Embed Size (px)
Citation preview

Ad
AD
a
ARRAA
KMCIEI
1
Sefosca
daarambafieg
0d
Electrochimica Acta 55 (2010) 1664–1669
Contents lists available at ScienceDirect
Electrochimica Acta
journa l homepage: www.e lsev ier .com/ locate /e lec tac ta
nalysis of C–F bond cleavages in methylfluorobenzoates—Fragmentation andimerization of anion radicals using convolution potential sweep voltammetry
. Muthukrishnan, M.V. Sangaranarayanan ∗,1
epartment of Chemistry, Indian Institute of Technology Madras, Chennai-600036, India
r t i c l e i n f o
rticle history:eceived 24 June 2009eceived in revised form 19 October 2009ccepted 19 October 2009vailable online 29 October 2009
a b s t r a c t
The electrochemical reduction of methylfluorobenzoates at glassy carbon electrodes is analyzed usingthe convolution potential sweep voltammetry (CPSV). The stabilization of the radical anion due to theelectron-withdrawing group is shown to lead to intra-molecular stepwise dissociative electron trans-fer. While methyl 2-fluorobenzoate (ortho isomer) follows EC mechanism, the methyl 4-fluorobenzoate
eywords:ethylfluorobenzoates
onvolution potential sweep voltammetryntra-molecular electron transferC-mechanism
(para-isomer) undergoes electro-dimerization prior to C–F bond cleavage. The first order rate constantfor the EC mechanism and the dimerization rate constant for the electro-dimerization are deduced fromthe classical as well as convolution potential sweep voltammetry. A plausible mechanism of dimerizationis suggested. The influence of the electron-withdrawing groups is illustrated by comparing the reductionbehaviour of 4-fluorobenzonitrile. The potential energy surfaces and electron density mapping employ-ing Gaussian 03 calculations provide further support for the validation of the mechanism pertaining to
on-radical interactions C–F bond cleavages.
. Introduction
The reductive cleavage of C–X bonds (where X = F, Cl, Br, I, O,, etc.) constitutes a fascinating area of research in the study oflectron transfer reactions in view of the crucial role played by theunctional group, dielectric properties of the solvent and magnitudef the driving force [1–3]. While there are compounds which followolely stepwise or concerted electron transfer pathways, there existases where the mechanism itself depends upon the driving forcend a transition from one mechanism to the other is feasible [4–8].
The C–F bond cleavage reactions in aromatic substrates is ren-ered difficult on account of the high activation energy barriersnd hence their study has been restricted to a smaller number ofromatic compounds. The reductive de-fluorination of 1,3 difluo-obenzene [9], pentafluoronitrobenzene [10] on mercury electrodend fluoromethylarenes [11] on glassy carbon electrode deservesention in this context. Alternately, the C–F bond reductions can
e accomplished with the help of suitable redox catalysts such asnthracene, phenanthrene, benzonitrile, chrysene, etc. [12]. A dif-
erent strategy for carrying out the reduction of C–F bonds consistsn introducing electron-withdrawing groups in the aromatic ring.g. fluorobenzonitrile [13,14]. Although electron-withdrawingroups such as –CN, –NO2, –COOCH3, etc. are expected to facilitate∗ Corresponding author. Tel.: +91 44 22574209; fax: +91 44 22570545.E-mail address: [email protected] (M.V. Sangaranarayanan).
1 ISE member.
013-4686/$ – see front matter © 2009 Elsevier Ltd. All rights reserved.oi:10.1016/j.electacta.2009.10.047
© 2009 Elsevier Ltd. All rights reserved.
C–F bond cleavages, a proper choice of the compounds is warrantedsince the para-substituted fluorocompounds may dimerize ratherthan undergoing simple reduction. Further, all methylhaloben-zoates (excluding the fluoro) undergo Ullmann type of coupling onreactive nickel surfaces [15], sacrificial anodes such as zinc, iron,aluminium, etc. [16,17]. It is hence of interest to investigate theelectron transfer processes in methylfluorobenzoates. In aromaticfluorocompounds, the radical anions dissociate to form the neu-tral radicals and corresponding ions. The neutral radicals lead todefluorinated compounds, upon abstraction of a proton. [18]. Inthe case of substrates undergoing dimerization, the logarithmicanalysis of the convoluted current provides the dimerization ratesand for acetophenone, the dimerization rate constant has beendeduced as 4 × 105 mol−1 l s−1 by Saveant and Tessier [19]. Sincethe C–CN bond breaking (instead of C–F bond cleavage) occurs inthe case of meta-fluorobenzonitrile [13], the ortho and para isomersof methylfluorobenzoates are chosen in the present study.
The convolution potential sweep voltammetry (CPSV) is a valu-able technique since (i) the entire data points of the voltammogramcan be made use of and (ii) no a priori assumption of the govern-ing rate laws is required. While the linear free energy relation isthe basis of the Butler–Volmer equation for simple electron trans-fer processes, the kinetics of dissociative electron transfer reactions
is customarily investigated using the quadratic activation-drivingforce relation via the Marcus–Hush formalism [20]. In this com-munication, we investigate the mechanism of C–F bond cleavagesin methyl 2-fluorobenzoate (OFMB) and methyl 4-fluorobenzoate(PFMB) using CPSV and density functional calculations.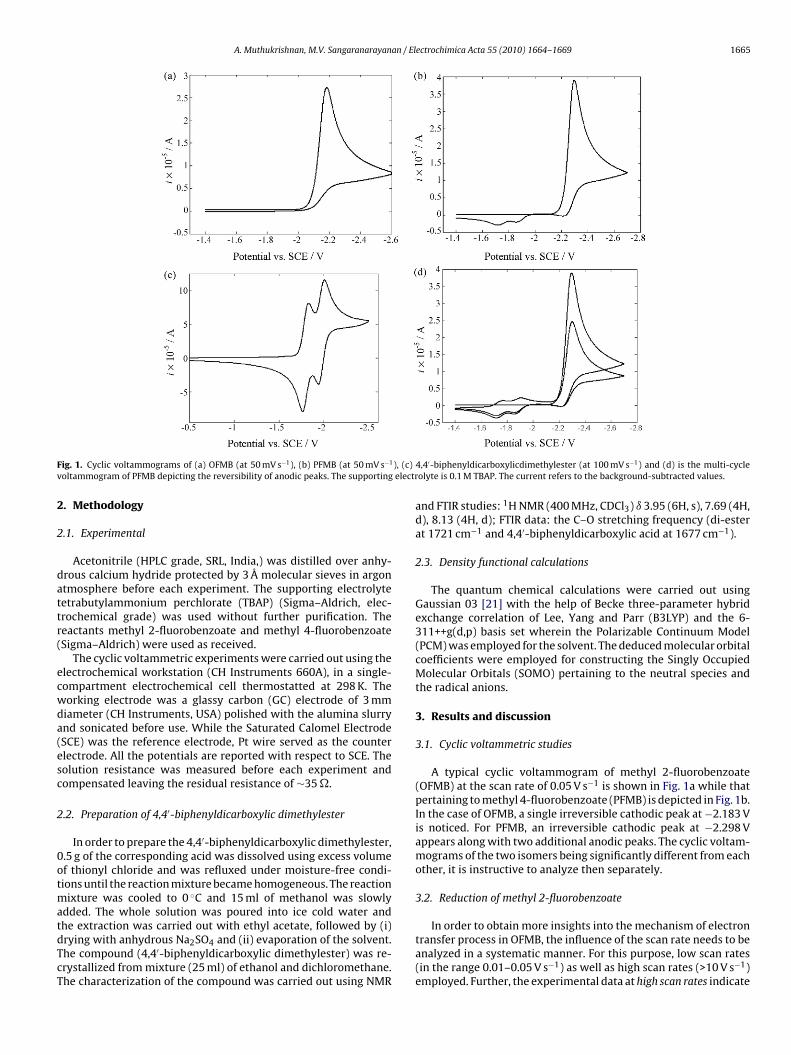
A. Muthukrishnan, M.V. Sangaranarayanan / Electrochimica Acta 55 (2010) 1664–1669 1665
F ), (c)v electr
2
2
dattr(
ecwda(esc
2
0otmatdTcT
ig. 1. Cyclic voltammograms of (a) OFMB (at 50 mV s−1), (b) PFMB (at 50 mV s−1
oltammogram of PFMB depicting the reversibility of anodic peaks. The supporting
. Methodology
.1. Experimental
Acetonitrile (HPLC grade, SRL, India,) was distilled over anhy-rous calcium hydride protected by 3 Å molecular sieves in argontmosphere before each experiment. The supporting electrolyteetrabutylammonium perchlorate (TBAP) (Sigma–Aldrich, elec-rochemical grade) was used without further purification. Theeactants methyl 2-fluorobenzoate and methyl 4-fluorobenzoateSigma–Aldrich) were used as received.
The cyclic voltammetric experiments were carried out using thelectrochemical workstation (CH Instruments 660A), in a single-ompartment electrochemical cell thermostatted at 298 K. Theorking electrode was a glassy carbon (GC) electrode of 3 mmiameter (CH Instruments, USA) polished with the alumina slurrynd sonicated before use. While the Saturated Calomel ElectrodeSCE) was the reference electrode, Pt wire served as the counterlectrode. All the potentials are reported with respect to SCE. Theolution resistance was measured before each experiment andompensated leaving the residual resistance of ∼35 �.
.2. Preparation of 4,4′-biphenyldicarboxylic dimethylester
In order to prepare the 4,4′-biphenyldicarboxylic dimethylester,.5 g of the corresponding acid was dissolved using excess volumef thionyl chloride and was refluxed under moisture-free condi-ions until the reaction mixture became homogeneous. The reaction
ixture was cooled to 0 ◦C and 15 ml of methanol was slowlydded. The whole solution was poured into ice cold water and
he extraction was carried out with ethyl acetate, followed by (i)rying with anhydrous Na2SO4 and (ii) evaporation of the solvent.he compound (4,4′-biphenyldicarboxylic dimethylester) was re-rystallized from mixture (25 ml) of ethanol and dichloromethane.he characterization of the compound was carried out using NMR4,4′-biphenyldicarboxylicdimethylester (at 100 mV s−1) and (d) is the multi-cycleolyte is 0.1 M TBAP. The current refers to the background-subtracted values.
and FTIR studies: 1H NMR (400 MHz, CDCl3) ı 3.95 (6H, s), 7.69 (4H,d), 8.13 (4H, d); FTIR data: the C–O stretching frequency (di-esterat 1721 cm−1 and 4,4′-biphenyldicarboxylic acid at 1677 cm−1).
2.3. Density functional calculations
The quantum chemical calculations were carried out usingGaussian 03 [21] with the help of Becke three-parameter hybridexchange correlation of Lee, Yang and Parr (B3LYP) and the 6-311++g(d,p) basis set wherein the Polarizable Continuum Model(PCM) was employed for the solvent. The deduced molecular orbitalcoefficients were employed for constructing the Singly OccupiedMolecular Orbitals (SOMO) pertaining to the neutral species andthe radical anions.
3. Results and discussion
3.1. Cyclic voltammetric studies
A typical cyclic voltammogram of methyl 2-fluorobenzoate(OFMB) at the scan rate of 0.05 V s−1 is shown in Fig. 1a while thatpertaining to methyl 4-fluorobenzoate (PFMB) is depicted in Fig. 1b.In the case of OFMB, a single irreversible cathodic peak at −2.183 Vis noticed. For PFMB, an irreversible cathodic peak at −2.298 Vappears along with two additional anodic peaks. The cyclic voltam-mograms of the two isomers being significantly different from eachother, it is instructive to analyze then separately.
3.2. Reduction of methyl 2-fluorobenzoate
In order to obtain more insights into the mechanism of electrontransfer process in OFMB, the influence of the scan rate needs to beanalyzed in a systematic manner. For this purpose, low scan rates(in the range 0.01–0.05 V s−1) as well as high scan rates (>10 V s−1)employed. Further, the experimental data at high scan rates indicate
1666 A. Muthukrishnan, M.V. Sangaranarayanan / El
Table 1Parameters derived from the cyclic voltammetric data.
Parameters Methyl 2-fluorobenzoate Methyl 4-fluorobenzoate
E0 vs SCE/V −2.190 −2.277
acpa
tpa2oias(
3
Scortii
I
T[ac
I
wcsbw
� being the time elapsed from the peak potential to the switch-
˛ 0.81 0.74D/cm2 s−1 0.83 × 10−5 2.50 × 10−5
Rate constants 10.87 s−1 1.38 × 106 mol−1 l s−1
reversible (diffusion-controlled) behaviour while a kineticallyontrolled behaviour was noticed at lower scan rates. The reversibleeak occurring at high scan rates is indicative of a stable radicalnion and thus the cyclic voltammogram depicted in Fig. 1a implies
he occurrence of EC mechanism (RX + e � RX•− k−→R• + X−). Theeak potential shifts cathodically with increase in the scan ratend the slope of ıEp,c vs ı log(v) plot i.e. ıEp,c/ı log(v) is estimated as5 mV consistent with the anticipated value of 29.6 mV for a firstrder chemical reaction. It is hence inferred that the electrochem-cal reduction occurs via the first order elimination of fluoride ionnd that the decomposition of radical anion is the rate determiningtep [22,23]. The transfer coefficient is deduced from peak widthEp/2 − Ep) value and equals 0.81 (Table 1).
.2.1. Convolution potential sweep voltammetryThe classical cyclic voltammetric analysis due to Nicholson and
hain [24] is valuable since tabular compilations and workingurves have been provided as diagnostic criteria for different typesf electron transfer processes. However, when a wide range of scanates is employed, the CPSV is more preferred since a priori assump-ion of kinetic laws is no longer required. The convolution current (I)s related to the voltammetric current (i) through the convolutionntegral [25] as
(t) = 1√�
∫ t
0
i(u)
(t − u)1/2du (1)
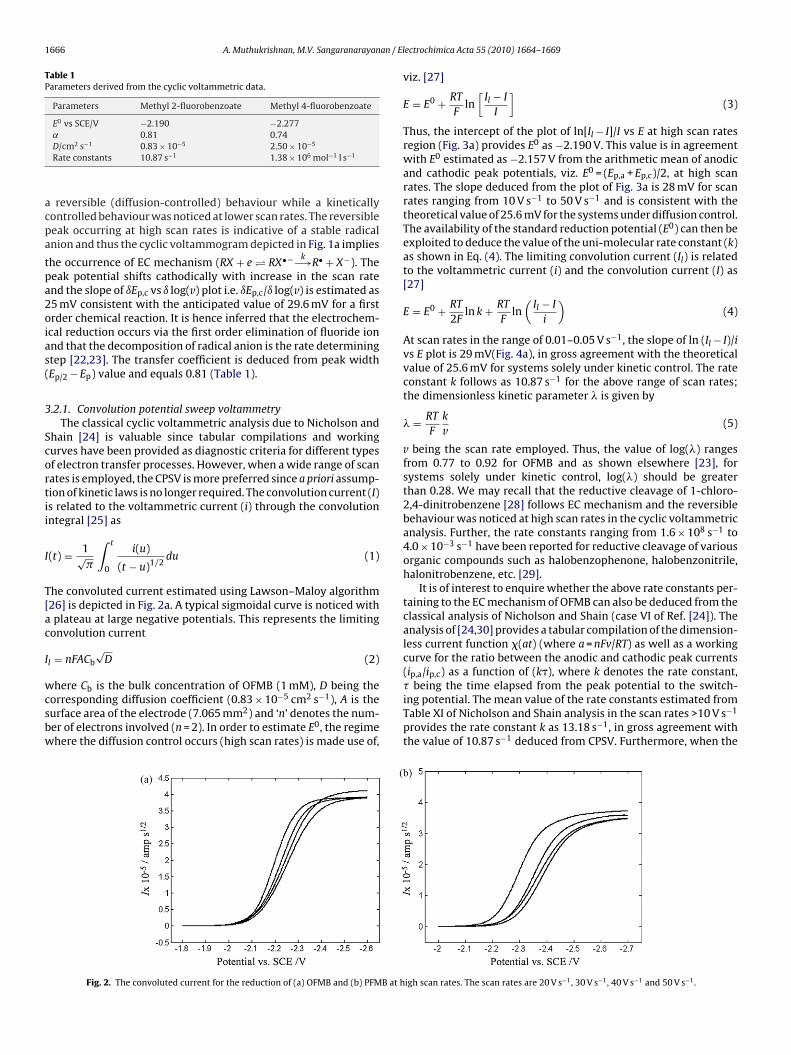
he convoluted current estimated using Lawson–Maloy algorithm26] is depicted in Fig. 2a. A typical sigmoidal curve is noticed withplateau at large negative potentials. This represents the limiting
onvolution current
l = nFACb√
D (2)
here Cb is the bulk concentration of OFMB (1 mM), D being the
orresponding diffusion coefficient (0.83 × 10−5 cm2 s−1), A is theurface area of the electrode (7.065 mm2) and ‘n’ denotes the num-er of electrons involved (n = 2). In order to estimate E0, the regimehere the diffusion control occurs (high scan rates) is made use of,Fig. 2. The convoluted current for the reduction of (a) OFMB and (b) PFMB at h
ectrochimica Acta 55 (2010) 1664–1669
viz. [27]
E = E0 + RT
Fln
[Il − I
I
](3)
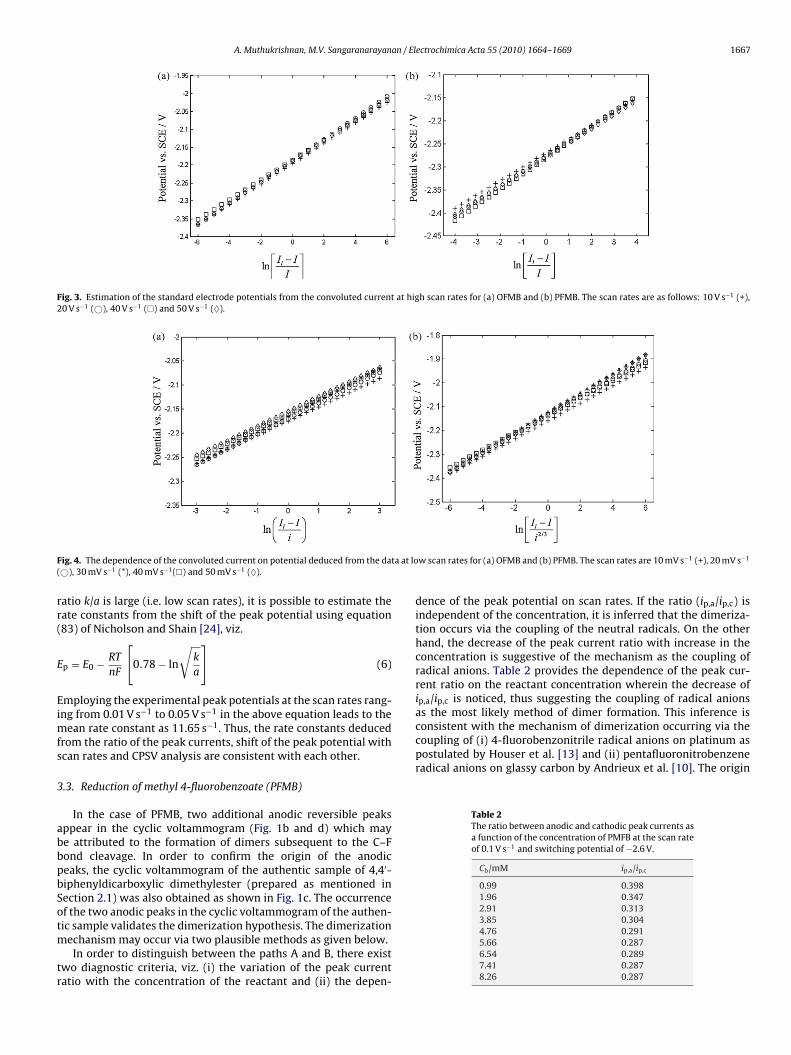
Thus, the intercept of the plot of ln[Il − I]/I vs E at high scan ratesregion (Fig. 3a) provides E0 as −2.190 V. This value is in agreementwith E0 estimated as −2.157 V from the arithmetic mean of anodicand cathodic peak potentials, viz. E0 = (Ep,a + Ep,c)/2, at high scanrates. The slope deduced from the plot of Fig. 3a is 28 mV for scanrates ranging from 10 V s−1 to 50 V s−1 and is consistent with thetheoretical value of 25.6 mV for the systems under diffusion control.The availability of the standard reduction potential (E0) can then beexploited to deduce the value of the uni-molecular rate constant (k)as shown in Eq. (4). The limiting convolution current (Il) is relatedto the voltammetric current (i) and the convolution current (I) as[27]
E = E0 + RT
2Fln k + RT
Fln
(Il − I
i
)(4)
At scan rates in the range of 0.01–0.05 V s−1, the slope of ln (Il − I)/ivs E plot is 29 mV(Fig. 4a), in gross agreement with the theoreticalvalue of 25.6 mV for systems solely under kinetic control. The rateconstant k follows as 10.87 s−1 for the above range of scan rates;the dimensionless kinetic parameter � is given by
� = RT
F
k
v(5)
v being the scan rate employed. Thus, the value of log(�) rangesfrom 0.77 to 0.92 for OFMB and as shown elsewhere [23], forsystems solely under kinetic control, log(�) should be greaterthan 0.28. We may recall that the reductive cleavage of 1-chloro-2,4-dinitrobenzene [28] follows EC mechanism and the reversiblebehaviour was noticed at high scan rates in the cyclic voltammetricanalysis. Further, the rate constants ranging from 1.6 × 108 s−1 to4.0 × 10−3 s−1 have been reported for reductive cleavage of variousorganic compounds such as halobenzophenone, halobenzonitrile,halonitrobenzene, etc. [29].
It is of interest to enquire whether the above rate constants per-taining to the EC mechanism of OFMB can also be deduced from theclassical analysis of Nicholson and Shain (case VI of Ref. [24]). Theanalysis of [24,30] provides a tabular compilation of the dimension-less current function �(at) (where a = nFv/RT) as well as a workingcurve for the ratio between the anodic and cathodic peak currents(ip,a/ip,c) as a function of (k�), where k denotes the rate constant,
ing potential. The mean value of the rate constants estimated fromTable XI of Nicholson and Shain analysis in the scan rates >10 V s−1
provides the rate constant k as 13.18 s−1, in gross agreement withthe value of 10.87 s−1 deduced from CPSV. Furthermore, when the
igh scan rates. The scan rates are 20 V s−1, 30 V s−1, 40 V s−1 and 50 V s−1.
A. Muthukrishnan, M.V. Sangaranarayanan / Electrochimica Acta 55 (2010) 1664–1669 1667
Fig. 3. Estimation of the standard electrode potentials from the convoluted current at high scan rates for (a) OFMB and (b) PFMB. The scan rates are as follows: 10 V s−1 (+),20 V s−1 (©), 40 V s−1 (�) and 50 V s−1 (♦).
F ta at lo(
rr(
E
Eimfs
3
abbpbSotm
tr
consistent with the mechanism of dimerization occurring via thecoupling of (i) 4-fluorobenzonitrile radical anions on platinum aspostulated by Houser et al. [13] and (ii) pentafluoronitrobenzeneradical anions on glassy carbon by Andrieux et al. [10]. The origin
Table 2The ratio between anodic and cathodic peak currents asa function of the concentration of PMFB at the scan rateof 0.1 V s−1 and switching potential of −2.6 V.
Cb/mM ip,a/ip,c
0.99 0.3981.96 0.3472.91 0.3133.85 0.304
ig. 4. The dependence of the convoluted current on potential deduced from the da©), 30 mV s−1 (*), 40 mV s−1(�) and 50 mV s−1 (♦).
atio k/a is large (i.e. low scan rates), it is possible to estimate theate constants from the shift of the peak potential using equation83) of Nicholson and Shain [24], viz.
p = E0 − RT
nF
[0.78 − ln
√k
a
](6)
mploying the experimental peak potentials at the scan rates rang-ng from 0.01 V s−1 to 0.05 V s−1 in the above equation leads to the
ean rate constant as 11.65 s−1. Thus, the rate constants deducedrom the ratio of the peak currents, shift of the peak potential withcan rates and CPSV analysis are consistent with each other.
.3. Reduction of methyl 4-fluorobenzoate (PFMB)
In the case of PFMB, two additional anodic reversible peaksppear in the cyclic voltammogram (Fig. 1b and d) which maye attributed to the formation of dimers subsequent to the C–Fond cleavage. In order to confirm the origin of the anodiceaks, the cyclic voltammogram of the authentic sample of 4,4′-iphenyldicarboxylic dimethylester (prepared as mentioned inection 2.1) was also obtained as shown in Fig. 1c. The occurrencef the two anodic peaks in the cyclic voltammogram of the authen-
ic sample validates the dimerization hypothesis. The dimerizationechanism may occur via two plausible methods as given below.In order to distinguish between the paths A and B, there exist
wo diagnostic criteria, viz. (i) the variation of the peak currentatio with the concentration of the reactant and (ii) the depen-
w scan rates for (a) OFMB and (b) PFMB. The scan rates are 10 mV s−1 (+), 20 mV s−1
dence of the peak potential on scan rates. If the ratio (ip,a/ip,c) isindependent of the concentration, it is inferred that the dimeriza-tion occurs via the coupling of the neutral radicals. On the otherhand, the decrease of the peak current ratio with increase in theconcentration is suggestive of the mechanism as the coupling ofradical anions. Table 2 provides the dependence of the peak cur-rent ratio on the reactant concentration wherein the decrease ofip,a/ip,c is noticed, thus suggesting the coupling of radical anionsas the most likely method of dimer formation. This inference is
4.76 0.2915.66 0.2876.54 0.2897.41 0.2878.26 0.287

1668 A. Muthukrishnan, M.V. Sangaranarayanan / Electrochimica Acta 55 (2010) 1664–1669
Fig. 5. The representation of SOMO for (a) OFMB and (b) PFMB. The molecu
ok
aov
wgTmd0
3c
FfapO[
l
E
idPal
ever, both the fragments (neutral radicals and fluoride ions) andthe �-anion complex become stable in the polar medium as canbe seen from the corresponding minimum in the potential energycalculations. The spike appearing in the potential energy may bedue to the high electronegativity of fluorine atoms.
2 In the case of stepwise mechanism, the cleaving bond length of the radical anion
Scheme 1. The two possible mechanisms for dimerization of PMFB.
f the concentration dependence is attributed to the second orderinetics arising in the second step of Scheme 1(A) given above.
The variation of the peak potential with scan rates also providesdiagnostic criterion for distinguishing between the two pathwaysf dimerization. In this case, the following equation depicting theariation Ep with log(v) has been postulated as
∂Ep
∂ log(v)= 1
1 + bln(10)
RT
F(7)
herein b refers to the order of the reaction. If the pathway as sug-ested by (A) occurs, b = 2 and hence ıEp/ı log(v) should be 19.7 mV.he experimental value for PFMB is 18 mV, thus supporting theechanism A wherein the rate determining step is the second order
imerization of radical anions. The transfer coefficient follows as.74 from peak width measurements.
.4. Standard reduction potentials and dimerization rateonstants
The potential dependent convoluted current is depicted inig. 2b. As in the earlier case, the standard reduction potential (E0)ollows from the high scan rate data as −2.277 V (Fig. 3b) and is ingreement with that obtained from the cathodic and anodic peakotentials (−2.279 V). Further, the standard reduction potentials ofFMB and PFMB are comparable to that of ethyl 3-fluorobenzoate
31].In order to estimate the dimerization rate constant (kd) the fol-
owing equation for the electrode potential [19]
= E0 + RT
3Fln
(2kd
3FA√
D
)+ RT
Fln
[Il − I
i2/3
](8)
s employed wherein the dimensionless kinetic parameter isefined as �d = (RT/F)(kdCb/v). The diffusion coefficient (D) forFMB is estimated as 2.50 × 10−5 cm2 s−1 using Eq. (2), Using thebove-mentioned value of E0 and the intercept from the plot ofn[(Il − I)/i2/3] vs E of Fig. 4b, a typical dimerization rate constant
lar orbital coefficients are obtained using the isovalue of 0.05 units.
(kd) is deduced as 1.38 × 106 mol−1 l s−1 and the correspondingdimensionless parameter (�d) as 2.95.
3.5. Estimation of bond lengths
In dissociative electron transfer reactions, two different typesof mechanism are customarily encountered, viz. stepwise and con-certed. The stepwise pathway refers to the formation of a stableradical anion subsequent to electron transfer. In order to infer thestepwise mechanism, bond lengths and partial atomic charges2
were calculated using Gaussian 03 version with the help of B3LYPand 6-311++g(d,p) as the basis set. The Polarizable ContinuumModel (PCM) was employed for the solvent. Fig. 5 depicts the SOMOof OFMB and PFMB radical anions. It is seen that the SOMO arepresent above the ester group and benzene ring. This implies thatthe electron is first transferred to the �* orbital of the reactants andsubsequently to �* orbital of the C–F bond.
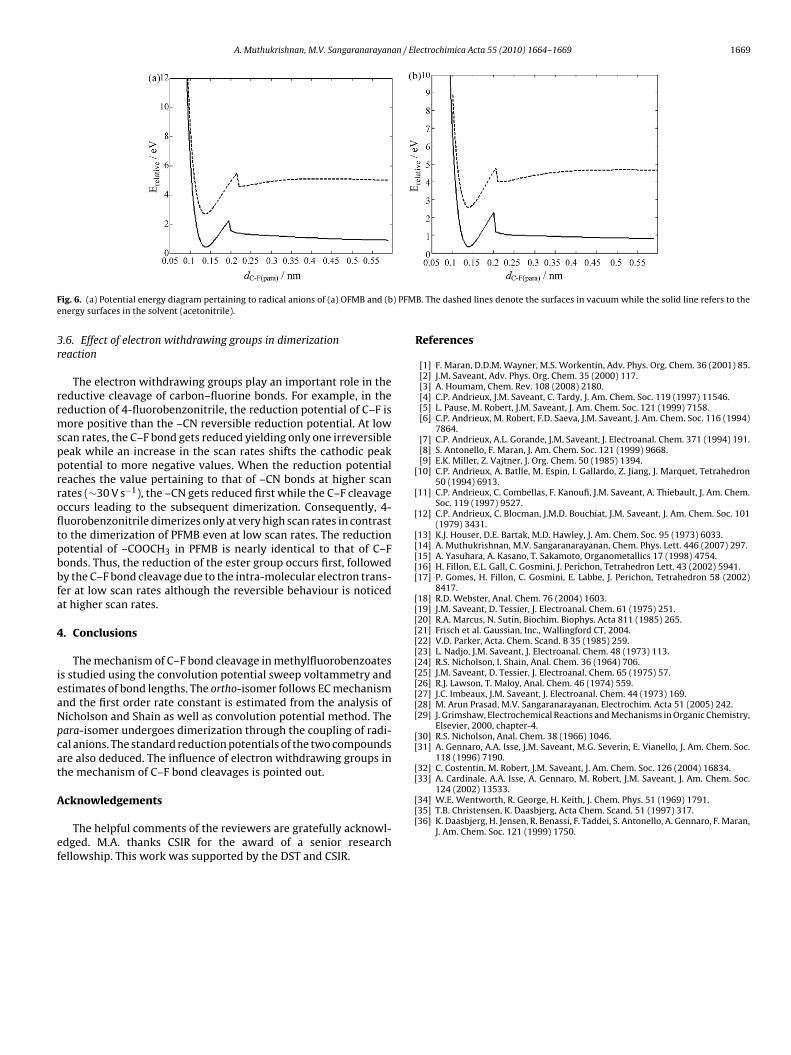
The computed potential energy surfaces in vacuum for theanionic radicals, exhibit two minima [32] indicating respectivelythe presence of (i) �-anion radical and (ii) �-anion radical asshown in Fig. 6. The first and deeper minimum denotes the strongstabilization of �-anion radical. The second minimum arises asa consequence of the ion-radical interactions. It is anticipatedthat electron-withdrawing groups will enhance the ion-radicalinteraction [33–36] on account of the attraction of the partial pos-itive charge (on carbon), towards the negatively charged F−. Fromthe investigation on the reduction of haloacetonitriles, Cardinaleet al. [33] demonstrated that the ion-radical interaction energydecreases from Cl− to I−. In general, the compounds having F− as thefunctional group should have higher ion-radical interactions. In thepresent study, both the isomers possess the electron-withdrawingcarbonyl group. In vacuum phase calculation, both the compoundsexhibit two minima and the ion-radical interaction energy typicallyvaries from 0.6 eV to 0.8 eV.
In the presence of the solvent (acetonitrile), the minimum per-taining to �-anion complex has vanished, thus indicating that theion-radical interaction energy is absent in the polar medium. How-
will not be significantly different from that of the reactant. From the quantum chem-ical calculations using Gaussian 03, it is found that the bond lengths of (a) OFMB(neutral: 1.35 Å and anioic radical: 1.38 Å) and (b) PFMB (neutral: 1.34 Å and anionicradical: 1.39 Å) are nearly the same. The Mulliken charge analysis yields the partialcharge of fluorine in the radical anionic forms as −0.25 (OFMB) and −0.28 (PFMB).Hence the bond is not broken after the electron transfer.
A. Muthukrishnan, M.V. Sangaranarayanan / Electrochimica Acta 55 (2010) 1664–1669 1669
F ) PFMe
3r
rrmspprrofltpbbfa
4
ieaNpcat
A
ef
[
[
[
[[[[[
[[[[[[[[[[[[
[[
[[33] A. Cardinale, A.A. Isse, A. Gennaro, M. Robert, J.M. Saveant, J. Am. Chem. Soc.
124 (2002) 13533.
ig. 6. (a) Potential energy diagram pertaining to radical anions of (a) OFMB and (bnergy surfaces in the solvent (acetonitrile).
.6. Effect of electron withdrawing groups in dimerizationeaction
The electron withdrawing groups play an important role in theeductive cleavage of carbon–fluorine bonds. For example, in theeduction of 4-fluorobenzonitrile, the reduction potential of C–F isore positive than the –CN reversible reduction potential. At low
can rates, the C–F bond gets reduced yielding only one irreversibleeak while an increase in the scan rates shifts the cathodic peakotential to more negative values. When the reduction potentialeaches the value pertaining to that of –CN bonds at higher scanates (∼30 V s−1), the –CN gets reduced first while the C–F cleavageccurs leading to the subsequent dimerization. Consequently, 4-uorobenzonitrile dimerizes only at very high scan rates in contrasto the dimerization of PFMB even at low scan rates. The reductionotential of –COOCH3 in PFMB is nearly identical to that of C–Fonds. Thus, the reduction of the ester group occurs first, followedy the C–F bond cleavage due to the intra-molecular electron trans-er at low scan rates although the reversible behaviour is noticedt higher scan rates.
. Conclusions
The mechanism of C–F bond cleavage in methylfluorobenzoatess studied using the convolution potential sweep voltammetry andstimates of bond lengths. The ortho-isomer follows EC mechanismnd the first order rate constant is estimated from the analysis oficholson and Shain as well as convolution potential method. Theara-isomer undergoes dimerization through the coupling of radi-al anions. The standard reduction potentials of the two compoundsre also deduced. The influence of electron withdrawing groups inhe mechanism of C–F bond cleavages is pointed out.
cknowledgements
The helpful comments of the reviewers are gratefully acknowl-dged. M.A. thanks CSIR for the award of a senior researchellowship. This work was supported by the DST and CSIR.
[[[
B. The dashed lines denote the surfaces in vacuum while the solid line refers to the
References
[1] F. Maran, D.D.M. Wayner, M.S. Workentin, Adv. Phys. Org. Chem. 36 (2001) 85.[2] J.M. Saveant, Adv. Phys. Org. Chem. 35 (2000) 117.[3] A. Houmam, Chem. Rev. 108 (2008) 2180.[4] C.P. Andrieux, J.M. Saveant, C. Tardy, J. Am. Chem. Soc. 119 (1997) 11546.[5] L. Pause, M. Robert, J.M. Saveant, J. Am. Chem. Soc. 121 (1999) 7158.[6] C.P. Andrieux, M. Robert, F.D. Saeva, J.M. Saveant, J. Am. Chem. Soc. 116 (1994)
7864.[7] C.P. Andrieux, A.L. Gorande, J.M. Saveant, J. Electroanal. Chem. 371 (1994) 191.[8] S. Antonello, F. Maran, J. Am. Chem. Soc. 121 (1999) 9668.[9] E.K. Miller, Z. Vajtner, J. Org. Chem. 50 (1985) 1394.10] C.P. Andrieux, A. Batlle, M. Espin, I. Gallardo, Z. Jiang, J. Marquet, Tetrahedron
50 (1994) 6913.11] C.P. Andrieux, C. Combellas, F. Kanoufi, J.M. Saveant, A. Thiebault, J. Am. Chem.
Soc. 119 (1997) 9527.12] C.P. Andrieux, C. Blocman, J.M.D. Bouchiat, J.M. Saveant, J. Am. Chem. Soc. 101
(1979) 3431.13] K.J. Houser, D.E. Bartak, M.D. Hawley, J. Am. Chem. Soc. 95 (1973) 6033.14] A. Muthukrishnan, M.V. Sangaranarayanan, Chem. Phys. Lett. 446 (2007) 297.15] A. Yasuhara, A. Kasano, T. Sakamoto, Organometallics 17 (1998) 4754.16] H. Fillon, E.L. Gall, C. Gosmini, J. Perichon, Tetrahedron Lett. 43 (2002) 5941.17] P. Gomes, H. Fillon, C. Gosmini, E. Labbe, J. Perichon, Tetrahedron 58 (2002)
8417.18] R.D. Webster, Anal. Chem. 76 (2004) 1603.19] J.M. Saveant, D. Tessier, J. Electroanal. Chem. 61 (1975) 251.20] R.A. Marcus, N. Sutin, Biochim. Biophys. Acta 811 (1985) 265.21] Frisch et al. Gaussian, Inc., Wallingford CT, 2004.22] V.D. Parker, Acta. Chem. Scand. B 35 (1985) 259.23] L. Nadjo, J.M. Saveant, J. Electroanal. Chem. 48 (1973) 113.24] R.S. Nicholson, I. Shain, Anal. Chem. 36 (1964) 706.25] J.M. Saveant, D. Tessier, J. Electroanal. Chem. 65 (1975) 57.26] R.J. Lawson, T. Maloy, Anal. Chem. 46 (1974) 559.27] J.C. Imbeaux, J.M. Saveant, J. Electroanal. Chem. 44 (1973) 169.28] M. Arun Prasad, M.V. Sangaranarayanan, Electrochim. Acta 51 (2005) 242.29] J. Grimshaw, Electrochemical Reactions and Mechanisms in Organic Chemistry,
Elsevier, 2000, chapter-4.30] R.S. Nicholson, Anal. Chem. 38 (1966) 1046.31] A. Gennaro, A.A. Isse, J.M. Saveant, M.G. Severin, E. Vianello, J. Am. Chem. Soc.
118 (1996) 7190.32] C. Costentin, M. Robert, J.M. Saveant, J. Am. Chem. Soc. 126 (2004) 16834.
34] W.E. Wentworth, R. George, H. Keith, J. Chem. Phys. 51 (1969) 1791.35] T.B. Christensen, K. Daasbjerg, Acta Chem. Scand. 51 (1997) 317.36] K. Daasbjerg, H. Jensen, R. Benassi, F. Taddei, S. Antonello, A. Gennaro, F. Maran,
J. Am. Chem. Soc. 121 (1999) 1750.





























