Embed Size (px)
Citation preview

An N-terminal variant of Trpv1 channel is requiredfor osmosensory transduction
Reza Sharif Naeini1, Marie-France Witty2, Philippe Seguela2 & Charles W Bourque1
Body fluid homeostasis requires the release of arginine-vasopressin (AVP, an antidiuretic hormone) from the neurohypophysis. This
release is controlled by specific and highly sensitive ‘osmoreceptors’ in the hypothalamus. Indeed, AVP-releasing neurons in the
supraoptic nucleus (SON) are directly osmosensitive, and this osmosensitivity is mediated by stretch-inhibited cation channels.
However, the molecular nature of these channels remains unknown. Here we show that SON neurons express an N-terminal splice
variant of the transient receptor potential vanilloid type-1 (Trpv1), also known as the capsaicin receptor, but not full-length Trpv1.
Unlike their wild-type counterparts, SON neurons in Trpv1 knockout (Trpv1–/–) mice could not generate ruthenium red–sensitive
increases in membrane conductance and depolarizing potentials in response to hyperosmotic stimulation. Moreover, Trpv1–/–
mice showed a pronounced serum hyperosmolality under basal conditions and severely compromised AVP responses to osmotic
stimulation in vivo. These results suggest that the Trpv1 gene may encode a central component of the osmoreceptor.
Recent studies have indicated that Trpv4, a channel homologous toTrpv1 (ref. 1), may contribute to the detection of osmotic signals2,3 andto the osmotic control of AVP release4,5. However, unlike the osmosen-sory transduction channel expressed in native AVP-releasing neurons,which is activated by hyperosmolality and inhibited by hypo-osmolality6–8, heterologously expressed Trpv4 channels are activatedby cell swelling and are insensitive to hypertonic stimuli2,3,9. A distinctchannel, or an additional accessory subunit, may therefore be requiredfor osmosensory transduction in AVP neurons. To our knowledge, onlyone molecularly defined cation channel has thus far been shown to haveproperties consistent with the native transduction channel of AVPneurons. This stretch-inhibited cation channel, termed SIC (ref. 10),is a chimeric channel composed of an N-truncated Trpv1 (exons 6–14)and the C-terminal domain of Trpv4. Although recent analysis has sug-gested that this chimera is an artifact that is not encoded by the mousegenome11, these observations nonetheless suggest the possibility thatTrpv1 variants may contribute to osmosensory transduction in nativeAVP neurons. In this study, we report that an N-terminal variant ofTrpv1 is essential for osmosensory transduction in mouse SON neurons.
RESULTS
An N-terminal variant of Trpv1 is expressed in the SON
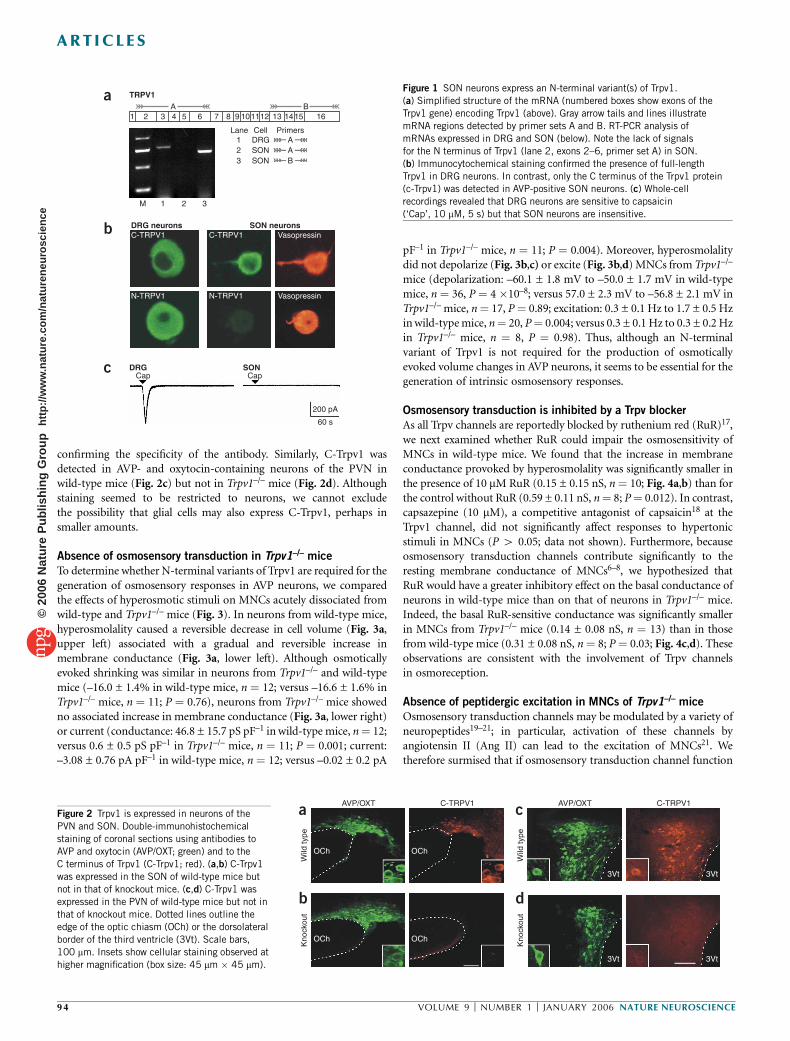
We isolated total RNA from the SON of adult mice and, using specificsets of primers, performed reverse transcription polymerase chainreaction (RT-PCR) to determine if variants of Trpv1 are expressed inthis tissue. Although the message encoding the N-terminal end of Trpv1(exons 2–6) was detected in dorsal root ganglia (DRG), which areknown to express the originally reported capsaicin receptor1, no signalcould be detected in the SON. In contrast, the C-terminal end of Trpv1
was present both in SON (Fig. 1a) and in DRG (data not shown). Todetermine whether Trpv1 gene products are translated in SON neurons,we probed isolated magnocellular neurosecretory cells (MNCs) withcombinations of antibodies to AVP and to the C or N terminus of theTrpv1 protein. AVP-containing MNCs could be stained with an anti-body to the C terminus of Trpv1 (C-Trpv1), but not by an antibody toits N terminus (N-Trpv1; Fig. 1b). In contrast, both antibodies stainedDRG neurons (Fig. 1b). These results indicate that MNCs express anN-truncated product of the Trpv1 gene. Because other splice variants ofTrpv1 have been reported to be insensitive to capsaicin12,13, a selectiveagonist of the capsaicin receptor1, we examined the effects of thiscompound on MNCs. These cells did not respond to capsaicin (mean ±s.e.m.: –0.16 ± 0.11 pA pF–1; P ¼ 0.13; n ¼ 9), whereas DRG neuronsshowed robust and reversible inward currents in response to the drug(–102.1 ± 18.8 pA pF–1; P ¼ 0.003; n ¼ 9; Fig. 1c). Resiniferatoxin,another Trpv1 agonist14, also elicited inward currents in DRG neurons(–79.7 ± 23.4 pA pF–1, n ¼ 7) but not in MNCs (–0.1 ± 0.1 pA pF–1,n ¼ 6; data not shown). Altogether, these data suggest that at least oneN-terminal variant of Trpv1 is expressed in SON neurons.
Products of Trpv1 gene are expressed in the SON and PVN
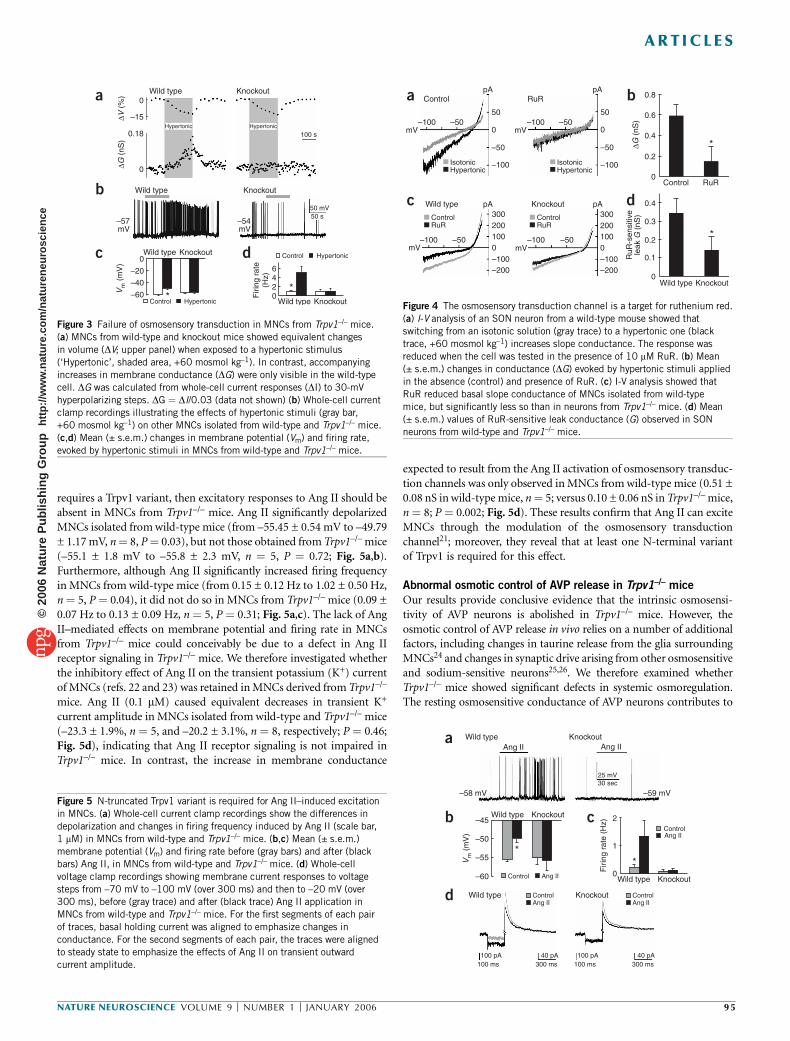
In addition to the SON, MNCs are also found in the paraventricularnucleus (PVN) of the hypothalamus. To determine whether Trpv1gene products can be found in both of these nuclei in situ, we stainedcoronal sections of the hypothalamus using an antibody specificto the C terminus of mouse Trpv1 (C-Trpv1) and a cocktail ofantibodies to AVP and oxytocin, peptides known to be contained inMNCs15. We found C-Trpv1 staining throughout the SON of wild-typemice (Fig. 2a) but not in the SON of Trpv1–/– mice16 (Fig. 2b),
Received 22 August; accepted 14 November; published online 4 December 2005; doi:10.1038/nn1614
1Centre for Research in Neuroscience, McGill University, 1650 Cedar Avenue, Montreal, Quebec H3G 1A4, Canada. 2Montreal Neurological Institute, McGill University,3801 University Street, Montreal, Quebec H3A 2B4, Canada. Correspondence should be addressed to C.W.B. ([email protected]).
NATURE NEUROSCIENCE VOLUME 9 [ NUMBER 1 [ JANUARY 2006 93
ART ICLES©
2006
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eneu
rosc
ienc
e
confirming the specificity of the antibody. Similarly, C-Trpv1 wasdetected in AVP- and oxytocin-containing neurons of the PVN inwild-type mice (Fig. 2c) but not in Trpv1–/– mice (Fig. 2d). Althoughstaining seemed to be restricted to neurons, we cannot excludethe possibility that glial cells may also express C-Trpv1, perhaps insmaller amounts.
Absence of osmosensory transduction in Trpv1–/– mice
To determine whether N-terminal variants of Trpv1 are required for thegeneration of osmosensory responses in AVP neurons, we comparedthe effects of hyperosmotic stimuli on MNCs acutely dissociated fromwild-type and Trpv1–/– mice (Fig. 3). In neurons from wild-type mice,hyperosmolality caused a reversible decrease in cell volume (Fig. 3a,upper left) associated with a gradual and reversible increase inmembrane conductance (Fig. 3a, lower left). Although osmoticallyevoked shrinking was similar in neurons from Trpv1–/– and wild-typemice (–16.0 ± 1.4% in wild-type mice, n ¼ 12; versus –16.6 ± 1.6% inTrpv1–/– mice, n ¼ 11; P ¼ 0.76), neurons from Trpv1–/– mice showedno associated increase in membrane conductance (Fig. 3a, lower right)or current (conductance: 46.8 ± 15.7 pS pF–1 in wild-type mice, n¼ 12;versus 0.6 ± 0.5 pS pF–1 in Trpv1–/– mice, n ¼ 11; P ¼ 0.001; current:–3.08 ± 0.76 pA pF–1 in wild-type mice, n ¼ 12; versus –0.02 ± 0.2 pA
pF–1 in Trpv1–/– mice, n ¼ 11; P ¼ 0.004). Moreover, hyperosmolalitydid not depolarize (Fig. 3b,c) or excite (Fig. 3b,d) MNCs from Trpv1–/–
mice (depolarization: –60.1 ± 1.8 mV to –50.0 ± 1.7 mV in wild-typemice, n ¼ 36, P ¼ 4 �10–8; versus 57.0 ± 2.3 mV to –56.8 ± 2.1 mV inTrpv1–/– mice, n¼ 17, P¼ 0.89; excitation: 0.3 ± 0.1 Hz to 1.7 ± 0.5 Hzin wild-type mice, n¼ 20, P¼ 0.004; versus 0.3 ± 0.1 Hz to 0.3 ± 0.2 Hzin Trpv1–/– mice, n ¼ 8, P ¼ 0.98). Thus, although an N-terminalvariant of Trpv1 is not required for the production of osmoticallyevoked volume changes in AVP neurons, it seems to be essential for thegeneration of intrinsic osmosensory responses.
Osmosensory transduction is inhibited by a Trpv blocker
As all Trpv channels are reportedly blocked by ruthenium red (RuR)17,we next examined whether RuR could impair the osmosensitivity ofMNCs in wild-type mice. We found that the increase in membraneconductance provoked by hyperosmolality was significantly smaller inthe presence of 10 mM RuR (0.15 ± 0.15 nS, n¼ 10; Fig. 4a,b) than forthe control without RuR (0.59 ± 0.11 nS, n¼ 8; P¼ 0.012). In contrast,capsazepine (10 mM), a competitive antagonist of capsaicin18 at theTrpv1 channel, did not significantly affect responses to hypertonicstimuli in MNCs (P 4 0.05; data not shown). Furthermore, becauseosmosensory transduction channels contribute significantly to theresting membrane conductance of MNCs6–8, we hypothesized thatRuR would have a greater inhibitory effect on the basal conductance ofneurons in wild-type mice than on that of neurons in Trpv1–/– mice.Indeed, the basal RuR-sensitive conductance was significantly smallerin MNCs from Trpv1–/– mice (0.14 ± 0.08 nS, n ¼ 13) than in thosefrom wild-type mice (0.31 ± 0.08 nS, n¼ 8; P¼ 0.03; Fig. 4c,d). Theseobservations are consistent with the involvement of Trpv channelsin osmoreception.
Absence of peptidergic excitation in MNCs of Trpv1–/– mice
Osmosensory transduction channels may be modulated by a variety ofneuropeptides19–21; in particular, activation of these channels byangiotensin II (Ang II) can lead to the excitation of MNCs21. Wetherefore surmised that if osmosensory transduction channel function
AVP/OXTa
b
c
d
OCh
OCh
OCh
3Vt
3Vt
3Vt
3Vt
OCh
Wild
type
Wild
type
Kno
ckou
t
Kno
ckou
t
C-TRPV1 AVP/OXT C-TRPV1
A
ADRGLane Cell Primers
123
SONSON
CapDRG SON
Cap
200 pA
60 s
AB
1 2
M 1 2 3
TRPV1
3 4 5 6 7 8 9 101112 13 1415 16B
C-TRPV1DRG neurons SON neurons
N-TRPV1
C-TRPV1
N-TRPV1
Vasopressin
Vasopressin
a
c
b
Figure 1 SON neurons express an N-terminal variant(s) of Trpv1.
(a) Simplified structure of the mRNA (numbered boxes show exons of the
Trpv1 gene) encoding Trpv1 (above). Gray arrow tails and lines illustrate
mRNA regions detected by primer sets A and B. RT-PCR analysis of
mRNAs expressed in DRG and SON (below). Note the lack of signals
for the N terminus of Trpv1 (lane 2, exons 2–6, primer set A) in SON.
(b) Immunocytochemical staining confirmed the presence of full-length
Trpv1 in DRG neurons. In contrast, only the C terminus of the Trpv1 protein(c-Trpv1) was detected in AVP-positive SON neurons. (c) Whole-cell
recordings revealed that DRG neurons are sensitive to capsaicin
(‘Cap’, 10 mM, 5 s) but that SON neurons are insensitive.
Figure 2 Trpv1 is expressed in neurons of the
PVN and SON. Double-immunohistochemical
staining of coronal sections using antibodies to
AVP and oxytocin (AVP/OXT; green) and to the
C terminus of Trpv1 (C-Trpv1; red). (a,b) C-Trpv1
was expressed in the SON of wild-type mice but
not in that of knockout mice. (c,d) C-Trpv1 was
expressed in the PVN of wild-type mice but not in
that of knockout mice. Dotted lines outline theedge of the optic chiasm (OCh) or the dorsolateral
border of the third ventricle (3Vt). Scale bars,
100 mm. Insets show cellular staining observed at
higher magnification (box size: 45 mm � 45 mm).
94 VOLUME 9 [ NUMBER 1 [ JANUARY 2006 NATURE NEUROSCIENCE
ART ICLES©
2006
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eneu
rosc
ienc
e
requires a Trpv1 variant, then excitatory responses to Ang II should beabsent in MNCs from Trpv1–/– mice. Ang II significantly depolarizedMNCs isolated from wild-type mice (from –55.45 ± 0.54 mV to –49.79± 1.17 mV, n¼ 8, P¼ 0.03), but not those obtained from Trpv1–/– mice(–55.1 ± 1.8 mV to –55.8 ± 2.3 mV, n ¼ 5, P ¼ 0.72; Fig. 5a,b).Furthermore, although Ang II significantly increased firing frequencyin MNCs from wild-type mice (from 0.15 ± 0.12 Hz to 1.02 ± 0.50 Hz,n¼ 5, P¼ 0.04), it did not do so in MNCs from Trpv1–/– mice (0.09 ±0.07 Hz to 0.13 ± 0.09 Hz, n ¼ 5, P ¼ 0.31; Fig. 5a,c). The lack of AngII–mediated effects on membrane potential and firing rate in MNCsfrom Trpv1–/– mice could conceivably be due to a defect in Ang IIreceptor signaling in Trpv1–/– mice. We therefore investigated whetherthe inhibitory effect of Ang II on the transient potassium (K+) currentof MNCs (refs. 22 and 23) was retained in MNCs derived from Trpv1–/–
mice. Ang II (0.1 mM) caused equivalent decreases in transient K+
current amplitude in MNCs isolated from wild-type and Trpv1–/– mice(–23.3 ± 1.9%, n ¼ 5, and –20.2 ± 3.1%, n ¼ 8, respectively; P ¼ 0.46;Fig. 5d), indicating that Ang II receptor signaling is not impaired inTrpv1–/– mice. In contrast, the increase in membrane conductance
expected to result from the Ang II activation of osmosensory transduc-tion channels was only observed in MNCs from wild-type mice (0.51 ±0.08 nS in wild-type mice, n¼ 5; versus 0.10 ± 0.06 nS in Trpv1–/– mice,n ¼ 8; P ¼ 0.002; Fig. 5d). These results confirm that Ang II can exciteMNCs through the modulation of the osmosensory transductionchannel21; moreover, they reveal that at least one N-terminal variantof Trpv1 is required for this effect.
Abnormal osmotic control of AVP release in Trpv1–/– mice
Our results provide conclusive evidence that the intrinsic osmosensi-tivity of AVP neurons is abolished in Trpv1–/– mice. However, theosmotic control of AVP release in vivo relies on a number of additionalfactors, including changes in taurine release from the glia surroundingMNCs24 and changes in synaptic drive arising from other osmosensitiveand sodium-sensitive neurons25,26. We therefore examined whetherTrpv1–/– mice showed significant defects in systemic osmoregulation.The resting osmosensitive conductance of AVP neurons contributes to
–57mV
–54mV
50 mV50 s
Wild type Knockout
0
0
–15
–20
–40
Firi
ng r
ate
(Hz)
–60
Control Hypertonic
Control Hypertonic
0642 *
* 0
0.18 100 sHypertonic Hypertonic
Wild type
Wild type Knockout
Wild type Knockout
Vm
(m
V)
Knockout
∆V (
%)
∆G (
nS)
c
b
a
d
Figure 3 Failure of osmosensory transduction in MNCs from Trpv1–/– mice.
(a) MNCs from wild-type and knockout mice showed equivalent changes
in volume (DV; upper panel) when exposed to a hypertonic stimulus
(‘Hypertonic’, shaded area, +60 mosmol kg–1). In contrast, accompanying
increases in membrane conductance (DG) were only visible in the wild-type
cell. DG was calculated from whole-cell current responses (DI) to 30-mV
hyperpolarizing steps. DG ¼ DI/0.03 (data not shown) (b) Whole-cell current
clamp recordings illustrating the effects of hypertonic stimuli (gray bar,
+60 mosmol kg–1) on other MNCs isolated from wild-type and Trpv1–/– mice.
(c,d) Mean (± s.e.m.) changes in membrane potential (Vm) and firing rate,
evoked by hypertonic stimuli in MNCs from wild-type and Trpv1–/– mice.
–100
Control
Wild type Knockout
Control RuR
RuR
∆G (
nS)
pA
pA pA
pA
mV
mV mV
mV–50 –100 –50
–50
Isotonic
ControlRuR
Hypertonic–100
50
300200100
–100
–100 –50 –100 –50
–200
0
300200100
–100–200
0
0
–50
–100
RuR
-sen
sitiv
ele
ak G
(nS
)
50
0.8
0.4
0.3
0.2
0.1
0.6
0.4
0.2
*
0
0
0
ControlRuR
IsotonicHypertonic
a
c d
b
Wild type Knockout
*
Figure 4 The osmosensory transduction channel is a target for ruthenium red.
(a) I-V analysis of an SON neuron from a wild-type mouse showed that
switching from an isotonic solution (gray trace) to a hypertonic one (black
trace, +60 mosmol kg–1) increases slope conductance. The response was
reduced when the cell was tested in the presence of 10 mM RuR. (b) Mean
(± s.e.m.) changes in conductance (DG) evoked by hypertonic stimuli applied
in the absence (control) and presence of RuR. (c) I-V analysis showed that
RuR reduced basal slope conductance of MNCs isolated from wild-type
mice, but significantly less so than in neurons from Trpv1–/– mice. (d) Mean
(± s.e.m.) values of RuR-sensitive leak conductance (G) observed in SON
neurons from wild-type and Trpv1–/– mice.
Ang II Ang II
25 mV30 sec
Wild type
Wild type
Control
100 pA 40 pA100 ms
100 pA100 ms300 ms
40 pA300 ms
Ang II
ControlAng II
Control
Firi
ng r
ate
(Hz)
Ang II
ControlAng II
Vm
(m
V)
Knockout
Knockout
Wild type Knockout
Wild type Knockout
–58 mV
–45 2
1
0
–50
–55
–60
–59 mV
**
a
b
d
cFigure 5 N-truncated Trpv1 variant is required for Ang II–induced excitation
in MNCs. (a) Whole-cell current clamp recordings show the differences in
depolarization and changes in firing frequency induced by Ang II (scale bar,
1 mM) in MNCs from wild-type and Trpv1–/– mice. (b,c) Mean (± s.e.m.)
membrane potential (Vm) and firing rate before (gray bars) and after (black
bars) Ang II, in MNCs from wild-type and Trpv1–/– mice. (d) Whole-cell
voltage clamp recordings showing membrane current responses to voltage
steps from –70 mV to –100 mV (over 300 ms) and then to –20 mV (over300 ms), before (gray trace) and after (black trace) Ang II application in
MNCs from wild-type and Trpv1–/– mice. For the first segments of each pair
of traces, basal holding current was aligned to emphasize changes in
conductance. For the second segments of each pair, the traces were aligned
to steady state to emphasize the effects of Ang II on transient outward
current amplitude.
NATURE NEUROSCIENCE VOLUME 9 [ NUMBER 1 [ JANUARY 2006 95
ART ICLES©
2006
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eneu
rosc
ienc
e

the basal electrical activity of these cells27,28, and thus to basal AVPrelease from the neurohypophysis29 and steady-state antidiuresis at thekidney30–33. As this conductance is absent in Trpv1–/– mice, wehypothesized that basal AVP release might be sufficiently compromisedto cause an increase in systemic osmolality. Indeed, under basalconditions, Trpv1–/– mice had a serum osmolality that was significantlygreater than that in wild-type mice (320.8 ± 1.1 mosmol kg–1 inTrpv1–/–
mice, n¼ 29; versus 311.2 ± 0.6 mosmol kg–1 in wild-type mice, n¼ 62;P ¼ 1 � 10–9). Finally, we hypothesized that the loss of intrinsicosmosensitivity in AVP neurons would severely impair the osmoticcontrol of AVP release in Trpv1–/– mice. As expected, linear regressionanalysis revealed that the slope of the relation between serum AVPconcentration (pg ml–1) and osmolality (mosmol kg–1) was significantlylower in Trpv1–/– mice than in wild-type mice (0.44 ± 0.18, n¼ 16, and1.79 ± 0.34, n¼ 28, respectively; Po 0.001; Fig. 6). Thus Trpv1–/– miceshow significant deficits in systemic osmoregulation.
DISCUSSION
Under hyperosmotic conditions, the cell shrinkage experienced by AVPneurons directly activates a stretch-inhibited osmosensory transductionchannel6, thereby causing a depolarization that contributes to excitationand AVP release. Trpv4 channels are required for the osmotic control ofAVP release4,5, and it has been proposed that the Trpv4 channel operatesas an osmosensor in vertebrates2,3. However, two lines of evidence argueagainst the notion that the osmosensor in AVP neurons is encoded as ahomomultimer of Trpv4. First, unlike the native osmosensory channelin mammalian AVP neurons, homomultimeric Trpv4 channels are notintrinsically mechanosensitive3; rather, swelling-induced increases inthe activity of these channels seems to be mediated indirectly throughthe production of intracellular signals34. Second, and also unlike thenative channel in mammalian AVP neurons, homomultimeric Trpv4channels expressed heterologously are activated by cell swelling2,3.Although experiments have suggested that Trpv4 may contribute tothe formation of channels required for hypotonic stimulus-inducednociception in rats9, Trpv4 has been shown to rescue the hypertonicityavoidance behavior defect of OSM-9 mutant worms35. These resultssuggest that the molecular environment in which Trpv4 subunits areexpressed can dictate whether they participate in the detection ofhypotonicity or hypertonicity36. Thus, the possibility that Trpv4 con-tributes to the formation of a mechano-gated, hypertonicity-activatedcation channel cannot be excluded. Although this issue requires furtherinvestigation, the evidence presented above indicates that Trpv4 alonecannot encode the osmoreceptor in AVP neurons. Another Trpvsubunit may therefore be required for osmoreception in these cells.
Because an artificial chimeric Trpv1-Trpv4 channel has been reported tooperate as a mechanically gated, stretch-inhibited cation channel10, weinvestigated the possibility that a product of the Trpv1 gene contributesto osmosensory transduction in AVP neurons.
Trpv1 N-terminal variants are needed for osmosensitivity
Probing the isolated SON with different sets of PCR primers revealedthat cells in this tissue express mRNA encoding large portions of Trpv1,but that regions encoded by exons 2–6 of the gene are absent. Inagreement with this finding, immunohistochemical analysis revealedthat, unlike DRG neurons, AVP-positive neurons in the SON could bestained by antibodies to the C terminus of the Trpv1 protein, but not byantibodies to its N terminus. The insensitivity of MNCs to capsaicin isalso consistent with recent studies showing that parts of the N terminusof the protein are required to mediate the effects of this drug12,13,37–39.Several N-terminal variants of Trpv1 have already beendescribed12,13,39, and others have been predicted to occur based ongenomic analysis11. Although we did not identify the nature of theTrpv1 variant(s) expressed in AVP neurons in this study, our dataindicate that their presence is essential for the intrinsic osmosensitivityof these cells. Indeed, although MNCs derived from Trpv1–/– mice stillshrank in response to hypertonic stimulation, they did not generateassociated increases in cation conductance, osmoreceptor potentials oraction potential discharges. Several mechanisms might explain the lackof intrinsic osmosensitivity in MNCs derived from Trpv1–/– mice. It ispossible that Trpv1 variants (i) are pore-forming subunits of thetransduction channel, (ii) are involved in maturation and targetingof the osmoreceptor or (iii) serve as accessory subunits that couplechanges in cell volume to changes in channel activity. Further work willbe required to investigate these issues. Finally, it is interesting to notethat acutely isolated DRG neurons, which contain the originallyreported capsaicin receptor (that is, ‘full-length Trpv1’), do not showincreases in cation current in response to osmotically induced shrink-ing (data not shown). This observation indicates that at least oneN-terminal variant of Trpv1 is required for osmosensitivity, but that thefull-length Trpv1 is not.
Trpv1 variants and the osmotic control of AVP release
The release of AVP during acute hyperosmolality in vivo is believed tobe triggered by a number of factors that together enhance the electricalactivity of MNCs24,27,29. Under these conditions, MNCs are believed toexperience changes in synaptic drive from osmosensitive40,41 andsodium (Na+)-sensitive neurons28,42,43 located in other parts of thebrain. Additionally, the membrane potential of MNCs is depolarized toenhance the probability of synaptic excitation28. In situ, this effect ismediated by the osmosensory transduction channels of AVP neurons6
and by osmotically regulated taurine release from glia surroundingthese cells24. The contributions of Na+-sensitive centers and glialtaurine release, however, are unlikely to depend on products of theTrpv1 gene. Indeed, sodium sensitivity is mediated by Nax
43, a memberof the voltage-gated sodium channel family44, and osmotically evokedtaurine release from glial cells depends on a volume-regulated anionchannel24. Given such redundancy, one might expect that mice lackingTrpv1 gene products could adapt to their condition and retain effectiveosmotically regulated AVP release. However, we found that the sensi-tivity of the relation between serum AVP and osmolality was decreasedby a factor of 4 in Trpv1–/– mice, indicating that loss of Trpv1 geneproducts significantly impairs this neuroendocrine reflex. Although itis tempting to conclude that the intrinsic osmosensitivity of AVPneurons is primarily responsible for this deficit, we cannot excludethe possibility that other osmosensory neurons in the brain also rely on
100
Wild type Knockout
80
1.787 0.440
60
300 320Osmolality (mosmol kg–1)
340 360 300 320Osmolality (mosmol kg–1)
340 360 380
Ser
um A
VP
(pg
ml–1
)
Ser
um A
VP
(pg
ml–1
)
40
20
0
100
80
60
40
20
0
Figure 6 Trpv1–/– mice show defects in systemic osmoregulation. Linear
regression analysis of the relation between serum AVP concentration and
serum osmolality in wild-type and Trpv1–/– mice. In each case, mice were
allowed to drink either water or 2% NaCl for 0–48 h and then were sampled
in order to examine the effects of a systemic osmotic stimulus. Numbers on
each graph show the slope of the best linear fit. The dotted line in the right
graph repeats the regression observed in wild-type mice.
96 VOLUME 9 [ NUMBER 1 [ JANUARY 2006 NATURE NEUROSCIENCE
ART ICLES©
2006
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eneu
rosc
ienc
e

Trpv1-dependent transduction channels. Further studies will berequired to investigate these issues.
Possible structure of the osmoreceptor
Our data indicate that one or more N-terminal variants of Trpv1 has acrucial role in the assembly of the stretch-inhibited channel thatunderlies osmoreception in AVP neurons and perhaps other types ofosmosensitive cells. As indicated earlier, Trpv1 variants could have avariety of roles in the formation of a functional osmoreceptor. Onepossibility is that the Trpv1 variants simply serve as molecular partnersinvolved in the maturation and/or targeting of the osmoreceptor45.Another possibility is that the Trpv1 variants are involved in couplingchanges in cell volume to changes in channel activity. However, ourresults show that in wild-type mice, Ang II can enhance the activity ofthis channel in the absence of osmotic stimulation. This observationsuggests that the variants may contribute to the formation of theosmosensory transduction channel itself, rather than simply operatingas accessory subunits involved in mechanical gating. This hypothesis isalso supported by the observation of a reduced RUR-sensitive basalcation conductance in AVP neurons from Trpv1–/– mice. Indeed, if theTrpv1 variants simply mediate stretch-inhibition, rather than beingpart of the pore-forming channel, then their absence in Trpv1–/– micewould be expected to result in a higher basal RUR-sensitive conduc-tance, which is not the case. A third possibility is that the Trpv1 variantsmay be a pore-forming subunit of the transduction channel. Thishypothesis is consistent with the observations described above and withthe blockade of osmoreceptor currents by RUR. Finally, it is possiblethat the native osmosensory transducer is a complex comprising morethan one subtype of Trpv channel. Indeed, in addition to Trpv1variants, AVP neurons are known to express Trpv246, a possible partnerof Trpv139, as well as Trpv4 (data not shown), a Trpv member alreadyimplicated in the osmotic control of AVP release4,5. Although Trpv1(ref. 47), Trpv2 (ref. 48) and Trpv4 (refs. 2,3) have all been reported tooperate as stretch-activated channels when expressed as homomulti-mers, it is possible, as suggested earlier, that a complex containing oneor more N-terminal variants of Trpv1 and one or both of the Trpv2 andTrpv4 subunits, or variants of these subunits, could form a functionalstretch-inhibited cation channel.
METHODSPreparation of isolated cells. We used 6- to 8-week-old male C57/BL mice
(Charles River Laboratories) or Trpv1–/– mice (BL126 S4, Jackson Laboratory).
We followed the guidelines of the Canadian Council on Animal Care and
protocols approved by the McGill University Animal Care Committee. Mice
were anesthetized using halothane (Sigma-Aldrich) and killed. Isolated SONs
were incubated for 30 min (room temperature) in an oxygenated PIPES
solution containing 130 mM NaCl, 5 mM KCl, 1 mM MgCl2, 20 mM PIPES,
1 mM CaCl2, 10 mM glucose, 0.5 mg ml–1 protease X and 0.5 mg ml–1 protease
XIV (Sigma-Aldrich). SONs were washed in protease-free PIPES solution,
triturated and plated on Petri dishes.
Cell perfusion and drug application. Cells were perfused with a HEPES-
buffered saline solution (pH 7.3) containing 150 mM NaCl, 3 mM KCl, 1 mM
MgCl2, 10 mM HEPES, 1 mM CaCl2, 10 mM glucose (312 mosmol kg–1 with
mannitol). We achieved perfusion at 200 ml min–1 via a three-barrel assembly
controlled by a fast stepper device (Warner Instruments). Ruthenium red
(10 mM) was dissolved in HEPES before every experiment. Capsaicin (10 mM
in ethanol) was diluted in HEPES to 10 mM. Ang II (20 mM in water) was
diluted in HEPES to 1 mM. Hypertonic stimuli were applied via HEPES
solution containing excess mannitol.
Measurement of changes in cell volume. Relative changes in cell volume (DV,
in %) were determined from maximal cell cross-sectional area (CSA) as
previously described6. For each image, we traced the cell perimeter using Scion
Image (Scion) and determined the CSA (in pixels). Values of averaged control
CSAs (CSA0) and of DV at different time points (DVt) were calculated from the
corresponding value of CSA (CSAt) using the equation: DV ¼ 100 � [(CSA01.5
– CSAt1.5)/CSA0
1.5].
Electrophysiology. Cells were patch-clamped with glass pipettes containing
120 mM potassium gluconate, 1 mM MgCl2, 1 mM EGTA, 10 mM HEPES,
4 mM ATP, 1 mM GTP and 14 mM phosphocreatine (280 mosmol kg–1; pH
7.34). We recorded from the cells using an Axopatch-200B amplifier (Axon
Instruments). Capacitance and series resistance were neutralized electronically,
and values of cell input capacitance (Ci) were noted. Voltages were not
corrected for an experimentally measured liquid junction error of –1 mV.
For steady-state current-voltage (I-V) analysis, voltage was ramped up from
–110 mV to –10 mV over 4 s, and brought back to the holding potential
(–70 mV). The slope of the I-V relation was measured between –80 mV and
–60 mV. In current-clamp experiments, membrane potential (excluding action
potentials and after-hyperpolarizations) and firing frequency were measured
during the last 10 s of the stimulus.
Measurement of serum AVP and osmolality. AVP levels were determined
using an enzyme-linked immunosorbant assay kit (R&D Systems). For each
mouse, 0.5 ml of blood was collected in a tube containing 500 kIU aprotinin
(Sigma-Aldrich) and centrifuged at 15,500g. The collected serum was assayed in
duplicates to obtain AVP measurements and to determine plasma osmolality
via a freezing point osmometer (Advanced Instruments).
Immunohistochemistry. Isolated cells from three different preparations were
fixed overnight with phosphate-buffered saline (PBS) containing 4% parafor-
maldehyde (Fisher Scientific) and washed with PBS at room temperature. After
a 1-h incubation with 1% normal goat serum (NGS) and 0.3% Triton-X, cells
were washed and incubated overnight at 4 1C with AVP monoclonal antibody
(PS41, 1:200 in PBS; gift from H. Gainer) and polyclonal antibodies (Neu-
romics Antibodies; 1:2000) to either the first 15 amino acids of the C terminus
or the last 18 amino acids of the N terminus of rat Trpv1. After being washed,
cells were incubated for 1 h with fluorescently labeled secondary antibodies
(1:200; Chemicon International). Images were acquired using a confocal
microscope (Perkin-Elmer). We used quantitative analysis of cytoplasmic
fluorescence to establish that MNCs are positively stained by antibody to the
C terminus of Trpv1 (n ¼ 53) but not by antibody to its N terminus (P ¼ 1 �10–8; n ¼ 35).
For staining of SON and PVN sections, anesthetized adult mice of both
genotypes were perfused with 4% paraformaldehyde, and their brains were
post-fixed for 2 h and placed in 30% sucrose overnight at 4 1C. Vibratome
sections (30 mm thick) containing the SON and PVN were placed in blocking
solution (0.1 M PBS, 0.2% Triton-X, 3% NGS) overnight, then incubated for
48 h with primary antibodies to AVP and oxytocin (1:400) and the C-terminal
domain of Trpv1 (Neuromics Antibodies, 1:500). After washing, sections were
incubated overnight with secondary antibodies (Molecular Probes: Alexa
Fluor 488 and Alexa Fluor 568, 1:200), coverslipped and imaged using a
confocal microscope.
RT-PCR. Total RNA was isolated from adult mouse SONs and DRGs using the
Trizol method. Random-primed cDNA synthesis was performed using the
SuperScript II reverse transcriptase (Invitrogen), then RT-cDNAs were PCR
amplified using 1-mM gene-specific primers and 3.5 units of Expand DNA
polymerase mix (Roche) during 40 thermal cycles (45 s at 94 1C, 45 s at 59 1C
and 1 min at 72 1C). Pairs of Trpv1-specific PCR primers corresponded to the
following sequences: (i) sense 5¢-CATCTTTGCTACCAGGAGTCG-3¢ and
reverse 5¢-TATCTGCCACCTCCACAAGG-3¢ (product ¼ 760 base pairs
(bp)); (ii) sense 5¢-AGTGACACTGATCGAGGATGG-3¢ and reverse 5¢-TTCAGCTGAACTTCTTCCGG-3¢ (product ¼ 660 bp).
Statistical analysis. All values are reported as mean ± s.e.m. Comparisons of
linear regressions and fits through the data were performed using Prism 4.0
(GraphPad Software). Comparison of means observed in different groups were
performed using a Student’s t-test or an analysis of variance (ANOVA), as
appropriate (SigmaStat 2.03; SPSS).
NATURE NEUROSCIENCE VOLUME 9 [ NUMBER 1 [ JANUARY 2006 97
ART ICLES©
2006
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eneu
rosc
ienc
e

ACKNOWLEDGMENTSWe thank H. Gainer (US National Institute of Neurological Disorders andStroke) for supplying the anti-AVP antibodies used in this study. We thank alsoS.H. Oliet, T. Stachniak, Z. Zhang, S. Ciura and E. Trudel for advice during thepreparation of the manuscript. This work was supported by operating grantsfrom the Canadian Institutes of Health Research (CIHR) to C.W.B. andP.S. Additional support was provided to C.W.B. through a CIHR SeniorInvestigator Award and a James McGill Research Chair. R.S.N. is a recipientof a CIHR Doctoral Award. P.S. is a Killam scholar.
COMPETING INTERESTS STATEMENTThe authors declare that they have no competing financial interests.
Published online at http://www.nature.com/natureneuroscience/
Reprints and permissions information is available online at http://npg.nature.com/
reprintsandpermissions/
1. Caterina, M.J. et al. The capsaicin receptor: a heat-activated ion channel in the painpathway. Nature 389, 816–824 (1997).
2. Liedtke, W. et al. Vanilloid receptor-related osmotically activated channel (VR-OAC), acandidate vertebrate osmoreceptor. Cell 103, 525–535 (2000).
3. Strotmann, R., Harteneck, C., Nunnenmacher, K., Schultz, G. & Plant, T.D. OTRPC4, anonselective cation channel that confers sensitivity to extracellular osmolarity. Nat. CellBiol. 2, 695–702 (2000).
4. Liedtke, W. & Friedman, J.M. Abnormal osmotic regulation in trpv4–/– mice. Proc. Natl.Acad. Sci. USA 100, 13698–13703 (2003).
5. Mizuno, A., Matsumoto, N., Imai, M. & Suzuki, M. Impaired osmotic sensation in micelacking TRPV4. Am. J. Physiol. 285, C96–C101 (2003).
6. Oliet, S.H. & Bourque, C.W. Mechanosensitive channels transduce osmosensitivity insupraoptic neurons. Nature 364, 341–343 (1993).
7. Oliet, S.H. & Bourque, C.W. Gadolinium uncouples mechanical detection and osmo-receptor potential in supraoptic neurons. Neuron 16, 175–181 (1996).
8. Oliet, S.H. & Bourque, C.W. Steady-state osmotic modulation of cationic conductance inneurons of rat supraoptic nucleus. Am. J. Physiol. 265, R1475–R1479 (1993).
9. Alessandri-Haber, N. et al. Hypotonicity induces TRPV4-mediated nociception in rat.Neuron 39, 497–511 (2003).
10. Suzuki, M., Sato, J., Kutsuwada, K., Ooki, G. & Imai, M. Cloning of a stretch-inhibitablenonselective cation channel. J. Biol. Chem. 274, 6330–6335 (1999).
11. Xue, Q., Yu, Y., Trilk, S.L., Jong, B.E. & Schumacher, M.A. The genomic organization ofthe gene encoding the vanilloid receptor: evidence for multiple splice variants. Geno-mics 76, 14–20 (2001).
12. Schumacher, M.A., Moff, I., Sudanagunta, S.P. & Levine, J.D. Molecular cloning of anN-terminal splice variant of the capsaicin receptor. Loss of N-terminal domainsuggests functional divergence among capsaicin receptor subtypes. J. Biol. Chem.275, 2756–2762 (2000).
13. Lu, G., Henderson, D., Liu, L., Reinhart, P.H. & Simon, S.A. TRPV1b, a functionalhuman vanilloid receptor splice variant. Mol. Pharmacol. 67, 1119–1127 (2005).
14. Szallasi, A. & Blumberg, P.M. Resiniferatoxin, a phorbol-related diterpene acts as anultrapotent analog of capsaicin, the irritant constituent in red pepper. Neuroscience 30,515–520 (1989).
15. Vandesande, F. & Dierickx, K. Identification of the vasopressin producing and of theoxytocin producing neurons in the hypothalamic magnocellular neurosecretroy system ofthe rat. Cell Tissue Res. 164, 153–162 (1975).
16. Caterina, M.J. et al. Impaired nociception and pain sensation in mice lacking thecapsaicin receptor. Science 288, 306–313 (2000).
17. Watanabe, H. et al. Anandamide and arachidonic acid use epoxyeicosatrienoic acids toactivate TRPV4 channels. Nature 424, 434–438 (2003).
18. Dickenson, A.H. & Dray, A. Selective antagonism of capsaicin by capsazepine: evidencefor a spinal receptor site in capsaicin-induced antinociception. Br. J. Pharmacol. 104,1045–1049 (1991).
19. Ster, J. et al. Insulin-like growth factor-1 inhibits adult supraoptic neurons via com-plementary modulation of mechanoreceptors and glycine receptors. J. Neurosci. 25,2267–2276 (2005).
20. Shibuya, I. et al. Patch-clamp analysis of the mechanism of PACAP-induced excitationin rat supraoptic neurones. J. Neuroendocrinol. 10, 759–768 (1998).
21. Chakfe, Y. & Bourque, C.W. Excitatory peptides and osmotic pressure modulatemechanosensitive cation channels in concert. Nat. Neurosci. 3, 572–579 (2000).
22. Nagatomo, T., Inenaga, K. & Yamashita, H. Transient outward current in adult ratsupraoptic neurones with slice patch-clamp technique: inhibition by angiotensin II.J. Physiol. (Lond.) 485, 87–96 (1995).
23. Li, Z. & Ferguson, A.V. Electrophysiological properties of paraventricular magnocellularneurons in rat brain slices: modulation of IA by angiotensin II. Neuroscience 71,133–145 (1996).
24. Hussy, N., Deleuze, C., Desarmenien, M.G. & Moos, F.C. Osmotic regulation of neuronalactivity: a new role for taurine and glial cells in a hypothalamic neuroendocrine structure.Prog. Neurobiol. 62, 113–134 (2000).
25. McKinley, M.J. et al. Vasopressin secretion: osmotic and hormonal regulation by thelamina terminalis. J. Neuroendocrinol. 16, 340–347 (2004).
26. Weisinger, R.S., Considine, P., Denton, D.A. & McKinley, M.J. Rapid effect of change incerebrospinal fluid sodium concentration on salt appetite. Nature 280, 490–491(1979).
27. Voisin, D.L. & Bourque, C.W. Integration of sodium and osmosensory signals invasopressin neurons. Trends Neurosci. 25, 199–205 (2002).
28. Bourque, C.W. Osmoregulation of vasopressin neurons: a synergy of intrinsic andsynaptic processes. Prog. Brain Res. 119, 59–76 (1998).
29. Poulain, D.A. & Wakerley, J.B. Electrophysiology of hypothalamic magnocellular neu-rones secreting oxytocin and vasopressin. Neuroscience 7, 773–808 (1982).
30. Robertson, G.L., Shelton, R.L. & Athar, S. The osmoregulation of vasopressin. KidneyInt. 10, 25–37 (1976).
31. Robertson, G.L. & Athar, S. The interaction of blood osmolality and blood volume inregulating plasma vasopressin in man. J. Clin. Endocrinol. Metab. 42, 613–620(1976).
32. Dunn, F.L., Brennan, T.J., Nelson, A.E. & Robertson, G.L. The role of blood osmolalityand volume in regulating vasopressin secretion in the rat. J. Clin. Invest. 52,3212–3219 (1973).
33. Schrier, R.W., Berl, T. & Anderson, R.J. Osmotic and nonosmotic control of vasopressinrelease. Am. J. Physiol. 236, F321–F332 (1979).
34. Vriens, J. et al. Cell swelling, heat, and chemical agonists use distinct pathways for theactivation of the cation channel TRPV4. Proc. Natl. Acad. Sci. USA 101, 396–401(2004).
35. Liedtke, W., Tobin, D.M., Bargmann, C.I. & Friedman, J.M. Mammalian TRPV4 (VR-OAC) directs behavioral responses to osmotic and mechanical stimuli in Caenorhabditiselegans. Proc. Natl. Acad. Sci. USA 100, 14531–14536 (2003).
36. Kung, C. A possible unifying principle for mechanosensation. Nature 436, 647–654(2005).
37. Jung, J. et al. Agonist recognition sites in the cytosolic tails of vanilloid receptor 1.J. Biol. Chem. 277, 44448–44454 (2002).
38. Hellwig, N., Albrecht, N., Harteneck, C., Schultz, G. & Schaefer, M. Homo- andheteromeric assembly of TRPV channel subunits. J. Cell Sci. 118, 917–928(2005).
39. Wang, C., Hu, H.Z., Colton, C.K., Wood, J.D. & Zhu, M.X. An alternative splicing productof the murine trpv1 gene dominant negatively modulates the activity of TRPV1 channels.J. Biol. Chem. 279, 37423–37430 (2004).
40. Richard, D. & Bourque, C.W. Synaptic control of rat supraoptic neurones during osmoticstimulation of the organum vasculosum lamina terminalis in vitro. J. Physiol. (Lond.)489, 567–577 (1995).
41. Leng, G., Brown, C.H. & Russell, J.A. Physiological pathways regulating the activity ofmagnocellular neurosecretory cells. Prog. Neurobiol. 57, 625–655 (1999).
42. Grob, M., Drolet, G. & Mouginot, D. Specific Na+ sensors are functionally expressed ina neuronal population of the median preoptic nucleus of the rat. J. Neurosci. 24,3974–3984 (2004).
43. Hiyama, T.Y. et al. Nax channel involved in CNS sodium-level sensing. Nat. Neurosci. 5,511–512 (2002).
44. Goldin, A.L. et al. Nomenclature of voltage-gated sodium channels. Neuron 28,365–368 (2000).
45. Tobin, D. et al. Combinatorial expression of TRPV channel proteins defines their sensoryfunctions and subcellular localization in. C. elegans neurons. Neuron 35, 307–318(2002).
46. Wainwright, A., Rutter, A.R., Seabrook, G.R., Reilly, K. & Oliver, K.R. Discrete expressionof TRPV2 within the hypothalamo-neurohypophysial system: implications for regulatoryactivity within the hypothalamic-pituitary-adrenal axis. J. Comp. Neurol. 474, 24–42(2004).
47. Birder, L.A. et al. Altered urinary bladder function in mice lacking the vanilloid receptorTRPV1. Nat. Neurosci. 5, 856–860 (2002).
48. Muraki, K. et al. TRPV2 is a component of osmotically sensitive cation channels inmurine aortic myocytes. Circ. Res. 93, 829–838 (2003).
98 VOLUME 9 [ NUMBER 1 [ JANUARY 2006 NATURE NEUROSCIENCE
ART ICLES©
2006
Nat
ure
Pub
lishi
ng G
roup
ht
tp://
ww
w.n
atur
e.co
m/n
atur
eneu
rosc
ienc
e





![[VII]. Regulation of Gene Expression Via Signal Transduction Reading List VII: Signal transduction Signal transduction in biological systems](https://img.pdfslide.us/doc/110x75/56649e385503460f94b28319/vii-regulation-of-gene-expression-via-signal-transduction-reading-list-vii.jpg)






















