Embed Size (px)
Citation preview

1
Allosteric inhibition of SHP2 Stimulates Anti-Tumor Immunity by
Transforming the Immunosuppressive Environment
Elsa Quintana1, Christopher J. Schulze
1, Darienne R. Myers
1, Tiffany J. Choy
1, Kasia Mordec
1,
David Wildes1, Nataliya Tobvis Shifrin
1, Amira Belwafa
1, Elena S. Koltun
2, Adrian L. Gill
2,
Mallika Singh1, Stephen Kelsey
1,2, Mark A. Goldsmith
1,2, Robert Nichols
1, Jacqueline A. M.
Smith1*
1 Department of Biology, Revolution Medicines, Inc., Redwood City, CA, USA.
2 Department of Chemistry, Revolution Medicines, Inc., Redwood City, CA, USA.
*e-mail: [email protected]
*Address: 700 Saginaw Drive, Redwood City, CA 94063
*Phone: 650-481-6920
Running Title
SHP2 in macrophages and tumor immunosuppression
The authors are full time employees of Revolution Medicines and declare no competing financial
interests.
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

2
Abstract The protein-tyrosine phosphatase SHP2 binds to phosphorylated signaling motifs on regulatory
immunoreceptors including PD-1, but its functional role in tumor immunity is unclear. Using
preclinical models, we show that RMC-4550, an allosteric inhibitor of SHP2, induces anti-tumor
immunity with effects equivalent to or greater than those resulting from checkpoint blockade. In
the tumor microenvironment, inhibition of SHP2 modulated T cell infiltrates similar to
checkpoint blockade. Additionally, RMC-4550 drove direct, selective depletion of pro-
tumorigenic M2 macrophages via attenuation of CSF-1 receptor signaling and increased M1
macrophages via a mechanism independent of CD8+T-cells or IFN-γ. These dramatic shifts in
polarized macrophage populations in favor of anti-tumor immunity were not seen with
checkpoint blockade. Consistent with a pleiotropic mechanism of action, RMC-4550 in
combination with either checkpoint or CSF-1R blockade caused additive anti-tumor activity with
complete tumor regressions in some mice; tumors intrinsically sensitive to SHP2 inhibition or
checkpoint blockade were particularly susceptible. Our preclinical findings demonstrate that
SHP2 thus plays a multifaceted role in inducing immune suppression in the tumor
microenvironment, through both targeted inhibition of RAS pathway-dependent tumor growth
and liberation of anti-tumor immune responses. Furthermore, these data suggest that inhibition
of SHP2 is a promising investigational therapeutic approach.
Significance Inhibition of SHP2 causes direct and selective depletion of pro-tumorigenic M2 macrophages
and promotes anti-tumor immunity, highlighting an investigational therapeutic approach for
some RAS pathway-driven cancers.
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

3
Introduction
Allosteric inhibition of the protein tyrosine phosphatase SHP2 (encoded by PTPN11), an
established signaling node in the RAS-MAPK growth and survival pathway, is a novel,
investigational therapeutic strategy for patients bearing tumors with specific oncogenic mutations
in this pathway (1-4). SHP2 is a positive transducer of receptor tyrosine kinase (RTK) signaling
(see Frankson et al. for a recent (5) review) but the molecular mechanism is still unclear. We and
others have shown that SHP2 acts upstream of RAS and promotes RTK-mediated RAS
nucleotide exchange and activation, likely through a scaffolding interaction with SOS1(1,6,7).
Clinical studies with investigational SHP2 inhibitors are ongoing and preliminary signs of
clinical activity in patients with non-small cell lung cancer (NSCLC) harboring KRAS
mutations, particularly KRASG12C
, have been reported (8). SHP2 is also widely expressed in
hematopoietic cells, including both lymphoid and myeloid cells, and there is emerging evidence
to support a role in anti-tumor immunity. The majority of reported studies have focused on
establishing a role for SHP2 in the regulation of T-cell function (9-11), although recently
myeloid-restricted deletion of SHP2 in mice was shown to suppress melanoma growth (12).
Tumor-associated myeloid cell infiltration is associated with clinical resistance to
immunotherapies (13) and correlates with a negative prognosis for most tumor types (14-22).
Identification of therapeutic strategies that can modulate the recruitment, survival and/or
reprograming of tumor-associated macrophages (TAMs) and improve the clinical response to
currently available immunotherapies is critical (23). Building a comprehensive understanding of
the impact, if any, of allosteric inhibition of SHP2 on innate and adaptive immunity, and how
this can influence the clinical response to checkpoint blockade, is fundamental to realizing the
full potential of this molecular targeted therapeutic strategy.
SHP2 may also be an important signaling node downstream of inhibitory receptors in
immune cells. SHP2 binds to tandem phosphorylated ITIM (immunoreceptor tyrosine-based
inhibition motif) and ITSM (immunoreceptor tyrosine-based switch motif) domains on
regulatory receptors in immune cells, including inhibitory immune receptors like PD-1 and
BTLA (24-26), and multiple reports have demonstrated a SHP2/PD-1 physical interaction in
vitro (25,27-33). Regulation of T-cell receptor signaling in vitro by SHP2 association with
CTLA4 has also been reported (34) although canonical ITIM/ITSM domains are not present in
CTLA4, so the significance of these reported associations is unclear (35). More recently, through
the application of cell-free biochemical experiments, it has been proposed that SHP2 transduces
the PD-1 inhibitory checkpoint signal by direct de-phosphorylation of the co-stimulatory
molecules CD28 and CD226 and, consequently, limits T-cell activation (28,36). Collectively
these studies have pointed to a role for SHP2 in regulation of T-cells. However, using a T-cell-
specific SHP2 deficient mouse model, Rota et al. concluded that SHP2 is dispensable for PD-1
signaling in T-cells in vivo, as well as for the global induction of T-cell exhaustion (11), a
process that PD-1 has been implicated in controlling. Furthermore, the control of immunogenic
tumors was not improved in these T-cell-SHP2-deficient mice, and the response to anti-PD-1
checkpoint blockade therapy was not affected (11). One plausible explanation for the apparent
discrepancy between these observations is that redundant mechanisms, such as the related
tyrosine phosphatase SHP1, can mediate PD-1 inhibitory signaling in the setting of SHP2
deficiency (37). The emergence of these types of compensatory signaling mechanisms highlights
the limitations of using genetically-engineered mouse models to interrogate the in vivo
mechanism(s) of action of SHP2. Moreover, the selective deletion of SHP2 protein from only a
subset of immune cells obscures the clinical implications of the findings thus far, as it does not
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

4
appropriately model the effects of pharmacological inhibition of SHP2 broadly in multiple
immune cell types in addition to tumor cells.
The recent availability of selective, orally-bioavailable small molecule allosteric
inhibitors of SHP2 provides an opportunity to interrogate the immunomodulatory mechanism(s)
of action of SHP2 in vivo using pharmacological tools that circumvent the various limitations
imposed by genetic approaches. Accordingly, Zhao et al. have reported that a selective, but low
potency, small molecule inhibitor of SHP2 decreases tumor burden by augmenting cytotoxic T-
cell-mediated anti-tumor immunity (9). However, in this study no evidence was provided to
substantiate a direct effect of the SHP2 inhibitor on T-cells in vivo, and further the impact of
SHP2 inhibition on the myeloid compartment was not evaluated (9).
In this study, we used the previously described potent and selective allosteric inhibitor of
SHP2, RMC-4550 (1), to generate an in-depth understanding of the integrated effects of SHP2
inhibition in vivo in the tumor microenvironment. Using syngeneic mouse models we reveal an
unanticipated impact of SHP2 on tumor immunity through modulation of both innate and
adaptive immune cells. Similar to immune checkpoint blockade, RMC-4550 caused an increase
in CD8+ T-cell tumor infiltrates. RMC-4550 also produced a direct and selective depletion of
pro-tumorigenic M2 macrophages through attenuation of CSF-1 receptor (CSF-1R) signaling.
The anti-tumor effects of RMC-4550 were additive with either immune checkpoint inhibitors or
an anti-CSF-1R antibody, consistent with a pleiotropic role for SHP2 in the tumor
microenvironment. Tumors that are intrinsically sensitive to SHP2 inhibition and also sensitive
to checkpoint blockade were particularly susceptible to RMC-4550 alone or the combination
treatment.
Collectively these findings highlight that SHP2 inhibition is a promising molecular
therapeutic strategy in cancer with potential dual activity: targeted suppression of tumor-intrinsic
RAS/MAPK dependent growth and promotion of anti-tumor immune responses through
transformation of the suppressive tumor immune microenvironment. Translation of the
preclinical combination advantages of a SHP2 inhibitor and checkpoint blockade into the clinical
setting would be a significant advance for patients bearing oncogenic RAS pathway alterations
and for whom current therapeutic options and benefits are limited.
Materials and Methods
Cell Lines and Reagents. All cell lines were obtained from ATCC except for MC38
(NTCCChina). Cells were grown in RPMI (CT26.WT, A20 and 4T1) or DMEM (MC38, EMT6,
B16-F10) supplemented with 10% heat inactivated fetal bovine serum and 1%
penicillin/streptomycin (Gibco). Cells were maintained at 37 °C in a humidified incubator at 5%
CO2. All cells were mycoplasma free and identity was confirmed by short tandem repeat
profile. Antibodies used for in vivo treatment were from BioXcell: anti-PD-L1 (10F.9G2), rat
IgG2b (LFT-2); anti-PD1 (RMP1-14), rat IgG2a (2A3), anti-CTLA4 (9D9), mIgG2b (MPC11);
anti-CSF-1R (AFS98), rat IgG2a (2A3), anti-CD4 (GK1.5) and anti-CD8 (2.43).
In vivo tumor challenge. All studies were compliant with all relevant ethical regulations
regarding animal research in accordance with approved institutional animal care and use
committee IACUC protocols at MI Bioresearch, Inc. (Ann Arbor, MI), WuXi Apptec. (Suzhou,
China) and HD Biosciences (San Diego, CA). Female (6-8 weeks old) immunocompetent mice
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

5
were implanted subcutaneously with 5E+05 CT26.WT cells, 5E+05 A20 cells or 5E+05 EMT6
cells (BALB/c, Envigo or Balb/c Rag2 ko/ko, Taconic), and 2E+05 MC38 or 5E+04 B16-F10
cells (C57BL/6J, SLAC Laboratory Animal Co., LTD.); or injected in the mammary fat pad with
5E+05 4T1 cells (BALB/c, Shanghai Lingchang Biological Technology Co., LTD.). Once
tumors reached an average size of 48 to 90 mm3, administration of RMC-4550 (30 mg kg−1, by
daily oral administration) or vehicle (2% HPMC in 50mM sodium citrate buffer), anti-PD-1,
anti-PD-L1 or anti-CTLA4 (10 mg kg−1, by intraperitoneal administration every three days), or
anti-CSF-1R (2 mg per mouse on day 1 followed by 0.2 mg per mouse 16 days after cell
implantation, by intraperitoneal administration), was initiated. Experiments with A20 or CT26
where anti-PD-L1 was investigated were not staged and treatment started 3 days after cell
implantation. The selective depletion of tissue macrophages by anti-CSF1R administration was
confirmed in liver by flow cytometry.
In vivo immune cell depletion experiments. Treatment began on day 7 at an overall mean
tumor burden of 72 mm3. Anti-CD4, anti-CD8 or combination were administered
intraperitoneally (0.5 mg per mouse on days 7, 8 and 9 followed by 0.2 mg per mouse on days 13
and 17). RMC-4550 was administered orally (30 mg kg−1 daily during 21 days starting at day
9). Depletion of immune cells in blood was confirmed by flow cytometry.
Immune phenotyping studies in tumors. Treatment with anti-PD-L1 (10 mg kg−1
intraperitoneal on days 3, 6, 10 and 13), RMC-4550 (30 mg kg−1, by oral daily on days 3 to 15)
or combination started on day 3 and tumors were processed for analysis on day 16 after cell
implantation. Treatment with anti-CTLA4 (10 mg kg−1, intraperitoneal on days 7, 10 and 14),
RMC-4550 (30 mg kg−1, daily oral on days 7 to 15) or the combination started on day 7 (79
mm3 tumors) and tumors were processed for analysis on day 16. Tumors were dissociated into
single cell suspension (GentleMACS C tubes and tumor dissociations Kit from Milteny Biotec,).
Antibodies used included CD3 (145-2C11, Biolegend), CD4 (RM4-5, BD Biosciences), CD8a
(53-6.7, BD Biosciences), CD45 (30-F11, Biolegend), CD25 (PC61, Biolegend), PD-1
(29F.1A12, Biolegend), FoxP3 (3G3, ThermoFisher) and MHC Class I (34-1-25, Biolegend),
CD11b (M1/70, BD Biosciences), Ly6C (HK1.4, Biolegend), F4/80 (BM8, Bbiolegend), MHC
Class II (proprietary from MI Bioresearch), CD45 (30-F11, BD Biosciences), CD206 (CO68C2,
Biolegend), CD11c (N418, ThermoFisher), Ly6G (1A8, BD Biosciences), CD19 (1D3, BD
Biosciences) and PD-L1 (B7H1, Biolegend), Ki67 (Biolegend, 16A8), AH1 Dextramer
(Immudex JG3294). ACK Lysing Buffer (Biolegend), Zombie Viability Dye (Biolegend), Fc
blocking agent (anti-CD16/32, Biolegend), FoxP3 Fix/Perm kit (eBiosciences), AbC Total
compensation (ThermoFisher), Cell staining buffer (BD Biosciences) were used. Samples were
run in an Attune NxT flow cytometer.
Immunohistochemistry detection for CD8a, F4/80, in mouse paraffin embedded tumors.
Anti-mouse CD8a (Cell Signaling, 98941, 1.6 µg/ml) or anti-F4/80 (Cell Signaling, 70076, 1.4
µg/ml) rabbit monoclonal antibodies were used with citrate-based pH 6.2 Heat-Induced Epitope
Retrieval. Sections (5 µm) were stained on the Biocare intelliPATH platform using the
manufacturer’s recommended settings. Antibody binding was detected with MACH4 HRP-
polymer Detection System followed by IntelliPATH FLX DAB chromogen and IntelliPATH
Hematoxylin kits. All reagents were from Biocare Medical, Pacheco CA. TissueScope LE whole
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

6
slide scanner (Huron Digital Pathology), Huron Viewer software and HALO® Image Analysis
software from Indica labs were used for analysis.
PD-1 NFAT Luciferase Reporter Assay. Engineered CHO-K1 cells (BPS Bioscience, 60536)
were incubated overnight in RPMI medium supplemented with 10% heat inactivated fetal bovine
serum and 1% penicillin/streptomycin (Gibco). Engineered Jurkat cells (BPS Bioscience, 60535)
were pre-incubated with RMC-4550 or anti-PD-1 for 30 minutes and added to CHO-K1 cells.
After 16 h, One-Step Luciferase Assay system (BPS Bioscience, 60690) was added according to
manufacturer’s instructions, and luminescence was measured on an EnVision Multilabel Plate
Reader (Perkin Elmer).
Staphylococcus aureus Enterotoxin B Superantigen (SEB) T cell stimulation assay. Human
buffy coat was obtained from San Diego Blood Bank. PBMC were isolated using SpMateTM
(Stemcells) and treated with anti-PD-1 (S228P, Invivogen), isotype control (Human IgG4,
Invivogen), RMC-4550 or vehicle (0.2% DMSO) at concentrations indicated. 30 min to 1h after
compound treatment, cells were stimulated with SEB (0.1 g ml−1, Toxin Technologies),
followed by incubation in presence of SEB and compounds at 37°C 5% CO2 incubator for 3
days. IL-2 content was analyzed in supernatant by standard ELISA (Abcam) with Perkin Elmer
Envision Microplate Reader.
Mixed Lymphocyte Reaction (MLR). Monocytes were isolated from fresh PBMC from healthy
donors (EasySep monocyte enrichment kit, Stemcell). Monocytes were differentiated (3 days)
and matured (3 days) into monocyte-derived dendritic cells (Mo-DCs) by using Milteny Biotec
reagents. Cells were immunophenotyped with CD14, CD209 and CD83 and purity was
confirmed to be >90%. Responder CD3+ T cells were prepared from a different donor using a
negative selection kit (Stemcell) to obtain untouched T cells. Cells were co-cultured at a final
ratio of T cells to Mo-DCs of 10:1. Anti-PD-1 (S228P, Invivogen), isotype control (Human
IgG4, Invivogen), RMC-4550 or vehicle were incubated for 5 days and supernatants were
assessed for IFN- by ELISA (Invitrogen).
In vitro studies with murine bone marrow derived macrophages (BMDM). Culture of
BMDMs: Bone marrow (BM) was isolated from the femurs of Balb/C mice. BM cells were
plated in complete Alpha-MEM media (Gibco) containing 10% heat-inactivated FBS (VWR)
and 1% Pen-Strep (Corning) and supplemented with CSF-1 (Peprotech) at 10 ng/mL or GM-CSF
(Peprotech) at 25 ng/mL. Growth inhibition and apoptosis assays: After 7 days of culture, BM
cells were dissociated (Gibco Cell Dissociation Buffer) and plated with media containing CSF-1
at 10 ng/mL or GM-CSF at 25 ng/mL. After 3 hours, cells were treated with either RMC-4550 or
BLZ-945 (Selleckchem). Cell proliferation was measured 72 h after compound treatment using
the CellTiterGlo reagent (Promega). Caspase activity was measured using the caspase 3/7 Glow
kit (Promega) 48 h after compound treatment. Polarization assay: BM cells were isolated as
above and cultured in 10 ng/mL CSF-1 for 6 days. The appropriate cytokine (R&D Systems) was
added (M1: IFN-gamma: 20 ng/mL, LPS: 100 ng/mL M2: IL-4: 20 ng/mL) and cells were
cultured for an additional 24 hours. Polarized BMDMs were treated with compound in the
presence of CSF-1 and the appropriate cytokine and analyzed for cell proliferation and caspase
3/7 activity as described. pERK assay: BM cells were isolated as above and cultured in either
CSF-1 or GM-CSF for 7 days. Growth factor was removed overnight and cells were treated with
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

7
compound for 1 hour. Cells were acutely stimulated with CSF-1 at 100 ng/mL or GM-CSF at 50
ng/mL for 10 minutes. pERK was analyzed using the AlphaLisa SureFire p-ERK1/2
Thr202/Tyr204 kit (PerkinElmer) according to the manufacturer’s instructions. pAkt was
assessed using the pAkt (Ser473) MSD kit (Meso Scale Diagnostics) according to the
manufacturer’s instructions.
Statistics. Quantitative data are presented as the mean ± standard deviation (s.d.) or the standard
error of the mean (s.e.m.), as specified in the figure legends. Statistical tests were performed
using GraphPad Prism 7.0. Two-sided Student’s t-tests were used for comparisons of the means
of data between two groups and one-way ANOVA with post-hoc Tukey’s test was used for
comparisons among multiple independent groups, unless otherwise specified. For animal studies,
animals were randomized before treatments, and all animals treated were included for the
analyses. P Value < 0,05 was considered significant.
Results
SHP2 inhibition induces anti-tumor immunity in vivo in checkpoint blockade-sensitive
tumors
We first examined the anti-tumor efficacy of the SHP2 inhibitor RMC-4550 in three syngeneic
tumor models that are partially sensitive to checkpoint blockade: A20 B-cell lymphoma and both
MC38 and CT26 colon carcinomas. RMC-4550 had a modest effect on growth of A20 cells in
3D in vitro culture but did not reduce the viability of MC38 or CT26 cancer cells at
concentrations achievable in vivo (1) (IC50 = 2 M, >10 M and 10 M respectively, Fig. S1A).
RMC-4550 did inhibit RAS-MAPK signaling, as measured by phosphorylated ERK (pERK)
levels, in A20 and MC38 cells (IC50 of 4 M and 22 nM, respectively, Fig. S1A) but not in CT26
cells (IC50 >10 M, Fig. S1A). Repeated oral daily dosing of RMC-4550 at 30 mg/kg
significantly slowed tumor growth in each of these models (Fig. 1A). No effect of RMC-4550 on
CT26 tumor growth was observed in vivo when tumors were established in RAG-2 deficient
mice, which lack T and B lymphocytes and are thus immunocompromised (Fig. 1B). These data,
together with the lack of in vitro effect on both viability and RAS-MAPK signaling in CT26 cells
(Fig. S1A), provided confidence that the observed efficacy in vivo was a function of SHP2-
mediated effects on immune cells in the tumor microenvironment. To corroborate these findings
we demonstrated that RMC-4550 did not inhibit tumor growth in immunocompetent mice when
both CD4+ and CD8+T-cells had been functionally depleted in vivo with blocking antibodies
(Fig. 1C). CD8+T-cell depletion alone completely abrogated the efficacy of RMC-4550,
indicating that these immune effector cells are essential for SHP2 inhibitor mediated anti-tumor
immunity (Fig. 1C). Depletion of CD4+T-cells inhibited tumor growth in vehicle-treated mice
and no further inhibition of growth was apparent with RMC-4550 (Fig. S1B).
SHP2 inhibition is additive in combination with a checkpoint inhibitor
RMC-4550 induced significant tumor growth inhibition of CT26 tumors that was superior to
anti-PD-L1 (Fig. 1D) and comparable to anti-CTLA4 (Fig. 1E). The combination of RMC-4550
with anti-PD-L1 demonstrated robust anti-tumor benefit as evidenced by a significant increase in
the time to reach endpoint tumor burden and by tumor regressions in 4 of 10 mice (Fig. 1D). In
contrast, RMC-4550 or anti-PD-L1 treatment alone did not result in any tumor-free animals.
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

8
Tumor-free survivors (TFS) remained tumor-free for at least 40 days and, importantly, were
resistant to tumor re-implantation, suggesting long-lasting adaptive immunity (Fig. S1C). All
treatments, including the combination, were well tolerated (Fig. S1D). Similar effects were
observed with anti-CTLA4, although the combination regimen was less well tolerated (Fig. 1E,
Fig. S1E).
SHP2 inhibition does not confer sensitivity to checkpoint blockade in PD-1 refractory
models
We tested the hypothesis that combination treatment with a SHP2 inhibitor could confer
sensitivity to checkpoint blockade using two syngeneic models that are refractory to anti-PD-1
treatment, 4T1 and B16F10. 4T1 breast carcinoma is a RAS/MAPK-dependent syngeneic line
sensitive to both MEK (trametinib, IC50 3D-growth = 5 nM) and SHP2 (RMC-4550, IC50 3D-
growth = 33 nM, Fig S1A) inhibition in vitro, while B16-F10 melanoma cells are insensitive to
SHP2 inhibition in vitro (Fig. S1A). RMC-4550 did not increase sensitivity to anti-PD-1 in vivo
in either of these models, irrespective of whether the cells were sensitive (4T1, Fig. S1F) or
insensitive (B16-F10, Fig. S1G) to the tumor intrinsic effects of SHP2 inhibition on RAS/MAPK
signaling.
SHP2 inhibition stimulates adaptive immunity
Analysis of the immune landscape of CT26 tumors demonstrated that RMC-4550 treatment
increased the percentage of CD3+T-cells by two-fold, from a baseline of 8±3% of CD45+ tumor-
infiltrating leukocytes (TILs) (Fig. S2A and B). CD8+T-cell frequency was increased in tumors
from RMC-4550-treated mice, while there was no change in CD4+T or T-regulatory (Treg) cell
frequency (Fig. 2A and B). Furthermore, the CD8+T-cells expressed less of the inhibitory
molecule PD-1 (Fig. 2B). The increase in CD8+T cell frequency was comparable to that
observed with checkpoint blockade, and the combination of RMC-4550 with either anti-PD-L1
or anti-CTLA4 exhibited additivity (Fig. 2A and B). The combinatorial effect with anti-CTLA4
on CD8+T cells frequency was statistically significant (Fig. 2B). RMC-4550 and anti-CTLA4
treatment-evoked increases in CD8+ T-cells were mostly localized to the border of the tumor and
while an increase relative to vehicle control was apparent for each of the single agent treatment
regimens only in the case of the combination of RMC-4550 with checkpoint blockade did the
increase reach statistical significance (Fig. 2C).
Following RMC-4550 treatment a higher frequency of the CD8+ tumor-infiltrating T
cells were specific for the tumor-associated antigen AH1 (analyzed by dextramer staining) (Fig.
S2C) and exhibited an activated profile, as they were proliferative (Ki67 staining) and expressed
the cytotoxic cytokine IFN (intracellular staining) (Fig. S2C). These changes did not reach
statistical significance but collectively are consistent with functional activation of the CD8+
tumor-infiltrating T cells.
RMC-4550 also increased the expression of class I MHC molecules and PD-L1 in CD45-
negative tumor cells, similarly to anti-CTLA4 (Fig. 2D). These effects were dependent on IFN
and CD8+T cells, as they were abrogated by depletion with the corresponding antibodies in vivo
(Fig. 2D). Consistent with the lack of intrinsic effects of RMC-4550 on proliferation of CT26
cells in vitro, the proliferation of CT26 cells in vivo was not affected, as measured by ki67
staining (Fig S2D). Finally, consistent with previous report (38), SHP2 inhibition did not affect
CT26 tumor vascularization as analyzed by CD31 tissue staining (Fig. S2E).
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

9
SHP2 inhibition does not phenocopy the effect of anti-PD-1 in T-Cells
The direct effects, if any, of SHP2 inhibition on T-cells were explored in vitro and in vivo and
were compared to those of immune checkpoint blockade. Focusing on the proposed role of SHP2
as a downstream transducer of PD-L1/PD-1 signaling (25,28) we obtained robust biochemical
evidence that the tandem phosphorylated ITIM and ITSM in PD-1 can activate the auto-inhibited
form of SHP2. Titration of purified full length SHP2 with a synthetic peptide that mimics the
PD-1 tandem phosphorylated ITIM/ITSM increased enzyme activity by 270-fold (EC50 of 3.2
nM) (Fig. S2F). The PD-1 peptide (10 nM) induced activation of the auto-inhibited form of
SHP2 was blocked by RMC-4550 with an IC50 of 7.1 nM. To monitor PD-L1/PD-1 signal
transduction in a cellular context we used a bioluminescent reporter assay in Jurkat T-cells.
These Jurkat cells were engineered to express human PD-1 and a luciferase reporter driven by an
NFAT response element, and were cocultured with a variant of CHO cells that can serve as
antigen-presenting cells (APCs). These APCs are CHO-K1 cells expressing human PD-L1 and
an engineered cell surface protein designed to activate cognate TCRs in an antigen-independent
manner. RMC-4550 caused a concentration-dependent activation of the NFAT luciferase
reporter with an apparent potency (EC50 = 3.5 nM) consistent with an on-mechanism effect for
the SHP2 inhibitor. However, the maximal signal induction was approximately four-fold lower
than that observed with anti-PD-1 (Fig. 2E). In human PBMC cultures both RMC-4550 and anti-
PD-1 enhanced IL-2 secretion in response to the superantigen SEB (staphylococcal enterotoxin
B), but the response to RMC-4550 was not prominent compared to that of anti-PD-1 (Fig. 2F
top). Furthermore, RMC-4550 (up to a test concentration of 5 M) did not elicit a response in
human T-cells during a mixed lymphocyte reaction, while anti-PD-1 produced a robust increase
in IFN release (Fig. 2F bottom).
Given the equivocal findings in vitro we elected to use an in vivo model to investigate the
role of SHP2 in PD-1 signaling. Checkpoint blockade has been shown to reduce CD8+ T-cell
exhaustion in different systems including the lymphocytic choriomeningitis virus (LCMV)
infection mouse model (39). A role for CD28-costimulation in CD8+T cell rescue in the LCMV
model has been confirmed (40) and we used this model to determine whether SHP2 inhibition
mimics the effects of anti-PD-1 on T-cell exhaustion and viral load reductions in vivo. In our
study, mice were challenged with LCMV clone 13 to establish a chronic infection, followed by
administration of RMC-4550 or anti-PD-L1. RMC-4550 induced a significant increase in the
frequency of CD8+T cells in the spleen, but this was not accompanied by a significant increase
in antigen-specific CD8+T cells. Ultimately, RMC-4550 failed to decrease viral titers in
peripheral organs (Fig. S2G). In contrast, anti-PD-L1 treatment did effectively increase antigenic
CD8+T cells, correlating with higher viral control in various organs (Fig. S2G).
In summary, while we cannot rule out a role for SHP2 downstream of PD-1 signaling we
have demonstrated, using various model systems in vitro and in vivo, that SHP2 inhibition and
PD-1 blockade are not equivalent with respect to direct modulation of T-cell function. Rather, it
seems likely that SHP2 inhibition can restrain PD-1 signaling to some extent, but that the full
downstream effects are blunted, potentially due to the recruitment of redundant signaling effector
molecules.
SHP2 inhibition modulates innate immunity, an effect not seen with checkpoint blockade
To explore additional mechanisms of SHP2 inhibitor action in vivo beyond transduction of
checkpoint signals, we focused on myeloid cells in the tumor microenvironment. CT26 tumors
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

10
are rich in myeloid cells; CD11b+ cells constitute 79±2% of CD45+ TILs and 64±2% of those
are F4/80+ tumor associated macrophages (TAMs). RMC-4550 treatment had a striking impact
on tumor myeloid infiltrates, in particular macrophages, inducing a 3-fold decrease in the
frequency of F4/80+ cells amongst CD45+ TILs (Fig. 3A-C and Fig. S3A). This finding was
confirmed by immunohistochemical staining (Fig. 3D); the decrease in macrophages was most
evident in the core of the tumor (Fig.3D, Fig. S3B).
TAMs are highly plastic and can acquire different phenotypes in the tumor
microenvironment ranging from pro-inflammatory M1 TAMs (MHCIIhigh
and CD206 negative/low
)
to pro-tumorigenic M2 TAMs (MHCIIlow
and CD206high
) (41). RMC-4550 induced a significant
decrease in the frequency of M2, the predominant population in CT26 tumors (>90% of TAMs,
Fig. 3A-C), and an increase in the frequency of M1 amongst CD45+ TILs; by extension the
M2/M1 ratio was dramatically reduced (Fig. 3B-C). Checkpoint blockade elicited only a modest
effect on TAM frequencies (Fig. 3B-C) but the combination of checkpoint blockade and RMC-
4550 drove an even deeper modulation of TAMs (Fig. 3B-C). Checkpoint blockade previously
has been shown to modulate TAM frequencies indirectly, via modulation of CD8+T-cell
frequency and IFNγ secretion in the tumor microenvironment (42,43). In contrast, RMC-4550-
mediated modulation of M2-TAM frequencies was unchanged by depletion of effector cells or
IFNγ cytokine (Fig. 3E). As expected, IFNγ or CD8+T-cell depletion decreased the overall
frequency of M1-TAMs; however a significant, RMC-4550-mediated increase was still apparent
(Fig. 3E). The expression of MHCI in M1 and M2-TAM was significantly increased with RMC-
4550 treatment and this effect was dependent of IFN and CD8+T cells (Fig. 3F). The expression
of PD-L1 in tumor associated macrophages was not changed by RMC-4550 treatment (Fig.
S3E).
Granulocytic myeloid-derived suppressor cells (MDSC) (gMDSC) and monocytic MDSC
(mMDSC) accounted for 7±1% and 14±1% of CD45+ TILs, respectively. Treatment with RMC-
4550 increased the frequency of mMDSCs but had no effect on gMDSCs (Fig. 3G and H). The
expression of MHCI or PD-L1 in MDSC was not changed upon RMC-4550 treatment (Fig.
S3E). To explore potential functional consequences of a SHP2 inhibitor-mediated increase in
mMDSC we used an in vitro suppression assay. Co-culture of human MDSC with T-cells
induced suppression of T-cell proliferation and IFN release (Fig. S3F). RMC-4550 alone had no
effect on T-cell proliferation or cytokine release (Fig, S3F) but was able to block the anti-
proliferative effects of MDSCs on CD8+ T-cells (Fig. S3F). A concomitant concentration-
dependent increase in IFN release was also observed (Fig. S3F). The viability of MDSCs in
vitro was not affected by RMC-4550 (92,5-93.5% viable compared to 93.3% viable in DMSO-
treated MDSCs, determined by Flow cytometry).
The frequency of myeloid cells in spleen or peripheral blood of tumor bearing mice was
unchanged with RMC-4550 treatment, suggesting that myelopoiesis was not affected at this
timepoint (Fig. S3C,D).
In summary, SHP2 inhibition produces a marked shift in polarized macrophage populations in
the tumor microenvironment in favor of antitumor immunity an effect that was not observed
upon checkpoint blockade. This selective effect of RMC-4550 on myeloid cells may underlie the
combination benefit of a SHP2 inhibitor and checkpoint blockade on tumor growth inhibition
(Fig. 1D, E).
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

11
SHP2 inhibition suppresses CSF-1R signaling and selectively affects viability of M2
macrophages
The prominent reduction in macrophage frequency observed in vivo following administration of
RMC-4550, and the apparent lack of dependence on effector lymphocytes or cytokines, is
consistent with a direct effect of SHP2 inhibition on macrophage viability. To evaluate this
possibility, bone marrow (BM) cells from BALB/c mice were differentiated with colony-
stimulating factor 1 (CSF-1) or granulocyte-macrophage colony stimulating factor (GM-CSF) in
vitro. CSF-1 differentiated bone marrow-derived macrophages (BMDMs) represent a population
of F4/80+, MHCIIlow
, CD11c- macrophages, while GM-CSF differentiated BM cells are
MHCIIhigh
, CD11c+, and likely represent a mixture of macrophages and dendritic cells (44).
RMC-4550 potently inhibited the growth of CSF-1-differentiated (IC50 = 13 nM) but not GM-
CSF-differentiated (IC50 > 1 M) BM cells (Fig. 4A). Additionally, SHP2 inhibition selectively
induced caspase3/7 activation, as a marker of apoptosis, in CSF-1 differentiated BMDMs (EC50
= 2.8 nM, Fig. 4B).
The colony stimulating factor 1 receptor (CSF-1R) is a receptor tyrosine kinase (RTK)
that controls the survival and proliferation of macrophages (45) and is the target of several
therapeutic agents in clinical development for cancer (46). The selective CSF-1R kinase inhibitor
BLZ945 (47) also showed selective growth inhibition and induction of apoptosis in CSF-1-
differentiated, but not GM-CSF-differentiated, BMDMs (Fig. S4A,B). The time course of growth
inhibition by BLZ945 or RMC-4550 was similar, and comparable to that caused by CSF-1
deprivation (Fig. S4C). Given these observations, together with the well-established role of
SHP2 as a positive signal transducer downstream of many RTKs, we hypothesized that SHP2
inhibition suppresses CSF-1R signaling. Indeed, we observed strong inhibition of ERK 1/2
phosphorylation by RMC-4550 after acute stimulation of BMDMs with CSF-1 (IC50 = 3 nM,
Fig. 4C). These results were recapitulated using a recombinant cell line that reports on CSF-1R
activation and signaling (Fig. S4D). SHP2 inhibition also decreased GM-CSF induced ERK 1/2
phosphorylation, albeit to a lesser extent (IC50 = 93 nM, Fig. 4C), an effect which was not
observed with BLZ-945 (Fig. S4E) and likely accounts for the moderate growth inhibitory effect
of RMC-4550 in these cells. Importantly, these in vitro results were recapitulated in monocytes
purified from human PBMCs, with SHP2 inhibition resulting in decreased ERK 1/2
phosphorylation and potent inhibition of growth (IC50 = 35 nM) (Fig. 4D and S4F). Moderate
suppression of AKT phosphorylation, another important signaling node for survival downstream
of CSF-1R, was observed with RMC-4550 in human monocytes but not in murine BMDMs (Fig.
S4G-I).
BMDMs were polarized to either an M1 (IFN-, LPS) or M2 (IL-4) phenotype to explore
the contribution of a selective intrinsic effect of SHP2 inhibition on M2 macrophages over M1 in
vitro. The M1 polarized macrophages expressed higher levels of iNOS, whereas M2 polarization
resulted in increased levels of CD206 and arginase (Fig. S4J). M2 macrophage viability was
sensitive to RMC-4550 (IC50 = 19 nM) but M1 polarized macrophages remained almost entirely
refractory to drug treatment (IC50 > 1 M) (Fig. 4E). Similarly, SHP2 inhibition selectively
induced caspase 3/7 activation in M2 but not M1 macrophages (Fig. 4F), which likely accounts
for the dramatic decrease in M2 frequency observed in vivo (Fig. 3B).
We were unable to determine the impact of SHP2 inhibition on macrophage
differentiation per se because RMC-4550 produced a significant decrease in monocyte viability
when present during the differentiation, precluding robust phenotypic characterization of the
differentiated cells.
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

12
Recent data have suggested that increased levels of IFN- and TNF- in the tumor
microenvironment, caused by infiltration of CD8+ T-cells, can trigger an adaptive response of
CSF-1 production by certain cancer cells (48). This in turn can promote recruitment and
proliferation of immunosuppressive TAMs, hampering the anti-tumor immune response to
checkpoint inhibitors. Treatment of CT26 cells in vitro with IFN- and TNF- did increase
production of CSF-1 mRNA (Fig. S5A). However, we propose that the ability of RMC-4550 to
inhibit CSF-1R signaling and decrease immunosuppressive TAM populations, as shown herein,
would negate any inhibitory effects of CSF-1 release by tumor cells.
SHP2 inhibition exhibits greater anti-tumor activity relative to CSF-1R inhibition in vivo
The contribution of SHP2-mediated blockade of the CSF-1R signaling pathway to the anti-tumor
efficacy of RMC-4550 in the CT26 model was examined by comparing the response to that of
CSF-1R blockade. Anti-CSF-1R treatment, in contrast to RMC-4550, did not induce any
significant tumor growth delay (Fig. 4G-H). These findings provide evidence that the in vivo
anti-tumor immunomodulatory effects of a SHP2 inhibitor reflect more than modulation of the
myeloid compartment alone.
The combination of anti-CSF-1R and RMC-4550 showed additive anti-tumor effects in the CT26
model (Fig. 4G-H). While unexpected, this result may reflect the differential mechanisms of
inhibition of CSF-1R signaling by these two agents. Activation of parallel signaling pathways
downstream of CSF-1R (e.g. PI3K/Akt) is insensitive to SHP2 blockade (Fig. S4I), while direct
receptor inhibition likely suppresses additional pro-survival signaling pathways. Given the role
of both SHP2 and CSF-1R as key signaling nodes in multiple cell types and the complexity of
the tumor microenvironment in vivo, further studies are required to elucidate the precise
mechanism(s) underlying this combinatorial effect.
SHP2 inhibition is additive in combination with checkpoint blockade in a SHP2 inhibitor-
sensitive syngeneic model
The combined tumor-intrinsic and immune-mediated anti-tumor effects of SHP2
inhibition have not been reported. EMT6 breast carcinoma is a RAS/MAPK dependent
syngeneic line sensitive to both MEK (trametinib, IC50 3D-growth = 47 nM) and SHP2 (RMC-
4550, IC50 3D-growth = 100 nM, Fig. S1A) inhibition in vitro. RMC-4550 alone significantly
inhibited growth of established EMT6 tumors in immunocompetent mice in vivo, an effect
superior to that of anti-PD-1 (Fig. 5A). The combination of RMC-4550 and anti-PD-1 resulted in
sustained tumor growth inhibition that greatly increased the time to reach endpoint (Fig. 5B).
This treatment also led to tumor regressions in 20% of mice, which were resistant to tumor re-
implantation, suggestive of long-lasting adaptive immunity (Fig. S5B). Treatment of EMT6
tumor bearing mice with RMC-4550 also induced a significant reduction in tumor cell
proliferation, as measured by Ki67 staining, analyzed 9 days after treatment (Fig. S5C). These
data corroborate the findings of cell intrinsic effects of RMC-4550 on proliferation of EMT6
cells in vitro (Fig. S1A).
Based on the collective observations presented here, we propose a model in which the
pleiotropic effects of SHP2 inhibition on both innate and adaptive immunity cooperate to
enhance tumor cell elimination (Fig. 5C). This study reveals a direct role for SHP2 in supporting
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

13
an immunosuppressive tumor microenvironment in addition to an impact on pro-inflammatory
macrophages, although the mechanism underlying the effect on M1 macrophages is unclear. We
have demonstrated that CD8+T cells are obligatory for the anti-tumor activity of SHP2
inhibition; however, the underlying mechanistic driver(s) of the augmented adaptive immune
response remains to be determined.
Discussion
In the present study, we demonstrate that SHP2 inhibition promotes anti-tumor immunity by
modulating both innate and adaptive immune cells. We propose that, although induction of anti-
tumor immunity by SHP2 inhibition is T-cell-dependent, a major driver of the response is
modulation of the macrophage compartment rather than a direct effect on T-cell signaling, thus
differentiating SHP2 inhibition from checkpoint blockade. Our data support a model in which
SHP2 inhibition has a direct impact on the viability of TAMs, thereby promoting a less
immunosuppressive tumor microenvironment. An appreciation of the tumor-extrinsic immune-
modulatory mechanisms of SHP2 should be instructive to the clinical evaluation of SHP2
allosteric inhibitors as a novel molecular therapeutic strategy in cancer patients.
Consistent with the proposed role of SHP2 as a downstream transducer of PD-1
checkpoint signaling in T cells (25,27-33,36,49) we have observed similarities between the in
vivo responses to SHP2 inhibition and immune checkpoint blockade in the tumor immune
microenvironment. We and others (9) have shown that an increase in tumor infiltrating CD8+T
cells is essential for SHP2 inhibitor-mediated control of established tumor growth and that these
T cells express less PD-1, suggesting that they are less exhausted in response to chronic antigen
exposure. However, while we have found a general concordance between the responses to anti-
PD-1 and SHP2 inhibition in various in vitro readouts of T-cell function, we have been unable to
demonstrate that pharmacological inhibition of SHP2 is equivalent to PD-1 blockade. In
particular, the disparate magnitude of the responses suggests that SHP2 is not the sole effector of
inhibitory PD-1 signaling in these model systems, as has been proposed previously (11,37). The
failure of RMC-4550 to phenocopy the effects of anti-PD-1 in the LCMV T-cell exhaustion
model in vivo also points to a greater complexity in PD-1 signaling than has perhaps been
appreciated thus far. In summary, while the present observations are consistent with a role for
SHP2 in PD-1 signal transduction and T-cell biology, the precise role for SHP2 in this pathway
vis a vis other redundant mechanisms, has yet to be elucidated.
More striking is the enhancement of tumor growth inhibition that we and others (9)
observe with the combination of global SHP2 inhibition and checkpoint blockade; this is
indicative of additional functions for SHP2 beyond checkpoint transduction in T-cells.
Significantly, we found that SHP2 inhibition had a profound impact on the survival and function
of suppressive monocytic immune cells such as TAMs and MDSCs. Here we demonstrate using
a pharmacological approach that SHP2 is a positive regulatory of ERK signaling downstream of
CSF-1R in human monocytes and murine BMDMs, which is in agreement with previous studies
using genetic deletion of PTPN11 (50). The inhibition of CSF-1R pro-survival signaling likely
accounts for the selective effects on M2 macrophage populations, as has been observed
previously with CSF-1R inhibitors (51-54), and is supported by the in vitro experiments in the
present study. The selective depletion of M2 macrophages in the tumor microenvironment after
SHP2 inhibition, without major effects on the M1 population, has important translational
implications. We did not observe effects of SHP2 inhibition on GM-CSF-differentiated
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

14
macrophages in vitro, suggesting that SHP2 does not play a role downstream of this receptor.
The GM-CSF receptor transduces pro-survival signals in M1 macrophages, which may be an
explanation of why SHP2 inhibition spares M1 cells and instead has a selective effect on M2s. In
addition to its role as a positive regulator of the RAS pathway, SHP2 has also been proposed to
negatively regulate STAT1 activation downstream of IFN signaling (55-57). As IFN/STAT1
signaling is important in M1 macrophage activation (58), inhibition of SHP2 may be supporting
a feed-forward loop for M1 macrophage polarization and survival, which encompasses not only
macrophage-intrinsic effects on signaling, but is influenced by the infiltration of IFN producing
CD8+ T-cells into the tumor. Consistent with this hypothesis, IFNγ or CD8+T cell depletion
induced an overall decrease in M1 frequency although a SHP2-inhibitor mediated increase was
still apparent. A role for SHP2 downstream of PD-1 in myeloid cells may also be possible. PD-1
signaling in myeloid cells can dampen anti-tumor immunity by regulating lineage fate
commitment and function of myeloid cells (59). Myeloid specific deletion of PD-1 in tumor
bearing mice resulted in a diminished accumulation of immature immunosuppressive cells and
an enhanced output of differentiated, inflammatory effector mMDSCs and phagocytic
macrophages, a phenotype similar to that of SHP2 inhibition.
Adaptive responses to signals in the tumor microenvironment are not restricted to the
immune compartment. There is compelling evidence that the infiltration of CD8+ T-cells can
induce production of CSF-1 by melanoma cells and other cancers by secretion of IFN- and
TNF-α (48,60), an effect we also observed in vitro in the colon CT26 model. Increased levels of
CSF-1 promote an increase in immunosuppressive M2 macrophages, via CSF-1R activation, and
a negative correlation with overall patient survival (48). The opposing effects of CD8+ T-cell
infiltration induced by checkpoint blockade could be counteracted by combination with anti-
CSF-1R therapies in a murine melanoma model (48). Intriguingly, our results suggest that SHP2
inhibition has the potential both to induce CD8+ T-cell infiltration and simultaneously to
counteract its negative consequences by suppressing CSF-1R signaling and therefore contract the
immunosuppressive macrophage population in the tumor microenvironment. This mechanism of
action may contribute to the enhanced anti-tumor activity we observed with RMC-4550 in
combination with checkpoint blockade. Correspondingly, it may account for the superior tumor
growth inhibition observed with the SHP2 inhibitor relative to anti-CSF-1R.
The potential for SHP2 inhibitors to provide therapeutic benefit in solid tumors bearing
SHP2-sensitive oncoproteins, in particular in non-small cell lung cancer (NSCLC), is the focus
of intensive clinical investigation. Multiple, rational combination strategies for a SHP2 inhibitor
with agents that target alternate nodes in the RAS-MAPK pathway (e.g. MEK (6); KRASG12C
(61) or RTK (62) inhibitors) or extra-proliferative functions of RAS (CDK4/6 (63)), have also
been proposed. The present data provide a strong rationale for a clinical combination strategy
with a SHP2 inhibitor and agents that target the immune system directly, such as anti-PD1 and
anti-CSF-1R. Patients bearing tumors that harbor oncogenic driver mutations sensitive to SHP2,
and with established clinical sensitivity to checkpoint inhibitors, for example KRASG12C
mutant-
NSCLC patients, could be particularly susceptible to this combination therapy. On the other hand
the present preclinical findings suggest that SHP2 inhibition seems unlikely to increase
sensitivity to an immune checkpoint inhibitor in checkpoint resistant tumors.
In summary, we have shown using preclinical models that SHP2 plays a central role in
inducing immune suppression in the tumor microenvironment both by inhibiting T-cells and
supporting the viability of pro-tumorigenic macrophages. SHP2 inhibition is an attractive
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

15
investigational therapeutic strategy with potential dual activity: targeted inhibition of RAS-
MAPK dependent tumor growth and liberation of anti-tumor immune responses by
transformation of the tumor microenvironment.
Acknowledgments. We would like to thank Dylan Daniel, Art Weiss and Cliff Lowell for
providing expert advice during the course of this work. We would also like to thank the
respective research teams at the following contract research organizations for the conduct of in
vitro and in vivo studies: MI Bioresearch (Ann Arbor, Michigan), HDB (San Diego, California),
WuXI Apptec (Shanghai, China), Ensigna (Brisbane, California), HistoTox Labs (Boulder,
Colorado) and PAIRimmune Inc (Québec, Canada). This work was supported in part by Sanofi,
Paris, France. The authors are full time employees of Revolution Medicines and declare no
competing financial interests.
References
1. Nichols RJ, Haderk F, Stahlhut C, Schulze CJ, Hemmati G, Wildes D, et al. RAS nucleotide cycling
underlies the SHP2 phosphatase dependence of mutant BRAF-, NF1- and RAS-driven cancers. Nat Cell
Biol 2018;20:1064-73
2. Ruess DA, Heynen GJ, Ciecielski KJ, Ai J, Berninger A, Kabacaoglu D, et al. Mutant KRAS-driven
cancers depend on PTPN11/SHP2 phosphatase. Nat Med 2018;24:954-60
3. Mainardi S, Mulero-Sanchez A, Prahallad A, Germano G, Bosma A, Krimpenfort P, et al. SHP2 is required
for growth of KRAS-mutant non-small-cell lung cancer in vivo. Nat Med 2018;24:961-7
4. Chen YN, LaMarche MJ, Chan HM, Fekkes P, Garcia-Fortanet J, Acker MG, et al. Allosteric inhibition of
SHP2 phosphatase inhibits cancers driven by receptor tyrosine kinases. Nature 2016;535:148-52
5. Frankson R, Yu ZH, Bai Y, Li Q, Zhang RY, Zhang ZY. Therapeutic Targeting of Oncogenic Tyrosine
Phosphatases. Cancer research 2017;77:5701-5
6. Fedele C, Ran H, Diskin B, Wei W, Jen J, Geer MJ, et al. SHP2 Inhibition Prevents Adaptive Resistance to
MEK Inhibitors in Multiple Cancer Models. Cancer Discov 2018;8:1237-49
7. Ahmed TA, Adamopoulos C, Karoulia Z, Wu X, Sachidanandam R, Aaronson SA, et al. SHP2 Drives
Adaptive Resistance to ERK Signaling Inhibition in Molecularly Defined Subsets of ERK-Dependent
Tumors. Cell Rep 2019;26:65-78.e5
8. Ou S-HI. The SHP2 inhibitor RMC-4630 in patients with KRAS-mutant non-small cell lung cancer:
Preliminary evaluation of a first-in-man phase 1 clinical trial. Sixth AACR-IASLC International Joint
Conference: Lung Cancer Translational Science from the Bench to the Clinic 2019:A12
9. Zhao M, Guo W, Wu Y, Yang C, Zhong L, Deng G, et al. SHP2 inhibition triggers anti-tumor immunity
and synergizes with PD-1 blockade. Acta PharmaceuticaSinicaB 2019;9:304-15
10. Liu W, Guo W, Shen L, Chen Z, Luo Q, Luo X, et al. T lymphocyte SHP2-deficiency triggers anti-tumor
immunity to inhibit colitis-associated cancer in mice. Oncotarget 2017;8:7586-97
11. Rota G, Niogret C, Dang AT, Barros CR, Fonta NP, Alfei F, et al. Shp-2 Is Dispensable for Establishing T
Cell Exhaustion and for PD-1 Signaling In Vivo. Cell Rep 2018;23:39-49
12. Xiao P, Guo Y, Zhang H, Zhang X, Cheng H, Cao Q, et al. Myeloid-restricted ablation of Shp2 restrains
melanoma growth by amplifying the reciprocal promotion of CXCL9 and IFN-gamma production in tumor
microenvironment. Oncogene 2018;37:5088-100
13. Pathria P, Louis TL, Varner JA. Targeting Tumor-Associated Macrophages in Cancer. Trends in
immunology 2019;40:310-27
14. Pedersen MB, Danielsen AV, Hamilton-Dutoit SJ, Bendix K, Norgaard P, Moller MB, et al. High
intratumoral macrophage content is an adverse prognostic feature in anaplastic large cell lymphoma.
Histopathology 2014;65:490-500
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

16
15. Jackute J, Zemaitis M, Pranys D, Sitkauskiene B, Miliauskas S, Vaitkiene S, et al. Distribution of M1 and
M2 macrophages in tumor islets and stroma in relation to prognosis of non-small cell lung cancer. BMC
immunology 2018;19:3
16. Shigeoka M, Urakawa N, Nakamura T, Nishio M, Watajima T, Kuroda D, et al. Tumor associated
macrophage expressing CD204 is associated with tumor aggressiveness of esophageal squamous cell
carcinoma. Cancer science 2013;104:1112-9
17. Kim KJ, Wen XY, Yang HK, Kim WH, Kang GH. Prognostic Implication of M2 Macrophages Are
Determined by the Proportional Balance of Tumor Associated Macrophages and Tumor Infiltrating
Lymphocytes in Microsatellite-Unstable Gastric Carcinoma. PLoS One 2015;10:e0144192
18. Bostrom MM, Irjala H, Mirtti T, Taimen P, Kauko T, Algars A, et al. Tumor-Associated Macrophages
Provide Significant Prognostic Information in Urothelial Bladder Cancer. PLoS One 2015;10:e0133552
19. Guo B, Cen H, Tan X, Ke Q. Meta-analysis of the prognostic and clinical value of tumor-associated
macrophages in adult classical Hodgkin lymphoma. BMC medicine 2016;14:159
20. Yin S, Huang J, Li Z, Zhang J, Luo J, Lu C, et al. The Prognostic and Clinicopathological Significance of
Tumor-Associated Macrophages in Patients with Gastric Cancer: A Meta-Analysis. PLoS One
2017;12:e0170042
21. Morita Y, Zhang R, Leslie M, Adhikari S, Hasan N, Chervoneva I, et al. Pathologic evaluation of tumor-
associated macrophage density and vessel inflammation in invasive breast carcinomas. Oncology letters
2017;14:2111-8
22. Zhang QW, Liu L, Gong CY, Shi HS, Zeng YH, Wang XZ, et al. Prognostic significance of tumor-
associated macrophages in solid tumor: a meta-analysis of the literature. PLoS One 2012;7:e50946
23. Cassetta L, Kitamura T. Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the
Response to Immune Checkpoint Inhibitors. Front Cell Dev Biol 2018;6:38
24. Gavrieli M, Watanabe N, Loftin SK, Murphy TL, Murphy KM. Characterization of phosphotyrosine
binding motifs in the cytoplasmic domain of B and T lymphocyte attenuator required for association with
protein tyrosine phosphatases SHP-1 and SHP-2. Biochemical and biophysical research communications
2003;312:1236-43
25. Sheppard KA, Fitz LJ, Lee JM, Benander C, George JA, Wooters J, et al. PD-1 inhibits T-cell receptor
induced phosphorylation of the ZAP70/CD3zeta signalosome and downstream signaling to PKCtheta.
FEBS Lett 2004;574:37-41
26. Marasco M, Berteotti A, Weyershaeuser J, Thorausch N, Sikorska J, Krausze J, et al. Molecular
mechanism of SHP2 activation by PD-1 stimulation. Science advances 2020;6:eaay4458
27. Chemnitz JM, Parry RV, Nichols KE, June CH, Riley JL. SHP-1 and SHP-2 Associate with
Immunoreceptor Tyrosine-Based Switch Motif of Programmed Death 1 upon Primary Human T Cell
Stimulation, but Only Receptor Ligation Prevents T Cell Activation. The Journal of Immunology
2004;173:945-54
28. Hui E, Cheung J, Zhu J, Su X, Taylor MJ, Wallweber HA, et al. T cell costimulatory receptor CD28 is a
primary target for PD-1-mediated inhibition. Science (New York, NY) 2017;355:1428-33
29. Latchman Y, Wood CR, Chernova T, Chaudhary D, Borde M, Chernova I, et al. PD-L2 is a second ligand
for PD-1 and inhibits T cell activation. Nat Immunol 2001;2:261-8
30. Peled M, Tocheva AS, Sandigursky S, Nayak S, Philips EA, Nichols KE, et al. Affinity purification mass
spectrometry analysis of PD-1 uncovers SAP as a new checkpoint inhibitor. Proc Natl Acad Sci U S A
2018;115:E468-e77
31. Yamamoto R, Nishikori M, Kitawaki T, Sakai T, Hishizawa M, Tashima M, et al. PD-1-PD-1 ligand
interaction contributes to immunosuppressive microenvironment of Hodgkin lymphoma. Blood
2008;111:3220-4
32. Okazaki T, Maeda A, Nishimura H, Kurosaki T, Honjo T. PD-1 immunoreceptor inhibits B cell receptor-
mediated signaling by recruiting src homology 2-domain-containing tyrosine phosphatase 2 to
phosphotyrosine. Proc Natl Acad Sci U S A 2001;98:13866-71
33. Yokosuka T, Takamatsu M, Kobayashi-Imanishi W, Hashimoto-Tane A, Azuma M, Saito T. Programmed
cell death 1 forms negative costimulatory microclusters that directly inhibit T cell receptor signaling by
recruiting phosphatase SHP2. J Exp Med 2012;209:1201-17
34. Marengere LE, Waterhouse P, Duncan GS, Mittrucker HW, Feng GS, Mak TW. Regulation of T cell
receptor signaling by tyrosine phosphatase SYP association with CTLA-4. Science (New York, NY)
1996;272:1170-3
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

17
35. Teft WA, Kirchhof MG, Madrenas J. A molecular perspective of CTLA-4 function. Annual review of
immunology 2006;24:65-97
36. Wang B, Zhang W, Jankovic V, Golubov J, Poon P, Oswald EM, et al. Combination cancer
immunotherapy targeting PD-1 and GITR can rescue CD8(+) T cell dysfunction and maintain memory
phenotype. Science immunology 2018;3
37. Celis-Gutierrez J, Blattmann P, Zhai Y, Jarmuzynski N, Ruminski K, Gregoire C, et al. Quantitative
Interactomics in Primary T Cells Provides a Rationale for Concomitant PD-1 and BTLA Coinhibitor
Blockade in Cancer Immunotherapy. Cell Rep 2019;27:3315-30.e7
38. Hao HX, Wang H, Liu C, Kovats S, Velazquez R, Lu H, et al. Tumor Intrinsic Efficacy by SHP2 and RTK
Inhibitors in KRAS-Mutant Cancers. Molecular cancer therapeutics 2019;18:2368-80
39. Barber DL, Wherry EJ, Masopust D, Zhu B, Allison JP, Sharpe AH, et al. Restoring function in exhausted
CD8 T cells during chronic viral infection. Nature 2006;439:682-7
40. Kamphorst AO, Wieland A, Nasti T, Yang S, Zhang R, Barber DL, et al. Rescue of exhausted CD8 T cells
by PD-1-targeted therapies is CD28-dependent. Science (New York, NY) 2017;355:1423-7
41. DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev
Immunol 2019
42. Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-Dimensional Analysis
Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint
Cancer Therapy. Cell 2018;175:1014-30.e19
43. Xiong H, Mittman S, Rodriguez R, Moskalenko M, Pacheco-Sanchez P, Yang Y, et al. Anti-PD-L1
Treatment Results in Functional Remodeling of the Macrophage Compartment. Cancer research
2019;79:1493-506
44. Na YR, Jung D, Gu GJ, Seok SH. GM-CSF Grown Bone Marrow Derived Cells Are Composed of
Phenotypically Different Dendritic Cells and Macrophages. Molecules and cells 2016;39:734-41
45. Pixley FJ, Stanley ER. CSF-1 regulation of the wandering macrophage: complexity in action. Trends in cell
biology 2004;14:628-38
46. Cannarile MA, Weisser M, Jacob W, Jegg AM, Ries CH, Ruttinger D. Colony-stimulating factor 1 receptor
(CSF1R) inhibitors in cancer therapy. J Immunother Cancer 2017;5:53
47. Pyonteck SM, Akkari L, Schuhmacher AJ, Bowman RL, Sevenich L, Quail DF, et al. CSF-1R inhibition
alters macrophage polarization and blocks glioma progression. Nat Med 2013;19:1264-72
48. Neubert NJ, Schmittnaegel M, Bordry N, Nassiri S, Wald N, Martignier C, et al. T cell-induced CSF1
promotes melanoma resistance to PD1 blockade. Sci Transl Med 2018;10
49. Lee KM, Chuang E, Griffin M, Khattri R, Hong DK, Zhang W, et al. Molecular basis of T cell inactivation
by CTLA-4. Science (New York, NY) 1998;282:2263-6
50. Wang L, Iorio C, Yan K, Yang H, Takeshita S, Kang S, et al. A ERK/RSK-mediated negative feedback
loop regulates M-CSF-evoked PI3K/AKT activation in macrophages. FASEB journal : official publication
of the Federation of American Societies for Experimental Biology 2018;32:875-87
51. Zhu Y, Knolhoff BL, Meyer MA, Nywening TM, West BL, Luo J, et al. CSF1/CSF1R blockade
reprograms tumor-infiltrating macrophages and improves response to T-cell checkpoint immunotherapy in
pancreatic cancer models. Cancer research 2014;74:5057-69
52. Ries CH, Cannarile MA, Hoves S, Benz J, Wartha K, Runza V, et al. Targeting tumor-associated
macrophages with anti-CSF-1R antibody reveals a strategy for cancer therapy. Cancer Cell 2014;25:846-59
53. Van Overmeire E, Stijlemans B, Heymann F, Keirsse J, Morias Y, Elkrim Y, et al. M-CSF and GM-CSF
Receptor Signaling Differentially Regulate Monocyte Maturation and Macrophage Polarization in the
Tumor Microenvironment. Cancer research 2016;76:35-42
54. Ao JY, Zhu XD, Chai ZT, Cai H, Zhang YY, Zhang KZ, et al. Colony-Stimulating Factor 1 Receptor
Blockade Inhibits Tumor Growth by Altering the Polarization of Tumor-Associated Macrophages in
Hepatocellular Carcinoma. Molecular cancer therapeutics 2017;16:1544-54
55. Tsai CC, Kai JI, Huang WC, Wang CY, Wang Y, Chen CL, et al. Glycogen synthase kinase-3beta
facilitates IFN-gamma-induced STAT1 activation by regulating Src homology-2 domain-containing
phosphatase 2. J Immunol 2009;183:856-64
56. Baron M, Davignon JL. Inhibition of IFN-gamma-induced STAT1 tyrosine phosphorylation by human
CMV is mediated by SHP2. J Immunol 2008;181:5530-6
57. Wu TR, Hong YK, Wang XD, Ling MY, Dragoi AM, Chung AS, et al. SHP-2 is a dual-specificity
phosphatase involved in Stat1 dephosphorylation at both tyrosine and serine residues in nuclei. The Journal
of biological chemistry 2002;277:47572-80
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

18
58. Hu X, Ivashkiv LB. Cross-regulation of signaling pathways by interferon-gamma: implications for immune
responses and autoimmune diseases. Immunity 2009;31:539-50
59. Strauss L, Mahmoud MAA, Weaver JD, Tijaro-Ovalle NM, Christofides A, Wang Q, et al. Targeted
deletion of PD-1 in myeloid cells induces antitumor immunity. Science immunology 2020;5:eaay1863
60. Cassetta L, Fragkogianni S, Sims AH, Swierczak A, Forrester LM, Zhang H, et al. Human Tumor-
Associated Macrophage and Monocyte Transcriptional Landscapes Reveal Cancer-Specific
Reprogramming, Biomarkers, and Therapeutic Targets. Cancer Cell 2019;35:588-602.e10
61. Christensen JG, Fell JB, Marx MA, Fischer J, Hallin J, Calinisan BB, et al. Insight towards therapeutic
susceptibility of KRAS mutant cancers from MRTX1257, a novel KRAS G12C mutant selective small
molecule inhibitor. Proceedings of the 110th Annual Meeting of the American Association for Cancer
Research; 2019 March 29-Apr 3; Atlanta GA 2019:Abstract LB-271
62. Dardaei L, Wang HQ, Singh M, Fordjour P, Shaw KX, Yoda S, et al. SHP2 inhibition restores sensitivity
in ALK-rearranged non-small-cell lung cancer resistant to ALK inhibitors. Nat Med 2018;24:512-7
63. Lee GL, Stahlhut C, Evans J, Reyes DF, Lorenzana EG, L. S, et al. Maximizing the therapeutic potential of
SHP2 inhibition with rational combination strategies in tumors driven by aberrant RAS-MAPK signaling.
Proceedings of the 110th Annual Meeting of the American Association for Cancer Research; 2019 March
29-Apr 3; Atlanta GA 2019:Abstract 1322
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

19
Figure Legends
Fig.1. SHP2 inhibition induces anti-tumor immunity in checkpoint blockade-sensitive
tumors and is additive in combination with a checkpoint inhibitor. (A-B) Oral daily
administration of 30 mg/kg RMC-4550 was used in all experiments. (A), Activity of RMC-4550
in A20, MC38 or CT26 tumor bearing immunocompetent mice. Kaplan-Meier plot showing
percentage of animals with tumor burden below 2000 mm3 for the duration of this study. (B),
Lack of activity of RMC-4550 in CT26 tumor bearing RAG-2 deficient immunocompromised
mice. (C), Activity of RMC-4550 in CT26 tumor bearing immunocompetent mice depleted of
CD4+ and CD8+T cells or CD8+T cells alone. Left two panels show tumor growth of individual
mice for each experimental group described, right panel shows Kaplan-Meier plot displaying
percentage of animals with tumor burden below 2000 mm3. (D), Activity of RMC-4550, anti-
PD-L1 (10 mg/kg, IP on days 3, 6, 10 and 13 post-implantation) or combination of both in CT26
tumor bearing immunocompetent mice. Dashed arrow indicates last day of treatment of RMC-
4550. Tumor growth of individual mice for each experimental group described and Kaplan–
Meier curves (right) are shown. (E) Same as D but using anti-CTLA4 (10 mg/kg, IP on days 7,
10, 14 and 17 post-implantation) as checkpoint inhibitor. Kaplan–Meier curves were compared
using the Mantel-Cox Log-Rank test (A-E, except for A20, used Logrank test for trend); n=10
animals per group (*P < 0.05; ***P < 0.001; ****P < 0.0001). TFS = Tumor-free survivors.
Fig. 2. SHP2 inhibition stimulates adaptive immunity in similar manner to checkpoint
inhibition. CT26 tumors derived from similar experiments as shown in Fig.1D or 1E, were taken
at day 16, after 13 or 9 days of treatment with anti-PD-L1 (A) or anti-CTLA4 (B, C, D)
respectively, and tumor immune cell infiltrates were analyzed by flow cytometry or
immunohistochemistry. (A, B), Quantification by flow cytometry of CD8+T or CD4+T cells in
CD45+TILs, ratio of CD8+T/Treg and PD-1 mean fluorescence intensity (MFI) of CD8+T cells.
(C), Quantification of CD8+T positive cells in CT26 tumors, as percentage of total number of
cells in each tumor section (graph in center) or in edge of tumor (right), determined by
immunohistochemistry. (D), Quantification by flow cytometry of MHC class I or PD-L1 MFI of
CD45 negative tumor cells and after in vivo depletion of IFN or CD8+T cells. (E), NFAT-
luciferase reporter gene PD-1/PD-L1 bioassay shows NFAT activation in response to increasing
concentrations of anti-PD-1 or RMC-4550. (F) SEB (top) or MLR (bottom) assay with human
cells from healthy donors shows secretion of IL-2 or IFN in response of increasing
concentrations of anti-PD-1 (1 ng/ml to 10 g/ml) or RMC-4550 (0.1 nM to 5 M). (A-C) Data
represent analysis of 5 mice per group, mean ± s.e.m. One-Way ANOVA followed by Holm-
Sidak test (A-B) or Tukey (C-D) (*P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001).
Fig. 3. SHP2 inhibition modulates innate immunity, an effect not seen with checkpoint
blockade. CT26 tumors derived from similar experiments as shown in Fig.1D-E, were taken at
day 16, after 13 or 9 days of treatment with anti-PD-L1 (B, G) or anti-CTLA4 (A, C, D, H)
respectively, and tumor myeloid cell infiltrates were analyzed by flow cytometry or
immunohistochemistry. (A), Representative flow cytometric analysis of MHC class II and
CD206 expression of tumor associated macrophages (TAMs) defined as CD45+/CD3-/CD19-
/CD11b+/Ly6G-/Ly6C low/F4/80+. Gates indicate strategy to define M1 and M2 TAMs. (B-C)
Quantification by flow cytometry of CD11b+, F4/80+, M1 or M2 of CD45+ TILs in each
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

20
experimental group as indicated. (D), Immunohistochemistry analysis for F4/80+ cells in
consecutive sections of same tumors analyzed in Figure 2C. Quantification of F4/80+ area, as
percentage of total area in each tumor section (core and border), is shown on right. (E) Analysis
by flow cytometry of the frequency of M1+ (top) or M2+ (bottom) tumor associated
macrophages upon in vivo depletion of IFN or CD8+T cells in CT26 tumors derived from the
same experiment as in Figure 2D. (F) MHC class I (MHCI) mean of fluorescence intensity
(MFI) analyzed by flow cytometry on M1 and M2 cells gated as in (E). (G, H), Quantification by
flow cytometry of mMDSC (CD45+/CD3-/CD19-/CD11b+/Ly-6C High/Ly-6G-) or gMDSC
(CD45+/CD3-/CD19-/CD11b+/Ly-6G+). (B-H) Data represent analysis of 5 mice per group,
mean ± s.e.m. One-Way ANOVA followed by Tukey (*P < 0.05; **P < 0.01; ***P < 0.001;
****P < 0.0001).
Fig. 4. SHP2 inhibition suppresses CSF-1R signaling and selectively affects viability of M2
macrophages. SHP2 inhibition exhibits greater anti-tumor activity relative to CSF-1R
inhibition in vivo. (A) The effect of RMC-4550 (72h) on proliferation of murine BM cells
grown in either CSF-1 (IC50 = 13 nM) or GM-CSF (IC50 not calculated due to shallow depth of
inhibition). (B) The effect of RMC-4550 (78h) on caspase 3/7 activation of murine BM cells. (C)
Effects of RMC-4550 on cellular pERK levels after acute stimulation of murine BM cells with
CSF-1 (IC50 = 3 nM) or GM-CSF (IC50 = 93 nM). (D) Western blot showing effects of RMC-
4550 and BLZ-945 on pERK levels of human monocytes acutely stimulated with CSF-1 for 5
minutes. (E) Murine BMDMs were polarized to M1 (IFN-, LPS) or M2 (IL-4) phenotypes for
24 h; Graph shows growth after teratment with RMC-4550 (72 h). (F) M1 and M2 polarized
BMDMs were treated with RMC-4550 (48h) and assayed for caspase 3/7 activation. (A-F) Data
represent the mean s.d. of n=2 or 3 independent biological experiments performed in technical
duplicate. (G-H), Activity of RMC-4550 (oral daily administration of 30 mg/kg), anti-CSF-1R (2
mg/mouse on staging day followed by 0.2 mg/mouse 6 days after staging) or combination of
both in CT26 tumor bearing immunocompetent mice. Tumor growth of individual mice for each
experimental group described (G) and Kaplan–Meier curves (H). Kaplan–Meier curves were
compared using the Mantel-Cox Log-Rank test; n=10 animals per group (*P < 0.05 **P < 0.01;
***P < 0.001; ****P < 0.0001).
Fig. 5. SHP2 inhibition is additive in combination with checkpoint inhibition in a SHP2
inhibitor-sensitive tumor model. (A-B), Activity of RMC-4550 (oral daily administration of 30
mg/kg), anti-PD-1 (10 mg/kg, IP every three days) or combination of both in EMT6 tumor
bearing immunocompetent mice. Tumor growth of individual mice for each experimental group
described (A) and Kaplan–Meier curves (B). Kaplan–Meier curves were compared using the
Mantel-Cox Log-Rank test; n=10 animals per group (*P < 0.05 **P < 0.01; ***P < 0.001; ****P
< 0.0001). TFS = Tumor-free survivors. (C), Working model for the effects of SHP2 inhibition
on anti-tumor immunity via modulation of both adaptive and innate mechanisms: blockade of
inhibitory signaling in CD8+T cells; direct and selective depletion of M2 pro-tumorigenic
macrophages through attenuation of CSF-1R signaling and induction of apoptotic cell death;
decrease of the suppressive potential of mMDSC; and increase of M1 macrophages via a
mechanism that is independent of CD8+T or IFN. Collectively these mechanisms contribute to
generate a less immunosuppressive environment and one that favors tumor cell elimination. In
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

21
those cancers with aberrant RAS/MAPK signaling, which are intrinsically sensitive to SHP2
inhibition, the ultimate impact on tumor cell growth will reflect integration of the both the direct,
targeted and anti-tumor immunity mechanisms.
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

Figure 1
A
D
B
C CT26, wild type mice, antibody-mediated immune cell depletion
10 20 30 40 50
Days post-implant
10 30 50 700
25
50
75
100
Days post-implant
% m
ice
with
tum
ors
< 2
00
0 m
m3
10 20 30 40
Days post-implant
CT26 colon MC38 colon A20 B cell
10 20 30 40 50
Days post-implant
CT26, RAG2-/- mice
Vehicle
RMC-4550
0
1000
2000
10 20 30 400
1000
2000
Days post-implant
10 20 30 40
Days post-implant
0
1000
2000
vehicle RMC-4550
Tum
or
volu
me
(mm
3)
Isotype control
CD4+/CD8+T
cell depletion
CD8+T cell
depletion
0
25
50
75
100
****
10 20 30 400
25
50
75
100
Days post-implant
0
25
50
75
100
% m
ice
wit
h t
um
ors
< 2
00
0 m
m3
E
* **** ****
10 20 30 40 500
25
50
75
100
Days post-implant
% m
ice
with
tum
ors
< 2
00
0 m
m3
10 20 30 40 500
1000
2000
Days post-implant
Tum
or
Volu
me (
mm
3)
10 20 30 40 50
Days post-implant
10 20 30 40 50
Days post-implant
10 20 30 40 50
Days post-implant
Isotype RMC-4550 Anti-PD-L1 Combination 4 TFS
*** ***
CT26 colon
10 20 30 40 500
25
50
75
100
Days post-implant
% m
ice
with
tum
ors
< 2
00
0 m
m3
10 20 30 40 500
1000
2000
Days post-implant
Tu
mo
r V
olu
me
(m
m3)
10 20 30 40 50
Days post-implant
10 20 30 40 50
Days post-implant
10 20 30 40 50
Days post-implant
Isotype RMC-4550 Anti-CTLA4 Combination 1 TFS
**** *** *
CT26 colon
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

Figure 2 A
Isotype
RMC-4550
Anti-CTLA4
Combination
0
5
10
15
20
CD
8+
T (
%C
D4
5+
)
***
*
*
*
*
0
20
40
60
CD
8+
T/T
reg
0
2
4
6
CD
4+
T (
%C
D4
5+
)0
2
4
6
8
10
PD
-1 o
f C
D8
+T
(MF
I, x
10
4)
*****
********
B
Isotype
RMC-4550
Anti-PD-L1
Combination
0
1
2
3
4
5
CD
4+
T (
%C
D4
5)
0
5
10
15
20
CD
8+
T/T
reg
0
5
10
15
CD
8+
T (
%C
D4
5)
0
2
4
6
8
10
PD
-1 o
f C
D8
+T
(MF
I, x
10
4)
D
0
2
4
6
8
MH
CI o
f
CD
45 n
ega
tive
cells
(M
FI,
x10
4)
-IFN -CD8+Isotype
******
1.0
1.2
1.4
1.6
PD
-L1 o
f
CD
45 n
ega
tive
ce
lls (
MF
I, x
10
3)
******
-IFN -CD8+Isotype
Vehicle RMC-4550 Anti-CTLA4
C Vehicle
RMC-4550
Anti-CTLA4
Combination
E
-9 -8 -7 -6 -50
2
4
6
8
log [Anti-PD-1 (g/ml)]
NF
AT
Lucife
rase
(AU
x10
4)
-10 -9 -8 -7 -6
log [RMC-4550 (M)]
IFN
(pg
/ml)
0
75
150
225
IL-2
(p
g/m
l)
0
500
1000
RMC-4550 Anti-PD-1
F
Vehicle
RMC-4550
Anti-CTLA4
Combination
0
5
10
15
20
25 *
Edgetumor area
0
5
10
15
CD
8+
T P
ositiv
e
ce
lls (
%)
*
Totaltumor area
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

Figure 3 A
D
C
0
20
40
60
F4
/80
+ (
%C
D4
5+
) ****
*******
**
0
5
10
M1
(%
CD
45
+) ****
**
**
****
*
0
20
40
60
M2
(%
CD
45
+)
****
********
******
0
10
20
30
40
50
M2
/M1
****
*******
****
Isotype
RMC-4550
Anti-CTLA4
Combination
0
20
40
60
F4
/80
(%
CD
45
)
*****
**
0
5
10
15
M1
(%
CD
45
)
******
0
20
40
60
M2
(%
CD
45
)
*****
****
0
25
50
M2
/M1
****
***
60
70
80
90
100
CD
11
b (
%C
D4
5)
**
Isotype
RMC-4550
Anti-PD-L1
Combination
B
Vehicle RMC-4550 Anti-CTLA4 Combination
0
10
20
30
40
F4
/80
Po
sitiv
e
Are
a (
%)
**
***
*
vehicle
RMC-4550
E F vehicle
RMC-4550
0.0
0.5
1.0
1.5
M1 (
%C
D45
) * ***
0
20
40
60
M2
(%
CD
45
)
-IFN -CD8+Isotype
*** *** ***
0
1
2
MH
CI of
M1+
(MF
I, x
10
4)
*
0
1
2
3
4
5
MH
CI
of
M2
+
(MF
I, x
10
4)
*
-IFN -CD8+Isotype
H G
0
10
20
30
40
mM
DS
C (
%C
D45
)
****
****
0
5
10
15
20
25
gM
DS
C (
%C
D4
5)
0
10
20
30
40
mM
DS
C (
%C
D4
5+
)
*******
****
**
0
10
20
30
gM
DS
C (
%C
D4
5+
)
Isotype
RMC-4550
Anti-CTLA4
Combination
Isotype
RMC-4550
Anti-PD-L1
Combination
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038
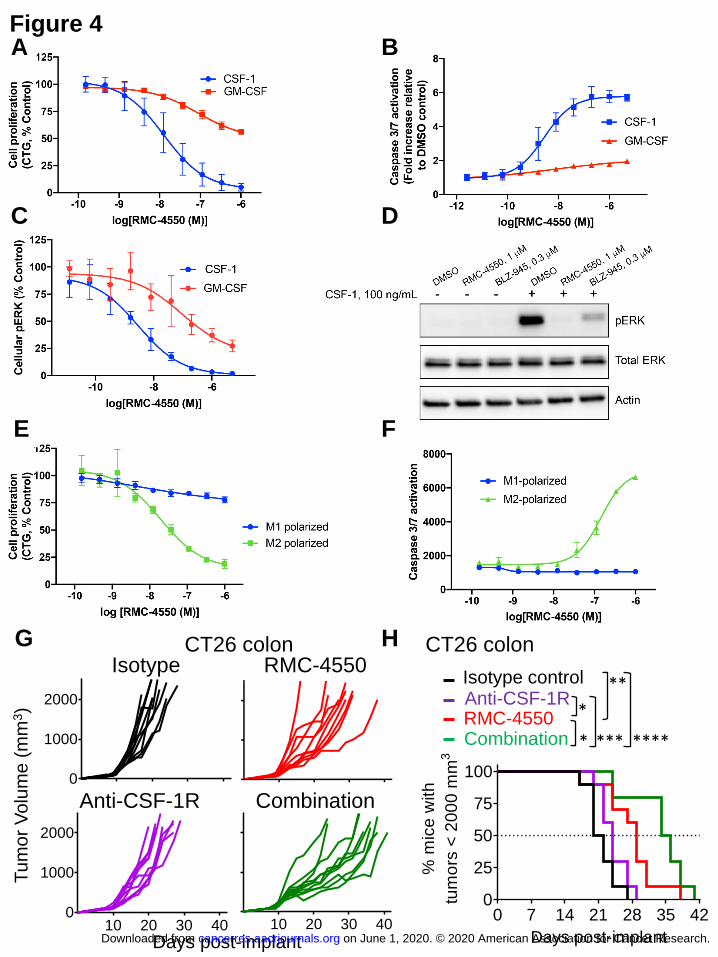
G H Isotype RMC-4550
0
1000
2000
10 20 30 400
1000
2000
10 20 30 40
Days post-implant
Tum
or
Volu
me
(m
m3)
Combination Anti-CSF-1R
0 7 14 21 28 35 420
25
50
75
100
Days post-implant
% m
ice
with
tum
ors
< 2
00
0 m
m3
RMC-4550 Anti-CSF-1R Isotype control
Combination
**
****
*
* ***
A
C
E
B Figure 4
D
F
CT26 colon CT26 colon
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

Figure 5 A B
25 50 750
25
50
75
100
Days post-implant
% m
ice w
ith t
um
ors
< 2
000
mm
30
1000
2000
7 14 21 280
1000
2000
7 14 21 28
1 TFS
Anti-PD-1
Isotype RMC-4550
Combination
2 TFS
Tum
or
Volu
me
(m
m3)
Days post-implant
RMC-4550 Anti-PD-1 Isotype control
Combination
**** ****
** *
EMT6 breast EMT6 breast
C
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038

Published OnlineFirst April 29, 2020.Cancer Res Elsa Quintana, Christopher J. Schulze, Darienne R. Myers, et al. transforming the immunosuppressive environmentAllosteric inhibition of SHP2 stimulates anti-tumor immunity by
Updated version
10.1158/0008-5472.CAN-19-3038doi:
Access the most recent version of this article at:
Material
Supplementary
http://cancerres.aacrjournals.org/content/suppl/2020/04/28/0008-5472.CAN-19-3038.DC1
Access the most recent supplemental material at:
Manuscript
Authoredited. Author manuscripts have been peer reviewed and accepted for publication but have not yet been
E-mail alerts related to this article or journal.Sign up to receive free email-alerts
Subscriptions
Reprints and
To order reprints of this article or to subscribe to the journal, contact the AACR Publications
Permissions
Rightslink site. Click on "Request Permissions" which will take you to the Copyright Clearance Center's (CCC)
.http://cancerres.aacrjournals.org/content/early/2020/04/28/0008-5472.CAN-19-3038To request permission to re-use all or part of this article, use this link
on June 1, 2020. © 2020 American Association for Cancer Research. cancerres.aacrjournals.org Downloaded from
Author manuscripts have been peer reviewed and accepted for publication but have not yet been edited. Author Manuscript Published OnlineFirst on April 29, 2020; DOI: 10.1158/0008-5472.CAN-19-3038







![CD8+ Tumor-Infiltrating T Cells Are Trapped in the Tumor … · 2016. 12. 19. · tumor cells induces immunogenic cross-presentation of dying tumor cells [4,5] or sensitizing tumor](https://img.pdfslide.us/doc/110x75/5fbd8f04c0953e25272e83ca/cd8-tumor-infiltrating-t-cells-are-trapped-in-the-tumor-2016-12-19-tumor-cells.jpg)





















