Embed Size (px)
Citation preview

of June 11, 2018.This information is current as
Periodontal Bone Loss-InducedPorphyromonas gingivalisagainst
and Adhesin Functional Epitopes Protects An Immune Response Directed to Proteinase
Ally, Robert N. Pike and Eric C. ReynoldsPaolini, Yu-Yen Chen, Paul D. Veith, Vivian Tam, Nafisa Neil M. O'Brien-Simpson, Rishi D. Pathirana, Rita A.
http://www.jimmunol.org/content/175/6/3980doi: 10.4049/jimmunol.175.6.3980
2005; 175:3980-3989; ;J Immunol
Referenceshttp://www.jimmunol.org/content/175/6/3980.full#ref-list-1
, 25 of which you can access for free at: cites 57 articlesThis article
average*
4 weeks from acceptance to publicationFast Publication! •
Every submission reviewed by practicing scientistsNo Triage! •
from submission to initial decisionRapid Reviews! 30 days* •
Submit online. ?The JIWhy
Subscriptionhttp://jimmunol.org/subscription
is online at: The Journal of ImmunologyInformation about subscribing to
Permissionshttp://www.aai.org/About/Publications/JI/copyright.htmlSubmit copyright permission requests at:
Email Alertshttp://jimmunol.org/alertsReceive free email-alerts when new articles cite this article. Sign up at:
Print ISSN: 0022-1767 Online ISSN: 1550-6606. Immunologists All rights reserved.Copyright © 2005 by The American Association of1451 Rockville Pike, Suite 650, Rockville, MD 20852The American Association of Immunologists, Inc.,
is published twice each month byThe Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
by guest on June 11, 2018
http://ww
w.jim
munol.org/
Dow
nloaded from

An Immune Response Directed to Proteinase and AdhesinFunctional Epitopes Protects against Porphyromonasgingivalis-Induced Periodontal Bone Loss1
Neil M. O’Brien-Simpson,* Rishi D. Pathirana,* Rita A. Paolini,* Yu-Yen Chen,*Paul D. Veith,* Vivian Tam,* Nafisa Ally,† Robert N. Pike,*† and Eric C. Reynolds2*
Porphyromonas gingivalis, a pathogen associated with periodontitis, bound to fibrinogen, fibronectin, hemoglobin, and collagentype V with a similar profile to that of its major virulence factor, the cell surface RgpA-Kgp proteinase-adhesin complex. Usingpeptide-specific, purified Abs in competitive inhibition ELISAs and epitope mapping assays, we have identified potential adhesinbinding motifs (ABMs) of the RgpA-Kgp complex responsible for binding to host proteins. The RgpA-Kgp complex and syntheticABM and proteinase active site peptides conjugated to diphtheria toxoid, when used as vaccines, protected against P. gingivalis-induced periodontal bone loss in the murine periodontitis model. The most efficacious peptide and protein vaccines were found toinduce a high-titer IgG1 Ab response. Furthermore, mice protected in the lesion and periodontitis models had a predominant P.gingivalis-specific IL-4 response, whereas mice with disease had a predominant IFN-� response. The peptide-specific Abs directedto the ABM2 sequence (EGLATATTFEEDGVA) protected against periodontal bone loss and inhibited binding of the RgpA-Kgpcomplex to fibrinogen, fibronectin, and collagen type V. Furthermore, the peptide-specific Abs directed to the ABM3 sequence(GTPNPNPNPNPNPNPGT) protected against periodontal bone loss and inhibited binding to hemoglobin. However, the mostprotective Abs were those directed to the active sites of the RgpA and Kgp proteinases. The results suggest that when theRgpA-Kgp complex, or functional binding motif or active site peptides are used as a vaccine, they induce a Th2 response thatblocks function of the RgpA-Kgp complex and protects against periodontal bone loss. The Journal of Immunology, 2005, 175:3980–3989.
C hronic periodontitis is an inflammatory disease of thesupporting tissues of the teeth leading to resorption ofalveolar bone and eventual tooth loss (1, 2). The disease
is a major public health problem in all societies and is estimated toaffect up to 15% of the adult dentate population, with severe formsaffecting 5–6% (3, 4). The development and progression ofchronic periodontitis has been associated with specific Gram-neg-ative bacteria in subgingival plaque (5). The presence of threeGram-negative bacterial species, Tannerella forsythia (formallyBacteroides forsythus), Treponema denticola, and Porphyromonasgingivalis, in subgingival plaque has been strongly associated withdisease (6). The persistence of P. gingivalis in subgingival plaquefrom periodontitis patients after treatment (scaling and root plan-ing) has been reported to be significantly associated with progres-sive alveolar bone loss (7). Moreover, an increase in P. gingivaliscell numbers in subgingival plaque has been shown to correlatewith disease severity as measured by attachment loss, periodontalpocket depth, and bleeding on probing (8). Furthermore, oral in-
fection with P. gingivalis has been shown to induce periodontalbone loss in mice, rats, and nonhuman primates (9–11).
A number of virulence factors have been reported to contributeto the pathogenicity of P. gingivalis including the following: LPS,fimbriae, hemagglutinin, hemolysin, and extracellular hydrolyticenzymes (especially the Arg- and Lys-specific proteinases) (12). Avirulence factor common to all strains of P. gingivalis is the cellsurface Arg-specific (RgpA) and Lys-specific (Kgp) proteinasesand their associated adhesins designated the RgpA-Kgp complex(13). Both outer membrane-associated and released forms of theproteinases are considered to be major virulence determinants inthe onset and progression of chronic periodontitis (14). P. gingi-valis wild-type cells treated with a protease inhibitor (N-�-p-tosyl-L-lysine chloromethyl ketone; TLCK)3 and spontaneous mutantswith reduced Arg-specific and Lys-specific activity have been re-ported to be less virulent in animal models (15). We have recentlyshown in the murine lesion model that P. gingivalis W50 isogenicmutants lacking either RgpA, RgpB (Arg-specific proteinase with-out associated adhesins), or Kgp exhibited significantly reducedpathogenicity when compared with the wild-type strain (16). Inthese experiments, the Kgp mutant was the least virulent (16). Wehave also shown that immunization with the RgpA-Kgp complexinduced Ag-specific, intraoral Abs and prevented subgingival col-onization of P. gingivalis and periodontal bone loss in the rodentperiodontitis model (11).
*Cooperative Research Centre for Oral Health Science, School of Dental Science,University of Melbourne, Victoria, Australia; and †Department of Biochemistry andMolecular Biology, Monash University, Clayton Campus, Victoria, Australia
Received for publication September 16, 2004. Accepted for publication July 7, 2005.
The costs of publication of this article were defrayed in part by the payment of pagecharges. This article must therefore be hereby marked advertisement in accordancewith 18 U.S.C. Section 1734 solely to indicate this fact.1 This work was supported by the Australian National Health and Medical ResearchCouncil (Project No. 990199) and the National Institutes of Health (Grant 1RO1DE14198-01). N.M.O.-S. is a C.R. Roper Fellow.2 Address correspondence and reprint requests to Dr. Eric C. Reynolds, CooperativeResearch Centre for Oral Health Science, School of Dental Science, University ofMelbourne, Victoria 3010, Australia. E-mail address: [email protected]
3 Abbreviations used in this paper: TLCK, N-�-p-tosyl-L-lysine chloromethyl ketone;DT, diphtheria toxoid; RT, room temperature; ABM, adhesin binding motif; CMC,carboxymethylcellulose; ABC, alveolar bone crest; PBST, PBS/0.05% Tween;PVDF, polyvinylidene difluoride; PMF, peptide mass fingerprinting; TFA, trifluoro-acetic acid; PAS, proteinase active site; CI, confidence interval.
The Journal of Immunology
Copyright © 2005 by The American Association of Immunologists, Inc. 0022-1767/05/$02.00
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

In this study, we show that P. gingivalis W50 whole cells ex-hibit the same binding pattern to fibrinogen, fibronectin, hemoglo-bin, and collagen type V as the RgpA-Kgp complex, which bindsto these proteins with nanomolar dissociation constants. We alsoshow that the adhesins of the RgpA-Kgp complex are important inproviding protection in the murine lesion and periodontitis modelswhen the complex is used as a vaccine and that the immune re-sponse is predominately a Th2 response. Furthermore, we showthat synthetic peptides corresponding to proteinase active site- andadhesin-binding sequences conjugated to diphtheria toxoid (DT)induce a Th2 Ab response that protects against P. gingivalis-in-duced bone loss in the murine periodontitis model. We also iden-tify the minimal Ab binding epitope of the protective adhesin pep-tides and show that these Abs inhibit the binding of the RgpA-Kgpcomplex to host proteins.
Materials and MethodsBacterial strains and growth conditions
P. gingivalis W50 or ATCC 33277 were grown and harvested as describedpreviously (8, 16).
Purification of the complexes
The isolation and purification of the RgpA complex and Kgp complex(from strain HG66) was conducted as described previously (17). The iso-lation and purification of the RgpA-Kgp complex (from strain W50) wasperformed as described previously (R. D. Pathirana, N. M. O’Brien-Simp-son, and E. C. Reynolds, unpublished data). Briefly, P. gingivalis strainW50 cells (2L) were grown to late exponential phase and harvested bycentrifugation (7500 � g, 30 min, 4°C) and washed twice with PG buffer(50 mM Tris-HCl, 150 mM NaCl, 5 mM CaCl2, and 5 mM cysteine-HCl;pH 8.0) in the anaerobic workstation. Cells were resuspended in PG buffer,total volume 60 ml, containing 0.5% v/v Triton X-114 and gently mixed atroom temperature (RT) for 45 min. The cell extract was centrifuged(7500 � g, 30 min, 4°C), and the collected supernatant was centrifuged(40,000 � g, 30 min, 4°C). The supernatant was filtered (0.2 �m) and thenapplied to an Arg-Sepharose column (Hiload XK 16/10 Q; Pharmacia),installed in a Pharmacia GP-250 FPLC system, in TC 50 buffer (50 mMTris-HCl, 50 mM NaCl, and 5 mM CaCl2; pH 7.4) at a flow rate of 1ml/min. Nonspecifically bound proteins were eluted with a linear gradientof 0–40% TC 500 buffer (500 mM NaCl, 50 mM Tris-HCl, and 5 mMCaCl2; pH 7.4) at a flow rate of 1.0 ml/min. The column was re-equili-brated with TC 50 buffer, and bound proteins were eluted with the samebuffer containing 500 mM arginine (pH 7.4) at a flow rate of 1 ml/min. Theeluant was monitored at 280 nm. All fractions were collected at 4°C andstored at �70°C before further processing.
Arginine-eluted fast protein liquid chromatography fractions were con-centrated using Vivaspin 20 concentrator (10,000 m.w. cutoff) (Sartorius)by centrifugation at 3000 � g for 15-min periods at 4°C until the eluantwas reduced to a volume of �1 ml. The filter membrane of the Cen-triprep-10 Concentrator (Sartorious) was then rinsed with 1 ml of TC 50buffer. Concentrated fractions were then desalted using a PD-10 columnper manufacturer’s instructions (Amersham Biosciences).
Immunization and murine lesion model
The murine lesion model experiments were approved by the University ofMelbourne Ethics Committee for Animal Experimentation and were con-ducted essentially as described previously (8). BALB/c mice 6–8 wk old(10 mice per group) were immunized s.c. (100 �l) with either 20 �g of theRgpA-Kgp complex, 20 �g of the RgpA complex, 20 �g of the Kgp com-plex, or 2 � 109 formalin-killed cells of P. gingivalis strain ATCC 33277;each Ag was emulsified in IFA. After 30 days, the mice were boosted withAg (s.c. injection, emulsified in IFA) and then, 12 days later, bled from theretrobulbar plexus. Two days after bleeding, mice were challenged with7.5 � 109 viable cells of P. gingivalis strain ATCC 33277 by s.c. injection(100 �l) in the abdomen, and lesion sizes were monitored daily over 7days. The P. gingivalis inocula were prepared using PG buffer in the an-aerobic workstation as described above. The number of viable cells in theinocula was verified by enumeration on HB agar.
Immunization and murine periodontitis model
The murine periodontitis experiments were based on the model of Baker etal. (9) and were approved by the University of Melbourne Ethics Com-
mittee for Animal Experimentation. BALB/c mice 6–8 wk old (10 miceper group) were immunized s.c. (100 �l) with either 20 �g of the RgpA-Kgp complex, 50 �g of peptide-DT conjugate (prepared as described ear-lier (8)), i.e., adhesin binding motif (ABM)-1 DT conjugate (ABM1-DT),ABM2-DT, ABM3-DT, protease active site 1 from RgpA proteinase DTconjugate (PAS1-R-DT), and PAS1-K-DT from the Kgp proteinase, 50 �gof DT, or PBS (pH 7.4) emulsified in IFA. After 30 days, the mice wereboosted with Ag (s.c. injection, emulsified in IFA) and then bled 12 dayslater from the retrobulbar plexus. After bleeding, mice received kanamycin(Sigma-Aldrich) at 1 mg/ml in deionized water ad libitum for 7 days. Threedays after the antibiotic treatment, mice were orally inoculated four times,2 days apart with 1 � 1010 viable P. gingivalis W50 cells (25 �l) in PGbuffer containing 2 g/100 ml carboxymethylcellulose (CMC; Sigma-Al-drich), and a control group was sham-infected with PG buffer containing 2g/100 ml CMC alone. Two weeks later, mice received another four doses(2 days apart) of 1 � 1010 viable P. gingivalis W50 cells (25 �l) in PGbuffer containing 2 g/100 ml CMC. The number of viable cells in eachinoculum was verified by enumeration on HB agar. Twenty-eight days afterthe second oral challenge, mice were killed, and the maxillae wereremoved.
Maxillae were boiled (1 min) in deionized water, mechanically de-fleshed, and immersed in 2 g/100 ml potassium hydroxide (16 h, 25°C).The maxillae were then washed (two times, deionized water) and immersedin 3% v/v hydrogen peroxide (6 h, 25-C). After washing, (two times, deion-ized water), the maxillae were stained with 0.1 g/100 ml aqueous methyl-ene blue, and a digital image of the buccal aspect of each half maxilla wascaptured with a Sound and Vision digital camera (SciTech) mounted on adissecting microscope, using Adobe Photoshop version 4.0 to assess hor-izontal bone loss. Horizontal bone loss is loss occurring in a horizontalplane, perpendicular to the alveolar bone crest (ABC) that results in areduction of the crest height. Each half-maxilla was aligned so that thebuccal and lingual molar cusps of each left or right image were superim-posed. A micrometer scale in plane with each half-maxilla was digitallyimaged at the same time so that measurements could be standardized foreach image. The area from the cementoenamel junction to the ABC foreach tooth was measured using Scion Image Beta 4.02 (Scion) imagingsoftware downloaded from the Scion Corporation web site (www.scion-corp.com/index.htm). Bone loss measurements were determined twice in arandom and blinded protocol by two standardized examiners.
ELISPOT assay
ELISPOT assay was performed using the Millipore Multiscreen 96-wellfiltration plates (MAHAS450; Millipore). Plates were coated with anti-mouse cytokine capture Abs (eBiosciences), specific for IL-4 (catalog no.14-7041; clone 11B11) and IFN-� (catalog no. 14-7312; clone R4-6A2), ata concentration of 4 �g/ml in 0.1 M sodium bicarbonate buffer (pH 9.5)and incubated overnight at 4°C. Plates were then washed with PBS andblocked with T cell medium (DMEM containing 10% v/v heat-inactivated(56°C, 30 min) FCS, 2 mM glutamine, 2 mM sodium pyruvate, 0.1 mM2-ME, 30 �g/ml gentamicin, 100 IU/ml penicillin, and 100 �g/ml strep-tomycin) for 1 h at 37°C. Lymph node (inguinal and popliteal for lesionmodel and submandibular for periodontitis model) T cells from Ag-immu-nized and control mice were first positively isolated/sorted using CD90�
beads and AutoMACS cell sorter (Miltenyi Biotec) per the manufacturer’sinstructions and were incubated (1 � 105/well) with gamma-radiated syn-geneic spleen cells as a source of APCs (2 � 105 cells/well) and 1 �g/mlof the Ag (RgpA-Kgp complex, Kgp complex, RgpA complex, or RgpBproteinase). Plates were incubated at 37°C in an atmosphere of 5% CO2 inair for 48 h in a humidified incubator. The plates were then washed inPBS/0.05% Tween (PBST) three times and once with Milli-Q water. Cy-tokine-specific biotinylated Abs (eBiosciences) specific for IL-4 (catalogno. 13-7042; clone BVD6-24G2) and IFN-� (catalog no. 13-7311; cloneXMG 1.2) were added at a concentration of 2 �g/ml in Dulbecco’s PBS/Tcell medium (1:1, v/v) and incubated at RT for 2 h. Plates were washed sixtimes in PBST, and Streptavidin-Alkaline Phosphatase conjugate (Roche)was added to the plates at a 1/1000 dilution in Dulbecco’s PBS/T cellmedium (1:1, v/v) and incubated for 1 h at RT. The plates were washed sixtimes in PBST and three times in PBS. One tablet of the substrate 5-bromo-4-chloro-3-indolyl phosphate/NBT (catalog no. B-565; Sigma-Aldrich)was dissolved in 10 ml of Milli-Q water, and 50 �l of this substrate bufferwas added per well. Spots were allowed to develop for 20–30 min, beforestopping the reaction by washing with tap water. The spots were countedusing EliSpot Reader Lite (version 2.9.; Autoimmun Diagnostika), and datawere expressed as spot-forming cells per million.
3981The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

ELISA
ELISAs were performed, as described previously (8, 18), in triplicate usinga solution (1 �g/ml) of the RgpA-Kgp complex in 0.1 M PBS (pH 7.4) tocoat wells of flat-bottom polyvinyl microtiter plates (Microtiter; DynatechLaboratories) overnight at 4°C.
Western blotting
Purified RgpA-Kgp complex was separated by SDS-PAGE in 12.5% acryl-amide gels (1 mm) by using the method of Laemmli (19) with a minigelsystem (Bio-Rad). Proteins were either stained in the gel using Coomassieblue or electrophoretically transferred onto polyvinylidene difluoride(PVDF) membrane using the method of Dashper et al. (20). After section-ing the membrane, the m.w. standards were stained with 0.1 g/100 ml ofCBB R250. The remaining sections were blocked for 1 h at 20°C with 5g/100 ml of nonfat skim milk powder in TN buffer (50 mM Tris-HCl (pH7.4) and 100 mM NaCl). Sections were subsequently incubated with mousesera diluted 1/25 with TN buffer. After 16 h at 20°C, the sections werewashed (4� TN buffer containing 0.05% v/v Tween 20 for 10 min) andthen incubated for 1 h at 20°C with HRP-conjugated goat Ig directedagainst mouse Ig (1/400 dilution) (Sigma-Aldrich). After washing (4� TNbuffer containing 0.05% v/v Tween 20 for 10 min), bound Ab was detectedwith 0.05 g/100 ml of 4-chloro-1-napthol in TN buffer containing 16.6%v/v methanol and 0.015 g/100 ml of H2O2. Color development was stoppedby rinsing the membranes with Milli-Q water.
In-gel digestion and peptide mass fingerprinting (PMF)
Protein bands were excised from the Coomassie blue-stained SDS-PAGEgel, and gel pieces were washed in 100 mM NH4HCO3/ethanol 1:1, re-duced and alkylated with DTT and iodoacetamide, and digested with se-quencing grade-modified trypsin (Promega) overnight at 37°C as publishedpreviously (21). The peptide extract containing 20 mM NH4HCO3 wasthen analyzed by MALDI-TOF MS using an Ultraflex TOF/TOF instru-ment (Bruker) in positive ion and reflectron mode. A saturated solution of4-hydroxy-�-cyanocinnamic acid was prepared in 97:3 acetone/0.1% v/vtrifluoroacetic acid (TFA). A thin layer was prepared by pipetting andimmediately removing 2 �l of this solution onto the 600-�m anchor chipsof the target plate. Sample (0.5 �l) was deposited on the thin layers with2.5 �l of 0.1% v/v TFA and allowed to adsorb for 5 min, after which thesample solution was removed, and the thin layers were washed once with10 �l of ice-cold 0.1% v/v TFA for 1 min. Spectra were calibrated by closeexternal calibration using a standard peptide mix. Proteins were identifiedby PMF against the P. gingivalis database (available from �www.tigr.org�)using an in-house Mascot search engine.
Binding of the RgpA-Kgp complex and P. gingivalis strain W50to host proteins
ELISA was used to determine the affinity of the RgpA-Kgp proteinaseadhesin complex and P. gingivalis strain W50 whole cell binding to fi-brinogen, fibronectin, collagen types I, III, IV, and V, and hemoglobin.Wells of flat-bottom polyvinyl microtiter plates (Microtiter; Dynatech Lab-oratories) were coated overnight at RT with 10 �g/ml of either fibrinogen,fibronectin, collagen types I, III, IV, and V, or hemoglobin in 0.1 M PBS(pH 7.2). After removing the coating solution, remaining uncoated plasticwas blocked with 1% (w/v) BSA in 0.1 M PBS for 1 h at RT. The blockingsolution was removed, and serial dilutions of a starting concentration of 2�g/ml RgpA-Kgp complex from P. gingivalis strain W50, 10 �g/ml (totalprotein) of P. gingivalis W50 whole cells in 0.1 M PBS containing 1 mMTLCK was added and incubated for 3 h at RT. After washing (four times,0.1 M PBS containing 0.1%, v/v Tween 20, PBST), dilutions of rabbitanti-RgpA-Kgp complex antisera or P. gingivalis W50 antisera (in 0.5%(w/v) BSA in 0.1 M PBS) was added to the plates and incubated for 2 h atRT. Plates were then washed (six times, PBST) and incubated with a1/2000 dilution of HRP-conjugated goat Ig directed against rabbit IgG(Sigma-Aldrich) in 0.5% (w/v) BSA in 0.1 M PBS for 1 h at RT. Wellswere then washed (six times, PBST), and bound Ab was detected as de-scribed above. ELISA results were used to derive the KD based on ananalog of the Michalis-Menten equation (22): OD � ODmax(protein)/KD �(protein). Where OD is the OD at a given protein concentration, ODmax isthe maximum OD for the ELISA plate reader, (protein) is the protein con-centration of the analyte, and KD is the apparent KD for a given OD at agiven protein concentration (ELISA data point). KD was calculated by aScatchard plot analysis of 1/(protein) vs 1/KD (ELISA data point): 1/KD �m(1/c), where m is the slope of the line and c � the intercept at the y-axis(22). OD415 was measured using a Bio-Rad 450 model microplate reader(Bio-Rad).
Purification of peptide-specific IgG
IgG was purified from antisera obtained from mice immunized withABM1, ABM2, and ABM3 peptide DT conjugates, or DT using the Im-munopure protein (A/G) IgG purification kit per the manufacturer’s in-structions (Pierce), and the protein concentrations of purified IgG sampleswere determined using the Bradford protein assay (Bio-Rad).
Competitive inhibition ELISA
Plates were coated overnight with 10 �g/ml fibrinogen, fibronectin, colla-gen V, or hemoglobin in 0.1 M PBS. After removal of the coating solution,remaining uncoated plastic was blocked with 1% w/v BSA in 0.1 M PBSfor 1 h at RT then washed (four times, PBST). Two-fold dilutions of pu-rified anti-ABM1, anti-ABM2, and anti-ABM3 IgG (in PBS containing0.5% w/v BSA), starting concentration of 100 nM, were added to eachplate followed by the addition of 0.5 �g/ml of the RgpA-Kgp complex (inPBS containing 0.5% BSA and 1 mM TLCK), with a final volume of 100�l. Plates were incubated for 3 h at RT and probed with a 1/3200 dilutionof rabbit anti-RgpA-Kgp complex antisera, and the plates were developedas described above. The percentage inhibition of the RgpA-Kgp complexbinding to host proteins by each of the purified ABM peptide-specific IgGwas calculated according to the following equation (22): percentage inhi-bition � 100 � (1 � ((OD with IgG � background)/(OD without IgG �background))), where background is the absorbance from wells with noRgpA-Kgp complex added. IC50 was calculated from the inhibition curves(percentage inhibition vs IgG concentration). OD415 was measured using aBio-Rad 450 model microplate reader (Bio-Rad).
Epitope analysis of the ABM and proteinase active site (PAS)peptide sequences
The Ab binding site for each of the ABM and PAS peptides was deter-mined by synthesizing overlapping eight or 10 residue peptides (offset byone) on a multipin peptide synthesis system (Chiron) using standard solidphase peptide synthesis protocols for Fmoc chemistry. Epitope mapping ofthe pin-bound peptides was conducted by ELISA as per Chiron Technol-ogies’ instructions, using mouse sera at a dilution of 1/1000 in 1% w/vnonfat skim milk powder in 0.1 M PBS (pH 7.4) containing 0.1% v/vTween 20. The bound Ab was detected by incubating the pins with 0.9 mMABTS in 80 mM citric acid (pH 4.0) buffer containing 0.005% v/v H2O2.OD415 was measured using a Bio-Rad 450 model microplate reader(Bio-Rad).
Statistical analysis
The maximum sizes of the lesions developed were statistically analyzedusing the Kruskal-Wallis test and Mann-Whitney U Wilcoxon rank sumtest with a Bonferroni correction for type 1 error (SPSS for Windows,Release 6.0; SPSS). The bone loss (mm2) data were statistically analyzedusing one-way ANOVA and Dunnett’s T3 test (SPSS for Windows, Re-lease 6.0; SPSS). Effect sizes, represented as Cohen’s (23) d were calcu-lated using the effect size calculator provided on-line by Evidence-BasedEducation U.K. web site (�http://cem.dur.ac.uk/ebeuk/research/effectsize/�).According to Cohen (23), a small effect size is d � 0.2 and �0.5, a mod-erate effect size is d � 0.5 � 0.8, and a large effect size is d � 0.8.
ResultsCharacterization of binding of P. gingivalis strain W50 wholecells and the RgpA-Kgp complex to fibrinogen, fibronectin,collagen types I, III, IV, and V, and hemoglobin
The binding of viable P. gingivalis whole cells of strain W50 andthe RgpA-Kgp complex to a variety of host proteins is shown inFig. 1, and the KD for each of the interactions was calculated usingScatchard plot analysis (Table I). P. gingivalis strain W50 wasfound to bind to all of the host proteins tested, with the strongestinteraction being toward fibrinogen followed by fibronectin, he-moglobin, collagen types I, III, IV, and then collagen type V (Ta-ble I). The RgpA-Kgp complex was found to bind in the nanomo-lar range to the host proteins fibrinogen, fibronectin, collagen typeV, and hemoglobin, with a similar pattern of binding as displayedby whole cells (Fig. 1, and Table I). However, unlike whole cells,the RgpA-Kgp complex did not bind to collagen types I, III, andIV. Peptide-specific IgG Abs against the synthetic peptides ABM1,ABM2, and ABM3 (Fig. 2) were used in a competitive inhibition
3982 PEPTIDES PROTECT AGAINST P. gingivalis-INDUCED BONE LOSS
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

ELISA to determine the inhibition constants (IC50) for any inhi-bition of binding of the RgpA-Kgp complex to the host proteins;fibrinogen, fibronectin, collagen type V, and hemoglobin. ABM1-specific IgG Abs did not inhibit the binding of the RgpA-Kgpcomplex to any of the host proteins tested. ABM2 IgG-specificAbs inhibited binding in the nanomolar range of the RgpA-Kgpcomplex to fibrinogen, fibronectin, and collagen type V, but not tohemoglobin (Table I). IgG-specific Abs for ABM3 inhibited bind-ing in the nanomolar range to hemoglobin but not to any of theother proteins tested (Table I).
Effect of immunizing mice with the RgpA-Kgp complex, RgpA,or Kgp on P. gingivalis-induced lesions in the murine lesionmodel
BALB/c mice were immunized with purified RgpA-Kgp complex,RgpA, Kgp, formalin-killed P. gingivalis cells (strain ATCC33277), or adjuvant alone, and then challenged s.c. with viable P.gingivalis cells (strain ATCC 33277). Lesion development wasmeasured over a 14-day period, and the maximum lesion sizes foreach group are shown in Fig. 3. Immunization with formalin-killedcells, the RgpA-Kgp complex, RgpA, or the Kgp significantly re-duced lesion development after P. gingivalis challenge. Of thethree protein Ags tested, the RgpA-Kgp complex had the biggesteffect on reducing lesion sizes (d � 6.00; 99% confidence interval(CI): �8.19, �3.39; p � 0.001), because mice immunized with the
RgpA-Kgp complex had significantly smaller lesions than miceimmunized with Kgp (d � 1.70; 99% CI: 0.42, 2.85; p � 0.01) orRgpA (d � 0.94; 99% CI: �0.20, 2.01) alone. Mice immunizedwith the Kgp alone tended to exhibit moderately larger lesions thanmice immunized with RgpA alone (d � 0.70; 99% CI: �0.40,1.76).
Effect of immunization with the RgpA-Kgp complex on P.gingivalis-induced alveolar bone loss in the murine periodontitismodel
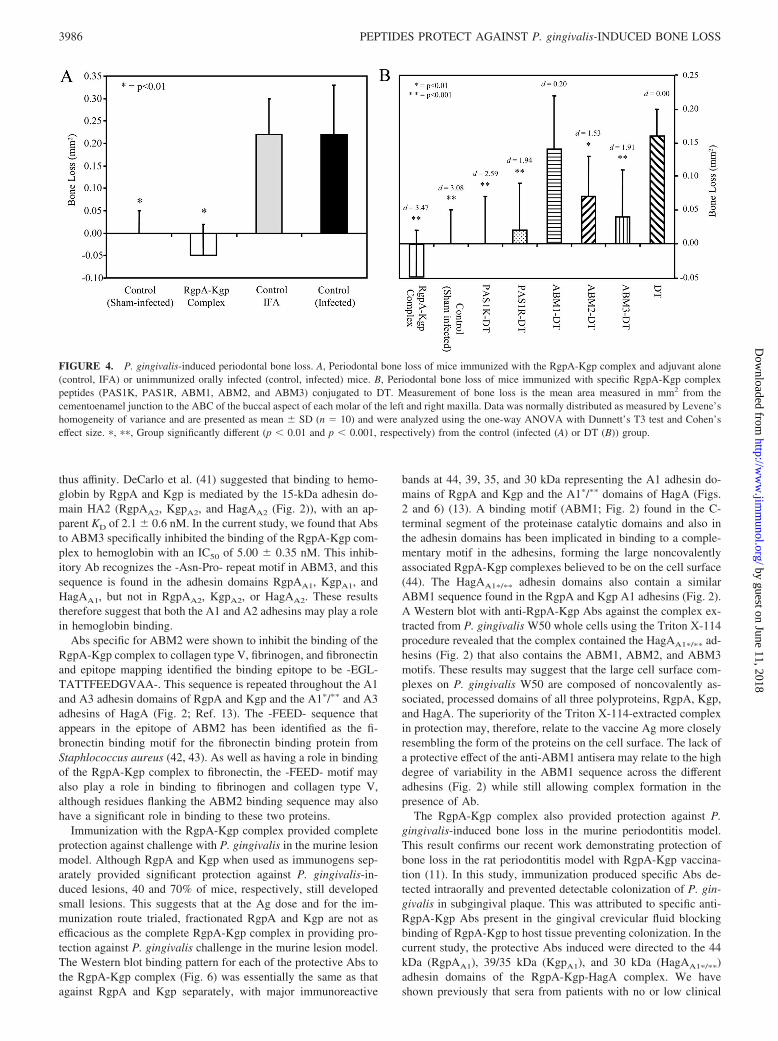
Mice were immunized (days 0 and 30) with the RgpA-Kgp com-plex or adjuvant alone and were then orally challenged with viableP. gingivalis W50. The RgpA-Kgp complex-immunized mice hadsignificantly ( p � 0.01) less bone loss than mice receiving injec-tions with adjuvant alone or control (infected) groups (Fig. 4A).There was no significant difference in bone loss between the con-trol (sham-infected) mice and the RgpA-Kgp complex-immunizedmice that were orally challenged with P. gingivalis, indicating thatimmunization with the RgpA-Kgp complex completely protectedagainst P. gingivalis-induced periodontal bone loss (Fig. 4A).
Ab subclass and cytokine responses induced by immunizationwith the RgpA-Kgp complex, RgpA, or Kgp in the murine lesionand periodontitis models
Before challenge with viable P. gingivalis cells either s.c. (lesionmodel) or intraorally (periodontal model), mice were bled, and thesera were collected by centrifugation. Fig. 5A shows the Ab sub-class reactivity to the RgpA-Kgp complex for each immunogen(RgpA-Kgp complex, RgpA, or Kgp) in the murine lesion andperiodontitis models. All of the proteins used to immunize mice inthe lesion model induced a high-titer IgG and low-titer IgM andIgA (IgA ELISA titers 100–300) response to the RgpA-Kgp com-plex. Furthermore, the predominant Ab subclass of each immuno-gen induced was IgG1, with only weakly immunoreactive IgG2a,IgG2b, and IgG3 RgpA-Kgp complex-specific Abs detected (Fig.5A). The same Ab subclass profile and titer of high IgG1 and lowIgG2a, IgG2b, and IgG3 was induced by the RgpA-Kgp complex-immunized mice that were protected against P. gingivalis-inducedbone loss in the periodontitis model (Fig. 5A). T cells isolated frompopliteal and inguinal (lesion model) or submandibular (periodon-titis model) lymph nodes from mice immunized and challengedwith viable P. gingivalis cells were stimulated with the respectiveAg (RgpA-Kgp complex, RgpA, or Kgp), and the cytokine re-sponse induced was determined by ELISPOT assay (Fig. 5, B andC). The predominant cytokine response in mice immunized andprotected against either lesion development or periodontal boneloss was IL-4 (Fig. 5, B and C). However, in the periodontitis
Table I. The apparent KD and inhibition constant (IC50) for the binding of P. gingivalis W50 and the RgpA-Kgp complex to hostproteins
W50 Cells RgpA-Kgp Complex Anti-ABM2 Anti-ABM3
Host protein KD (�g/mL) KD (nM) IC50 (nM) IC50 (nM)Fibrinogen 0.25 0.09 1.86 0.69 28.75 5.86 b
Fibronectin 0.83 0.19 3.79 1.22 18.75 4.34 b
Collagen type I 2.15 0.58 a b b
Collagen type III 2.31 0.29 a b b
Collagen type IV 1.50 0.32 a b b
Collagen type V 6.99 0.63 3.67 1.16 20.6 3.84 b
Hemoglobin 1.28 0.38 7.88 1.23 b 5.00 0.35
a No binding detected.b No inhibition detected.
FIGURE 1. Binding of P. gingivalis strains W50 (f) and the RgpA-Kgp complex (u) to a range of connective tissue and blood proteins. Con-nective tissue and blood proteins were coated at 10 �g/ml, and the ELISAwas conducted as described in Materials and Methods. Values are shownas the averages of at least three experiments.
3983The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

model, control mice that exhibited disease (bone loss) had a pre-dominant IFN-� cytokine response to the RgpA-Kgp complex(Fig. 5C).
Western blot analysis of the murine Ab responses
Antisera from mice immunized with RgpA, Kgp, and the RgpA-Kgp complex (from the lesion and periodontitis models) were usedto probe the RgpA-Kgp complex in a Western blot (Fig. 6A). Theprotective Abs to the RgpA-Kgp complex from both the periodon-titis and lesion models were immunoreactive with the same pro-teins of molecular mass 44, 39, 35, and 30 kDa. These areas of theCoomassie blue-stained SDS-PAGE gel of the RgpA-Kgp com-plex were excised and subjected to in-gel digestion and PMF. The48, 45, 44, and 39 kDa Coomassie blue-stained bands of the com-plex were identified as Kgpcat, RgpAcat, RgpAA1, and KgpA1, re-spectively (Figs. 2 and 6), as identified previously (24). The majorband at 30 kDa was identified as HagAA1�/�� (Figs. 2 and 6), andthe faint band at 35 kDa was identified as a truncated KgpA1. Absinduced by RgpA alone and Kgp alone were also immunoreactivewith complex proteins of molecular mass 44, 39, 35, and 30 kDa(Fig. 6A). A faint immunoreactive band at 45 kDa was also de-tected by antisera induced by RgpA, Kgp, and the RgpA-Kgp com-plex (Fig. 6A). To determine whether this response was directed tothe catalytic domain of the RgpA proteinase, the RgpB proteinase(97% sequence identity to the RgpA proteinase) was probed with
Abs raised against the RgpA-Kgp complex and the RgpB protein-ase (Fig. 6B). The RgpB proteinase Abs were strongly immuno-reactive with RgpB proteinase; however, the RgpA-Kgp complexAbs did not recognize the RgpB proteinase, suggesting that thefaint band detected at 45 kDa was also derived from the A1adhesins.
Effect of immunization with synthetic peptides on thedevelopment of P. gingivalis-induced bone loss in the murineperiodontitis model
Mice were immunized (days 0 and 30) with synthetic peptides(Fig. 2) representing sequences from the RgpA-Kgp complex con-jugated to DT and then orally challenged with viable P. gingivalisW50 cells. Fig. 4B shows the level of alveolar bone loss inducedin immunized mice after challenge with P. gingivalis. Mice im-munized with peptides PAS1K, PAS1R, ABM2, and ABM3 (Fig.2), and the RgpA-Kgp complex exhibited significantly less boneloss than control DT mice. The RgpA-Kgp complex was signifi-cantly more effective than control DT (d � 3.47; 99.9% CI: 5.43,1.27), ABM1 (d � 2.54; 99.9% CI: 0.86, 4.09), and ABM2 (d �1.95; 99.9% CI: 0.43, 3.36) in protecting mice against P. gingiva-lis-induced bone loss (Fig. 4B). Mice immunized with the syn-thetic peptide-DT conjugates PAS1K-DT, PAS1R-DT, andABM3-DT displayed significantly ( p � 0.001) less bone loss com-pared with control DT-infected mice. Furthermore, immunization
FIGURE 2. Diagrammatic representation of RgpA, Kgp, and HagA showing the relative positions of the PAS and ABM epitopes. indicates putativeLPS attachment site.
3984 PEPTIDES PROTECT AGAINST P. gingivalis-INDUCED BONE LOSS
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

with PAS1K-DT was found to be significantly more effective atprotecting against bone loss than immunization with ABM1-DT(d � 1.91; 99.9% CI: 0.59, 3.15), ABM2-DT (d � 1.12; 99% CI:0.19, 1.99), and control DT (d � 2.59; 99.9% CI: 0.88, 4.01). Theeffect sizes suggested that PAS1K-DT was slightly more effectiveat protecting against bone loss than PAS1R-DT (d � 0.4; 99.9%CI: �0.73, 1.50) and moderately more effective compared withABM3-DT (d � 0.59; 99.9% CI: �0.49, 1.66). PAS1R-DT andABM3-DT immunizations were found to be significantly ( p �0.01) more effective (d � 1.43; 99.9% CI: 0.14, 2.66 and d � 1.38;99.9% CI: 0.15, 2.55, respectively) at protecting against P. gingi-valis-induced bone loss compared with ABM1-DT immunizationand moderately more effective than ABM2-DT immunization (d �0.63; 99.9% CI: �0.54, 1.77 and d � 0.50; 99.9% CI: �0.61, 1.59,respectively). Mice immunized with ABM2-DT exhibited signif-icantly ( p � 0.01) less bone loss than control DT-infected mice(d � 1.53; 99.9% CI: 0.11, 2.87); however, ABM1-DT-immunizedmice were not protected from P. gingivalis-induced bone loss.
Ab subclass responses induced by immunization with syntheticpeptides in the murine periodontitis model
Before oral challenge with viable P. gingivalis cells, mice werebled, and the sera were collected by centrifugation. Fig. 7 showsthe Ab subclass reactivity to the RgpA-Kgp complex for each ofthe peptide Ags. PAS1K, PAS1R, and ABM3 induced high-titerIgG Abs (predominantly IgG1) that recognized the RgpA-Kgpcomplex. The PAS1K peptide induced a slightly higher IgG1 Abtiter than the PAS1R and ABM3 peptides. The Ab titers ofPAS1K, PAS1R, and ABM3 were significantly ( p � 0.001) higherthan that induced by ABM1 and ABM2. Furthermore, the ABM2peptide induced IgG1 Abs that displayed a higher titer ( p � 0.01)of binding to the RgpA-Kgp complex than the IgG1 Abs inducedby ABM1.
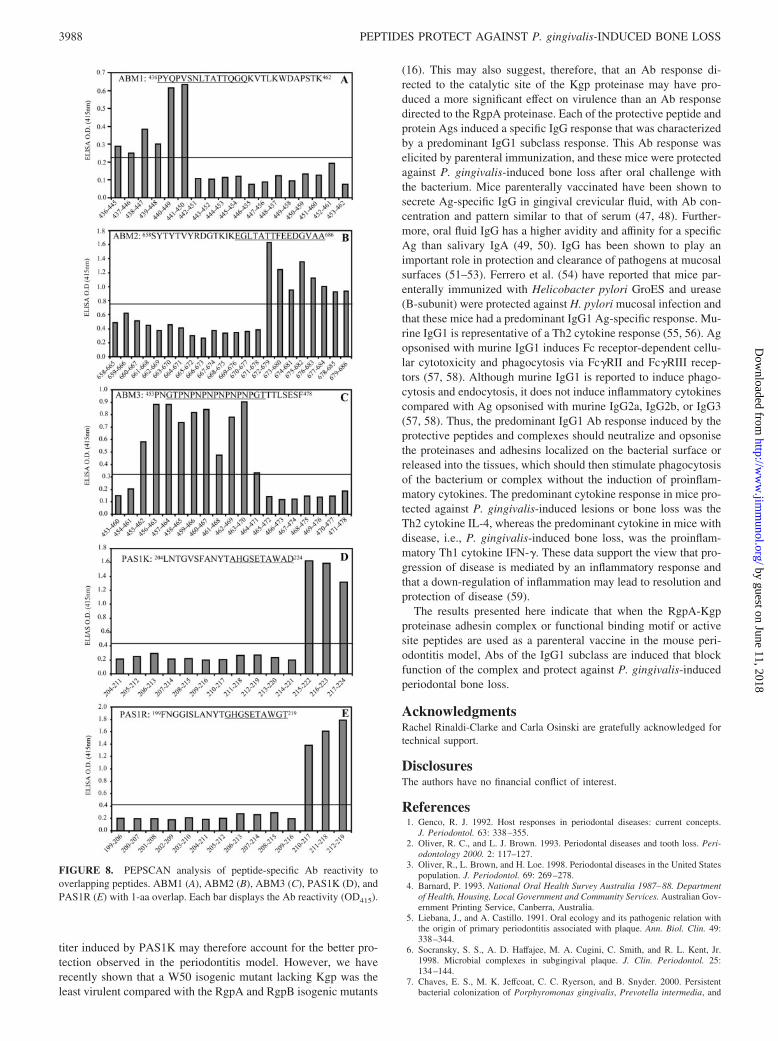
Epitope mapping of PAS1K, PAS1R, ABM1, ABM2, and ABM3
Overlapping eight residue peptides (offset by one) for PAS1K,PAS1R, ABM2, and ABM3 and overlapping 10 residue peptides (off-set by one) for ABM1 were synthesized on resin pins. The minimal
Ab binding epitope was then identified for each peptide, using thecorresponding peptide-specific sera (Fig. 8). A 2-fold increase in OD(415 nm) above background was considered as a positive Ab re-sponse. The minimal Ab binding sites for each peptide were as fol-lows: PAS1K, 215AHGSETAWAD224; PAS1R, 210GHGSETAWGT219; ABM1, 436PYQPVSNLTATTQGQ450; ABM2, 672EGLTATTFEEDGVAA686; and ABM3, 714GTPNPNPNPNPNPNPGT737.
DiscussionA major virulence factor of P. gingivalis is the proteinase-adhesincomplex, the RgpA-Kgp complex, that has been shown to be abun-dant on the cell surface by mass spectrometric analysis of outermembrane preparations separated by two-dimensional gel electro-phoresis (24, 25). In this study, we demonstrate a similar pattern ofbinding of P. gingivalis W50 whole cells to the host proteins fi-brinogen, fibronectin, hemoglobin, and collagen type V as the pu-rified RgpA-Kgp complex. Furthermore, we identified ABMscommon to RgpA, Kgp, and HagA that are likely to be responsiblefor binding to fibrinogen, fibronectin, hemoglobin, and collagentype V. This together with the abundance of RgpA, Kgp, andHagA on the cell surface suggests that these proteins may be themajor mechanism of W50 whole cell binding to these host pro-teins. This suggestion is supported by the work of Shi et al. (26)who have shown that a mutant lacking RgpA, Kgp, and HagA wasdefective in hemoglobin binding.
The RgpA-Kgp complex bound with high affinities to fibrino-gen, fibronectin, hemoglobin, and collagen type V; however, un-like whole cells, the complex did not bind significantly to collagentypes I, II, and IV, suggesting that other cell surface adhesins areresponsible for cellular binding to these host proteins. Both W50whole cells and the RgpA-Kgp complex displayed high-affinitybinding to collagen type V. Collagen type V has been shown to bean integral part of basement membrane of human aortas, arteries,atherosclerotic plaques, smooth muscle cells, interstitial connec-tive tissue of human oral mucosa, and the placenta, and is a majorcomponent of the amniotic sac (27–31). Because RgpA has beenshown to hydrolyze collagen types I, III, IV, and V (32, 33), theability of the RgpA-Kgp complex to adhere to collagen type Vmay target the RgpA-Kgp complex and P. gingivalis cells to theendothelium of arterial walls as well as placental and connectivetissues. These tissues may then be degraded by the RgpA protein-ase causing tissue and vascular disruption. Recently, Lin et al. (34,35) demonstrated that the fetuses of pregnant mice challenged s.c.with P. gingivalis were growth restricted and that there was anincrease in the placental Th1/Th2 cytokine ratio with significantincreases in proinflammatory (Th1) cytokines IFN-�, TNF-�, IL-2,and IL-12 and decreases in the anti-inflammatory (Th2) cytokinesIL-4 and IL-10. Furthermore, P. gingivalis DNA was detected inall of the placentas where the fetuses were growth restricted. Arecent study by Jain et al. (36) showed that rabbits orally chal-lenged with P. gingivalis developed periodontitis and had signif-icantly greater lipid deposition in the aorta than control animals. Infact, there was a positive correlation (r2 � 0.9501) between P.gingivalis-induced bone loss (disease severity) and the extent oflipid deposition. As well as enhancing colonization of host tissue,the binding to collagen type V may be an important factor in theassociation of periodontitis with coronary heart disease (37) andpreterm births and low birth weights (38, 39).
Pike et al. (40) have previously shown that RgpA and Kgpbound to fibrinogen with KD values of 8.5 nM and 4.0 nM, re-spectively. Although these data are in the nanomolar range, theyare 2- to 4-fold higher than the KD value for the whole RgpA-Kgpcomplex, suggesting that combining RgpA and Kgp to form acomplex increases the valency of binding sites for fibrinogen and
FIGURE 3. Maximum lesion size of mice challenged with P. gingivalisstrain ATCC 33277. BALB/c mice were challenged s.c. with 7.5 � 109
viable P. gingivalis ATCC 33277 cells. Animals were monitored over a14-day period for lesion size. Data presented represent mean SD (n �10) and were analyzed using the nonparametric Kruskall-Wallis test withthe Mann-Whitney U Wilcoxon rank sum test and Cohen’s effect size. �,��, Group significantly different (p � 0.01 and p � 0.001, respectively)from the control (IFA) group.
3985The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
thus affinity. DeCarlo et al. (41) suggested that binding to hemo-globin by RgpA and Kgp is mediated by the 15-kDa adhesin do-main HA2 (RgpAA2, KgpA2, and HagAA2 (Fig. 2)), with an ap-parent KD of 2.1 0.6 nM. In the current study, we found that Absto ABM3 specifically inhibited the binding of the RgpA-Kgp com-plex to hemoglobin with an IC50 of 5.00 0.35 nM. This inhib-itory Ab recognizes the -Asn-Pro- repeat motif in ABM3, and thissequence is found in the adhesin domains RgpAA1, KgpA1, andHagAA1, but not in RgpAA2, KgpA2, or HagAA2. These resultstherefore suggest that both the A1 and A2 adhesins may play a rolein hemoglobin binding.
Abs specific for ABM2 were shown to inhibit the binding of theRgpA-Kgp complex to collagen type V, fibrinogen, and fibronectinand epitope mapping identified the binding epitope to be -EGL-TATTFEEDGVAA-. This sequence is repeated throughout the A1and A3 adhesin domains of RgpA and Kgp and the A1�/�� and A3adhesins of HagA (Fig. 2; Ref. 13). The -FEED- sequence thatappears in the epitope of ABM2 has been identified as the fi-bronectin binding motif for the fibronectin binding protein fromStaphlococcus aureus (42, 43). As well as having a role in bindingof the RgpA-Kgp complex to fibronectin, the -FEED- motif mayalso play a role in binding to fibrinogen and collagen type V,although residues flanking the ABM2 binding sequence may alsohave a significant role in binding to these two proteins.
Immunization with the RgpA-Kgp complex provided completeprotection against challenge with P. gingivalis in the murine lesionmodel. Although RgpA and Kgp when used as immunogens sep-arately provided significant protection against P. gingivalis-in-duced lesions, 40 and 70% of mice, respectively, still developedsmall lesions. This suggests that at the Ag dose and for the im-munization route trialed, fractionated RgpA and Kgp are not asefficacious as the complete RgpA-Kgp complex in providing pro-tection against P. gingivalis challenge in the murine lesion model.The Western blot binding pattern for each of the protective Abs tothe RgpA-Kgp complex (Fig. 6) was essentially the same as thatagainst RgpA and Kgp separately, with major immunoreactive
bands at 44, 39, 35, and 30 kDa representing the A1 adhesin do-mains of RgpA and Kgp and the A1�/�� domains of HagA (Figs.2 and 6) (13). A binding motif (ABM1; Fig. 2) found in the C-terminal segment of the proteinase catalytic domains and also inthe adhesin domains has been implicated in binding to a comple-mentary motif in the adhesins, forming the large noncovalentlyassociated RgpA-Kgp complexes believed to be on the cell surface(44). The HagAA1�/�� adhesin domains also contain a similarABM1 sequence found in the RgpA and Kgp A1 adhesins (Fig. 2).A Western blot with anti-RgpA-Kgp Abs against the complex ex-tracted from P. gingivalis W50 whole cells using the Triton X-114procedure revealed that the complex contained the HagAA1�/�� ad-hesins (Fig. 2) that also contains the ABM1, ABM2, and ABM3motifs. These results may suggest that the large cell surface com-plexes on P. gingivalis W50 are composed of noncovalently as-sociated, processed domains of all three polyproteins, RgpA, Kgp,and HagA. The superiority of the Triton X-114-extracted complexin protection may, therefore, relate to the vaccine Ag more closelyresembling the form of the proteins on the cell surface. The lack ofa protective effect of the anti-ABM1 antisera may relate to the highdegree of variability in the ABM1 sequence across the differentadhesins (Fig. 2) while still allowing complex formation in thepresence of Ab.
The RgpA-Kgp complex also provided protection against P.gingivalis-induced bone loss in the murine periodontitis model.This result confirms our recent work demonstrating protection ofbone loss in the rat periodontitis model with RgpA-Kgp vaccina-tion (11). In this study, immunization produced specific Abs de-tected intraorally and prevented detectable colonization of P. gin-givalis in subgingival plaque. This was attributed to specific anti-RgpA-Kgp Abs present in the gingival crevicular fluid blockingbinding of RgpA-Kgp to host tissue preventing colonization. In thecurrent study, the protective Abs induced were directed to the 44kDa (RgpAA1), 39/35 kDa (KgpA1), and 30 kDa (HagAA1�/��)adhesin domains of the RgpA-Kgp-HagA complex. We haveshown previously that sera from patients with no or low clinical
FIGURE 4. P. gingivalis-induced periodontal bone loss. A, Periodontal bone loss of mice immunized with the RgpA-Kgp complex and adjuvant alone(control, IFA) or unimmunized orally infected (control, infected) mice. B, Periodontal bone loss of mice immunized with specific RgpA-Kgp complexpeptides (PAS1K, PAS1R, ABM1, ABM2, and ABM3) conjugated to DT. Measurement of bone loss is the mean area measured in mm2 from thecementoenamel junction to the ABC of the buccal aspect of each molar of the left and right maxilla. Data was normally distributed as measured by Levene’shomogeneity of variance and are presented as mean SD (n � 10) and were analyzed using the one-way ANOVA with Dunnett’s T3 test and Cohen’seffect size. �, ��, Group significantly different (p � 0.01 and p � 0.001, respectively) from the control (infected (A) or DT (B)) group.
3986 PEPTIDES PROTECT AGAINST P. gingivalis-INDUCED BONE LOSS
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

signs of periodontitis that have a high IgG4 and low IgG2 subclassAb titer to the RgpA-Kgp complex recognized the RgpAA1 andKgpA1 adhesins in a Western blot (8). Patients with mild to severedisease with a low IgG4 and high IgG2 subclass Ab titer to theRgpA-Kgp complex recognized only a 44-kDa protein band (Rg-pAA1). Using epitope mapping analysis, we localized the protec-tive immunoreactive sequences in RgpAA1 and KgpA1 to a 120-aasequence that was common to both adhesins and contained thethree ABMs (ABM1, ABM2, and ABM3). Subgingival applica-tion in humans of a mAb directed to a sequence within this 120-aaspan of the A1 adhesin prevented recolonization of P. gingivalisfor up to 9 mo at sites with severe attachment loss (45). Nakagawaet al. (46) have shown that Abs directed to adhesin components ofRgpA and Kgp enhanced polymorphonuclear leukocyte-mediatedphagocytosis of all four P. gingivalis serotypes (33277, serotypeA; A7A1-28, serotype B; W50, serotype C; and 381, serotype D)tested.
Immunization with the peptide epitopes PAS1K, PAS1R,ABM3, and AMB2 induced protection against P. gingivalis-in-duced bone loss. The protective effect of immunization withPAS1K appeared to be moderately better than the protection in-duced by PAS1R, ABM3, and AMB2, as demonstrated by theeffect sizes (Fig. 4). Immunization with PAS1R and ABM3 pro-duced similar effects. The protection elicited by immunization withthe RgpA-Kgp complex was significantly ( p � 0.01) greater thanall of the peptide vaccines except for PAS1K. The difference inbone loss induced by P. gingivalis in mice immunized with ABM1compared with PAS1K, PAS1R, and ABM3 may be attributed tothe difference in Ab response induced. ABM1 induced signifi-cantly less ( p � 0.001) Ab compared with PAS1K, PAS1R, andABM3. The total IgG and IgG1 Ab titers induced by PAS1K werelarger than those induced by PAS1R and ABM3. This higher Ab
FIGURE 6. Western blot analysis of the RgpA-Kgp complex (A) andthe RgpB proteinase (B) probed with proteinase-adhesin antisera. TheRgpA-Kgp complex (A) was separated by SDS-PAGE, transferred ontoPVDF membrane, and probed with anti-RgpA-Kgp antisera (1:50 TNbuffer) (lane 1, from the lesion model), preimmune sera (lane 2), RgpA(lane 3), Kgp (lane 4), and RgpA-Kgp complex (lane 5, antisera from theperiodontitis model); lane 6 represents the protein blot of the RgpA-Kgpcomplex. The RgpB proteinase (B) was separated by SDS-PAGE, trans-ferred onto PVDF membrane, and probed with antisera (1:50 TN buffer) tothe RgpA-Kgp complex (lane 2) and RgpB proteinase (lane 3); lane 1represents the protein blot of the RgpB proteinase stained with Coomassieblue. Molecular mass markers are shown in kilodaltons.
FIGURE 7. Serum Ab subclass responses of mice immunized with thePAS and ABM peptides. Sera from mice immunized with PAS1K, PAS1R,ABM1, ABM2, and ABM3 were used in the ELISA, with the RgpA-Kgpcomplex as the absorbed Ag. Ab responses total IgG (f), IgG1 (u), IgG2a(�), IgG2b (p), and IgG3 (o) are expressed as the ELISA titer OD415
obtained minus double the background level, with each titer representingthe mean SD of three values.
FIGURE 5. Serum Ab subclass and cytokine responses of immunized mice in the lesion and periodontitis models. A, Sera from mice (lesion model)immunized with RgpA-Kgp complex (u), RgpA complex (f), Kgp complex (z), and sera from mice (periodontitis model) immunized with the RgpA-Kgpcomplex (�) were used in the ELISA, with the RgpA-Kgp complex as the absorbed Ag. Ab responses are expressed as the ELISA titer A415 obtained minusdouble the background level, with each titer representing the mean SD of three values. B, Popliteal and inguinal lymph nodes from mice (lesion model)immunized with RgpA-Kgp complex, RgpA complex, and Kgp complex, and submandibular lymph nodes from mice (periodontitis model) immunized withthe RgpA-Kgp complex, and control mice were used in an ELISPOT assay for the detection of RgpA-Kgp complex-specific IL-4 (�) and IFN-� (f) Tcell responses. Cytokine responses are expressed as spot-forming cells per million obtained minus the background, with each ELISPOT representing themean and SD of four values.
3987The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from
titer induced by PAS1K may therefore account for the better pro-tection observed in the periodontitis model. However, we haverecently shown that a W50 isogenic mutant lacking Kgp was theleast virulent compared with the RgpA and RgpB isogenic mutants
(16). This may also suggest, therefore, that an Ab response di-rected to the catalytic site of the Kgp proteinase may have pro-duced a more significant effect on virulence than an Ab responsedirected to the RgpA proteinase. Each of the protective peptide andprotein Ags induced a specific IgG response that was characterizedby a predominant IgG1 subclass response. This Ab response waselicited by parenteral immunization, and these mice were protectedagainst P. gingivalis-induced bone loss after oral challenge withthe bacterium. Mice parenterally vaccinated have been shown tosecrete Ag-specific IgG in gingival crevicular fluid, with Ab con-centration and pattern similar to that of serum (47, 48). Further-more, oral fluid IgG has a higher avidity and affinity for a specificAg than salivary IgA (49, 50). IgG has been shown to play animportant role in protection and clearance of pathogens at mucosalsurfaces (51–53). Ferrero et al. (54) have reported that mice par-enterally immunized with Helicobacter pylori GroES and urease(B-subunit) were protected against H. pylori mucosal infection andthat these mice had a predominant IgG1 Ag-specific response. Mu-rine IgG1 is representative of a Th2 cytokine response (55, 56). Agopsonised with murine IgG1 induces Fc receptor-dependent cellu-lar cytotoxicity and phagocytosis via Fc�RII and Fc�RIII recep-tors (57, 58). Although murine IgG1 is reported to induce phago-cytosis and endocytosis, it does not induce inflammatory cytokinescompared with Ag opsonised with murine IgG2a, IgG2b, or IgG3(57, 58). Thus, the predominant IgG1 Ab response induced by theprotective peptides and complexes should neutralize and opsonisethe proteinases and adhesins localized on the bacterial surface orreleased into the tissues, which should then stimulate phagocytosisof the bacterium or complex without the induction of proinflam-matory cytokines. The predominant cytokine response in mice pro-tected against P. gingivalis-induced lesions or bone loss was theTh2 cytokine IL-4, whereas the predominant cytokine in mice withdisease, i.e., P. gingivalis-induced bone loss, was the proinflam-matory Th1 cytokine IFN-�. These data support the view that pro-gression of disease is mediated by an inflammatory response andthat a down-regulation of inflammation may lead to resolution andprotection of disease (59).
The results presented here indicate that when the RgpA-Kgpproteinase adhesin complex or functional binding motif or activesite peptides are used as a parenteral vaccine in the mouse peri-odontitis model, Abs of the IgG1 subclass are induced that blockfunction of the complex and protect against P. gingivalis-inducedperiodontal bone loss.
AcknowledgmentsRachel Rinaldi-Clarke and Carla Osinski are gratefully acknowledged fortechnical support.
DisclosuresThe authors have no financial conflict of interest.
References1. Genco, R. J. 1992. Host responses in periodontal diseases: current concepts.
J. Periodontol. 63: 338–355.2. Oliver, R. C., and L. J. Brown. 1993. Periodontal diseases and tooth loss. Peri-
odontology 2000. 2: 117–127.3. Oliver, R., L. Brown, and H. Loe. 1998. Periodontal diseases in the United States
population. J. Periodontol. 69: 269–278.4. Barnard, P. 1993. National Oral Health Survey Australia 1987–88. Department
of Health, Housing, Local Government and Community Services. Australian Gov-ernment Printing Service, Canberra, Australia.
5. Liebana, J., and A. Castillo. 1991. Oral ecology and its pathogenic relation withthe origin of primary periodontitis associated with plaque. Ann. Biol. Clin. 49:338–344.
6. Socransky, S. S., A. D. Haffajee, M. A. Cugini, C. Smith, and R. L. Kent, Jr.1998. Microbial complexes in subgingival plaque. J. Clin. Periodontol. 25:134–144.
7. Chaves, E. S., M. K. Jeffcoat, C. C. Ryerson, and B. Snyder. 2000. Persistentbacterial colonization of Porphyromonas gingivalis, Prevotella intermedia, and
FIGURE 8. PEPSCAN analysis of peptide-specific Ab reactivity tooverlapping peptides. ABM1 (A), ABM2 (B), ABM3 (C), PAS1K (D), andPAS1R (E) with 1-aa overlap. Each bar displays the Ab reactivity (OD415).
3988 PEPTIDES PROTECT AGAINST P. gingivalis-INDUCED BONE LOSS
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from

Actinobacillus actinomycetemcomitans in periodontitis and its association withalveolar bone loss after 6 months of therapy. J. Clin. Periodontol. 27: 897–903.
8. O’Brien-Simpson, N., R. Paolini, and E. Reynolds. 2000. RgpA-Kgp peptide-based immunogens provide protection against Porphyromonas gingivalis chal-lenge in a murine lesion model. Infect. Immun. 68: 4055–4063.
9. Baker, P. J., R. T. Evans, and D. C. Roopenian. 1994. Oral infection with Por-phyromonas gingivalis and induced alveolar bone loss in immunocompetent andsevere combined immunodeficient mice. Arch. Oral Biol. 39: 1035–1040.
10. Holt, S. C., J. Ebersole, J. Felton, M. Brunsvold, and K. S. Korman. 1988. Im-plantation of Bacteroides gingivalis in nonhuman primates initiates progressionof periodontitis. Science 239: 55–57.
11. Rajapakse, P. S., N. M. O’Brien-Simpson, N. Slakeski, B. Hoffmann, andE. C. Reynolds. 2002. Immunization with the RgpA-Kgp proteinase-adhesincomplexes of Porphyromonas gingivalis protects against periodontal bone loss inthe rat periodontitis model. Infect. Immun. 70: 2480–2486.
12. Mayrand, D., and S. C. Holt. 1988. Biology of asaccharolytic black-pigmentedBacteroides species. Microbiol. Rev. 52: 134–152.
13. O’Brien-Simpson, N. M., P. D. Veith, S. G. Dashper, and E. C. Reynolds. 2003.Porphyromonas gingivalis gingipains: the molecular teeth of a microbial vam-pire. Curr. Protein Pept. Sci. 4: 409–426.
14. Kadowaki, T., K. Nakayama, K. Okamoto, N. Abe, A. Baba, Y. Shi,D. Ratnayake, and K. Yamamoto. 2000. Porphyromonas gingivalis proteinases asvirulence determinants in progression of periodontal diseases. J. Biochem. 128:153–159.
15. Kesavalu, L., S. C. Holt, and J. L. Ebersole. 1997. Porphyromonas gingivalisvirulence in a murine lesion model: effects of immune alterations. Microb.Pathog. 23: 317–326.
16. O’Brien-Simpson, N. M., R. A. Paolini, B. Hoffmann, N. Slakeski, S. G. Dashper,and E. C. Reynolds. 2001. Role of RgpA, RgpB, and Kgp proteinases in virulenceof Porphyromonas gingivalis W50 in a murine lesion model. Infect. Immun. 69:7527–7534.
17. Pike, R., W. McGraw, J. Potempa, and J. Travis. 1994. Lysine- and arginine-specific proteinases from Porphyromonas gingivalis. J. Biol. Chem. 269:406–411.
18. O’Brien-Simpson, N. M., C. L. Black, P. S. Bhogal, S. M. Cleal, N. Slakeski,T. J. Higgins, and E. C. Reynolds. 2000. Serum IgG and IgG subclass responsesto the RgpA-Kgp proteianse-adhesin complex of P. gingivalis in adult periodon-titis. Infect. Immun. 68: 2704–2712.
19. Laemmli, U. K. 1970. Cleavage of structural proteins during the assembly of thehead of bacteriophage T4. Nature 227: 680–685.
20. Dashper, S. G., N. M. O’Brien-Simpson, P. S. Bhogal, A. D. Franzmann, andE. C. Reynolds. 1998. Purification and characterization of a putative fimbrialprotein/receptor of Porphyromonas gingivalis. Aust. Dent. J. 43: 99–104.
21. Mortz, E., T. N. Krogh, H. Vorum, and A. Gorg. 2001. Improved silver stainingprotocols for high sensitivity protein identification using matrix-assisted laserdesorption/ionization-time of flight analysis. Proteomics 1: 1359–1363.
22. Qiu, X., P. Schroeder, and D. Bridon. 1996. Identification and characterization ofa C(K/R)TC motif as a common epitope present in all subtypes of hepatitis Bsurface antigen. J. Immunol. 156: 3350–3356.
23. Cohen, J. 1969. Statistical Power Analysis for the Behavioral Sciences. AcademicPress, New York.
24. Veith, P. D., G. H. Talbo, N. Slakeski, S. G. Dashper, C. Moore, R. A. Paolini,and E. C. Reynolds. 2002. Major outer membrane proteins and proteolytic pro-cessing of RgpA and Kgp of Porphyromonas gingivalis W50. Biochem. J. 363:105–115.
25. O’Brien-Simpson, N. M., P. D. Veith, S. G. Dashper, and E. C. Reynolds. 2004.Antigens of bacteria associated with periodontitis. Periodontology 2000. 35: 101–134.
26. Shi, Y., D. B. Ratnayake, K. Okamoto, N. Abe, K. Yamamoto, and K. Nakayama.1999. Genetic analyses of proteolysis, hemoglobin binding, and hemagglutinationof Porphyromonas gingivalis: construction of mutants with a combination ofrgpA, rgpB, kgp, and hagA. J. Biol. Chem. 274: 17955–17960.
27. Kerenyi, T., B. Voss, J. Rauterberg, H. G. Fromme, and H. Jellinek. 1984. Pres-ence of connective tissue proteins on the endothelium of the rat aorta. Exp. Mol.Pathol. 40: 380–390.
28. Schuppan, D., J. Becker, H. Boehm, and E. G. Hahn. 1986. Immunofluorescentlocalization of type-V collagen as a fibrillar component of the interstitial con-nective tissue of human oral mucosa, artery and liver. Cell Tissue Res. 243:535–543.
29. Voss, B., and J. Rauterberg. 1986. Localization of collagen types I, III, IV and V,fibronectin and laminin in human arteries by the indirect immunofluorescencemethod. Pathol. Res. Pract. 181: 568–575.
30. Merker, H. J., D. Bremer, H. J. Barrach, and R. Gossrau. 1987. The basementmembrane of the persisting maternal blood vessels in the placenta of Callithrixjacchus. Anat. Embryol. 176: 87–97.
31. Polzin, W. J., E. G. Lockrow, and W. K. Morishige. 1997. A pilot study identi-fying type V collagenolytic activity in human amniotic fluid. Am. J. Perinatol. 14:103–106.
32. Bedi, G. S., and T. Williams. 1994. Purification and characterization of a colla-gen-degrading protease from Porphyromonas gingivalis. J. Biol. Chem. 269:599–606.
33. Houle, M. A., D. Grenier, P. Plamondon, and K. Nakayama. 2003. The collage-nase activity of Porphyromonas gingivalis is due to Arg-gingipain. FEMS Mi-crobiol. Lett. 221: 181–185.
34. Lin, D., M. A. Smith, C. Champagne, J. Elter, J. Beck, and S. Offenbacher. 2003.Porphyromonas gingivalis infection during pregnancy increases maternal tumor
necrosis factor �, suppresses maternal interleukin-10, and enhances fetal growthrestriction and resorption in mice. Infect. Immun. 71: 5156–5162.
35. Lin, D., M. A. Smith, J. Elter, C. Champagne, C. L. Downey, J. Beck, andS. Offenbacher. 2003. Porphyromonas gingivalis infection in pregnant mice isassociated with placental dissemination, an increase in the placental Th1/Th2cytokine ratio, and fetal growth restriction. Infect. Immun. 71: 5163–5168.
36. Jain, A., E. L. Batista, Jr., C. Serhan, G. L. Stahl, and T. E. Van Dyke. 2003. Rolefor periodontitis in the progression of lipid deposition in an animal model. Infect.Immun. 71: 6012–6018.
37. Beck, J. D., S. Offenbacher, R. Williams, P. Gibbs, and R. Garcia. 1998. Peri-odontitis: a risk factor for coronary heart disease? Ann. Periodontol. 3: 127–141.
38. Offenbacher, S., V. Katz, G. Fertik, J. Collins, D. Boyd, G. Maynor, R. McKaig,and J. Beck. 1996. Periodontal infection as a possible risk factor for preterm lowbirth weight. J. Periodontol. 67: 1103–1113.
39. Offenbacher, S., H. L. Jared, P. G. O’Reilly, S. R. Wells, G. E. Salvi,H. P. Lawrence, S. S. Socransky, and J. D. Beck. 1998. Potential pathogenicmechanisms of periodontitis associated pregnancy complications. Ann. Periodon-tol. 3: 233–250.
40. Pike, R. N., J. Potempa, W. McGraw, T. H. Coetzer, and J. Travis. 1996. Char-acterization of the binding activities of proteinase-adhesin complexes from Por-phyromonas gingivalis. J. Bacteriol. 178: 2876–2882.
41. DeCarlo, A. A., M. Paramaesvaran, P. L. Yun, C. Collyer, and N. Hunter. 1999.Porphyrin-mediated binding to hemoglobin by the HA2 domain of cysteine pro-teinases (gingipains) and hemagglutinins from the periodontal pathogen Porphy-romonas gingivalis. J. Bacteriol. 181: 3784–3791.
42. Signas, C., G. Raucci, K. Jonsson, P. E. Lindgren, G. M. Anantharamaiah,M. Hook, and M. Lindberg. 1989. Nucleotide sequence of the gene for a fi-bronectin-binding protein from Staphylococcus aureus: use of this peptide se-quence in the synthesis of biologically active peptides. Proc. Natl. Acad. Sci. USA86: 699–703.
43. McGavin, M. J., G. Raucci, S. Gurusiddappa, and M. Hook. 1991. Fibronectinbinding determinants of the Staphylococcus aureus fibronectin receptor. J. Biol.Chem. 266: 8343–8347.
44. Slakeski, N., P. S. Bhogal, N. M. O’Brien-Simpson, and E. C. Reynolds. 1998.Characterisation of a second cell-associated Arg-specific cysteine proteinase ofPorphyromonas gingivalis and identification of an adhesin binding motif in-volved in association of the PrtR and PrtK proteinases and adhesins into largecomplexes. Microbiology 144: 1583–1592.
45. Booth, V., F. P. Ashley, and T. Lehner. 1996. Passive immunization with mono-clonal antibodies against Porphyromonas gingivalis in patients with periodontitis.Infect. Immun. 64: 422–427.
46. Nakagawa, T., T. Sims, Q. Fan, J. Potempa, J. Travis, L. Houston, and R. C. Page.2001. Functional characteristics of antibodies induced by Arg-gingipain(HRgpA) and Lys-gingipain (Kgp) from Porphyromonas gingivalis. Oral Micro-biol. Immunol. 16: 202–211.
47. Maticic, M., M. Poljak, K. Seme, and U. Skaleric. 2003. The IgG antibody profileto various antigen regions of hepatitis C virus differs in oral fluid and serum ofpatients with chronic hepatitis C. Oral Microbiol. Immunol. 18: 176–182.
48. Parry, J. V., K. R. Perry, and P. P. Mortimer. 1987. Sensitive assays for viralantibodies in saliva: an alternative to tests on serum. Lancet 2: 72–75.
49. Cartry, O., P. Moja, A. Quesnel, B. Pozzetto, F. R. Lucht, and C. Genin. 1997.Quantification of IgA and IgG and specificities of antibodies to viral proteins inparotid saliva at different stages of HIV-1 infection. Clin. Exp. Immunol. 109:47–53.
50. Stiles, B. G., A. R. Garza, R. G. Ulrich, and J. W. Boles. 2001. Mucosal vacci-nation with recombinantly attenuated staphylococcal enterotoxin B and protec-tion in a murine model. Infect. Immun. 69: 2031–2036.
51. Senda, S., E. Cheng, and H. Kawanishi. 1989. IgG in murine intestinal secretions:aging effect and possible physiological role. Scand. J. Immunol. 29: 41–47.
52. Barkon, M. L., B. L. Haller, and H. W. Virgin. 1996. Circulating immunoglobulinG can play a critical role in clearance of intestinal reovirus infection. J. Virol. 70:1109–1116.
53. O’Neal, C. M., G. R. Harriman, and M. E. Conner. 2000. Protection of the villusepithelial cells of the small intestine from rotavirus infection does not requireimmunoglobulin A. J. Virol. 74: 4102–4109.
54. Ferrero, R. L., J. M. Thiberge, I. Kansau, N. Wuscher, M. Huerre, andA. Labigne. 1995. The GroES homolog of Helicobacter pylori confers protectiveimmunity against mucosal infection in mice. Proc. Natl. Acad. Sci. USA 92:6499–6503.
55. Vitetta, E. S., J. Ohara, C. D. Myers, J. E. Layton, P. H. Krammer, andW. E. Paul. 1985. Serological, biochemical, and functional identity of B cell-stimulatory factor 1 and B cell differentiation factor for IgG1. J. Exp. Med. 162:1726–1731.
56. Lai, Y. H., and T. R. Mosmann. 1999. Mouse IL-13 enhances antibody produc-tion in vivo and acts directly on B cells in vitro to increase survival and henceantibody production. J. Immunol. 162: 78–87.
57. Ravetch, J. V., and J. P. Kinet. 1991. Fc receptors. Annu. Rev. Immunol. 9:457–492.
58. Hazenbos, W. L., I. A. Heijnen, D. Meyer, F. M. Hofhuis, C. R. Renardel deLavalette, R. E. Schmidt, P. J. Capel, J. G. van de Winkel, J. E. Gessner,T. K. van den Berg, and J. S. Verbeek. 1998. Murine IgG1 complexes triggerimmune effector functions predominantly via Fc� RIII (CD16). J. Immunol. 161:3026–3032.
59. Van Dyke, T. E., and C. N. Serhan. 2003. Resolution of inflammation: a newparadigm for the pathogenesis of periodontal diseases. J. Dent. Res. 82: 82–90.
3989The Journal of Immunology
by guest on June 11, 2018http://w
ww
.jimm
unol.org/D
ownloaded from





























