Embed Size (px)
Citation preview

1
An Ab Initio Study of the Relative Stabilities of the Isomers of
CH2N2 and SiH2N2
Colin Thomson and Christopher Glidewell Department of Chemistry, University of S t . Andrews, S t . Andrews, Fife KY16 9ST, Scotland Received 14 April 1982; accepted 17May 1982
A b inztio SCF calculations of the equilibrium geometries have been carried out on nine possible isomers of MHzN2, where M = C or Si, and compared with the results of MNDO calculations. The results for the carbon compounds are in good agreement with available experimental data, but in the case of the sili- con compounds, the molecules are predicted to be unstable with respect to decomposition to SiHz and Nz. The inclusion of electron correlation at the MP3 level does not alter the order of the relative stabili- ties, although the importance of the correlation contribution varies substantially between the different isomers.
INTRODUCTION
There has been considerable experimentall,2 and theoretical3 interest during the last few years in the structures and relative thermodynamic stabilities of organosilicon compounds, particu- larly silahydrocarbons. Substitution of silicon for carbon stabilizes some of the possible isomers, but on the other hand, it seems that compounds which contain multiple bonds between carbon and silicon are less stable than the alternative structures containing a silicon atom which is less than tetra- valent.:+'
In previous theoretical work, the stability of various species on the CSiH2,s-y CSiH3+,6 CSiH4,6-15 C2SiH4,16-19 and C ~ S ~ H S ~ O hypersur- faces have been determined, several of the more recent studies via SCF calculations employing gradient techniques. These studies have yielded valuable information on these unusual species, only a few of which have been characterized ex- perimentally.' Finally, there have been two studies of the isomers of Si2H4.21*22
However, calculations on the silicon analog of compounds containing C, N, and H have not yet been reported in the literature, although there has been some experimental evidence for sila-carbo- diimide intermediates. In this article we present the results of a study of the realtive stabilities of the isomers of CH2N2 and SiH2N2. A comparison between these two series of compounds is of some interest, in view of the large difference in stability of the carbon isomers.23 There has been some previous theoretical work on the CH2N2 isomers,24
Journal of' Computational Chemistry, Vol. 4, No. 1, 1-8 (1983) 0 1983 by .John Wiley & Sons, Inc.
but no comprehensive study using gradient tech- niques.
Experimentally, of the possible isomers of CH2N2, diazomethane(I), cyanamide(II), and di- azirine(II1) are well known and have been char- acterized both chemically and spectroscopically.23 Very recently isocyanamide(1V) has also been confirmed experimentally via the millimeter-wave spectrum.25 Derivatives of nitrilimine(V) and carbodiimide(V1) are known, but there is doubt concerning the observation of the parent com- pounds. There have been no reports on the prep- aration of isodiazirine(VI1) and its derivatives, nor of the carbene(VII1). A detailed discussion of the chemistry and relative stabilities of these mole- cules is given by M ~ f f a t . ~ ~
H. H, N=N \
\ N-C=N \ / H/c=N=N H/ CH,
(1) (11) (111)
\ H-C=N=N-H H
H/N-N=C
(IV) (V) H-N=C=N-H €3-N-N H-N-N-H
\ / (VI) ( V W (VIII)
c: \I CH
Substitution of carbon for silicon in these species might be expected to alter appreciably the relative stabilities of the isomers, since there are substantial variations in the bond types to carbon and silicon in these compounds. Certain of these species were suggested as intermediates in early
CCC 0192-8651/83/010001-08$02.60

2 Thomson and Glidewell
studies of the ammonolysis of tetrachlorosilane (see ref. 1 ) but there has been no definitive evi- dence for any of these species in recent experi- mental work, with the exception of the sila-car- bodiimide referred to above.26
METHOD OF CALCULATION
All geometry optimizations were carried out using the split-valence 3-21G basis set,27 and the analytical gradient method incorporated in the GAUSSIAN 80 program system.28 Convergence difficulties were found with several of the silicon- containing species, and damping routines were added to the SCF routines in GAUSSIAN 80 to overcome this problem.
To obtain more reliable relative energies and charge distributions for these molecules, single point calculations were performed at the 3-21G geometries including electron correlation by the MP3 method29 with the extended 6-31G* basis set.27-30 These are denoted MP3/6-31G* // 3-21G following Pople's notation.27 In a limited number of molecules, reoptimization of the geometry was carried out using the 4-31G* basis set.
We have also carried out MND031 calculations of the geometry and relative stabilities of these molecules in order to compare the predictions of this widely used semiempirical procedure with a6 initio methods in the case of silicon-containing compounds.
RESULTS OF CALCULATIONS
It is convenient to present the ab initio results for each CH2N2 isomer and its silicon analog to- gether and to compare the two species; we examine the relative stabilities of the two series of isomers in the Discussion section, where we also compare the results of the MNDO calculations with both the ab initio results and the experimental struc- tures.
Diazomethane and Siladiazomethane
The calculated geometrical parameters of di- azomethane are in reasonable agreement with earlier w0rk,2~-3~ and experiment33 and are given in Table I(i). The molecule is calculated to be planar, as observed.
However, the structure of the silicon analog computed with the 3-21G basis set is quite differ- ent. The molecule is nonplanar, the planar form analogous to diazomethane lying 0.065 a.u. higher in energy. The Si-N-N fragment is essentially
linear, but the bond angles H-Si-N are ca. 85". Furthermore, the H-Si-H bond angle is 95". The Si-N bond is rather short, as is the N-N bond. Examination of the orbital composition shows large differences from diazomethane. In the latter, the highest orbitals are delocalized over the C-N-N fragment, but in H2SiN2, the HOMO is largely localized on Si, with small contributions from the symmetrical combination of 1s orbitals on the H atoms. We note, however, that this mol- ecule is isoelectronic with H2PCN where the cal- culated H-P-C and H-P-H bond angles are ca. 96", and 96.4", respectively. A detailed exam- ination of the wavefunction, however, shows rel- atively little bonding between Si and N, quite unlike the bonding in CH2N2, and stretching the Si-N bond to 4 A increases the energy by only ca. 0.01 a.u. We thus conclude that this species is a loose complex of distorted H2Si and N2, and in fact, the N-N distance is close to that found in N2.
G ~ d d a r d ~ ~ has examined the bonding in diazo- methane with the GVB method and both minimal and DZ basis sets, and concluded that the SCF de- scription of the bonding is rather poor, with the molecule being better described as biradical-like. However, the calculated geometry with the 3-21C basis set agrees well wit,h experiment in both our work and that of M ~ f f a t . ~ ~ The computed geom- etry is also in reasonable agreement with the MNDO results, although the MNDO Si-N bond length of 1.91 A is somewhat shorter than the ab initio value.
Cyanamide and Silacyanamide
The computed structural qarameters for these molecules are given in Table I(ii) together with the experimental values.35 Our results on cyanamide parallel those of other calculations a t the SCF level, predicting a planar molecule. However, the STO-3G basis predicts a nonplanar ~pecies .3~ Vincent and D y k ~ t r a ~ ~ have presented the results of a detailed study of the cyanamide-isocyanamide rear- rangement and reviewed earlier work. It is clear from this study that SCF calculations without po- larization functions predict a planar structure, addition of polarization functions results in a nonplanar structure, but the inversion barrier is small. Correlation effects (calculated by the SCEP r n e t h ~ d ~ ~ , ~ ~ ) significantly alter the energy differ- ence between cyanamide and isocyanamide, sta- bilizing the cyanide isomer more. In the light of these results, we also carried out geometry opti-
Isomers of CH2N2 and SiH2N2 3
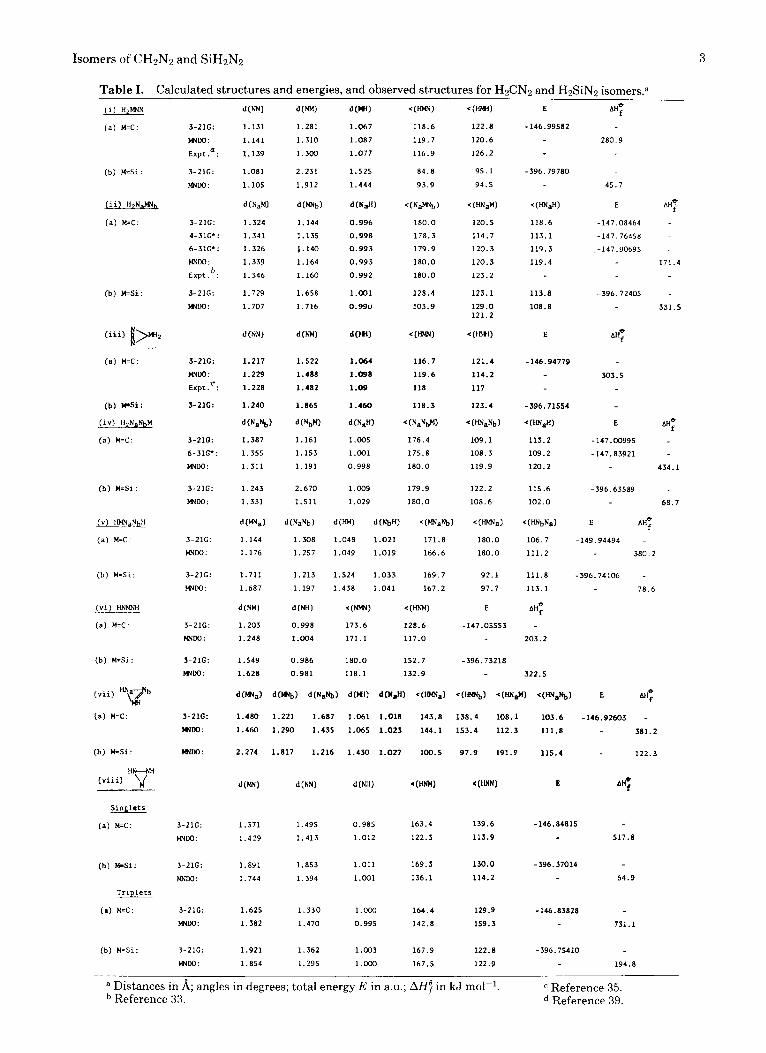
Table 1. Calculated structures and energies, and observed structures for H&N2 and H2SiN2 is0mers.a dWN1
1.131
1.141
1.139
1.081
1.105
d(NaM)
1.324
1.341
1.326
1.339
1.346
1.729
1.707
dWN)
1.217
1.229
1.228
1.240
d(Na%)
1.387
1.355
1.311
1.243
1.331
d W a )
1.144
1.176
1.711
1.687
d(NW
1.203
1.248
1.549
1.628
d(NW
1.281
1.310
1.300
2.231
1.912
d(mb)
1.144
1.135
1.140
1.164
1.160
1.658
1.716
d " M )
1.522
1.488
1.482
1.865
d (NbH)
1.161
1.153
1.191
2.670
1.511
d ( W
1.067
1.087
1.077
1.525
1.444
d ( b H )
0.996
0.998
0.993
0.993
0.992
1.001
0.990
d OW
1.064
1.098
1-09
1.460
d(N,Hl
1.005
1.001
0.998
1.009
1.029
'(M)
122.8
120.6
126.2
95. 1
94.5
<(t'JJaH)
120.5
114.7
120.3
120.3
123.2
123.1
129.0 121.2
<(Hb*l)
121.4
114.2
117
123.4
<(maNb)
109.1
108.3
119.9
122.2
108.6
< ( m a )
180.0
180.0
92.1
97.7
E
E
-146.99582
-396.79780
<W,H)
118.6
113.1
119.3
119.4
113.8
108.8
E
-146.94779
-596.71554
<(m,H)
113.2
109.2
120.2
115.6
102.0
<(t'JJbNa)
106.7
111.2
111.8
113.1
AH:
203.2
322.5
( i ) HzmN
(a) M=C: 280.9
45.7
E
-147.08464
-147.76458
-147.90695
-396.72405
AH;
303.5
E
-1 47.00995
-147.83921
- 396.63589
E AH;
-149.94494 -
380.2
-396.74106 - 78.6
3-ZIG:
moo: Expt.":
3-21G:
moo:
3-21G:
4-31c9:
6-31G.:
ENDO:
Expt.b:
3-21G:
moo:
3-21G:
mm: Expt.=:
3-2lG:
3-210:
6-31G.:
"Do:
3-21C:
moo:
3-21G:
moo:
3-21G:
moo:
3-21G:
moo:
3-2IG:
moo:
3-21G:
mw:
mw:
3-21C:
MNW:
3-21G:
moo:
3-21G:
moo:
3-21G:
mm:
118.6
119.7
116.9
84.8
93.9
< (NaMh' )
180.0
178.3
179.9
180.0
180.0
128.4
103.9
<(W)
116.7
119.6
118
118.3
<(NaNbH)
176.4
175.8
180.0
179.9
180.0
(b) I l=Si :
AH':
171.4
331.5
4
034.1
68.7
( i i ) HpN,mk
(a) H=C:
(b) W=Si:
(a) H=C:
(b) psi: ( i v ) H z N , N ~ H
(a) H=C:
(b) H=Si :
d(NaNb) d ( W d(h'H) <(maNb)
1.308 1.048 1.021 171.8
1.257 1.049 1.019 166.6
1.213 1.524 1.033 169.7
1.197 1.438 1.041 167.2
d ( W <(") <(W)
( v ) tCWnNbH
(a ) H=C
(b) M=Si:
1 v i ) H N W
( a ) H=C: 0.998 173.6 128.6 -147.05553
1.004 171.1 117.0
0.986 180.0 152.7 -396.73218
0.981 118.1 132.9
(b) H=SI :
d(MNa) d(%) d(NaNb) d ( W dDIaW < ( m a ) <(mb) <(man) <(ma%)
1.480 1.221 1.687 1.061 1.018 143.8 138.4 108.1 103.6
1.4M) 1.290 1.435 1.065 1.023 144.1 153.4 112.3 111.8
2.274 1.817 1.216 1.430 1.027 100.5 97.9 191.9 115.4
E 4 -146.92603 -
381.2
122.3
AH:
Singlets
(a) H=C: 1.371
1.429
1.495
1.413
0.985
1.012
163.4
122.3
139.6
113.9
-146.84815
517.8
(b) H-Si: 1.691
1.744
1.853
1.394
1.011 1.W1
169.3
136.1
130.0
114.2
-396.37014
64.9
T r i p l e t s
(a) M=C: 1.625
1.382
1.330
1.470
l.m
0.995
164.4
142.8
129.9
159.3
-146.83828
731.1
(b) M-Si: 1.921
1.854
1.362
1.235
1.003
1 .m 167.9
167.5
122.8
122.9
-396.75410
194.8
a Distances in 8; angles in degrees; total energy E in a.u.; AH7 in kJ mol-'. c Reference 35. Reference 33. Reference 39.

4 Thomson and Glidewell
mizations for cyanamide using the 4-31G* basis set, and a more limited study using the 6-31G* basis set. The aim of this investigation was to see if the nonplanar structure could be reproduced with a smaller basis set than double zeta. Table I(ii) shows that the deviation from planarity is appreciable only for the 4-31G* geometry, but the bond lengths in this case are rather out of line. The energy difference between planar and nonplanar forms is very small, a point emphasized by Dykstra et al.36 It is clear that this molecule (and isocy- anamide) are systems where the 3-216 basis set fails to give a reliable geometry. This has also been noted in the simpler molecule HNC0.38
The 3-21G geometry of the silicon analogue is shown in Table I(ii). Even with the 3-21G basis set, this molecule is nonplanar, and with the 6-31G* basis set even more so, although a full optimization of the geometry was not attempted. However, in addition to this marked difference in geometry, the molecule has an extremely large dipole moment.
There are considerable differences between the MNDO and ab initio results for this molecule (un- like the case of cyanamide, where the two methods are in excellent agreement). Although cyanamide is linear at the C, in silacyanamide, the NSiN angle is calculated as 128.4' (3-21G) or 103.9' (MNDO), definitely not 180'. However, the linear form is very close in energy and the barrier is only ca. 5 kJ mol-'. The discrepancy in the bond lengths and angles as calculated by different methods is probably due to the very shallow nature of the surface.
Diazirine and Sila-Diazirine
Diazirine has been the subject of many theo- retical investigations (see ref. 23 for details), and we report the results of our work for completeness and comparison with the silicon-containing analog. Table I(iii) gives the computed geometries, and the experimental geometry for diazirine.39 The agreement with other work for diazirine is satis- factory, and the structure of the sila-diazirine is not substantially different to that of the carbon compound. However, the charges on the atoms are quite different, the silicon bearing a positive charge of 0.87 compared to the carbon charge of -0.23. This of course reflects the usual trends with silicon, but it leads in this ring system to the ni- trogen atoms carrying a negative charge about twice that in the carbon compound. The N-N bond length is increased somewhat, but the mo-
lecular orbital composition is similar to that in diazirine.
Isocyanamide and Sila-Isocyanamide
The results for isocyanamide a t the 3-21G level parallel those for cyanamide, except that the SCF calculations in this case give a small deviation from planarity. The calculated structure of these mol- ecules is given in Table I(iv). The structure is in good agreement with that of Moffat32 and Vincent and D y k ~ t r a , ~ ~ but the latter authors also found the geometry to be sensitive both to the basis set and to correlation effects. An optimization of the geometry a t the 6-31G* level gave rather similar values for the parameters.
The sila-isocyanamide, like sila-diazomethane, has a very large Si-N bond length, and exami- nation of the molecular orbitals show this molecule to be a loose complex between H2N2 and Si, since there is no significant bonding interaction between the central N atom and Si. It is, however, clear from the work of Davis and G ~ d d a r d , ~ ~ that the SCF method is unable to describe this system well, and we plan to further investigate this system using correlated wavefunctions.
Nitrilimine and Sila-Nitrilimine
Nitrilimine is not known in the free state4l al- though it has been postulated following UV pho- tolysis of d i a ~ o m e t h a n e . ~ ~ Derivatives are, how- ever, kn0wn*"~1 but there is no structural infor- mation available on the geometry of the backbone. In fact, it is believed that the earlier reports of its preparation were in error41; the substance actually prepared was isocyanamide.
The structural parameters given in Table I(v) are in reasonable agreement with those of Mof- fat."' It is noteworthy that the HCN linkage is strictly linear in our calculations, and the molecule is planar. STO-3G calculations predict a nonplanar system.
In contrast, the sila-nitrilimine is W-shaped with an H-Si-N bond angle of 92'. The NH bond is a t an angle of 111.8 with the next N atom but is below the Si-N-N plane.
Carbodiimide and Sila-Carbodiimide
Although there is an extensive literature on substituted carb~diimides:~ the parent compound does not seem to have been prepared. Some diffi-
Isomers of CH2N2 and SiH2Nz 5
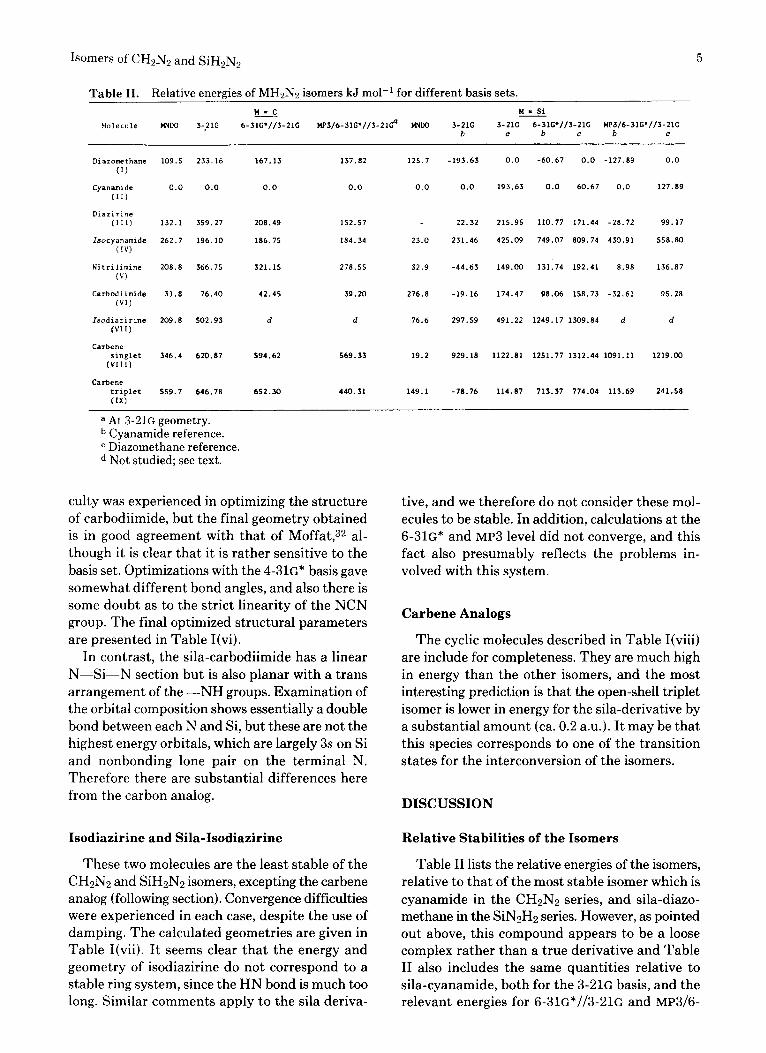
Table 11. Relative energies of MH2N2 isomers kJ mo1-I for different basis sets. W - C W = Si
Holecule W O O 3-,2lC 6-31G*//3-21G W3/6-31G4//3-21dI MNW 3-2lG 3-21G 6-316*//3-21G HPd/6-31G*//3-21G b C b C b C
Diazomethane 109.5 233.16 167.13 (1)
(11)
( I l l ) 132.1 359.27 208.49
Isocyanamide 262.7 196.10 186.75
Cyananide 0 .0 0.0 0 .0
Diazirine
( I V )
N i t r i l imine 208.8 366.75 321.15 ( V )
Carbodiimide 31.8 76.40 42.45 ( V I )
(VII) I sod iaz ir ine 209.8 502.93 d
Carbene s i n g l e t 346.4 620.87 594.62
( V I 1 1 )
Carbene t r i p l e t 559.7 646.78 652.30 (1x1
137.82
0.0
152.57
184.34
278.55
39.20
d
569.33
440.31
125.7
0.0
23 .0
3 2 . 9
276.8
76.6
19.2
149.1
-193.63
0.0
22.32
231.46
-44.63
-19.16
297.59
929.18
-78.76
0.0
193.63
215.95
425.09
149.00
174.47
491.22
1122.81
114.87
-60.67 0 .0 -127.89
0.0 60.67 0.0
110.77 171.44 -28.72
749.07 809.74 430.91
131.74 192.41 8.98
98.06 158.73 -32.61
1249.17 1309.84 d
1251.77 1312.44 1091.11
713.37 774.04 113.69
0.0
127.89
99.17
558.80
136.87
95.28
d
1219.00
241.58
a At 3-21G geometry. Cyanamide reference. Diazomethane reference. Not studied; see text.
culty was experienced in optimizing the structure of carbodiimide, but the final geometry obtained is in good agreement with that of Moffat,32 al- though it is clear that it is rather sensitive to the basis set. Optimizations with the 4-31G* basis gave somewhat different bond angles, and also there is some doubt as to the strict linearity of the NCN group. The final optimized structural parameters are presented in Table I(vi).
In contrast, the sila-carbodiimide has a linear N=Si=N section but is also planar with a trans arrangement of the -NH groups. Examination of the orbital composition shows essentially a double bond between each N and Si, but these are not the highest energy orbitals, which are largely 3s on Si and nonbonding lone pair on the terminal N. Therefore there are substantial differences here from the carbon analog.
Isodiazirine and Sila-Isodiazirine
These two molecules are the least stable of the CH2N2 and SiH2N2 isomers, excepting the carbene analog (following section). Convergence difficulties were experienced in each case, despite the use of damping. The calculated geometries are given in Table I(vii). It seems clear that the energy and geometry of isodiazirine do not correspond to a stable ring system, since the HN bond is much too long. Similar comments apply to the sila deriva-
tive, and we therefore do not consider these mol- ecules to be stable. In addition, calculations at the 6 - 3 1 G * and MP3 level did not converge, and this fact also presumably reflects the problems in- volved with this system.
Carbene Analogs
The cyclic molecules described in Table I(viii) are include for completeness. They are much high in energy than the other isomers, and the most interesting prediction is that the open-shell triplet isomer is lower in energy for the sila-derivative by a substantial amount (ca. 0.2 a.u.). I t may be that this species corresponds to one of the transition states for the interconversion of the isomers.
DISCUSSION
Relative Stabilities of the Isomers
Table I1 lists the relative energies of the isomers, relative to that of the most stable isomer which is cyanamide in the CHzN2 series, and sila-diazo- methane in the SiN2H2 series. However, as pointed out above, this compound appears to be a loose complex rather than a true derivative and Table I1 also includes the same quantities relative to sila-cyanamide, both for the 3-21G basis, and the relevant energies for 6-31G*//3-21G and MP3/6-
6 Thomson and Glidewell
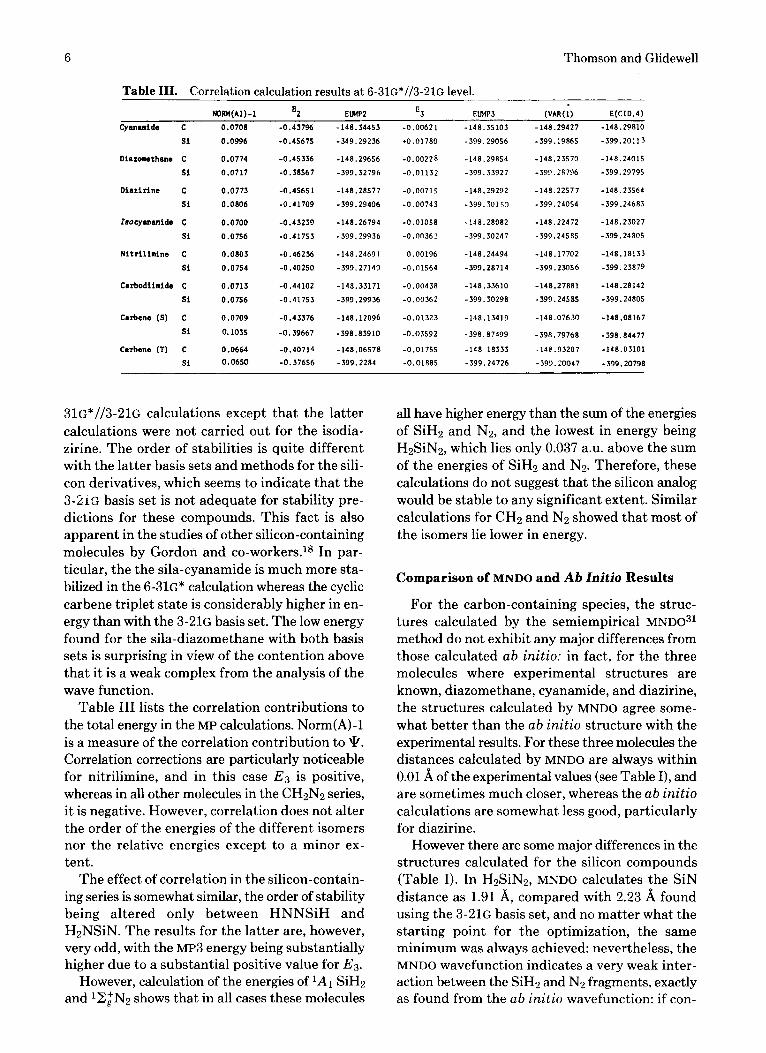
Table 111. Correlation calculation results at 6-31G*//3-21G level.
NORM(A1)-1 E2 EWPZ E3 EUMPS (VAR(1) E(CI0,I)
Cyanamide C 0.0708 -0.43796 -148.34453 -0.0062 1 -148.35103 -148.29427 -148 29810
Si 0.0996 -0.45675 -349.29236 +O .01780 -399,29056 -399.19865 -399.2011 3
Oiaroaethane C 0.0774 -0.45336 -148.29656 -0.00228 -148.29854 -148.23570 -148.24015 si 0.0717 -0.58567 -399.52796 -0.01132 -399.33927 -399.28796 -399.29795
Diazirine C 0.0773 -0.45651 -148.28577 -0.00715 -148.29292 -148.22577 -148.23564 si 0.0806 -0.41709 -399.29406 -0.00743 -399.30150 -399.24054 -399.24683
I8OCyanmide C 0.0700 -0.43239 -148.26794 -0.01058 -148.28082 -148.22472 -148.23027 si 0.0756 -0.41755 -399.29936 -0 .On362 -399.30247 -399.24585 -399.24805
N i t r i l i n i n e C n.0803 -0.46236 -148.24691 0.00196 -148.24494 -148.17702 -148.18133 si 0.0754 -0.40250 -399.27149 -0.01564 -399.28714 -599.23056 -399.23879
Carbodiinide C 0.0715 -0.44102 -148.33171 -0.00438 -148.33610 -148.27881 -148.28142 si 0.0756 -0 A1753 -399.29936 -0 .O0362 -399.30298 -399.24535 -399.24805
Carbene (S) C 0.0709 -0.43376 -148.12096 -0.01323 -148.13419 -148.07630 -148.08167
0.1035 -0.39667 -398.83910 -0.03592 -398.87499 -398.79768 -398,84477
Carbene (T) C 0.0664 -0.40714 -148.06578 -0.01755 -148 18333 -148.03207 -148.03101 si 0,0650 -0.37656 -399.2284 -0.01885 -399.24726 -j9!).20047 -399.20798
si
31G*//3-21G calculations except that the latter calculations were not carried out for the isodia- zirine. The order of stabilities is quite different with the latter basis sets and methods for the sili- con derivatives, which seems to indicate that the 3-21s basis set is not adequate for stability pre- dictions for these compounds. This fact is also apparent in the studies of other silicon-containing molecules by Gordon and co-workers.la In par- ticular, the the sila-cyanamide is much more sta- bilized in the 6-31G* calculation whereas the cyclic carbene triplet state is considerably higher in en- ergy than with the 3-21G basis set. The low energy found for the sila-diazomethane with both basis sets is surprising in view of the contention above that it is a weak complex from the analysis of the wave function.
Table I11 lists the correlation contributions to the total energy in the MP calculations. Norm(A)-1 is a measure of the correlation contribution to \k. Correlation corrections are particularly noticeable for nitrilimine, and in this case E3 is positive, whereas in all other molecules in the CH2N2 series, i t is negative. However, correlation does not alter the order of the energies of the different isomers nor the relative energies except to a minor ex- tent.
The effect of correlation in the silicon-contain- ing series is somewhat similar, the order of stability being altered only between HNNSiH and H2NSiN. The results for the latter are, however, very odd, with the MP3 energy being substantially higher due to a substantial positive value for EB.
However, calculation of the energies of lA 1 SiH2 and 'Z;N2 shows that in all cases these molecules
all have higher energy than the sum of the energies of SiH2 and Nz, and the lowest in energy being H2SiN2, which lies only 0.037 a.u. above the sum of the energies of SiHz and N2. Therefore, these calculations do not suggest that the silicon analog would be stable to any significant extent. Similar calculations for CH2 and N2 showed that most of the isomers lie lower in energy.
Comparison of MNDO and Ab Initio Results
For the carbon-containing species, the struc- tures calculated by the semiempirical MND031 method do not exhibit any major differences from those calculated ab initio: in fact, for the three molecules where experimental structures are known, diazomethane, cyanamide, and diazirine, the structures calculated by MNDO agree some- what better than the ab initio structure with the experimental results. For these three molecules the distances calculated by MNDO are always within 0.01 8, of the experimental values (see Table I), and are sometimes much closer, whereas the ab initio calculations are somewhat less good, particularly for diazirine.
However there are some major differences in the structures calculated for the silicon compounds (Table I). In H2SiN2, MNDO calculates the SiN distance as 1.91 A, compared with 2.23 8, found using the 3-21G basis set, and no matter what the starting point for the optimization, the same minimum was always achieved: nevertheless, the MNDO wavefunction indicates a very weak inter- action between the SiH2 and N2 fragments, exactly as found from the ab initio wavefunction: if con-

Isomers of CH2N2 and SiH2N2 7
strained to be planar, the H2SiN2 molecule disso- ciated to SiH2 + N2. A similar large difference in calculated SiN distances occurs in sila-isocyana- mide HBNNSi, where MNDO calculates a value of 1.51 A, very much shorter than the ab initio value of 2.6'7 A. Whereas the sila-carbodiimide, HNSiNH, is calculated to have C2h symmetry in the 3-21G basis, with a linear NSiN fragment, the structure calculated by MNDO is W-shaped, of CzU symmetry with an N-Si-N angle of only 117.1'. On the other hand, the structures calculated by MNDO and by the 3-21G basis set for H2NSiN and for sila-nitrilimine, HSiNNH, although different in detail do not exhibit such gross discrepancies. Sila-diazirine was calculated to rearrange to the diazosilane H2SiN2.
Turning to the relative energies, the order cal- culated for the carbon species a t the MP3/6- 3 1 ~ * / / 3 - 2 1 ~ level is (11) < (VI) < (I) < (111) < (IV) < (v) < (VIII) < ( Ix) . MNDO correctly orders six of the isomers, thus: (11) < (VI) < (I) < (111) < (V) < (VIII), but exaggerates the stability of (VII), placing it between (I) and (III), and also places (IV) above (V) rather than below it. Quantitatively also, the agreement between the relative energies calculated by MNDO and a t the MP3 level is rea- sonably satisfactory for the isomers (II), (VI), (I), and (111).
CONCLUSIONS
We have compared the results of geometry predictions by MNDO with those of ab initio cal- culations using the 3-21G basis set. For the carbon analog, the different methods are in good agree- ment with each other and with experiment.
In the case of the silicon-containing species, there are serious disagreements, suggesting that the MNDO parameterization is inadequate for this type of molecule, and according to all the ab initio calculations, all of the H2SiN2 species are likely to represent only weakly bound local minima o n the surface.
The authors thank the University of St. Andrews Com- puting Laboratory for a generous amount of computer time, and Professor J. A. Pople for a copy of the GAUSSIAN 80 program system, which was modified for use with the VMS 2.0 operating system by one of the authors (C.T.).
References
1. L. E. Guselnikov and N. S. Namethin, Chem. Reu., 79,
2. P. P. Gaspar, React. Zntermed., 1,229 (1978). 529 (1979).
3. J. D. Goddard, Y. Yoshioka, and H. F. Schaefer 111, J . Am. Chem. Soc., 102,7644 (1980).
4. M. H. Lien and A. C. Hopkinson, Chem. Phys. Lett., 80,114 (1981).
5. J. N. Murrell, H. W. Kroto, and M. F. Guest, J . Chem. SOC. Chem. Commun., 619 (1977).
6. A. C. Hopkinson and M. H. Lien, J . Chem. SOC., Chem. Commun., 107 (1980).
7. M. S. Gordon and J. A. Pople, J . Am. Chem. SOC., 103, 2945 (1981).
8. H. B. Schlegel, S. Wolfe, and K. Mislow, J . Chem. SOC. Chem. Commun., 246 (1975).
9. 0. P. Strausz, L. Gammie, G. Theodorakoupoulos, P. G. Mezey, and I. G. Csizmadia, J . Am. Chem. SOC., 98, 1622 (1976).
10. R. Ahlrichs and R. Heinzmann, J . Am. Chem. Soc., 99, 7452 (1977).
11. M. S. Gordon, Chem. Phys. Lett., 54,9 (1978). 12. D. M. Hood and H. F. Schaefer 111, J . Chem. Phys., 68,
2985 (1978). 13. 0. P. Strausz, R. K. Gosavi, G. Theodorakoupoulos,
and I. G. Csizmadia, Chem. Phys. Lett., 58, 43 (1978).
14. R. K. Gosavi, H. E. Gunning, and 0. P. Strausz, Chem. Phys. Lett., 59,321 (1978).
15. K. Vasudevan and F. Grein, Chem. Phys. Lett., 75,75 (1980).
16. J. C. Barthelat, G. Trinquier, and G. Bertrand, J . Am. Chem. SOC., 101,3785 (1979).
17. M. H. Lien and A. C. Hopkinson, Chem. Phys. Lett., 80,114 (1981).
18. M. S. Gordon and R. D. Koob, J . Am. Chem. SOC., 103, 2939 (1981).
19. M. S. Gordon, J . Am. Chem. SOC., 102,7419 (1980). 20. M. S. Gordon, Chem. Phys. Lett., 76,163 (1980). 21. L. C. Snyder and Z. R. Wasserman, J. Am. Chem. SOC.,
101,5222 (1979). 22. R. A. Poirier and J. D. Goddard, Chem. Phys. Lett., 80,
37 (1981). 23. J. B. Moffat, in Chemistry of the Diazonium and Diazo
Groups, S. Patai, Ed., Interscience, London, 1977, Chap. 1.
24. B. T. Hart, Austr. J. Chem., 26,461,477 (1973). 25. E. Schafer, M. Winnewisser, and J. J. Christiansen,
Chem. Phys. Lett., 81,380 (1981). 26. W. Audo, H. Tsumaki, and M. Ikeno, J . Chem. SOC.
Chem. Commun., 597 (1981). 27. J. S. Binkley, J. A. Pople, and W. J. Hehre, J . Am.
Chem. SOC., 102,939 (1980). 28. J. S. Binkley, R. A. Whiteside, R. Krishnan, R. Seeger,
D. J. DeFrees, H. B. Schlegel, S. Topiol, L. R. Kahn, and J. A. Pople, GAUSSIAN 80, Carnegie-Mellon Uni- versity, 1980.
29. J. A. Pople, J. S. Binkley, and R. Seeger, Znt. J . Quantum Chem. Symp., 10,l (1976).
30. P. C. Hariharan and J. A. Pople, Chem. Phys. Lett., 16, 217 (1972).
31. M. J. S. Dewar and W. Thiel, J . Am. Chem. SOC., 99, 4899 (1977).
32. J. B. Moffat, J. Mol. Struct., 52, 275 (1979). 33. A. P. Cox, L. F. Thomas, and J. Sheridan, Nature
34. S. P. Walch and W. A. Goddard 111, J . Am. Chem. SOC.,
35. J. K. Tyler, L. F. Thomas, and J. Sheridan, J . Chem.
36. M. A. Vincent and C. E. Dykstra, J . Chem. Phys., 73,
37. C. E. Dykstra, H. F. Schaefer 111, and W. Meyer, J .
(London), 181,1000 (1958).
97,5319 (1975).
SOC. Chem. Commun., 155 (1959).
3838 (1980).
Chem. Phys., 65,2740 (1976).

8 Thomson and Glidewell
38. C. Glidewell and C. Thomson, Chem. Phys. Lett., 86,
39. L. Pierce and V. Dobyns, J. Am. Chem. SOC., 84,2651
40. J. H. Davis and W. A. Goddard 111, J . Am. Chem. SOC.,
99,7111 (1977). 340 (1982).
(1962).
41. J. P. Anselme, J . Chem. Ed., 54,296 (1977). 42. J. F. Ogilvie, J . Mol. Struct., 3,513 (1969). 43. K. Wagner, K. Findeisen, W. Schafer, and W. Dietrich,
Angew. Chem. Znt., 20,819 (1981).





























