Embed Size (px)
Citation preview

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 1/49
Amino Acid Synthesis &
Degradation

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 2/49
12/25/2012 2
Introduction:
Amino acid degradation involves two steps:
1. Relaease of α-amino group (NH3 release)
2. α – ketoacids formation: that enters into
intermediary pathway and liberates energy.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 3/49
12/25/2012 3
Essential Amino Acids:
The essential amino acids cannot be
synthesized (or produced in sufficient
amounts) by the body and, therefore, must be
obtained from the diet in order for normalprotein synthesis to occur.
e.g. Cystein, Tyrosine, Histidine, Valine.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 4/49
12/25/2012 4
Non-Essential Amino Acids:
Nonessential amino acids can be synthesized
in sufficient amounts from the intermediates
of metabolism or, as in the case of cysteine
and tyrosine, from essential amino acids. E.g. Alanine, Arginine, Tyrosine etc

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 5/49
12/25/2012 5
Classification of Essential & Non-
Essential Amino Acids

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 6/49
12/25/2012 6
A. Glucogenic amino acids
Amino acids whose catabolism yields
pyruvate or one of the intermediates of the
citric acid cycle are termed glucogenic or
glycogenic. These intermediates are substrates for
gluconeogenesis.
E.g. Alanine, Arginine, Histidine, Methionineetc

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 7/49
12/25/2012 7
B. Ketogenic Amino Acids:
Amino acids whose catabolism yields either
acetoacetate or one of its precursors (acetyl
CoA or acetoacetyl CoA) are termed as
ketogenic. E.g. Leucine, Lysine.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 8/49
12/25/2012 8
Amino Acids Degradation:
Classification
A. Amino acids that form Oxaloacetate
B. Amino acids that form α-ketoglutarate
C. Amino acids that form PyruvateD. Amino acids that form Fumarate
E. Amino acids that form Succinyl CoA
F. Amino acids that form Acetyl CoA or Acetoacetyl CoA.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 9/49
12/25/2012 9
A. Amino acids that form Oxaloacetate
• Leukemic cells are unable to synthesize
sufficient asparagine to support their
growth.
• Which therefore require asparagine from
the blood.
• Asparaginase, which hydrolyzes
asparagine to aspartate, can be
administered systemically to treat leukemic
patients.• It lowers the level of asparagine in the
plasma and, therefore, deprives cancer
cells of a required nutrient.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 10/49
12/25/2012 10
B. Amino acids that form α-ketoglutarate
1. Glutamine:
Glutamine ------------------ Glutamate
Glutamate ----------------- α-ketoglutarate
2. Proline:
Proline --------------
Glutamate Glutamate ---------- α-ketoglutarate
Glutaminase
Oxidative
Deamination
Transamination
Oxidation

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 11/49
12/25/2012 11
B. Amino acids that form α-ketoglutarate
3. Arginine:
Arginine -------------------- Ornithine
Ornithine ------------------- α-ketoglutarate
4. Histidine:

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 12/49
12/25/2012 12
C. Amino acids that form Pyruvate
1. Alanine:
2. Serine:
3. Glycine:

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 13/49
12/25/2012 13
C. Amino acids that form Pyruvate
4. Cystine:
Cystine ---------------- Cysteine
Cysteine -------------- Pyruvate
5. Threonine:
Threonine ---------- Pyruvate
Threonine ----------
α
-ketoButyrate
NADH+
Desulfuration
Succinyl Co-A

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 14/49
12/25/2012 14
D. Amino acids that form Fumarate
Tyrosin
Phenylalnoin

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 15/49
12/25/2012 15
E. Amino acids that form Succinyl Co-A.
1. Valine & Isoleucine: These branched-
chain amino acids generate propionyl CoA,
which is converted to succinyl CoA by
biotin- and vitamin B12 –requiring reactions.2. Threonine: This amino acid is dehydrated
to α-ketobutyrate, which is converted to
propionyl CoA and then to succinyl CoA.3. Mehtionine: Mthionine degradation and re-
synthesis pathway is given as below:

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 16/49
12/25/2012 16
E. Amino acids that form Acetyl CoA
or Acetoacetyl CoA.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 17/49
12/25/2012 17
F. Amino acids that form Acetyl CoA
or Acetoacetyl CoA.
1. Leucine: This amino acid is exclusively ketogenic in itscatabolism, forming acetyl CoA and acetoacetate.
2. Isoleucine: This amino acid is both ketogenic andglucogenic, because its metabolism yields acetyl CoA
and propionyl CoA.3. Lysine: An exclusively ketogenic amino acid, this
amino acid is unusual in that neither of its amino groupsundergoes transamination as the first step in catabolism.Lysine is ultimately converted to acetoacetyl CoA.
4. Tryptophan: This amino acid is both glucogenic andketogenic because its metabolism yields alanine andacetoacetyl CoA.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 18/49
12/25/2012 18
Catabolism Of The Branched-chain Amino Acids
The branched-chain amino acids, isoleucine,
leucine, and valine, are essential amino
acids.
In contrast to other amino acids, they aremetabolized primarily by the peripheral
tissues (particularly muscle), rather than by
the liver. Their degradative pathway steps are as
follows:

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 19/49
12/25/2012 19
Steps:
1. Transamination: Removal of the aminogroups of all three amino acids is catalyzedby a single, vitamin B6 –requiring enzyme,α
- amino acid aminotransferase .2. Oxidative Decarboxylation: Removal of
the carboxyl group is catalyzed by a singlemultienzyme complex, α -keto acid
dehydrogenase complex. It uses thiamine pyrophosphate, lipoic acid,
FAD, NAD+, and CoA as its coenzymes.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 20/49
12/25/2012 20
Steps: (continued……)
3. Dehydrogenation: Oxidation of the products
formed in the above reaction yields α-β-
unsaturated acyl CoA derivatives.
4. End Products: The catabolism of isoleucine yields acetyl CoA
(ketogenic) and succinyl CoA (glucogenic).
Valine yields succinyl CoA (glucogenic). Leucine is ketogenic, being metabolized to
acetoacetate and acetyl CoA.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 21/49
12/25/2012 21
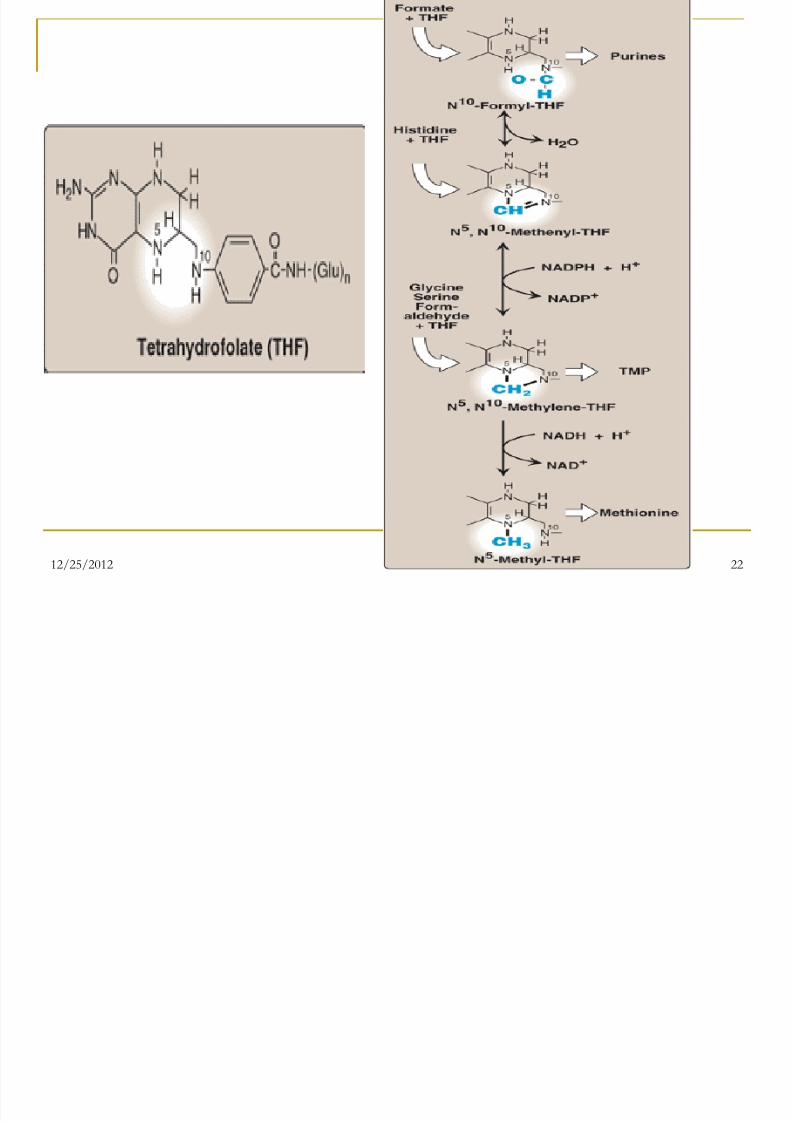
Folic acid: a carrier of one-carbon units
The active form of folic acid, tetrahydrofolic
acid (THF), is produced from folate by
Dihydrofolate Reductase in a two-step
reaction requiring two moles of NADPH. The carbon unit carried by THF is bound to
nitrogen N5 or N10, or to both N5 and N10.
THF allows one-carbon compounds to berecognized and manipulated by biosynthetic
enzymes.
7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 22/49
12/25/2012 22

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 23/49
12/25/2012 23
Biosynthesis of Nonessential Amino Acids
These are synthesized from intermediates of
metabolism or, as in the case of tyrosine and
cysteine, from the essential amino acids
phenylalanine and methionine, respectively. The synthetic reactions for the nonessential
amino acids are described below:

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 24/49
12/25/2012 24
A. Synthesis From α-keto Acids:
Alanine, aspartate, and
glutamate are
synthesized by transfer
of an amino group to
the α-keto acids
pyruvate, oxaloacetate,
and α-ketoglutarate,
respectively.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 25/49
12/25/2012 25
B. Synthesis By Amidation
1. Glutamate: This amino
acid, which contains an
amide linkage with
ammonia is formed from
glutamate . The reaction is driven by
the hydrolysis of ATP.
2. Asparagine: This is
formed from aspartate by
asparagine synthetase ,
using glutamine as the
amide donor.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 26/49
12/25/2012 26
C. Proline
Glutamate is converted to proline by
cyclization and reduction reactions.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 27/49
12/25/2012 27
Serine & Glycine Synthesis:
Serine: Serine can alsobe formed from glycinethrough transfer of ahydroxymethyl group by
serine hydroxymethyl transfease.
Glycine: This amino acidis synthesized fromserine by removal of a
hydroxymethyl group,also by serine hydroxymethyl transfease.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 28/49
12/25/2012 28
Cysteine:
This amino acid is
synthesized by two
consecutive reactions:
1. Homocysteinecombines with serine,
forming cystathionine.
2. Which then is
hydrolyzed to α-ketobutyrate and
cysteine.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 29/49
12/25/2012 29
Tyrosine:
Tyrosine is formed fromphenylalanine byphenylalanine hydroxylase .
The reaction requiresmolecular oxygen and thecoenzymetetrahydrobiopterin (BH4).
Tyrosine, like cysteine, isformed from an essentialamino acid and is,
therefore, nonessential onlyin the presence of adequatedietary phenylalanine.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 30/49
12/25/2012 30
Metabolic Defects in Amino Acid
Metabolism
Phenylketonuria
Maple Syrup Urine Disease
Albinism
Homocystinuria
Alkaptonuria

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 31/49
12/25/2012 31
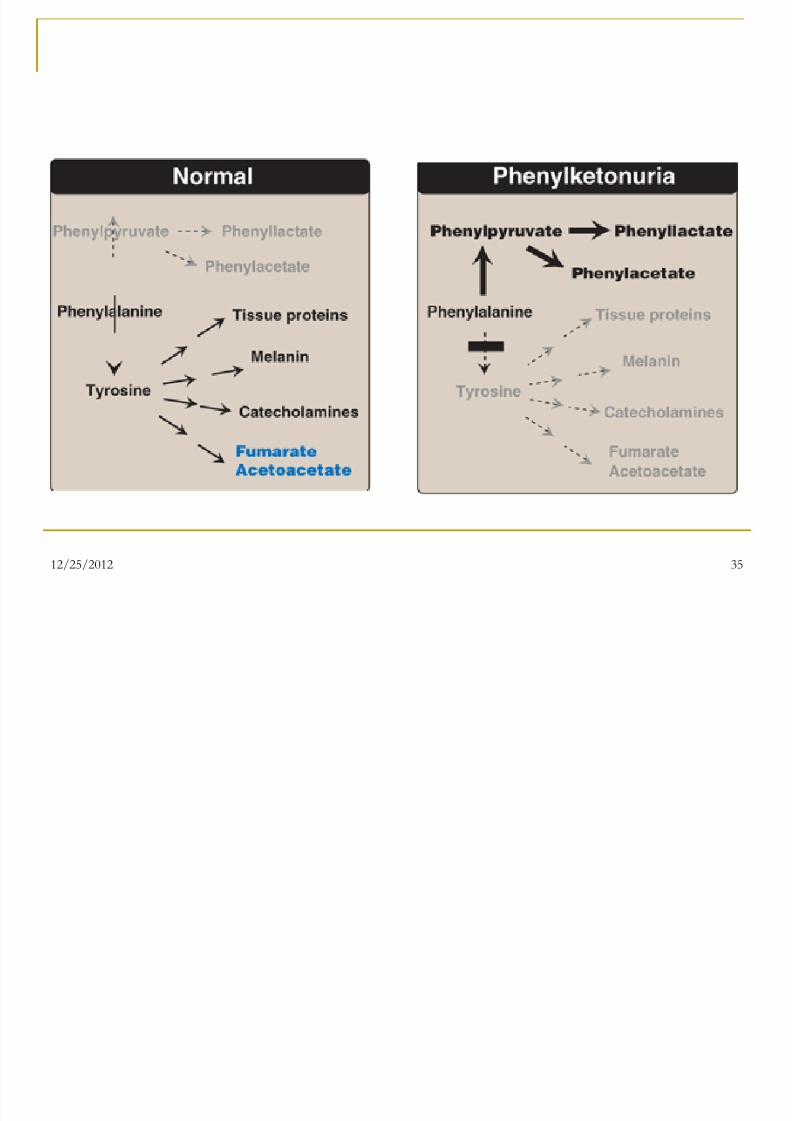
1. Phenylketonuria ( PKU )
Autosomal recessive genetic disorder
Caused by hepatic phenylalanine
hydroxylase deficiency
When PAH is deficient, phenylalanine
accumulates and is converted into
phenylpyruvate which is detected in the
urine(musty odor urine)

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 32/49
12/25/2012 32
Hyperphenylalaninemia may also be caused by
Deficiencies in dihydropteridine (BH2)
reductase , which regenerates BH4 from BH2.
BH4 is also required for tyrosine hydroxylase
and tryptophan hydroxylase , which catalyze
reactions leading to the synthesis of
neurotransmitters, such as serotonin andcatecholamines.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 33/49
12/25/2012 33
Hyperphenylalaninemia:

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 34/49
12/25/2012 34
Characteristics of PKU
Hyperphenylalaninemia
CNS symptoms: Mental retardation, failure to walk
or talk, seizures, hyperactivity, tremor, microcephaly,
and failure to grow. Hypopigmentation:
Fair hair, light skin color, and blue eyes
The hydroxylation of tyrosine by tyrosinase , which is
the first step in the formation of the pigmentmelanin, is competitively inhibited by the high levels
of phenylalanine present in PKU.
7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 35/49
12/25/2012 35

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 36/49
12/25/2012 36
Treatment:
Avoid diet rich in phenylalanine.
Use synthetic amino acid preparations
containing low contents of phenylalanine.
But
Phenylalanine is an essential amino acid.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 37/49
12/25/2012 37
2. Maple Syrup Urine Disease
A rare (1:185,000), autosomal recessive disorder
Characterized by a partial or complete deficiency in
branched-chain α-keto acid dehydrogenase , an
enzyme complex that decarboxylates leucine,isoleucine, and valine
Disease is characterized by feeding problems,
vomiting, dehydration, severe metabolic acidosis,
and a characteristic maple syrup odor to the urine. If untreated, the disease leads to mental
retardation, physical disabilities, and even death.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 38/49
12/25/2012 38
Classes of MSUD:
a. Classic form: The most common type of MSUD
Leukocytes or cultured skin fibroblasts show
little or no branched-chain α-keto acid dehydrogenase activity
Infants show symptoms within the firstseveral days of life.
If not diagnosed and treated, is lethal in thefirst weeks of life.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 39/49
12/25/2012 39
Classes of MSUD: (continued…..)
b. Intermediate Form: A higher level of enzyme activity
(approximately 3 –15% of normal).
The symptoms are milder and show an onsetfrom infancy to adulthood.
c. Thiamine Responsive Form:
Increased activity of branched-chain α-keto acid dehydrogenase if given large doses of this vitamin.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 40/49
12/25/2012 40
Treatment:
A synthetic formula that contains limited
amounts of leucine, isoleucine, and valine
Early diagnosis and lifelong dietary treatment
is essential if the child with MSUD is todevelop normally.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 41/49
12/25/2012 41
3. Albinism
A defect in tyrosine metabolism results in a
deficiency in the production of melanin.
Result is the partial or full absence of pigment
from the skin, hair, and eyes.
It may be of different types like:
Autosomal recessive (primary mode),
Autosomal dominant, or X-linked.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 42/49
12/25/2012 42
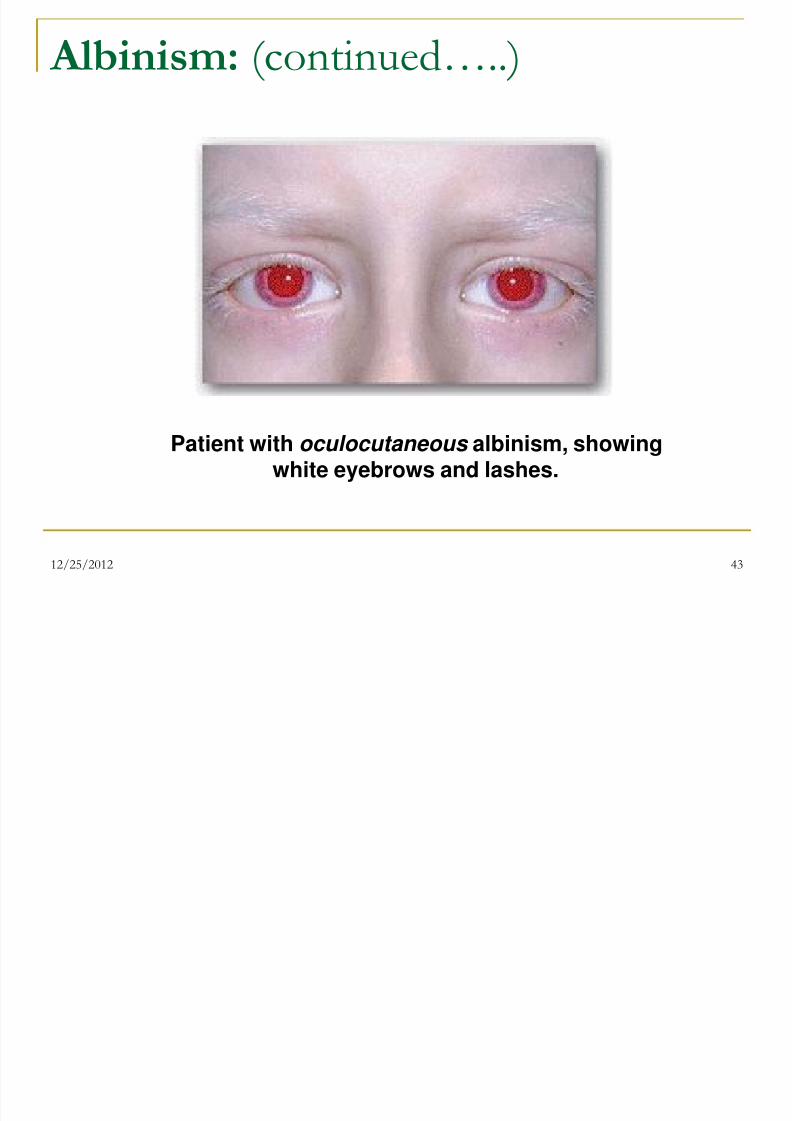
Albinism: (continued…..)
Complete albinism (also called tyrosinase -negative oculocutaneous albinism) resultsfrom a deficiency of tyrosinase activity,causing a total absence of pigment from thehair, eyes, and skin
It is the most severe form of the condition. Inaddition to hypopigmentation, affected
individuals have vision defects andphotophobia (sunlight hurts their eyes). Theyare at increased risk for skin cancer.
7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 43/49
12/25/2012 43
Albinism: (continued…..)
Patient with oculocutaneous albinism, showingwhite eyebrows and lashes.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 44/49
12/25/2012 44
4. Homocystinuria
Defects in the metabolism of homocysteine
Autosomal recessive illnesses
Characterized by high plasma and urinary
levels of homocysteine and methionine and
low levels of cysteine.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 45/49
12/25/2012 45
Causes:
The most common
cause of
homocystinuria is a
defect in the enzyme
cystathionine β-
synthase , which
converts homocysteine
to cystathionine.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 46/49
12/25/2012 46
Homocystinuria: (continued)
Symptoms include:
Ectopia lentis (displacement of the lens of the eye)
Skeletal abnormalities
Premature arterial disease Osteoporosis
Mental retardation
Patients can be responsive or nonresponsive to oral
administration of pyridoxine (vitamin B6)—a
coenzyme of cystathionine β-synthase.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 47/49
12/25/2012 47
Treatment:
Restriction of methionine intake and
supplementation with vitamins B6, B12, and
folate.

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 48/49
12/25/2012 48
5. Alkaptonuria
A rare metabolic disease involving a deficiency in
homogentisic acid oxidase
Symptoms include:
Homogentisic aciduria, Large joint arthritis
Black ochronotic pigmentation of cartilage and
collagenous tissue

7/30/2019 Amino Acid Synthesis & Degradation
http://slidepdf.com/reader/full/amino-acid-synthesis-degradation 49/49
Treatment:
Diets low in protein—especially in
phenylalanine and tyrosine—help reduce the
levels of homogentisic acid, and decrease the
amount of pigment deposited in body tissues.





























