Embed Size (px)
Citation preview

Eur. J. Biochem. 123, 267-274 (1982) 0 FEBS 1982
Affinity Labelling of Yeast Phenylalanyl-tRNA Synthetase with a 3'-Oxidised tRNAPhe Isolation and Sequence of the Labelled Peptide
Michel RENAUD, Franco FASIOLO, Mireille BALTZINGER, Yves BOULANGER, and Pierre REMY
Institut de Biologie Moltculaire et Cellulaire du Centre National de la Recherche Scientifique, Strasbourg
(Received November 9, 1981)
Yeast phenylalanyl-tRNA synthetase was specifically labelled with a 3'-oxidised tRNAPhe. Stoichiometric inactivation was achieved with the incorporation of 2 mol oxidised tRNAPh"/mol enzyme which corresponds exactly to the stoichiometry of tRNA binding. The labelled peptide has been isolated using a quick chromato- graphic procedure which can be applied to any covalent complex formed between a tRNA and an aminoacyl- tRNA synthetase. The isolated peptide (18 amino acids) was found to encompass the unique cysteine sequence of the smaller /? subunit of the enzyme.
Aminoacyl-tRNA synthetases have been extensively stud- ied over the past few years from many viewpoints but so far little is known as to the amino acids involved in their catalytic site. The effect of chemical modification of some residues such as cysteine or histidine has been reported [l -41 but no clear demonstration of their involvement in catalysis has been given yet. Only recently the photochemical cross-linking of ATP to isoleucyl-tRNA synthetase [5] provided some struc- tural data on the active site of the enzyme. Unfortunately the high extent of enzyme denaturation and the low yield of photochemical cross-linking are the most serious drawbacks of this photoaffinity labelling method.
More recently a new affinity labelling method has been proposed [6 - 81 : it involves a Schiff s base formation between the 3' end of an oxidised tRNA (tRNA,,) and the &-amino group of a lysine residue in (or near) the active site of the aminoacyl-tRNA synthetase and its subsequent reduction by cyanoborohydride derivatives. This reaction proceeds with a fairly high yield thus allowing isolation and analysis of the labelled peptide.
Previous studies carried out in our laboratory showed that yeast phenylalanyl-tRNA synthetase is a tetramer of the a2 p2 type (a, M , = 73000, /?, M , = 62000). It binds 1 mol of each substrate (ATP, phenylalanine and tRNAPhe) per mol a/? and the binding is anticooperative with respect to the amino acid [9]; tRNAPhe interacts mainly, if not exclusively, with the smaller /? subunit which carries the lysine residue able to bind covalently the 3' end of oxidised tRNAPhe [6].
In this paper we describe the isolation and analysis of the peptide containing this labelled amino acid residue. This procedure can be applied to any covalent complex of tRNA,, and an aminoacyl-tRNA synthetase, involving a Schiff s base formation and its subsequent reduction by cyanoborohydride derivatives. The amino acid composition and sequence of the
Abbreviation. tRNA,,, tRNA oxidised by periodate on its 3'-ter- minal ribose.
Enzymes. Phenylalanyl-tRNA synthetase (EC 6.1 .I .20) ; tRNA nu- cleatidyltransferase (EC 2.7.7.25); trypsin (EC 3.4.21.4); pancreatic ribo- nuclease (EC 3.1.27.5); TI ribonuclease (EC 3.1.27.3); carboxypeptidase A (EC 3.4.17.1); carboxypeptidase Y (EC 3.4.17.4); staphylococcal protease (EC 3.4.21.19).
peptide covalently linked to tRNAF:e will be discussed in relation to the structural and catalytic data available for yeast phenylalanyl-tRNA synthetase.
EXPERIMENTAL PROCEDURE
Materials
Phenylalanyl-tRNA synthetase was purified according to [lo]. Its activity and molar concentration were determined as reported [lo, 111. Yeast tRNA-nucleotidyltransferase was pre- pared as previously published [12]. Trypsin was from Worth- ington (Freehold, NJ, USA) and was purified by affinity chromatography on two successive columns of UMP-Se- pharose to remove pancreatic ribonuclease [I 31 and Kunitz- inhibitor - Sepharose to eliminate chymotrypsin and residual trace amounts of ribonuclease [14]. Pancreatic and T1 ribo- nucleases were from Worthington (Freehold, NJ, USA) and Sankyo (Tokyo, Japan) respectively. Carboxypeptidase A and Y were from Worthington also staphylococcal protease was a gift from Dr G. R. Drapeau [15].
Sephadex G-50 was obtained from Pharmacia (Uppsala, Sweden) and DEAE-cellulose from Whatman (Springfield Mill, Maidstone, Kent, UK). Radiochemicals were from the Radiochemical Centre (Amersham, UK). All chemicals were from Merck (Darmstadt, FRG), analytical grade.
Per ioda te Oxidation of t R N A
Yeast tRNAPhe was purified by counter-current distribu- tion [16]; its accepting capacity was about 1600 pmol phenyl- alanine/A260 unit. Its 3 ' - & ~ was specifically labelled by exchange with either [3H]ATP or [I4C]ATP using a technique derived from [17, 181. Periodate oxidation of tRNAPhe-C-C- [3H]AoH was carried out in the dark for 15 min at 0°C in 0.1 M potassium acetate buffer pH 5.0 containing 1 mM sodium periodate. The reaction was stopped by addition of glycerol. The mixture was then dialysed exhaustively against deionised water. The specific radioactivity of the tRNA used in this experiment was about 30 counts 3H min-' pmol-' or 3 counts 14C min-' pmol-'. After periodate oxidation more than 97 % of the tRNA could no longer be aminoacylated.

268
Scliiff s Base Formation and Reduction
The reaction between tRNAz!e and phenylalanyl-tRNA synthetase was carried out at 37 "C either in 25 mM potassium phosphate buffer at various pH values (7.0, 7.5 and 8.0) or in 25 mM barbital buffer pH 8.5, both containing 10 mM MgCl2 and 5 mM sodium cyanoborohydride. For a typical experi- ment the incubation mixture contained 6 pM enzyme and 20 pM [3H]tRNA!te (z 30 counts min-' pmol-'). At various time intervals aliquots of 25 pI were pipetted out, diluted in 50 mM Tris/HCl buffer pH 7.5 containing 1 M NaCl and 100 mM glycine and filtered onto cellulose membranes to estimate the amount of covalent complex formed ; the mem- branes were washed twice in 5 ml of the same buffer and three times in 5 ml H20, dried and subjected to scintillation counting.
Yield of' Inactivation
The yield of enzyme inactivation was determined by mea- suring its residual aminoacylation activity : at various time intervals aliquots of 5 pl were pipetted out and diluted in I ml of 50 mM Tris/HCI buffer pH 7.5 containing 30 mM KC1, 0.1 mM EDTA, 2.5 mM reduced glutathion, 0.1 mg/ml bovine serum albumin; 5 pl of this dilution was added to 200 pl of an aminoacylation mixture containing [14C]phenyl- alanine (50 pCi/mol) [I I].
The residual aminoacylation activity was compared to that of phenylalanyl-tRNA synthetase incubated with un- modified tRNAPhe under the same conditions.
The reaction between tRNAz!' and the synthetase was performed in the absence and presence of the small substrates (ATP 10 mM, phenylalanine 0.1 mM).
Isolation of the Peptide Covalently Linked to tRNAf4'
The covalent complex (= 40 nmol enzyme) was extensively dialysed against 0.1 M Tris/HCl buffer pH 8.0 and hydrolysed with purified trypsin (see Experimental Procedure) at a ratio trypsin/synthetase of 1 : 8 (w/w). Digestion was carried out in 1 % NH4HC03 (w/v) for 4 h at 37 "C. The tryptic digest was loaded onto a column of DEAE-cellulose (5 x 1 cm) equilibrated with 0.1 M Tris/HCl buffer pH 7.0. The column was washed with 50 ml of the same buffer containing 0.2 M NaC1, then eluted with a linear gradient of NaCl in the same buffer, from 0.2 M to 1 M (total volume of the gradient (200 ml). The elution was monitored by radioactivity mea- surements on aliquots (50 pl) of each fraction. Radioactive fractions were pooled, dialysed against deionised water and lyophilised. The lyophilised material was digested with pan- creatic ribonuclease (250 pg) for 2 h at 37 "C and subjected to fingerprinting techniques or to further enzymatic digestions.
Fingerprinting Techniques
Aliquots ( z 10 nmol) were subjected to thin-layer electro- phoresis and chromatography on cellulose plates. Electro- phoresis was run in the first dimension, in a Camag apparatus, at pH 2 (8 % acetic acid/2 % formic acid, v/v), pH 4.4 (2 % pyridine/4% acetic acid/l5% acetone, v/v) or pH 6.5 (10% pyridine/0.5 % acetic acid, v/v) for 1.5 h at 400 V. The plates were dried in a fume cupboard and run in the second dimen- sion in butan-I-ol/acetic acid/water/pyridine (15 : 3 : 12 : 10, v/v>.
Peptides were stained with 0.3% ninhydrin in ethanol (containing 3 % collidine and 10 % acetic acid), or with fluo-
rescamine (first spray with 3 %, vjv, collidine in acetone, the second with 0.01 %, w/v, fluorescamine in acetone). Peptides containing tryptophan were detected with the Ehrlich reagent (1 g p-dimethylaminobenzaldehyde, 10 ml 12 M HCl, 90 ml acetone). Radioactive peptides were detected by autoradiog- raphy of the map (or with the beta Camera 6 F 290 bE).
Elution of the Peptides
For amino acid analysis the spots were directly eluted from the plate with 6 M HCI: the cellulose was scratched off the plate and transferred into an Eppendorf plastic tube (1 ml); 300 pl 6 M HCI were added and the mixture was allowed to stand at room temperature for about 1 h with occasional stirring. The tube was centriduged and the super- natant was pipetted out. The operation was repeated with a further 300 pl of 6 M HCl and the combined supernatants were thoroughly spun down and transferred to glass tubes for hydrolysis (24 h at 105 "C under nitrogen). For sequence studies the elution was carried out in a similar way with the replacement of 6 M HCI by 50 % (v/v) acetic acid. The super- natant was lyophilised.
Staphylococcal Protease and Therinolysin Digestions
The radioactive fractions eluted from the DEAE-cellulose column (25 nmol peptide) were dissolved in 1 % NH4HC03 (w/v); the enzyme was added at a ratio 1 : 20 (w/w) and the mixture was incubated at 37 "C for 24 h. The resulting digests were lyophilised.
Curhoxypeptidase A and Y Digestions
The commercial suspension of carboxypeptidase A (z 50 mg/ml) was diluted 10-fold in 10% LiCl (w/v) whereas the lyophilised carboxypeptidase Y was dissolved in water (1 mg/ml); 5 p1 of each solution was added to 1-5 nmol peptide. Digestions were carried out at 37 "C for 1 - 4 h either in 1 % NH4HC03 (w/v) (carboxypeptidase A) or in 0.1 M pyridine acetate buffer pH 5.5 (carboxypeptidase Y). The re- sulting digests were lyophilised prior to amino acid analysis.
Amino Acid Analysis
analyser. Amino acid analyses were performed on a Durrum D500
Succinylation of Thiol Groups with Maleic Acid
The covalent complex between enzyme and unlabelled tRNAPhe (about 25 nmol enzyme) was labelled with [2,3-14C]- maleic acid (zz 10 counts min-' pmol-') in the presence of 8 M urea under the conditions used for the native enzyme [19]. After exhaustive dialysis to remove the excess of reagent it was treated as described above.
RESULTS
Schiffs Base Formation and Reduction
A high increase in both the velocity and yield of the reaction between tRNA:? and phenylalanyl-tRNA synthetase was observed when the pH was raised from 7.0 to 8.5. This reflects the increase in concentration of the unprotonated amino group involved on the one hand and the higher selec-
,_..... .> c .-;g-@A+b;H n- b-
- ." 75 A
" c .- ._ 50
2 U
a, a I
25
-v- I
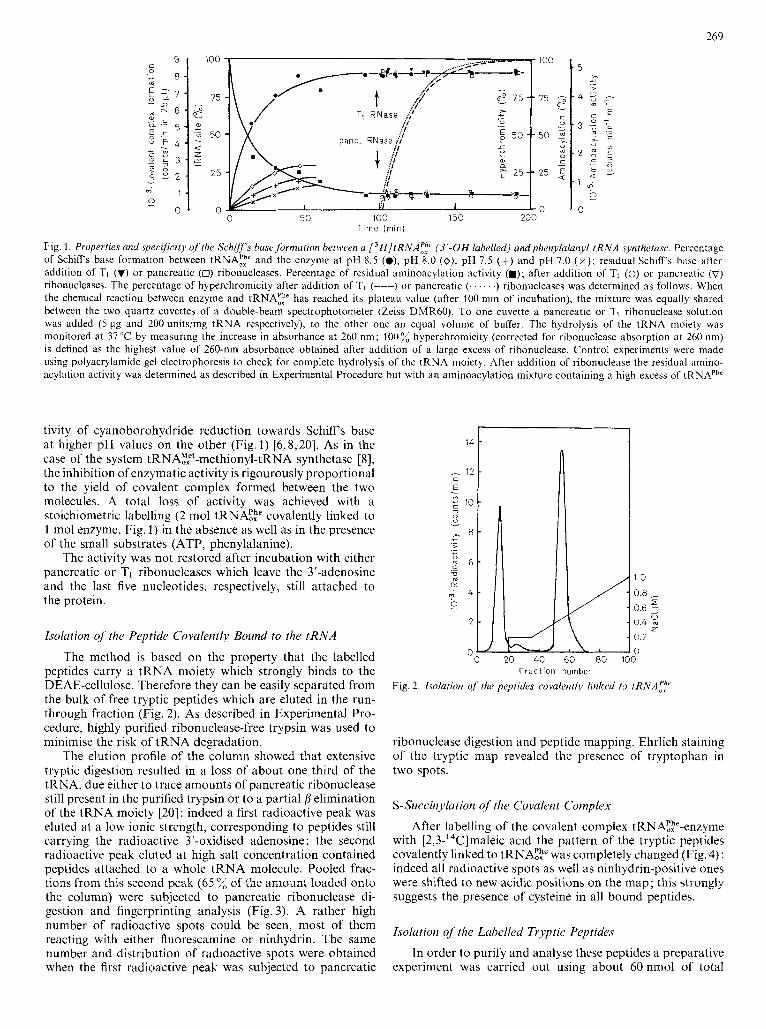
Fig. 1. Properties and specifiirity o f the Schiff's base formation between a L 3 H ] t RNA:: (3'-OH labelled) and phenylulunyl-tRNA synthetase. Percentage of Schiffs base formation between tRNA;: and the enzyme at pH 8.5 (a), pH 8.0 (o), pH 7.5 (+) and pH 7.0 ( x ) ; residual Schiff's base aftcr addition o f TI (v) or pancreatic (0) ribonucleases. Percentage of residual aminoacylation activity (m); after addition of TI (0) or pancreatic (v) ribonucleases. The percentage of hyperchromicity after addition of TI (---) or pancreatic (. . . ' . .) ribonucleases was determined as follows. When the chemical reaction between enzyme and tRNA:,h' has reached its plateau value (after 100 min of incubation), the mixture was equally shared between the two quartz cuvettes of a double-beam spectrophotometer (Zeiss DMR60). To one cuvette a pancreatic or TI ribonuclease solution was added ( 5 pg and 200 units/mg tRNA respectively), to the other one an equal volume of buffer. The hydrolysis of the tRNA moiety was monitored at 37°C by measuring the increase in absorbance at 260 nm; 100% hyperchromicity (corrected for ribonuclease absorption at 260 nm) is defined as the highest value of 260-nm absorbance obtained after addition of a large excess of ribonuclease. Control experiments were made using polyacrylamide gel electrophoresis to check for complete hydrolysis of the tRNA moiety. After addition of ribonuclease the residual amino- acylation activity was determined as described in Experimental Procedure but with an aminoacylation mixture containing a high excess of tRNAPhe
1 100
-- 75
--50
-- 25
0
tivity of cyanoborohydride reduction towards Schiff s base at higher pH values on the other (Fig. 1) [6,8,20]. As in the case of the system tRNA!:,"'-methionyl-tRNA synthetase [8], the inhibition of enzymatic activity is rigourously proportional to the yield of covalent complex formed between the two molecules. A total loss of activity was achieved with a stoichiometric labelling (2 mol tRNAE;' covalently linked to 1 mol enzyme, Fig. 1) in the absence as well as in the presence of the small substrates (ATP, phenylalanine).
The activity was not restored after incubation with either pancreatic or TI ribonucleases which leave the 3'-adenosine and the last five nucleotides, respectively, still attached to the protein.
Isolation of the Peptide Covalently Bound to the tRNA
The method is based on the property that the labelled peptides carry a tRNA moiety which strongly binds to the DEAE-cellulose. Therefore they can be easily separated from the bulk of free tryptic peptides which are eluted in the run- through fraction (Fig. 2 ) . As described in Experimental Pro- cedure, highly purified ribonuclease-free trypsin was used to minimise the risk of tRNA degradation.
The elution profile of the column showed that extensive tryptic digestion resulted in a loss of about one third of the tRNA, due either to trace amounts of pancreatic ribonuclease still present in the purified trypsin or to a partial p elimination of the tRNA moiety [20] : indeed a first radioactive peak was eluted at a low ionic strength, corresponding to peptides still carrying the radioactive 3'-oxidised adenosine; the second radioactive peak eluted at high salt concentration contained peptides attached to a whole tRNA molecule. Pooled frac- tions from this second peak (65 of the amount loaded onto the column) were subjected to pancreatic ribonuclease di- gestion and fingerprinting analysis (Fig. 3). A rather high number of radioactive spots could be seen, most of them reacting with either fluorescamine or ninhydrin. The same number and distribution of radioactive spots were obtained when the first radioactive peak was subjected to pancreatic
1.0
0.8 I 5
0.6 : 0.4 % 0.2 0
z
" 0 20 40 60 80 100 F r a c t i o n number
Fig. 2. Isolation qf the peptides covalentlj linked to tRNAf:
ribonuclease digestion and peptide mapping. Ehrlich staining of the tryptic map revealed the presence of tryptophan in two spots.
S-Succinylution qf the Covalent Complex
After labelling of the covalent complex tRNAEE'-enzyme with [2,3-'4C]maleic acid the pattern of the tryptic peptides covalently linked to tRNAEp was completely changed (Fig. 4) : indeed all radioactive spots as well as ninhydrin-positive ones were shifted to new acidic positions on the map; this strongly suggests the presence of cysteine in all bound peptides.
Isolation of the Labelled Tryptic Peptides
In order to purify and analyse these peptides a preparative experiment was carried out using about 60 nmol of total
270
c
I- I.. _..-.. 3 t C:;!e7 :*.-
5 4
8 L.
9e. ... . . .. C
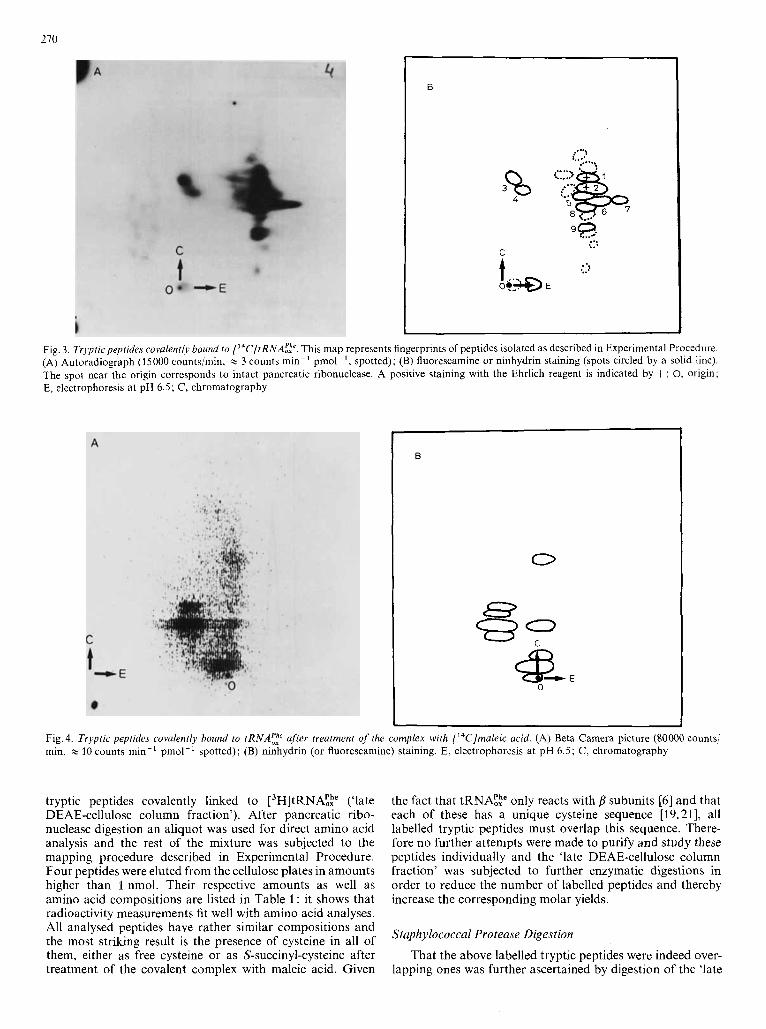
Fig. 3. Trypfic peptides covalenrly bound fo ['"C]tRNA;?. This map represents fingerprints of peptides isolated as described in Experimental Procedure. (A) Autoradiograph (15000 counts/min, x 3 counts min-' pmol-', spotted); (B) fluorescamine or ninhydrin staining (spots circled by a solid line). The spot near the origin corresponds to intact pancreatic ribonuclease. A positive staining with the Ehrlich reagent is indicated by +; 0, origin; E, electrophoresis a t pH 6.5; C, chromatography
0
0
Fig. 4. Tryptic peptides covalently bound to tRNA;: after treatment of the complex with ['4C]maleic acid. (A) Beta Camera picture (SO000 counts/ min, x 10 counts min-' pmol-' spotted); (B) ninhydrin (or fluorescamine) staining. E, electrophoresis at pH 6.5; C, chromatography
tryptic peptides covalently linked to ['H]tRNA!te ('late DEAE-cellulose column fraction'). After pancreatic ribo- nuclease digestion an aliquot was used for direct amino acid analysis and the rest of the mixture was subjected to the mapping procedure described in Experimental Procedure. Four peptides were eluted from the cellulose plates in amounts higher than 1 nmol. Their respective amounts as well as amino acid compositions are listed in Table 1 : it shows that radioactivity measurements fit well with amino acid analyses. All analysed peptides have rather similar compositions and the most striking result is the presence of cysteine in all of them, either as free cysteine or as S-succinyl-cysteine after treatment of the covalent complex with maleic acid. Given
the fact that tRNA!,h" only reacts with subunits [6] and that each of these has a unique cysteine sequence [19,21], all labelled tryptic peptides must overlap this sequence. There- fore no further attempts were made to purify and study these peptides individually and the 'late DEAE-cellulose column fraction' was subjected to further enzymatic digestions in order to reduce the number of labelled peptides and thereby increase the corresponding molar yields.
Staphylococcal Protease Digestion
That the above labelled tryptic peptides were indeed over- lapping ones was further ascertained by digestion of the 'late
21 1
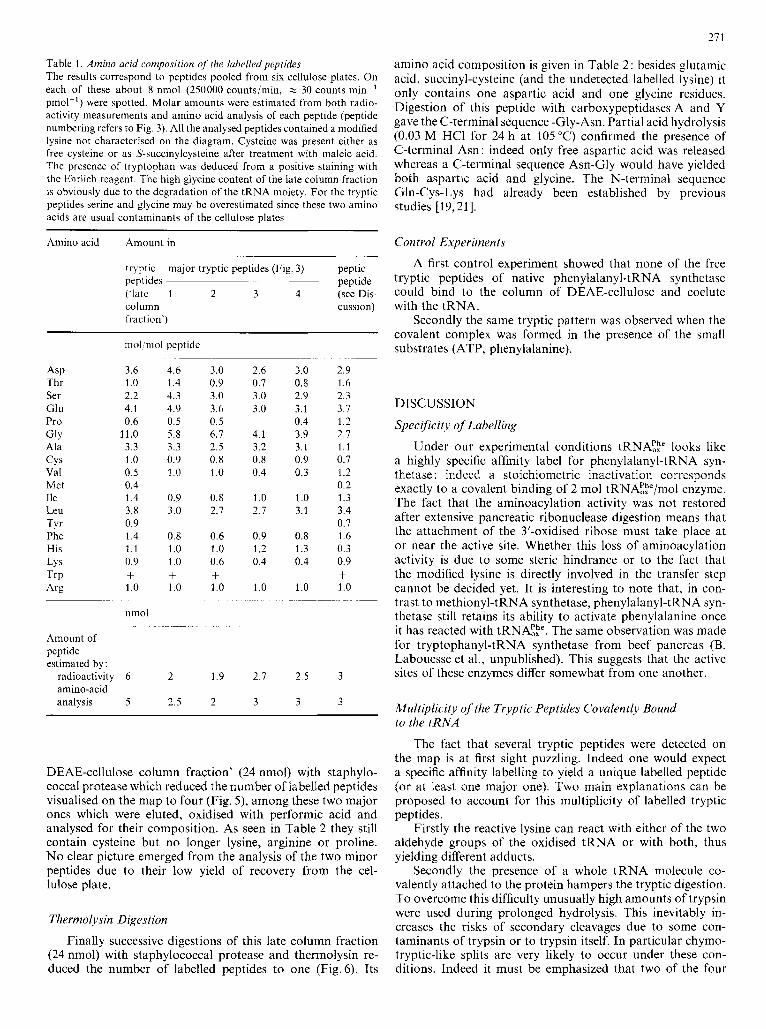
Table 1. Amino acid composition of' the lubelled peptides The results correspond to peptides pooled from six cellulose plates. On each of these about 8 nmol (250000 counts/min, Y 30 counts min-' pmol-') were spotted. Molar amounts were estimated from both radio- activity measurements and amino acid analysis of each peptide (peptide numbering refers to Fig. 3). All the analysed peptides contained a modified lysine not characterised on the diagram. Cysteine was present either as free cysteine or as S-succinylcysteine after treatment with maleic acid. The presence of tryptophan was deduced from a positive staining with the Ehrlich reagent. The high glycine content of the late column fraction is obviously due to the degradation of the tRNA moiety. For the tryptic peptides serine and glycine may be overestimated since these two amino acids are usual contaminants of the cellulose plates
Amino acid Amount in -~
tryptic major tryptic peptides (Fig 3) peptic peptides peptide ('ldte 1 2 3 4 (see Dis- column cussion) fraction')
ASP Thr Ser Glu Pro GIY Ala CYS Val Met Ile Leu TYr Phe His LY s TrP Arg
mol mol peptide
3 6 4 6 1 0 1 4 2 2 4 3 4 1 4 9 0 6 0 5
1 1 0 5 8 3 3 3 3 1 0 0 9 0 5 1 0 0 4 1 4 0 9 3 8 3 0 0 9 1 4 0 8 1 1 1 0 0 9 1 0 + + 1 0 1 0
nmol
~
~~~
~
3.0 0.9 3.0 3.6 0.5 6.1 2.5 0.8 1 .o
0.8 2.1
0.6 1.0 0.6 + 3 .o -.
Amount of peptide estimated by:
radioactivity 6 2 1.9 amino-acid analysis 5 2.5 2
~
2 6 0 1 3 0 3 0
4 1 3 2 0 8 0 4
1 0 2 1
0 9 1 2 0 4
1 0
~
2 1
3
__ 3 0 2 9 0 8 1 6 2 9 2 3 3 1 3 7 0 4 1 2 3 9 2 1 3 1 1 1 0 9 0 7 0 3 1 2
0 2 1 0 1 3 3 1 3 4
0 1 0 8 1 6 1 3 0 3 0 4 0 9
1 0 1 0 +
2.5 3
3 3
DEAE-cellulose column fraction' (24 nmol) with staphylo- coccal protease which reduced the number of labelled peptides visualised on the map to four (Fig. 5), among these two major ones which were eluted, oxidised with performic acid and analysed for their composition. As seen in Table 2 they still contain cysteine but no longer lysine, arginine or proline. No clear picture emerged from the analysis of the two minor peptides due to their low yield of recovery from the cel- lulose plate.
Thermolysin Digestion
Finally successive digestions of this late column fraction (24 nmol) with staphylococcal protease and thermolysin re- duced the number of labelled peptides to one (Fig.6).. Its
amino acid composition is given in Table 2: besides glutamic acid, succinyl-cysteine (and the undetected labelled lysine) it only contains one aspartic acid and one glycine residues. Digestion of this peptide with carboxypeptidases A and Y gave the C-terminal sequence -Gly-Asn. Partial acid hydrolysis (0.03 M HC1 for 24 h at 105 "C) confirmed the presence of C-terminal Asn: indeed only free aspartic acid was released whereas a C-terminal sequence Asn-Gly would have yielded both aspartic acid and glycine. The N-terminal sequence Gln-Cys-Lys had already been established by previous studies [19,21].
Control Experiments
A first control experiment showed that none of the free tryptic peptides of native phenylalanyl-tRNA synthetase could bind to the column of DEAE-cellulose and coelute with the tRNA.
Secondly the same tryptic pattern was observed when the covalent complex was formed in the presence of the small substrates (ATP, phenylalanine).
DISCUSSION
Specificity of Labelling
Under our experimental conditions tRNAFp looks like a highly specific affinity label for phenylalanyl-tRNA syn- thetase : indeed a stoichiometric inactivation corresponds exactly to a covalent binding of 2 mol tRNAFp/mol enzyme. The fact that the aminoacylation activity was not restored after extensive pancreatic ribonuclease digestion means that the attachment of the 3'-oxidised ribose must take place at or near the active site. Whether this loss of aminoacylation activity is due to some steric hindrance or to the fact that the modified lysine is directly involved in the transfer step cannot be decided yet. It is interesting to note that, in con- trast to methionyl-tRNA synthetase, phenylalanyl-tRNA syn- thetase still retains its ability to activate phenylalanine once it has reacted with tRNA;,h". The same observation was made for tryptophanyl-tRNA synthetase from beef pancreas (B. Labouesse et al., unpublished). This suggests that the active sites of these enzymes differ somewhat from one another.
Multiplicity of the Tryptic Peptides Covalently Bound to the tRNA
The fact that several tryptic peptides were detected on the map is at first sight puzzling. Indeed one would expect a specific affinity labelling to yield a unique labelled peptide (or at least one major one). Two main explanations can be proposed to account for this multiplicity of labelled tryptic peptides.
Firstly the reactive lysine can react with either of the two aldehyde groups of the oxidised tRNA or with both, thus yielding different adducts.
Secondly the presence of a whole tRNA molecule co- valently attached to the protein hampers the tryptic digestion. To overcome this difficulty unusually high amounts of trypsin were used during prolonged hydrolysis. This inevitably in- creases the risks of secondary cleavages due to some con- taminants of trypsin or to trypsin itself. In particular chymo- tryptic-like splits are very likely to occur under these con- ditions. Indeed it must be emphasized that two of the four

212
0
Fig. 5. Autoradiograph of labelled peptides after staphylococcal protease digestion. In this experiment ['4C]tRNA:te was used as in Fig. 3. (A) Auto- radiograph (35000 counts/min, % 3 counts min-' pmol-', spotted); (B) ninhydrin or fluorescamine staining. E, electrophoresis at pH 4.4; C, chromatography
Table 2. Amino acid composition of the labelled peptides For each experiment (staphylococcal protease and successive digestions) about 24 nmol labelled peptides were used and spotted on the cellulose plates (12 nmol per plate, see Fig. 5 and 6). Cysteine was present either as cysteic acid (peptides 1 and 2) after performic acid oxidation or as S-succinyl-cysteine (last column) after treatment with maleic acid. Amounts of peptide were estimated from both amino acid analysis and radioactivity measurements
Amino acid Amount in
peptides produced peptide produced by staphylococcal by successive protease digestion digestions with (Fig. 5 ) staphylococcal
1 2 thermolysin ~~~ protease and
(Fig. 6)
mol/mol peptide
ASP Thr Ser GI u Pro GlY Ala c y s Val Met Ile Leu TY Phe His LYS TrP '4%
2.8 1.9 0.7
0.6 0.6 1 .o 1 .o 1 .o
2.5 1 .o 1 .o 1.1 0.7 0.4 0.9 1 .o
1 .o 0.7
0.8 0.3
nmol -
Amount of peptide 3.2 2.8 13.7
Fig. 6. Autoradiograph of the labelled peptide afier successive digestions with staphylococcal protease and thermolysin. For this last experiment unlabelled tRNAzp was used but the complexe was treated with ['"CI- maleic acid (120000 counts/min, zz 10 counts min-' pmol-', spotted). E, electrophoresis at pH 4.4; C, chromatography
labelled tryptic peptides listed in Table 1 contain tryptophan (peptides 1 and 2) whereas the two other ones do not (pep- tides 3 and 4) which suggests some partial cleavage of trypto- phyl bonds. Finally, amino acid analyses, S-succinylation of the covalent complex and further digestions with other pro- teases unambiguously showed that all labelled tryptic peptides overlap the same sequence containing a cysteine residue (and the modified lysine). Previous structural studies have shown that the f l subunit of phenylalanyl-tRNA synthetase contains a unique cysteine sequence which is present twice in the polypeptide chain [19,21] : Tyr-Asn-Trp-Lys-Pro-Glu-Glu- Gln-Cys-Lys. Therefore it must be common to all labelled tryptic peptides and tyrosine should be present in some of

213
those listed in Table 1 since it was found in the analysis of the 'late DEAE-cellulose fraction'. However it must be recalled that for isolation of these peptides from cellulose plates tritiated tRNA was used and peptides were visualised by both fluorescamine and ninhydrin stainings. Under these conditions N-terminal residues are practically destroyed. As shown in Table 1 the yields of elution are rather low for these peptides since altogether the four major ones represent 20% of the initial amounts spotted on the plates. This is another good reason to use further proteolysis in order to reduce both the number and sizes of the labelled peptides and thereby increase their final yield.
Staphylococcal Protease and Thermolysin Digestions
Analysis of the tryptic peptides arising from the covalent complex shows that they must all contain this unique but does not tell which of its two lysines has reacted with the 3'-oxidised ribose. Since the two major radioactive peptides produced by digestion with staphylococcal protease (SP) still contain cysteine but not proline, the modified lysine must be adjacent to the cysteine residue. Given the results of digestions with staphylococcal protease (SP), thermolysin (TH) and carboxy- peptidases A and Y the sequence around the modified lysine can be written as follows :
the /l subunit are present in this radioactive spot, in par- ticular proline, tyrosine and tryptophan (Table I ) .
However, the quoted structural studies [19,21] have shown that no cysteine-containing peptides were released upon mild tryptic digestion of native phenylalanyl-tRNA synthetase since the two cysteine residues of the B subunit were found in the B' subunit of the trypsin-modified enzyme (ff', M , = 40000). Therefore if the mild tryptic treatment of the covalent complex adenosine,,-phenylalanyl-t RNA synthetase had proceeded in the same way one should have found no radioactivity associated with the dialysable fraction using this alternative isolation procedure. The results reported in this study show that, under the conditions of mild proteolysis, this cysteine-containing peptide was partially released from the covalent adenosine,,-enzyme complex. This unexpected finding at least suggests that one of the two copies of the cysteine peptide (also containing the modified lysine) is not very far away from the peptide bond normally cleaved by trypsin upon mild proteolysis of the native enzyme. A slight conformational change or steric hindrance caused by the co- valent binding of the 3'-oxidised adenosine could therefore result in a further although partial digestion of the azfll enzyme. Of course this hypothesis will have to be tested by further structural work on native and trypsin-modified phenylalanyl-tRNA synthetases.
SP TH SP SP
. . . Tyr-Asn-Trp-Lys-Pro-Glu-Glu-Gln-Cys-L ys-Gl! -Am-(Ala,Leu)-Asp-(Gly, His)-Asp- . 1 1 1 I
I 3'-Adenosine
As seen in Table 2 the yield of elution increases as the size and number of labelled peptides are reduced by further digestions: over 50% for the final therniolytic peptide as compared to the above 20 % for the tryptic ones.
An Alternative Isolation Procedure for the Labelled Peptides
The labelled peptide was also obtained and partially puri- fied using an alternative method which is specific to phenyl- alanyl-tRNA synthetase. It consisted in an extensive digestion of the covalent complex ['4C]tRNA!!e-enzyme (about 40 nmol) with pancreatic ribonuclease followed by mild proteolysis with trypsin under the conditions previously described [22]. The peptides released by trypsin were separated from the trypsin-modified ct2 enzyme by ultrafiltration through a PM 10 Amicon membrane and were found to contain 70% of the total radioactivity incorporated into the protein. After exhaustive digestion with trypsin (at 37°C for 24 h with a ratio trypsin/peptides of 1 : 10) this dialysable fraction was applied to a column of Sephadex G-50 (100 x 0.8 cm) equili- brated and eluted with 0.1 NH4HC03 (w/v). Fractions of 4 ml were collected at a flow rate of 16 ml/h. One main radioactive peak was obtained and the resulting fraction was digested with pepsin for 24 h at 37°C with a ratio pepsin/ peptides of 1 : 20. This final digest was subjected to peptide mapping as reported in Experimental Procedure. One major radioactive spot was found on the cellulose plate; it was eluted, hydrolysed and analysed. Of course, unlike the other method which readily selects the covalently bound peptides, this alternative approach is obviously insufficient to yield a pure peptide from such a complex mixture after only two chromatographic steps but it is worth noticing that all the amino acids expected from the unique cysteine sequence of
Some Properties of the Labelled Peptides In a previous paper [2] it was shown that thiol groups
of phenylalanyl-tRNA synthetase may play a part in the transfer step and that under certain conditions an interchain ct-p disulphide bridge could be formed. It must be pointed out that the labelled peptide contains a cysteine residue adjacent to the modified lysine. Obviously this cysteine must be in the vicinity of the 3'-adenosine in the complex tRNAPhe- phenylalanyl-tRNA synthetase. Besides this it could also be close to the ci subunit, although at this stage it is not known which of the two copies of the cysteine peptide of f l is involved in this interchain disulphide bridge formation. This would further support the idea that the two active sites of the enzyme are close to contact areas between ci and f l subunits [6].
It is also interesting to note the presence in the labelled sequence of acidic (Glu), basic (Lys) and aromatic residues (Tyr, Trp). Other studies have emphasized the importance and possible role of such peptide sequences in RNA-protein interactions [23 - 251.
In conclusion the method developed in this study, to label specifically the active site of a given aminoacyl-tRNA syn- thetase with subsequent isolation and analysis of the labelled peptide, has two main advantages: firstly it gives some insight into the binding site of a crucial part of the tRNA, the CCA- end; secondly it is a useful tool to look for sequence homologies in what could be a common site to all aminoacyl- tRNA synthetases.
Similar experiments are now under way in our laboratory using cognate and non-cognate systems.
The authors wish to thank Mrs N. Menard, G. Nussbaum and C. Rether for their excellent technical assistance as well as Mr M. Schlegel

274
for her valuable help in tRNA purification and also Mr H. Bacha for heiping us with kinetic studies. We are also grateful to Professor J. P. Ebel for his constant interest in this work. Finally we thank the Management of the Levurerie FALA, Strasbourg for their generous gift of yeast cells. This work was partly supported by a grant from the Centre National de la Recherche Scientijlque and the Delegation Ginirale a la Recherche Scientifique et Technique.
REFERENCES 1.
2.
3. 4.
5 . 6.
7.
8.
9.
10.
M.
MacElroy, W. D., Delvea, M. & Travis, J . (1967) Science (Wush.
Murayama, A,, Raffin, J. P., Remy, P. & Ebel, J. P. (1975) FEBS
Hennecke, H. & Bock, A. (1974) Eur. J . Biochem. 50, 157- 166. Raffin, J. P. & Remy, P. (1978) Biochim. Biophys. Actu, 520, 164-
Yue, U. T. & Schimmel, P. R (1977) Biochemistry, 16,4678-4684. Baltzinger, M., Fasiolo, F. & Remy, P. (1979) Eur. J . Biochern. 97,
Fayat, G., Hountondji, C. & Blanquet, S. (1979) Eur. J . Biochem.
Hountondji, C., Fayat, G. & Blanquet, S. (1979) Eur. J . Biochem.
Fasiolo, F., Ebel, J. P. & Lazdunski, M. (1977) Eur. J . Biochem.
Fasiolo, F. & Ebel, J. P. (1974) Eur. J. Biochem. 49, 257-263.
DC) 157,151 - 160.
Lett. 53, 15-22.
174.
87 - 92.
96, 87-92.
102, 247 - 250.
73. 7-15.
11. Fasiolo, F., Remy, P., Pouyet, J. & Ebel, J. P. (1974) Eur. J . Bio-
12. Rether, B., Bonnet, J. & Ebel, J. P. (1974) Eur. J . Biochem. 50,
13. Brison, 0. & Chambon, P. (1976) Anal. Biochem. 75,402-409. 14. Johnson, D. A. & Travis, J. (1976) Anal. Biochem. 72, 573 - 576. 15. Houmard, J. & Drapeau, G. R. (1972) Proc. Nut1 Acad. Sci. USA,
16. Dirheimer, G. & Ebel, J. P. (1967) Bull. Soc. Chim. Biol. 49, 1679-
17. Renaud, M., Dietrich, A., Giege, R., Rtmy, P. & Ebel, J. P. (1979)
18. Sternbach, H. & Cramer, F. (1977) Hoppe-Seyler’s 2. Physiol. Chem.
19. Robbe-Saul, S., Fasiolo, F. & Boulanger, Y. (1977) FEBS Lett. 84,
20. Borch, R. F., Bernstein, M. D. & Durst, H. D. (1971) J . Am. Chem.
21. Potier, S., Robbe-Saul, S. & Boulanger, Y. (1980) Biochim. Biophys.
22. Fasiolo, F., Boulanger, Y. & Ebel, J. P. (1975) Eur. J . Biochem. 53,
23. Sellini, H., Maurizot, J. C., Dimicoli, J. L. & Helene, C. (1973)
24. Toulme, J. J., Charlier, M. & Helene, C. (1974) Proc. Nail Acad.
25. Lefivre, J. F., Ehrlich, R., Kilhofer, M. C. & RCmy, P. (1980) FEBS
chem. 50,227-236.
281 - 288.
69,3506- 3509.
1687.
Eur. J . Biochem. 101,475-483.
358, 312.
57 - 62.
SOC. 93,2897 - 2904.
Acta, 624, 130-141.
487 - 492.
FEBS Lett. 30, 219-224.
Sci. USA, 71, 3185-3188.
Lett. 114,219-224.
Renaud, F. Fasiolo, M. Baltzinger, Y. Boulanger, and P. Remy, Institut de Biologie MolCculaire et Cellulaire du Centre National de la Recherche Scientifique, 15 Rue Rene-Descartes, Esplanade, F-67084 Strasbourg-Cedex, Bas-Rhin, France