Embed Size (px)
Citation preview

Introduction to Translational Bioinformatics
Atul Butte, MD, PhDChief, Division of Systems Medicine,
Department of Pediatrics,Department of Medicine, and, by courtesy, Computer Science
Center for Pediatric Bioinformatics, LPCHStanford University
[email protected] @atulbutte
Disclosures• Scientific founder and advisory board membership
– Genstruct– NuMedii– Personalis
• Past or present consultancy– Lilly– NuMedii– Johnson and Johnson– Genstruct– Tercica– Ansh Labs– Prevendia
• Honoraria– Lilly– Siemens
Agenda• Why Translational Bioinformatics?• Basic Introduction to Molecular Biology• How these molecules get measured• Ontologies• Software• Common challenges between medical and bio-informatics
• Genomic Nosology and Drug Repositioning• Discovering biomarkers from public moleuclar data• Patients presenting with whole genome sequences
• Conclusions
Kilo KiloMega
1

KiloMegaGiga
KiloMegaGigaTera
KiloMegaGigaTeraPeta
KiloMegaGigaTeraPetaExa
KiloMegaGigaTeraPetaExa
Zetta
2

What is translational bioinformatics?• Translational bioinformatics
– Development of analytic, storage, and interpretive methods – Optimize the transformation of increasingly voluminous genomic and
biological data into diagnostics and therapeutics for the clinician
• Includes– Research on the development of novel techniques for the integration of
biological and clinical data – Evolution of clinical informatics methodology to encompass biological
observations
• End product of translational bioinformatics – Newly found knowledge from these integrative efforts that can be
disseminated to a variety of stakeholders, including biomedical scientists, clinicians, and patients
Why translational bioinformatics?Eight reasons for excitement1. There is an increasing call for translational medicine:
Universities, Congress, NIH, and elsewhere: “What did we get for our money?”
2. Many tools now exist that enable the large-scale parallel quantitative assessment of molecular state- Premier example is RNA expression detection microarray- High-bandwidth measurement tools; not just big, but nearly
comprehensive- Low-cost
- Quantitating every gene in the genome: ~$200 per sample- Measuring 1.8 million human polymorphisms: ~$300 per sample- Knocking out every gene in the worm: ~$4500 for the kit- Sequencing entire human genome: $3000
Sequencing Excitement• 454/Roche, Life Technologies• Pacific Biosystems: sequence
human genome in 15 minutes• Run times in minutes
at a cost of hundreds of dollars• 20 TB in 15 minutes• Helicos: $30k genome• Complete Genomics:
80 genomes/day• Illumina: $3500 per
genome• Ion Torrent
3

Nature, October 2010.
Why translational bioinformatics?3. Incredible amounts of publicly-available data
- GenBank: Hundreds of organisms have been completely sequenced, including man and mouse; 310,000+ species have had SOME sequence measured
- GEO has 625,000+ samples today from 25000+ experiments- ArrayExpress has 170,000+ samples today from 5200+ experiments- EBI Pride: 14,800+ samples yielding over 111 million mass spectra- NCBI dbGAP (genotype and phenotype): 250 genetic studies across
850,000 individuals
4

Total 800,000 microarrays availableDoubles every two years
Butte AJ. Translational Bioinformatics: coming of age. JAMIA, 2008.
Total 800,000 microarrays availableDoubles every two years
Intel Science Talent Search semi-finalists• Rohan Chakicherla (2009)• Denzil Sikka (2009)• Tony Ho (2010)Intel and Siemens Competition finalist• Andrew Liu (2010)
Intel Science Talent Search semi-finalists• Rohan Chakicherla (2009)• Denzil Sikka (2009)• Tony Ho (2010)Intel and Siemens Competition finalist• Andrew Liu (2010)
Butte AJ. Translational Bioinformatics: coming of age. JAMIA, 2008.
5

6

Validation methods are increasingly commoditized
7

Translational Pipeline
Clinical and Molecular Measurements
Translational Question or Trial
Statistical/Computational methods
Validating drug or biomarker
Translational Pipeline
Clinical and Molecular Measurements
Translational Question or Trial
Statistical/Computational methods
Validating drug or biomarker
Com
mod
ity
Com
mod
ity
Why translational bioinformatics?4. New community of sharing: tools, data, publications
- Journals require this; NIH is starting to require this- Along with communities comes contention and agreement:
“What is the name for this gene?”- Increasing standardization in names, abbreviations, codes,
and file formats- Where standards have not been reached, there is at least the
understanding that it must be reached
Why translational bioinformatics?5. Change in role of the bioinformatician from service provider to
question asker– Each high-bandwidth measure yields sizeable amount of raw data– Distilling raw data and filtering out noise through the proper use of
bioinformatics– Bioinformatics clearly plays a role in the storage, retrieval, and sharing
of measurements, and relating to clinical outcomes– However, role for bioinformatics in genomic medicine is now beyond
that of providing a service, and in enabling new and interesting questions to be asked in biomedical research
– A researcher, whether clinical, experimental, or theoretical, can ask questions no one else can ask today, when powered by bioinformatics
Why translational bioinformatics?6. Increased funding for this line of work
- NIH Roadmap started in May 2002; Dr. Zerhouni’sroadmap for medical research in the 21st century
- ”At no other time has the need for a robust, bidirectional information flow between basic and translational scientists been so necessary.”
- Three major themes- New Pathways to Discovery: addresses Bioinformatics
and Computational Biology- Research Teams of the Future: cell biologists and
computer programmers working to accelerate movement of scientific discoveries from the bench to the bedside
- Re-engineering the Clinical Research Enterprise: transforming basic research discoveries into drugs, treatments, methods for prevention.
- Clinical and Translational Science Award (CTSA): 60 funded at ~$30M- RFA mentions “informatics” 38 times- Recognition that the problem of translational medicine will not go away
without the help of informatics
0
20
40
60
80
100
120
140
160
180
1995
1996
1997
1998
1999
2000
2001
2002
2003
2004
2005
2006
2007
2008
2009
2010
0%
5%
10%
15%
20%
25%
30% Why translational bioinformatics?7. It’s incredible how much bioinformatics you need to know
just to read the New England Journal of Medicine!- “Shrunken centroid”, “Unsupervised cluster analyses”,
“gene-expression signature”- “global scaling, or normalization”- “q value for each gene represents the probability that it
is falsely called significantly deregulated”- “class-prediction analyses”, “10-fold cross validation”- “implemented in the PLINK tool set as a Cochran-Mantel-Haenszel
stratified analysis”
8. Paucity of people trained to make use of these resources
8

Basic Biology• Organisms need to produce proteins for a variety of
functions over a lifetime– Enzymes to catalyze reactions– Structural support– Hormone to signal other parts of the organism
• Problem: how to reliably and reproducibly build proteins– How to encode the instructions for making a specific protein?
• Step one: nucleotides
Basic Biology
• Naturally form double helixes• Redundant information in each strand
• Complementary nucleotides form base pairs• Base pairs are put together in chains (strands)
5’
3’
3’
5’
Chromosomes• We do not know exactly how strands of DNA wind up to make a
chromosome• Each chromosome has a single double-strand of DNA• 22 human chromosomes are paired• In human females, there are two X chromosomes• In males, one X and one Y
What does a gene look like?• Genes contain biological information• Most encode instructions to make a one or more proteins• DNA before a gene is called upstream, and can contain
regulatory elements• Most genes are discontinuous: introns and exons• There is a code for the start and end of the protein
coding portion• Theoretically, the biological system can determine
promoter regions and intron-exon boundaries using the sequence syntax alone
Area between genes• Human genome contains 3.2 billion
base pairs (3200 Mb) but only 35k genes• Coding region 90 Mb = 1.5% of genome• 50+% of genome are repeated sequences
– Long interspersed nuclear elements
– Short interspersed nuclear elements
– Long terminal repeats– Microsatellites
• On average, one microsatellitedifference every 2,000 basepairs
Genome size• We’re the smartest, so we must have the
largest genome, right?• Not quite• Our genome contains
3200 Mb (~750 megabytes)
• E. coli has 4 Mb• Yeast has 12 Mb• Pea has 4800 Mb• Maize has 5000 Mb• Wheat has 17000 Mb
9

Genomes of other organisms• Plasmodium falciparum chromosome 2
Gardner M, et al. Science; 282: 1126 (1998).
mRNA • mRNA can be transcribed at up to several hundred
nucleotides per minute• Some eukaryotic genes can take many hours to
transcribe– Dystrophin takes 20 hours to transcribe
• Most mRNA ends with poly-A, so it is easy to pick out• Can look for the presence of specific mRNA using the
complementary sequence
mRNA is made from DNA • Genes encode
instructions to make proteins
• The design of a protein needs to be duplicable
• mRNA is transcribed from DNA within the nucleus
• mRNA moves to the cytoplasm, where the protein is formed
• Central Dogma:DNA RNA Protein
Protein
Digitizing amino acid codes
• Proteins are made of 20 (22) amino acids
• Yet each position can only be one of 4 nucleotides
• Nature evolved into using 3 nucleotides to encode a single amino acid
• A chain of amino acids is made from mRNA
•Selenocysteine
•Pyrrolysine
Genetic Code
Nature; 409: 860 (2001).
Protein targeting
• The first few amino acids may serve as a signal peptide
• Works in conjunction with other cellular machinery to direct protein to the right place
10

Transcriptional Regulation• Amount of protein is roughly governed by RNA level• Transcription into RNA can be activated or repressed by
transcription factors
What starts the process?
• Transcriptional programs can start from– Hormone action on receptors– Shock or stress to the cell– New source of, or lack of
nutrients– Internal derangement of cell
or genome– Many, many other internal
and external stimuli
Periodic Table for Biology• Knowing all the genes
is the equivalent of knowing the periodic table of the elements
• Instead of a table, our periodic table may read like a tree
More Information• Department of Energy Primer on
Molecular Genetics http://www.ornl.gov/hgmis/publicat/primer/primer.pdf
• T. A. Brown, Genomes 3nd edition,John Wiley and Sons, 2006.
Gene Measurement TechniquesDNA• Sequencing• PolymorphismsRNA• DNA Microarrays• WafersProtein• 2D-PAGE• Mass spectrometry• Protein interactionsAntibodies• Arrays
Cytogenetic Maps• Chromosomal banding (Giemsa staining, and others) known for
decades• Help in mapping STS• FISH: fluorescence-in-situ-hybridization• DNA complementing unique regions, tagged by STS
Kirsch I, et al. Nature Genetics 24:339 (2000).
11

Genetic Maps• Genome map where polymorphic loci are positioned relative to
one another on basis of recombination during meiosis• Unit centimorgans (cM) or 1% chance of recombination• Genetic distance was known to roughly correlate with physical
distance• Mapping between genetic disease
and physical distance
Van Etten W, et al. Nature Genetics 22:384 (1999).
Mapping to Diseases• Array of markers can be evaluated together• Linkage evaluated by log-odds (LOD) score• LOD score over 3 is significant• Now with so many
markers and loci being tested, significance isharder
Sequencing Reactions• Polymerase Chain Reaction
• Sanger Reactions• Four color fluorescence base
sequence detection• Laser detector• Automated process
Sequencing Reactions• PHRED: base-quality score
for each base, based on probability of erroneous call
• PHRED quality score of X means error probability of 10-x/10
• PHRED score of 30 means 99.9% accuracy for base call
Buetow KH, et al. Nature Genetics 21:323 (1999).
Sequencing Reactions• PHRAP: assembles sequence data
using base-quality scores into sequence contigs
• Assembly-quality scores
• Most of the genome was sequenced over 12 months
• Highest throughput center at Whitehead: 100,000 sequencing reactions per 12 hours
• Robots pick 100,000 colonies, sequence 60 million nucleotides per day
12

Assembly• Contamination from non-human sequences removed• Clones overlaid on physical map• High-quality semiautomatic sequencing from both ends of very
large numbers of numbers of human genome fragments• Finding the overlaps takes memory: Drosophila 600 GB RAM
Genome Browsers• Genome browsers: University of California at Santa Cruz and
EnsEMBL• Overlap sequence, cytogenetic, SNP, genetic maps• Overlap annotations, disease genes
Transposable Elements• LINE, SINE, LTR retroposons and DNA transposons• Fools machinery into transcribing RNA and making into
proteins, which then work to copy the RNA into DNA somewhere else in the genome
Protein-Coding Genes• 1.5% genome is genes• Between 30,500 and 35,500 genes (and dropping)• Largest gene dystrophin 2.4 Mb• Longest coding sequence titin 80,780 bp, 178 exons• Gene has average 2.6 distinct transcripts• Human has twice as many genes as fly or worm• Differences: spreadout, alternativing splicing• 223 proteins with significant homology to bacteria, but not
eukaryotes
Many More Protein Domains• Each domain is present in more genes• Some domains and combinations of domains unique to human
Single Nucleotide Polymorphisms• A single nucleotide position in the genome sequence for which
there are two or more alternative alleles• Alleles present at frequency typically over 1% in human
population• SNP rate 1 per 1300 bases• dbSNP has over 34 million (4.6 million in genes)• Typical gene has 2-4 common variants
(effective number of alleles under 2)• Consistent with small founding population under 10,000• Left Africa 5,000 years ago with this distribution• Any village represents 70% of variation in the world• Common disease-common variant hypothesis: common diseases
occur due to segregation of common alleles, not occurrence of rate mutations
13

Single Nucleotide Polymorphisms
• One SNP per 1300 nucleotides on average
• Three step approach• First, find the genes
you are interested in• Second, catalog all the
polymorphisms in a gene (by sequencing)
• Third, measure those polymorphisms in a larger population
Clinical use of SNPs• New publication with
association of SNP with disease is almost a daily occurrence
Gao, X. et al. Effect of a single amino acid change in MHC class I molecules on the rate of progression to AIDS. N Engl J Med 344, 1668-75 (2001).
HapMap Genome Wide Association Studies
• Low-cost of genotyping has enabled surveys of the genome for changes
• ~2 million differences in the genome sequenced for ~$400
• Assumption: ancestral genome with variant associated with susceptibility to disease or condition
• Variant has spread throughout the population• Case-control study, more of a variant than expected• Surveyed variants are markers for the “true” variant
14

• Welcome Trust surveyed 14,000 cases of 7 diseases and 5,000 control subjects, including type 2 diabetes mellitus
• Survey of 500,000 markers• Strong association of a few alleles• Replicated/validated in many other populations
WTCCC, Nature 447:661, 2007.
SNPs and pharmacogenomics
• Genes will help us determine which drugs to use in particular disease subtypes
• Genes will help us predict those who get side-effects
Sesti F. PNAS 97:10613, 2000
• Myopathy is a 1:30k side-effect of statins• Survey of 300,000 markers in 85 subjects with statin-
associated (80 mg) myopathy, 90 controls• Strong association at a single allele• Replicated/validated in second population of 17k patients
SEARCH Group. NEJM 2008, 359:789.
15

RNA expression detection microarrays
Butte AJ. Nature Reviews Drug Discovery (2002).Schena M, et al. PNAS 93:10614 (1996).
Nature Genetics, 21: supplement (Jan 1999).
• Genome-wide, quantitative• Commodity items• International repositories of data
Experiment Design• Quantitate specific RNA
expression before and after an intervention
• Compare expression between two tissue types
• Compare expression between different strains or constructed organisms
• Compare expression between neighboring cells
Luo L, et al. Nature Medicine; 5: 117 (1999).
Validation
• In situ hybridization• Real-time Polymerase
Chain Reaction
2D-PAGE
• Two axis = two properties of proteins: pH versus mass
• Global view of proteins
• Patterns can be scanned, saved and searched
• Spots need to be picked for identification
• Unfortunately, not very quantitative
Gygi, S. P., Rochon, Y., Franza, B. R. & Aebersold, R. Correlation between protein and mRNA abundance in yeast. Mol Cell Biol 19, 1720-30 (1999). Gygi, S. P. & Aebersold, R. Proteomics: A Trends Guide. (2000).
16

Clinical uses for proteomics
• Petricoin, et al., used this technique on serum
• Finding markers distinguishing ovarian cancer versus non-neoplasia
• Quest for biomarkers
Petricoin, E. F. et al. Use of proteomic patterns in serum to identify ovarian cancer. Lancet 359, 572-7. (2002).
Quantitative proteomics• The examples so far
demonstrate identification, not quantification
• One can take advantage of the extreme sensitivity of detection of mass spectrometry
• Add to the proteins a known amount of label
Protein chips
• Detection vs. Function
• Kinase chips
Williams, D. M. & Cole, P. A. Kinase chips hit the proteomics era. Trends Biochem Sci 26, 271-3 (2001).
Protein Detection• Specific
antibodies• Antibodies need
to be available
Protein-protein interactions
• Synthesize human proteins with extra pieces
• If proteins interact, extra pieces join, and cause signal in yeast
• Can also use mass-spec to id binding partners, complexes
Rual, JF., et al. Nature 437:1173 (2005).
Interactions explain disease
K. Lage, et al. Nature Biotechnology 25:309 (2007).
17

Antigen arrays
• Spotted human GST-fusion proteins
• Can be used to probe human plasma for antibody binding
• “Antibody-ome”
Michael E. Hudson, Irina Pozdnyakova, Kenneth Haines, Gil Mor, Michael Snyder. PNAS 104:17494, 2007.
Existing clustering techniques• Several algorithms have already been
developed for knowledge discovery and data-mining of RNA expression data sets
• Hierarchical clustering: Eisen MB, PNAS 95:14863. Look at the neighbors of unknown genes/samples.
• Self-organizing maps: Tamayo P, PNAS 96:2907. Similar items are near each other.
• Relevance networks: Butte AJ, PNAS 97:12182. Networks of positive and negative association.
• Principal components: Raychaudhuri S, PSB 2000. How can I best spread my samples?
• Nearest neighbors: Golub TR, Science286:531. What genes best predict my outcome individually?
• Support vector machines: Brown MP, PNAS 97:262. What combination of genes best splits my outcome? Tell Me MoreButte AJ. Nature Reviews Drug Discovery, 1:951.
Cluster• An absolute standard tool in microarray
analysis• http://rana.lbl.gov/EisenSoftware.htm• Performs a variety of types of cluster
analysis and other types of processing on large microarray datasets– hierarchical clustering– self-organizing maps (SOMs)– k-means clustering– principal component analysis
• For Windows 95/98/NT only
GeneCluster 2.0• Whitehead Institute• http://www-genome.wi.mit.edu/cancer/software/
genecluster2/gc2.html
• Java based– Nearest neighbor– Self-organizing maps (SOMs)– Marker analysis
TIGR MEV• Multi-experiment viewer• http://www.tigr.org/softlab• Extensive
documentation• Java-based
GenMAPP• Gene Microarray Pathway Profiler• Paints pathway pictures in the context of microarray
measurements• http://www.genmapp.org• Can help
translate list of genes into implicated pathways
18

Kyoto Encyclopedia of Genes and Genomics
R / Bioconductor• Open source version of SPLUS, a well-established
statistical system originally from Bell Labs– http://www.r-project.com– http://www.bioconductor.org
Accession Numbers
Genbank Unigene
NCBI EntrezGene ID
M63603 Hs.85050
5350
Gene Ontology
HUGO Gene Nomenclature RefSeq
OMIMdbSNP
Chip
228202_at
Looking up results• Gene Expression Omnibus
– http://www.ncbi.nlm.nih.gov/geo
• Database Referencing of Array Genes Online– http://pevsnerlab.kennedykrieger.org/
• Resourcerer– http://compbio.dfci.harvard.edu/tgi/cgi-
bin/magic/r1.pl
• Netaffx– http://www.netaffx.com
• AILUN– http://ailun.stanford.edu
Biomarkers• A characteristic that is objectively measured and evaluated
as an indicator of normal biologic processes, pathogenic processes, or pharmacologic responses to a therapeutic intervention
• Type 0: natural history of a disease• Type 1: effects of an intervention in accordance with the
mechanism of action of the drug, even though the mechanism might not be known to be associated with clinical outcome
• Type 2: surrogate endpoints, change in marker predicts clinical benefit
R Frank and R Hargreaves. Nature Reviews Drug Discovery 2003, 2:566.
Adding plausibility
• Body of evidence needed to support classification of biomarkers– Plausibility– Correlation / Association– Predictive power (prognostic value)– Cause
• Systems biology provides data for correlation and predictive power
• But without a priori knowledge, you cannot ascertain plausibility or hypothesize the cause
19

Development of Type 1 Biomarkers
• A priori validation of type 1 biomarkers is impossible for novel targets without a positive control
• The more innovative the target, the less validated the biomarkers
• Biomarkers are validated in parallel with drug candidates
How many samples do we need?• To prove (p < 0.05) an 8% difference in transplant
rejection rate between two drugs, is it easier to study 10 patients or 100 patients?
• To make a list of genes that differentiate patients with early rejection from long-term non-rejection, is it easier to use 1 sample of each, or 100 samples of each?
Modified from Yeoh, et al. Cancer Cell 2002, 1: 133.
RejectNon-reject
Genes
…and much more about modeling the variation of the condition
With microarray diagnostics, sample size is less about power…
Reject
Non-reject
Genes
How do we avoid overfitting?• In other words, with too few samples, it is too easy to
overfit the measurements, especially when measuring 20 to 30 thousand genes
• We have techniques like support vector machines that even further expand the number of features
• And even the ones we get wrong, we later find they’re been misclassified, or define a new subgroup…
Yeoh, et al. Cancer Cell 2002, 1: 133.
Cross-validation• Random permutation and cross-validation are
commonly used in evaluating strategies for picking diagnostic genes
• These can help reduce the danger of overfitting• But only additional samples will allow algorithms
to learn the variation in disease• This reduces false positives
• But how do we report with such high bandwidth?• How do we report genes without a reason?
Pan W. Genome Biology 2002. http://genomebiology.com/2002/3/5/research/0022Wolfinger RD. Journal of Computational Biology 2001, 8:625.
Gene Ontology
Ashburner, et al. Nature Genetics 25:25, 2000.
20

Unified Medical Language System• How to model and compare experimental context, phenotypes?• Label using the Unified Medical Language System• Largest unification of 148 biomedical vocabularies
– 1.5M+ concepts, 7.4M terms, 32M+ relations
• Scalable– Human and model organisms– Molecular, physiological,
pathological, epidemiology, ecology
• Already has Gene Ontology, NCBI taxonomy, HUGO, OMIM,ICD-9, SNOMED-CT, MeSH, NCI
• UMLS enables integration– Joining with hospital databases of
patients or samples
Related, possiblysynonymous
MeSH HeadingD017667
SNOMEDT-1A013
CRISP0600-1092
UMLS ConceptC0872094Adipogenesis
Co-occurence
UMLS ConceptC0206131AdipocytesAdipocyteMature fat cellFat Cell
UMLS ConceptC0011849Diabetes Mellitusdm
ICD-9250
Ontologies in biology
• Open Biological Ontologies (OBO)– http://obo.sourceforge.net/– 53 ontologies
• National Center for Biomedical Ontology– Mark Musen, PI– http://www.bioontology.org/
Clinical Bioinformatics Ontology• How do we store these
genomic test results in medical records?
• Curated controlled vocabulary for current molecular diagnostic and cytogenic techniques
Common Challenges• High bandwidth data collection
– Physiological measurements with high sample rates– Higher density microarrays
• Data storage– 15% US population = 200 million multiGB images– Raw sequencing trace files for one human = 300 terabytes
Kohane I. JAMIA; 7: 512 (2000).
Common Challenges• Measurement Noise
– Artifacts in physiological measures– Poor expression measurement
reproducibility
• Data Models– Lack of standards in medical records
• HL7, HIPAA
– Too many standards in bioinformatics• Gene Expression Markup Language (GEML)• Gene Expression Omnibus (GEO)• Microarray Markup Language (MAML)
– Medical record as sample annotation
Common Challenges• Many frequencies and phase shifts
– Clinical endocrinology spans seconds to decades– What are the naturally occurring genomic frequencies?
• What is the relevant source for data?– What is the functional tissue for sleep apnea, hypertension,
diabetes?
21

Common Challenges• Comparing new signals to old
Common Challenges• Continued development of
controlled vocabularies
HL7
Common Challenges• Security
HL7
• Privacy• Ethics
Using Genomics to Diagnose
• Difficulty distinguishing between leukemias
• Microarrays can find genes that help make the diagnosis easier
Golub TR. Science 286:531, 1999.
Using Genomics to Predict
• Patients with seemingly the same B-cell lymphoma
• Looking at pattern of activated genes helped discover two subsets of lymphoma
• Big differences in survival
Alizadeh AA. Nature 403:503, 2000.Nature Medicine 9:9.
Using Genomics to Treat
• Genes will help us determine which drugs to use in particular disease subtypes
• Genes will help us predict those who get side-effects
Sesti F. PNAS 97:10613, 2000
22

Using Genomics to Find New Drugs
• The human genome project and genomics will help us find new drugs
• The entire pharmaceutical industry currently targets 500 cellular targets; this will grow to 3,000 to 10,000
Scherf, U. Nature Genetics 24:236.Butte, AJ. PNAS 97:12182.
Gene Expression
Genetics
Haplotypes
Proteomics Mass spectrometry
Phenotypes
Quantitative Trait Loci
Algorithms to integrate genetic, genomic, proteomic, animal model, and clinical measurements are needed for the next step in addressing complex disorders
Combining Clinical (CT) and Genomic Data
• 28 human hepatocellular cancer: 32 imaging
E Segal. Nature Biotechnology; 25: 675 (2007).
32
23

Genomic nosology• Linnaeus 1707-1778• Promoted binomial
nomenclature for taxonomy– Homo sapiens,
Mus musculus
• Because we can sequence DNA, we commonly re-organize species in taxonomy based on their genome– Pneumocystis jiroveci and
Pneumocystis carinii 300 year old pine (1709)Hamarikyu Garden
Tokyo, Japan
Genomic nosology• Linnaeus also co-founder
of systematic nosology– Nosology = classification of
disease
– Genera Morborum (1763)
• Why not classify diseases based on genomics?– Could reshuffle thinking
about diseases and drugs
– Public molecular data: ~800,000+ arrays, ~23000+ experiments, grows 2-3x/yr
Exanthematic Feverish, with skin eruptions
Critical Feverish, with urinary problems
Phlogistic Feverish, with heavy pulse and topical pain
Dolorous Painful
Mental With alienation of judgment
Quietal With loss of movement
Motor With involuntary motion
Suppressorial With impeded motions
Evacuatorial With evacuation of liquids
Deformities Changed appearance of solid parts
Blemishes External and palpable
Bramley M. Coding Matters 2001, 8:1.
• 39 Cancer of the buccal cavity• 40 Cancer of stomach and liver• 41 Cancer of peritoneum, intestines, rectum• 42 Cancer of female genital organs• 43 Cancer of breast• 44 Cancer of skin• 45 Cancer of other organs or not specified
Lung is an "other organ“; Brain is an "other organ"
• 50 Diabetes– No type 1 or type 2
• Endocrine diseases were under General Diseases• 88 Disease of the thyroid body
– Under Disease of the Respiratory System
• 5 Smallpox, 13 Cholera, 15 Plague, 21 Glanders, 22 Anthrax– All bioterroristic today
• 189 Visitation from God
AILUN: Extracting GEO gene lists• GEO has 12.6+ billion
measurements across ~4000 platforms
• Decoding measured gene is a challenge– Varied use of identifiers– Identifiers change meaning
• We have ~100 million mappings to NCBI Gene ids
• We mapped 67% of GEO platforms to NCBI identifiers
Gene Identifier Gene Identifier Vocabulary
AI262683 GenBank
NM_000015 GenBank
Hs.2 UniGene
NP_000006 Protein
P11245 Protein
NAT2 NCBI Gene official symbols
AAC2 NCBI Gene all symbols
IMAGE:1870937 IMAGE clone
UI-H-FG1-bgl-g-02-0-UI
University of Iowa clone
IMAGp998I184581_Institute of Molecular Biology
and Genetics Ukraine clone
10286060 GenBank GI
TC110817 OriGene Technologies Clone
HIE06837r Gunma University Clone
CMPD10049 University of Padova Clone
3NHC3746Institute of Medical Science
Japan Clone
Human N-acetyltransferase 2
Chen R, Butte AJ. Nature Methods, November 2007.
Unified Medical Language System• How to model and compare experimental context, phenotypes?• Label using the Unified Medical Language System (UMLS)• Largest unification of 160+ biomedical vocabularies
– 10M+ concepts, 64M+ relations
• Scalable– Human and model organisms– Molecular, physiological,
pathological, epidemiology, ecology
• Already has Gene Ontology, NCBI taxonomy, OMIM, ICD-9,SNOMED-CT, MeSH, NCI
• UMLS enables integration– Joining with hospital databases of
patients or samples
Related, possiblysynonymous
MeSH HeadingD017667
SNOMEDT-1A013
CRISP0600-1092
UMLS ConceptC0872094Adipogenesis
Co-occurence
UMLS ConceptC0206131AdipocytesAdipocyteMature fat cellFat Cell
UMLS ConceptC0011849Diabetes Mellitusdm
ICD-9250
Butte AJ, Kohane IS. Nature Biotechnology, 2006, 24:55.
24

~300 Diseases and Conditions
20k+ Genes
Blue: gene goes down in diseaseYellow: gene goes up in disease
Human Disease Gene Expression Collection
Butte AJ, Kohane IS. Nature Biotechnology, 2006, 24:55.Butte AJ, Chen R. Proc AMIA Fall Symposium, 2006.Chen R, Butte AJ. Nature Methods, 2007.Dudley J, Tibshirani R, Deshpande T, Butte AJ. Molecular
Systems Biology, 2009.Shen-Orr S, ... Davis MM, Butte AJ. Nature Methods, 2010.
Joel Dudley• How believable is
the public disease data?
• Which samples look more like each other?• Two diseases in
the same tissue?• Two tissues for
the same disease?
Dudley J, Tibshirani R, Deshpande T, Butte AJ. Molecular
Systems Biology, 2009.
Joel Dudley
• Regardless of whose data, normalizing/analysis methods:• Samples for disease resemble each other, across labs• Two tissues for the same disease are more similar than
two diseases in the same tissueDudley J, Tibshirani R, Deshpande T, Butte AJ. Molecular Systems Biology, 2009.
Joel Dudley
Dudley J, Butte AJ. Pacific Symposium on Biocomputing, 2008, 2009.Suthram S, et al. PLoS Computational Biology, 2010.
Dudley J, Tibshirani R, Deshpande T, Butte AJ. Molecular Systems Biology, 2009.
• The Gene-expression Nosology of Medicine (GNOMED)
Silpa Suthram
Dudley J, Tibshirani R, Butte AJ. Molecular Systems Biology, 2009.Suthram S, et al. PLoS Computational Biology, 2010.
• 68 protein-interaction networks are in common across all diseases
• Common signature of disease
• Drugs that target proteins in this common signature treat significantly more diseases than drugs that target other proteins
Silpa Suthram
Sirota M, Dudley JT, ..., Pasricha J, Butte AJ. Science Translational Medicine, 2011.
Marina SirotaJoel Dudley
25

Candidate anti-seizure drug against inflammatory bowel disease
Marina SirotaJoel Dudley
Sirota M, Dudley JT, ..., Pasricha J, Butte AJ. Science Translational Medicine, 2011.
Anti-seizure drug works against a rat model of inflammatory bowel disease
Dudley JT, Sirota M, ..., Pasricha J, Butte AJ. Science Translational Medicine, 2011.
Marina SirotaJoel Dudley
Mohan M Shenoy Jay Pasricha
Anti-ulcer drug works for lung adenocarcinoma
• Human lung adenocarcinoma cell lines explanted into mouse models
• Followed growth over 11 days• Positive-control dose of
doxorubicin grew to 2x original volume
• Tumors in mice treated with vehicle grew to 3.25x original volume
• Not only did our compound work statistically better than control, it worked in a dose-dependent manner
• Tumors in mice treated with 50 mg/kg/day grew 2.8x
• Those treated with 100 mg/kg/day grew only 2.3x.
Joel DudleyMarina Sirota
Julien SageSirota M, Dudley JT,..., Sage J, Butte AJ. Science Translational Medicine, 2011,
Control With Cimetidine
Rong Chen
Integrating data sets to find a blood protein marker for cross-organ acute rejection
Disease Tissue RNA Predict Serum ProteinDo any of these 10 candidate serum proteins
distinguish acute rejection?
Chen R, ..., Sarwal M, Butte AJ. PLoS Computational Biology, 2010.
Three out of 10 are new serum protein markers for kidney acute rejection
Rong ChenTara SigdelMinnie Sarwal
Chen R, ..., Sarwal M, Butte AJ. PLoS Computational Biology, 2010.
Rong ChenTara SigdelNeeraja KambhamMinnie Sarwal
One of the markers stains
higher in kidney acute
rejection
Chen R, ..., Sarwal M, Butte AJ. PLoS Computational Biology, 2010.
26

One of the markers stains
higher in kidney acute
rejection...and heart
and liver rejection
Rong ChenTara SigdelNeeraja KambhamMinnie Sarwal
Chen R, ..., Sarwal M, Butte AJ. PLoS Computational Biology, 2010.
The same three protein markers work for heart transplant acute rejection
Rong ChenKiran K. KhushHannah
Valantine
Chen R, ..., Sarwal M, Butte AJ. PLoS Computational Biology, 2010.
173
Published online August 10, 2009
Lancet, May 1, 2010.
175
Patient zero40 year old male in
good health presents to his doctor with his whole genome
No symptomsExercises regularlyTakes no medicationsFamily history of
aortic aneurysmFamily history of
sudden death
Presents with 2.8 million SNPs
752 copy number variants
Variants predisposing to cardiac risk
Ashley et al (2010), Lancet375:1525
• Rare variants in 3 genes clinically associated with sudden cardiac death: TMEM43, DSP, and MYBPC3
• Variant in LPA consistent with a family history of coronary artery disease
Euan Ashley and team
27

Variants predisposing to cardiac risk
Ashley et al (2010), Lancet375:1525
• Rare variants in 3 genes clinically associated with sudden cardiac death: TMEM43, DSP, and MYBPC3
• Variant in LPA consistent with a family history of coronary artery disease
Euan Ashley and team Ashley EA*, Butte AJ*, Wheeler MT, Chen R, Klein TE, Dewey FE, Dudley JT, Ormond KE, Pavlovic A, Hudgins L, Gong L, Hodges LM, Berlin DS, Thorn CF, Sangkuhl K, Hebert JM, Woon M, Sagreiya H, Whaley R, Morgan AA, Pushkarev D, Neff
NF, Knowles W, Chou M, Thakuria J, Rosenbaum A, Zaranek AW, Church G, Greely HT*, Quake SR*, Altman RB*. Clinical evaluation incorporating a personal genome. Lancet, 2010.
Russ Altman and team
Pharmacogenomics predictions• Heterozygous null mutation in CYP2C19 clopidogrel resistance?• Variants associated with positive response to lipid-lowering therapy• CYP4F2 and VKORC1 variants low initial warfarin dose
Existing SNP-disease databases are too limited for application to a human genome
Genome-wide association studies• NHGRI GWAS Catalog
– 1032 papers 5050 SNPs for 557 diseases (6280 records), but 26% without OR, 33% without risk/protective alleles
Individual candidate-gene associations• NIH Genetic Association Database
– 56,000 papers, 130,000 records, ~2000 genes, only 4% with dbSNP ids, 1706 with alleles, none with risk/protective
• Online Mendelian Inheritance in Man– no dbSNP ids, monogenic
• Human Genome Mutation Database– 113247 mutations, most Mendelian disease, few SNPs, no
genotypes, or odds ratios
• Study published in 2008 in Inflammatory Bowel Disease
• Crohn’s Disease and Ulcerative Colitis
• Investigated 9 loci in 700 Finnish IBD patients
• We record 100+ items
– GWAS, non‐GWAS papers
– Disease, Phenotype
– Population, Gender
– Alleles and Genotypes
– p‐value (and confidence)
– Odds ratio (and confidence)
– Technology, Study design
– Genetic model
• Mapped to UMLS conceptsRong ChenOptra Systems
• Study published in 2008 in Inflammatory Bowel Disease
• Crohn’s Disease and Ulcerative Colitis
• Investigated 9 loci in 700 Finnish IBD patients
• We record 100+ items
– GWAS, non‐GWAS papers
– Disease, Phenotype
– Population, Gender
– Alleles and Genotypes
– p‐value (and confidence)
– Odds ratio (and confidence)
– Technology, Study design
– Genetic model
• Mapped to UMLS concepts
• Study published in 2009 in Rheumatology
• Ankylosing spondylitis
• Investigated 8 SNPs in IL23R in 2000 UK case‐control patients
• Tables can be rotated• NLP is hard
28

• Study published in 2009 in Rheumatology
• Ankylosing spondylitis
• Investigated 8 SNPs in IL23R in 2000 UK case‐control patients
• Tables can be rotated• NLP is hard
• Study published in 2009 in Rheumatology
• Ankylosing spondylitis
• Investigated 8 SNPs in IL23R in 2000 UK case‐control patients
• Tables can be rotated• NLP is hard
What are the alleles for rs1004819?
Alleles for rs1004819 are C and T
~11% of records reported genotypes in the negative strand
29

Number of papers curated
Distinct SNPs
Diseases and phenotypes
Narrow phenotypes
Records
5,478 67,678 1563 11,911 191,526
Rong ChenOptra Systems
VARIMED: Variants Informing Medicine
Chen R, Davydov EV, Sirota M, Butte AJ. PLoS One. 2010 October: 5(10): e13574.
Moving from OR to LR
Odds ratioRatio of odds of test positivity in cases over
odds of test positivity in non-cases
Likelihood ratio (+)The probability of test positive in cases, over the
probability of test positive in non-cases
Very similar, but different...Morgan A, Chen R, Butte AJ. Genomic Medicine, 2010.
Post-test probability is calculated with likelihood ratio
Pre-test odds x likelihood ratio Post-test odds
Pre-test odds x LR1 x LR2 x LR3 Post-test odds
Morgan A, Chen R, Butte AJ. Genomic Medicine, 2010.
30
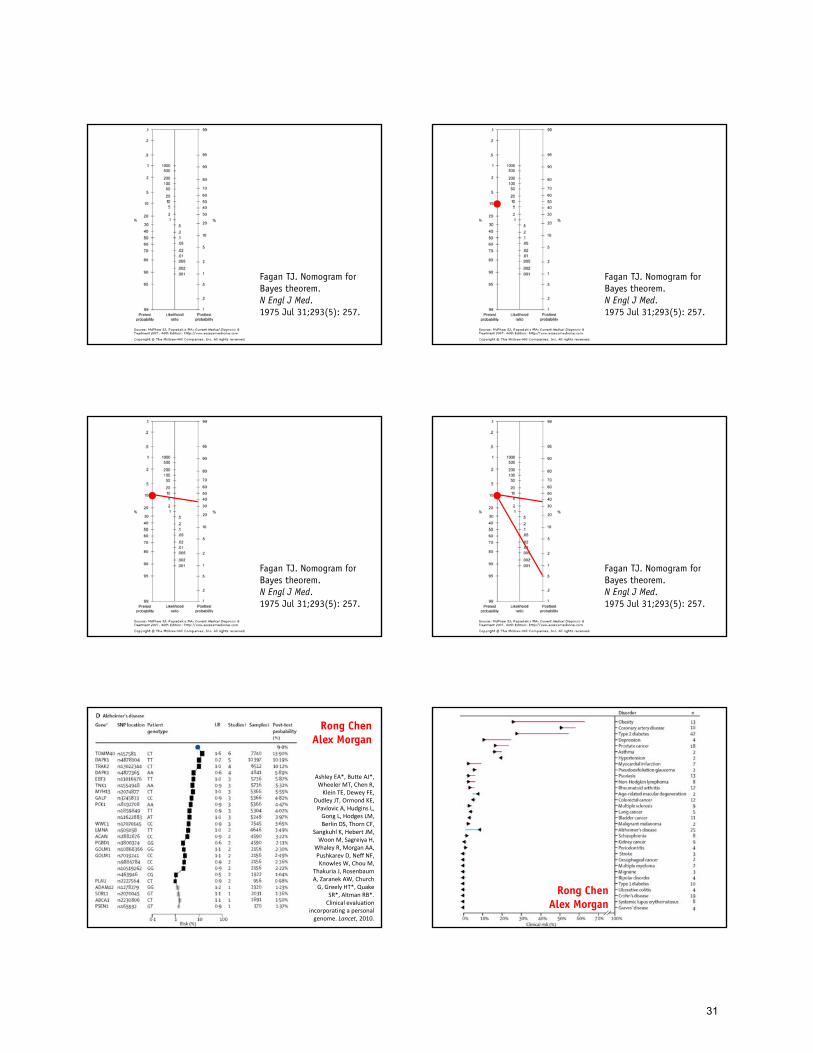
Fagan TJ. Nomogram for Bayes theorem. N Engl J Med. 1975 Jul 31;293(5): 257.
Fagan TJ. Nomogram for Bayes theorem. N Engl J Med. 1975 Jul 31;293(5): 257.
Fagan TJ. Nomogram for Bayes theorem. N Engl J Med. 1975 Jul 31;293(5): 257.
Fagan TJ. Nomogram for Bayes theorem. N Engl J Med. 1975 Jul 31;293(5): 257.
Ashley EA*, Butte AJ*, Wheeler MT, Chen R, Klein TE, Dewey FE,
Dudley JT, Ormond KE, Pavlovic A, Hudgins L, Gong L, Hodges LM, Berlin DS, Thorn CF,
Sangkuhl K, Hebert JM, Woon M, Sagreiya H,
Whaley R, Morgan AA, Pushkarev D, Neff NF, Knowles W, Chou M,
Thakuria J, Rosenbaum A, Zaranek AW, Church G, Greely HT*, Quake
SR*, Altman RB*. Clinical evaluation
incorporating a personal genome. Lancet, 2010.
Rong ChenAlex Morgan
Rong ChenAlex Morgan
31

Alzheimer’s Disease: All family members have relative genetic protective, except the son
Father Mother
Son Daughter
Rong Chen
R Dewey, R Chen, ..., AJ Butte, E Ashley. PLoS Genetics, 2011.
Plotting family disease risk
Rong Chen
FatherMother
SonDaughter
Both the son and daughter are protective from obesity while both parents are at risk.
Both parents are at slight risk on esophagitis, and the daughter is at high risk and the son has protection.
Both parents are protective from Alzheimer’s disease. The son is at high risk and the daughter is protective although less protective than the parents.
Both the son and the daughter have higher risk of Crohn’s disease than the parents, but all family members are at the protective side.
All in the family... Not.Rong Chen
Dewey R, Chen R, ..., AJ Butte, E Ashley. PLoS Genetics, 2011.
Visualizing family psoriasis risk compared to background HapMap individuals
f Father > 72%ilem Mother > 98%ile
s Son > 79%iled Daughter > 79%ile
Rong ChenDewey R, Chen R, ..., AJ Butte, E Ashley. PLoS Genetics, 2011.
Disease risk comparing with HapMap CEU individuals
Population risk was calculated from 174 HapMap individuals on 4.03M SNPs.
Plotting individual or family risk against ethnic-matched background
Rong ChenDewey R, Chen R, ..., AJ Butte, E Ashley. PLoS Genetics, 2011.
32

So what can we do about the risk?
• Diseases with higher post-test probabilities• How to alter the influence of genetics?
• Diseases are caused by genes and environment
• We need a simple “prescription” for environmental change for a genome-enabled patient
• How do we compensate for our genomes?
Rong ChenAlex MorganJoel Dudley
Many physicians do not know how to use the genome Take Home Points
• The patients and samples are there. The data are there. Tools to automate integrative biology are needed, but we cannot ignore the biology.
• Integration of data across modalities may be fruitful. Representing phenotype, environ-mental, and experimental context is possible.
• The bioinformatician can ask and answer important questions because of our ability to implement methods
Microarrays for an Integrative Genomics• One of the first books on microarray analysis and
experimental design• Barnes and Noble, Borders, Amazon: US$32-40
Use and Analysis of Microarray Data
• Butte AJ. Nature Reviews Drug Discovery 2002, 1:651.
AMIA Summit on Translational
Bioinformatics 2012www.amia.org/jointsummits2012
March 2012
Translational Bioinformatics
Biomedical Informatics 217
CS 275
Spring 2012
bmi217.stanford.edu
scpd.stanford.edu
33

Diseases
Vantage
Translational Bioinformatics
Irene Liu: etiological nosology
Annie Chiang: drugs repurposing
David Chen: probing diseases via clinical and genomic data
Alex Morgan: meta-analysis methods
Andy Kogelnik: use clinician order entry to predict harm
Keiichi Kodama: T1DM, integration for T2DM Rong Chen: disease SNPs and
biomarkers Joel Dudley: diseases and
evolution
Marina Sirota: autoimmune informatics
Shiv Venkatasubrahmanyam: stem cell informatics
Shai Shen-Orr: immunology bioinformatics
Sangeeta English: integration for obesity
Staff bioinformatics Post-doc Medical student Ph.D. student Masters student Undergraduate student High school student
Gavi Kohlberg: AML biomarkers
Erik Corona: SNPs and evolution
Adam Grossman: Physiology and disease
Silpa Suthram: protein interactions in disease Purvesh Khatri: pathway
informatics
David Ruau: pain informatics Chirag Patel: environment-wide studies Linda Liu: gender differences Yannick Pouliot: adverse events Rob Tirrell: aging
Sanchita Bhattacharya:human immunity monitoring
Yuri Kim: lung cancer biomarkers
Daniel Li: cancer methylation
Ryo Kita: immunology
Collaborators• Takashi Kadowaki, Momoko Horikoshi, Kazuo Hara, Hiroshi Ohtsu / U Tokyo• Kyoko Toda, Satoru Yamada, Junichiro Irie / Kitasato Univ and Hospital• Shiro Maeda / RIKEN• Alejandro Sweet-Cordero, Julien Sage / Pediatric Oncology• Mark Davis, C. Garrison Fathman / Immunology• Russ Altman, Steve Quake / Bioengineering• Euan Ashley, Joseph Wu, Tom Quertermous / Cardiology• Mike Snyder, Carlos Bustamante, Anne Brunet / Genetics• Jay Pasricha / Gastroenterology• Rob Tibshirani, Brad Efron / Statistics• Hannah Valantine, Kiran Khush/ Cardiology• Ken Weinberg / Pediatric Stem Cell Therapeutics• Mark Musen, Nigam Shah / National Center for Biomedical Ontology• Minnie Sarwal / Nephrology• David Miklos / Oncology
Support• Lucile Packard Foundation for Children's Health• NIH: NLM, NIGMS, NCI, NIAID; NIDDK, NHGRI, NIA, NHLBI, NCRR• March of Dimes• Hewlett Packard• Howard Hughes Medical Institute• California Institute for Regenerative Medicine• PhRMA Foundation• Stanford Cancer Center, Bio-X
• Tarangini Deshpande• Alan Krensky, Harvey Cohen• Hugh O’Brodovich• Isaac Kohane
Admin and Tech Staff• Susan Aptekar• Meelan Phalak• Camilla Morrison• Alex Skrenchuk
34





























