Embed Size (px)
Citation preview

Akt promotes cisplatin resistance in human ovarian cancer cells through
inhibition of p53 phosphorylation and nuclear function
Michael Fraser1,2,3, Tao Bai1,2,3 and Benjamin K. Tsang1,2,3*
1Reproductive Biology Unit and Division of Gynecologic Oncology, Department of Obstetrics and Gynecology,University of Ottawa, Ottawa, ON, Canada2Department of Cellular and Molecular Medicine, University of Ottawa, Ottawa, ON, Canada3Hormones, Growth and Development Program, Ottawa Health Research Institute, The Ottawa Hospital (Civic Campus),Ottawa, ON, Canada
Resistance to cisplatin-based chemotherapy is a major cause oftreatment failure in human ovarian cancer. Wild-type TP53 statusis often, but not always, associated with cisplatin sensitivity, sug-gesting that additional factors may be involved. Overexpression/activation of the phosphatidylinositol-3-kinase/Akt pathway iscommonly observed in ovarian cancer, and Akt activation is a de-terminant of chemoresistance in ovarian cancer cells, an effectthat may be due, in part, to its inhibitory actions on p53-depend-ent apoptosis. To that end, we examined the role and regulation ofp53 in chemosensitive ovarian cancer cells, as well as in their che-moresistant counterparts, and investigated if and how Akt influen-ces this pathway. Cisplatin induced apoptosis in chemosensitive,but not chemoresistant cells, and this was inhibited by downregu-lation of p53. Cisplatin upregulated PUMA in a p53-dependentmanner, and the presence of PUMA was necessary, but not suffi-cient for cisplatin-induced apoptosis. p53 was phosphorylated onnumerous N-terminal residues, including Ser15, Ser20, inresponse to cisplatin in chemosensitive, but not chemoresistantcells. Furthermore, activation of Akt inhibited the cisplatin-induced upregulation of PUMA, and suppressed cisplatin-inducedp53 phosphorylation, while inhibition of Akt increased total andphospho-p53 contents and sensitized p53 wild-type, chemoresist-ant cells to cisplatin-induced apoptosis. Finally, mutation of Ser15and/or Ser20, but not of Ser37, to alanine significantly attenuatedthe ability of p53 to facilitate CDDP-induced apoptosis, and thiswas independent of PUMA expression. These results support thehypothesis that p53 is a determinant of CDDP sensitivity, and sug-gest that Akt contributes to chemoresistance, in part, by attenuat-ing p53-mediated PUMA upregulation and phosphorylation ofp53, which are essential, but independent determinants of sensitiv-ity to CDDP-induced apoptosis.' 2007 Wiley-Liss, Inc.
Key words: ovarian cancer; apoptosis; p53; Akt; cisplatin
Resistance to cisplatin (CDDP: cis-diaminedichloroplatinum)-based chemotherapy is a major cause of treatment failure in humanovarian cancer. Chemoresistance is a multifactorial phenomenon,the molecular mechanisms of which are poorly understood. Whilealterations in DNA platination do not appear to play a significantrole in chemoresistance,1 induction of apoptosis is a key effect ofCDDP chemotherapy,2 and alterations in the apoptotic capacity arefrequently observed and are important determinants of chemosensi-tivity.3–5 Indeed, we and others have shown that key regulatorsof the apoptotic response to CDDP, including X-linked inhibitorof apoptosis protein (XIAP), FLIP, and the MAP kinases (p38,JNK, ERK), are dysregulated in chemoresistant ovarian cancercells,1,3,4,6–8 relative to their sensitive counterparts.
The normal function of the p53 tumor suppressor is associatedwith chemosensitivity and improved clinical outcome in ovariancancer.3,9–11 Indeed, our previous data suggest that p53 is a deter-minant of CDDP sensitivity in ovarian cancer cells. However,wild-type tumor protein 53 (TP53) alone is not a direct predictorof chemotherapeutic response,3,4 suggesting that additional mech-anisms, unrelated to TP53 genotype, play important roles in regu-lating CDDP sensitivity.
p53-mediated apoptosis occurs via transcriptional upregulationof gene products such as Bax,12 p53-Upregulated Modulator of
Apoptosis (PUMA) and NOXA,13,14 transcriptional repression ofgene products such as Bcl-212 and survivin,15,16 and via a tran-scription-independent mechanism whereby p53 directly binds toBcl-2 and Bcl-XL at the mitochondria.17,18 Recent evidence sug-gests that upregulation of PUMA is an important mechanism ofCDDP-induced apoptosis.19 However, whether PUMA is suffi-cient for CDDP-induced apoptosis is not known. Additional tran-scription-independent mechanisms of p53-induced apoptosis havealso been proposed.20 Moreover, p53-mediated apoptosis is de-pendent upon the phosphorylation of several residues, includingSer15, Ser20 and Ser37.21–23
Akt is a serine/threonine kinase that is activated in a phosphati-dylinositol-3-OH-kinase (PI3K)-dependent manner by growth fac-tors and cytokines, and is implicated in cell proliferation and sur-vival. The PI3K/Akt pathway is frequently overexpressed/acti-vated in ovarian cancers,24–27 and activation of Akt promotes achemoresistant phenotype, whereas inhibition of Akt sensitizeschemoresistant cells to CDDP-induced apoptosis.3,5,7,28 However,our previous data also suggest a functional relationship betweenAkt and p53 in this regard.3
While Akt modulates p53 content by phosphorylating murinedouble minute-2 (MDM2), thus promoting the ubiquitin-depend-ent proteolysis of p53,29–32 Akt can also regulate p53 function in-dependently of changes in p53 content or subcellular localiza-tion.33 Since phosphorylation of p53 is required for its proapop-totic effects, it is possible that Akt may suppress apoptosis via thisprocess.
We investigated the hypothesis that activated Akt inhibitsCDDP-induced apoptosis and confers CDDP resistance in culturedovarian cancer cells, in part, by attenuating p53 phosphorylationand activation of p53-responsive gene products, independently ofp53 content. We show that p53 is required for CDDP-induced apo-ptosis in ovarian cancer cells, and that this is dependent upon theinduction of PUMA. Moreover, CDDP induces phosphorylation ofp53 on multiple residues in chemosensitive ovarian cancer cells,but not the respective chemoresistant variants. Akt attenuated bothPUMA and phospho-p53 upregulation, and conferred chemoresist-
Grant sponsor: National Cancer Institute of Canada, Canadian CancerSociety; Grant number: 013335; Grant sponsor: Canadian Institutes ofHealth Research; Grant number: MOP-15691.*Correspondence to: Ottawa Health Research Institute, 725 Parkdale
Avenue, Ottawa, Ontario, Canada K1Y 4E9. Fax:1613-761-4403.E-mail: [email protected] 13 May 2007; Accepted after revision 27 July 2007DOI 10.1002/ijc.23086Published online 4 October 2007 in Wiley InterScience (www.interscience.
wiley.com).
Abbreviations: CDDP, cis-diaminedichloroplatinum; DMEM, Dulbec-co’s modified Eagle medium; DN-Akt, dominant-negative Akt; GAPDH,glyceraldehyde phosphate dehydrogenase; MDM2, murine double minute-2; PI3K, phosphatidylinositol-3-OH-kinase; PMSF, phenylmethylsulfonylfluoride; PUMA, p53-upregulated modulator of apoptosis; RPMI, RoswellPark Memorial Institute; RT-PCR, reverse transcriptase polymerase chainreaction; SDS-PAGE, sodium dodecyl sulfate–polyacrylamide gel electro-phoresis; siRNA, small inhibitory RNA; TP53, tumor protein 53; Xiap, X-linked inhibitor of apoptosis protein.
Int. J. Cancer: 122, 534–546 (2008)' 2007 Wiley-Liss, Inc.
Publication of the International Union Against Cancer

ance. Finally, we show that p53 hypophosphorylation is associatedwith chemoresistance, and that phosphorylation of Ser15 andSer20, but not of Ser37, are required for maximal CDDP-inducedapoptosis.
Taken together, these data have important implications for ourunderstanding of the molecular mechanisms of chemoresistance inovarian cancer cells, and in particular, the role of Akt and p53 inthis process. Since chemoresistance limits treatment success forthis disease, it is critical to understand how resistant cells evadethe normal execution of apoptosis.
Material and methods
Reagents
Cisplatin (CDDP), Hoechst 33258, phenylmethylsulfonyl fluo-ride (PMSF), sodium orthovanadate (Na3VO4) and aprotinin werepurchased from Sigma (St. Louis, MO). Mouse monoclonal anti-bodies to p53 (DO-1) and Bax (2D2) were from Santa Cruz Bio-technologies (San Diego, CA). Rat monoclonal anti-HA was pur-chased from Roche (clone 3F10, Palo Alto, CA). Mouse monoclo-nal anti-MDM2 was from Calbiochem (Ab-1, San Diego, CA).Mouse monoclonal anti-phospho-p53 (Ser15; clone 16G8), rabbitpolyclonal anti-phospho-p53 (Ser6, Ser20, Ser33, Ser37, Ser46)and rabbit polyclonal anti-PARP antibodies were from Cell Sig-naling Technology (Beverly, CA). Rabbit polyclonal anti-PUMAwas from Sigma. Mouse anti-glyceraldehyde phosphate dehydro-genase (GAPDH) (ab8245) was from Abcam (Cambridge, UK).Small inhibitory RNA (siRNA) to p53 was purchased from CellSignaling Technology. siRNA to PUMA was purchased fromSanta Cruz Biotechnologies. Control siRNA was from Dharmacon(Lafayette, CO). Ribojuice siRNA transfection reagent was fromNovagen (San Diego, CA). Pre-stained sodium dodecyl sulfate–polyacrylamide gel electrophoresis (SDS-PAGE) standards werefrom BioRad (Hercules, CA). Adenoviral dominant-negative Aktwas a generous gift from Dr. Kenneth Walsh (CardiovascularResearch, St. Elizabeth’s Medical Centre, Boston, MA). HA-tagged wild-type p53 in pcDNA3.1 was a generous gift from Dr.Jin Q. Cheng, Moffitt Cancer Center, University of South Florida,Tampa, FL.
Cell lines and cell culture
Chemosensitive ovarian cancer cells (A2780s and OV2008) andtheir respective chemoresistant variants (A2780cp and C13*) werecultured as previously reported3,4,7,34 in Dulbecco’s modified Eaglemedium (DMEM)/F12 and RPMI 1640. OVCAR-3 cells were cul-tured in RPMI 1640 media. All media were supplemented with10% FBS, streptomycin (50 g/ml), penicillin (50 U/ml), fungizone(0.625 g/ml; Life Technologies BRL, Carlsbad, CA) and nonessen-tial amino acids (1%). Cells were plated at a density of 5 3 104
cells/cm2 on 6-well plates or 60-mm dishes 18 hr prior to the initia-tion of treatment. At the time of treatment, cell density was<85%.
Site-directed mutagenesis
HA-tagged wild-type p53 in pcDNA3 was used as a templatefor site-directed mutagenesis, using the QuickChange Site-Directed Mutagenesis Kit from Stratagene (La Jolla, CA). Primersused to mutate p53 to alanine at the indicated site(s) were asfollows: Ser15: Forward 50-GTCGAGCCCCCTCTGGCACAGGAAACATTTTCAGACC-30, Reverse 50-GGTCTGAAAATGTTTCCTGTGCCAGAGGGGGCTCGAC-30; Ser20: Forward 50-CTGAGTCAGGAAACATTTGCAGACCTATGGAAACTACTT-30,Reverse 50-AAGTAGTTTCCATAGGTCTGCAAATGTTTCCTGACTCAG-30; Ser37: Forward 50-TGTCCCCCTTGCCGGCA-CAAGCAATGGATGATTTG-30, Reverse 50-CAAATCATCCATTGCTTGTGCCGGCAAGGGGGACA; Ser15/Ser20: Forward50-GTCGCACAGGAAACATTTGCAGACCTATGGAAACTACTT-3, Reverse: 50-AAGTAGTTTCCATAGGTCTGCAAATGTTTCCTGTGCCAG-30. The template plasmid DNA was amplifiedusing Pfu Polymerase (Fermentas, Hanover, MD) according to
the manufacturer’s instructions for 16 cycles, digested with DpnI(Fermentas) for 1 hr at 37�C, and then transformed into XL10Gold cells (Stratagene) and plated onto LB plates containing 100lg/ml ampicillin. The following day, colonies were picked,amplified overnight at 37�C in LB broth containing 100 lg/mlampicillin and the plasmid DNA was extracted using the PlasmidMini Kit from Qiagen (Valencia, CA). The presence of mutationswas confirmed by direct sequencing at the OHRI sequencing fa-cility.
Hoechst 33258 staining
At the end of the culture period, cells attached to the growthsurface were removed by trypsin treatment [trypsin (0.05%),EDTA (0.53 mM); 37�C, 1 min]. Attached and floating cells werepooled, pelleted by centrifugation and resuspended in phosphate-buffered formalin (10%) containing Hoechst 33258 (12.5 ng/ml).Cells were spotted onto slides for microscopy. Nuclear stainingwas observed using a Zeiss fluorescence microscope (magnifica-tion 4003). Cells with typical apoptotic nuclear morphology (nu-clear shrinkage, condensation and fragmentation) were identifiedand counted as previously reported,3,4,7,35 using randomly selectedfields. A minimum of 200 cells were counted in each treatmentgroup. The counter was ‘‘blinded’’ to sample identity to avoid ex-perimental bias. Data are expressed as the percentage of total cellsshowing apoptotic morphology.
Protein extraction and western blot analysis
Cells were pelleted and lysed in ice-cold lysis buffer (pH 7.4)containing 50 mM Hepes, 150 mM NaCl, 1.5 mM MgCl2, 1 mMEGTA, 100 mM NaF, 10 mM NaPPi, 10% Glycerol and 1% Tri-ton X-100. Protease inhibitors PMSF (1 mM) and aprotonin (10 g/l), as well as 1 mM Na3VO4 were added to the lysis buffer freshly.Cell lysates were sonicated briefly, incubated on ice for 1 hr andpelleted by centrifugation (15,000g; 20 min). The supernatant wastaken as whole-cell lysate and stored at 220�C for subsequentanalyses. Protein concentration was determined using Bio-Rad DCprotein assay kit. Equal amounts of proteins (30–70 lg) wereloaded and resolved by 10% SDS-PAGE and electrotransferred(30 V, 16 hr) onto nitrocellulose membranes (Bio-Rad). Mem-branes were blocked (room temperature, 1 hr) with 5% Blotto[Tris-HCl (10 mM; pH 8.0), NaCl (150 mM), Tween 20 (0.05%,v/v; TBS-Tween 20) containing skim milk (5%; w/v)], then incu-bated overnight with primary antibodies [p53 (1:1,000), Bax(1:1,000), PUMA (1:1,000), MDM2 (1:2,000) or GAPDH(1:30,000)] and subsequently with the appropriate horseradish per-oxidase (HRP)-conjugated secondary antibody [1:2,000 in 5%Blotto; room temperature, 1 hr; 1:20,000 for GAPDH]. Peroxidaseactivity was visualized with an Enhanced Chemiluminescence Kit(Amersham Pharmacia Biotech, Arlington Heights, IL) after 3washes (15 min/wash) with TBS-Tween 20. Signal intensity wasdetermined densitometrically using Scion Image software, version4.02, from Scion Corporation (Frederick, MD). All quantifiedwestern blot data were corrected for loading using the anti-GAPDH blots. Western blots shown in figures are representativeof at least 3 independent experiments.
Immunoprecipitation–western blots
Cultured cells were transfected with HA-tagged p53 constructsfor 24 hr (as indicated) and then treated with CDDP for a further24 hr. The cells were lysed in standard lysis buffer (as earlier) for1 hr on ice, then transferred to a 1.5-ml microcentrifuge tube andcentrifuged for 20 min at 14,000g to remove cellular debris. Thesupernatants were analyzed for total protein content, and 300 lgof total protein was incubated with 15 ll of agarose-immobilizedgoat polyclonal anti-HA antibody (Bethyl Laboratories, Mont-gomery, TX) in a final volume of 300 ll, adjusted with lysisbuffer. Immunoprecipitation was carried out with gentle rocking,overnight at 4�C. The agarose beads were pelleted by centrifuga-tion at 500g for 2 min, and then washed 3 times with 1 ml lysis
535REGULATION OF p53 BY Akt IN OVARIAN CANCER CELLS

buffer, with each wash followed by a 2-min centrifugation at500g. After the final wash, 30 ll of 23 SDS sample buffer wasadded to the beads, the samples were boiled and then loaded onto12% SDS-PAGE gels. Following protein transfer to nitrocellulose,phospho-p53, total p53 and exogenous HA-p53 were detected bywestern blotting as described earlier.
Reverse transcriptase polymerase chain reaction
Cultured cells were harvested as earlier, and cell pellets werestored at 280�C until further processing. Total RNA wasextracted using the RNeasy Mini Kit from Qiagen. An aliquot oftotal RNA from each sample was subjected to DNase I treatmentto remove genomic DNA contamination using the DNA-free kitfrom Ambion (Austin, TX). mRNA was reverse-transcribed usingoligo(dT) primers with Moloney murine leukemia virus (M-MuLV) reverse transcriptase via the Retroscript kit from Ambion.PCR primers were from Invitrogen (Burlington, ON) as follows:PUMA sense: 50-TGTGACCACTGGCATTCATT-30; PUMAantisense: 50-CCTGTAAGATACTGTATATGCGCTGC-30; b-actin sense: 50-GGACTTCGAGCAAGAGATGG-30; b-actin anti-sense: 50-CACCTTCACCGTTCCAGTTT-30. To ensure linearityof the results, the cycle number was optimized by performingPCR reactions at 20–45 cycles. All subsequent PCR reactionswere performed within the linear range of amplification. PCRswere performed using HotStarTaq Polymerase from Qiagen. PCRconditions following activation (15 min; 95�C) were: denaturation(94�C) for 45 sec, annealing (PUMA: 56�C, b-actin: 54�C) for 45sec, extension (72�C) for 30 sec, for 37 or 25 cycles for PUMAand b-actin, respectively. Ten microliters of each PCR productwas separated on a 1.5% agarose–ethidium bromide gel andvisualized by ultraviolet transillumination using a BioRad GelDocsystem.
Genomic DNA sequencing
To confirm TP53 genotype, total genomic DNA from each cellline was extracted using the DNeasy Tissue Kit (Qiagen) and wassubjected to exon-specific PCR amplification using primers fromthe SNPCapture p53 Mutation Screening Kit from Panomics (Fre-mont, CA). PCR products, corresponding to TP53 genomic DNAfrom exons 5 to 9 (which encode the DNA binding domain ofp53), were visualized by 2% agarose gel electrophoresis andcloned into the pCR4-TOPO sequencing vector from Invitrogen,according to the manufacturer’s instructions. Following the trans-formation and growth of chemically competent E. coli, the plas-mid DNA was purified by the Plasmid Miniprep Kit from Qiagen.Isolated plasmid DNA was sequenced by the Dye Terminatormethod at the Ottawa Genome Centre. Sequences were comparedwith the published human TP53 sequence using NCBI Blast2.TP53 status was as follows: A2780s-WT; A2780cp-V172F,R260S; OV2008-WT; C13*-WT; OVCAR-3-Q317R.
Adenoviral infection
Cells were infected with an HA-tagged ‘‘triple-A’’ (K179A,T308A, S473A) dominant-negative Akt (DN-Akt) or LacZ cDNAat various multiplicities of infection (MOI) as indicated in the textand previously described.8 As previously reported, adenovirusinfection efficiency at MOI of 5, as determined by an X-gal stain-ing assay against LacZ construct infected cells, was >90%.3,4
DN-Akt expression was confirmed by western blot analysisagainst the HA epitope tag.
RNA interference
Six microliters of transfection reagent (Novagen) was added to244 ll of DMEM/F12 without serum. The mixture was vortexedand incubated for 5 min at room temperature. Following incuba-tion, 7.5 ll of 10 lM stock siRNA construct was added. The mix-ture was incubated at room temperature for a further 15 min. Dur-ing this period, the culture media was removed from the cells andthe cells were washed once with phosphate-buffered saline. The
siRNA mixture was added to each well with an additional 1,250 llof complete (10% FBS) media. The cells were returned to the in-cubator and the media was removed 6 hr later and replaced withfresh, complete media for the duration of the culture (24–48 hr).Downregulation was confirmed by western blot analysis.
Transient transfection
Cells were cultured overnight in 6-well plates and then trans-fected with 1 lg of pcDNA3.1-derived vectors (empty vector con-trol) using Lipofectamine Plus (Invitrogen) in 1 ml serum-free me-dium according to the manufacturer’s instructions. Three hourspost-transfection, each well was supplemented with 1 ml of me-dium containing 20% FBS. Twenty-four hours post-transfection,media were removed and the cells were harvested or treated asrequired for a further 24 hr.
DNA binding assay
For the assessment of p53-DNA binding capacity, nuclearlysates of CDDP-treated cells were obtained using the NE-PERNuclear/Cytoplasmic Extraction kit from Pierce (Rockford, IL),according to the manufacturer’s instructions and as previouslyreported in our laboratory,36 and total nuclear protein contentswere determined (as earlier). To assess p53-DNA binding, weused the p53 TransAM kit from ActiveMotif (Carlsbad, CA).Briefly, equivalent amounts of nuclear protein were incubated for1 hr at room temperature in separate wells of a 96-well plate con-taining an immobilized p53 consensus oligonucleotide (50-GGA-CATGCCCGGGCATGTCC-30). After 1 hr, plates were washed33 in fresh 13 wash buffer (supplied with kit), and then incu-bated for 1 hr at room temperature with anti-p53 antibody. Theplates were washed a further 33 with 13 wash buffer, and thenincubated 1 hr with HRP-conjugated anti-rabbit antibody. After afurther wash step, HRP was detected using 13 developing solution(supplied with kit), and the reaction was stopped with 13 stopsolution (supplied with kit) when a light blue color was observed.Plates were immediately read using a UV spectrophotometer at450 nm. In addition to sample wells, the assay was performed on 3wells containing only lysis buffer as a negative control. Assayspecificity was confirmed by preincubation with free mutant orwild-type oligonucleotide.
Statistical analysis
Results are expressed as the mean 6 SEM of at least 3 inde-pendent experiments. Statistical analysis was carried out by one-or two-way ANOVA or by Student’s t test (where appropriate)using PRISM software (Version 3.0; GraphPad, San Diego, CA).Differences between multiple experimental groups were deter-mined by the Bonferroni or Tukey post-hoc tests. Statistical signif-icance was inferred at p < 0.05.
Results
p53 is required for CDDP-induced apoptosis in humanovarian cancer cells
While wild-type TP53 is frequently associated with chemosen-sitivity in human ovarian cancer cells and tumors, ovarian tumorsand cell lines bearing wild-type TP53 are often chemoresistant,suggesting that p53 is necessary, but not sufficient, for chemosen-sitivity, and that additional mechanisms are likely.3,4,10
To determine the involvement of p53 in CDDP-induced apopto-sis, p53 wild-type, chemosensitive (OV2008) ovarian cancer cellswere transfected with siRNA targeting p53 (or control; 0–100 nM;24 hr) and treated with CDDP (0, 5 lM; 24 hr). As shown in Fig-ure 1a, in the presence of the control siRNA, CDDP upregulatedp53 and the p53-responsive gene product PUMA (lane 1 vs. lane2) and significantly increased the percentage of cells undergoingapoptosis (1.24% 6 0.25% vs. 29.9% 6 3.97%, p < 0.001).By contrast, p53 siRNA downregulated p53 and attenuatedthe CDDP-induced upregulation of PUMA. Furthermore,
536 FRASER ET AL.
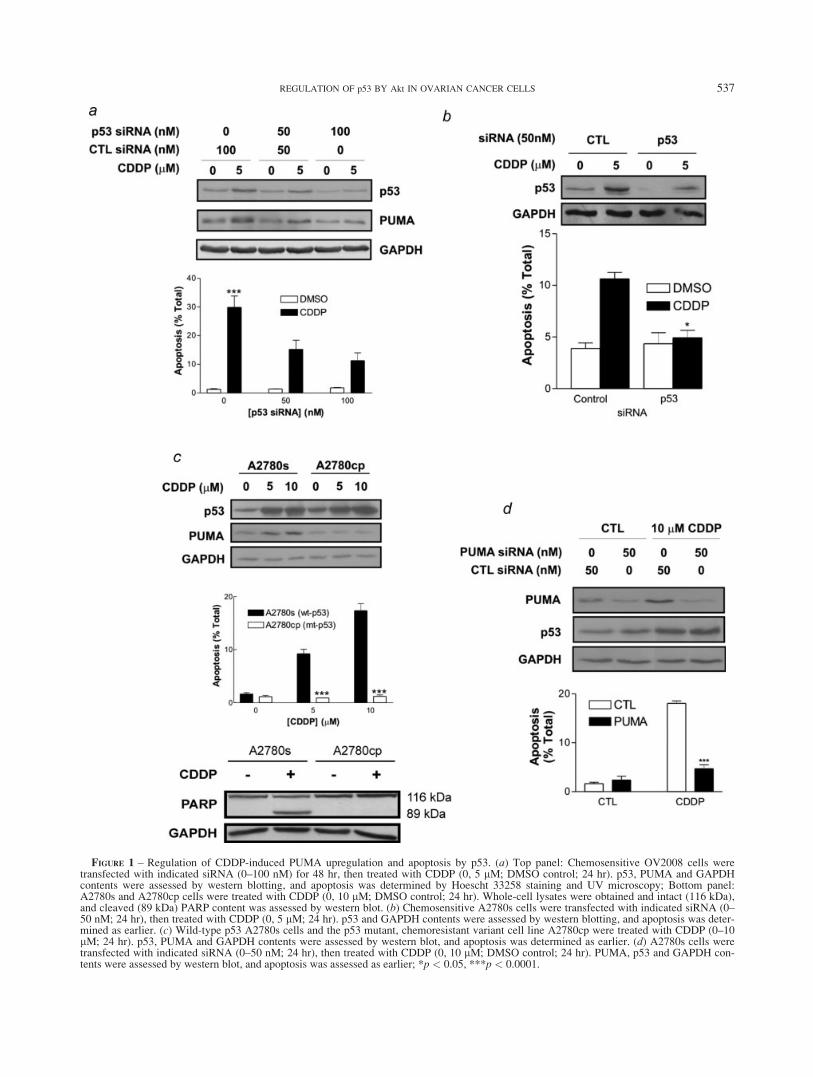
FIGURE 1 – Regulation of CDDP-induced PUMA upregulation and apoptosis by p53. (a) Top panel: Chemosensitive OV2008 cells weretransfected with indicated siRNA (0–100 nM) for 48 hr, then treated with CDDP (0, 5 lM; DMSO control; 24 hr). p53, PUMA and GAPDHcontents were assessed by western blotting, and apoptosis was determined by Hoescht 33258 staining and UV microscopy; Bottom panel:A2780s and A2780cp cells were treated with CDDP (0, 10 lM; DMSO control; 24 hr). Whole-cell lysates were obtained and intact (116 kDa),and cleaved (89 kDa) PARP content was assessed by western blot. (b) Chemosensitive A2780s cells were transfected with indicated siRNA (0–50 nM; 24 hr), then treated with CDDP (0, 5 lM; 24 hr). p53 and GAPDH contents were assessed by western blotting, and apoptosis was deter-mined as earlier. (c) Wild-type p53 A2780s cells and the p53 mutant, chemoresistant variant cell line A2780cp were treated with CDDP (0–10lM; 24 hr). p53, PUMA and GAPDH contents were assessed by western blot, and apoptosis was determined as earlier. (d) A2780s cells weretransfected with indicated siRNA (0–50 nM; 24 hr), then treated with CDDP (0, 10 lM; DMSO control; 24 hr). PUMA, p53 and GAPDH con-tents were assessed by western blot, and apoptosis was assessed as earlier; *p < 0.05, ***p < 0.0001.
537REGULATION OF p53 BY Akt IN OVARIAN CANCER CELLS

CDDP-induced apoptosis was significantly inhibited by p53siRNA (p < 0.05). This was also observed in the unrelated wild-type p53 chemosensitive ovarian cancer cell line A2780s, in whichp53 siRNA downregulated p53 in both DMSO- and CDDP-treatedcells and significantly inhibited CDDP-induced apoptosis (10.6%6 0.63% vs. 3.89%6 0.54%, p < 0.05; Fig. 1b).
To further examine the role of p53 in CDDP sensitivity,A2780s and their mutant p53 (V172, R260S) chemoresistant coun-terparts, A2780cp, were cultured in the presence of CDDP (0–10lM; 24 hr). As shown in Figure 1c, CDDP upregulated p53 andPUMA and induced apoptosis in the A2780s cells, whereas thiswas significantly attenuated in the A2780cp cells (p < 0.001). Thedifferential induction of apoptosis was confirmed by examiningthe CDDP-induced cleavage of the caspase-3 substrate poly(ADP)ribose polymerase (PARP).37 A2780s and A2780cp cells were cul-tured in the presence or absence of CDDP (0, 10 lM; 24 hr).CDDP induced cleavage of intact 116 kDa PARP into the 89 kDaform in the chemosensitive A2780s cells, but not in the chemore-sistant A2780cp cells (Fig. 1c; bottom panel). Consistent with thepresence of mutant p53 in the latter cell line, total p53 levels werehigher in the absence of CDDP compared with the wild-typeA2780s cells, likely due to loss of MDM2-mediated negative feed-back against p53, which has been previously reported.4,38
To evaluate whether PUMA is required for CDDP-induced apo-ptosis, we treated A2780s cells with PUMA or control siRNA (0–50 nM; 48 hr) and CDDP (0, 10 lM; 24 hr). As shown in Figure1d, PUMA siRNA markedly downregulated PUMA content, with-out affecting p53, and significantly attenuated CDDP-induced apo-ptosis (18.1% 6 0.48% vs. 4.68% 6 0.81%, p < 0.001, alleffects). These data suggest that PUMA is necessary for CDDP-induced apoptosis.
Taken together, these results demonstrate that p53 is requiredfor sensitivity to CDDP-induced apoptosis in ovarian cancer cells,and that PUMA is necessary for this effect.
PUMA is not sufficient for CDDP-induced apoptosis
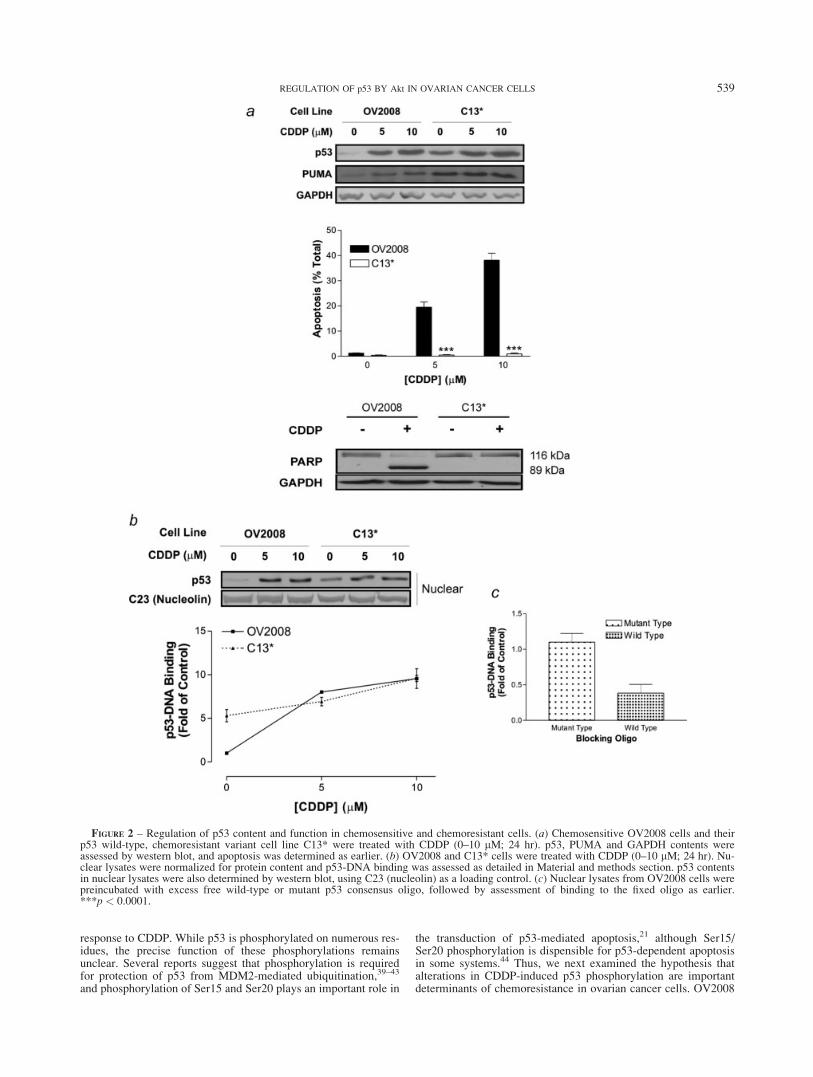
To evaluate whether PUMA is sufficient for CDDP-induced ap-optosis, we treated chemosensitive OV2008 cells and their chemo-resistant, wild-type p53 counterparts, C13*, with CDDP (as ear-lier) and examined p53 and PUMA contents as well as apoptosis.As shown in Figure 2a, basal p53 and PUMA levels were elevatedin the C13* cells, relative to the isogenic, chemosensitive OV2008cells. However, CDDP upregulated p53 and PUMA and inducedapoptosis in the OV2008 cells but not in the C13* cells. We thenconfirmed this differential induction of apoptosis by examiningthe CDDP-induced cleavage of PARP. CDDP induced the cleav-age of PARP in OV2008 cells but not in C13* cells (Fig. 2a; bot-tom panel). These results suggest that the presence of PUMA isnot sufficient for CDDP-induced apoptosis.
To further evaluate the basal and CDDP-induced nuclear func-tion of p53, we assessed p53-DNA binding. As shown in Figure2b, CDDP caused the nuclear accumulation of p53 in OV2008 (p< 0.01) and C13* cells (p < 0.05) in response to CDDP, althoughbasal nuclear p53 content was higher in the C13* cells (p < 0.05).Likewise, basal p53-DNA binding was �5-fold higher in theC13* cells relative to the OV2008 cells, which is consistent withthe high basal levels of PUMA in these cells. However, CDDPupregulated p53-DNA binding to a significantly greater extent inOV2008 than in C13* (p < 0.01). Thus, the change in p53-DNAbinding was far greater in the chemosensitive cells than in their re-sistant variants. The specificity of the assay for p53 was confirmedby introduction of free wild-type p53 consensus oligonucleotide,which competed out p53 binding to the immobilized oligo. Bycontrast, mutated p53 consensus oligonucleotide did not affectp53 binding to the immobilized oligo (Fig. 2c). Thus, C13* cellsare resistant to CDDP, despite having high basal p53-DNA bind-ing activity and PUMA content, suggesting that p53 nuclear activ-ity and PUMA upregulation are insufficient to support a CDDP-sensitive phenotype. Thus, it is likely that additional mechanisms
of CDDP-induced, p53-dependent apoptosis are at play in thesecells.
Activation of Akt attenuates CDDP-induced p53nuclear function and apoptosis
The PI3K/Akt pathway is frequently activated in human ovariancancers24–27 and is a determinant of chemoresistance in humanovarian cancer cells.3,5,7,28 Furthermore, our previous data suggestthat Akt-mediated chemoresistance may, in part, be mediatedthrough alterations in p53-mediated apoptosis.3 Thus, we nextexamined the effects of Akt activation on CDDP-induced p53-mediated gene expression, using A2780s cells stably transfectedwith empty pcDNA3 vector (A2780-CTL) or pcDNA3 containingconstitutively active Akt2 (A2780-AAkt2). The phenotype ofthese cells has been extensively characterized.3,5,28 As shown inFigure 3a, CDDP markedly upregulated p53 and PUMA andinduced apoptosis in the control-transfected cells. In contrast,CDDP only slightly upregulated p53 in the A2780-AAkt2 cells,although basal p53 levels were higher in these cells. Despite thepresence of p53, active Akt attenuated the CDDP-induced upregu-lation of PUMA, and significantly inhibited CDDP-inducedapoptosis (15.5% 6 0.97% vs. 1.53% 6 0.22% at 10 lM CDDP,p < 0.001). To confirm the antiapoptotic effects of Akt activation,we examined CDDP-induced PARP cleavage in A2780-CTL andA2780-AAkt2 cells. As shown in Figure 3a (bottom panel),CDDP induced PARP cleavage in A2780-CTL cells, and this wascompletely attenuated in A2780-AAkt2 cells.
To evaluate the kinetics of CDDP-induced PUMA upregulationand the effects of Akt activation on this parameter, we next per-formed a time-course analysis on PUMA protein content inresponse to CDDP in A2780-CTL or A2780-AAkt2 cells. We alsoexamined the protein content of MDM2 over the same duration.As shown in Figure 3b, PUMA upregulation occurred between 3and 6 hr post-CDDP in the A2780-CTL cells. Upregulation ofMDM2 occurred even earlier, with marked upregulation within 1–3 hr post-CDDP. Upregulation of these gene products was mark-edly lower in the A2780-AAkt2 cells, which is consistent with aninactivation of p53-mediated gene transcription in these cells.
We next evaluated the effects of Akt activation on PUMAmRNA content by reverse transcriptase polymerase chain reaction(RT-PCR). A2780-CTL and A2780-AAkt2 cells were cultured inthe presence of CDDP (10 lM; 6 hr) and PUMA and b-actinmRNA contents were assessed. As expected, CDDP significantlyupregulated the PUMA mRNA in A2780-CTL cells (Fig. 3c; p <0.05) but not in the A2780-AAkt2 cells. Six hours of culture in theabsence of CDDP (DMSO alone) in A2780-CTL cells did notchange PUMA mRNA content, relative to the 0 hr group (lane 1vs. lane 2). This result is consistent with the hypothesis that Aktattenuates the CDDP-induced upregulation of PUMA gene tran-scription.
To further evaluate the effects of Akt activation on p53 biologi-cal function, we evaluated p53 nuclear accumulation and DNAbinding in A2780-AAkt2 cells and their control-transfected coun-terparts. CDDP significantly increased nuclear p53 localization(p < 0.05) and this was not significantly affected by activation ofAkt. Furthermore, CDDP induced p53 binding to its consensus oli-gonucleotide in the A2780-CTL cells, and this was significantlyattenuated in the A2780-AAkt2 (Fig. 3d, p < 0.001). Taken to-gether, these data suggest that Akt may in part confer resistancethrough inhibition of the nuclear functions of p53, including theexpression of p53-dependent gene products such as MDM2 andPUMA.
CDDP-induced p53 phosphorylation is attenuatedin chemoresistant cells
p53-mediated upregulation of PUMA is necessary, but not suffi-cient, for CDDP-induced apoptosis. We therefore evaluated thehypothesis that phosphorylation of p53, which is required for p53-mediated apoptosis,21 is dysregulated in chemoresistant cells in
538 FRASER ET AL.
response to CDDP. While p53 is phosphorylated on numerous res-idues, the precise function of these phosphorylations remainsunclear. Several reports suggest that phosphorylation is requiredfor protection of p53 from MDM2-mediated ubiquitination,39–43
and phosphorylation of Ser15 and Ser20 plays an important role in
the transduction of p53-mediated apoptosis,21 although Ser15/Ser20 phosphorylation is dispensible for p53-dependent apoptosisin some systems.44 Thus, we next examined the hypothesis thatalterations in CDDP-induced p53 phosphorylation are importantdeterminants of chemoresistance in ovarian cancer cells. OV2008
FIGURE 2 – Regulation of p53 content and function in chemosensitive and chemoresistant cells. (a) Chemosensitive OV2008 cells and theirp53 wild-type, chemoresistant variant cell line C13* were treated with CDDP (0–10 lM; 24 hr). p53, PUMA and GAPDH contents wereassessed by western blot, and apoptosis was determined as earlier. (b) OV2008 and C13* cells were treated with CDDP (0–10 lM; 24 hr). Nu-clear lysates were normalized for protein content and p53-DNA binding was assessed as detailed in Material and methods section. p53 contentsin nuclear lysates were also determined by western blot, using C23 (nucleolin) as a loading control. (c) Nuclear lysates from OV2008 cells werepreincubated with excess free wild-type or mutant p53 consensus oligo, followed by assessment of binding to the fixed oligo as earlier.***p < 0.0001.
539REGULATION OF p53 BY Akt IN OVARIAN CANCER CELLS
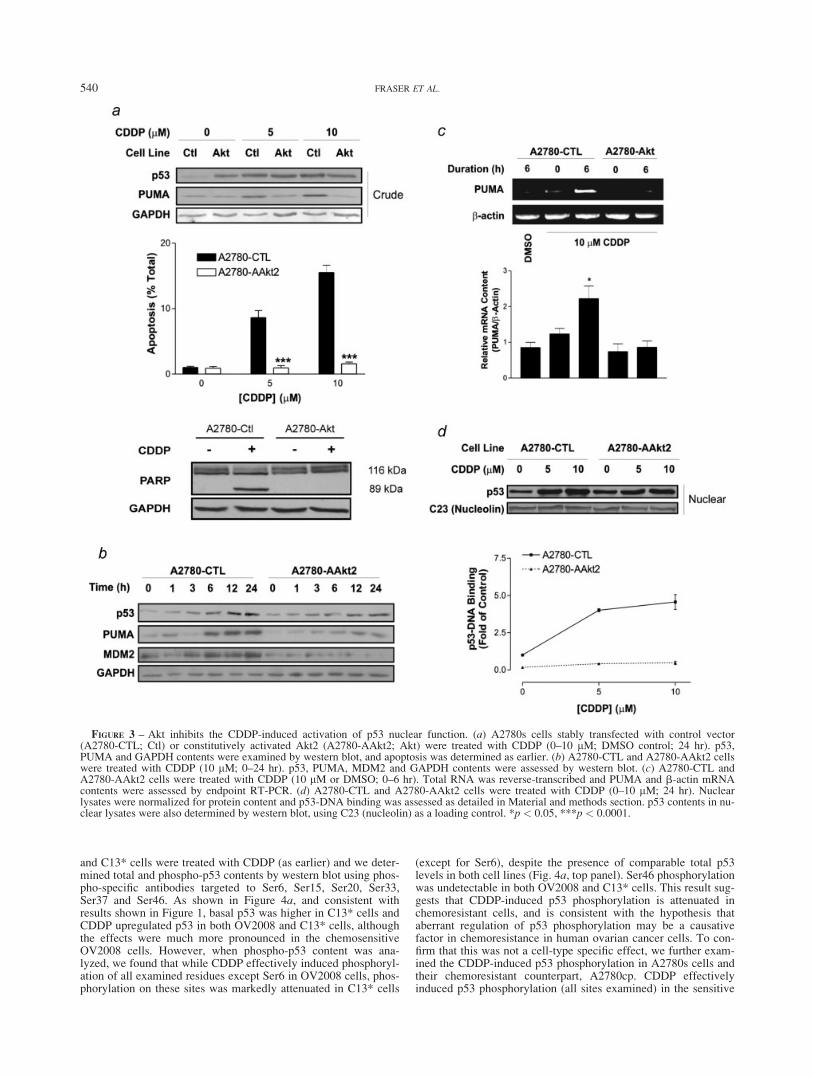
and C13* cells were treated with CDDP (as earlier) and we deter-mined total and phospho-p53 contents by western blot using phos-pho-specific antibodies targeted to Ser6, Ser15, Ser20, Ser33,Ser37 and Ser46. As shown in Figure 4a, and consistent withresults shown in Figure 1, basal p53 was higher in C13* cells andCDDP upregulated p53 in both OV2008 and C13* cells, althoughthe effects were much more pronounced in the chemosensitiveOV2008 cells. However, when phospho-p53 content was ana-lyzed, we found that while CDDP effectively induced phosphoryl-ation of all examined residues except Ser6 in OV2008 cells, phos-phorylation on these sites was markedly attenuated in C13* cells
(except for Ser6), despite the presence of comparable total p53levels in both cell lines (Fig. 4a, top panel). Ser46 phosphorylationwas undetectable in both OV2008 and C13* cells. This result sug-gests that CDDP-induced p53 phosphorylation is attenuated inchemoresistant cells, and is consistent with the hypothesis thataberrant regulation of p53 phosphorylation may be a causativefactor in chemoresistance in human ovarian cancer cells. To con-firm that this was not a cell-type specific effect, we further exam-ined the CDDP-induced p53 phosphorylation in A2780s cells andtheir chemoresistant counterpart, A2780cp. CDDP effectivelyinduced p53 phosphorylation (all sites examined) in the sensitive
FIGURE 3 – Akt inhibits the CDDP-induced activation of p53 nuclear function. (a) A2780s cells stably transfected with control vector(A2780-CTL; Ctl) or constitutively activated Akt2 (A2780-AAkt2; Akt) were treated with CDDP (0–10 lM; DMSO control; 24 hr). p53,PUMA and GAPDH contents were examined by western blot, and apoptosis was determined as earlier. (b) A2780-CTL and A2780-AAkt2 cellswere treated with CDDP (10 lM; 0–24 hr). p53, PUMA, MDM2 and GAPDH contents were assessed by western blot. (c) A2780-CTL andA2780-AAkt2 cells were treated with CDDP (10 lM or DMSO; 0–6 hr). Total RNA was reverse-transcribed and PUMA and b-actin mRNAcontents were assessed by endpoint RT-PCR. (d) A2780-CTL and A2780-AAkt2 cells were treated with CDDP (0–10 lM; 24 hr). Nuclearlysates were normalized for protein content and p53-DNA binding was assessed as detailed in Material and methods section. p53 contents in nu-clear lysates were also determined by western blot, using C23 (nucleolin) as a loading control. *p < 0.05, ***p < 0.0001.
540 FRASER ET AL.

A2780s cells. By contrast, while Ser6, Ser33 and Ser37 werephosphorylated in A2780cp cells to similar extent as in the chemo-sensitive A2780s cells, phosphorylation of Ser15 and Ser20 wasmarkedly attenuated in the resistant cells, despite high total p53content (Fig. 4, bottom panel). Ser46 phosphorylation wasslightly, although not completely, attenuated in the chemoresistantcells. Taken together, these results demonstrate that p53 phospho-rylation, particularly of Ser15 and Ser20, is markedly attenuatedin chemoresistant ovarian cancer cells, and support the hypothesisthat CDDP resistance may, in part, be mediated through aberrantregulation of phosphorylation at these sites.
Akt attenuates CDDP-induced p53 phosphorylation
While Akt activation can attenuate p53-mediated gene expres-sion and promote a chemoresistant phenotype, whether p53 phos-
phorylation is affected is unknown. To test this possibility, wecultured A2780-CTL and A2780-AAkt2 cells in the absence andpresence of CDDP (as earlier) and phospho- and total p53 con-tents were analyzed by western blot. As shown in Figure 5a,while CDDP upregulated total and phospho-p53 contents (Ser6,Ser15, Ser20, Ser33 and Ser37) in a concentration-dependentmanner in the control A2780-CTL cells, phosphorylation at thesesites (except Ser6) was markedly attenuated in the A2780-AAkt2cells. This result demonstrates that Akt activation inhibits p53phosphorylation, suggesting a novel mechanism of Akt-mediatedcell survival. Furthermore, these findings are consistent with thehypothesis that downregulation of p53 phosphorylation may, inpart, contribute to Akt-mediated chemoresistance in ovarian can-cer cells.
To further examine the role of Akt on p53 phosphorylation, che-moresistant, p53 wild-C13* cells were infected with adenoviralDN-Akt (MOI 5 0–80, 48 hr; LacZ control). This adenovirus enc-odes a kinase-dead mutant form of Akt (K179A), which also can-not be activated by phosphorylation (T308A, S473A). We previ-ously demonstrated that expression of DN-Akt sensitizes chemo-resistant cells to CDDP-induced apoptosis in a p53-dependentmanner.3 DN-Akt expression was confirmed by western blotagainst the HA epitope (Fig. 5b). Expression of DN-Akt upregu-lated total p53, and this p53 was phosphorylated on Ser15, Ser20and Ser37. Moreover, while DN-Akt upregulated total p53 by 1.5-fold (1.5 6 0.02 at MOI 5 80 relative to MOI 5 0), phospho-p53(Ser15) was upregulated by nearly 10-fold (9.8 6 1.2 at MOI 580 relative to MOI 5 0), which represents a significant increase inthe ratio of phospho/total p53 of nearly 7-fold (6.8 6 0.84 at MOI5 80 relative to MOI 5 0, p < 0.0001).
To further confirm the effects of DN-Akt on phospho-p53 con-tent, chemoresistant A2780cp cells were infected with DN-Akt(MOI 5 0, 80; as earlier) and total and phospho-p53 (Ser15) con-tents were evaluated by western blot. Since Akt can induce anMDM2-dependent downregulation of p53,29–32 and since mutantp53 does not transactivate the MDM2 gene, resulting in reducedMDM2 protein content,4 we surmised that DN-Akt should notincrease total p53 in p53 mutant cells, thus providing an ideal sys-tem to study the effects of Akt inhibition on p53 phosphorylation.Expression of DN-Akt was confirmed by detection of the HA epi-tope. As expected, DN-Akt did not upregulate p53 in these cells,but increased phosphorylation of Ser15 was observed in cellsinfected with DN-Akt, relative to the LacZ-infected control cells(Fig. 5c, upper panel). Indeed, DN-Akt increased the ratio of phos-pho/total p53 by greater than 2.5-fold, relative to the LacZ-infected cells (Fig. 5c, lower panel, p < 0.01), demonstratingthat suppression of endogenous Akt activity upregulates p53phosphorylation.
Taken together, these results suggest that Akt inhibits p53 phos-phorylation and offer a novel paradigm for Akt-mediated chemo-resistance and cell survival.
Phosphorylation of Ser15 and Ser20 is required for optimalCDDP-induced, p53-dependent apoptosis
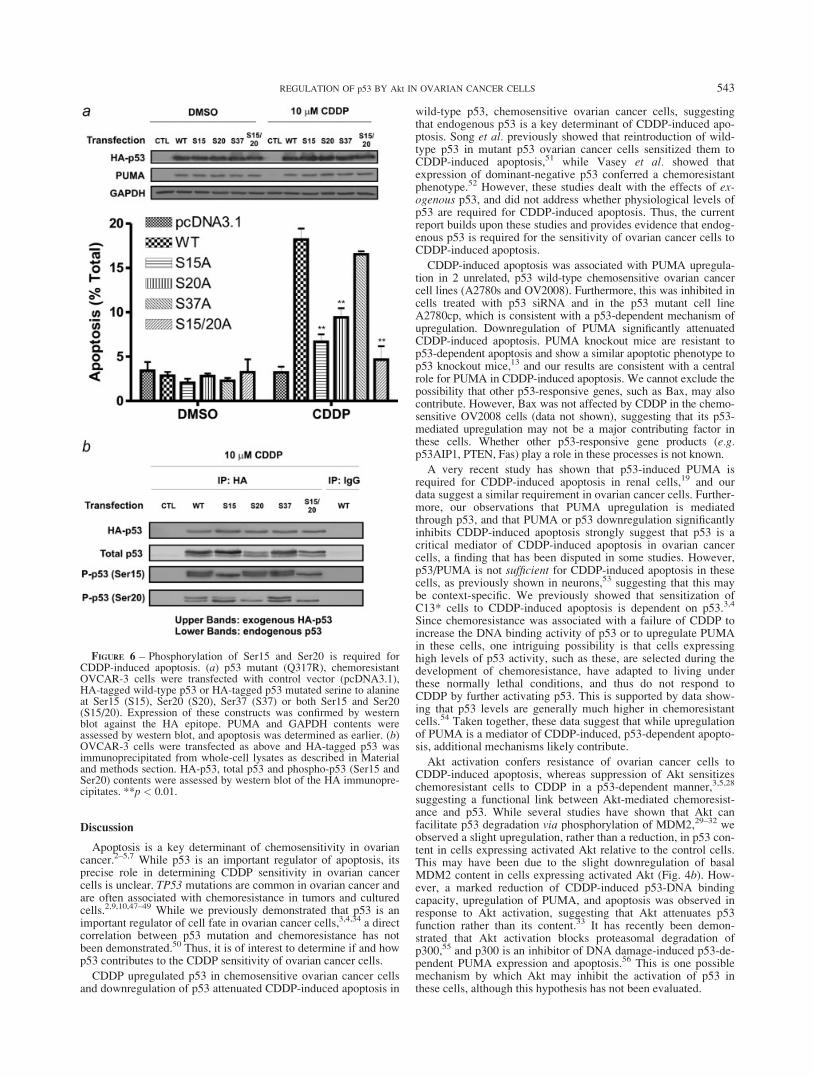
To test whether p53 phosphorylation is a determinant of CDDP-induced apoptosis, we mutated p53 such that it could not undergophosphorylation (Ser to Ala) at key sites (Ser15, Ser20 or Ser37).We then transiently transfected CDDP-resistant, p53 mutant(Q317R)45 OVCAR-3 cells with HA-tagged WT, S15A, S20A,S37A-p53 or a combined S15A/S20A-p53 (pcDNA3.1 empty vec-tor control; 24 hr) and then treated the cells with CDDP (10 lM;24 hr). OVCAR-3 cells were chosen because a previous reportsuggested that reconstitution of wild-type p53 was sufficient tosensitize these cells to CDDP,46 whereas we have demonstratedthat this is not the case in other p53 mutant ovarian cancer celllines, such as A2780cp.3,4 All of the p53 constructs wereexpressed to equivalent levels in the absence of CDDP, as mea-sured by western blot against the HA epitope, and this was notaffected by treatment with CDDP (Fig. 6a, top panel). CDDP
FIGURE 4 – CDDP-induced p53 phosphorylation is attenuated inchemoresistant cells. (a) OV2008 and C13* or (b) A2780s andA2780cp cells were treated with CDDP (0–10 lM; 24 hr) and totaland phospho-p53 contents were assessed by western blot using anti-bodies directed against total p53 (DO-1) or against specific phospho-rylated residues on p53, as indicated.
541REGULATION OF p53 BY Akt IN OVARIAN CANCER CELLS
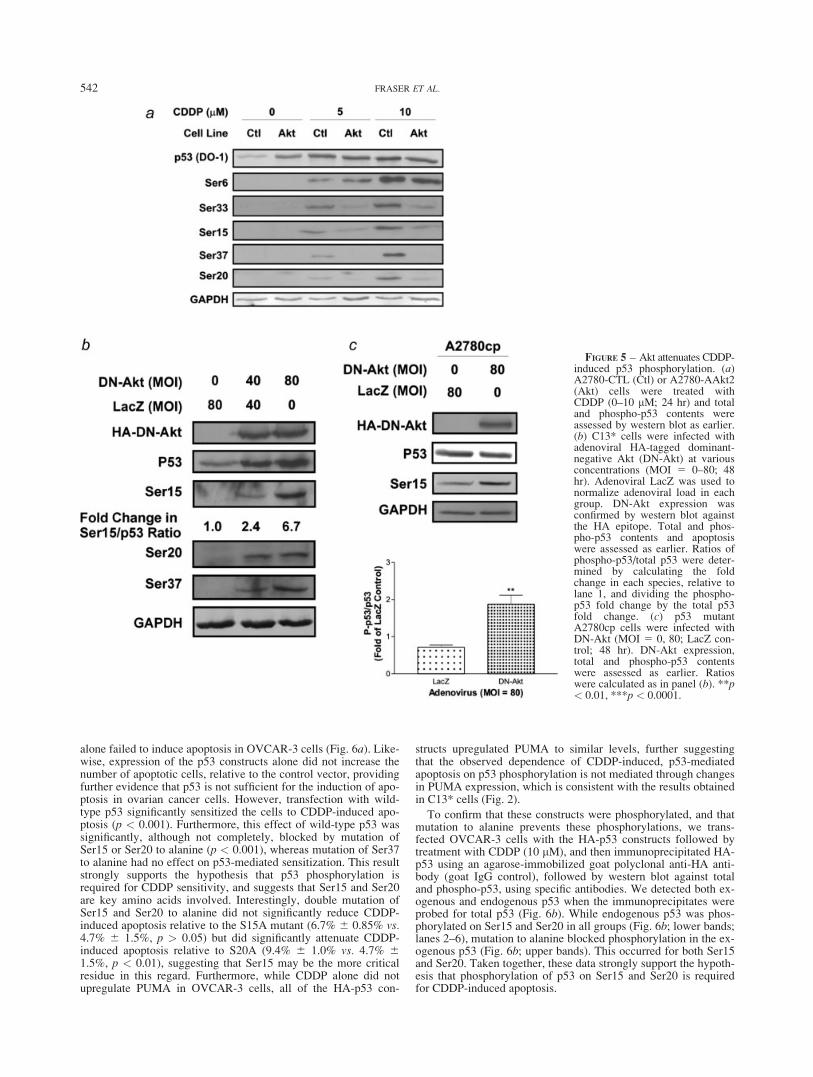
alone failed to induce apoptosis in OVCAR-3 cells (Fig. 6a). Like-wise, expression of the p53 constructs alone did not increase thenumber of apoptotic cells, relative to the control vector, providingfurther evidence that p53 is not sufficient for the induction of apo-ptosis in ovarian cancer cells. However, transfection with wild-type p53 significantly sensitized the cells to CDDP-induced apo-ptosis (p < 0.001). Furthermore, this effect of wild-type p53 wassignificantly, although not completely, blocked by mutation ofSer15 or Ser20 to alanine (p < 0.001), whereas mutation of Ser37to alanine had no effect on p53-mediated sensitization. This resultstrongly supports the hypothesis that p53 phosphorylation isrequired for CDDP sensitivity, and suggests that Ser15 and Ser20are key amino acids involved. Interestingly, double mutation ofSer15 and Ser20 to alanine did not significantly reduce CDDP-induced apoptosis relative to the S15A mutant (6.7% 6 0.85% vs.4.7% 6 1.5%, p > 0.05) but did significantly attenuate CDDP-induced apoptosis relative to S20A (9.4% 6 1.0% vs. 4.7% 61.5%, p < 0.01), suggesting that Ser15 may be the more criticalresidue in this regard. Furthermore, while CDDP alone did notupregulate PUMA in OVCAR-3 cells, all of the HA-p53 con-
structs upregulated PUMA to similar levels, further suggestingthat the observed dependence of CDDP-induced, p53-mediatedapoptosis on p53 phosphorylation is not mediated through changesin PUMA expression, which is consistent with the results obtainedin C13* cells (Fig. 2).
To confirm that these constructs were phosphorylated, and thatmutation to alanine prevents these phosphorylations, we trans-fected OVCAR-3 cells with the HA-p53 constructs followed bytreatment with CDDP (10 lM), and then immunoprecipitated HA-p53 using an agarose-immobilized goat polyclonal anti-HA anti-body (goat IgG control), followed by western blot against totaland phospho-p53, using specific antibodies. We detected both ex-ogenous and endogenous p53 when the immunoprecipitates wereprobed for total p53 (Fig. 6b). While endogenous p53 was phos-phorylated on Ser15 and Ser20 in all groups (Fig. 6b; lower bands;lanes 2–6), mutation to alanine blocked phosphorylation in the ex-ogenous p53 (Fig. 6b; upper bands). This occurred for both Ser15and Ser20. Taken together, these data strongly support the hypoth-esis that phosphorylation of p53 on Ser15 and Ser20 is requiredfor CDDP-induced apoptosis.
FIGURE 5 – Akt attenuates CDDP-induced p53 phosphorylation. (a)A2780-CTL (Ctl) or A2780-AAkt2(Akt) cells were treated withCDDP (0–10 lM; 24 hr) and totaland phospho-p53 contents wereassessed by western blot as earlier.(b) C13* cells were infected withadenoviral HA-tagged dominant-negative Akt (DN-Akt) at variousconcentrations (MOI 5 0–80; 48hr). Adenoviral LacZ was used tonormalize adenoviral load in eachgroup. DN-Akt expression wasconfirmed by western blot againstthe HA epitope. Total and phos-pho-p53 contents and apoptosiswere assessed as earlier. Ratios ofphospho-p53/total p53 were deter-mined by calculating the foldchange in each species, relative tolane 1, and dividing the phospho-p53 fold change by the total p53fold change. (c) p53 mutantA2780cp cells were infected withDN-Akt (MOI 5 0, 80; LacZ con-trol; 48 hr). DN-Akt expression,total and phospho-p53 contentswere assessed as earlier. Ratioswere calculated as in panel (b). **p< 0.01, ***p < 0.0001.
542 FRASER ET AL.
Discussion
Apoptosis is a key determinant of chemosensitivity in ovariancancer.2–5,7 While p53 is an important regulator of apoptosis, itsprecise role in determining CDDP sensitivity in ovarian cancercells is unclear. TP53 mutations are common in ovarian cancer andare often associated with chemoresistance in tumors and culturedcells.2,9,10,47–49 While we previously demonstrated that p53 is animportant regulator of cell fate in ovarian cancer cells,3,4,34 a directcorrelation between p53 mutation and chemoresistance has notbeen demonstrated.50 Thus, it is of interest to determine if and howp53 contributes to the CDDP sensitivity of ovarian cancer cells.
CDDP upregulated p53 in chemosensitive ovarian cancer cellsand downregulation of p53 attenuated CDDP-induced apoptosis in
wild-type p53, chemosensitive ovarian cancer cells, suggestingthat endogenous p53 is a key determinant of CDDP-induced apo-ptosis. Song et al. previously showed that reintroduction of wild-type p53 in mutant p53 ovarian cancer cells sensitized them toCDDP-induced apoptosis,51 while Vasey et al. showed thatexpression of dominant-negative p53 conferred a chemoresistantphenotype.52 However, these studies dealt with the effects of ex-ogenous p53, and did not address whether physiological levels ofp53 are required for CDDP-induced apoptosis. Thus, the currentreport builds upon these studies and provides evidence that endog-enous p53 is required for the sensitivity of ovarian cancer cells toCDDP-induced apoptosis.
CDDP-induced apoptosis was associated with PUMA upregula-tion in 2 unrelated, p53 wild-type chemosensitive ovarian cancercell lines (A2780s and OV2008). Furthermore, this was inhibited incells treated with p53 siRNA and in the p53 mutant cell lineA2780cp, which is consistent with a p53-dependent mechanism ofupregulation. Downregulation of PUMA significantly attenuatedCDDP-induced apoptosis. PUMA knockout mice are resistant top53-dependent apoptosis and show a similar apoptotic phenotype top53 knockout mice,13 and our results are consistent with a centralrole for PUMA in CDDP-induced apoptosis. We cannot exclude thepossibility that other p53-responsive genes, such as Bax, may alsocontribute. However, Bax was not affected by CDDP in the chemo-sensitive OV2008 cells (data not shown), suggesting that its p53-mediated upregulation may not be a major contributing factor inthese cells. Whether other p53-responsive gene products (e.g.p53AIP1, PTEN, Fas) play a role in these processes is not known.
A very recent study has shown that p53-induced PUMA isrequired for CDDP-induced apoptosis in renal cells,19 and ourdata suggest a similar requirement in ovarian cancer cells. Further-more, our observations that PUMA upregulation is mediatedthrough p53, and that PUMA or p53 downregulation significantlyinhibits CDDP-induced apoptosis strongly suggest that p53 is acritical mediator of CDDP-induced apoptosis in ovarian cancercells, a finding that has been disputed in some studies. However,p53/PUMA is not sufficient for CDDP-induced apoptosis in thesecells, as previously shown in neurons,53 suggesting that this maybe context-specific. We previously showed that sensitization ofC13* cells to CDDP-induced apoptosis is dependent on p53.3,4
Since chemoresistance was associated with a failure of CDDP toincrease the DNA binding activity of p53 or to upregulate PUMAin these cells, one intriguing possibility is that cells expressinghigh levels of p53 activity, such as these, are selected during thedevelopment of chemoresistance, have adapted to living underthese normally lethal conditions, and thus do not respond toCDDP by further activating p53. This is supported by data show-ing that p53 levels are generally much higher in chemoresistantcells.54 Taken together, these data suggest that while upregulationof PUMA is a mediator of CDDP-induced, p53-dependent apopto-sis, additional mechanisms likely contribute.
Akt activation confers resistance of ovarian cancer cells toCDDP-induced apoptosis, whereas suppression of Akt sensitizeschemoresistant cells to CDDP in a p53-dependent manner,3,5,28
suggesting a functional link between Akt-mediated chemoresist-ance and p53. While several studies have shown that Akt canfacilitate p53 degradation via phosphorylation of MDM2,29–32 weobserved a slight upregulation, rather than a reduction, in p53 con-tent in cells expressing activated Akt relative to the control cells.This may have been due to the slight downregulation of basalMDM2 content in cells expressing activated Akt (Fig. 4b). How-ever, a marked reduction of CDDP-induced p53-DNA bindingcapacity, upregulation of PUMA, and apoptosis was observed inresponse to Akt activation, suggesting that Akt attenuates p53function rather than its content.33 It has recently been demon-strated that Akt activation blocks proteasomal degradation ofp300,55 and p300 is an inhibitor of DNA damage-induced p53-de-pendent PUMA expression and apoptosis.56 This is one possiblemechanism by which Akt may inhibit the activation of p53 inthese cells, although this hypothesis has not been evaluated.
FIGURE 6 – Phosphorylation of Ser15 and Ser20 is required forCDDP-induced apoptosis. (a) p53 mutant (Q317R), chemoresistantOVCAR-3 cells were transfected with control vector (pcDNA3.1),HA-tagged wild-type p53 or HA-tagged p53 mutated serine to alanineat Ser15 (S15), Ser20 (S20), Ser37 (S37) or both Ser15 and Ser20(S15/20). Expression of these constructs was confirmed by westernblot against the HA epitope. PUMA and GAPDH contents wereassessed by western blot, and apoptosis was determined as earlier. (b)OVCAR-3 cells were transfected as above and HA-tagged p53 wasimmunoprecipitated from whole-cell lysates as described in Materialand methods section. HA-p53, total p53 and phospho-p53 (Ser15 andSer20) contents were assessed by western blot of the HA immunopre-cipitates. **p < 0.01.
543REGULATION OF p53 BY Akt IN OVARIAN CANCER CELLS

p53 is phosphorylated in response to various cell stresses, andp53 phosphorylation protects p53 from MDM2-mediated ubiquiti-nation and proteasomal degradation. Phosphorylation of p53 onSer15 and/or Ser20 is required for p53-induced apoptosis,21
although this does not hold true under all circumstances.44 CDDPinduced phosphorylation on numerous p53 residues, includingSer15 and Ser20, in 2 chemosensitive ovarian cancer cell lines,but not in their respective chemoresistant variant cell lines, sug-gesting that altered p53 phosphorylation may contribute to thechemoresistance of these cells. Moreover, these sites are requiredfor CDDP-induced apoptosis. To our knowledge, this is the firstdemonstration that p53 phosphorylation is altered in chemoresist-ant cells and is required for CDDP-induced apoptosis, and pro-vides compelling evidence that aberrant regulation of phosphoryl-ation may be a critical determinant of the sensitivity of humanovarian cancer cells to CDDP-induced apoptosis. We cannotexclude the possibility that phosphorylation of sites other thanSer15 and Ser20 (including those in the p53 C-terminal) may alsobe implicated in this process, although it appears that Ser37 doesnot play a major role in CDDP-induced apoptosis. Indeed, Ser46is an important phosphorylation site for the regulation of p53-induced gene expression and apoptosis.57 However, Ser46 phos-phorylation was undetectable in OV2008 or C13* cells exposed toCDDP. Moreover, while Ser46 phosphorylation was detected inA2780s cells, it was also observed in the chemoresistant A2780cpcells. These data suggest that Ser46 may not be required forCDDP-induced, p53-dependent apoptosis in ovarian cancer cells.
The mechanism by which Ser15/Ser20 phosphorylation altersthe apoptotic capacity of p53 remains unclear, though alterationsin the phosphorylation of these sites did not affect p53-inducedPUMA upregulation. Unger et al. showed similar results withrespect to Bax activation.21 However, the possibility that phospho-rylation of these sites may alter the expression of additional p53-responsive genes, thereby promoting p53-mediated apoptosis,requires further investigation.
While our results suggest that Ser15 phosphorylation may not berequired for p53-dependent transactivation in ovarian cancer cells,our results agree with the observation that Ser15 is not required formaintenance of p53 steady-state levels.58 We recently demonstratedthat CDDP induces the direct targeting of p53 to the mitochondriain chemosensitive cells, but not in their resistant counterparts,59 andthat p53 targeted to the mitochondria was able to induce apoptosismore rapidly than nuclear-targeted p53. Thus, phosphorylation maybe implicated in the mitochondrial translocation of p53. Whilerecent data suggest that phosphorylation is not the determining fac-tor for the alternate translocation of p53 to the nucleus or the mito-chondria,60 whether phosphorylation is required for mitochondrialtranslocation has not been evaluated. p53 also directly activatesBax, an event that requires its PUMA-dependent liberation from ap53-Bcl-XL complex.20 Thus, it is possible that this processrequires phosphorylation of p53 on 1 or more residues. Thesehypotheses are currently under investigation in our laboratory.
Akt activation reduced the CDDP-induced phosphorylation ofp53 on several residues while dominant-negative Akt upregulatedp53 and induced p53 phosphorylation independently of changes incontent. Interestingly, Yamaguchi et al. did not observe a change inp53 phosphorylation in response to Akt activation.33 However, theyused a [32P]-orthophosphate labeling assay, which may not be sensi-tive enough to detect changes in phosphorylation at individualamino acids in polyphosphorylated molecules such as p53. In con-trast, our western blot assay permits an analysis of phosphorylationstatus at specific residues. While this manuscript was in preparation,Limesand et al. reported that activated Akt1 in murine salivary aci-nar cells attenuates p53 phosphorylation at Ser18 (equivalent tohuman Ser15).61 We have significantly extended these findings bydemonstrating that activated Akt inhibits the phosphorylation ofnumerous p53 residue (in addition to Ser15), and, importantly, thatendogenous Akt inhibits p53 phosphorylation.
The mechanism by which Akt regulates p53 phosphorylation isunclear. CDDP-induced phosphorylation of Ser6 was not affected
by Akt activation, suggesting that the actions of Akt in this respectmay be distal to DNA damage itself. Moreover, the phosphoryla-tions attenuated by Akt activation are regulated by diverse kinases,suggesting that Akt likely acts at a downstream step. One candi-date target molecule is ATR, which is activated by CDDP, andwhich directly and indirectly facilitates the phosphorylation ofp53 on numerous residues, including Ser15, Ser20 and Ser37.Intriguingly, ATR contains a putative Akt phosphorylation site(RRRLSS436). As such, Akt may phosphorylate ATR, therebyattenuating its activation, and reducing ATR-dependent p53 phos-phorylation. We are currently evaluating this hypothesis in ourlaboratory.
The contribution of p53 to chemosensitivity remains unclear,although several studies2,9,10,49,62 have demonstrated a strong corre-lation between wild-type TP53 and sensitivity to CDDP and pro-longed survival in human ovarian cancer. In addition, p53 isrequired for CDDP-induced apoptosis in ovarian cancer cell cultureand xenografts.3,11,51 However, other studies fail to show a correla-tion between wild-type p53 and CDDP sensitivity.63–65 Many ofthese studies have relied upon nonspecific ablation of p53 function(e.g. expression of HPV-E6) and/or did not specifically distinguishbetween CDDP-induced apoptosis and necrosis. This is of concernsince apoptosis is a primary effect of CDDP-centered chemotherapyin ovarian cancer patients and in xenograft models of human ovar-ian cancer.2,48 Moreover, because specific TP53 mutations mayaffect p53 function differently, a detailed analysis of the relation-ship between particular mutations and chemoresistance is war-ranted, and may shed light upon the role of p53 in chemoresistance.
The current study suggests that CDDP upregulates PUMA viaactivation of p53, thereby facilitating apoptosis. Furthermore,CDDP induces p53 phosphorylation on several residues, includingSer15 and Ser20, which are not phosphorylated in chemoresistantcells and are required for efficient CDDP-induced apoptosis. Akteffectively blocks these processes, thereby conferring resistance toCDDP-induced apoptosis. Either mutation of the TP53 gene,which inhibits the upregulation of PUMA, or failure to phospho-rylate wild-type p53, or both, results in a chemoresistant pheno-type; both events are required for the full apoptotic response toCDDP. This is consistent with the observation that wild-typeTP53 status is not always correlated with chemosensitivity. Inaddition, while other p53-independent cellular events, includingthe downregulation of Xiap, may be required for CDDP-inducedapoptosis, our evidence suggests that effective activation andphosphorylation of p53 is essential. Thus, it will be of interest tostudy the effects of chemotherapy on total and phospho-p53 andPUMA contents in human ovarian tumors with respect to treat-ment outcomes. Moreover, since Akt attenuates both processes, itis important to study the relationship between activation/overex-pression of Akt in ovarian tumors and sensitivity to CDDP.
In summary, we have demonstrated that p53 is essential forCDDP-induced apoptosis in human ovarian cancer cells, and thatthis is mediated, at least in part, through the upregulation ofPUMA and the phosphorylation of p53 on Ser15 and Ser20. Fur-thermore, p53 phosphorylation is attenuated in chemoresistantcells, suggesting a novel mechanism of chemoresistance. Activa-tion of Akt confers resistance by blocking p53-mediated transacti-vation and p53 phosphorylation. A more thorough understandingof the molecular mechanisms underling chemoresistance in humanovarian cancer may ultimately improve treatment outcomes forthis disease.
Acknowledgements
The authors thank Dr. Mohammed Abedini for the immunopre-cipitation protocols. B.K.T. is the recipient of a grant from theNational Cancer Institute of Canada (013335), with funds from theCanadian Cancer Society, and from the Canadian Institutes ofHealth Research (MOP-15691). M.F. was the recipient of aCanada Graduate Scholarship Doctoral Research Award from theCanadian Institutes of Health Research.
544 FRASER ET AL.

References
1. Mansouri A, Ridgway LD, Korapati AL, Zhang Q, Tian L, Wang Y,Siddik ZH, Mills GB, Claret FX. Sustained activation of JNK/p38MAPK pathways in response to cisplatin leads to Fas ligand inductionand cell death in ovarian carcinoma cells. J Biol Chem 2003;278:19245–56.
2. Sato S, Kigawa J, Minagawa Y, Okada M, Shimada M, Takahashi M,Kamazawa S, Terakawa N. Chemosensitivity and p53-dependent apo-ptosis in epithelial ovarian carcinoma. Cancer 1999;86:1307–13.
3. Fraser M, Leung BM, Yan X, Dan HC, Cheng JQ, Tsang BK. p53 is adeterminant of X-linked inhibitor of apoptosis protein/Akt-mediatedchemoresistance in human ovarian cancer cells. Cancer Res 2003;63:7081–8.
4. Sasaki H, Sheng Y, Kotsuji F, Tsang BK. Down-regulation of X-linked inhibitor of apoptosis protein induces apoptosis in chemoresist-ant human ovarian cancer cells. Cancer Res 2000;60:5659–66.
5. Dan HC, Sun M, Kaneko S, Feldman RI, Nicosia SV, Wang HG,Tsang BK, Cheng JQ. Akt phosphorylation and stabilization of X-linked inhibitor of apoptosis protein (XIAP). J Biol Chem 2004;279:5405–12.
6. Abedini MR, Qiu Q, Yan X, Tsang BK. Possible role of FLICE-likeinhibitory protein (FLIP) in chemoresistant ovarian cancer cells invitro. Oncogene 2004;23:6997–7004.
7. Asselin E, Mills GB, Tsang BK. XIAP regulates Akt activity andcaspase-3-dependent cleavage during cisplatin-induced apoptosis inhuman ovarian epithelial cancer cells. Cancer Res 2001;61:1862–8.
8. Li J, Feng Q, Kim JM, Schneiderman D, Liston P, Li M, VanderhydenB, Faught W, Fung MF, Senterman M, Korneluk RG, Tsang BK.Human ovarian cancer and cisplatin resistance: possible role of inhibi-tor of apoptosis proteins. Endocrinology 2001;142:370–80.
9. Perego P, Giarola M, Righetti SC, Supino R, Caserini C, Delia D,Pierotti MA, Miyashita T, Reed JC, Zunino F. Association betweencisplatin resistance and mutation of p53 gene and reduced bax expres-sion in ovarian carcinoma cell systems. Cancer Res 1996;56:556–62.
10. Righetti SC, Della Torre G, Pilotti S, Menard S, Ottone F, ColnaghiMI, Pierotti MA, Lavarino C, Cornarotti M, Oriana S, Bohm S, Bres-ciani GL, et al. A comparative study of p53 gene mutations, proteinaccumulation, and response to cisplatin-based chemotherapy inadvanced ovarian carcinoma. Cancer Res 1996;56:689–93.
11. Song K, Cowan KH, Sinha BK. In vivo studies of adenovirus-medi-ated p53 gene therapy for cis-platinum-resistant human ovarian tumorxenografts. Oncol Res 1999;11:153–9.
12. Miyashita T, Krajewski S, Krajewska M, Wang HG, Lin HK, Lieber-mann DA, Hoffman B, Reed JC. Tumor suppressor p53 is a regulatorof bcl-2 and bax gene expression in vitro and in vivo. Oncogene1994;9:1799–805.
13. Villunger A, Michalak EM, Coultas L, Mullauer F, Bock G, Ausser-lechner MJ, Adams JM, Strasser A. p53- and drug-induced apoptoticresponses mediated by BH3-only proteins puma and noxa. Science2003;302:1036–8.
14. Wong HK, Fricker M, Wyttenbach A, Villunger A, Michalak EM,Strasser A, Tolkovsky AM. Mutually exclusive subsets of BH3-onlyproteins are activated by the p53 and c-Jun N-terminal kinase/c-Junsignaling pathways during cortical neuron apoptosis induced by arsen-ite. Mol Cell Biol 2005;25:8732–47.
15. Hoffman WH, Biade S, Zilfou JT, Chen J, Murphy M. Transcriptionalrepression of the anti-apoptotic survivin gene by wild type p53. J BiolChem 2002;277:3247–57.
16. Mirza A, McGuirk M, Hockenberry TN, Wu Q, Ashar H, Black S,Wen SF, Wang L, Kirschmeier P, Bishop WR, Nielsen LL, PickettCB, et al. Human survivin is negatively regulated by wild-type p53and participates in p53-dependent apoptotic pathway. Oncogene2002;21:2613–22.
17. Erster S, Mihara M, Kim RH, Petrenko O, Moll UM. In vivo mito-chondrial p53 translocation triggers a rapid first wave of cell death inresponse to dna damage that can precede p53 target gene activation.Mol Cell Biol 2004;24:6728–41.
18. Mihara M, Erster S, Zaika A, Petrenko O, Chittenden T, Pancoska P,Moll UM. p53 has a direct apoptogenic role at the mitochondria. MolCell 2003;11:577–90.
19. Jiang M, Wei Q, Wang J, Du Q, Yu J, Zhang L, Dong Z. Regulationof PUMA-a by p53 in cisplatin-induced renal cell apoptosis. Onco-gene 2006;25:4056–66.
20. Chipuk JE, Bouchier-Hayes L, Kuwana T, Newmeyer DD, Green DR.PUMA couples the nuclear and cytoplasmic proapoptotic function ofp53. Science 2005;309:1732–5.
21. Unger T, Sionov RV, Moallem E, Yee CL, Howley PM, Oren M,Haupt Y. Mutations in serines 15 and 20 of human p53 impair its apo-ptotic activity. Oncogene 1999;18:3205–12.
22. Bulavin DV, Saito S, Hollander MC, Sakaguchi K, Anderson CW,Appella E, Fornace AJ, Jr. Phosphorylation of human p53 by p38kinase coordinates N-terminal phosphorylation and apoptosis inresponse to UV radiation. EMBO J 1999;18:6845–54.
23. Shono T, Tofilon PJ, Schaefer TS, Parikh D, Liu TJ, Lang FF. Apo-ptosis induced by adenovirus-mediated p53 gene transfer in humanglioma correlates with site-specific phosphorylation. Cancer Res2002;62:1069–76.
24. Bellacosa A, de Feo D, Godwin AK, Bell DW, Cheng JQ, AltomareDA, Wan M, Dubeau L, Scambia G, Masciullo V, Ferrandina G, Ben-edetti Panici P, et al. Molecular alterations of the AKT2 oncogene inovarian and breast carcinomas. Int J Cancer 1995;64:280–5.
25. Cheng JQ, Godwin AK, Bellacosa A, Taguchi T, Franke TF, HamiltonTC, Tsichlis PN, Testa JR. AKT2, a putative oncogene encoding a mem-ber of a subfamily of protein-serine/threonine kinases, is amplified inhuman ovarian carcinomas. Proc Natl Acad Sci USA 1992;89:9267–71.
26. Sun M, Wang G, Paciga JE, Feldman RI, Yuan ZQ, Ma XL, ShelleySA, Jove R, Tsichlis PN, Nicosia SV, Cheng JQ. AKT1/PKBa kinaseis frequently elevated in human cancers and its constitutive activationis required for oncogenic transformation in NIH3T3 cells. Am JPathol 2001;159:431–7.
27. Yuan ZQ, Sun M, Feldman RI, Wang G, Ma X, Jiang C, Coppola D,Nicosia SV, Cheng JQ. Frequent activation of AKT2 and induction ofapoptosis by inhibition of phosphoinositide-3-OH kinase/Akt pathwayin human ovarian cancer. Oncogene 2000;19:2324–30.
28. Yuan ZQ, Feldman RI, Sussman GE, Coppola D, Nicosia SV, ChengJQ. AKT2 inhibition of cisplatin-induced JNK/p38 and Bax activationby phosphorylation of ASK1: implication of AKT2 in chemoresist-ance. J Biol Chem 2003;278:23432–40.
29. Gottlieb TM, Leal JF, Seger R, Taya Y, Oren M. Cross-talk betweenAkt, p53 and Mdm2: possible implications for the regulation of apo-ptosis. Oncogene 2002;21:1299–303.
30. Mayo LD, Donner DB. A phosphatidylinositol 3-kinase/Akt pathwaypromotes translocation of Mdm2 from the cytoplasm to the nucleus.Proc Natl Acad Sci USA 2001;98:11598–603.
31. Ogawara Y, Kishishita S, Obata T, Isazawa Y, Suzuki T, Tanaka K,Masuyama N, Gotoh Y. Akt enhances Mdm2-mediated ubiquitinationand degradation of p53. J Biol Chem 2002;277:21843–50.
32. Zhou BP, Liao Y, Xia W, Zou Y, Spohn B, Hung MC. HER-2/neuinduces p53 ubiquitination via Akt-mediated MDM2 phosphorylation.Nat Cell Biol 2001;3:973–82.
33. Yamaguchi A, Tamatani M, Matsuzaki H, Namikawa K, Kiyama H,Vitek MP, Mitsuda N, Tohyama M. Akt activation protects hippocam-pal neurons from apoptosis by inhibiting transcriptional activity ofp53. J Biol Chem 2001;276:5256–64.
34. Fraser M, Chan SL, Chan SS, Fiscus RR, Tsang BK. Regulation ofp53 and suppression of apoptosis by the soluble guanylyl cyclase/cGMP pathway in human ovarian cancer cells. Oncogene 2006;25:2203–12.
35. Sasaki H, Kotsuji F, Tsang BK. Caspase 3-mediated focal adhesionkinase processing in human ovarian cancer cells: possible regulationby X-linked inhibitor of apoptosis protein. Gynecol Oncol 2002;85:339–50.
36. Wang Y, Chan S, Tsang BK. Involvement of inhibitory nuclear factor-jB (NFjB)-independent NFjB activation in the gonadotropic regula-tion of X-linked inhibitor of apoptosis expression during ovarian follic-ular development in vitro. Endocrinology 2002;143:2732–40.
37. Tewari M, Quan LT, O’Rourke K, Desnoyers S, Zeng Z, Beidler DR,Poirier GG, Salvesen GS, Dixit VM. Yama/CPP32b, a mammalianhomolog of CED-3, is a CrmA-inhibitable protease that cleaves thedeath substrate poly(ADP-ribose) polymerase. Cell 1995;81:801–9.
38. Honda R, Tanaka H, Yasuda H. Oncoprotein MDM2 is a ubiquitinligase E3 for tumor suppressor p53. FEBS Lett 1997;420:25–7.
39. Ashcroft M, Kubbutat MH, Vousden KH. Regulation of p53 functionand stability by phosphorylation. Mol Cell Biol 1999;19:1751–8.
40. Ashcroft M, Taya Y, Vousden KH. Stress signals utilize multiplepathways to stabilize p53. Mol Cell Biol 2000;20:3224–33.
41. Chehab NH, Malikzay A, Stavridi ES, Halazonetis TD. Phosphoryla-tion of Ser-20 mediates stabilization of human p53 in response toDNA damage. Proc Natl Acad Sci USA 1999;96:13777–82.
42. Fuchs SY, Adler V, Pincus MR, Ronai Z. MEKK1/JNK signaling sta-bilizes and activates p53. Proc Natl Acad Sci USA 1998;95:10541–6.
43. Shieh SY, Ikeda M, Taya Y, Prives C. DNA damage-induced phos-phorylation of p53 alleviates inhibition by MDM2. Cell 1997;91:325–34.
44. Chao C, Herr D, Chun J, Xu Y. Ser18 and 23 phosphorylation isrequired for p53-dependent apoptosis and tumor suppression. EMBOJ 2006;25:2615–22.
45. Yaginuma Y, Westphal H. Abnormal structure and expression of thep53 gene in human ovarian carcinoma cell lines. Cancer Res1992;52:4196–9.
46. Mujoo K, Zhang L, Klostergaard J, Donato NJ. Emergence of cisplatin-resistant cells from the OVCAR-3 ovarian carcinoma cell line withp53 mutations, altered tumorigenicity, and increased apoptotic sensi-tivity to p53 gene replacement. Int J Gynecol Cancer 2000;10:105–14.
545REGULATION OF p53 BY Akt IN OVARIAN CANCER CELLS

47. Akeshima R, Kigawa J, Takahashi M, Oishi T, Kanamori Y, ItamochiH, Shimada M, Kamazawa S, Sato S, Terakawa N. Telomerase activ-ity and p53-dependent apoptosis in ovarian cancer cells. Br J Cancer2001;84:1551–5.
48. Kigawa J, Sato S, Shimada M, Takahashi M, Itamochi H, KanamoriY, Terakawa N. p53 gene status and chemosensitivity in ovarian can-cer. Hum Cell 2001;14:165–71.
49. Reles A, WenWH, Schmider A, Gee C, Runnebaum IB, Kilian U, JonesLA, El-Naggar A, Minguillon C, Schonborn I, Reich O, KreienbergR, et al. Correlation of p53 mutations with resistance to platinum-based chemotherapy and shortened survival in ovarian cancer. ClinCancer Res 2001;7:2984–97.
50. Lavarino C, Pilotti S, Oggionni M, Gatti L, Perego P, Bresciani G,Pierotti MA, Scambia G, Ferrandina G, Fagotti A, Mangioni C, Luc-chini V, et al. p53 gene status and response to platinum/paclitaxel-based chemotherapy in advanced ovarian carcinoma. J Clin Oncol2000;18:3936–45.
51. Song K, Li Z, Seth P, Cowan KH, Sinha BK. Sensitization of cis-plat-inum by a recombinant adenovirus vector expressing wild-type p53gene in human ovarian carcinomas. Oncol Res 1997;9:603–9.
52. Vasey PA, Jones NA, Jenkins S, Dive C, Brown R. Cisplatin, campto-thecin, and taxol sensitivities of cells with p53-associated multidrugresistance. Mol Pharmacol 1996;50:1536–40.
53. Cregan SP, Arbour NA, Maclaurin JG, Callaghan SM, Fortin A,Cheung EC, Guberman DS, Park DS, Slack RS. p53 activation do-main 1 is essential for PUMA upregulation and p53-mediated neuro-nal cell death. J Neurosci 2004;24:10003–12.
54. Brown R, Clugston C, Burns P, Edlin A, Vasey P, Vojtesek B, KayeSB. Increased accumulation of p53 protein in cisplatin-resistant ovar-ian cell lines. Int J Cancer 1993;55:678–84.
55. Chen J, Halappanavar SS, St-Germain JR, Tsang BK, Li Q. Role ofAkt/protein kinase B in the activity of transcriptional coactivatorp300. Cell Mol Life Sci 2004;61:1675–83.
56. Iyer NG, Chin SF, Ozdag H, Daigo Y, Hu DE, Cariati M, Brindle K,Aparicio S, Caldas C. p300 regulates p53-dependent apoptosis afterDNA damage in colorectal cancer cells by modulation of PUMA/p21levels. Proc Natl Acad Sci USA 2004;101:7386–91.
57. Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T,Nishimori H, Tamai K, Tokino T, Nakamura Y, Taya Y. p53AIP1, apotential mediator of p53-dependent apoptosis, and its regulation bySer-46-phosphorylated p53. Cell 2000;102:849–62.
58. Dumaz N, Meek DW. Serine15 phosphorylation stimulates p53 trans-activation but does not directly influence interaction with HDM2.EMBO J 1999;18:7002–10.
59. Yang X, Fraser M, Moll UM, Basak A, Tsang BK. Akt-mediated cis-platin resistance in ovarian cancer: modulation of p53 action oncaspase-dependent mitochondrial death pathway. Cancer Res 2006;66:3126–36.
60. Nemajerova A, Erster S, Moll UM. The post-translational phosphoryl-ation and acetylation modification profile is not the determining factorin targeting endogenous stress-induced p53 to mitochondria. CellDeath Differ 2005;12:197–200.
61. Limesand KH, Schwertfeger KL, Anderson SM. MDM2 is requiredfor suppression of apoptosis by activated Akt1 in salivary acinar cells.Mol Cell Biol 2006;26:8840–56.
62. Petty R, Evans A, Duncan I, Kurbacher C, Cree I. Drug resistance inovarian cancer—the role of p53. Pathol Oncol Res 1998;4:97–102.
63. Mi RR, Ni H. MDM2 sensitizes a human ovarian cancer cell line.Gynecol Oncol 2003;90:238–44.
64. Pestell KE, Hobbs SM, Titley JC, Kelland LR, Walton MI. Effect ofp53 status on sensitivity to platinum complexes in a human ovariancancer cell line. Mol Pharmacol 2000;57:503–11.
65. Mano Y, Kikuchi Y, Yamamoto K, Kita T, Hirata J, Tode T, IshiiK, Nagata I. Bcl-2 as a predictor of chemosensitivity and prognosisin primary epithelial ovarian cancer. Eur J Cancer 1999;35:1214–19.
546 FRASER ET AL.




















![The Antiproliferative Effect of Cyclodipeptides from ...suppressor signals such as PI3K, Akt, Ras, Raf, TRK, NF1, LKN1, PTEN, p53, and TSC1 and TSC2 have largely involved [16,17]](https://img.pdfslide.us/doc/110x75/5e6f16bf38db12762825828e/the-antiproliferative-effect-of-cyclodipeptides-from-suppressor-signals-such.jpg)








