Embed Size (px)
Citation preview
AMBER

AMBER 7
• What is AMBER?– A collective name for a suite of programs
that allow users to carry out molecular dynamic simulations.
– And a set of molecular mechanical force fields for the simulation of biomolecules
– Has a number of useful programs…• Where is it?
– Available on /applic/amber7/
Basic Information Flow In Amber
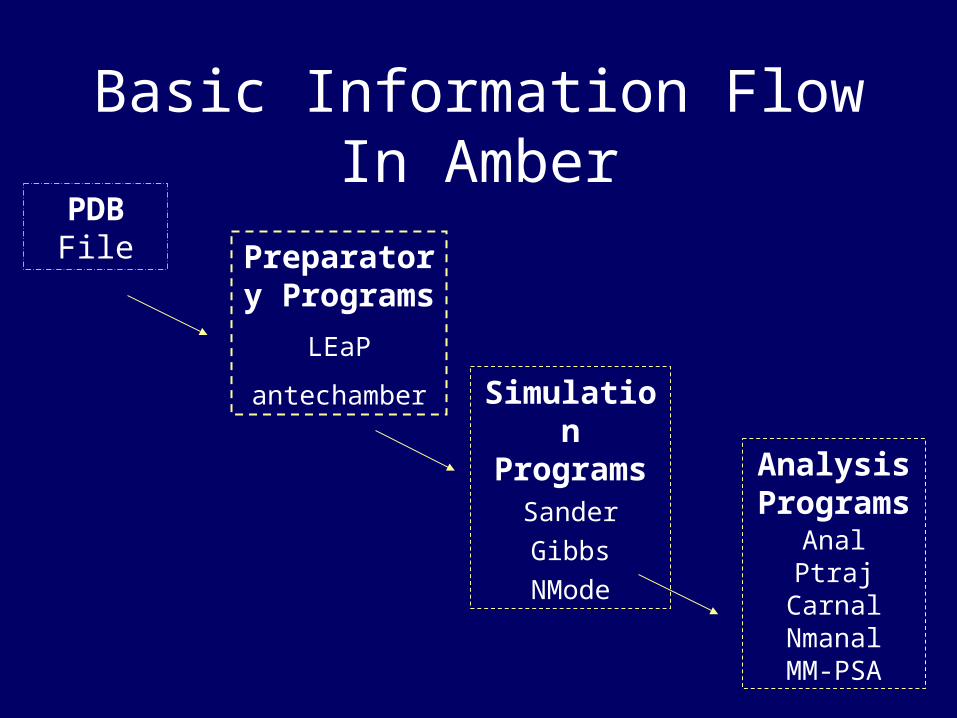
PDB File Preparator
y Programs
LEaP
antechamber
Simulation
ProgramsSander
Gibbs
NMode
Analysis Program
sAnalPtraj
CarnalNmanalMM-PSA

Basic Information Flow In Amber
PDB File Preparator
y Programs
LEaP
antechamber
Simulation
ProgramsSander
Gibbs
NMode
Analysis Program
sAnalPtraj
CarnalNmanalMM-PSA

Preparatory Programs• Leap (used for proteins/nucleic acids)
– A generic name given to xleap and tleap– Xleap has a graphical user interface– Used to prepare the input for the AMBER
molecular mechanics program.
• What does it do?– Used to manipulate objects– Examples of objects; atoms, residues etc.– Can be used to set atom charge, identify the
position of disulphide bridges, delete bonds, addition of atoms, ions etc…
Preparatory Programs - Leap
Basic Information Flow In Amber
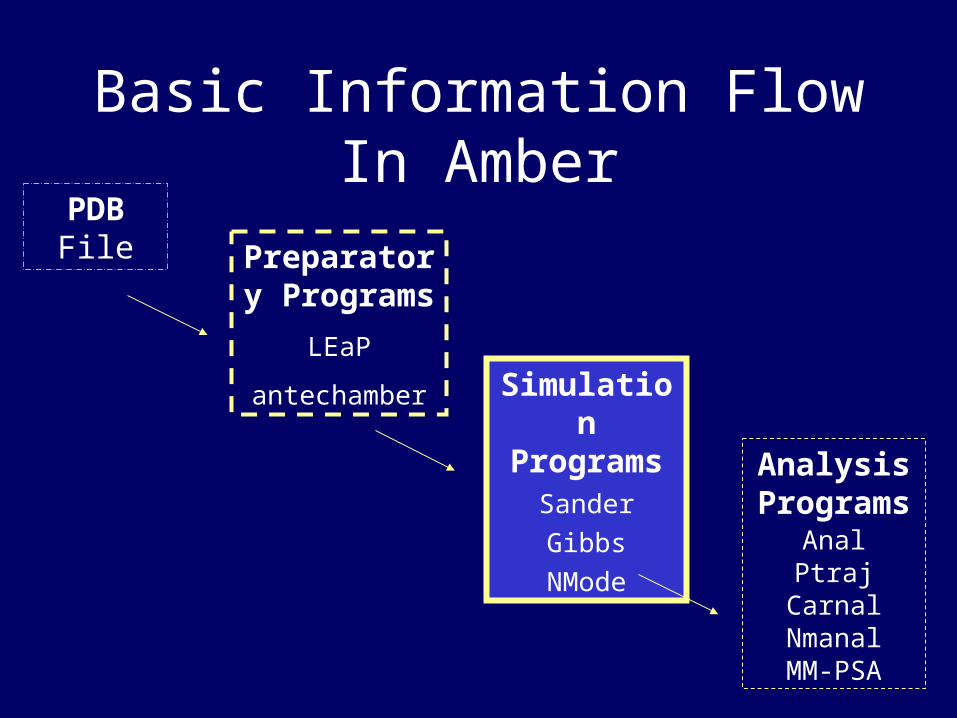
PDB File Preparator
y Programs
LEaP
antechamber
Simulation
ProgramsSander
Gibbs
NMode
Analysis Program
sAnalPtraj
CarnalNmanalMM-PSA

Simulation Programs
• Sander (Simulated Annealing with NMR-Derived Energy Restraints)
– Energy minimization, molecular dynamics and NMR refinements
– Provides direct support for several force fields for proteins and nucleic acids, and for several water models and other organic solvents.

Sander
Minimize Structure
&cntrl
imin=1, maxcyc=1000,
ntb=0,
ntpr=10,
&end
Restrain residues 1-100
1.0
RES 1 100
END
END
Eof

Simulation Programs
• Gibbs– Concerned with free energy
calculations.
– Calculates the free energy difference, G, between two states “0” and “1”

Simulation Programs
• Nmode– Performs molecular mechanics
calculations on proteins and nucleic acids.
– It uses first and second derivative information to find local minima, transition states, and to perform vibrational analysis.

Basic Information Flow In Amber
PDB File Preparator
y Programs
LEaP
antechamber
Simulation
ProgramsSander
Gibbs
NMode
Analysis Program
sAnalPtraj
CarnalNmanalMM-PSA

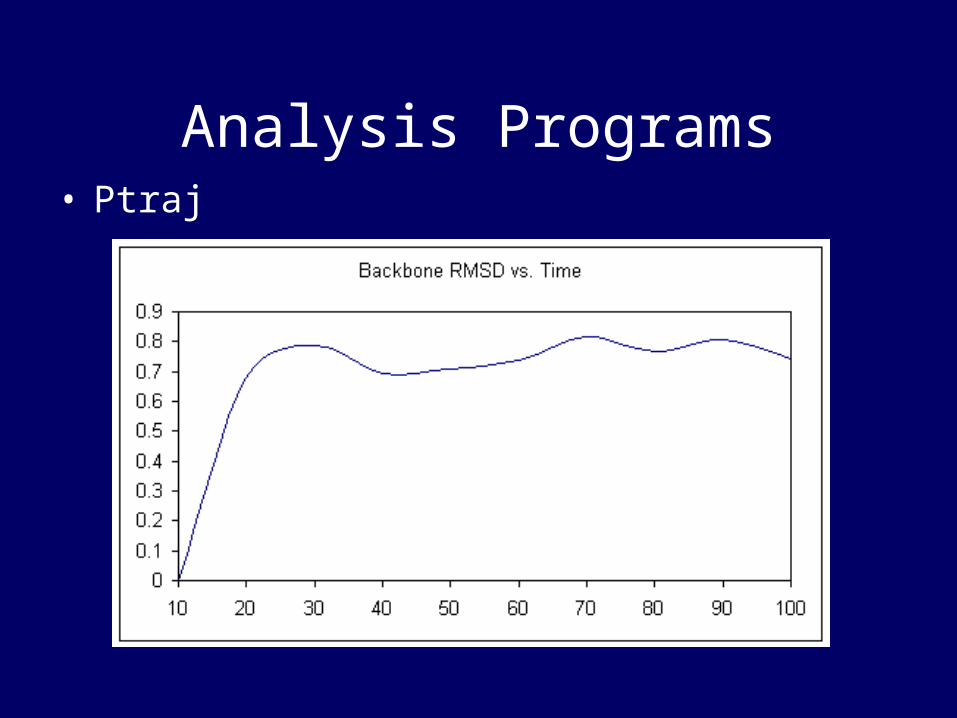
Analysis Programs• Ptraj
• A program to process/analyse 3-D coordinates/trajectories outputted from the AMBER programs.
• Includes•Calculate angles between atoms,•Compute and average structure over
all configurations read in.•Calculate pair distances in selected
atoms•Look at hydrogen bond
donors/acceptors
Analysis Programs• Ptraj

Analysis Programs• Anal
• Energy analysis module of AMBER, its key function is the decomposition of the energy among different groups of atoms in order to find the interaction energies between different parts of the structure
• Carnal• A coordinate analysis program• In addition to conventional trajectory
measurements it allows comparisons between multiple streams of coordinates
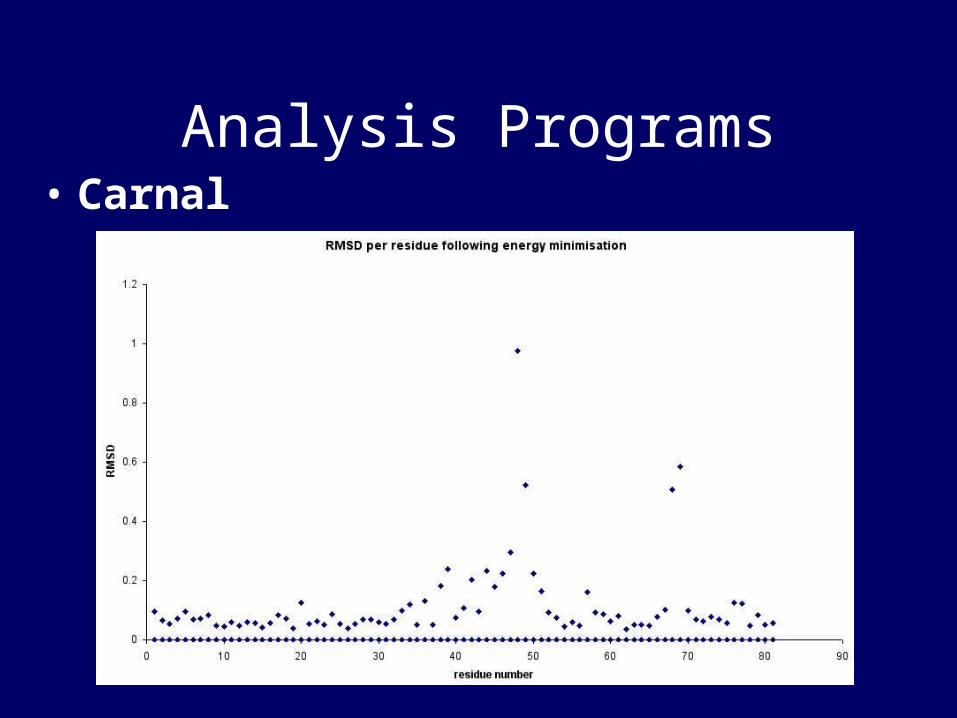
Analysis Programs• Carnal

Analysis Programs• Carnal

Other Programs•Protonate
–Add protons to a heavy-atom protein or DNA PDB file
•Ambpdb
–Produce the PDB file from the amber output.
•Pol_h
–Set positions of polar hydrogens in proteins.
•Gwh
–Set positions of polar hydrogens onto water oxygen positions.

Other Programs •MM-PBSA
•Post-proccessing method to evaluate free energies of binding of molecules in solution.
•Profec (Pictorial Representation Of Free Energy Changes)
•software tools for carrying out and displaying extraploative free energy calculations.
•Resp (Restrained ElectroStatic Potential)
•fits the quantum mechanically calculated electrostatic potential at molecular surfaces using an atom-centered point charge model.

Useful Links
• General– http://amber.scripps.edu/
• Tutorials– http://amber.scripps.edu/tutorial/index.html
• The amber archive– http://structbio.vanderbilt.edu/search.phtml

An Example Loop Dynamics of the HIV-1 Integrase
Core DomainUse AMBER to conduct molecular dynamics
simulations.There is a disordered surface loop near to the
active site and because surface loops are often involved in catalysis, understanding the conformational dynamics of the loop is important in understanding the catalytic mechanism.

An Example
PDB file – 1qs4
PREPARE
tleap add a water box and counter ions to complete the model system ANALYSIS
Ptraj
MOVIEVMD
SIMULATION
Sander
Energy minimisation, molecular dynamics
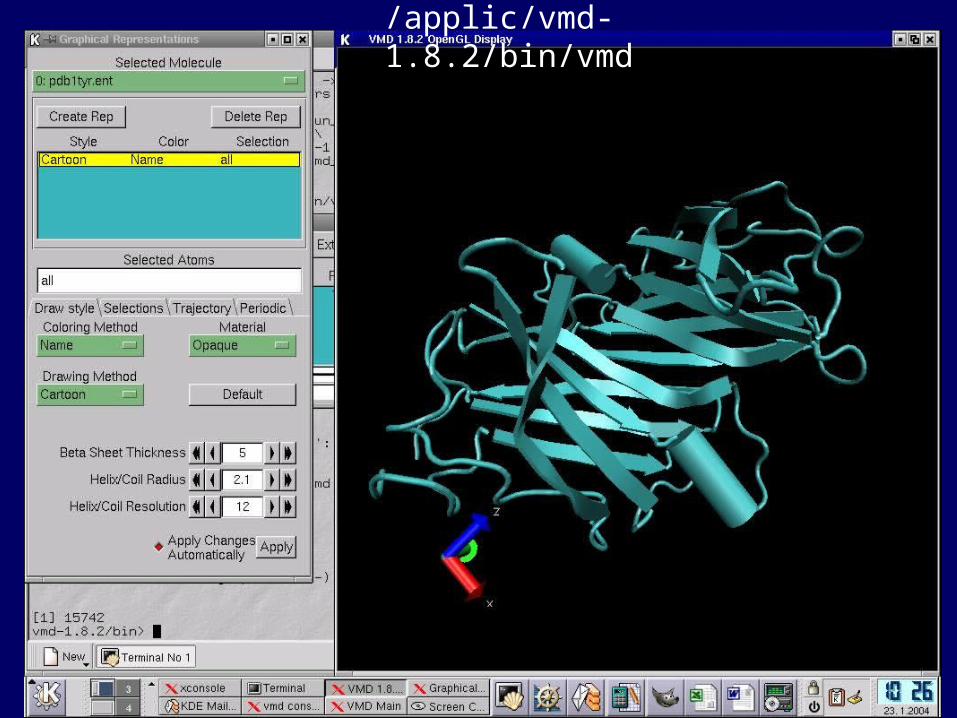
VMD
/applic/vmd-1.8.2/bin/vmd





























