Embed Size (px)
Citation preview

AE
NNa
b
c
ARRA
KLRIIO
1
dfactaeaileb[
lSi
eT
0h
Enzyme and Microbial Technology 52 (2013) 325– 330
Contents lists available at SciVerse ScienceDirect
Enzyme and Microbial Technology
jou rn al hom epage: www.elsev ier .com/ locate /emt
n efficient in vitro refolding of recombinant bacterial laccase inscherichia coli
asrin Mollaniaa, Khosro Khajehb,∗, Bijan Ranjbarb, Fatemeh Rashnob,eda Akbari c, Mehrnoosh Fathi-Roudsarib
Department of Biology, Faculty of Basic Sciences, Hakim Sabzevari University, Sabzevar, IranDepartment of Biochemistry and Biophysics, Faculty of Biological Sciences, Tarbiat Modares University, Tehran, IranDepartment of Microbiology, Faculty of Sciences, Islamic Azad University, Arak Branch, Arak, Iran
a r t i c l e i n f o
rticle history:eceived 14 March 2012eceived in revised form 22 February 2013ccepted 6 March 2013
eywords:
a b s t r a c t
Laccases (benzenediol oxygen oxidoreductases, EC 1.10.3.2) are important multicopper enzymes thatare used in many biotechnological processes. A recombinant form of laccase from Bacillus sp. HR03 wasoverexpressed in Escherichia coli BL-21(DE3). Inclusion body (IB) formation happens quite often duringrecombinant protein production. Hence, developing a protocol for efficient refolding of proteins frominclusion bodies to provide large amounts of active protein could be advantageous for structural and
accaseefolding
nclusion bodyntrinsic fluorescenceverexpression
functional studies. Here, we have tried to find an efficient method of refolding for this bacterial enzyme.Solubilization of inclusion bodies was carried out in phosphate buffer pH 7, containing 8 M urea and 4 mM�-mercaptoethanol and refolding was performed using the dilution method. The effect of different addi-tives was investigated on the refolding procedure of denaturated laccase. Mix buffer (phosphate bufferand citrate buffer, 100 mM) containing 4 mM ZnSO4 and 100 mM sorbitol was selected as an optimized
etic p
refolding buffer. Also Kin. Introduction
Laccases (monophenol, dihydroxyphenylalanine: oxygen oxi-oreductase, EC 1.14.18.1) are members of the multicupper oxidaseamily of enzymes that catalyze the oxidation of a variety ofromatic substances to less hazardous compounds [1–3]. Lac-ases are promising enzymes for industrial applications due toheir broad spectrum of phenolic and non-phenolic substratesnd the wide range of reactions that can be catalyzed by thesenzymes [4,5]. Although high-level expression of laccases has beenchieved in Escherichia coli, accumulation of recombinant proteinsn the form of cytoplasmic inclusion bodies is particularly prob-ematic [6]. Therefore, one of the major challenges in bioprocessngineering is efficient conversion of these inactive and insolu-le protein aggregates into soluble and correctly folded enzymes7,8].
As we have previously reported, a novel laccase gene was iso-
ated from a local Bacillus sp. HR03 (accession number FJ663050).equence analysis of the isolated gene showed highest similar-ty to the thermostable laccase (CotA) from Bacillus subtilis. The∗ Corresponding author at: Department of Biochemistry, Faculty of Biological Sci-nces, Tarbiat Modares University, P.O. Box 14115-154, Tehran, Iran.el.: +98 21 82884717; fax: +98 21 82884717.
E-mail address: [email protected] (K. Khajeh).
141-0229/$ – see front matter © 2013 Elsevier Inc. All rights reserved.ttp://dx.doi.org/10.1016/j.enzmictec.2013.03.006
arameters of soluble and refolded laccase were analyzed.© 2013 Elsevier Inc. All rights reserved.
sequences of Bacillus species were obtained from National Centerfor Biotechnology Information (http://www.ncbi.nlm.nih.gov) andmultiple sequence alignment was performed with ClustalW ver-sion 1.82. Studies on the native enzyme, revealed a number ofunusual biochemical properties [9]. Compared to other bacteriallaccases, this enzyme showed a different profile of thermoin-activation. This isolated laccase was active when incubated athigh temperature, which makes the enzyme interesting for thebiotechnological applications. Unfortunately, enzyme expressionin E. coli yields a small fraction of active protein, which seems tobe inadequate for many structural and biochemical studies, while aremarkable fraction aggregates in inclusion bodies. Although therehave been relatively few successful efforts for refolding of fun-gal laccase from inclusion body (IB), but other studies have foundthat bacterial laccases (e.g. Bacillus subtilis) cannot be recoveredfrom the IBs and only a soluble fraction representing about 10% ofthe total heterologous laccase can be purified [10]. Expression oflaccase from Bacillus HR03 also resulted in a small fraction of sol-uble enzyme which seems to be inadequate for many structureland biochemical studies. Here, we have tried to refold the laccasefrom Bacillus sp. HR03 to gain more active enzyme molecules afterheterologous expression. Based on our knowledge, this is the first
report on optimization of refolding procedure of a bacterial lac-case from inclusion bodies. Intrinsic fluorescence intensity was alsoused to investigate the conformational changes during the processof refolding.
3 icrobia
2
2
gE(w
2
i(dc1w
2
piTtwpt
2
sbab
2
utptosamyds
Fu
26 N. Mollania et al. / Enzyme and M
. Materials and methods
.1. Materials
Isopropyl-�-D-thiogalactopyranoside (IPTG), dithiothreitol (DTT) and oxidizedlutathione (GSSG) were purchased from Sigma Aldrich (St. Louis, MO, USA).scherichia coli BL21 (DE3) cells, pET21a (+) vector were purchased from NovagenUSA). All other reagents were purchased from Merck (Darmstadt, Germany) andere of analytical grade.
.2. Protein expression
The recombinant plasmid pET21a (+) containing laccase gene was transformednto E. coli BL21 (DE3). A selected lac+ transformant strain was grown in Luria-BertaniLB) medium supplemented with 100 �g/mL ampicillin at 37 ◦C. When the opticalensity at 600 nm (OD600) reached about 0.5, IPTG and CuSO4 were added to a finaloncentration of 0.1 mM and 2 mM, respectively. The temperature was reduced to8 ◦C. After 4 h shaker was turned off to provide a micro aeration condition. Cellsere subsequently harvested by centrifugation (8000 × g, 15 min, 4 ◦C).
.3. Separations of inclusion bodies
Bacterial pellets were resuspended in the 100 mM potassium phosphate bufferH 7 (buffer A). The solution was sonicated for six pulses of 40 s. The soluble and
nsoluble protein fractions were separated by centrifugation at 8000 × g for 30 min.he pellet was subsequently washed with buffer B (buffer A containing 2 M urea)hat allow efficient removal of nonspecific proteins from inclusion bodies. The finalashed pellet was solubilized and denatured in 5 mL of buffer C (100 mM potassiumhosphate pH 7, containing 8 M urea and 4 mM �-mercaptoethanol) and then cen-rifuged at 8000 × g for 30 min. The supernatant in this stage was used for refolding.
.4. Purification
Unfolded protein was loaded on Ni-NTA agarose column (Amersham Bio-ciences) that had been equilibrated with buffer C. Bound proteins were eluted withuffer C containing 200 mM imidazole and the purity was ensured by SDS-PAGEccording to the Laemmli method [11]. The gel was stained by Coomassie brilliantlue R-250. Protein concentration was determined by the Bradford method [12].
.5. In vitro refolding of laccase using dilution procedure
The refolding procedure was carried out through gradual removing of the denat-rant, via dilution and dialysis. The solubilized laccase (0.06 mg/mL) was addedo the refolding medium and incubated at 4 ◦C for 6 h. To optimize the refoldingrocess, small-scale assays were carried out in 1 mL reactions, where concentra-ion of different components and reaction conditions were variable (following therder indicated below) glycerol (0–15%), Triton X100 (0–3%), PEG (0–3 mg/mL),orbitol, sodium chloride, glucose, lactose, trehalose, immidazole, glycine and L-
rginine (0–400 mM), time (0–24 h), protein (0.02–0.15 mg/mL), pH (5–9) andetals (0–6 mM) were used to find the optimal condition of refolding. Refoldingield is formally defined as the percentage of soluble protein following dilution refol-ing, as assessed by enzyme activity measurements and compared with activity ofoluble form.
ig. 1. SDS-PAGE analysis of laccase from E. coli. (A) Lane 1 marker, lanes 2 pellet after inrea and purification. (B) Lane 1 soluble enzyme after expression, lane 2 purified refolded
l Technology 52 (2013) 325– 330
2.6. In vitro refolding of laccase using one-step dialysis strategies
Denatured-reduced protein (2 mL at 0.15 mg/mL) was loaded into a dialysis baghaving a membrane molecular weight cutoff of 10 kDa, and then dialyzed againstrefolding buffer under stirring condition at 4 ◦C. The optimization of refoldingmedium was also performed using different types of buffer, pH, additives and diva-lent ions as mentioned in Section 2.4.
2.7. Spectroscopic studies
The purification of refolded enzyme was carried out. Purity of the protein wasconfirmed by using SDS-PAGE according to Laemmli method. Protein concentra-tion was determined using Bradford method. Fluorescence of refolded laccase wasmeasured on a Perkin Elmer luminescence spectrometer LS 55. The excitation wave-length was set at 280 nm and the emission spectra were recorded from 300 to400 nm. The excitation and emission slit were both set to 10 nm.
2.8. Laccase activity
Laccase activity was determined spectrophotometrically at 525 nm by followingthe oxidation of 0.05 mM syringaldazine in 100 mM potassium phosphate buffer pH7.0 (ε = 65,000 M−1 cm−1) Syringaldazine was dissolved in absolute methanol [13].All assays were performed in triplicates. Furthermore, the purity of the refoldedprotein was analyzed using SDS–PAGE. Then coomassie brilliant blue staining wasperformed according to Laemmli method. The protein concentration was deter-mined by the Bradford assay using BSA as the standard [12]. For comparison ofcatalytic parameters of the soluble and refolded laccase forms, the soluble laccasewas obtained as previously reported [9].
3. Results and discussion
3.1. Expression of the recombinant laccase in E. coli BL21 (DE3)
Recombinant laccase from Bacillus HR03 was expressed in E.coli cells. A remarkable amount of this enzyme was expressed isan insoluble inactive form within inclusion bodies. Therefore, dif-ferent refolding methods for large-scale in vitro activation wereinvestigated [7]. E. coli cells were sediment from 500 mL mediumculture and lysed by sonication. Since the IBs have higher densities,high-speed centrifugation results in separation of these aggregatesfrom contaminating cellular fragments/proteins. The wash step isnecessary to remove contaminants, especially proteins, which mayhave absorbed onto the hydrophobic surface of the inclusion bodiesduring the procedure.
3.2. Dependence of the IB solubilization efficiency to appliedconditions
As a starting and crucial step for each refolding reaction, thesolubilization strategy has to be considered. The washed and puri-fied inclusion bodies were resuspended and incubated in a buffercontaining a strong denaturant (Fig. 1A). The IB solubilization was
duction, lane 3 soluble inclusion bodies before and lane 4 after washing with 2 M enzyme and lane 4 marker.
N. Mollania et al. / Enzyme and Microbial Technology 52 (2013) 325– 330 327
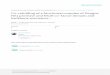
Fig. 2. Effect of various concentrations of �-mercaptoethanol (�ME) as a reducingagent on IB solubilization and yield of protein refolding. Denaturation was per-formed in 100 mM potassium phosphate (pH 7) containing 8 M urea and reducinga
saS8sHsrwi8b(ordtwt
F(
in small-scale experiments using dilution method at 4 ◦C. The sol-ubilized proteins were poured into the folding mixtures in whichthe various buffer conditions were tested separately. First, enzymerefolding was studied toward different buffer systems (data not
gent.
tudied as a function of the urea concentration at room temper-ture, and then the effect of pH on this process was monitored.olubilization improvement of IBs was observed by incorporating
M urea in 100 mM potassium phosphate buffer at pH 7 (data nothown). Laccase has four cysteine residues and one disulfide bond.ence the influence of the presence of additional reducing agents
uch as �-mercaptoethanol and dithiothreitol (DTT) in the foldingeaction was investigated. The optimization of solubilizing mediumas performed by changing the type and concentration of reduc-
ng agents in 100 mM potassium phosphate buffer pH 7 containing M urea (Fig. 2). Results showed that the highest levels of solu-ilized IBs were obtained using 8 M urea under neutral conditions100 mM potassium phosphate buffer pH 7) and in the presencef a reducing agent (4 mM �-mercaptoethanol). The addition of aeducing agent keeps all cysteines in the reduced state and breaksisulfide bonds, formed during the preparation. After solubilizationhe solution was centrifuged to remove the remaining aggregateshich could act as the nuclei to trigger further aggregation during
he refolding.
ig. 3. Investigation the effect of pH 6, 7, 8 and 9, on refolding with dilution methodfor more details see Section 2).
Fig. 4. Effect of different concentration of ZnSo4 as a cofactor on the refolding recov-ery yield. ZnSo4 was added to the refolding buffer and mixture was incubated at 4 ◦Cfor 6 h.
3.3. In vitro activation of the laccase
Among the methods used for in vitro activation of the enzymesthat are usually used in the enzyme refolding, dilution and dialysisprocedures were tested here [14]. As a part of this study, the refol-ding parameters of laccase from Bacillus sp. HR03 were optimized
Fig. 5. Effect of protein concentration (a) and time of incubation at 4 ◦C (b) onrefolding. Refolding was carried out in 100 mM Mix buffer (pH 7) containing 4 mMZnSO4.

3 icrobial Technology 52 (2013) 325– 330
sodpbomstbrcieibmTbcswc
aactlcitbtpo
mttgiti(oftrotwoiTtodh2isfoth
Table 1Effect of various additives on laccase refolding.
Additives Specific activity (U/mg)
Blank (without additives) 0.11Sorbitol (0.1 M) 0.25PEG (2 mg/mL) 0.21Glycerol (5% v/v) 0.19Imidazole (0.05 M) 0.16NaCl (100 mM) 0.07Glucose (0.1 M) 0.05Lactose (0.1 M) 0.05Trehalose (0.1 M) 0.05Tween 80 (2%) 0.05Glycine (0.1 M) 0.04Arginine (0.1 M) 0.02Triton X100 (1%) 0.02[HMIm] [Cl] (2%) 0.006[BMIm] [Cl] (2%) 0.005
28 N. Mollania et al. / Enzyme and M
hown). Then the effect of pH on the refolding of laccase was studiedver the range of 5–9 (Fig. 3). The maximum recovery of refol-ing was obtained at mix buffer (containing sodium citrate andhosphate buffer) at the pH between 7 and 8. Time-course of incu-ation showed that increasing the time up to 6 h improves the yieldf refolding. Some buffers represent a suitable refolding environ-ent via stabilizing the native conformation of the enzyme while
ome of the buffer interactions destabilize the protein conforma-ion. The pH of the refolding mixture may influence the disulfideond formation and the laccase stability. For an efficient proteinefolding process of a copper-binding protein, the presence of theofactors was demonstrated to be essential [15–17]. Since laccases also a copper-binding protein, we performed in vitro refoldingxperiments using sulphate salts of copper and zinc. No refoldingmprovement was detected in the presence of copper, which cane due to the fact that free copper acts as a reducing agent in theedium and subsequently reduces the copper centers in laccase.
herefore, the free copper was removed by dialysis against mixuffer, pH 7 and then the laccase refolding was measured indi-ated by its oxidative activity on syringaldazine as the reducingubstrate and its specific activity. No considerable improvementas observed in this condition compared to the control (without
ofactor).The effect of zinc on the refolding of laccase was also determined
s the atomic radii of copper and zinc are similar. When zinc wasdded to the selected buffer at the beginning of the refolding pro-edure, the refolding efficiency was slightly increased. Fig. 4 showshe effect of various concentration of ZnSO4 on the refolding ofaccase. In vitro laccase activation was promoted by increasing theoncentration of ZnSO4 up to 4 mM. A reduction in laccase refoldingn higher concentrations of this cofactor was observed. It is likelyhat the cofactors may interact with their corresponding proteinsefore the folding of the polypeptide chain, and therefore enhanceshe folding reaction or may serve as a nucleation site that directsolypeptide folding. Also cofactors often stabilize the native statesf the proteins [15,18].
Protein refolding is a complex process that often depends onany factors including protein concentration or additives during
he refolding process [19,20]. Therefore, it can be suggested thathe formation of a soluble enzyme by in vitro refolding does notuarantee the formation of a fully active laccase from IB. Follow-ng increasing the refolding efficiency, different concentrations ofhe unfolded protein were used. Total activity improved by increas-ng the concentration up to 0.06 mg/mL after 5 h incubation at 4 ◦CFig. 5a and b). Due to the kinetic competition between higher orderf aggregation and first-order folding process, yields of correctlyolded proteins was decreased when the initial protein concen-ration was increased. The protein aggregation during differentefolding steps is a bottle neck in recovering the high amountsf proteins [21,22]. After optimization of the protein concentra-ion for preventing further protein aggregation, several additivesere selected to block aggregation. In addition, the effect of numer-
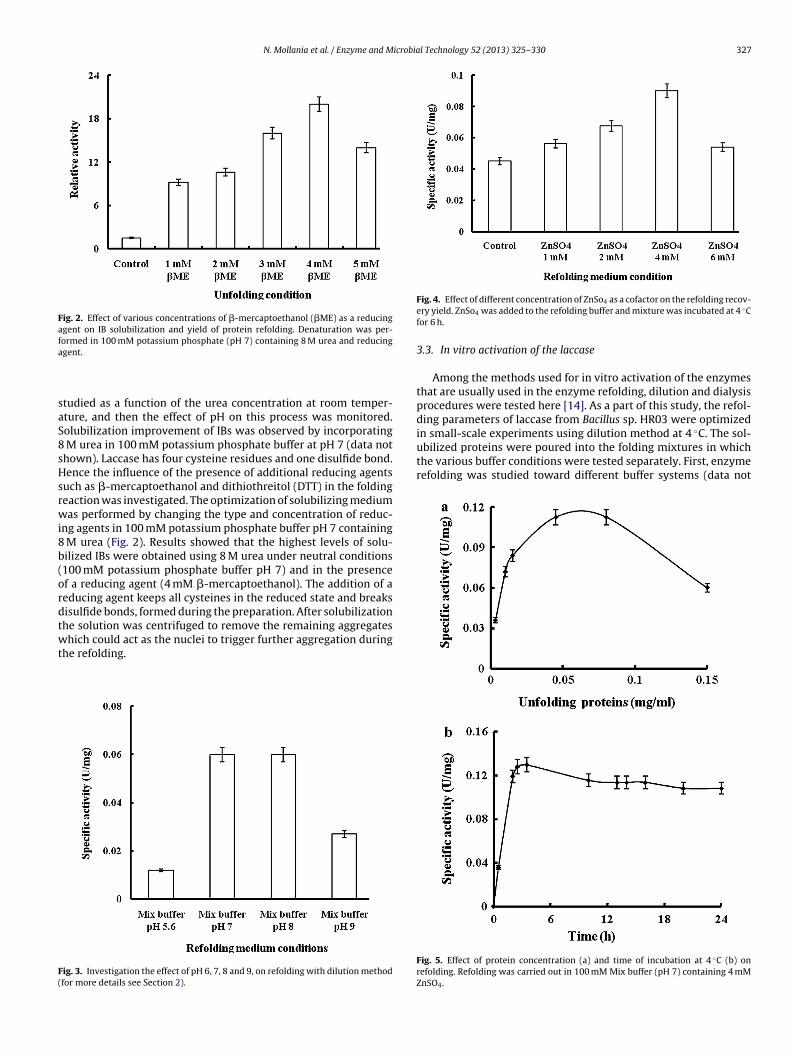
us solution additives on the refolding yields of the native proteinn optimized buffer containing 4 mM ZnSO4 have been tested.he chosen additives, in this work, are listed in Table 1. Amonghese, sorbitol, polyethylene glycol (PEG), glycerol and imidaz-le had significant effect on this process [23,24]. Subsequently,ifferent concentrations of these additives were examined. Theighest recovered activity was obtained at 100 mM sorbitol and
mg/mL PEG. Sorbitol increased the refolding recovery by enhanc-ng protein-protein interactions and stability. PEG was used touppress aggregation because it reduces side-chain interaction of
olding intermediates without interfering with refolding processr enhances the solubility of refolding intermediates. Furthermore,he results showed that 5% of glycerol and 50 mM imidazole couldave significant effect on the refolding improvement (Fig. 6a–d).Refolding mixture used contained 4 mM ZnSO4, in 100 mM Mix buffer (pH 7). Exper-iments were performed at least in triplicate and the standard deviations were ±5%.
In addition, the one-step dialysis strategy for the refolding ofinclusion bodies has been used. The excess amount of the denatu-rant was first removed by overnight dialysis against various buffersand the laccase activity was measured through its oxidative activityon syringaldazine as the substrate. The effect of ZnSO4 as a cofactorand also the effect of the pH on laccase refolding were studied overthe range of 1–4 mM and pH 5–9, respectively. To analyze laccaserefolding, different concentrations of additives were added to thedialysis medium. The best condition for the optimum refolding wasachieved in the potassium phosphate buffer, pH 7, without anyadditives or cofactors and only by increasing the time of the dia-lyzing up to 9 h will result in more refolding of the enzyme. Alsothe effect of the temperature within the hours of dialyzing showedhigher amounts of the refolded enzyme in 4 ◦C. Using differentprotein concentrations it was found that increasing the unfoldedprotein concentration up to 0.15 mg/mL enhances the refoldingyield. The specific activity and kcat can be used as the indicator ofrefolding percentage in two aforesaid methods. The results showedthat between the two procedures utilized, laccase can be recoveredwith higher efficiency using the drop dilution approach. The effi-ciency of dilution method is 6 folds greater than that of dialysismethod at the same condition.
3.4. Spectroscopic studies
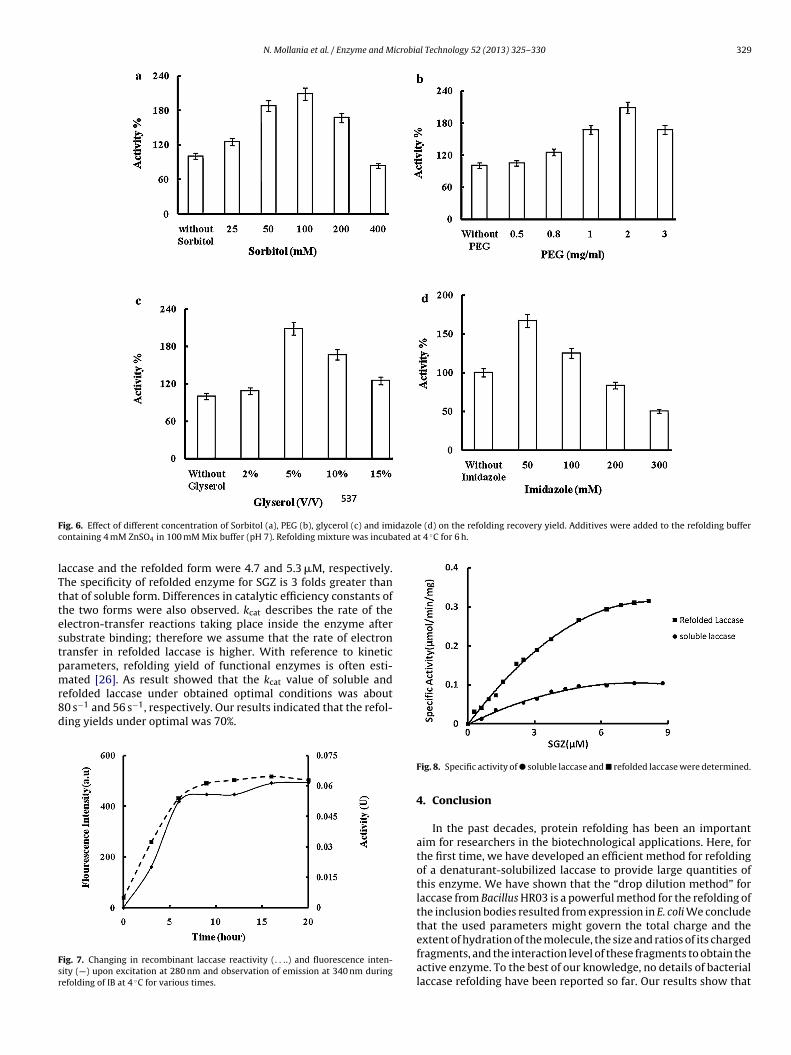
Purification of the folded laccase was performed in order tomonitor structural changes during the refolding procedure usingfluorescence spectroscopy (Fig. 1B). Enzymes obtained after refol-ding through the two optimized procedures mentioned above wereused for spectroscopic studies. Samples were taken every two hoursfor 20 h. The change in fluorescence intensity of laccase for dilutionmethod as a function of incubation time is shown in Fig. 7. Thereis an increase in the intensity over 0–6 h and no further changeswere seen as the time increased to 20 h indicating that completefolding might have been reached within the first 6 h. Fluorescenceemission has been established to study denaturation and renatu-ration of proteins. Since the fluorescence emission of a protein isthe result of its intrinsic fluorophores like Trp and Tyr residues theintensity of emission is related to the protein conformation whichmay expose or burry the internal Trp and Tyr residues [25].
3.5. Catalytic comparison of soluble and refolded laccase
Kinetic parameters of soluble and refolded laccase were quanti-tatively determined with SGZ as a substrate at pH 7.0 and then kcat
and KM values were calculated (Fig. 8). The KM values for soluble
N. Mollania et al. / Enzyme and Microbial Technology 52 (2013) 325– 330 329
F idazole (d) on the refolding recovery yield. Additives were added to the refolding bufferc ated at 4 ◦C for 6 h.
lTttestpmr8d
Fsr
ig. 6. Effect of different concentration of Sorbitol (a), PEG (b), glycerol (c) and imontaining 4 mM ZnSO4 in 100 mM Mix buffer (pH 7). Refolding mixture was incub
accase and the refolded form were 4.7 and 5.3 �M, respectively.he specificity of refolded enzyme for SGZ is 3 folds greater thanhat of soluble form. Differences in catalytic efficiency constants ofhe two forms were also observed. kcat describes the rate of thelectron-transfer reactions taking place inside the enzyme afterubstrate binding; therefore we assume that the rate of electronransfer in refolded laccase is higher. With reference to kineticarameters, refolding yield of functional enzymes is often esti-
ated [26]. As result showed that the kcat value of soluble andefolded laccase under obtained optimal conditions was about0 s−1 and 56 s−1, respectively. Our results indicated that the refol-ing yields under optimal was 70%.
ig. 7. Changing in recombinant laccase reactivity (. . ..) and fluorescence inten-ity (—) upon excitation at 280 nm and observation of emission at 340 nm duringefolding of IB at 4 ◦C for various times.
Fig. 8. Specific activity of � soluble laccase and � refolded laccase were determined.
4. Conclusion
In the past decades, protein refolding has been an importantaim for researchers in the biotechnological applications. Here, forthe first time, we have developed an efficient method for refoldingof a denaturant-solubilized laccase to provide large quantities ofthis enzyme. We have shown that the “drop dilution method” forlaccase from Bacillus HR03 is a powerful method for the refolding ofthe inclusion bodies resulted from expression in E. coli We concludethat the used parameters might govern the total charge and the
extent of hydration of the molecule, the size and ratios of its chargedfragments, and the interaction level of these fragments to obtain theactive enzyme. To the best of our knowledge, no details of bacteriallaccase refolding have been reported so far. Our results show that
3 icrobia
tcgts
R
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
[
30 N. Mollania et al. / Enzyme and M
he method we have introduced here for the refolding of enzymeould be used in renaturation of other bacterial laccase, which oftenet trapped in inclusion bodies. In future further investigations onhe characterization and comparison of the refolded protein andoluble form of enzyme will be carried out.
eferences
[1] Majeau JA, Brar SK, Tyagi RD. Laccases for removal of recalcitrant and emergingpollutants. Bioresource Technology 2010;101:2331–50.
[2] Couto SR, Industrial Herrera JLT. biotechnological applications of laccases: areview. Biotechnology Advances 2006;24:500–13.
[3] Pieper DH, Reineke W. Engineering bacteria for bioremediation. Current Opin-ion in Biotechnology 2000;11:262–70.
[4] Leedjarv A, Ivask A, Virta M, Kahru A. Analysis of bioavailable phenols fromnatural samples by recombinant luminescent bacterial sensors. Chemosphere2006;64:1910–9.
[5] Ryan D, Leukes W, Burton S. Improving the bioremediation of phenolic wastew-aters by Trametes versicolor. Bioresource Technology 2007;98:579–87.
[6] Pérez-Boada M, Doyle WA, Ruiz-Duenas FJ, Martínez MJ, Martínez AT, SmithAT. Expression of Pleurotus eryngii versatile peroxidase in Escherichia coliand optimisation of in vitro folding. Enzyme and Microbial Technology2002;30:518–24.
[7] Vallejo LF, Rinas U. Strategies for the recovery of active proteins throughrefolding of bacterial inclusion body proteins. Microbial Cell Factories2004;3:11–22.
[8] Tsumoto K, Ejima D, Kumagai I, Arakawa T. Practical considerations in refol-ding proteins from inclusion bodies. Protein Expression and Purification2003;28:1–8.
[9] Mohammadian M, Fathi-Roudsari M, Mollania N, Badoei-Dalfard A, Khajeh K.Enhanced expression of a recombinant bacterial laccase at low temperatureand microaerobic conditions: purification and biochemical characterization.Journal of Industrial Microbiology and Biotechnology 2010;37:863–9.
10] Martins LO, Soares CM, Pereira MM, Teixeira M, Costa T, Jones GH, Henriques
AO. Molecular and biochemical characterization of a highly stable bacteriallaccase that occurs as a structural component of the Bacillus subtilis endosporecoat. Journal of Biological Chemistry 2002;277:18849–59.11] Laemmli UK. Cleavage of structural proteins during the assembly of the headof bacteriophage T4. Nature 1970;227:680–5.
[
l Technology 52 (2013) 325– 330
12] Bradford MM. A rapid and sensitive method for the quantitation of microgramquantities of protein utilizing the principle of protein–dye binding. AnalyticalBiochemistry 1976;72:248–54.
13] Harkin JH, Obst JR. Syringaldazine, an effective reagent for detecting laccaseand peroxidase in fungi. Experientia 1973;37:381–7.
14] Sørensen HP, Sperling-Petersen HU, Mortensen KK. Dialysis strategies for pro-tein refolding: preparative streptavidin production. Protein Expression andPurification 2003;31:149–54.
15] Leckner J, Bonander N, Wittung-Stafshede P, Malmstrom BG, Karlsson BG. Theeffect of the metal ion on the folding energetics of azurin: a comparison ofthe native, zinc and apoprotein. Biochimica et Biophysica Acta 1997;1342:19–27.
16] Wittung-Stafshede P. Role of cofactors in protein folding. Accounts of ChemicalResearch 2002;35:201–8.
17] Wittung-Stafshede P. Role of cofactors in folding of the blue-copper proteinazurin. Inorganic Chemistry 2004;43:7926–33.
18] Pozdnyakova I, Guidry J, Wittung-Stafshede P. Copper stabilizes azurin bydecreasing the unfolding rate. Archives of Biochemistry and Biophysics2001;390:146–8.
19] Dong XY, Huang Y, Sun Y. Refolding kinetics of denatured-reduced lysozymein the presence of folding aids. Journal of Biotechnology 2004;114:135–42.
20] Yasuda M, Murakami Y, Sowa A, Ogino H, Ishikawa H. Effect of addi-tives on refolding of denatured protein. Biotechnology Progress 1998;14:601–6.
21] Hevehan DL, Clark EDB. Oxidative renaturation of lysozyme at high concentra-tion. Biotechnology and Bioengineering 1997;54:221–30.
22] Kiefhaber T, Rudolph R, Kohler HH, Buchner J. Protein aggregation in vivo: aquantitative model of the kinetic competition between folding and aggregation.Biotechnology (NY) 1991;9:825–9.
23] Meng FG, Park YD, Zhou HM. Role of proline, glycerol, and heparin as proteinfolding aids during refolding of rabbit muscle creatine kinase. InternationalJournal of Biochemistry and Cell Biology 2001;33:701–9.
24] Aghashe VR, Hart FU. Roles of molecular chaperons in cytoplasmic proteinfolding. Seminars in Cell & Developmental Biology 2000;11:15–25.
25] Brunet JE, Gonzalez GA, Sotomayor CP. Intramolecular tryptophan heme
energy transfer in horseradish peroxidase. Photochemistry and Photobiology1983;38:253–4.26] Ho JG, Middelberg AP. Estimating the potential refolding yield of recombi-nant proteins expressed as inclusion bodies. Biotechnology and Bioengineering2004;87:584–92.