Embed Size (px)
Citation preview

An Introduction toMultiple Sequence
AlignmentsCédric Notredame

chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
Manguel M, Samaniego F.J., Abraham Wald’s Work on Aircraft Suvivability, J. American Statistical Association. 79, 259-270, (1984)

Our Scope
How Can I Use My Alignment?
How Does The Computer Align The Sequences?
How Can I Assemble a Mult. Aln?
What are the Difficulties?

Outline
-Why Do We Need Multiple Sequence Alignment ?
-The progressive Alignment Algorithm
-A possible Strategy…
-Potential Difficulties

Pre-requisite
-How Do Sequences Evolve?
-How can We COMPARE Sequences ?
-How can We ALIGN Sequences ?

Why Do We Need Multiple Sequence
Alignment ?

Sometimes Two Sequences Are Not Enough…
The man with TWO watches NEVER knows the time

chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
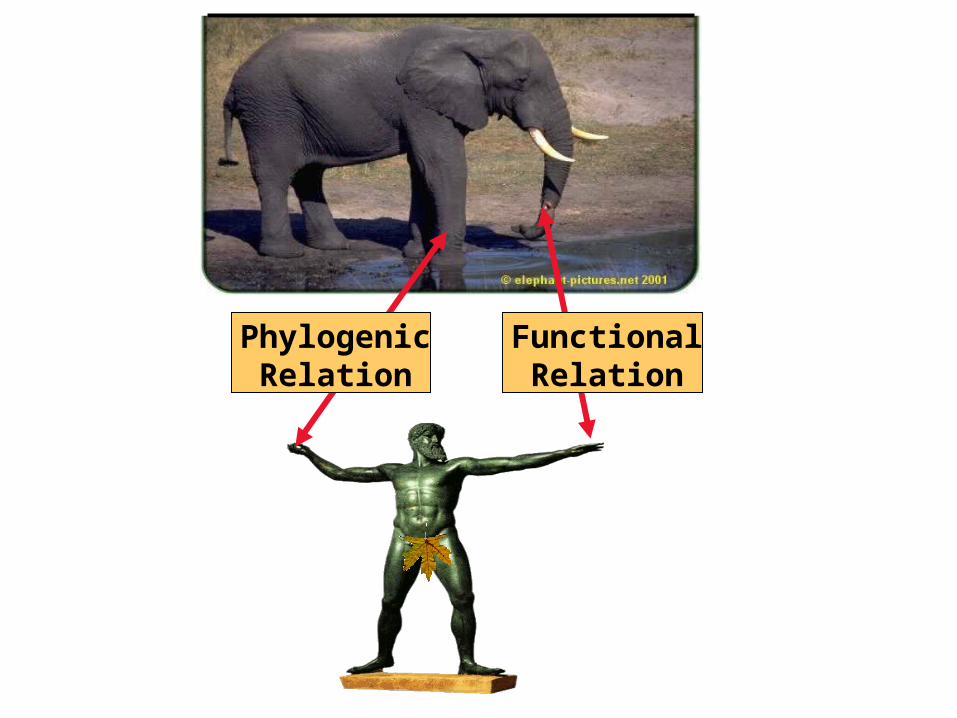
What is A Multiple Sequence Alignment?
Structural Criteria:Residues are arranged so that those playing a similar role end up in the same column.
Evolution Criteria:Residues are arranged so that those having the same ancestor end up in the same column.
PhylogenicRelation
FunctionalRelation
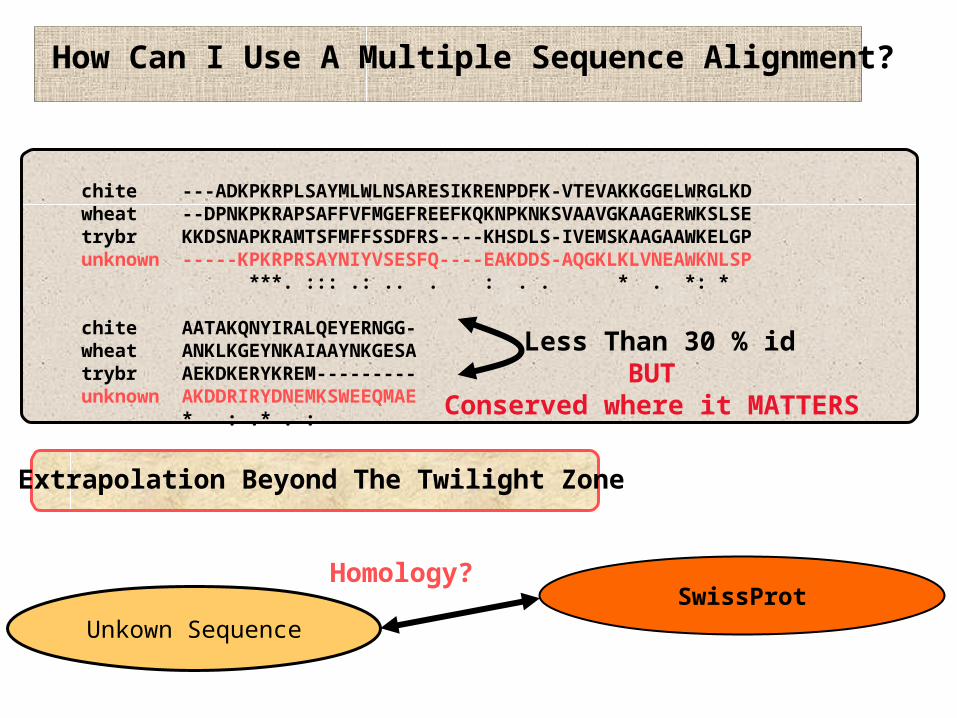
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPunknown -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------unknown AKDDRIRYDNEMKSWEEQMAE * : .* . :
How Can I Use A Multiple Sequence Alignment?
Extrapolation Beyond The Twilight Zone
SwissProtUnkown Sequence
Homology?
Less Than 30 % idBUT
Conserved where it MATTERS
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
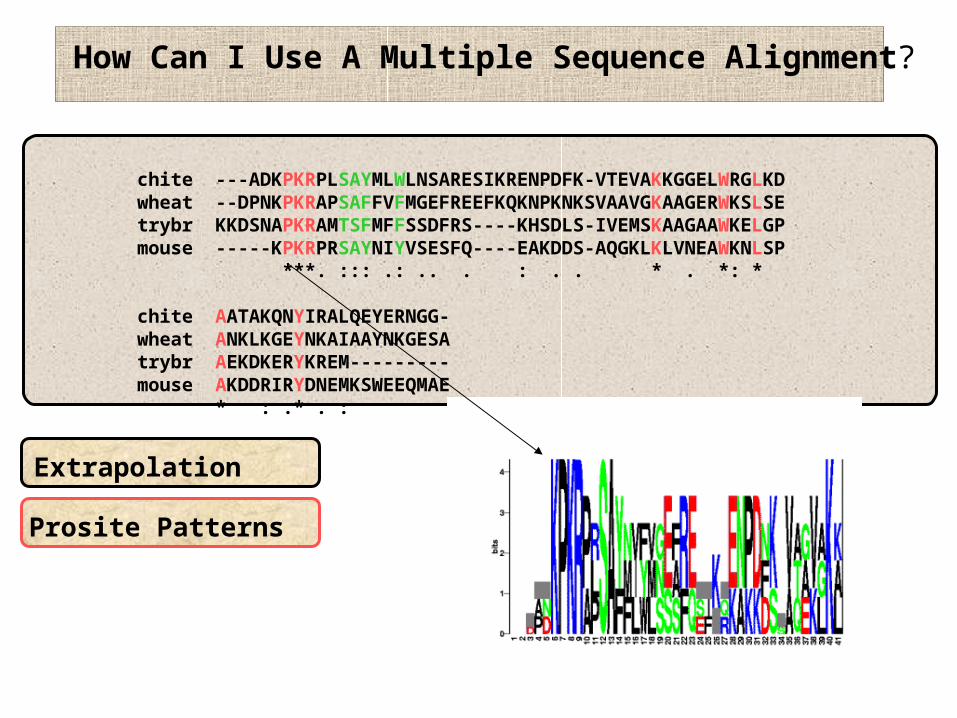
How Can I Use A Multiple Sequence Alignment?
Extrapolation
Prosite Patterns

chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
How Can I Use A Multiple Sequence Alignment?
Extrapolation
Prosite PatternsP-K-R-[PA]-x(1)-[ST]…
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
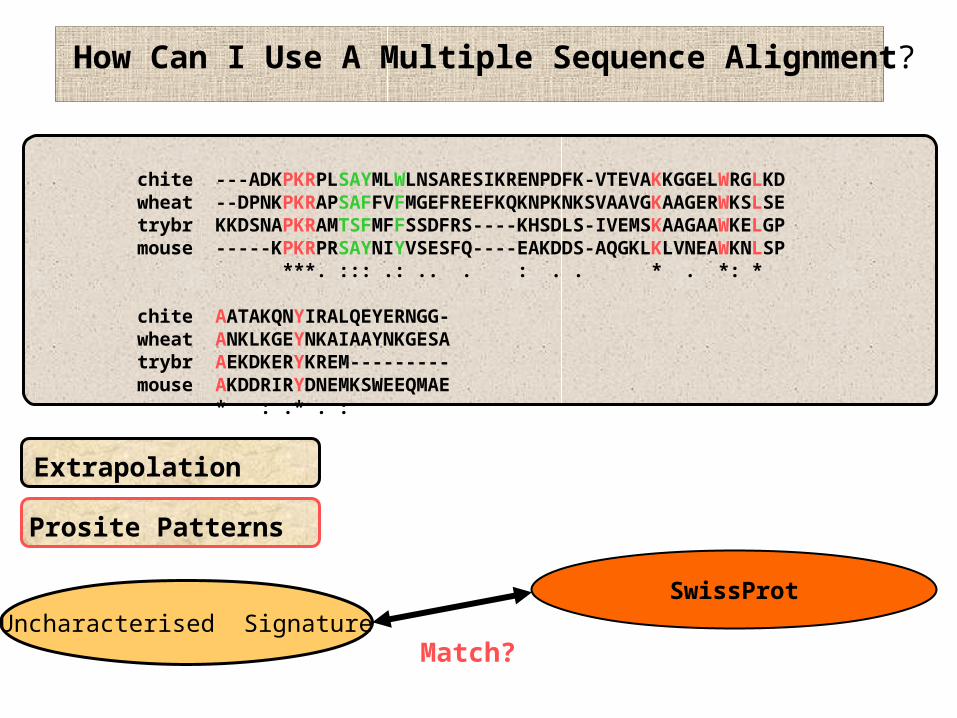
How Can I Use A Multiple Sequence Alignment?
Extrapolation
Prosite Patterns
SwissProtUncharacterised Signature
Match?
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-IQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
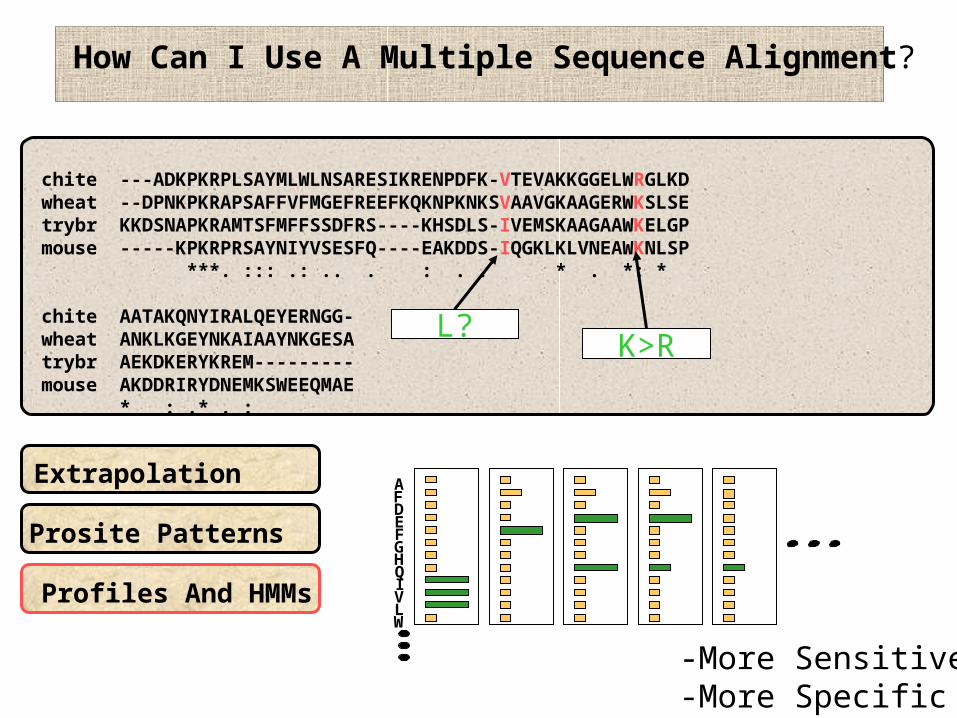
How Can I Use A Multiple Sequence Alignment?
Extrapolation
Prosite Patterns
Profiles And HMMs
-More Sensitive-More Specific
L?K>R
AFDEFGHQIVLW
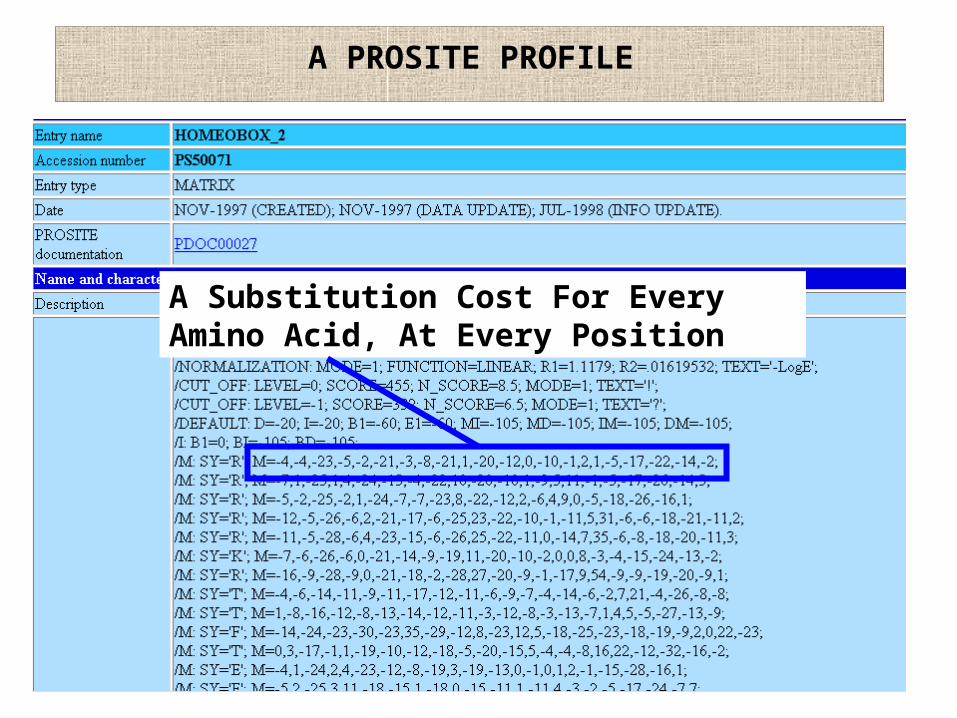
A PROSITE PROFILE
A Substitution Cost For Every Amino Acid, At Every Position
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
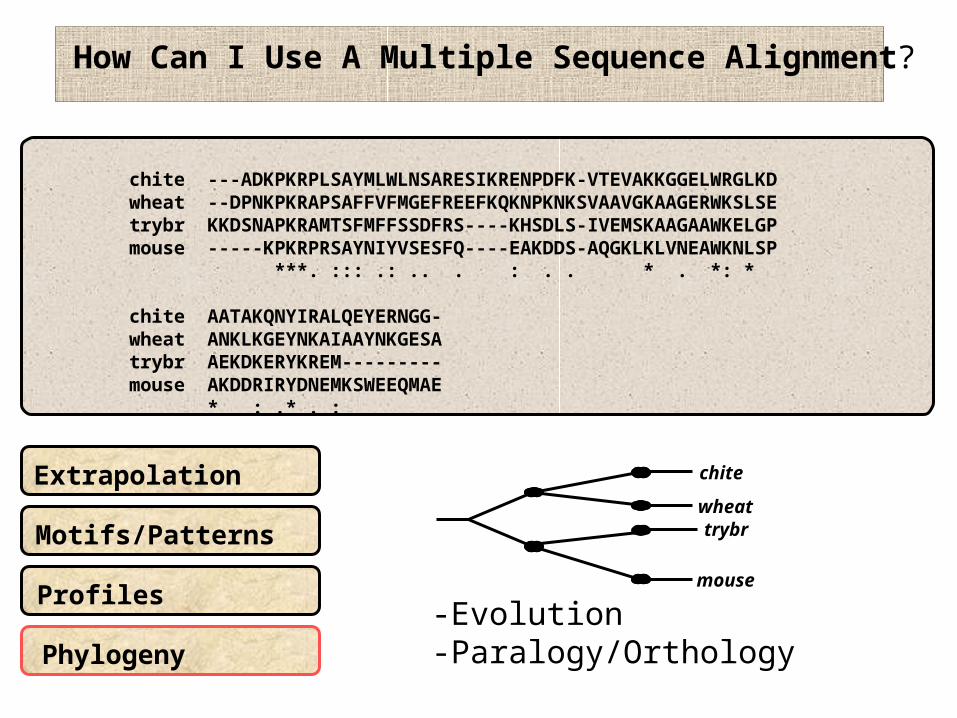
How Can I Use A Multiple Sequence Alignment?
Extrapolation
Motifs/Patterns
Phylogeny
chite
wheattrybr
mouse
-Evolution-Paralogy/Orthology
Profiles
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
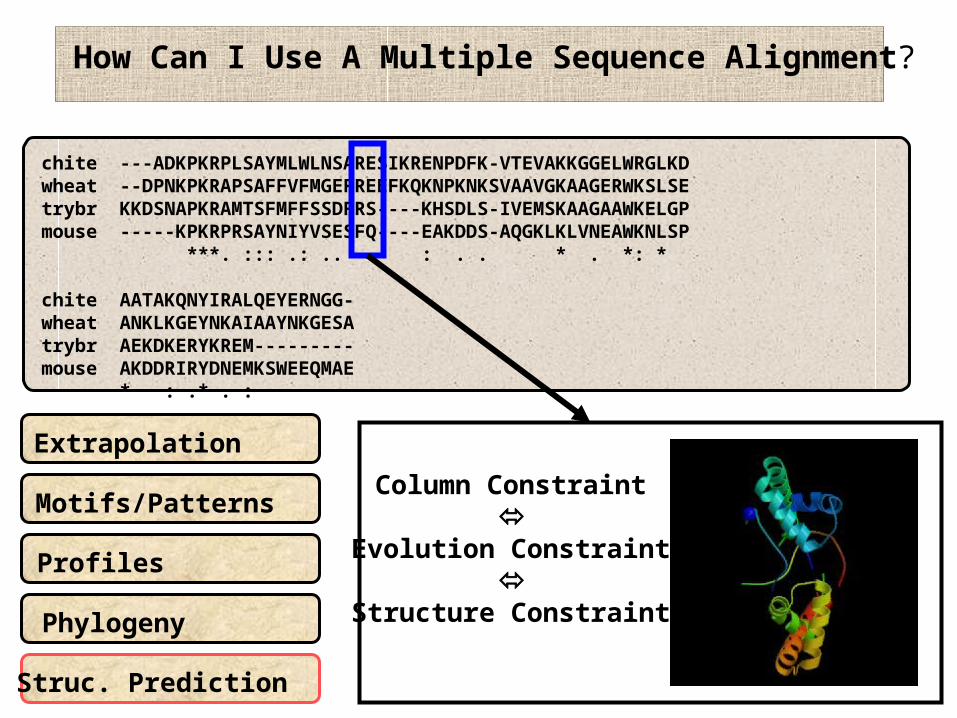
How Can I Use A Multiple Sequence Alignment?
Extrapolation
Motifs/Patterns
Phylogeny
Profiles
Struc. Prediction
Column Constraint
Evolution Constraint
Structure Constraint
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
How Can I Use A Multiple Sequence Alignment?
Extrapolation
Motifs/Patterns
Phylogeny
Profiles
Struc. Prediction
PsiPred OR PhD For secondary Structure Prediction: 75% Accurate.
Threading: is improving but is not yet as good.
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
How Can I Use A Multiple Sequence Alignment?
Automatic MultipleSequence Alignment methodsare not always perfect…
You know better…With your big BRAIN

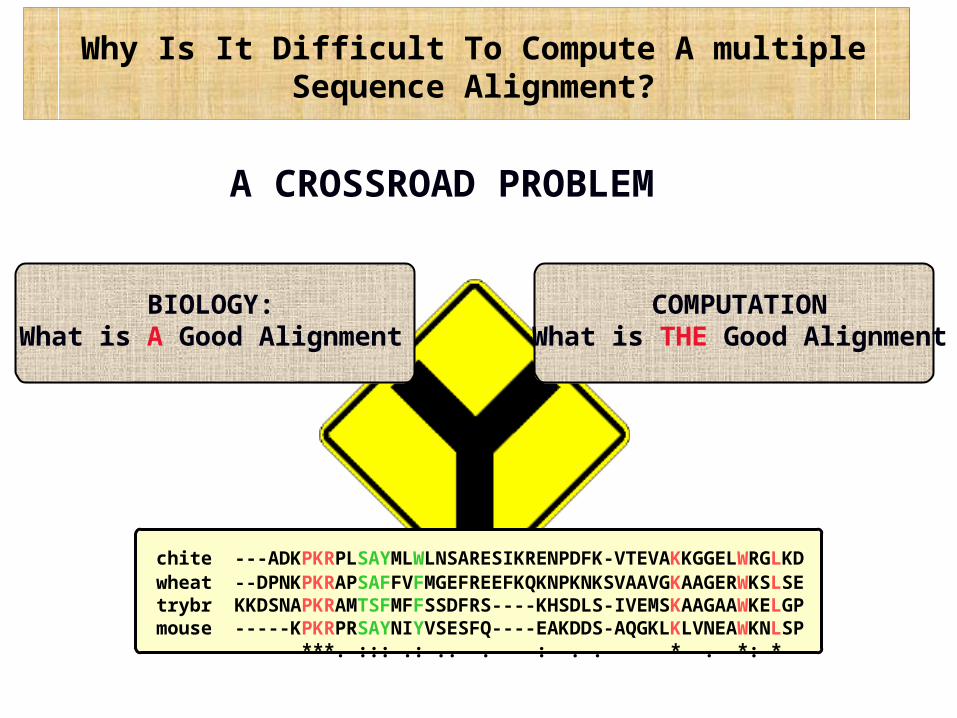
Why Is It Difficult To Compute A multiple Sequence Alignment?
A CROSSROAD PROBLEM
BIOLOGY:What is A Good Alignment
COMPUTATIONWhat is THE Good Alignment
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *

The Biological Problem.
Same as PairWise Alignment Problem
We do NOT know how Sequences Evolve.
We do NOT understand the Relation Between Structures and Sequences.
We would NOT recognize the Correct Alignment if we had it IN FRONT of our eyes…

The Biological Problem.The Charlie Chaplin Paradox

The Biological Problem.How to Evaluate an Alignment
-Substitution Matrix (Blosum)
-An Evaluation Function
AAACC
-Gap Penalties.
-A nice set of Sequences
A
A
A CSums of Pairs: Cost=6
C
Over-estimation of the Substitutions
Easy to compute

The COMPUTATIONAL Problem.Producing the Alignment
-Substitution Matrix (Blosum)
-An Evaluation Function
-Gap Penalties.
-A nice set of Sequences
-An Alignment Algorithm
GLOBAL Alignment
Will It Work?

HOW CAN I ALIGN MANY SEQUENCES
2 Globins =>1 Min

3 Globins =>2 hours
HOW CAN I ALIGN MANY SEQUENCES

4 Globins => 10 days
HOW CAN I ALIGN MANY SEQUENCES

5 Globins => 3 years
HOW CAN I ALIGN MANY SEQUENCES
6 Globins =>300 years
HOW CAN I ALIGN MANY SEQUENCES
!DHEA
Loaded
7 Globins =>30. 000 years
HOW CAN I ALIGN MANY SEQUENCES
Solidified Fossil,Old stuff

8 Globins =>3 Million years
HOW CAN I ALIGN MANY SEQUENCES

The Progressive Multiple Alignment
Algorithm(Clustal W)


Making An Alignment
Any Exact Method would be TOO SLOW
We will use a Heuristic Algorithm.
Progressive Alignment Algorithm is the most Popular
-Fast
-ClustalW
-Greedy Heuristic (No Guarranty).
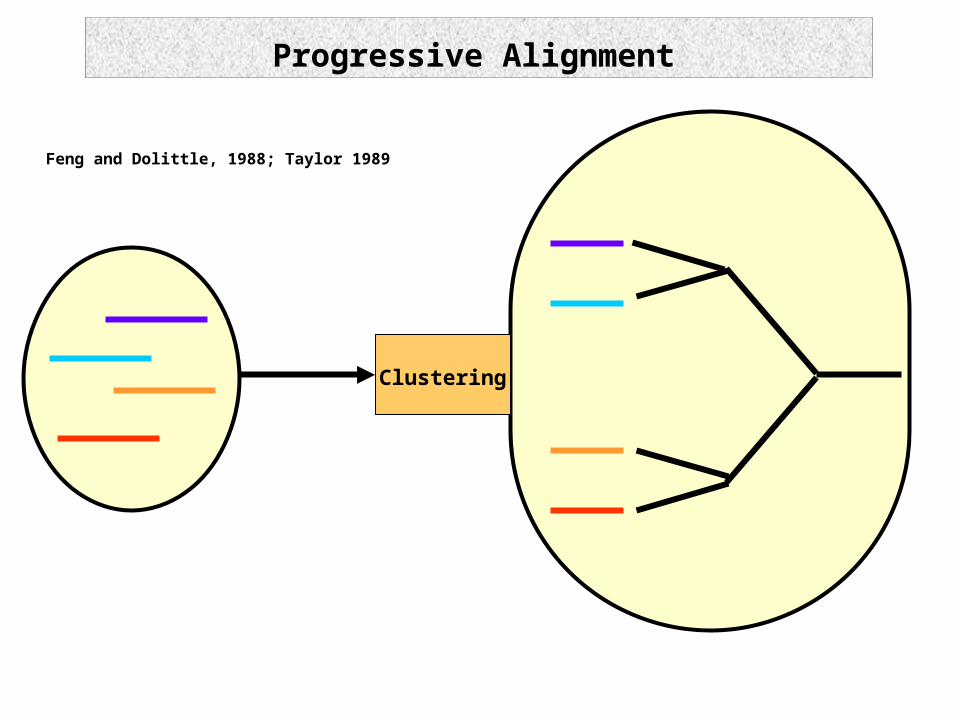
Progressive Alignment
Feng and Dolittle, 1988; Taylor 1989
Clustering
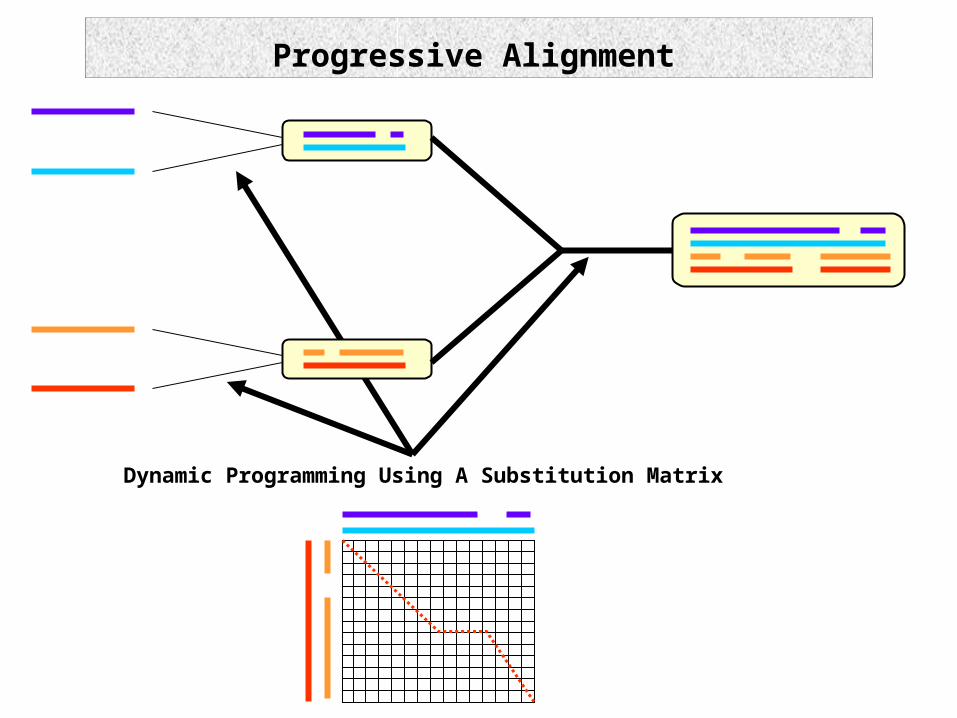
Dynamic Programming Using A Substitution Matrix
Progressive Alignment
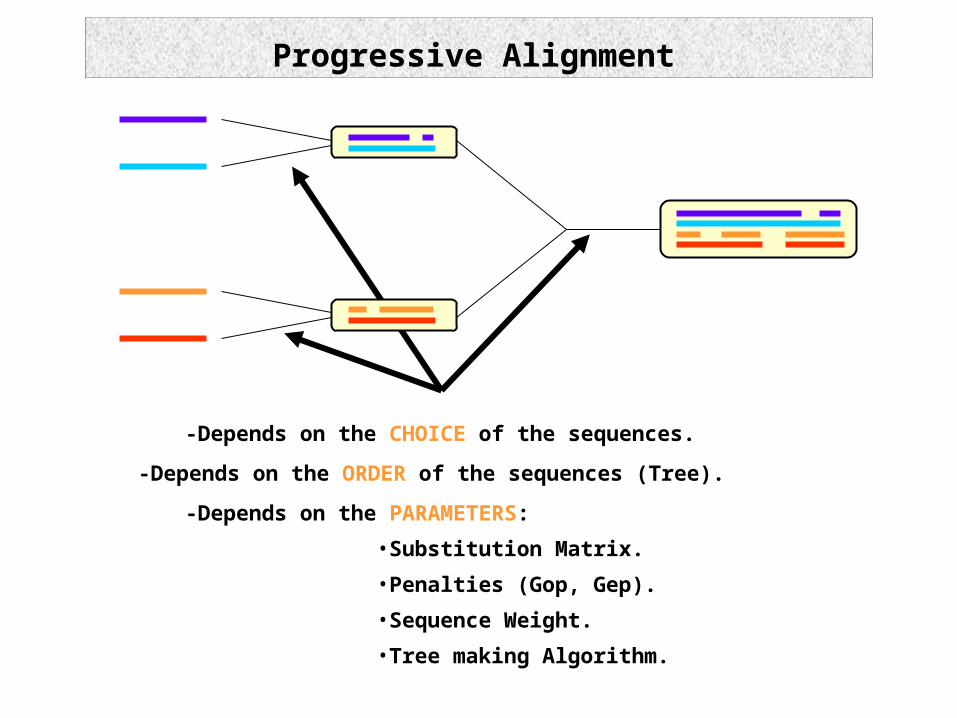
Progressive Alignment
-Depends on the ORDER of the sequences (Tree).
-Depends on the CHOICE of the sequences.
-Depends on the PARAMETERS:
•Substitution Matrix.
•Penalties (Gop, Gep).
•Sequence Weight.
•Tree making Algorithm.

Progressive AlignmentWhen Does It Work
Works Well When Phylogeny is Dense
No outlayer Sequence.
Image: River Crossing

SeqA GARFIELD THE LAST FA-T CATSeqB GARFIELD THE FAST CA-T ---SeqC GARFIELD THE VERY FAST CATSeqD -------- THE ---- FA-T CAT
CLUSTALW (Score=20, Gop=-1, Gep=0, M=1)
SeqA GARFIELD THE LAST FA-T CATSeqB GARFIELD THE FAST ---- CATSeqC GARFIELD THE VERY FAST CATSeqD -------- THE ---- FA-T CAT
CORRECT (Score=24)
Progressive AlignmentWhen Doesn’t It Work
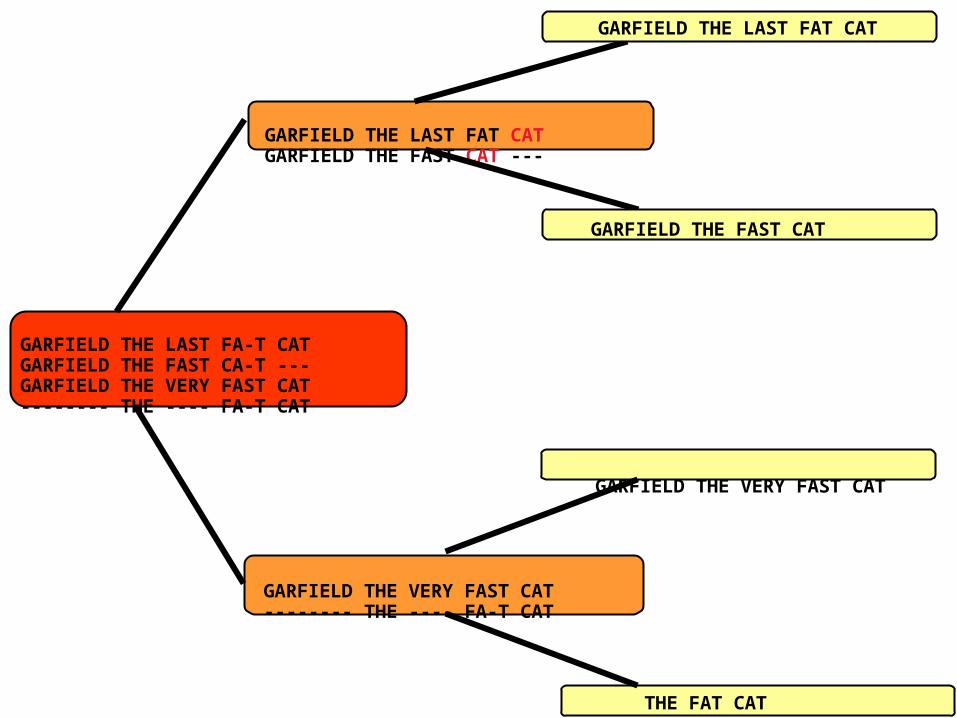
GARFIELD THE LAST FAT CATGARFIELD THE FAST CAT ---
GARFIELD THE LAST FAT CAT
GARFIELD THE FAST CAT
GARFIELD THE VERY FAST CAT
THE FAT CAT
GARFIELD THE VERY FAST CAT-------- THE ---- FA-T CAT
GARFIELD THE LAST FA-T CATGARFIELD THE FAST CA-T ---GARFIELD THE VERY FAST CAT-------- THE ---- FA-T CAT

Building the Right Multiple Sequence
Alignment.
Recognizing The Right Sequences When you Meet Them…

Gathering Sequences: BLAST

Common Mistake:Sequences Too Closely Related
PRVA_MACFU SMTDLLNAEDIKKAVGAFSAIDSFDHKKFFQMVGLKKKSADDVKKVFHILDKDKSGFIEEPRVA_HUMAN SMTDLLNAEDIKKAVGAFSATDSFDHKKFFQMVGLKKKSADDVKKVFHMLDKDKSGFIEEPRVA_GERSP SMTDLLSAEDIKKAIGAFAAADSFDHKKFFQMVGLKKKTPDDVKKVFHILDKDKSGFIEEPRVA_MOUSE SMTDVLSAEDIKKAIGAFAAADSFDHKKFFQMVGLKKKNPDEVKKVFHILDKDKSGFIEEPRVA_RAT SMTDLLSAEDIKKAIGAFTAADSFDHKKFFQMVGLKKKSADDVKKVFHILDKDKSGFIEEPRVA_RABIT AMTELLNAEDIKKAIGAFAAAESFDHKKFFQMVGLKKKSTEDVKKVFHILDKDKSGFIEE :**::*.*******:***:* :****************..::******:***********
PRVA_MACFU DELGFILKGFSPDARDLSAKETKTLMAAGDKDGDGKIGVDEFSTLVAESPRVA_HUMAN DELGFILKGFSPDARDLSAKETKMLMAAGDKDGDGKIGVDEFSTLVAESPRVA_GERSP DELGFILKGFSSDARDLSAKETKTLLAAGDKDGDGKIGVEEFSTLVSESPRVA_MOUSE DELGSILKGFSSDARDLSAKETKTLLAAGDKDGDGKIGVEEFSTLVAESPRVA_RAT DELGSILKGFSSDARDLSAKETKTLMAAGDKDGDGKIGVEEFSTLVAESPRVA_RABIT EELGFILKGFSPDARDLSVKETKTLMAAGDKDGDGKIGADEFSTLVSES :*** ******.******.**** *:************.:******:**
-IDENTICAL SEQUENCES BRING NO INFORMATION FOR THE MULTIPLE SEQUENCE ALIGNMENT
-MULTIPLE SEQUENCE ALIGNMENTS THRIVE ON DIVERSITY…
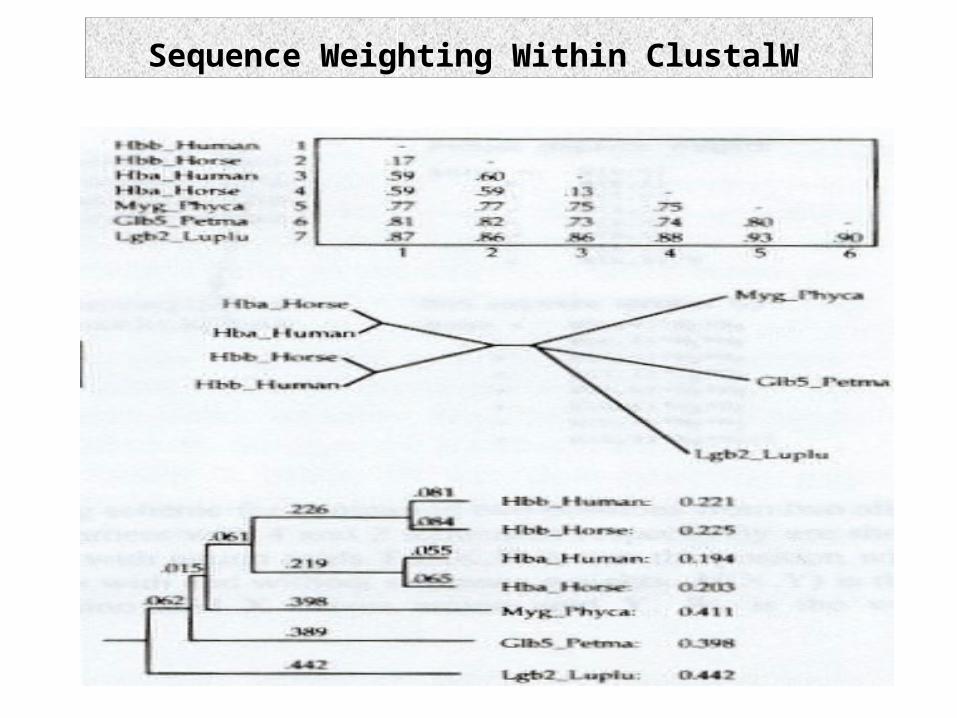
Sequence Weighting Within ClustalW

Selecting Diverse Sequences (Opus II)
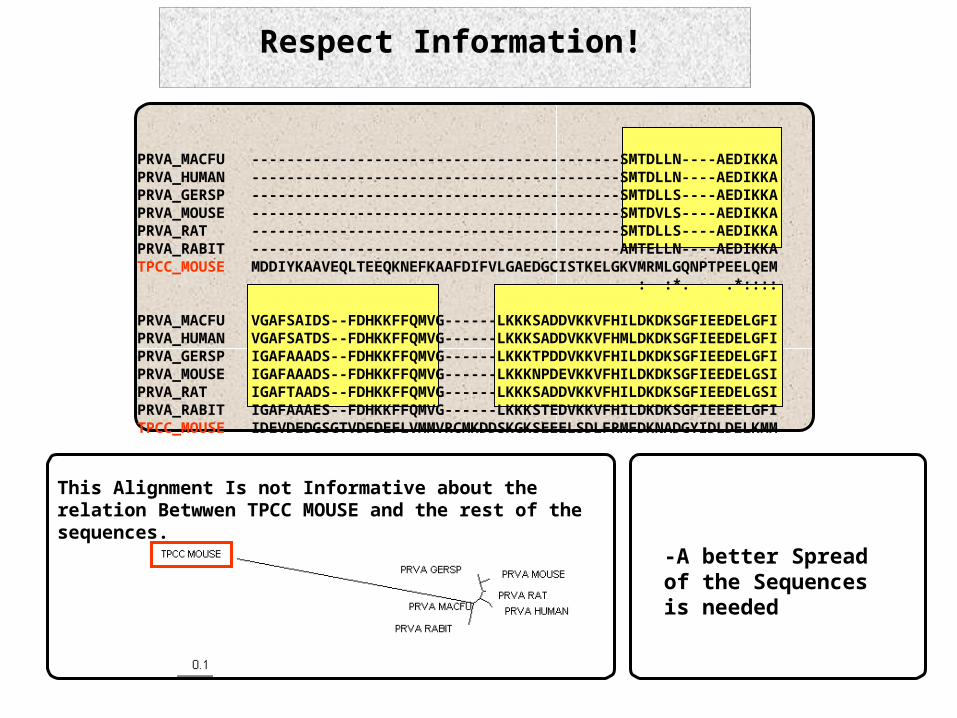
Respect Information!
This Alignment Is not Informative about the relation Betwwen TPCC MOUSE and the rest of the sequences.
PRVA_MACFU ------------------------------------------SMTDLLN----AEDIKKAPRVA_HUMAN ------------------------------------------SMTDLLN----AEDIKKAPRVA_GERSP ------------------------------------------SMTDLLS----AEDIKKAPRVA_MOUSE ------------------------------------------SMTDVLS----AEDIKKAPRVA_RAT ------------------------------------------SMTDLLS----AEDIKKAPRVA_RABIT ------------------------------------------AMTELLN----AEDIKKATPCC_MOUSE MDDIYKAAVEQLTEEQKNEFKAAFDIFVLGAEDGCISTKELGKVMRMLGQNPTPEELQEM : :*. .*::::
PRVA_MACFU VGAFSAIDS--FDHKKFFQMVG------LKKKSADDVKKVFHILDKDKSGFIEEDELGFIPRVA_HUMAN VGAFSATDS--FDHKKFFQMVG------LKKKSADDVKKVFHMLDKDKSGFIEEDELGFIPRVA_GERSP IGAFAAADS--FDHKKFFQMVG------LKKKTPDDVKKVFHILDKDKSGFIEEDELGFIPRVA_MOUSE IGAFAAADS--FDHKKFFQMVG------LKKKNPDEVKKVFHILDKDKSGFIEEDELGSIPRVA_RAT IGAFTAADS--FDHKKFFQMVG------LKKKSADDVKKVFHILDKDKSGFIEEDELGSIPRVA_RABIT IGAFAAAES--FDHKKFFQMVG------LKKKSTEDVKKVFHILDKDKSGFIEEEELGFITPCC_MOUSE IDEVDEDGSGTVDFDEFLVMMVRCMKDDSKGKSEEELSDLFRMFDKNADGYIDLDELKMM
-A better Spread of the Sequences is needed

Selecting Diverse Sequences (Opus II)
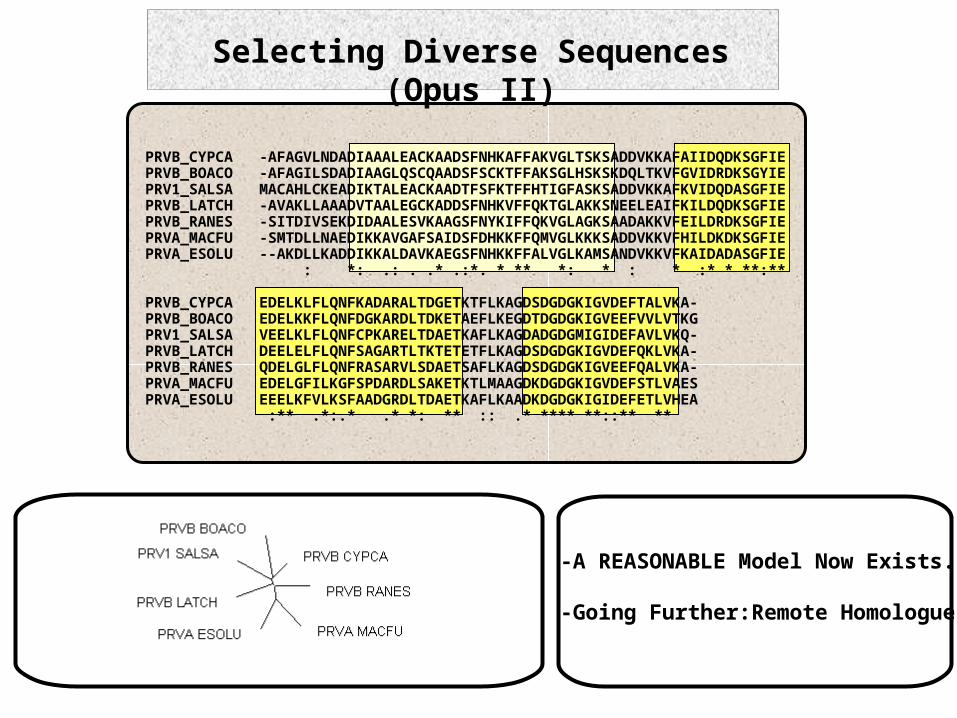
Selecting Diverse Sequences (Opus II)
PRVB_CYPCA -AFAGVLNDADIAAALEACKAADSFNHKAFFAKVGLTSKSADDVKKAFAIIDQDKSGFIEPRVB_BOACO -AFAGILSDADIAAGLQSCQAADSFSCKTFFAKSGLHSKSKDQLTKVFGVIDRDKSGYIEPRV1_SALSA MACAHLCKEADIKTALEACKAADTFSFKTFFHTIGFASKSADDVKKAFKVIDQDASGFIEPRVB_LATCH -AVAKLLAAADVTAALEGCKADDSFNHKVFFQKTGLAKKSNEELEAIFKILDQDKSGFIEPRVB_RANES -SITDIVSEKDIDAALESVKAAGSFNYKIFFQKVGLAGKSAADAKKVFEILDRDKSGFIEPRVA_MACFU -SMTDLLNAEDIKKAVGAFSAIDSFDHKKFFQMVGLKKKSADDVKKVFHILDKDKSGFIEPRVA_ESOLU --AKDLLKADDIKKALDAVKAEGSFNHKKFFALVGLKAMSANDVKKVFKAIDADASGFIE : *: .: . .* .:*. * ** *: * : * :* * **:**
PRVB_CYPCA EDELKLFLQNFKADARALTDGETKTFLKAGDSDGDGKIGVDEFTALVKA-PRVB_BOACO EDELKKFLQNFDGKARDLTDKETAEFLKEGDTDGDGKIGVEEFVVLVTKGPRV1_SALSA VEELKLFLQNFCPKARELTDAETKAFLKAGDADGDGMIGIDEFAVLVKQ-PRVB_LATCH DEELELFLQNFSAGARTLTKTETETFLKAGDSDGDGKIGVDEFQKLVKA-PRVB_RANES QDELGLFLQNFRASARVLSDAETSAFLKAGDSDGDGKIGVEEFQALVKA-PRVA_MACFU EDELGFILKGFSPDARDLSAKETKTLMAAGDKDGDGKIGVDEFSTLVAESPRVA_ESOLU EEELKFVLKSFAADGRDLTDAETKAFLKAADKDGDGKIGIDEFETLVHEA :** .*:.* .* *: ** :: .* **** **::** **
-A REASONABLE Model Now Exists.
-Going Further:Remote Homologues.

Aligning Remote Homologues
PRVA_MACFU ------------------------------------------SMTDLLNA----EDIKKAPRVA_ESOLU -------------------------------------------AKDLLKA----DDIKKAPRVB_CYPCA ------------------------------------------AFAGVLND----ADIAAAPRVB_BOACO ------------------------------------------AFAGILSD----ADIAAGPRV1_SALSA -----------------------------------------MACAHLCKE----ADIKTAPRVB_LATCH ------------------------------------------AVAKLLAA----ADVTAAPRVB_RANES ------------------------------------------SITDIVSE----KDIDAATPCS_RABIT -TDQQAEARSYLSEEMIAEFKAAFDMFDADGG-GDISVKELGTVMRMLGQTPTKEELDAITPCS_PIG -TDQQAEARSYLSEEMIAEFKAAFDMFDADGG-GDISVKELGTVMRMLGQTPTKEELDAITPCC_MOUSE MDDIYKAAVEQLTEEQKNEFKAAFDIFVLGAEDGCISTKELGKVMRMLGQNPTPEELQEM : ::
PRVA_MACFU VGAFSAIDS--FDHKKFFQMVG------LKKKSADDVKKVFHILDKDKSGFIEEDELGFIPRVA_ESOLU LDAVKAEGS--FNHKKFFALVG------LKAMSANDVKKVFKAIDADASGFIEEEELKFVPRVB_CYPCA LEACKAADS--FNHKAFFAKVG------LTSKSADDVKKAFAIIDQDKSGFIEEDELKLFPRVB_BOACO LQSCQAADS--FSCKTFFAKSG------LHSKSKDQLTKVFGVIDRDKSGYIEEDELKKFPRV1_SALSA LEACKAADT--FSFKTFFHTIG------FASKSADDVKKAFKVIDQDASGFIEVEELKLFPRVB_LATCH LEGCKADDS--FNHKVFFQKTG------LAKKSNEELEAIFKILDQDKSGFIEDEELELFPRVB_RANES LESVKAAGS--FNYKIFFQKVG------LAGKSAADAKKVFEILDRDKSGFIEQDELGLFTPCS_RABIT IEEVDEDGSGTIDFEEFLVMMVRQMKEDAKGKSEEELAECFRIFDRNADGYIDAEELAEITPCS_PIG IEEVDEDGSGTIDFEEFLVMMVRQMKEDAKGKSEEELAECFRIFDRNMDGYIDAEELAEITPCC_MOUSE IDEVDEDGSGTVDFDEFLVMMVRCMKDDSKGKSEEELSDLFRMFDKNADGYIDLDELKMM : . .: .. . *: * : * :* : .*:*: :** .
PRVA_MACFU LKGFSPDARDLSAKETKTLMAAGDKDGDGKIGVDEFSTLVAES-PRVA_ESOLU LKSFAADGRDLTDAETKAFLKAADKDGDGKIGIDEFETLVHEA-PRVB_CYPCA LQNFKADARALTDGETKTFLKAGDSDGDGKIGVDEFTALVKA--PRVB_BOACO LQNFDGKARDLTDKETAEFLKEGDTDGDGKIGVEEFVVLVTKG-PRV1_SALSA LQNFCPKARELTDAETKAFLKAGDADGDGMIGIDEFAVLVKQ--PRVB_LATCH LQNFSAGARTLTKTETETFLKAGDSDGDGKIGVDEFQKLVKA--PRVB_RANES LQNFRASARVLSDAETSAFLKAGDSDGDGKIGVEEFQALVKA--TPCS_RABIT FR---ASGEHVTDEEIESLMKDGDKNNDGRIDFDEFLKMMEGVQTPCS_PIG FR---ASGEHVTDEEIESIMKDGDKNNDGRIDFDEFLKMMEGVQTPCC_MOUSE LQ---ATGETITEDDIEELMKDGDKNNDGRIDYDEFLEFMKGVE :: .. :: : :: .* :.** *. :** ::

SomeGuideline
s…

Do Not Use Two Many Sequences…

Reading Your Alignment
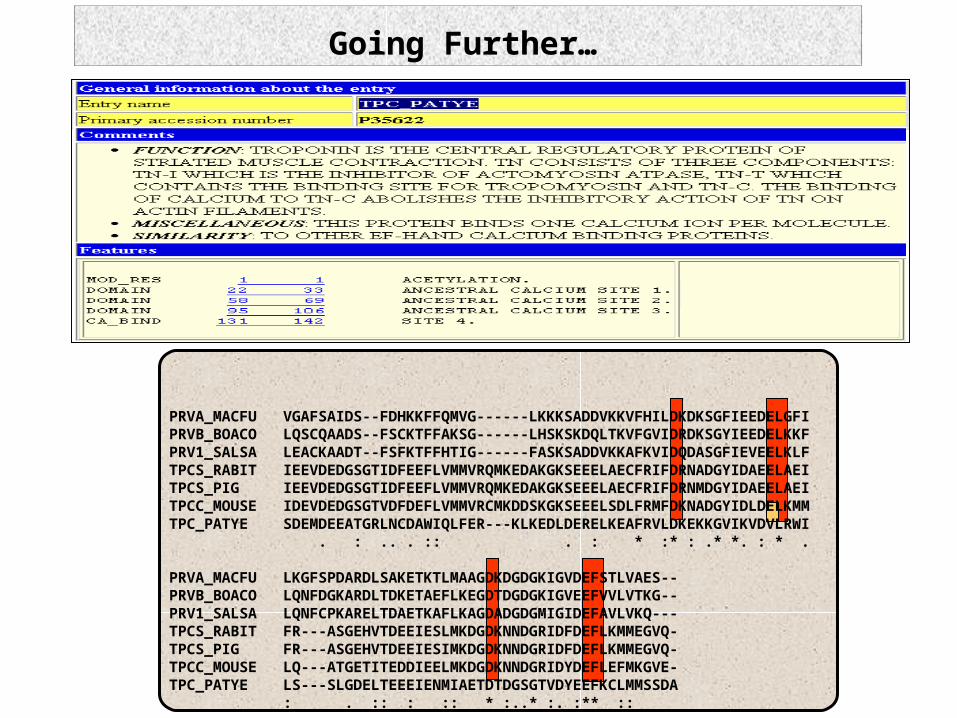
Going Further…
PRVA_MACFU VGAFSAIDS--FDHKKFFQMVG------LKKKSADDVKKVFHILDKDKSGFIEEDELGFIPRVB_BOACO LQSCQAADS--FSCKTFFAKSG------LHSKSKDQLTKVFGVIDRDKSGYIEEDELKKFPRV1_SALSA LEACKAADT--FSFKTFFHTIG------FASKSADDVKKAFKVIDQDASGFIEVEELKLFTPCS_RABIT IEEVDEDGSGTIDFEEFLVMMVRQMKEDAKGKSEEELAECFRIFDRNADGYIDAEELAEITPCS_PIG IEEVDEDGSGTIDFEEFLVMMVRQMKEDAKGKSEEELAECFRIFDRNMDGYIDAEELAEITPCC_MOUSE IDEVDEDGSGTVDFDEFLVMMVRCMKDDSKGKSEEELSDLFRMFDKNADGYIDLDELKMMTPC_PATYE SDEMDEEATGRLNCDAWIQLFER---KLKEDLDERELKEAFRVLDKEKKGVIKVDVLRWI . : .. . :: . : * :* : .* *. : * .
PRVA_MACFU LKGFSPDARDLSAKETKTLMAAGDKDGDGKIGVDEFSTLVAES--PRVB_BOACO LQNFDGKARDLTDKETAEFLKEGDTDGDGKIGVEEFVVLVTKG--PRV1_SALSA LQNFCPKARELTDAETKAFLKAGDADGDGMIGIDEFAVLVKQ---TPCS_RABIT FR---ASGEHVTDEEIESLMKDGDKNNDGRIDFDEFLKMMEGVQ-TPCS_PIG FR---ASGEHVTDEEIESIMKDGDKNNDGRIDFDEFLKMMEGVQ-TPCC_MOUSE LQ---ATGETITEDDIEELMKDGDKNNDGRIDYDEFLEFMKGVE-TPC_PATYE LS---SLGDELTEEEIENMIAETDTDGSGTVDYEEFKCLMMSSDA : . :: : :: * :..* :. :** ::

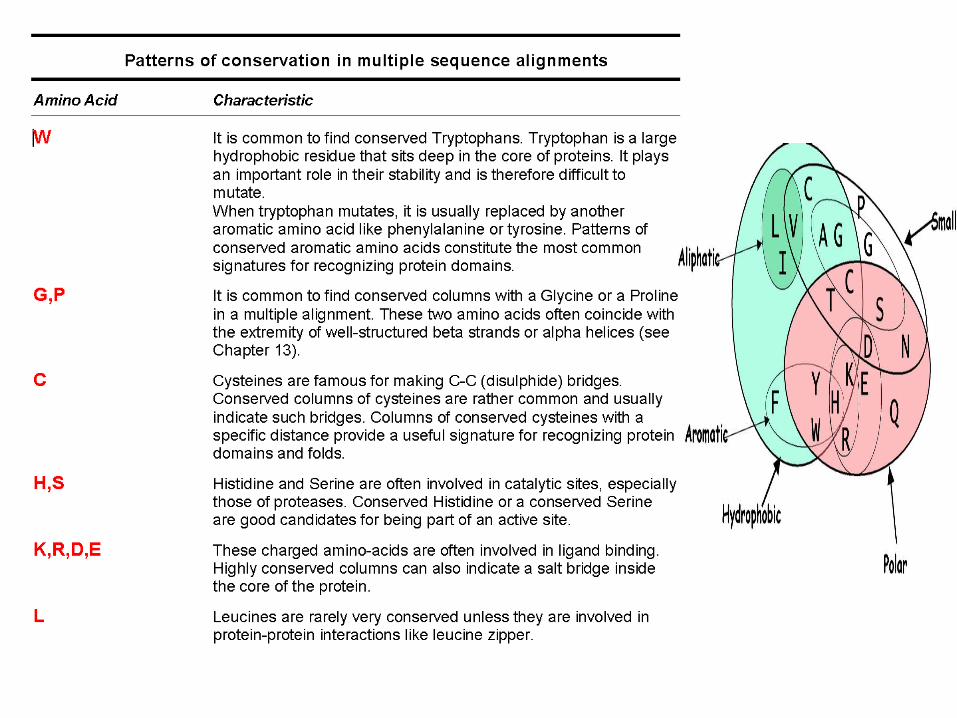
WHAT MAKES A GOOD ALIGNMENT…
-THE MORE DIVERGEANT THE SEQUENCES, THE BETTER
-THE FEWER INDELS, THE BETTER
-NICE UNGAPPED BLOCKS SEPARATED WITH INDELS
-DIFFERENT CLASSES OF RESIDUES WITHIN A BLOCK:
•Completely Conserved•Conserved For Size and Hydropathy•Conserved For Size or Hydropathy
-THE ULTIMATE EVALUATION IS A MATTER OF PERSONNAL JUDGEMENT AND KNOWLEDGE.


Potential Difficulties

DO NOT OVERTUNE!!!
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :
DO NOT PLAY WITH PARAMETERS IF YOU KNOW THE ALIGNMENT YOU WANT: MAKE IT YOURSELF!
chite ---ADKPKRPL-SAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAP-SAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPR-SAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. :*: .: .. . : . . * . *: *
chite AATAKQNYIRALQEYERNGG-wheat ANKLKGEYNKAIAAYNKGESAtrybr AEKDKERYKREM---------mouse AKDDRIRYDNEMKSWEEQMAE * : .* . :

TUNING or NOT TUNING!!!
-MOST METHODS ARE TUNED FOR WORKING WELL ON AVERAGE
-PARAMETERS BEHAVIOUR DO NOT NECESSARILY FOLLOW THE THEORY (i.e. Substitution Matrices).
-A GOOD ALIGNMENT IS USUALLY ROBUST(i.e. Changes little).
-TUNE IF YOU WANT TO CONVINCE YOURSELF.
-PARAMETERS TO TUNE USUALLY INCLUDE:•GOP/ GEP•MATRIX•SENSITIVITY Vs SPEED
GOP
GEP
Substitution Matrices (Etzold and al. 1993)
Gonnet 61.7 %Blosum50 59.7 %
Pam250 59.2 %

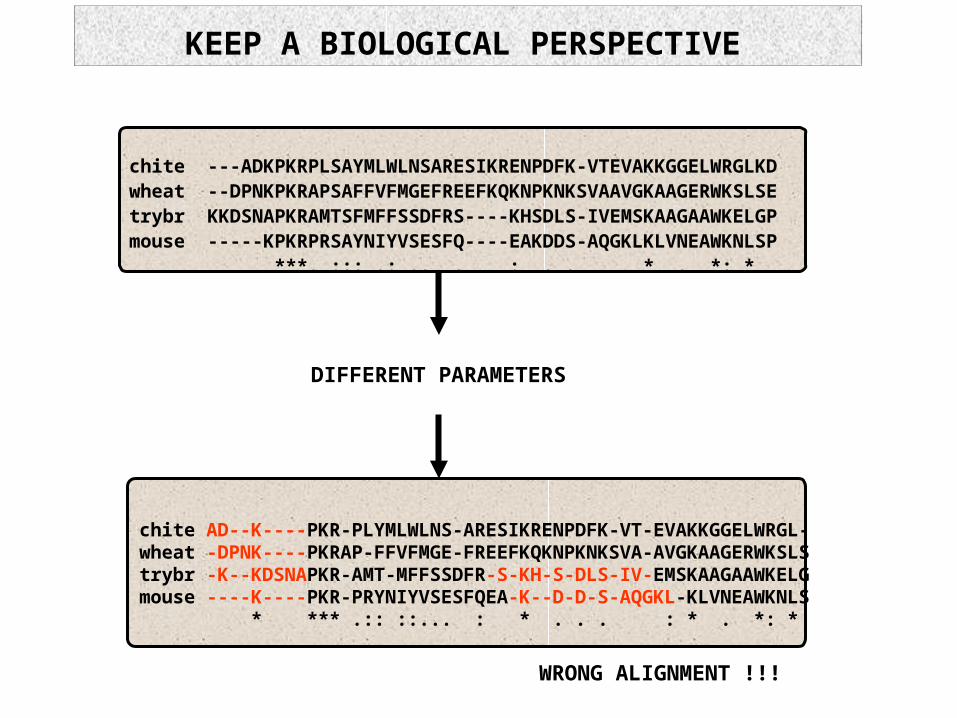
KEEP A BIOLOGICAL PERSPECTIVE
chite ---ADKPKRPLSAYMLWLNSARESIKRENPDFK-VTEVAKKGGELWRGLKDwheat --DPNKPKRAPSAFFVFMGEFREEFKQKNPKNKSVAAVGKAAGERWKSLSEtrybr KKDSNAPKRAMTSFMFFSSDFRS----KHSDLS-IVEMSKAAGAAWKELGPmouse -----KPKRPRSAYNIYVSESFQ----EAKDDS-AQGKLKLVNEAWKNLSP ***. ::: .: .. . : . . * . *: *
chite AD--K----PKR-PLYMLWLNS-ARESIKRENPDFK-VT-EVAKKGGELWRGL- wheat -DPNK----PKRAP-FFVFMGE-FREEFKQKNPKNKSVA-AVGKAAGERWKSLStrybr -K--KDSNAPKR-AMT-MFFSSDFR-S-KH-S-DLS-IV-EMSKAAGAAWKELG mouse ----K----PKR-PRYNIYVSESFQEA-K--D-D-S-AQGKL-KLVNEAWKNLS * *** .:: ::... : * . . . : * . *: *
DIFFERENT PARAMETERS
WRONG ALIGNMENT !!!

REPEATS
THERE IS A PROBLEM WHEN TWO SEQUENCES DO NOT CONTAIN THE SAME NUMBER OF REPEATS
IT IS THEN BETTER TO MANUALLY EXTRACT THE REPEATS AND TO ALIGN THEM. INDIVIDUAL REPEATS CAN BE RECOGNIZED USING DOTTER

Naming Your Sequences The Right Way

What Are The AvailableMethods
???
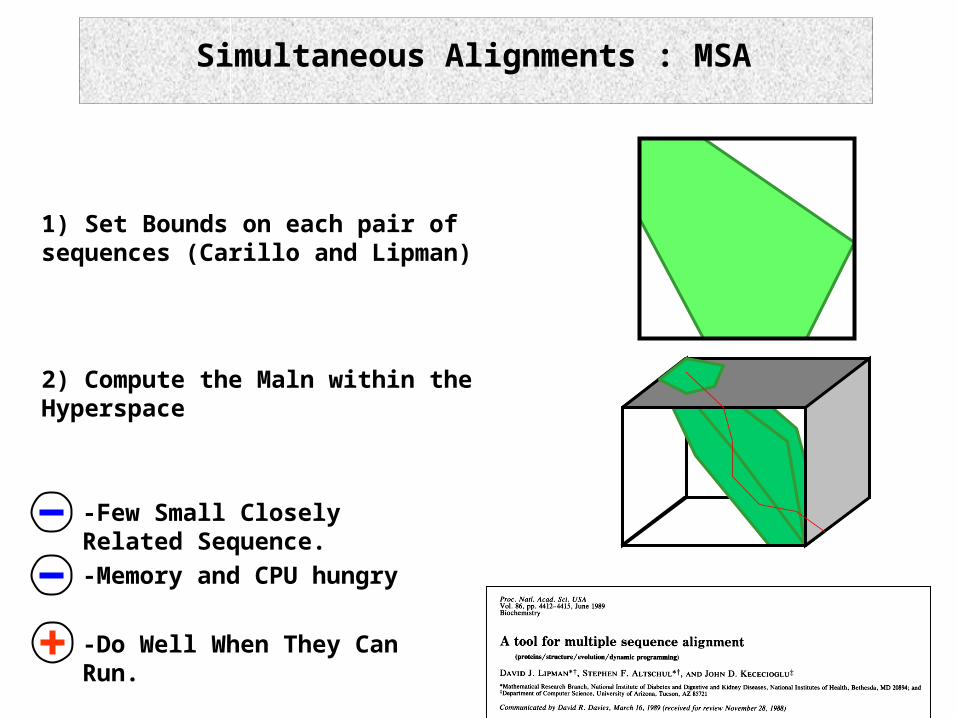
Simultaneous Alignments : MSA
1) Set Bounds on each pair of sequences (Carillo and Lipman)
2) Compute the Maln within the Hyperspace
-Few Small Closely Related Sequence.
-Do Well When They Can Run.
-Memory and CPU hungry
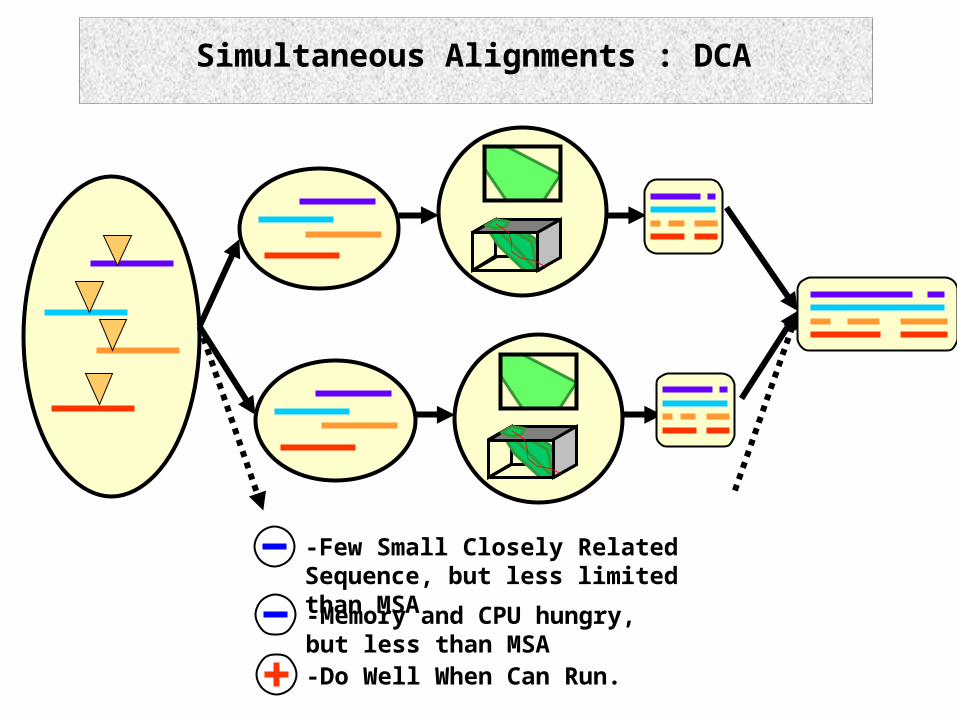
Simultaneous Alignments : DCA
-Few Small Closely Related Sequence, but less limited than MSA
-Do Well When Can Run.
-Memory and CPU hungry, but less than MSA
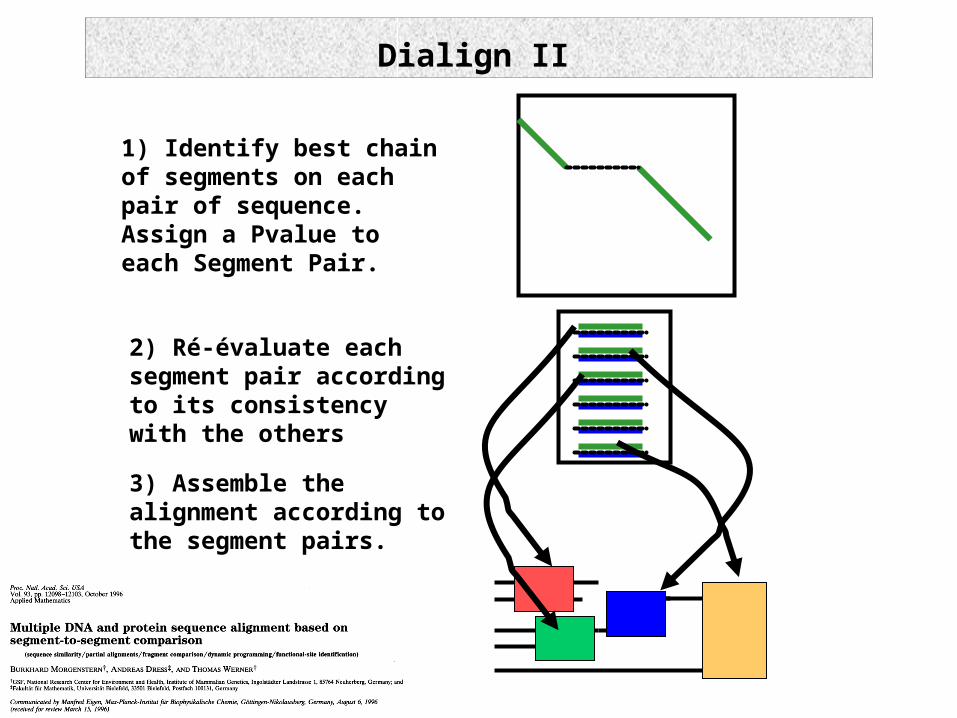
Dialign II
1) Identify best chain of segments on each pair of sequence. Assign a Pvalue to each Segment Pair.
3) Assemble the alignment according to the segment pairs.
2) Ré-évaluate each segment pair according to its consistency with the others
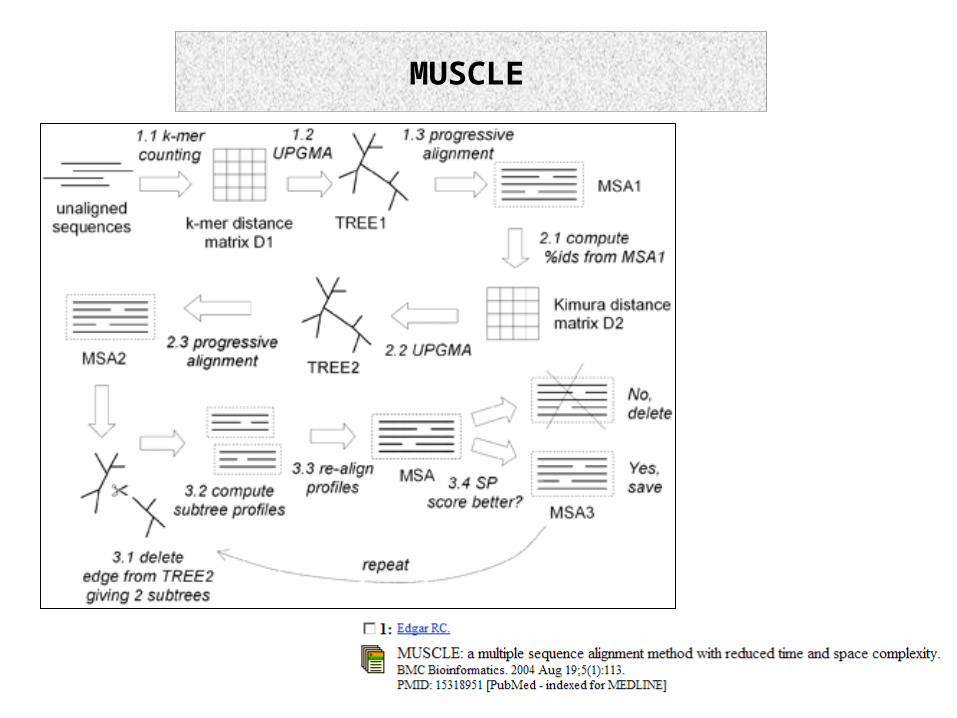
Muscle
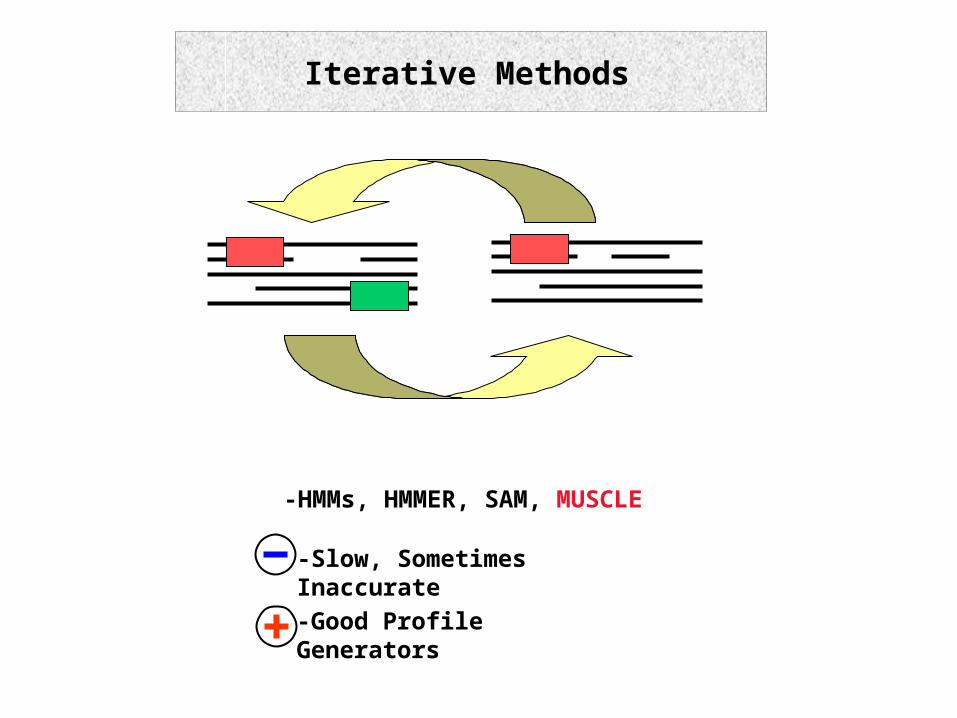
7.16.1 ProgressiveIterative Methods
-HMMs, HMMER, SAM, MUSCLE
-Slow, Sometimes Inaccurate-Good Profile Generators
7.16.1 Progressive
MUSCLE
phylogenomics.berkeley.edu/cgi-bin/muscle/input_muscle.py
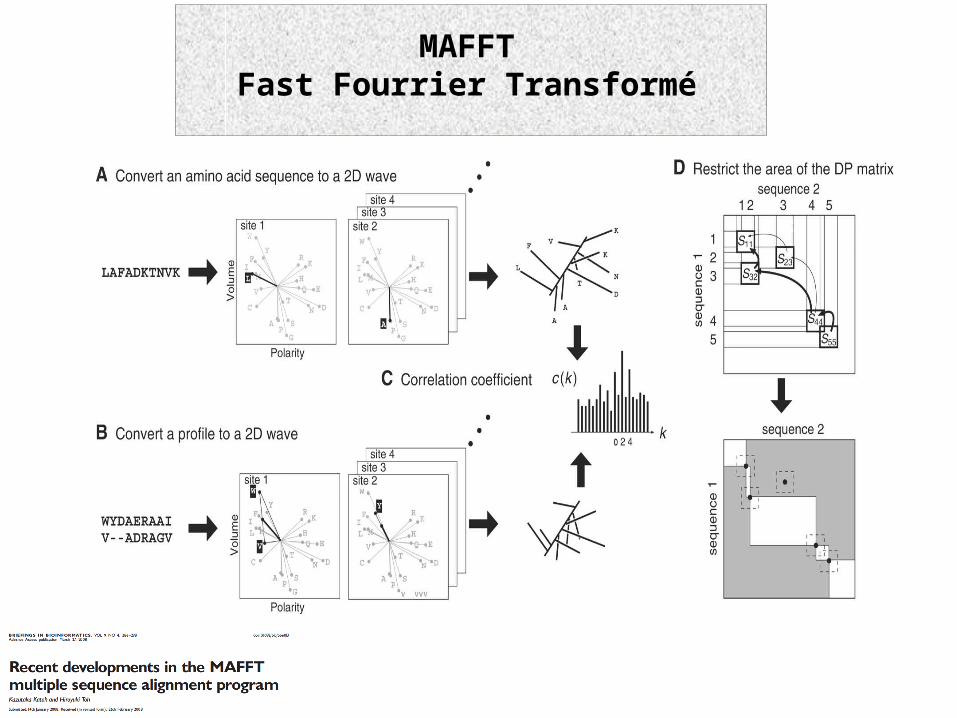
MAFFTFast Fourrier Transformé
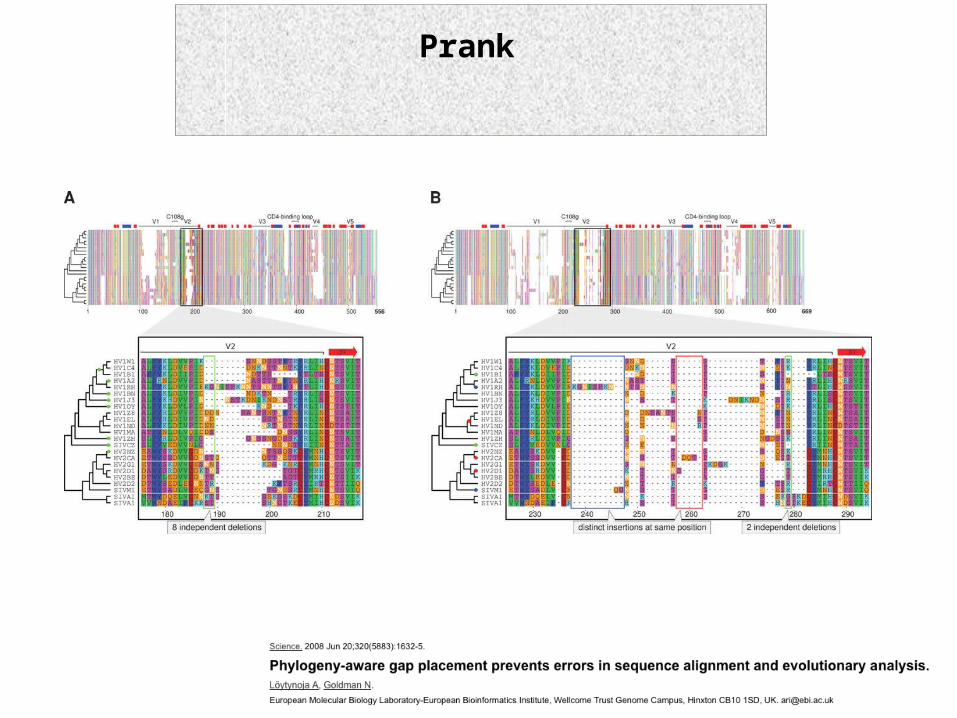
Prank
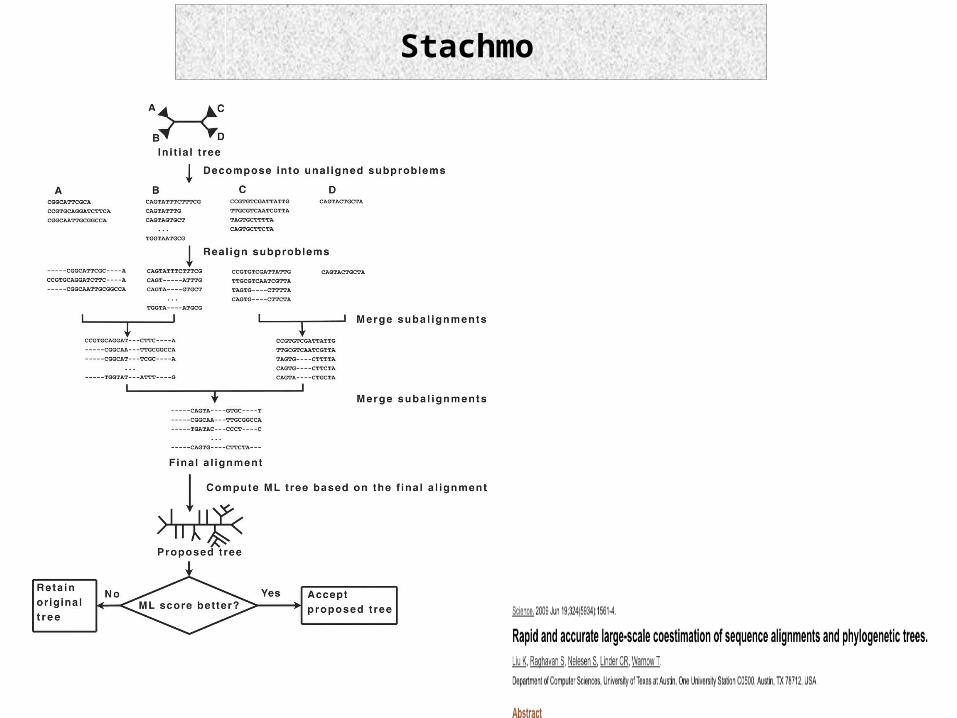
Stachmo
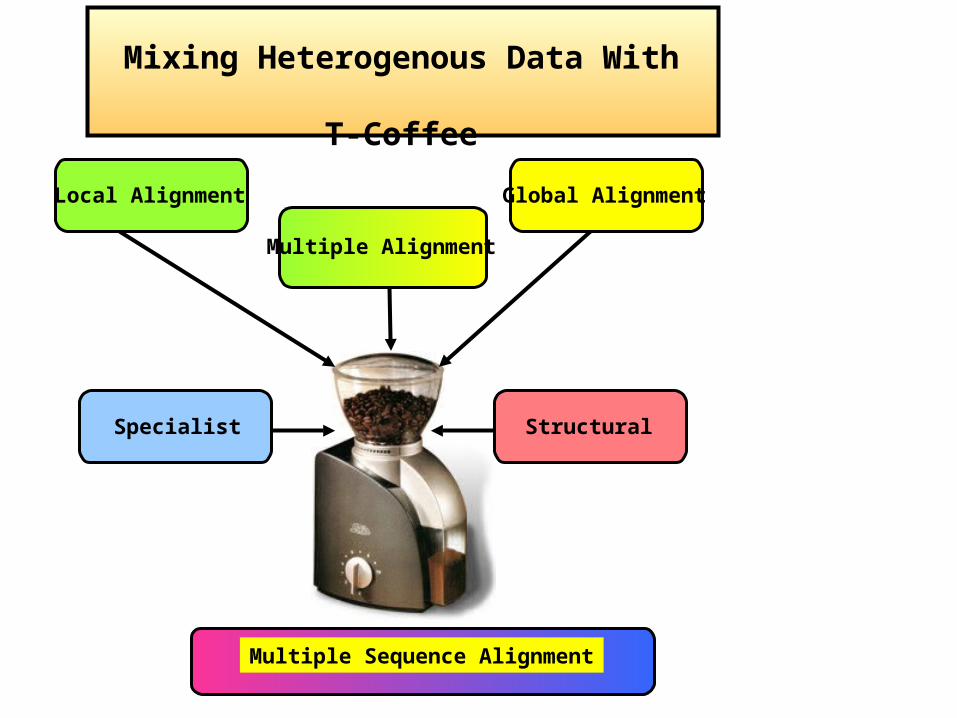
Mixing Heterogenous Data With
T-Coffee
Local Alignment Global Alignment
Multiple Sequence Alignment
Multiple Alignment
StructuralSpecialist
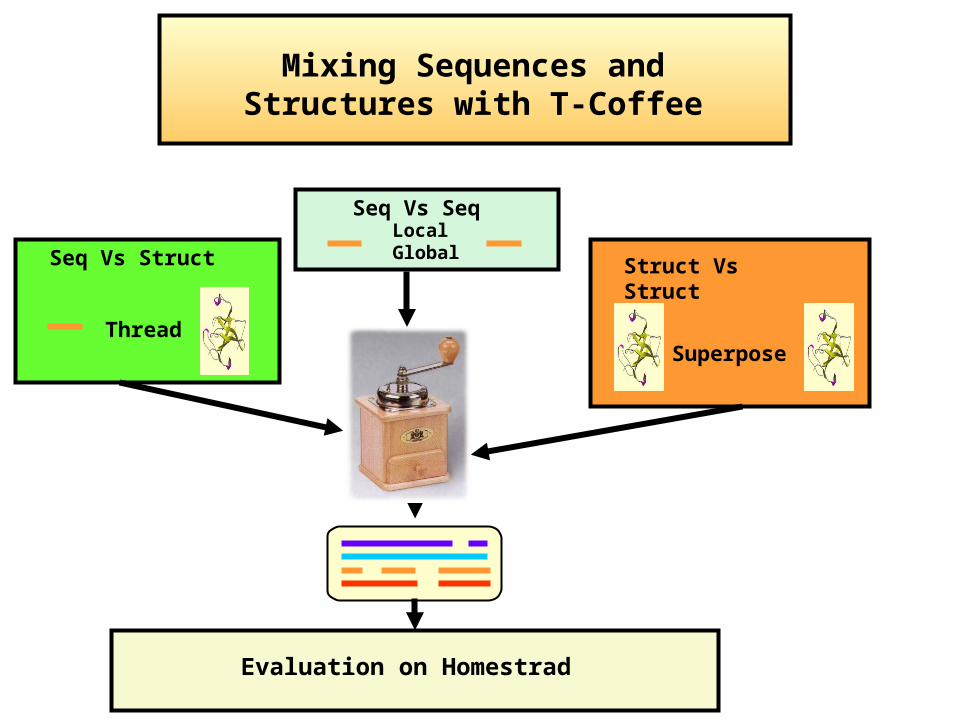
Struct Vs StructSeq Vs Struct
Thread
Evaluation on Homestrad
Superpose
Seq Vs SeqLocalGlobal
Mixing Sequences and Structures with T-Coffee
www.tcoffee.org

What is The Best Method
?
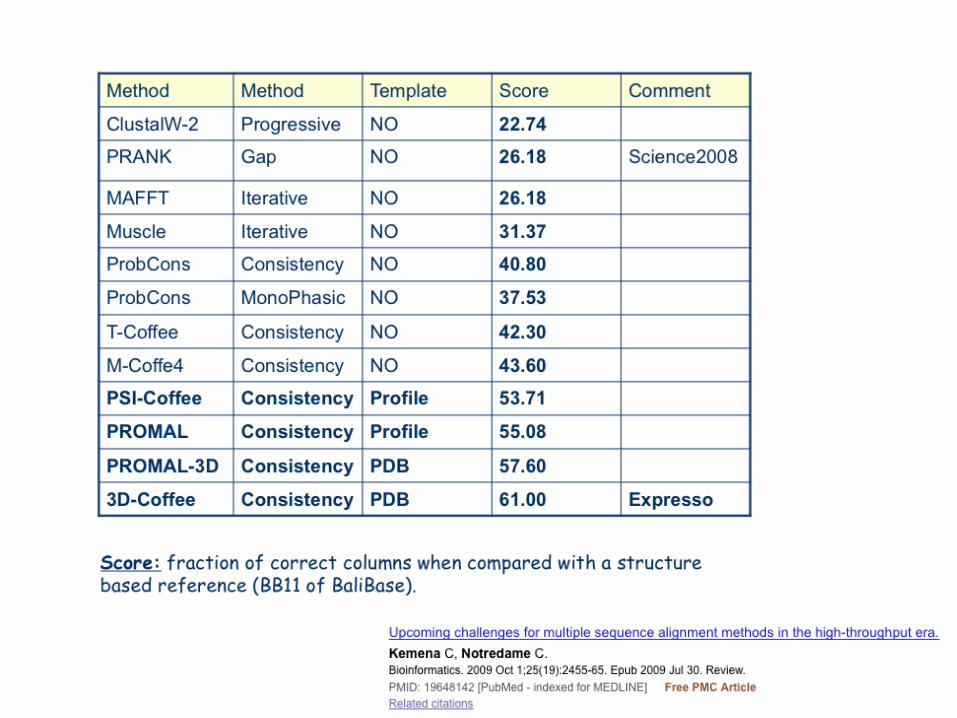

A better Question…
• What is the Best Alignment ?• What is the best bit of my
alignment ?
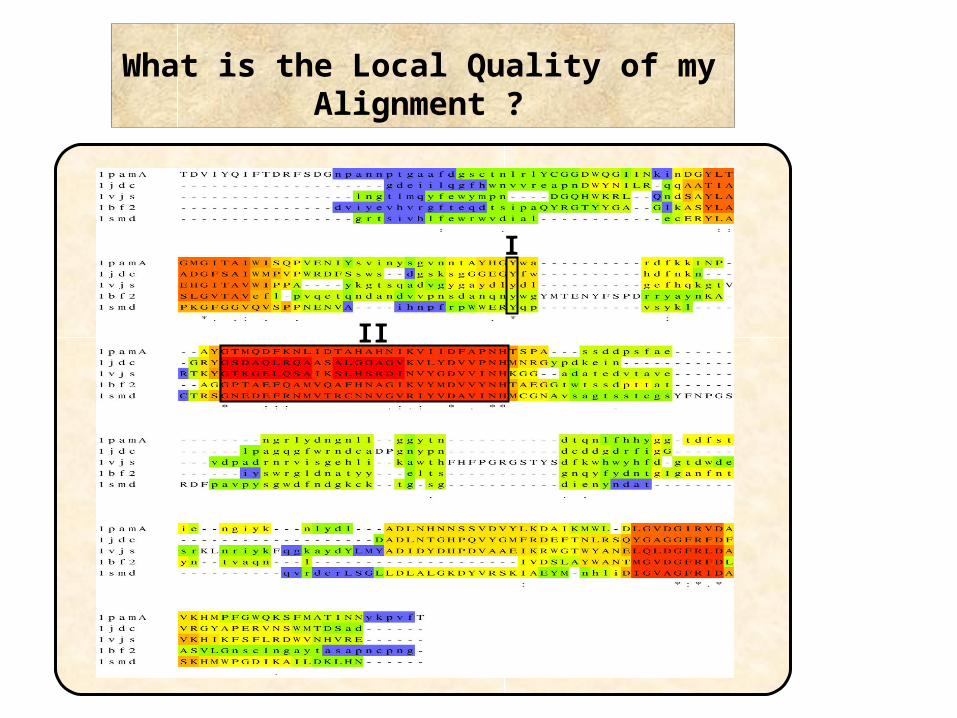
What is the Local Quality of my Alignment ?
II
I

Choosing the right method
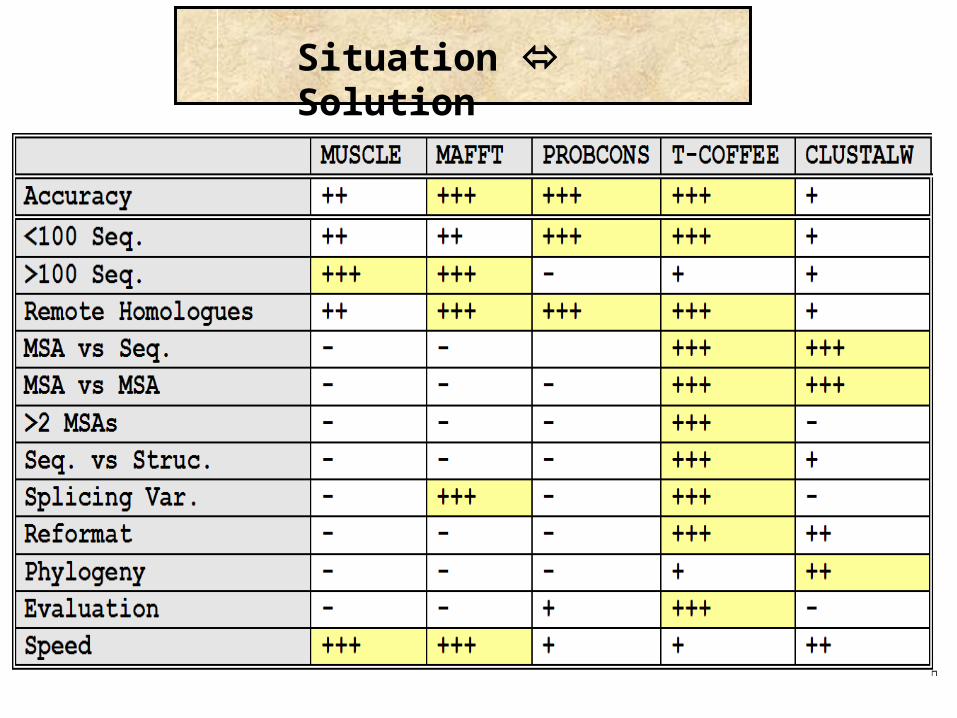
Situation Solution
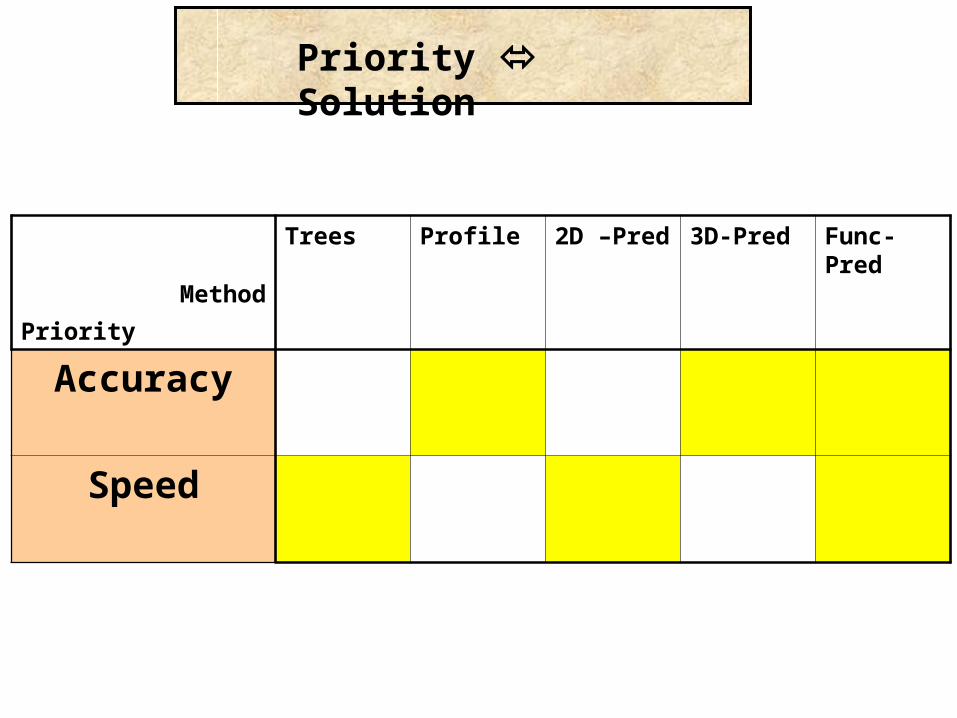
Priority Solution
Method
Priority
Trees Profile 2D –Pred 3D-Pred Func-Pred
Accuracy
Speed
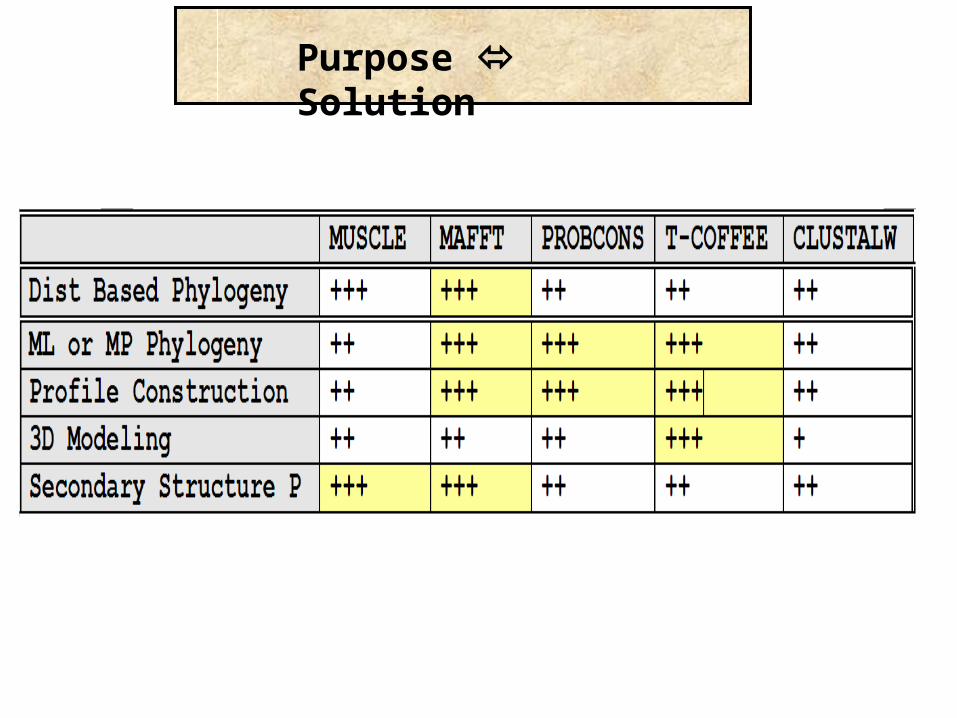
Purpose Solution

Conclusion
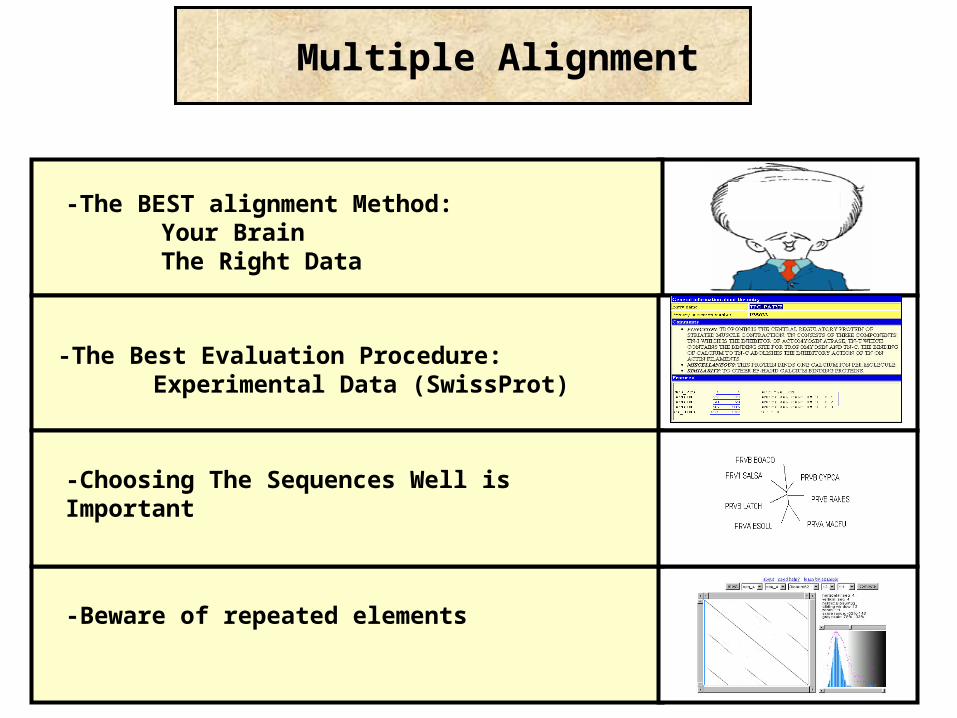
-The BEST alignment Method: Your BrainThe Right Data
-Beware of repeated elements
Multiple Alignment
-The Best Evaluation Procedure:Experimental Data (SwissProt)
-Choosing The Sequences Well is Important

Know Your Problem: What do you want to do with your MSA
Multiple Alignment
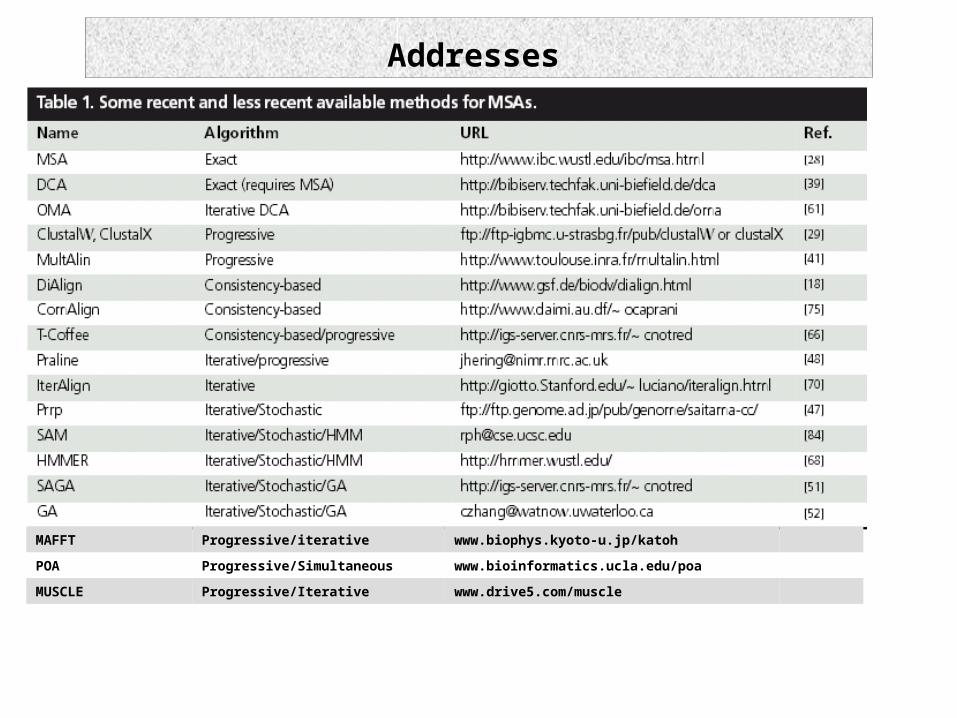
Addresses
MAFFT Progressive/iterative www.biophys.kyoto-u.jp/katoh
POA Progressive/Simultaneous www.bioinformatics.ucla.edu/poa
MUSCLE Progressive/Iterative www.drive5.com/muscle