Embed Size (px)
Citation preview

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 1 of 54 Template version date: 3-5-2015
An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects with Breast Cancer
Lead Org. ID HCI103657 /IRB# 103657
Principal Investigator Adam Cohen, MD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT 84112 801-587-4024 [email protected] Alana Welm, PhD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected]
Sub-investigator(s) Saundra Buys, MD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] Anna Beck, MD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] John Ward, MD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] Theresa Werner, MD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected]

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 2 of 54 Template version date: 3-5-2015
Lynn Henry, MD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] Elizabeth Prystas, MD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] Angela Gerrard, APRN Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] Rosie Conder, APRN Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] Chanteel Ballard, APRN Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] Jutta Deininger, APRN Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT 84112 [email protected] Jeff Yap, PhD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected]

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 3 of 54 Template version date: 3-5-2015
John Hoffman, MD Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected] Ken Boucher, PhD (Statistician) Huntsman Cancer Institute 2000 Circle of Hope Salt Lake City, UT, 84112 [email protected]
Drug Manufacturer Eli Lilly and Company Donald Thornton, MD Senior Director Eli Lilly and Company Lilly Corporate Center DC 2146 Indianapolis, IN 46285 [email protected] P: 317-277-1798 F: 317-277-5912
Investigational agent
Merestinib
IND Number 136622
Historical Protocol Versions Version 1: Version 2: Version 3:

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 4 of 54 Template version date: 3-5-2015
TABLE OF CONTENTS
Page
1 LIST OF ABBREVIATIONS .................................................................................... 6 2 PROTOCOL SIGNATURE .................................................................................... 10
3 STUDY SUMMARY ............................................................................................... 11 1 OBJECTIVES .......................................................................................................... 15
1.1 Primary Objectives and Endpoint .................................................................. 15 1.2 Secondary Objectives and Endpoint ............................................................ 15
2 BACKGROUND ...................................................................................................... 15
2.1 Background on Breast Cancer Bone Metastasis ........................................ 15 2.2 Background on MSP and RON ..................................................................... 16
2.3 Background on Merestinib ............................................................................. 17 2.4 Background on Sodium Fluoride F 18 Injection .......................................... 18
2.5 Rationale for Conducting this Study ............................................................. 18 2.6 Risk/Benefit Assessment ................................................................................ 19
2.7 Primary Hypothesis ......................................................................................... 19
2.8 Rationale for population and endpoints ....................................................... 19
2.9 Preliminary data (pre-clinical) ........................................................................ 20
2.10 Preliminary data (clinical)............................................................................ 22 3 DRUG INFORMATION .......................................................................................... 22
3.1 Mechanism of Action ....................................................................................... 22
3.2 Nonclinical Pharmacokinetics ........................................................................ 23
3.3 Absorption ......................................................................................................... 23
3.4 Distribution ........................................................................................................ 23 3.5 Metabolism ....................................................................................................... 24
3.6 Excretion ........................................................................................................... 24 3.7 Adverse Effects ................................................................................................ 24
3.8 Pregnancy and Lactation ................................................................................ 25 3.9 Drug Interactions ............................................................................................. 26
3.10 Dosing and Administration.......................................................................... 26
3.11 Preparation, Storage and Reconciliation of Supplies ............................. 26 4 STUDY DESIGN..................................................................................................... 27
4.1 Description ........................................................................................................ 27 4.2 Dose Limiting Toxicity ..................................................................................... 27
4.3 Number of Patients .......................................................................................... 28 4.4 Stopping Rules ................................................................................................. 28
4.5 Number of Study Centers ............................................................................... 29
4.6 Study Duration ................................................................................................. 29 5 ELIGIBILITY CRITERIA ........................................................................................ 29
5.1 Inclusion Criteria for Urinary Screening ....................................................... 29 5.2 Inclusion Criteria for Treatment ..................................................................... 29
5.3 Exclusion Criteria ............................................................................................. 30

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 5 of 54 Template version date: 3-5-2015
6 STRATIFICATION FACTORS ............................................................................. 33 7 TREATMENT PLAN............................................................................................... 33
7.1 Merestinib Administration Schedule ............................................................. 33 7.2 Merestinib Treatment ...................................................................................... 34
7.3 Sodium Fluoride F 18 Administration ........................................................... 35 7.4 Prohibited Concomitant Medications ............................................................ 35
7.5 Duration of Therapy ........................................................................................ 36 8 TOXICITIES AND DOSEAGE MODIFICATION ................................................ 37
8.1 Dose Modifications .......................................................................................... 37
8.2 Supportive Care ............................................................................................... 37 9 STUDY CALENDAR .............................................................................................. 38
10 STUDY PROCEDURES ..................................................................................... 40
10.1 Screening ...................................................................................................... 40
10.2 Visit 2 (Week 1, Day 1) ............................................................................... 40 10.3 Visit 3 ............................................................................................................. 41
10.4 Visit 4 ............................................................................................................. 41
10.5 Visit 5 ............................................................................................................. 42 10.6 Visit 6 (Week 13/End of Treatment (+/-7 days)) ..................................... 42
11 CRITERIA FOR EVALUATION AND ENDPOINT.......................................... 43 11.1 Efficacy .......................................................................................................... 43
11.2 Safety ............................................................................................................. 44 12 STATISTICAL CONSIDERATIONS ................................................................. 44
13 REGISTRATION GUIDELINES ........................................................................ 45
14 DATA SUBMISSION SCHEDULE .................................................................... 46
14.1 Pathology review .......................................................................................... 46
14.2 Correlative Studies ...................................................................................... 46 15 ETHICAL AND REGULATORY CONSIDERATIONS ................................... 47
15.1 Informed consent ......................................................................................... 47 15.2 Institutional Review ...................................................................................... 47 15.3 Data and Safety Monitoring Plan ............................................................... 47
15.4 Adverse Events / Serious Adverse Events .............................................. 47 15.5 SAE Reporting Requirements .................................................................... 50
15.6 Reporting of Pregnancy .............................................................................. 51
15.7 Protocol Amendments ................................................................................. 52
15.8 Protocol Deviations ...................................................................................... 52 15.9 FDA Annual Reporting ................................................................................ 52 15.10 Clinical Trials Data Bank ............................................................................. 52
15.11 Record Keeping............................................................................................ 52 16 BIBLIOGRAPHY .................................................................................................. 53
Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 6 of 54 Template version date: 3-5-2015
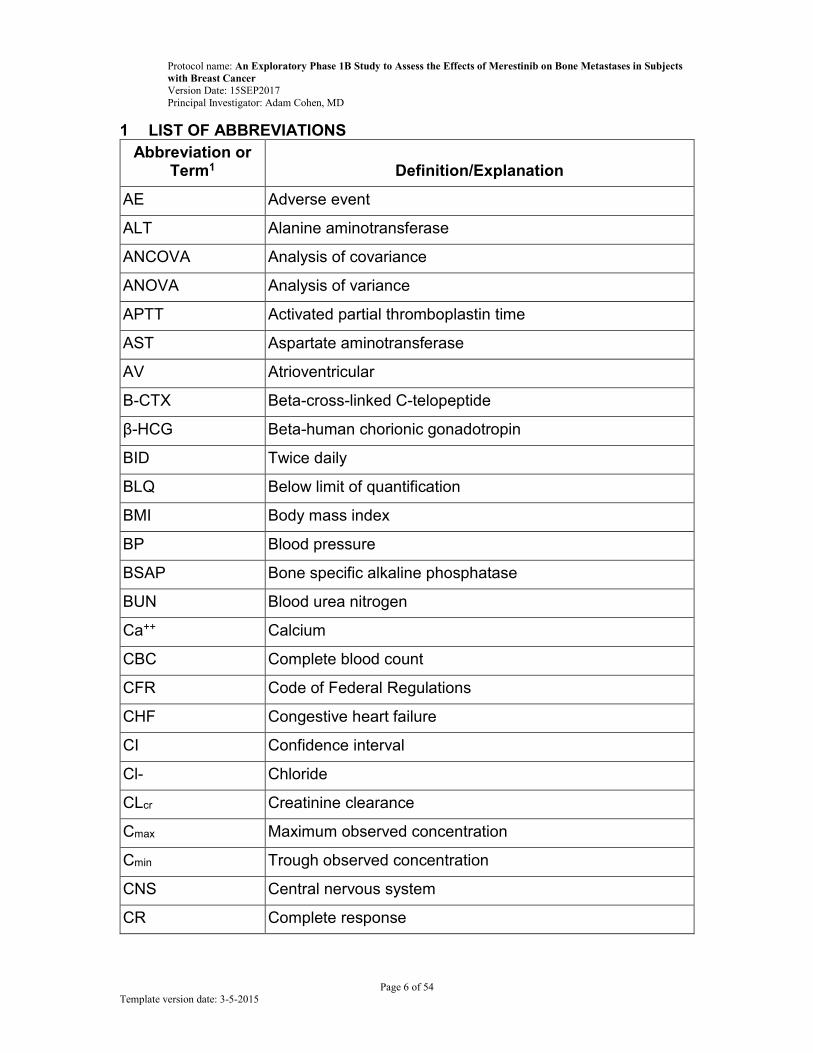
1 LIST OF ABBREVIATIONS
Abbreviation or Term1 Definition/Explanation
AE Adverse event
ALT Alanine aminotransferase
ANCOVA Analysis of covariance
ANOVA Analysis of variance
APTT Activated partial thromboplastin time
AST Aspartate aminotransferase
AV Atrioventricular
Β-CTX Beta-cross-linked C-telopeptide
β-HCG Beta-human chorionic gonadotropin
BID Twice daily
BLQ Below limit of quantification
BMI Body mass index
BP Blood pressure
BSAP Bone specific alkaline phosphatase
BUN Blood urea nitrogen
Ca++ Calcium
CBC Complete blood count
CFR Code of Federal Regulations
CHF Congestive heart failure
CI Confidence interval
Cl- Chloride
CLcr Creatinine clearance
Cmax Maximum observed concentration
Cmin Trough observed concentration
CNS Central nervous system
CR Complete response

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 7 of 54 Template version date: 3-5-2015
Abbreviation or Term1 Definition/Explanation
CRF Case report form
CT Computed tomography
CTCAE Common Toxicity Criteria for Adverse Events
CV Coefficient of variation
CYP Cytochrome P450
D/C Discontinue
ECOG Eastern Cooperative Oncology Group
eCRF Electronic case report form
DLT Dose Limiting Toxicity
ECG Electrocardiogram
Eg Exempli gratia (for example)
FACS Fluorescence Activated Cell Sorting
FDA Food and Drug Administration
FDG-PET Fluorodeoxyglucose (FDG)-positron emission tomography (PET)
GCP Good Clinical Practice
GFR Glomerular filtration rate
GGT Gamma glutamyl transferase
GLP Good laboratory practice
HBsAg Hepatitis B surface antigen
HBV Hepatitis B virus
HCO3- Bicarbonate
HCV Hepatitis C virus
HIV Human immunodeficiency virus
HR Heart rate
hr Hour or hours
IC50 Half maximal inhibitory concentration

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 8 of 54 Template version date: 3-5-2015
Abbreviation or Term1 Definition/Explanation
i.e. Id est (that is)
IEC Independent ethics committee
INR International normalized ratio
IRB Institutional review board
IU International unit
IV Intravenous, intravenously
LDH Lactate dehydrogenase
LLQ Lower limit of quantitation
MedRA Medical Dictionary for Drug Regulatory Activities
MRI Magnetic resonance imaging
MRSD Maximum recommended starting dose
MSP Macrophage stimulating protein
MTD Maximum tolerated dose
NaF-PET/CT Sodium Fluoride (NaF) positron emission tomography (PET)/Computed Tomography(CT)
NOAEL No-observed-adverse-effect level
NOEL No-observed-effect-level
NTX Cross-linked N-telopeptide
P1NP Procollagen type 1 intact N-terminal propeptide
PD Pharmacodynamic(s)
PFS Progression Free Survival
PK Pharmacokinetic(s)
PO Per os (administered by mouth)
PR Partial response
PT Prothrombin time
PTT Partial thromboplastin time
QC Quality control

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 9 of 54 Template version date: 3-5-2015
Abbreviation or Term1 Definition/Explanation
RBC Red blood cell
QD Once daily
QTc QT interval corrected
QTcF QT interval corrected using Frederichia equation
SAE Serious adverse event
SD Standard deviation or stable disease
T1/2 Terminal elimination half-life
T3 Triiodothyronine
T4 Thyroxine
Tmax Time of maximum observed concentration
TID Three times daily
TSH Thyroid-stimulating hormone
ULN Upper limit of normal
ULQ Upper limit of quantitation
UV Ultraviolet
WBC White blood cell
WOCBP Women of childbearing potential
WONCBP Women of nonchildbearing potential
All of these abbreviations may or may not be used in protocol.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 10 of 54 Template version date: 3-5-2015
2 PROTOCOL SIGNATURE I confirm that I have read this protocol, and I will conduct the study as outlined herein and according to the ethical principles stated in the latest version of the Declaration of Helsinki, the applicable ICH guidelines for good clinical practice, and the applicable laws and regulations of the federal government. I will promptly submit the protocol to the IRB for review and approval. Once the protocol has been approved by the IRB, I understand that any modifications made during the course of the study must first be approved by the IRB prior to implementation except when such modification is made to remove an immediate hazard to the subject. I will provide copies of the protocol and all pertinent information to all individuals responsible to me who assist in the conduct of this study. I will discuss this material with them to ensure that they are fully informed regarding the study treatment, the conduct of the study, and the obligations of confidentiality. Note: This document is signed electronically through submission and approval by the Principal Investigator in the University of Utah IRB Electronic Research Integrity and Compliance Administration (ERICA) system.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 11 of 54 Template version date: 3-5-2015
3 STUDY SUMMARY
Title An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects with Breast Cancer
Short Title N/A
Protocol Number 103657
IND 136622
Phase 1B
Design Single Arm
Study Duration 18 months
Study Center(s) Single site: Huntsman Cancer Institute
Objectives Primary: • To assess the tolerability of
Merestinib in combination with standard breast cancer therapies
• To measure the change in urinary N-telopeptide level after 12 weeks of therapy
Secondary: To measure the absolute and
percentage change in serum β–CTX, TRAP-5b, P1NP, and BSAP at time points from baseline to end of study (week 12)
To evaluate time to skeletal-related event(s) following initiation of merestinib dosing
To evaluate the change in pain scores and pain medication usage for patients with bone pain
To evaluate changes in NaF PET scan uptake with 12 weeks of merestinib therapy
Exploratory: To measure MSP levels in
serum/plasma and tumor (when available) and determine whether MSP levels correlate with response to merestinib
To measure RON and pRON levels in tumor biopsy samples (when available) and determine whether

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 12 of 54 Template version date: 3-5-2015
RON or pRON levels correlate with response to merestinib.
To collect peripheral blood leukocytes at time points for future immunophenotyping, to assess markers of immune response.
To collect tumor biopsy samples (when available) for future assessment of tumor-infiltrating lymphocytes (TILs) and determine whether TILs correlate with response to merestinib and to compare TIL clones using single cell sequencing.
Number of Subjects 16 evaluable (8 on bisphosphonate and 8 on denosumab)
Diagnosis and Main Eligibility Criteria Indication: Breast Cancer patients with bone metastases. Inclusion Criteria:
Subjects 18 years of age or older at the time of written informed consent is obtained
Subjects with a histological diagnosis of breast cancer
Subjects with presence of metastatic bone disease
o At least 1 osteolytic bone metastases must be present
Urinary N-telopeptide level above the ULN for age at ARUP
Archived or freshly biopsied primary and/or bone metastatic tumor tissue available in paraffin-embedded blocks or slides
Current and/or prior use of chemotherapy and/or hormonal therapy is allowed
At least one prior therapy for metastatic breast cancer
Concurrent treatment with bisphosphonates or denosumab is required
Exclusion criteria (abbreviated here; please see full protocol)

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 13 of 54 Template version date: 3-5-2015
History of osteonecrosis of the jaw
Prohibited treatments and/or therapies:
Patients receiving medications that are known to be substrates of CYP2C8 (including paclitaxel), CYP2C9, or CYP2C19 or to be oral substrates of CYP3A with narrow therapeutic window. Subjects who have discontinued any of these medications must have a wash-out period of at least 5 days or 5 half-lives of the drug (whichever is longer) prior to the first dose of merestinib
Exposure to any investigational drug or placebo within 4 weeks of enrollment
History of diseases with influence on bone metabolism, such as Paget’s disease, osteogenesis imperfecta, hyperparathyroidism, and untreated primary or secondary hyperthyroidism within 12 months prior to study entry
Study Product, Dose, Route, Regimen
Merestinib 20mg PO daily for 2-4 weeks then 40mg PO Qday for 2-4 weeks then 80mg PO Qday
Duration of administration 12 weeks (drug will continue to be provided longer term if there is benefit)
Reference therapy None
Statistical Methodology Rates of AEs will be described. The percent of patients with each potential MTD will be reported. The primary endpoint (change in urinary N-telopeptide levels) will be assessed with a paired t-test. These analyses will be conducted in accordance with the intention-to-treat principle. Secondary analyses will evaluate the per-protocol population. The time to

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 14 of 54 Template version date: 3-5-2015
first on-study Skeletal Related Event (SRE) will be evaluated using Kaplan-Meier product-limit survival methods and Cox proportional hazards.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 15 of 54 Template version date: 3-5-2015
1 OBJECTIVES
1.1 Primary Objectives and Endpoint
1.1.1 To assess the tolerability of merestinib in combination with standard breast cancer therapies.
1.1.2 To measure the change in urinary N-telopeptide level after 12 weeks of therapy with merestinib
1.2 Secondary Objectives and Endpoint
1.2.1 To measure the absolute and percentage change in serum β–CTX, TRAP-5b, P1NP, and BSAP at time points from baseline to 12 weeks.
1.2.2 To evaluate determine time to skeletal-related event(s) following initiation of merestinib dosing
1.2.3 To evaluate change in pain (as measured by pain scores and narcotic use) during and after 12 weeks of merestinib treatment
1.2.4 To evaluate the change in bone lesion uptake on NaF PET scan after 12 weeks of merestinib treatment.
1.2.5 To measure MSP levels in serum/plasma and tumor (when available) and determine whether MSP levels correlate with response to merestinib.
1.2.6 To measure RON and pRON levels in pretreatment tumor biopsy samples (when available) and determine whether RON or pRON levels correlate with response to merestinib.
1.2.7 To collect peripheral blood leukocytes at time points for future immunophenotyping, to assess markers of immune response.
1.2.8 To collect tumor biopsy samples (when available) for future assessment of tumor-infiltrating lymphocytes (TILs) and determine whether TILs correlate with response to merestinib and to compare TIL clones using single cell sequencing.
2 BACKGROUND
2.1 Background on Breast Cancer Bone Metastasis Osteolytic bone metastasis is a problem associated with metastatic breast, lung, thyroid, and kidney cancers, as well as multiple myeloma. A characteristic feature of these metastases is their osteolytic nature, which causes remarkable bone loss, fractures, severe pain, hypercalcemia, and other detrimental effects. Approximately 70% of breast cancer patients with advanced disease develop at least one bone lesion [1, 2]. The prognosis of breast cancer patients with bone

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 16 of 54 Template version date: 3-5-2015
metastases is poor, with a median survival time from detection of lesions of about 1 year. [3] Bone lesions cause morbidity through skeletal related events (SREs). SREs include pathologic fractures, pain or instability requiring focal radiation or surgery, spinal cord compression, or hypercalcemia. Standard treatment for preventing SREs involves the use of anti-osteoclastic agents, such as intravenous bisphosphonates or the anti-RANKL antibody denosumab. However, even with the use of these, ~40% of women with breast cancer bone metastases will have an SRE in a 2 year period. [4] Therefore, better treatment of bone metastases is needed.
2.2 Background on MSP and RON The Macrophage Stimulating Protein (MSP), its receptor RON, and the protease that activates MSP, MT-SP1, are concurrently expressed in up to 20% of breast cancer patients. The overexpression of this pathway has been shown to be an independent prognostic indicator for metastasis and poor survival [5]. Breast cancer patients whose tumor expresses high levels of MSP, the MSP activator matriptase, and/or RON exhibit increased metastasis to lung, liver, brain, and bone, with bone being the most frequent site of metastasis. RON is expressed in osteoclasts, and expression of RON increases dramatically during osteoclast differentiation [6]. In addition, MSP has been shown to cause activation of osteoclasts in vitro [7, 8]. This suggests that the ability of tumors that either express MSP or cause activation of serum-derived MSP at the tumor site [9] may drive osteolytic bone metastasis due to a favorable interaction of active MSP with differentiated, mature osteoclasts. Treatment with a RON inhibitor, ASLAN002, successfully inhibits the development and progression of osteolytic lesions caused by breast cancer cells in an animal model (Figure 1). ASLAN002 has been studied in a Phase I monotherapy clinical trial in cancer patients and has demonstrated a relatively safe and reversible toxicity profile that can be easily monitored (NCT01721148). In addition, analysis of markers of bone turnover in these Phase I patients demonstrates favorable changes in bone turnover markers after treatment with ASLAN002, especially in women (decreased CTX and increased BSAP; Figure 2). ASLAN002 is no longer available. Nonetheless, this data suggests that osteoclasts expressing RON could be a potentially useful therapeutic target in patients with osteolytic bone metastasis, particularly when the patients’ tumor expresses the RON ligand, MSP. RON is also expressed in approximately 50% of breast tumors and in resident macrophages [5]. Our data also indicate that inhibiting RON has additional benefit by inhibiting growth of RON-expressing tumors and by inducing an improved anti-tumor immune response that is especially effective in metastatic

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 17 of 54 Template version date: 3-5-2015
outgrowth [10, 11]. Thus, we believe that RON inhibition can have a dual anti-tumor and anti-osteolytic effect in patients with bone metastasis.
2.3 Background on Merestinib
Merestinib (formerly LY2801653) is an oral tyrosine kinase inhibitor with close chemical similarity to ASLAN002 that is also a potent inhibitor of the RON kinase (IC50 12nM in biochemical assays and 11nM in cellular assays). [12] Merestinib also inhibits 15 other kinases (MET, AXL, ROS1, PDGFRA, FLT3, MERTK, TYR03, TEK, DDR1, DDR2, neurotrophic tyrosine receptor kinase [NTRK]-1/2/3, MKNK1/2). Merestinib has been investigated in patients with advanced cancer including advanced or metastatic solid tumors, adenocarcinoma of the colon or rectum, head and neck squamous cell carcinoma (HNSCC), cholangiocarcinoma (CCA), or uveal melanoma with liver metastasis. As of 20 August 2016, 204 patients and 52 healthy subjects have been enrolled in clinical trials of merestinib in doses ranging from 5mg per day to 240mg per day. All doses above 10mg per day achieve serum concentrations of at least 50ng/ml, which is three times the IC50 of RON. (See Figure 6.1 from Investigational Brochure)
As a single agent, 120mg per day is the recommended phase 2 dose, and 80mg has been recommended for combination studies. Merestinib is well tolerated with the primary toxicity being elevated liver enzymes. (See Table 6.6 from Investigator’s Brochure)

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 18 of 54 Template version date: 3-5-2015
2.4 Background on Sodium Fluoride F 18 Injection
Sodium Fluoride F 18 Injection is a FDA approved positron-emitting radiopharmaceutical containing radioactive fluoride 18F. The imaging agent is used for diagnostic purposes in conjunction with positron emission tomography (PET) imaging. Fluoride 18F ions decay by positron emission with a half-life of 109.7 minutes. Fluoride 18F ions usually accumulate in the skeleton in an even fashion, with greater deposition in the axial skeleton (e.g., vertebrae and pelvis) than in the appendicular skeleton, and greater deposition in the bones around joints than in the shafts of long bones. Increased deposition of fluoride 18F around joints can occur in arthritis or following trauma. Sodium Fluoride F 18 Injection is indicated as a bone imaging agent to define areas of altered osteogenic activity. The tendency for fluoride 18F to accumulate in the vicinity of primary and metastatic malignancy in bone has proven clinically useful in detection of such lesions [18, 19, 20, 21, 22, 23]. Studies have shown sodium fluoride F 18 (18F-NaF) PET and 18F-NaF PET/computerized axial tomography (CT) to be superior to conventional methods, such as whole-body magnetic resonance imaging (MRI) and 99mTc-methylene diphosphonate (MDP) bone scans, for accurate and sensitive detection of bone metastases [18, 19, 20, 22, 23].
2.5 Rationale for Conducting this Study
There is a great need for new therapies for metastatic cancers that have progressed on or are unresponsive to current therapy. There are no approved

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 19 of 54 Template version date: 3-5-2015
RON targeted agents in standard clinical use. In the population of subjects with advanced cancer with osteolytic bone metastasis and/or whose disease is unresponsive to standard treatment, the potential for benefit from merestinib outweighs the potential risks for toxicity.
2.6 Risk/Benefit Assessment
Potential Risks: Patients undergoing treatment with merestinib may suffer side effects from this treatment (see Section 3.0). Patients will also undergo the physical risks of an additional biopsy and additional blood draws and tests. These risks are not significantly increased compared to patients who participate in other research trials or those that are treated with investigational drugs. Protection against Risks: Potential risks may arise due to treatment with an investigational drug. However participants will be carefully monitored by the co-investigator physicians participating in this trial. Standard adverse event criteria and reporting mechanisms will be followed. Potential Benefits: Treatment with merestinib may provide additional benefits in several aspects of patient care including: a reduction in bone destruction caused by tumor lesions, reduction of bone pain and, potentially, a reduction of metastases in sites other than bone. Because the anticipated benefits to participants are substantial, the risks to subjects are reasonable in relation to the anticipated benefits.
2.7 Primary Hypothesis Our central hypothesis is that patients with osteolytic bone metastasis/es with high levels of bone turnover despite standard therapy (including bisphosphonates or denosumab) will exhibit less bone destruction after treatment with the RON inhibitor merestinib. Measurable endpoints of the study will include levels of serum bone resorption markers. Patients who achieve benefit, as determined by their clinician, will be given the opportunity to remain on merestinib until such time as they progress or are considered to no longer benefit from treatment with merestinib. This study will also measure time to skeletal-related events, time to detection of a new metastasis in bone or any other site, as well as evaluation of any existing visceral metastases by RECIST criteria.
2.8 Rationale for population and endpoints Markers of bone turnover are strong predictors of SREs. Commonly used markers of bone turnover include the cross-linked N-telopeptide of type 1 collagen (NTX), which can be measured in the serum or urine, the beta-cross-linked C-telopeptide of type 1 collagen (CTX), N-terminal propeptide of type 1

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 20 of 54 Template version date: 3-5-2015
procollagen (P1NP), and bone specific alkaline phosphatase (BSAP), all measured in the serum. Elevated urine NTX in cancer patients not taking anti-resorptive medications increased the risk of SRE by 150%. [13] In women with breast cancer bone metastases taking bisphosphonates, having a persistent elevation in urinary NTX levels doubles the rate of SREs. [14] Therefore, women who have elevated urinary NTX levels despite standard therapy are a population in need of better therapy for bone metastases. In prior studies of therapies for bone metastases, various thresholds for success have been used. In osteoporosis, a decrease in serum NTX of 30% or of urine NTX by 50% is considered evidence for effect of anti-resoprtive therapy. [15] In cancer studies of denosumab or bisphosphonates, 65% decrease [14, 16] in urine NTX has been used as an endpoint.
2.9 Preliminary data (pre-clinical)
In preclinical studies, overexpression of MSP in tumor cells driven by the polyomavirus middle T antigen under the control of the mouse mammary tumor virus promoter (MMTV-PyMT) led to spontaneous metastasis to bone and a significant increase in metastasis to other organs [5]. Breast cancer cells injected directly into the tibia of mice cells develop into osteolytic lesions which are visible by X-ray. PyMT-MSP cells are primary mouse mammary tumor cells that have been engineered to overexpress MSP. DU4475 is a human cell line that was derived from a patient with triple negative breast cancer. DU4475 express high levels of MSP endogenously. Treatment of these mouse models with the RON inhibitor ASLAN002 in an adjuvant setting, shortly after injection of tumor cells, was able to prevent a significant amount of bone destruction normally caused by these lesions (Figure 1 A,B). In similar mouse studies, the RON inhibitor ASLAN002 was also able to prevent growth of some primary tumors expressing RON [11] and growth of lung metastases through activation of a robust anti-tumor immune response [10]. Thus, in this study we will also follow response of metastases outside of the bone (when applicable) to the RON inhibitor merestinib.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 21 of 54 Template version date: 3-5-2015
Figure 1. A. B.
Figure 1. RON inhibition reduces MSP tumor-induced osteolysis. (A) X-ray images of PyMT-MSP bone lesions from mice treated with the RON inhibitor ASLAN002. Treatment began 3 days post tumor cell injection. Mice were sacrificed 21 days post injection for analysis. The graph represents quantification of the osteolytic area in PyMT-MSP bone lesions from mice treated with the RON inhibitor ASLAN002 (n=4-5 per group). (B) Representative X-ray images of DU4475-induced bone lesions from mice treated with the RON inhibitor ASLAN002. Treatment began 3 days post tumor cell injection and mice were sacrificed 28 days post injection for analysis. The graph represents quantification of the osteolytic area in DU4475 bone lesions from mice in each experimental group (n=5).
Human tumor cells
Primary mouse tumor cells
PyMT-MSP tumor cells

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 22 of 54 Template version date: 3-5-2015
2.10 Preliminary data (clinical)
The RON inbhibitor ASLAN002 has been studied in a Phase I clinical trial and has demonstrated a relatively safe and reversible toxicity profile that can be easily monitored (NCT01721148). In addition, analysis of markers of bone turnover in these Phase I patients demonstrates a significant change in bone turnover markers after treatment with the RON inhibitor ASLAN002, especially in women (Figure 2). Taken together, this data suggests that osteoclasts expressing RON could be a potentially useful therapeutic target in patients with osteolytic bone metastasis, particularly when the patients’ tumor expresses the RON ligand, MSP. Figure 2.
Figure 2. Serum levels of β-CTX in Phase I patients treated with ASLAN002. Levels of a bone turnover marker, β-CTX, were measured in patients with solid tumors in a Phase I trial. The bar graphs represent the greatest percent change in β-CTX levels normalized to the individual patients’ baseline. The graph on the left represents the percent change in females while the graph on the right represents the percent change in males.
3 DRUG INFORMATION
3.1 Mechanism of Action Merestinib inhibits RON phosphorylation. RON is a receptor tyrosine kinase whose ligand is MSP. Activation of RON leads to the binding and phosphorylation of adaptor proteins and subsequent activation of signal transducers, such as PI3K, AKT, and ERK.
-80
-60
-40
-20
0
20
40
Greatest % change in CTX level from baseline -
females
-40
-20
0
20
40
Greatest % change in CTX level from baseline -
males

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 23 of 54 Template version date: 3-5-2015
3.2 Nonclinical Pharmacokinetics Nonclinical metabolism and PK studies of merestinib were conducted in rats and dogs either in conjunction with toxicology studies or as separate PK investigations. These nonclinical studies were conducted toward the initial assessment of the absorption, distribution, metabolism, and excretion of merestinib as follows.
3.3 Absorption In rats, exposure to merestinib generally increased proportionally with dose over the dose range to 0.2 to 3 mg/kg (data from 4-week study), but greater than dose-proportional over the dose range 3 to 30 mg/kg (data from 13-week study). Exposures were consistent between the 2 studies, but potential accumulation was observed after dosing for 13 weeks in rats. Exposures in male rats trended slightly lower than in females, but with no clear consistent difference observed (<2-fold difference between groups at all doses). In dogs, exposure increased proportionally with increasing dose from 1 to 75 mg/kg (data from both the 4-week and the 13-week study). Exposures were consistent between the 2 studies, and potential accumulation was observed after multiple dosing in dogs. Exposures to merestinib were similar in male and female dogs. Absorption, distribution, metabolism, and excretion of merestinib in rats and dogs have been determined following single oral doses of [14C] merestinib. Parent merestinib is absorbed and is the main component circulating in the plasma of both species. Based on average values estimated in the nonclinical species, the predicted absorption rate constant was 0.433 hr-1, and the bioavailability was 60%.
3.4 Distribution In preliminary studies, merestinib was administered via gavage at 50 mg/kg to nude mice bearing S114 tumors (derived from ovary cells). Merestinib was distributed into tumors with a tumor-to-plasma ratio of 0.55 in mice at 2 hours postdose. A quantitative whole-body autoradiographic disposition study was conducted in male and female pigmented Long Evans rats and male nonpigmented Sprague Dawley rats following a single 10-mg/kg oral dose of [14C]merestinib. Radioactivity related to [14C]merestinib was extensively distributed into tissues and was selectively associated with melanin-containing tissues. Maximum concentrations of radioactivity for Long Evans rats were observed at 4 hours postdose, and concentrations generally reduced over time. The liver, uveal tract of the eye, epididymis (male), pigmented skin, small intestine, and thymus (female) were the tissues with the highest concentrations. Very low levels of radioactivity crossed the blood–brain barrier and blood–testis barrier. Merestinib shows almost complete absorption after oral administration with the enabled formulation. Maximum plasma concentration occurs after approximately 5

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 24 of 54 Template version date: 3-5-2015
hours. There is extensive variability in the rate of absorption. Healthy subject data indicate that a high-fat meal may enhance merestinib exposure to more than twice that observed in the fasted state.
3.5 Metabolism The metabolism of merestinib was studied in vitro in rat, dog, and human cryopreserved hepatocytes, and in vivo in rats and dogs. Parent compound undergoes oxidative metabolism producing 2 predominant metabolites LSN2800870 (N-desmethylation) and LSN2887652 (oxidation of methylpyridone). LSN2800870 and LSN2887652 are both pharmacologically active metabolites. The metabolism of merestinib was examined in intact or biliary cannulated male Sprague Dawley rats following a single 10-mg/kg oral dose of [14C]merestinib or male Beagle dogs following a single 1-mg/kg intravenous or 10-mg/kg oral dose of [14C]merestinib. More metabolites were observed in rat feces, bile, and plasma than in dog feces and plasma. Urine was not profiled due to the low renal excretion of total radioactivity. The primary metabolic pathways observed in rats involved N-desmethylation of the N-methylindazole followed by oxidation and/or glucuronide conjugation on either the indazole or pyrazole moiety, and oxidation of the methyl substituent on the methylpyridone. An additional pathway that involved direct N-linked glucuronide conjugation was observed in dogs.
3.6 Excretion Excretion studies using [14C]merestinib were conducted following administration of a single oral or intravenous dose to rats or dogs. Table 5.6 summarizes the percentage of radiolabeled dose eliminated. The majority of the dose was excreted in the bile or feces. Renal excretion is negligible
3.7 Adverse Effects 6.2.1.2.2. Serious Adverse Events

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 25 of 54 Template version date: 3-5-2015
Table 6.5 summarizes SAEs that were considered possibly related to study drug treatment in Study JSBA. As of 20 August 2016, a total of 76 SAEs in 41 patients have been reported in Study JSBA. Ten patients experienced a total of 14 possibly treatment-related SAEs.
Overall, 177 of 181 patients (97.8%) reported at least 1 TEAE in Study JSBA. Across all study parts and grades, the most frequent TEAEs occurring patients and believed to be related to study drug treatment were fatigue (28.2%), ALT increased (23.2%), AST increased (20.4%), nausea (18.8%), edema peripheral (12.2%), and vomiting (11.6%). Three patients in Study JSBA discontinued treatment due to merestinib AEs of AST increased, left ventricular systolic dysfunction, and blood bilirubin increased. The AE of left ventricular dysfunction occurred in the setting of antecedent concentric left ventricular hypertrophy in a patient who mistakenly increased his daily dose to a dose greater than was assigned. The AE of blood bilirubin increased in 1 patient occurred in the setting of a disease-related biliary tract obstruction. Consequently, the investigator assessed this event as related to both the biliary tract obstruction and study drug treatment.
3.8 Pregnancy and Lactation No information is available on the use of merestinib in pregnant or nursing women. Merestinib causes loss of pregnancy in pregnant female rats treated during the period of organogenesis. Women of childbearing potential and their sexual partners should be instructed to use an approved, double barrier method of

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 26 of 54 Template version date: 3-5-2015
contraception throughout treatment with merestinib and for a minimum of 3 months thereafter.
3.9 Drug Interactions On the basis of in vitro data, there is a possible risk of drug-drug interaction when merestinib is coadministered with substrates of CYP2C8, CYP2C9, and CYP2C19 that have a narrow therapeutic range, such as paclitaxel, warfarin, phenytoin, tricyclic antidepressants, and tolbutamide, as concentrations of these drugs may be increased. Concomitant use with paclitaxel is currently excluded in clinical trials. There is also a possible risk of drug–drug interactions when merestinib is coadministered with strong CYP3A4 inhibitors or inducers. It is recommended that strong CYP3A4 inhibitors and inducers be avoided within 2 weeks before the start of study drug or during the study unless necessary to treat an emergent or life-threatening medical need. Investigators should consider other medications that are not CYP3A4 inhibitors or inducers and, when this is not possible, assess the risks with concomitant administration in their decision making. Furthermore, although there was no evidence of mechanism-based inhibition of CYP3A by merestinib at up to 12 µM, because concentrations in the intestine could be >12 µM, the potential for merestinib to inhibit CYP3A metabolism in the intestine cannot be completely ruled out. This could theoretically affect orally administered concomitant medications that are substrates of CYP3A and have a narrow therapeutic index. In vitro studies to assess the potential for transporter-mediated drug–drug interactions with merestinib suggest merestinib may increase the concentrations of P-gp substrates. Until the clinical evaluation of this interaction is complete, digoxin therapeutic drug monitoring is recommended for patients taking digoxin.
3.10 Dosing and Administration An intrapatient dose escalation design will be used. Eligible patients will receive merestinib at 20mg daily for 2-4 weeks followed by 40mg daily for 2-4 weeks followed by 80mg daily for 4-8 weeks. Merestinib will be administered daily with water (approximately 200 mL) with food, approximately at the same time (±2 hours), each day.
3.11 Preparation, Storage and Reconciliation of Supplies Merestinib is supplied for clinical trial use as tablets. Each tablet contains 5, 20, 40, or 80 mg of merestinib. The 40 mg strength tablet is available as a blue color, nonagon shape tablet or a beige color round tablet. The 5 mg (round shape), 20 mg (capsule shaped), and 80 mg (modified oval shaped) are all beige in color.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 27 of 54 Template version date: 3-5-2015
The tablets are composed of merestinib and the following inactive ingredients:
silicon dioxide
croscarmellose sodium
hypromellose acetate succinate
mannitol
microcrystalline cellulose
sodium lauryl sulfate
sodium stearyl fumarate The proprietary color mix contains blue hypromellose, titanium dioxide, triacetin, and FD&C Blue 2 indigo carmine. The product should be stored at room temperature. It is the responsibility of the Investigator to ensure that a current record of investigational product disposition is maintained at study site where investigational product is inventoried and disposed. Records or logs must comply with applicable regulations and guidelines.
4 STUDY DESIGN
4.1 Description This is a single arm, open label, pharmacodynamics, intrapatient dose escalation phase 1B study.
4.2 Dose Limiting Toxicity DLTs will be assessed beginning with the first dose through week 12. Because this is an intrapatient dose escalation trial, the MTD for each person will be determined. The MTD for each person will be defined as either the highest dose at which no DLT is experienced for that person or 80mg per day, whichever is lower. DLTs will cause dose de-escalation, but women will be taken off study only for DLTs that occur at dose level 0.
AEs will be assessed using CTCAEv4. AEs will be attributed as definitely, probably, possibly, or unlikely to be related to the investigational drug and to standard of care medication.
DLTs will be defined as any of the following events if they are possibly, probably, or definitely related to merestinib:
Any death not clearly due to the underlying disease or extraneous causes
Non- hematologic toxicity:

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 28 of 54 Template version date: 3-5-2015
o Grade 3 or higher (may exclude grade 3 fatigue if duration < 1 week)
o Hy's law o AST or ALT > 3 times the upper limit of normal AND o Total bilirubin > 2 times the upper limit of normal AND o Alkaline phosphatase < 2 times the upper limit of normal AND o No other reason for liver injury
Hematologic toxicity o Grade 4 neutropenia or thrombocytopenia >7 days o ≥Grade 3 thrombocytopenia with bleeding o Neutropenic fever
Any intolerable grade 2 toxicity, i.e., a grade 2 toxicity for which the patient stops the drug for more than 7 days or is unwilling to continue at the same dose despite appropriate supportive care, unless the PI or a sub-investigator deems it probably or definitely due to the standard of care medications or the cancer.
Any AE that causes delay in anti-cancer therapy by more than 4 weeks unless the PI or a sub-investigator deems it definitely unrelated to the investigational drug.
The following will never be considered AEs: Abnormal lymphocyte counts, abnormal cholesterol, obesity.
4.3 Number of Patients
16 evaluable patients will be enrolled. Patients will be considered evaluable if
they have not permanently discontinued therapy at the 12 weeks evaluation and
have urinary NTX levels measured at the beginning of therapy and after 12
weeks. Non-evaluable patients will be replaced to ensure 8 evaluable patients
per cohort. Patients will be stratified based on whether they are on
bisphosphonate or denosumab. 8 patients on bisphosphonate and 8 patients on
denosumab will be enrolled. Patients who discontinue bisphosphonate or
denosumab will not be replaced given the long half-lives of these drugs.
4.4 Stopping Rules
If 3 of the first 10 subjects has a DLT on dose level 0 and has to stop the drug for toxicity that cannot be controlled with standard supportive care, then the trial will be stopped. With this rule, if the true percent of women who cannot tolerate dose level 0 is 35%, which is the standard stopping point for phase 1 studies, there is a 74% chance of stopping the study. If, as is more likely, the true percent of

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 29 of 54 Template version date: 3-5-2015
women who cannot tolerate dose level 0 is 10%, there is a 93% chance of not stopping the study.
4.5 Number of Study Centers
This is a single site trial at Huntsman Cancer Institute.
4.6 Study Duration
Participants will be on treatment for 12 weeks. Participants who are
considered to be benefitting from merestinib will have the option of continuing
on merestinib as long as they are benefitting. From enrollment of the first
patient until the end of the primary endpoint evaluation for the last patient is
expected to be 15 months.
5 ELIGIBILITY CRITERIA
This eligibility checklist is used to determine patient eligibility and filed with signature in the patient research chart. Patient No. ______________________ Patient’s Initials: (L,F,M) _________________
5.1 Inclusion Criteria for Urinary Screening Yes/No (Response of “no” = patient ineligible)
5.1.1 _____ 18 years of age or older at the time of written informed consent is obtained
5.1.2 _____ Female at birth
5.1.3 _____ Post-menopausal or undergoing ovarian suppression
5.1.4 _____ Histological diagnosis of breast cancer
5.2 Inclusion Criteria for Treatment
5.2.1 _____ At least 1 osteolytic bone metastases must be present
5.2.2 _____ Urinary N-telopeptide level above the ULN for age measured at ARUP
5.2.3 _____ Archived or freshly biopsied primary and/or bone metastatic

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 30 of 54 Template version date: 3-5-2015
tumor tissue available in paraffin-embedded blocks or slides that is expected to yield 9 slides
5.2.4 _____ Life expectancy of ≥ 6 months
5.2.5 _____ Toxicity related to prior treatments must either have resolved to grade 1 or less, returned to baseline, or be deemed irreversible
5.2.6 ______Eastern Cooperative Oncology Group (ECOG) performance status of 0 or 1 (within 28 days prior to enrollment)
5.2.7 ______ Planning to remain on current breast cancer therapy for at least 12 weeks.
5.2.8 ______ At least one prior line of therapy for metastatic breast cancer
5.2.9 ______ Concurrent treatment with bisphosphonates or denosumab is required.
5.3 Exclusion Criteria Yes/No (Response of “yes” = patient ineligible)
5.3.1 _____ Unable to swallow or take anything orally
5.3.2 _____ ECG abnormalities:
Prolonged QTc (Bazette’s or Fredericia’s correction) interval on screening ECG (≥ 450 msec)
QRS ˃ 120 msec
PR ˃ 210 msec
Any prior history, or current evidence of second- or third-degree heart block
Heart rate ˂ 40 beats per minute at screening
ECG second degree heart block (Mobitz’s Type 2 or Wenckebach)
Complete heart block
Left bundle branch block or bifascicular block (right bundle branch block and left anterior hemiblock together)
Episodes of ventricular tachycardia

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 31 of 54 Template version date: 3-5-2015
5.3.3 _____ Any known prior malignancy (not including non-melanoma skin cancers), unless treated with curative intent
5.3.4 _____ A serious uncontrolled medical disorder or active infection, which would impair the ability of the subject to receive protocol therapy
5.3.5 _____ Current or recent (within 3 months) gastrointestinal disease that could impact the absorption (i.e., unmanageable diarrhea or malabsorption at the time of screening)
5.3.6 _____ Inadequate bone marrow function defined as:
Absolute neutrophil count (ANC) ˂ 1,500 cells/mm3
Platelet count ˂ 100,000 cells/mm3
Hemoglobin ˂ 9 g/dL
5.3.7 _____ Inadequate hepatic function defined as:
Total bilirubin ˃ 1.5 x institutional upper limit of normal (IULN) (Unless due to diagnosis of Gilbert’s Syndrome)
Alanine aminotransaminase (ALT) and aspartate aminotransaminase (AST) ˃ 2.5 x IULN
5.3.8 _____ Inadequate renal function defined as: Serum creatinine ˃ 1.5 x ULN
5.3.9 _____ Prothrombin time (PT)/partial thromboplastin time (PTT) ˃ 1.5 times the ULN
5.3.10 _____ Serum sodium, potassium, and calcium levels equivalent to Grade 1 AE values as defined by Common Terminology Criteria for Adverse Events (CTCAE) version 4.0
5.3.11 _____ Any atrophic macular condition including intermediate or advanced age-related macular degeneration
5.3.12 _____ Patients receiving medications that are known to be substrates of CYP2C8 (including paclitaxel), CYP2C9, or CYP2C19 or to be oral substrates of CYP3A with narrow therapeutic window (listed on http://medicine.iupui.edu/clinpharm/ddis/main-table). Subjects who have discontinued any of these medications must have a wash-out period of at least 5 days or 5 half-lives of the drug (whichever is longer) prior to the first dose of merestinib

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 32 of 54 Template version date: 3-5-2015
5.3.13 _____ Exposure to any investigational drug or placebo within 4 weeks of enrollment
5.3.14 _____ Any other sound medical, psychiatric, and/or social reasons as determined by the investigator
5.3.15 _____ History of diseases with influence on bone metabolism, such as Paget’s disease, osteogenesis imperfecta, active primary or secondary hyperparathyroidism, and primary or secondary hyperthyroidism within 12 months prior to study entry
5.3.16 _____ Patients with known symptomatic brain metastasis. Subjects with controlled brain metastasis (no radiographic progression at least 4 weeks following radiation and/or surgical treatment and no neurological signs or symptoms) will be allowed
5.3.17 _____ History of allergy to merestinib or chemically related compounds
5.3.18 _____ History of osteonecrosis of the jaw
5.3.19 _____ Change in chemotherapy or hormone therapy within 8 weeks of the start of the study.
5.3.20 _____ Active gout or inflammatory arthritis requiring treatment
5.3.21 _____ Use within 28 days of registration of calcitonin, recombinant parathyroid hormone-related peptides, mithramycin, radium, strontium ranelate, or gallium nitrate.
5.3.22 _____ Adult patients who require monitored anesthesia for PET scanning due to claustrophobia.
I certify that this patient meets all inclusion and exclusion criteria for enrollment onto this study. ______________________________ _________ _______ Investigator Signature Date Time

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 33 of 54 Template version date: 3-5-2015
6 STRATIFICATION FACTORS Women will be stratified based on the anti-resorptive medication being used at baseline. Half of the population will be using bisphosphonates and half denosumab.
7 TREATMENT PLAN
7.1 Merestinib Administration Schedule The merestinib dose escalation timing will depend on the schedule of the other anticancer regimen.
7.1.1 For medications given continuously, such as tamoxifen or aromatase inhibitors (with or without ovarian suppression), or on a cycle length of 2 weeks or less, such as weekly adriamycin:
Subjects will receive merestinib 20mg PO QDay (dose level 0) for 2 weeks followed by 40mg PO Qday (dose level 1) for 2 weeks followed by 80mg PO daily (dose level 2) for 8 weeks. Dose escalation will occur at the week 3 and 5 visit if there have been no DLTs, as defined in section 4.2. If a DLT occurs, merestinib will be held until the toxicity resolves to grade 1 or less. Merestinib will then be restarted at the next lowest dose level. If a DLT occurs at dose level 0, merestinib will be stopped and the patient will be withdrawn from the study. Merestinib will be administered daily with water (approximately 200 mL) following a meal, approximately at the same time (±2 hours) each day.
7.1.2 For medications given intermittently on cycle lengths of 3 weeks: Subjects will receive merestinib 20mg PO QDay (dose level 0) for 3 weeks followed by 40mg PO Qday (dose level 1) for 3 weeks followed by 80mg PO daily (dose level 2) for 6 weeks. Dose escalation will occur at week 4 and 7 visits if there have been no DLTs, as defined in section 4.2. If a DLT occurs, merestinib will be held until the toxicity resolves to grade 1 or less. Merestinib will then be restarted at the next lowest dose level. If a DLT occurs at dose level 0, merestinib will be stopped and the patient will be withdrawn from the study. Merestinib will be administered daily with water (approximately 200 mL) following a meal, approximately at the same time (±2 hours) each day.
7.1.3 For medications given intermittently on cycle lengths of 4 weeks: Subjects will receive merestinib 20mg PO QDay (dose level 0) for 4 weeks followed by 40mg PO Qday (dose level 1) for 4 weeks followed by 80mg PO daily (dose level 2) for 4 weeks. Dose escalation will occur at week 5 and 9 visits if there have been no DLTs, as defined in section 4.2. If a DLT occurs, merestinib will be held until the toxicity resolves to grade 1 or less. Merestinib will then be

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 34 of 54 Template version date: 3-5-2015
restarted at the next lowest dose level. If a DLT occurs at dose level 0, merestinib will be stopped and the patient will be withdrawn from the study. Merestinib will be administered daily with water (approximately 200 mL) following a meal, approximately at the same time (±2 hours) each day. The week 3 visit will still occur to allow for safety monitoring.
7.2 Merestinib Treatment
7.2.1 How Supplied, Stored, Packaged and Labeled Eli Lilly will be responsible for the active ingredient and will be responsible for the final formulation and packaging of the investigational medicinal product (IMP). The physicochemical properties and the pharmaceutical specifications of the IMP for this study are provided in the Investigator’s Brochure (IB). Eli Lilly is responsible for shipping the IMP to study site and strict inventory control for the drug accountability will be implemented. Merestinib tablets should be stored in tightly closed original containers at temperatures between 15°C to 25°C (at controlled room temperature) and protected from light. The Investigator should ensure that the IMP is stored in accordance with the environmental conditions (temperature, light, and humidity) as determined by Eli Lilly. If concerns regarding the quality or appearance of the IMP arise, do not dispense the IMP and contact Eli Lilly immediately.
7.2.2 Preparation and Administration
Merestinib will be stored and dispensed by the Huntsman Cancer Institute Investigational Pharmacy. Enough medicine will be dispensed to last until the next scheduled study visit.
7.2.3 Accountability and Compliance
Sites will be required to keep a temperature log to establish a record of compliance with these storage conditions. It is the responsibility of the Investigator to ensure that a current record of IMP disposition is maintained at study site where IMP is inventoried and disposed. Records or logs must comply with applicable regulations and guidelines.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 35 of 54 Template version date: 3-5-2015
7.3 Sodium Fluoride F 18 Administration
7.3.1 Patient Preparation No specific preparation is required. The participant’s height and weight will be measured using calibrated and medically approved devices in order to determine accurate SUV measurements.
7.3.2 Injection Procedure Prior to injection, qualified site personnel will assay the dose in the dose calibrator and record the assay reading and time. After the injection of Sodium Fluoride F 18, the syringe will be re-assayed for residual activity and the assay reading and time recorded. An IV catheter is placed to allow for injection of the Sodium Fluoride F 18. The participant is then injected with 8-10 mCi. This is followed by an uptake period of 60 (55-70 allowed) minutes. Prior to placing the participant on the PET/CT scanner the participant should void their bladder. It is critical that the same uptake time be used for each of the 18F-NaF PET/CT scans obtained on the research participant.
7.3.3 NaF-PET/CT Imaging Procedure
The participant will be imaged in a supine position, with both arms positioned alongside their torso. An initial Scout Scan: 120-140 kV; 10-30 mA will be obtained. The non-diagnostic attenuation and localization scan will be obtained at 120 kVp, 0.5s rotation speed, 500 mA tube current, 64 x 1.25 mm collimation for the axial scans of the head and the legs. A pitch of 1.35 will be used for the helical scan of the torso and larger patients will use 140 kVp rather than 120. Scan FOV is vertex to feet. The participant should use shallow breathing. Time/bed position will be 4 minutes.
7.3.4 NaF-PET/CT Post-imaging
The participant should void the bladder after completing the scan. In addition, the participant should drink as much water as possible and void frequently after the scan. This will reduce the bladder radiation dose. Walking or similar physical activity post injection also appears to help reduce background activity.
7.4 Prohibited Concomitant Medications The following medications are prohibited during the study:
Oral substrates of CYP3A4 with narrow therapeutic index as listed on http://medicine.iupui.edu/clinpharm/ddis/main-table.
Substrates of CYP2C8, CYP2C9, and 2C19 with narrow therapeutic index, such as those listed on http://medicine.iupui.edu/clinpharm/ddis/main-table.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 36 of 54 Template version date: 3-5-2015
Calcitonin, recombinant parathyroid hormone-related peptides, mithramycin, radium, strontium ranelate, or gallium nitrate.
7.5 Duration of Therapy Subjects must be withdrawn from the study treatment for the following reasons:
Disease Progression
Change in anti-cancer therapy (stopping or taking a break from an anticancer therapy will not be considered a change requiring withdrawal)
Change in anti-resorptive therapy
Unacceptable Toxicity
Subject withdraws consent from the study treatment and/or study procedures. A subject must be removed from the trial at his/her own request or at the request of his/her legally acceptable representative. At any time during the trial and without giving reasons, a subject may decline to participate further. The subject will not suffer any disadvantage as a result.
Subject is lost to follow-up.
Death. Subjects may be withdrawn from the study for the following reasons:
The subject is non-compliant with study drug, trial procedures, or both; including the use of anti-cancer therapy not prescribed by the study protocol.
If, in the investigator's opinion, continuation of the trial would be harmful to the subject's well-being.
The development of a second cancer.
Development of an intercurrent illness or situation which would, in the judgment of the investigator, significantly affect assessments of clinical status and trial endpoints.
Deterioration of ECOG performance status to 4.
Subjects who withdraw from treatment before 4 weeks will be asked to participate in a final visit 4 weeks after discontinuation of therapy. All subject will be followed through 30 days after drug discontinuation for adverse events
Subjects who withdraw or are withdrawn before the 12 week urine NTX assessment will be replaced.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 37 of 54 Template version date: 3-5-2015
8 TOXICITIES AND DOSEAGE MODIFICATION This study will utilize the CTCAE (NCI Common Terminology Criteria for Adverse Events) Version 4.0 for adverse event and serious adverse event reporting. A copy of the CTCAE Version 4.0 can be downloaded: (http://safetyprofiler-ctep.nci.nih.gov/CTC/CTC.aspx).
8.1 Dose Modifications
Dose level Dose
0 20mg PO daily
1 40mg PO daily
2 80mg PO daily
As discussed in Section 7.1, patients will increase the dose to the next level at the week 3 and 5 visits for continuously or weekly dosed anti-cancer agents, the week 4 and 7 visits for anti-cancer agents doses every 3 weeks, or the week 5 and 9 visits for anti-cancer agents dosed every 4 weeks, unless there have been DLTs. Drug can be held for up to 7 days at one time or 14 days total for any reason. Drug must be taken for at least 3 consecutive days immediately before the week 12 urine NTX.
8.2 Supportive Care
8.2.1 All supportive measures consistent with optimal patient care will be given throughout the study.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 38 of 54 Template version date: 3-5-2015
9 STUDY CALENDAR For continuous, weekly, or every 4 week dosed anti-cancer agents:
Event Screening 12 weeks Treatment Phase
Follow-up
Study Day
Days -28 to Day -1
Day 1, Week 1 (First dose)
Week 3
+/- 3 days
Week 5
+/- 3 days
Week 9
+/- 3 days
Week 13/ End of Treatment
(EOT)
+/- 7 days
30 days
+/- 7 days
Step
1 Step
2 Visit 2 Visit 3 Visit 4 Visit 5 Visit 6
Sign Screening Informed Consent
X
Sign Protocol Specific Informed Consent/Enrollmenta
X
Medical History X
Height and Weight X X X X X X
Physical Examination X X X X X X
ECOG Performance Status/Vital Signs
X X X X X X
Standard 12-lead ECG X
CBC, CMP X X X X X
Coagulation Profile X X
Tumor Assessments (CT Chest/Abd/Pelvis or PET/CT)d X X
Merestinib administration Dosing will occur daily beginning Day 1
Monitor for Serious Adverse Eventsc
X X X X X X
Monitor for Non-Serious Adverse Events
X X X X X X
Recording of Concomitant Medications
X X X X X X X
Urine NTX X X X X X
Blood collection for bone turnover markers (β-CTX, serum NTX, P1NP, and BSAP) and exploratory endpoints (MSP, PBMC for immunophenotyping)
X (Prior to first dose)
X X X X
Pain VAS X X X X X
Opioid dose X X X X X X
NaF-PET/CT Xb X
Archival or fresh tissue from a metastasis (preferably bone) or primary
X

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 39 of 54 Template version date: 3-5-2015
ECOG=Eastern Cooperative Oncology Group; ECG=electrocardiogram; EOT=End-of-Treatment, IP= investigational product
a A subject is considered enrolled only when a protocol specific informed consent is signed b Within 7 days of Week 1 Day 1 (first dose) c Adverse event collection starts with administration of the first dose of the IP and continues until the last protocol related contact or final follow up d A diagnostic (with IV contrast) CT Chest/Abd/Pelvis or PET/CT will be used for tumor assessments.
For anti-cancer agents dosed every 3 weeks:
Event Screening 12 weeks Treatment Phase
Follow-up
Study Day
Days -28 to Day -1
Day 1, Week 1 (First dose)
Week 4
+/- 3 days
Week 7
+/- 3 days
Week 10
+/- 3 days
Week 13/ End of Treatment
(EOT)
+/- 7 days
30 days
+/- 7 days
Step
1 Step
2 Visit 2 Visit 3 Visit 4 Visit 5 Visit 6
Sign Screening Informed Consent
X
Sign Protocol Specific Informed Consent/Enrollmenta
X
Medical History X
Height and Weight X X X X X X
Physical Examination X X X X X X
ECOG Performance Status/Vital Signs
X X X X X X
Standard 12-lead ECG X
CBC, CMP X X X X X
Coagulation Profile X X
Tumor Assessments (CT Chest/Abd/Pelvis or PET/CT)d X X
Merestinib administration Dosing will occur daily beginning Day 1
Monitor for Serious Adverse Eventsc
X X X X X X
Monitor for Non-Serious Adverse Events
X X X X X X
Recording of Concomitant Medications
X X X X X X X
Urine NTX X X X X X
Blood collection for bone turnover markers (β-CTX, serum NTX, P1NP, and BSAP) and exploratory endpoints (MSP, PBMC for immunophenotyping)
X (Prior to first dose)
X X X X
Pain VAS X X X X X
Opioid dose X X X X X X
NaF-PET/CT Xb X

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 40 of 54 Template version date: 3-5-2015
10 STUDY PROCEDURES
10.1 Screening Within 28 days of registration, all patients will have a screening evaluation. The screening evaluation will occur in 2 steps. Step 1 Screening
1. Sign and date an IRB-approved protocol specific screening informed consent
2. Urine for urine NTX Subjects with a urine NTX above the upper limit of normal for age will then proceed to Step 2 screening. Subjects with a urine NTX at or below the upper limit of normal for age will be noted as screen failures. Step 2 Screening
1. Sign and date an IRB-approved protocol specific informed consent 2. Medical History 3. Height and Weight 4. Vital signs (blood pressure, pulse rate, respiratory rate, and
temperature) 5. Physical Examination 6. ECOG Performance Status 7. Standard 12-lead ECG 8. CBC 9. CMP 10. PT and PTT 11. Diagnostic CT Chest/Abd/Pelvis or PET/CT 12. Opioid dose 13. Recording of concomitant medications 14. Archival or fresh tissue availability confirmation
10.2 Visit 2 (Week 1, Day 1)
1. Pre-treatment Blood draw for MSP, bone turnover markers and exploratory endpoints
Archival or fresh tissue from a metastasis (preferably bone) or primary
X

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 41 of 54 Template version date: 3-5-2015
1. Pre-treatment β–CTX, NTX, P1NP, and BSAP. 2. Recording of Concomitant Medications 3. Begin Merestinib administration 4. Pain VAS 5. Opioid dose 6. NaF-PET/CT (before first dose)
10.3 Visit 3 The following procedures will be performed:
1. Height and Weight 2. Vital signs blood pressure, pulse rate, respiratory rate, and
temperature) 3. Physical Examination 4. ECOG Performance Status 5. Urine for urine NTX 6. CBC 7. CMP 8. Monitor for Serious Adverse Events 9. Monitor for Non-Serious Adverse Events 10. Recording of Concomitant Medications 11. Blood collection for bone turnover markers and exploratory
endpoints 12. β-CTX, serum NTX, P1NP, and BSAP. 13. Pain VAS 14. Opioid dose
10.4 Visit 4
The following procedures will be performed:
1. Height and Weight 2. Vital signs (blood pressure, pulse rate, respiratory rate, and
temperature) 3. Physical Examination 4. ECOG Performance Status 5. Urine for urine NTX 6. CBC 7. CMP 8. Monitor for Serious Adverse Events 9. Monitor for Non-Serious Adverse Events 10. Recording of Concomitant Medications 11. Blood collection for bone turnover markers and exploratory
endpoints

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 42 of 54 Template version date: 3-5-2015
12. β-CTX, serum NTX, P1NP, and BSAP 13. Pain VAS 14. Opioid dose
10.5 Visit 5
After taking merestinib for 8 weeks, subjects will be seen. The following procedures will be performed:
1. Height and Weight 2. Vital signs (sitting blood pressure, pulse rate, respiratory rate, and
temperature) 3. Physical Examination 4. ECOG Performance Status and Vital Signs 5. Urine for urine NTX 6. CBC 7. CMP 8. Monitor for Serious Adverse Events 9. Monitor for Non-Serious Adverse Events 10. Recording of Concomitant Medications 11. Blood collection for bone turnover markers and exploratory
endpoints 12. β-CTX, serum NTX, P1NP, and BSAP 13. Pain VAS 14. Opioid dose
10.6 Visit 6 (Week 13/End of Treatment (+/-7 days))
After taking merestinib for 12 weeks, subjects will be seen for the end of treatment visit. The following procedures will be performed:
1. Height and Weight 2. Vital signs (sitting blood pressure, pulse rate, respiratory rate, and
temperature) 3. Physical Examination 4. ECOG Performance Status and Vital Signs 5. Urine for urine NTX 6. CBC 7. CMP 8. PT and PTT 9. Diagnostic CT Chest/Abd/Pelvis or PET/CT 10. Monitor for Serious Adverse Events 11. Monitor for Non-Serious Adverse Events 12. Recording of Concomitant Medications

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 43 of 54 Template version date: 3-5-2015
13. Blood collection for bone turnover markers and exploratory endpoints
14. Pain VAS 15. Opioid dose 16. NaF-PET/CT
10.7 Follow-Up
1. Height and Weight 2. Vital signs (sitting blood pressure, pulse rate, respiratory rate, and
temperature) 3. Physical Examination 4. ECOG Performance Status and Vital Signs 5. Monitor for Serious Adverse Events 6. Monitor for Non-Serious Adverse Events 7. Recording of Concomitant Medications
11 CRITERIA FOR EVALUATION AND ENDPOINT
11.1 Efficacy Primary endpoint: Bone turnover will be assessed by measuring the level of urine NTX at baseline and 12 weeks. Women will be evaluable if they are still taking merestinib at the end of study assessment. The outcome will be measured as the percent change from baseline. Secondary endpoints: Bone turnover will be assessed by measuring the level of serum beta-CTX, TRAP-5b, P1NP, and BSAP at the beginning of week 3, 5, 9 and 13. Outcome is given as percent change from baseline. Skeletal related events (SREs) will be defined as pathologic fractures, bone pain or instability requiring focal radiation or surgery, spinal cord compression, or hypercalcemia. Bone pain score will be defined as maximum pain in the prior 24 hours will be assessed using a numeric scale from 0-10. Analgesic use: Total opioid use will be calculated and converted to morphine equivalents using (http://www.globalrph.com/narcotic.cgi), with 0 correction for cross-tolerance. NaF-PET/CT: One target lesion per patient will be chosen at the beginning of the study. The difference of SUVmax between the end of study and beginning of study scans in the target lesions will be the outcome.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 44 of 54 Template version date: 3-5-2015
Overall tumor assessment using the Diagnostic CT Chest/Abd/Pelvis or PET/CT will be done using RECIST 1.1
11.2 Safety
11.2.1 Safety Parameters and Definitions Safety assessments will consist of monitoring and recording adverse events, including serious adverse events and non-serious adverse events of special interest; measurement of protocol-specified safety laboratory assessments; measurement of protocol-specified vital signs; and other protocol-specified tests that are deemed critical to the safety evaluation of the study. Collection and reporting requirements for AE/SAE discussed in section 15.4 and 15.5
12 STATISTICAL CONSIDERATIONS In all analyses the bisphosphonate cohort and the denosumab cohort will be analyzed separately. For the primary endpoint, nonevaulable patients will be replaced to ensure 8 evaluable patients per cohort. For the secondary endpoints, patients with missing data for an endpoint will be excluded from analysis of that endpoint but will be included in all other analyses. Primary endpoint: Bone turnover will be assessed by measuring the level of urine NTX at baseline and 12 weeks using a paired t-test. Women will be evaluable if they are still taking merestinib at the end of study assessment. The outcome will be measured as the difference in level from baseline. Based on published papers, we estimate the standard error of the change from baseline in urine NTX at 3 months to be 8.7 percentage points if there is no effect and 15.6 percentage points if there is one, so averaging the square of these standard errors to estimate the standard deviation, with alpha of 0.05 and beta (1-power) of 0.15, we estimate a sample size of 8 patients per group to detect an average 60% decrease in urine NTX over the 12 weeks. [17] To account for drop out, we will recruit 10 people per group. Secondary endpoints: Bone turnover will be assessed by measuring the level of serum beta-CTX, TRAP-5b, P1NP, and BSAP. Outcome will be reported as percent change from baseline at each timepoint (beginning of

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 45 of 54 Template version date: 3-5-2015
week 3, 5, 9, and 13). Comparisons between baseline and week 13 will be performed using a paired t-test. Skeletal related events (SREs) will be defined as pathologic fractures, bone pain or instability requiring focal radiation or surgery, spinal cord compression, or hypercalcemia. SREs will be reported as the percent of people with an SRE during the study. Confidence intervals will be calculated using the normal approximation to the binomial confidence interval. Bone pain score Maximum pain in the prior 24 hours will be assessed using a numeric scale from 0-10. The change in pain scale from beginning of study to the end in people completing 12 weeks will be reported. Statistical significance of the change in pain will be assessed using the Mann-Whitney test. Analgesic use: Total opioid use will be calculated and converted to morphine equivalents using (http://www.globalrph.com/narcotic.cgi), with 0 correction for cross-tolerance. The change in total opioid use from the beginning of study to the end will be reported. Statistical significance of the change in opiod use will be determined using the paired t-test. NaF PET/CT: One target lesion per patient will be chosen at the beginning of the study. The difference of SUVmax between the end of study and beginning of study scans will be the outcome. Extrapolating from FDG-PET data, we assume a baseline mean SUV of 6.3 with standard deviation 3.4 and a coefficient of variation of 56%. With a 2-sided alpha of 0.05, 8 people gives an 80% power to detect a 60% mean decrease in SUV using a paired t-test.
13 REGISTRATION GUIDELINES Patients must meet all of the eligibility requirements listed in Section 5 prior to registration. Study related screening procedures can only begin once the patient has signed a consent form. Patients must not begin protocol treatment prior to registration. Treatment should start within five working days after registration. To register eligible patients on study, complete a Clinical Trials Office Patient Registration Form and submit to: [email protected].

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 46 of 54 Template version date: 3-5-2015
14 DATA SUBMISSION SCHEDULE The Case Report Forms (CRFs) are a set of (electronic or paper) forms for each patient that provides a record of the data generated according to the protocol. CRF’s should be created prior to the study being initiated and updated (if applicable) when amendments to the protocol are IRB approved. Data capture should be restricted to endpoints and relevant patient information required for planned manuscripts. These forms will be completed on an on-going basis during the study. The medical records will be source of verification of the data. During the study, the CRFs will be monitored for completeness, accuracy, legibility and attention to detail by a member of the Research Compliance Office. The CRFs will be completed by the Investigator or a member of the study team as listed on the Delegation of Duties Log. The data will be reviewed no less than annually by the Data and Safety Monitoring Committee. The Investigator will allow the Data and Safety Monitoring Committee or Research Compliance Office personnel access to the patient source documents, clinical supplies dispensing and storage area, and study documentation for the above-mentioned purpose. The Investigator further agrees to assist the site visitors in their activities.
14.1 Pathology review
14.1.1 Required material 8 unstained slides will be sent to the lab of Dr. Alana Welm. 1 H and E slide will go to Dr. Rachel Factor to confirm the presence of tumor.
14.1.2 Time points for submission Pretreatment, either archive tissue or a fresh biopsy
14.2 Correlative Studies
14.2.1 Timepoints Pretreatment blood samples will be collected for MSP levels. Pretreatment, week 3, week 5, week 9, and week 13 blood samples will be collected for PBMC for immunophenotyping and for assessing serum β–CTX, NTX, TRAP-5b, P1NP, and BSAP.
14.2.2 Samples needed Pretreatment: 2 CPT tubes, 2 serum tubes
14.2.3 Processing CPT tubes and 1 serum tube will be brought to Dr. Welm’s lab at the time of collection for processing for MSP, immunophenotyping, and serum TRAP-5b. Other serum tubes will be sent to ARUP for serum β-CTX, serum NTX, P1NP, and BSAP.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 47 of 54 Template version date: 3-5-2015
15 ETHICAL AND REGULATORY CONSIDERATIONS
15.1 Informed consent Informed consent will be obtained from all research participants prior to performing any study procedures using the most recent IRB approved version.
15.2 Institutional Review Study will be approved by the Institutional Review Board of University of Utah.
15.3 Data and Safety Monitoring Plan A Data and Safety Monitoring Committee (DSMC) is established at Huntsman Cancer Institute (HCI) and approved by the NCI to assure the well-being of patients enrolled on Investigator Initiated Trials that do not have an outside monitoring review. Roles and responsibilities of the DSMC are set forth in the NCI approved plan. The activities of this committee include a quarterly review of adverse events including SAEs, important medical events, significant revisions or amendments to the protocol, and approval of cohort/dose escalations. If the DSMC and/or the PI have concerns about unexpected safety issues, the study will be stopped and will not be resumed until the issues are resolved. The DSMC also reviews and approves audit reports generated by the Research Compliance Office. This is a Phase I study. For each phase I study using an agent of potential
risk a member of the DSMC will be assigned as the primary medical monitor.
The primary medical monitor will be notified immediately of all SAEs. A formal
report should be submitted to the primary medical monitor within 10 days. All
serious adverse events (SAEs) occurring in patients treated at HCI or its
affiliates will also be reviewed by the full DSMC quarterly. The full committee
will also review all grade 3 or greater toxicities for patients on treatment and
within 30 day follow-up window (only if possibly, probably or definitely
related).
15.4 Adverse Events / Serious Adverse Events This study will utilize the CTCAE (NCI Common Terminology Criteria for Adverse Events) Version 4.0 for AE and SAE reporting. An electronic copy of the CTCAE Version 4.0 can be downloaded from: http://safetyprofiler-ctep.nci.nih.gov/CTC/CTC.aspx
15.4.1 Adverse Events (AE) An adverse event is the appearance or worsening of any undesirable sign, symptom, or medical condition occurring after starting the study drug even if the event is not considered to be related to study drug. For the purposes of this study, the terms toxicity and adverse event are used

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 48 of 54 Template version date: 3-5-2015
interchangeably. Medical conditions/diseases present before starting study drug are only considered adverse events if they worsen after starting study drug. Abnormal laboratory values or test results constitute adverse events only if they induce clinical signs or symptoms, are considered clinically significant, or require therapy. Adverse events will be collected from the time of first protocol mandated procedure, until subject completes study participation or until 30 days after subject prematurely withdraws (without withdrawing consent) or is withdrawn from the study. Information about all adverse events, whether volunteered by the subject, discovered by investigator questioning, or detected through physical examination, laboratory test or other means, will be collected and recorded and followed as appropriate.
The occurrence of adverse events should be sought by non-directive questioning of the patient at each visit or phone contact during the study. Adverse events also may be detected when they are volunteered by the patient during or between visits or through physical examination, laboratory test, or other assessments. As far as possible, each adverse event should be evaluated to determine:
1. the severity grade based on CTCAE v.4 (grade 1-5)
2. its relationship to the study drug(s) (definite, probable, possible, unlikely, not related)
3. its duration (start and end dates or if continuing at final exam)
4. action taken (no action taken; study drug dosage adjusted/temporarily interrupted; study drug permanently discontinued due to this adverse event; concomitant medication taken; non-drug therapy given; hospitalization/prolonged hospitalization)
5. whether it constitutes an SAE All adverse events will be treated appropriately. Such treatment may include changes in study drug treatment as listed in the dose modification section of this protocol (see section 8 for guidance). Once an adverse event is detected, it should be followed until its resolution, and assessment should be made at each visit (or more frequently, if necessary) of any changes in severity, the suspected relationship to the study drug, the interventions required to treat it, and the outcome. Information about common side effects already known about the investigational drug is described in the Drug Information (section 3) and the most recent Investigator Brochure. This information will be included in

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 49 of 54 Template version date: 3-5-2015
the patient informed consent and will be discussed with the patient during the study as needed.
All adverse events will be immediately recorded in the patient research chart.
15.4.2 Serious Adverse Event (SAE)
Information about all serious adverse events will be collected and recorded. A serious adverse event is an undesirable sign, symptom or medical condition which:
is fatal or life-threatening
results in persistent or significant disability/incapacity
is medically significant, i.e., defined as an event that jeopardizes the patient or may require medical or surgical intervention to prevent one of the outcomes listed above
causes congenital anomaly or birth defect
requires inpatient hospitalization or prolongation of existing hospitalization, unless hospitalization is for:
o routine treatment or monitoring of the studied indication, not associated with any deterioration in condition (procedures such as central line placements, paracentesis, pain control)
o elective or pre-planned treatment for a pre-existing condition that is unrelated to the indication under study and has not worsened since the start of study drug
o treatment on an emergency outpatient basis for an event not fulfilling any of the definitions of a SAE given above and not resulting in hospital admission
o social reasons and respite care in the absence of any deterioration in the patient’s general condition
Serious Adverse events will be collected from the time of first protocol mandated procedure, until subject completes study participation or until 30 days after subject prematurely withdraws (without withdrawing consent) or is withdrawn from the study. Any death from any cause while a patient is receiving treatment on this protocol or up to 30 days after the last dose of protocol treatment, or any death which occurs more than 30 days after protocol treatment has ended but which is felt to be treatment related, must be reported.
Toxicities which fall within the definitions listed above must be reported as an SAE regardless if they are felt to be treatment related or not. Toxicities

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 50 of 54 Template version date: 3-5-2015
unrelated to treatment that do NOT fall within the definitions above, must simply be documented as AEs in the patient research chart.
15.5 SAE Reporting Requirements SAEs must be reported to the DSMC, the FDA, the IRB, and the drug manufacturer, according to the requirements described below: A MedWatch 3500A form must be completed and submitted to [email protected] and to the Lilly Local Safety Representative (FAX 866-644-1697) as soon as possible, but no later than 10 days of first knowledge or notification of event (5 days for fatal or life threatening event).
DSMC Notifications:
An HCI Research Compliance Officer (RCO) will process and submit the MedWatch form to the proper DSMC member as necessary for each individual study.
The RCO will summarize and present all reported SAEs according to the Data and Safety Monitoring Plan at the quarterly DSMC meeting.
FDA Notifications:
Adverse events occurring during the course of a clinical study that meet the following criteria will be promptly reported to the FDA:
Serious
Unexpected
Definitely, Probably or Possibly Related to the investigational drug
Fatal or life-threatening events that meet the criteria above will be reported within 7 calendar days after first knowledge of the event by the investigator; followed by as complete a report as possible within 8 additional calendar days.
All other events that meet the criteria above will be reported within 15 calendar days after first knowledge of the event by the investigator.
The RCO will review the MedWatch report for completeness, accuracy and applicability to the regulatory reporting requirements.
The RCO will ensure the complete, accurate and timely reporting of the event to the FDA.
The Regulatory Coordinator will submit the report as an amendment to the IND application.
All other adverse events and safety information not requiring expedited reporting that occur or are collected during the course of the study will be summarized and reported to the FDA through the IND Annual

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 51 of 54 Template version date: 3-5-2015
Report.
IRB Notification:
Events meeting the University of Utah IRB reporting requirements (http://www.research.utah.edu/irb/) will be submitted through the IRB’s electronic reporting system within 10 working days.
Drug Manufacturer Notifications:
The SAE reporting will begin with administration of the first dose of study medication. Any SAE occurring prior to study medication administration that the investigator believes may have been caused by a protocol procedure must be reported within 24 hours to the IRB, regulatory authority and Eli Lilly as per the local requirement. Any SAE, regardless of causality assessment, must be collected to the completion of the study (including the follow-up visit) or patient withdrawal from the study, whichever is longer. All SAEs must be followed to resolution or, if resolution is unlikely, to stabilization. Any SAE event judged by the investigator to be related to merestinib should be reported to Eli Lilly regardless of the length of time that has passed since study completion.
All SAEs, regardless of their relationship to study medication, must be reported on the SAE page of the eCRF as soon as possible but no later than 24 hours of the Investigator and/or Institution receiving notification of any serious adverse event experienced by a patient participating in the study and receiving study drug. Fax SAEs to the local safety representative using the following Fax number: 866-644-1697.
*Medwatch 3500A form can be found at http://www.fda.gov/Safety/MedWatch/HowToReport/DownloadForms/ucm2007307.htm
15.6 Reporting of Pregnancy Although pregnancy is not considered an adverse event, it is the responsibility of investigators or their designees to report any pregnancy or lactation in a subject, including the pregnancy of a male subjects’ female partner as an SAE. Pregnancies or lactation that occurs during the course of the trial or with 30 days of completing the trial or starting another new anticancer therapy, whichever is earlier, must be reported to the DSMC, IRB, FDA, and the sponsor as applicable. All subjects and female partners who become pregnant must be followed to the completion/termination of the pregnancy. Pregnancy outcomes of spontaneous abortion, missed abortion, fetal death, intrauterine death, miscarriage and stillbirth must be reported as serious events.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 52 of 54 Template version date: 3-5-2015
15.7 Protocol Amendments Any amendments or administrative changes in the research protocol during the period, for which the IRB approval has already been given, will not be initiated without submission of an amendment for IRB review and approval. These requirements for approval will in no way prevent any immediate action from being taken by the investigator in the interests of preserving the safety of all patients included in the trial.
15.8 Protocol Deviations A protocol deviation (or violation) is any departure from the defined procedures and treatment plans as outlined in the protocol version submitted and previously approved by the IRB. Protocol deviations have the potential to place participants at risk and can also undermine the scientific integrity of the study thus jeopardizing the justification for the research. Protocol deviations are unplanned and unintentional events. Because some protocol deviations pose no conceivable threat to participant safety or scientific integrity, reporting is left to the discretion of the PI within the context of the guidelines below. The IRB requires the prompt reporting of protocol deviations which are:
Exceptions to eligibility criteria. Intended to eliminate apparent immediate hazard to a research
participant or Harmful (caused harm to participants or others, or place them at
increased risk of harm - including physical, psychological, economic, or social harm), or
Possible serious or continued noncompliance
15.9 FDA Annual Reporting An annual progress report will be submitted to the FDA within 60 days of the anniversary of the date that the IND went into effect. (21 CFR 312.33).
15.10 Clinical Trials Data Bank The study will be registered on http://clinicaltrials.gov and the NCI CTRP (Clinical Trials Reporting Program) by the Clinical Trials Office.
15.11 Record Keeping Per 21 CFR 312.57, Investigator records shall be maintained for a period of 2 years following the date a marketing application is approved; or, if no application is filed or the application is not approved, until 2 years after the investigation is discontinued and the FDA is notified.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 53 of 54 Template version date: 3-5-2015
16 BIBLIOGRAPHY Literature Cited
1. Brown, J.E., et al., Bone resorption predicts for skeletal complications in
metastatic bone disease. Br J Cancer, 2003. 89(11): p. 2031-7.
2. Coleman, R.E., Future directions in the treatment and prevention of bone
metastases. Am J Clin Oncol, 2002. 25(6 Suppl 1): p. S32-8.
3. Cetin, K., et al., Survival in patients with breast cancer with bone metastasis: a
Danish population-based cohort study on the prognostic impact of initial stage of
disease at breast cancer diagnosis and length of the bone metastasis-free interval.
BMJ Open, 2015. 5(4): p. e007702.
4. Stopeck, A.T., et al., Denosumab compared with zoledronic acid for the treatment
of bone metastases in patients with advanced breast cancer: a randomized,
double-blind study. J Clin Oncol, 2010. 28(35): p. 5132-9.
5. Welm, A.L., et al., The macrophage-stimulating protein pathway promotes
metastasis in a mouse model for breast cancer and predicts poor prognosis in
humans. Proc Natl Acad Sci U S A, 2007. 104(18): p. 7570-5.
6. Yang, G., et al., Functional grouping of osteoclast genes revealed through
microarray analysis. Biochem Biophys Res Commun, 2008. 366(2): p. 352-9.
7. Kurihara, N., et al., Macrophage-stimulating protein activates STK receptor
tyrosine kinase on osteoclasts and facilitates bone resorption by osteoclast-like
cells. Blood, 1996. 87(9): p. 3704-10.
8. Kurihara, N., et al., Macrophage-stimulating protein (MSP) and its receptor,
RON, stimulate human osteoclast activity but not proliferation: effect of MSP
distinct from that of hepatocyte growth factor. Exp Hematol, 1998. 26(11): p.
1080-5.
9. Bhatt, A.S., et al., Coordinate expression and functional profiling identify an
extracellular proteolytic signaling pathway. Proc Natl Acad Sci U S A, 2007.
104(14): p. 5771-6.
10. Eyob, H., et al., Inhibition of ron kinase blocks conversion of micrometastases to
overt metastases by boosting antitumor immunity. Cancer Discov, 2013. 3(7): p.
751-60.
11. Bieniasz, M., et al., Preclinical Efficacy of Ron Kinase Inhibitors Alone and in
Combination with PI3K Inhibitors for Treatment of sfRon-Expressing Breast
Cancer Patient-Derived Xenografts. Clin Cancer Res, 2015. 21(24): p. 5588-600.
12. Yan, S.B., et al., LY2801653 is an orally bioavailable multi-kinase inhibitor with
potent activity against MET, MST1R, and other oncoproteins, and displays anti-
tumor activities in mouse xenograft models. Invest New Drugs, 2013. 31(4): p.
833-44.
13. Brown, J.E., et al., Bone turnover markers as predictors of skeletal complications
in prostate cancer, lung cancer, and other solid tumors. J Natl Cancer Inst, 2005.
97(1): p. 59-69.
14. Lipton, A., et al., Normalization of bone markers is associated with improved
survival in patients with bone metastases from solid tumors and elevated bone
resorption receiving zoledronic acid. Cancer, 2008. 113(1): p. 193-201.

Protocol name: An Exploratory Phase 1B Study to Assess the Effects of Merestinib on Bone Metastases in Subjects
with Breast Cancer
Version Date: 15SEP2017
Principal Investigator: Adam Cohen, MD
Page 54 of 54 Template version date: 3-5-2015
15. Eastell, R., Management of osteoporosis due to ovarian failure. Med Pediatr
Oncol, 2003. 41(3): p. 222-7.
16. Lipton, A., et al., Zoledronic acid and survival in breast cancer patients with bone
metastases and elevated markers of osteoclast activity. Oncologist, 2007. 12(9): p.
1035-43.
17. Bekker, P.J., et al., A single-dose placebo-controlled study of AMG 162, a fully
human monoclonal antibody to RANKL, in postmenopausal women. J Bone Miner
Res, 2004. 19(7): p. 1059-66.
18. Schirrmeister H, Guhlmann A, Elsner K, et al. Sensitivity in detecting osseous
lesions depends on anatomic localization: planar bone scintigraphy versus 18F
PET. J Nucl Med. 1999;40:1623–1629.
19. Schirrmeister H, Guhlmann A, Kotzerke J, et al. Early detection and accurate
description of extent of metastatic bone disease in breast cancer with fluoride ion
and positron emission tomography. J Clin Oncol. 1999;17:2381–2389.
20. Schirrmeister H, Glatting G, Hetzel J, et al. Prospective evaluation of the clinical
value of planar bone scans, SPECT, and (18)F-labeled NaF PET in newly
diagnosed lung cancer. J Nucl Med. 2001;42:1800–1804.
21. Department of Health and Human Services. Food and Drug Administration.
Positron Emission Tomography Drug Products; Safety and Effectiveness of
Certain PET Drugs for Specific Indications. Notice. March 10, 2000. Federal
Register.65:12999-13009.
22. Hetzel M, Arslandemir C, Konig HH, et al. F-18 NaF PET for detection of bone
metastases in lung cancer: accuracy, cost-effectiveness, and impact on patient
management. J Bone Miner Res. 2003;18:2206–2214.
23. Even-Sapir E, Metser U, Flusser G, et al. Assessment of malignant skeletal
disease: initial experience with 18F-fluoride PET/CT and comparison between
18F-fluoride PET and 18F-fluoride PET/CT. J Nucl Med. 2004;45:272–278.