997;49:151–155.
25. Steiner MS, Barrack ER. Transforming growth factor-beta1 over-production in prostate cancer: effects on growth in vivo and invitro. Mol Endocrinol 1992;6:15–25.
26. Thompson TC, Troung LD, Timme TL, Kadmon D, McCune BK,Flanders KC, Scardino PT, Park SH. Transforming growth factorbeta1 as a biomarker for prostate cancer. J Cell Biochem [Suppl]1992;16:54–61.
27. Steiner M, Zhou Z-Z, Tonb DC, Barrack ER. Expression of trans-forming growth factor-beta1 in prostate cancer. Endrocinology1994;135:2240–2247.
28. Tu H, Jacobs SC, Borkowski A, Kyprianou N. Incidence of ap-optosis and cell proliferation in prostate cancer: relationshipwith TGF-beta1 and bcl-2 expression. Int J Cancer 1996;69:357–363.
29. Hering S, Surig D, Freystadt D, Schatz H, Pfeiffer A. Regulationof transforming growth factor beta by sex steroids. Horm MetabRes 1995;27:345–351.
30. Zhuang SH, Burnstein K. Antiproliferative effect of 1alpha,25-dihydroxyvitamin D3 in human prostate cancer cell line LNCaPinvolves reduction of cyclin-dependent kinase 2 activity andpersistent G1 accumulation. Endocrinology 1998;139:1197–1207.
31. Liu M, Lee MH, Cohen M, Bommakanti M, Freedman LP. Tran-scriptional activation of the Cdk inhibitor p21 by vitamin D3leads to the induced differentiation of the myelomonocytic cellline U937. Genes Dev 1996;10:142–153.
32. Lee WH, Morton RA, Epstein JI, Brooks JD, Campbell PA, BovaGS, Hsieh WS, Isaacs WB, Nelson WG. Cytidine methylation ofregulatory sequences near the p-class glutathione S-transferasegene accompanies human prostatic carcinogenesis. Proc NatlAcad Sci USA 1994;91:11733–11737.
33. Lee WH, Isaacs WB, Bova GS, Nelson WG. CG island methyl-ation changes near the GSTP1 gene in prostatic carcinoma cellsdetected using the polymerase chain reaction: a new prostatecancer biomarker. Cancer Epidemiol Biomarkers Prev 1997;6:443–450.
34. Nelson JB, Chan-Tack K, Hedican SP, Magnuson SR, OpgenorthTJ, Bova GS, Simons JW. Endothelin-1 production and de-creased endothelin B receptor expression in advanced prostatecancer. Cancer Res 1996;56:663–668.
35. Nelson JB, Lee WH, Nguyen SH, Jarrard DF, Brooks JD, Mag-nuson SR, Opgenorth TJ, Nelson WG, Bova GS. Methylation ofthe 58 CpG island of the endothelin B receptor gene is commonin human prostate cancer. Cancer Res 1997;57:35–37.
36. Chomczynski P, Sacchi N. Single-step method of RNA isolationby acid guanidinium thiocyanate-phenol-chloroform extraction.Anal Biochem 1987;162:156–159.
37. Gonzalgo ML, Hayashida T, Bender CM, Pao MM, Tsai YC,Gonzales FA, Nguyen HD, Nguyen TT, Jones PA. The role ofDNA methylation in expression of the p19/p16 locus in humanbladder cancer cell lines. Cancer Res 1998;58:1245–1252.
38. Gonzalgo M, Jones PA. Rapid quantitation of methylation dif-ferences at specific sites using methylation-sensitive singlenucleotide primer extension (Ms-SNuPE). Nucleic Acids Res1997;25:2529–2531.
39. Gonzalgo ML, Bender CM, You EH, Glendening M, Flores JF,Walker GJ, Hayward NK, Jones PA, Fountain JW. Low fre-quency of p16/CDKN2A methylation in sporadic melanoma:comparative approaches for methylation analysis of primarytumors. Cancer Res 1997;57:5336–5347.
40. Xiong Z, Laird PW. COBRA: a sensitive and quantitative DNAmethylation assay. Nucleic Acids Res 1997;25:2532–2534.
41. Snedecor GW, Cochran WG. Statistical methods. Sixth edition.Ames, IA: Iowa State University Press; 1967.
CDK Inhibitor and Methylation Patterns 241

42. Meikle AW, Stanish WM. Familial prostatic cancer risk and lowtestosterone. J Clin Endocrinol Metab 1982;54:1104–1108.
43. Narod SA, Dupont A, Cusan L, Diamond P, Gomez J-L, SuburuR, Labrie F. The impact of family history on early detection ofprostate cancer. Nat Med 1995;1:99–101.
44. Lukas J, Parry D, Aagaard L, Mann DJ, Bartkova J, Strauss M,Peters G, Bartek J. Retinoblastoma-protein-dependent cell-cycleinhibition by the tumour suppressor p16. Nature 1995;375:503–506.
45. Herman JG, Jen J, Merlo A, Baylin SB. Hypermethylation-associated inactivation indicates a tumor suppressor role forp15INK4B. Cancer Res 1996;56:722–727.
46. Halvorsen OJ, Haukaas S, Hoisaeter PA, Akslen LA. Expressionof p16 protein in prostatic adenocarcinomas, intraepithelial neo-plasia, and benign/hyperplastic glands. Urol Oncol 1997;3:59–66.
47. Wang E. Senescent human fibroblasts resist programmed celldeath and failure to suppress bcl2 is involved. Cancer Res 1995;55:2284–2292.
48. Campisi J. Aging and cancer: the double-edged sword of repli-cative senescence. J Am Geriatr Soc 1997;45:482–488.
49. Hara E, Smith R, Parry D, Tahara H, Stone S, Peters G. Regula-tion of p16CDKN2 expression and its implications for cell im-mortalization and senescence. Mol Cell Biol 1996;16:859–867.
50. McConnell BB, Starborg M, Brookes S, Peters G. Inhibitors ofcyclin-dependent kinases induce features of replicative senes-cence in early passage human diploid fibroblasts. Curr Biol1998;8:351–354.
51. Shapiro GI, Edwards CD, Kobzik L, Godleski H, Richards W,Sugarbaker DJ, Rollins BJ. Reciprocal Rb inactivation andp16INK4 expression in primary lung cancers and cell lines. Can-cer Res 1995;55:505–509.
52. Knudsen ES, Pazzagli C, Born TL, Bertolaet BL, Knudsen KE,Arden KC, Henry RR, Feramisco JR. Elevated cyclins and cyclin-dependent kinase activity in the rhabdomyosarcoma cell lineRD. Cancer Res 1998;58:2042–2049.
53. Yeager T, Stadler W, Belair C, Puthenveettil J, Olopade O, Rezni-koff C. Increased p16 levels correlate with pRB alterations inhuman urothelial cells. Cancer Res 1995;55:493–497.
54. Parry D, Bates S, Man DJ, Peters G. Lack of cyclin D-Cdk com-plexes in Rb-negative cells correlates with high levels ofp16INK4/MTS1 tumour suppressor gene product. EMBO J1995;14:503–511.
55. Kubota Y, Fujinami K, Uemura H, Dobashi Y, Miyamoto H,Iwasaki Y, Kitamura H, Shuin T. Retinoblastoma gene muta-tions in primary human prostate cancer. Prostate 1995;27:314–320.
56. Bookstein R, Rio P, Madreperla SA, Hong F, Allred C, GrizzleWE, Lee W-H. Promoter deletion and loss of retinoblastomagene expression in human prostate carcinoma. Proc Natl AcadSci USA 1990;87:7762–7766.
57. Phillips SMA, Barton CM, Lee SJ, Morton DG, Wallace DMA,Lemoine NR, Neoptolemos JP. Loss of the retinoblastoma sus-ceptibility gene (RB1) is a frequent and early event in prostatictumorigenesis. Br J Cancer 1994;70:1252–1257.
58. Brooks JD, Bova S, Isaacs WB. Allelic loss of the retinoblastomagene in primary human prostatic adenocarcinomas. Prostate1995;26:35–39.
59. Zion M, Ben-Yehuda D, Avraham A, Cohen O, Wetzler M, Mel-loul D, Ben-Neriah Y. Progressive de novo DNA methylation atthe bcr-abl locus in the course of chronic myelogenous leukemia.Proc Natl Acad Sci USA 1994;91:10722–10726.
60. Graff JR, Herman JG, Lapidus RG, Chopra H, Xu R, Jarrard DF,Isaacs WB, Pitha PM, Davidson NE, Baylin SB. E-cadherin ex-pression is silenced by DNA hypermethylation in human breastand prostate carcinomas. Cancer Res 1995;55:5195–5199.
61. Whang YE, Wu X, Suzuki H, Reiter RE, Tran C, Vessella RL,Said JW, Isaacs WB, Sawyers CL. Inactivation of the tumor sup-pressor PTEN/MMAC1 in advanced human prostate cancerthrough loss of expression. Proc Natl Acad Sci USA 1998;95:5246–5250.
242 Nguyen et al.





























