Embed Size (px)
Citation preview

Advances in Mass Spectrometry for the Analysis of Emerging Persistent Organic Pollutants

Dedication To my grandmother, Judy Beaulieu
The true "beauty-on-duty" and Michael O'Leary, a teacher and friend

Örebro Studies in Chemistry 24
LAUREN MULLIN
Advances in Mass Spectrometry for the Analysis of Emerging Persistent Organic Pollutants

© Lauren Mullin, 2019
Title: Advances in Mass Spectrometry for the Analysis of Emerging Persistent Organic Pollutants
Publisher: Örebro University 2019 www.publications.oru.se
Print: Örebro University, Repro 09/2019
ISSN 1651-4270 ISBN 978-91-7529-303-5

Abstract Lauren Mullin (2019): Advances in Mass Spectrometry for the Analysis of Emerging Persistent Organic Pollutants. Örebro Studies in Chemistry 24. Mass spectrometry (MS) is a technique widely implemented for the measurement of environmental pollutants. A critical tool for the analysis of persistent organic pollutants (POPs) over several decades, MS as coupled with liquid and gas chroma-tography (LC and GC) techniques enables the analysis of emerging POPs. The aim of this thesis was to investigate the use of alternative MS-based techniques to assist specific analytical challenges including separation of stereoisomers using supercriti-cal fluid chromatography (SFC), reduced ionization competition with appropriate mobile phase additives, and applied rotationally averaged collision-cross section (CCS) of ions via ion mobility measurements of emerging POPs.
Chromatographic efficiency improvements for the brominated flame retardant, hexabromocyclododecane (HBCDD), were implemented through the development of two supercritical fluid chromatography (SFC) methods. Based on the inherent qualities of supercritical fluids, separation of both predominant diastereomers and respective enantiomers was performed in a shorter time with wider chromato-graphic resolution using SFC than existing LC methods.
Turning next to MS ionization considerations, the emerging perfluoroalkyl sub-stance hexafluoropropylene oxide-dimer acid (HFPO-DA) was investigated. Fol-lowing a survey of analytical methodologies for HFPO-DA, the challenge of ex-treme dimer formation, in-source fragmentation and very low [M-H]- production was described. Method development using alternative mobile phase additives in currently used LC-MS acquisition techniques was deployed.
Finally, ion mobility spectrometry (IMS) was implemented in a non-targeted ac-quisition study of indoor dust samples. This study used IMS coupled with quadru-pole time-of-flight MS to identify a wide range of contaminant classes, including emerging POPs. Identification confidence is a challenge currently facing non-targeted studies, and the use of prediction mechanisms of analyte IMS gas-phase separations was explored.
Through applying diverse alternative techniques, increased method performance was explored for emerging POPs analyses.
Keywords: Mass Spectrometry; Liquid Chromatography; Supercritical Fluid Chromatography; Ion Mobility; POPs; Electrospray Ionization Lauren Mullin, School of Science and Technology Örebro University, SE-701 82 Örebro, Sweden


List of Papers
Paper I: Mullin, L., Burgess, J.A., Jogsten, I.E., Geng, D., Aubin, A. and van Bavel, B. 2015. Rapid separation of hexabromocy-clododecane diastereomers using a novel method combin-ing convergence chromatography and tandem mass spec-trometry. Analytical Methods, 7(7), 2950-2958. https://doi.org/10.1039/C4AY02923B
Paper II: Riddell, N., Mullin, L., van Bavel, B., Ericson Jogsten, I., McAlees, A., Brazeau, A., Synnott, S., Lough, A., McCrin-dle, R. and Chittim, B. 2016. Enantioselective analytical-and preparative-scale separation of hexabromocyclododec-ane stereoisomers using packed column supercritical fluid chromatography. Molecules, 21(11), 1509-1520. https://doi.org/10.3390/molecules21111509
Paper III: Mullin, L., Katz, D., Riddell, N., Plumb, R., Burgess, J.A., Yeung, L.W. and Jogsten, I.E. 2019. Analysis of hex-afluoropropylene oxide-dimer acid (HFPO-DA) by Liquid Chromatography-Mass Spectrometry (LC-MS): Review of Current Approaches and Environmental Levels. Trends in Analytical Chemistry, 118, 828-839. https://doi.org/10.1016/j.trac.2019.05.015
Paper IV: Ionization Enhancement of Hexafluoropropylene oxide dimer acid (HFPO-DA) Through Mobile Phase Additive Selection. To be submitted to Rapid Communications in Mass Spec-trometry
Paper V: Liquid Chromatography-Ion Mobility-High Resolution Mass Spectrometry for Analysis of Pollutants in Indoor Dust: Identification and Predictive Capabilities. To be submitted to Analytica Chimica Acta


List of Abbreviations
ABPR AOP
APCI
APGC
APPI
Ar
CCS
CDT
CI
CID
Cl
CO2
Da
DDE
DDT
DIA
DL-PCBs
DSSTox
DT-IMS
EDA
EF
EI
ESCi™
ESI
eV
FAIMS
FOSA
FTOHs
FWHM
GC
GCxGC
H.E.T.P.
Automated back pressure regulator
Adverse Outcome Pathway
Atmospheric pressure chemical ionisation
Atmospheric pressure gas chromatography
Atmospheric pressure photoionisation
Argon
Collision-cross section
Cyclododeca-1,5,9-triene
Chemical ionisation
Collision-induced dissociation
Chlorine
Carbon dioxide
Dalton
1,1-Dichloro-2,2-bis(p-chlorophenyl) ethylene
Dichlorodiphenyl trichloroethane
Data independent analysis
Dioxin-like polychlorinated biphenyls
Distributed Structure-Searchable Toxicity database
Drift time ion mobility spectrometry
Effect-directed analysis
Enantiomeric fraction
Electron ionisation
Electrospray/chemical ionization switching
Electrospray ionisation
Electron volts
Field-assymetric waveform ion mobility spectrometry
Perfluoro-1-octanesulfonamide
Fluorotelomer alcohols
Full width at half-maximum height
Gas chromatography
Dual column gas chromatography
Height-equivalent of a theoretical plate

HBCDD Hexabromocyclododecane
HCB Hexachlorobenzene
HFPO-DA Hexafluoropropylene-oxide dimer acid
HRGC High resolution gas chromatography
HRMS High resolution mass spectrometry
IMS Ion mobility spectrometry
LC Liquid chromatography
LOD Limit-of-detection
LOQ Limit-of-quantification
LRT Long range transport
m/z Mass to charge ratio
MeOH-PBDEs Methylhydroxylated polybrominated diphenyl ethers
MRM Multiple reaction monitoring
MS Mass spectrometry
m Meter
ms Milliseconds
MS/MS Tandem mass spectrometry
N Nitrogen
NBFRs Novel brominated flame retardants
OCs Organochlorine pesticides
OH-PCBs Hydroxylated polychlorinated biphenyl ethers
OPFRs Organophosphate flame retardants
OPs Organophosphorus compounds
PAHs Polyaromatic hydrocarbons
PBDEs Polybrominated diphenyl ethers
PBT Persistent, bioaccumulative and toxic
PCA Principal component analysis
PCB Polychlorinated biphenyls
PCDD Polychlorinated dibenzo-p-dioxins
PCDF Polychlorinated dibenzofurans
PFAS Per-/polyfluoroalkyl substances
PFECAs Perfluoroethercarboxylic acids
PFESAs Perfluoroethersulfonic acids
PFHxS Perfluorohexanesulfonic acid

PFOA Perfluorooctanoic acid
PFOS Perfluorooctane sulfonic acid
POPs Persistent organic pollutants
PPCPs Pharmaceuticals and personal care products
QTOF Quadrupole time-of-flight
R Mass resolution
RPLC Reversed-phase liquid chromatography
RS Chromatographic resolution
RT Retention time
SFC Supercritical fluid chromatography
SFE Supercritical fluid extraction
SIM Single ion monitoring
SPE Solid-phase extraction
TIC Total ion current
TIMS Trapped ion mobility spectrometry
TOF-MS Time-of-flight mass spectrometry
TQ-MS Tandem quadrupole mass spectrometry
TW-IMS Traveling wave ion mobility spectrometry
USEPA United States Environmental Protection Agency


Table of Contents
1.0 INTRODUCTION ............................................................................ 151.1 Persistent organic pollutants (POPs) overview ................................... 15
1.1.1 Physico-chemical characteristics of legacy POPs ......................... 161.1.2 Toxicity of legacy POPs ............................................................. 191.1.3 Emerging POPs .......................................................................... 201.1.4 Consideration and challenges in POPs monitoring ..................... 21
1.2 Mass spectrometry-based analysis methods for POPs ........................ 221.2.1 Chromatography techniques for POPs analysis .......................... 221.2.2 Why Mass Spectrometry for POPs? ............................................ 24
1.3 Types of MS used for POPs analysis .................................................. 281.3.1 Magnetic sector .......................................................................... 281.3.2 Tandem quadrupole ................................................................... 291.3.3 Time-of-flight ............................................................................. 30
2.0 ALTERNATIVE TECHNIQUES FOR MASS SPECTROMETRY-BASED ANALYSIS OF POPS .................................................................. 332.1 Supercritical Fluid Chromatography ................................................. 332.2 Atmospheric Pressure Gas Chromatography ..................................... 372.3 Ion Mobility Spectrometry ................................................................ 38
3.0 AIM OF THE THESIS ...................................................................... 41
4.0 IMPLEMENTATION OF ALTERNATIVE TECHNIQUES IN EMERGING POPS ANALYSIS ............................................................... 434.1 Supercritical fluid chromatography for hexabromocyclododecane (HBCDD) ................................................................................................ 43
4.1.1 Diastereomer separation using SFC ............................................ 454.1.2 Enantiomer separation using SFC .............................................. 47
4.2 Hexafluoropropylene oxide-dimer acid (HFPO-DA) ......................... 494.2.2 Improvement to the ionization of HFPO-DA ............................. 52
4.3 Non-targeted acquisition of dust samples using TOF-MS ................. 544.3.1 Application of Ion Mobility Spectrometry .................................. 56
5.0 CONCLUSIONS AND FUTURE WORK ......................................... 61
6.0 ACKNOWLEDGEMENTS ............................................................... 63
7.0 REFERENCES .................................................................................. 65


LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs 15
1.0 Introduction Use of mass spectrometry as an analytical tool to measure the molecular mass of compounds and perform structural elucidation has aided numerous scientific fields of study. Development of modern mass spectrometers can be traced through several key advances occurring through the mid- to later parts of the twentieth century (Griffiths, 2008). These advances have in-cluded the development of gas-phase ionization sources, quadrupole mass filters, high-resolution mass measurements and numerous others, all driven by the analytical needs of scientists to accurately and precisely determine a wide range of compound classes in various matrices. One field that has sig-nificantly benefited from these advances is that of environmental chemistry. Though health-impacting xenobiotic chemical exposure has occurred for many years, the high use in the 19th and 20th centuries of human-created chemicals found to aid industrial and consumer processes ultimately led to the release of highly toxic compounds away from their sites of production. Use of mass spectrometry, as described in this thesis, was instrumental in identifying these chemicals in both biological and environmental compart-ments. This continues today, as numerous emerging environmental contam-inants are identified and quantified to drive mitigating actions which protect human and environmental health. As both fields of mass spectrometry and environmental chemistry have grown, they have done so in an intertwined way, such that necessities of environmental analysis which include increased sensitivity, selectivity and wider breadth of analyte coverage have benefitted from advances in mass spectrometry. Extending from this, the continued analytical challenges that emerging contaminants present, along with ana-lytes from all fields of scientific measurement, now inform the needs of mass spectrometry development. In order to embark on an exploration of alter-native mass spectrometry approaches for environmental analysis, it is nec-essary to first lay the groundwork of current methods and their capabilities.
1.1 Persistent organic pollutants (POPs) overview As modern society has advanced, so has the use of industrial processes and synthesized chemicals to produce and enhance consumer products. Alt-hough many of these chemicals were employed initially for the betterment of human life, their use over time revealed unintended consequences. The case of dichlorodiphenyl trichloroethane (DDT), a widely used insecticide, and its ethylene metabolite DDE causing severe down-stream impacts on the North American bird population was highlighted by Rachel Carson in

16
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
the 1962 book Silent Spring (Jones and de Voogt, 1999). In the years since, the public has increasingly been aware of both biological and environmental impacts of intentionally and unintentionally produced man-made chemi-cals. As a result, a key list of organohalogenated pollutants, referred to as persistent organic pollutants (POPs), was identified as being persistent in the environment, bioaccumulative, and toxic (PBT), and capable of wide-spread distribution away from the site of emission (Jones and de Voogt, 1999, Stockholm Convention, All POPs, 2019). In 2001, an international treaty in the form of the Stockholm Convention was established with the intention of eliminating or severely restricting the use of these chemicals in order to protect human and environmental health. The 12 compounds orig-inally delineated were referred to as the “dirty dozen,” and are listed in Table 1, as well as the additional compounds which have been added since 2001 (Stockholm Convention, All POPs, 2019).
1.1.1 Physico-chemical characteristics of legacy POPs The qualities of POPs (persistence in the environment, bioaccumulative, toxic (PBT) and potential for long range transport (LRT)) are a result of their physico-chemical properties (Jones and de Voogt, 1999). The chemical structures across the legacy POPs can be generalized as halogenated, with multiple rings or highly branched. The carbon-halogen bond, observed in all of these compounds, is highly stable to hydrolysis, and thus results in persistence in biological and environmental systems (Ritter et al., 1995). Moreover, the incidence of a carbon-halogen bond on a benzene ring, as can be seen with polychlorinated biphenyls (PCBs), polychlorinated dibenzo-p-dioxins/furans (PCDD/Fs), DDT and hexachlorobenzene (HCB) increases the stability of this bond (Ritter et al., 1995, Guo and Kannan, 2015). Halogenated compounds in general are also lipophilic, and easily partition into the fatty deposits of mammals (Ritter et al., 1995). They then experience bioaccumulation and biomagnification being transported through the food chain (Schecter, 1994).
The initial migration of POPs into the environment is either due to unin-tentional release, such as the formation of PCDD/Fs as by-products of com-bustion processes (Schecter, 1994) or intentional usage and subsequent leaching of compounds from intended materials or locations, such as the polybrominated diphenyl ethers (PBDEs) (Alaee et al., 2003) and organo-chlorine pesticides (OCs) (Stockholm Convention, All POPs, 2019). Upon release, the fate of POPs in the environment and biota is dependent on their aqueous solubility, vapor pressure and three partitioning coefficients (Jones

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
17
and de Voogt, 1999). These coefficients are water-octanol (KOW), which has been used as a measure for lipophilicity, air-water (KAW), and octanol-air (KOA) (Jones and de Voogt, 1999, Wania and Mackay et al., 1999). Table 1.1 summarizes a selection of these properties across the original dirty dozen, which are characterized by a preferential organic phase partitioning (log KOW and KOA values), organic phases represented by octanol in these coefficients include aerosols, soil, vegetables and animal tissue (Wania and Mackay et al., 1999).
The persistence of POPs is defined by the length of degradation half-lives of the compounds in various environmental compartments (Jones and de Voogt, 1999, Wania and Mackay et al., 1999). Specifically, half-lives in excess of 2-5 days in air, 2-6 months in water and 6-12 months in soil and sediment are indicative of a persistent compound (Wania and Mackay et al., 1999). From a risk assessment and legislative perspective, the determi-nation of half-lives should be approached with the consideration that mul-tiple environmental factors will impact these degradation half-life results. Thus, the use of various models to describe these processes have been uti-lized in these contexts (Rodan et al., 1999).

18
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Table 1: Summary of physico-chemical properties (derived from EPI Suite) of POPs delineated in the Stockholm convention. The original 12 “dirty dozen” are bold, while the more recently added POPs are italicized.
POP CAS no. vapour pres-sure (mm
Hg)
Log KAW Log KOW Log KOA water solu-bility
(mg/L) Aldrin 309-00-2 1.20E-04 * -2.75 * 6.5 8.80 * 0.0017 *
Chlordane 57-74-9 9.98E-06 * -1.7 * 6.16 to 6.22
8.92 * 0.056 *
DDT 50-29-3 1.60E-07 * -3.47 * 6.91 9.82 * 0.0055 *
Dieldrin 60-57-1 3.00E-06* -3.86 * 5.20 to 5.40
8.13 * 0.20 to 0.25 *
Endrin 72-20-8 3.00E-06* -3.38 * 5.20 to 5.40
8.13 * 0.20-0.25 *
Heptachlor 76-44-8 4.00E-04 * -1.92 * 5.47 to 6.1 7.64 * 0.18 *
Hexachlorobenzene 118-74-1 1.80E-05 * -1.16 * 5.73 7.83 * 0.0062 *
Mirex 2385-85-5 8.0E-05 * -1.48 * 6.89 NA 0.085 *
Toxaphene 8001-35-2 3.44E-6 *,**
-2.81 ** 6.75 ** 9.56 ** 0.0044 *,**
PCBs+ -- 5.81E-07 to 1.64E-05 *
-2.55** to -3.42 *
6.63 to 8.27**
9.7 to 10.51
0 to 0.0015 *
PCDFs++ -- 1.50E-09 -2.69 6.8 10.05 0
PCDDs++ -- 2.21E-06 -3.30** 6.63** NA 0.0019**
PFOS 1763-23-1 0.0064 *,** 0.35 ** 4.49 ** None; 4.84 **
0.10 *,**; 4.02 **,+++
HBCDD 3194-55-6 4.70E-07 * -2.73 7.74 ** None; 10.47 **
0.0086 *
Decabromodiphenyl ether
1163-19-5 6.23E-10** -6.31 ** 12.11 ** None; 18.42 **
2.84E-11 **
Chlordecone 143-50-0 2.25E-07* -5.66 5.41 None; 11.07 **
2.70*
Hexabromobiphenyl 82865-89-2
2.49E-09 ** -4.17 ** 9.10 ** None; 13.27 **
1.80E-06 **
Hexa- and heptabro-modiphenyl ether##
-- 2.87E-09 *,** to 3.10E-10*,**
-4.72 to -5.11 **
8.55 to 9.44 **
13.27 to 14.55 **
2.16E-07 to 4.15E-06
**
Hexachlorobutadi-ene
87-68-3 2.20E-01 * -0.37 * 4.78 None; 5.16 **
3.2 *
alpha-Hexachlorocy-clohexane
119911-70-5
3.52E-05 * -4.76 * 4.14 8.84 0.24-8.0 *
beta-Hexachlorocy-clohexane
319-85-7 3.52E-05 * -4.76 * 4.14 8.84 0.24-8.0 *
Lindane (gamma-Hexaxhlorocyclo-
hexane)
58-89-9 3.52E-05 * -4.76 * 4.14 8.84 0.24-8.0 *
Pentachlorobenzene 608-93-5 1.01E-03 * -1.54 * 5.17 6.49 0.83 *
Pentachlorophenol and its salts/esters
87-86-5 1.10E-04 * -6.00 * 5.12 None; 11.12 **
14.0 *
Polychlorinated napthalenes#
-- 2.10E-06 * -2.32 ** 6.39 ** 9.25 0.043 **

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
19
Short-chain chlorin-ated parraffins
(SCCPs)
-- -- -- -- -- --
technical Endosulfan and isomers
115-29-7 6.00E-07 * -2.8 * 3.83 8.64 0.33~
Tetra- and pentabro-modiphenyl ether###
-- 3.10E-08 to 7.00E-08 *
-3.92 to -4.32 **
6.77 ** to 6.84
10.53 to 11.31
0.0004 to 0.001 **
~ @ 22 degrees C
* @ 25 degrees C
** predicted
+ PCBs 77, 138, 153, 169, 180
++ 2,3,7,8-TCDD and 2,3,7,8-TCDF
+++ potassium salt PFOS
# PCN 52
## BDE-154 and 183 ### BDE-47 and 99
1.1.2 Toxicity of legacy POPs As mentioned previously, POPs are lipophilic, and bioaccumulate as a result of their resistance to hydrolysis and strength of chemical bonds (Ritter et al., 1995). The primary route of exposure to the legacy POPs for humans is via diet, largely through meat and fish products (Schecter, 1994). Biomag-nification of certain POPs (apart from endrin and lindane) has been ob-served (Schecter, 1994), tending to accumulate in adipose tissue (Geyer et al., 2000). Toxicity of POPs have been and continue to be investigated in both laboratory and case-studies of acute exposure (Schecter, 1994). Gen-erally, health impacts including neurotoxicity, cancer, reproductive, immu-nological and endocrine disruption and skin rashes (known as “chlor-acne,” resulting from prolonged or high-level dermal exposure to chlorinated com-pounds) are reported (Schecter, 1994). Health effects vary depending on the POP(s) in question, and, in the case of the multiple congener group of PCDD/Fs and PCBs, stereo and positional chemistry. Specifically, the 2,3,7,8-substituted PCDD/F congeners are the most toxic (Schecter, 1994). In the case of PCBs, the non-ortho-substituted congeners exhibit coplanar structures akin to PCDD/Fs and are thus referred to as dioxin-like PCBs (DL-PCBs) and exhibit the same physiological mechanism of action (Schec-ter, 1994). Levels of POPs are monitored in human populations by the

20
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Centers for Disease Control, the European Commission, other national and international organizations and in various academic studies (Schecter, 1994, Fiedler, 2003, Salihovic et al., 2012).
To better understand the toxic effects that combinations of POPs and other chemicals of concern have, the application of effect-directed analysis (EDA) assesses the biological activity of a total environmental sample fol-lowed by chemical analysis to identify the toxic constituents which are then tested for direct biological activity (Brack et al., 2007, 2016). The bioassays employed involve both in-vitro and in-vivo tests that look at cellular re-sponse and organism response for fish, Daphnia and algae and are intended to follow the Adverse Outcome Pathway (AOP) such that results of the bi-oassays can be translated to meaningful toxicological impacts (Brack et al., 2016). Following and during testing, samples are fractionated to afford iso-lation and partitioning of compound classes for more specific assessment (Brack et al., 2016). Though numerous challenges exist with isolation of specific compound effects one implication of this approach is the ability to incorporate emerging chemicals of concern (Brack et al., 2016), which are discussed in the following section.
1.1.3 Emerging POPs Monitoring for additional persistent, bio-accumulative and toxic chemicals has continued since the implementation of the Stockholm Convention (Muir and Howard, 2006). A study conducted in 2006 and revised in 2010 by Environment Canada (Muir and Howard, 2006, Howard and Muir, 2010) assessed 22,263 listed chemicals in commerce and industrial use for struc-tural indicators of persistence, bioaccumulation and long-range transport (Muir and Howard, 2006, Howard and Muir, 2010). The most recent study identified 610 chemicals with high suspected potential for persistence and bioaccumulation (Howard and Muir, 2010). Clearly, further assessments of compound toxicity are required, though the findings highlight that com-monly used high-production compounds potentially pose similar threats as legacy POPs. Hexabromocyclododecane (HBCDD) and per- and poly-fluoroalkyl substances (PFAS) such as perfluorooctanesulfonic acid (PFOS) represent some of the later-adopted compounds to be legislated for by the Stockholm convention, along with twelve additional compounds listed in Table 1. One important consideration for emerging POPs is possible differ-ences in terms of physico-chemical properties typical to legacy POPs (Table 1). This is exemplified by PFOS which has considerably higher vapor pres-

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
21
sure, air-water partitioning coefficient, and water solubility for the potas-sium salt PFOS which has implications for environmental distribution that may be different than legacy POPs. In 2019, another PFAS, perfluorooc-tanoic acid (PFOA) has been listed under the Stockholm Convention; per-fluorohexanesulfonic acid (PFHxS), which is under current consideration for future inclusion in the Stockholm Convention (Stockholm Convention, Chemicals proposed for listing under the Convention, 2019). Other emerg-ing contaminants discussed in the literature but not considered yet by the Stockholm Convention include novel brominated and organophosphate flame retardants (NBFRs and OPFRs, respectively) (Lorenzo et al., 2018), which in some cases may be used as replacements for previously banned PBDEs (Howard and Muir, 2010). Indeed, a number of these OPFRs also differ from legacy POPs in that they do not contain halogens, while the class exhibits concerning characteristics such as ubiquitous contamination in the environment and potential toxicity and carcinogenicity (Stubbings et al., 2017). Overall, continued assessment of chemicals for their possible threat to environmental and biological health is necessary.
1.1.4 Consideration and challenges in POPs monitoring Study of the fate and behavior of POPs in the environment and biota follow the basic workflow, described by Guo and Kannan (2015). The steps of this workflow include initial sampling of environmental or biological matrices, transportation and storage of the samples to mitigate contamination alter-ation of samples, and finally analysis (Guo and Kannan, 2015). Sample analysis is comprised of sample extraction, purification of extracts, separa-tion, identification and, in many cases, quantification, and reporting (Guo and Kannan, 2015). Modern approaches for the detection and quantifica-tion of legacy and emerging POPs rely largely on mass spectrometry (MS) following some form of chromatographic separation and will be discussed in the coming sections. Though not all studies follow this approach com-pletely (eg. some studies provide qualitative, not quantitative assessment), there exist commonly recognized considerations and challenges. POPs anal-ysis encompasses diverse environmental phases (air, water, soil) which re-quire specific sample treatment prior to detection (Guo and Kannan, 2015, Lorenzo et al., 2018). Numerous matrix constituents can interfere with the selectivity, specificity and sensitivity of POPs analysis, and their removal is necessary though not always complete (Guo and Kannan, 2015, Lorenzo et al., 2018). Concentrations of POPs can span several orders of magnitude across samples (Megson et al., 2016), requiring detection techniques with a

22
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
wide quantitative range. Additionally, sub-ppt level concentrations in envi-ronmental and biological samples demand methods which can accurately and precisely measure at low levels (Guo and Kannan, 2015, Lorenzo et al., 2018).
Emerging POPs monitoring studies share the above considerations and challenges, while also presenting unique ones. Both HBCDD and PFOS (along with most other ionic PFAS) in their native state require liquid chro-matography (LC) separation (Covaci et al., 2007, Guo and Kannan, 2015, Lorenzo et al., 2018). This is divergent from the traditional use of gas chro-matography (GC), discussed later in this manuscript and widely imple-mented in the analysis of legacy POPs. HBCDD and PFOS additionally ex-hibit complex stereochemistry (Heeb et al., 2005, Langlois, 2007), and, though not unique among the POPs, is a consideration from the chromato-graphic method perspective. Prior to and during detection, contamination from equipment used for monitoring can be introduced from emerging POPs used in numerous consumer goods including laboratory items (Lo-renzo et al., 2018). This is true for PFAS and OPFRs, and analysts currently replace contaminating materials and/or use trapping mechanisms nested in the analytical equipment prior to sample injection (Lorenzo et al., 2018). Outside of the Stockholm Convention listing, emerging POPs or contami-nants of concern may not have authentic standards making absolute quan-tification impossible (Muir and Howard, 2006, Howard and Muir, 2010, Lorenzo et al., 2018), or if available may lack isotopically labelled standards which have been found to improve quantitative methods (Muir and How-ard, 2006, Guo and Kannan, 2015). Perhaps the grandest challenge for emerging POPs is posed by Muir and Howard (2006) and Howard and Muir (2010): with the thousands of chemicals in use currently, only a small percentage are monitored in targeted methods (Muir and Howard, 2006, Howard and Muir, 2010). Effectively detecting and confirming the identity of environmental contaminants require innovative approaches not used in routine monitoring of POPs (Muir and Howard, 2006).
1.2 Mass spectrometry-based analysis methods for POPs
1.2.1 Chromatography techniques for POPs analysis Prior to MS detection, chromatographic separation of analytes is a critical step of the analytical process. In the case of the legacy and many emerging POPs, GC historically has been the most widely utilized method. Legacy POPs are well suited to GC analysis due to their volatility (refer to Section 1). GC

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
23
typically employs a temperature gradient, during which an injected analyte travels through a capillary column coated with a stationary phase aided by the flow of a carrier gas (McMaster, 2008). Volatized sample is introduced to the column and gaseous analytes interact with the stationary phase to varying degrees. This results in the focusing of the analytes into chromato-graphic peaks which are directed from the column outlet to the detector (McMaster, 2008). Although POPs analyses are routinely performed using GC, there have been cases of co-elutions for some PCB and PCDD/F conge-ners on generic GC columns (Reiner et al., 2006). The use of specialized proprietary stationary phases such as BPX-DXN (SGE Analytical Science) and Rtx-Dioxin2 (Restek Corporation) columns can help improve separa-tions for compound classes (Reiner et al., 2006). Additional studies on PCDD/Fs utilized high resolution gas chromatography (HRGC), such as dual column gas chromatography (GCXGC) or 60 m columns for enhanced chromatographic resolution (Reiner et al., 2006, Hajšlová et al., 2007).
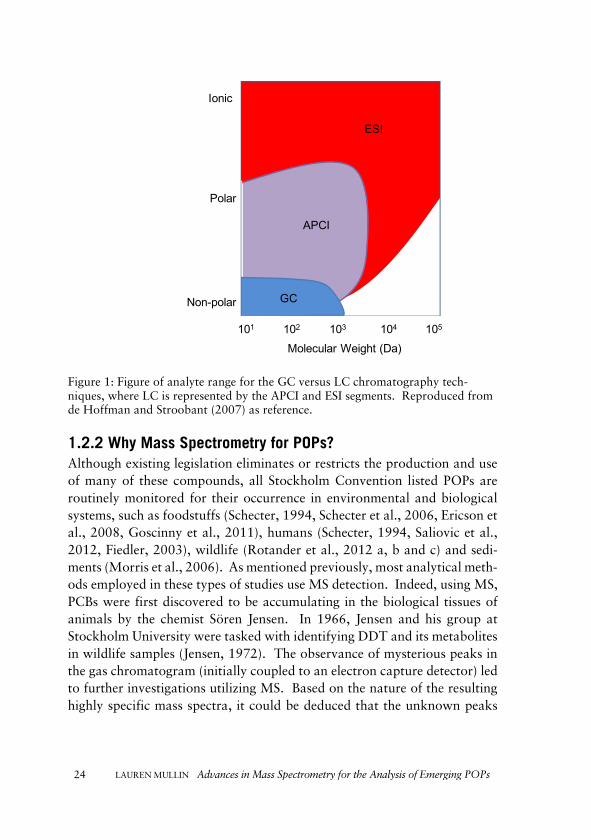
LC is a separation technique more suited to non-volatile and more polar compounds, which cannot, in their native form, be converted to the gas phase for GC analysis (de Hoffmann and Stroobant, 2007, Watson and Sparkman, 2007). Reversed-phase liquid chromatography (RPLC) utilizes a non-polar stationary phase, and a polar mobile phase to separate analytes based on their interaction with the stationary phase. The composition of the mobile phase determines analyte retention and their subsequent elution off the column (Dorsey and Dill, 1989). Typically, the columns are packed with spherical silica particles, and a very wide array of surface derivatives afford options for separations of diverse compounds (Dorsey and Dill, 1989). The interfacing of LC with MS was slightly more challenging than in GC initially, as a result of the large volume of liquid introduced affecting the gas pressure of the MS source (Watson and Sparkman, 2007). However, a variety of ionization techniques have been developed and coupled with LC to make it well suited to MS, which include electrospray ionization (ESI), atmospheric pressure chemical ionization (APCI) and atmospheric pressure photoionization (APPI) which are described in Section 1.2.2 (de Hoffmann and Stroobant, 2007, Watson and Sparkman, 2007). As com-pared to GC, LC has greatly increased the number of compounds which can be introduced into the MS following separation, as displayed by Figure 1 (de Hoffmann and Stroobant, 2007). Further advances in coupling APCI ionization with GC and SFC are discussed in Sections 2.2 and 4.1.1, respec-tively, later in this thesis.
24
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Figure 1: Figure of analyte range for the GC versus LC chromatography tech-niques, where LC is represented by the APCI and ESI segments. Reproduced from de Hoffman and Stroobant (2007) as reference.
1.2.2 Why Mass Spectrometry for POPs? Although existing legislation eliminates or restricts the production and use of many of these compounds, all Stockholm Convention listed POPs are routinely monitored for their occurrence in environmental and biological systems, such as foodstuffs (Schecter, 1994, Schecter et al., 2006, Ericson et al., 2008, Goscinny et al., 2011), humans (Schecter, 1994, Saliovic et al., 2012, Fiedler, 2003), wildlife (Rotander et al., 2012 a, b and c) and sedi-ments (Morris et al., 2006). As mentioned previously, most analytical meth-ods employed in these types of studies use MS detection. Indeed, using MS, PCBs were first discovered to be accumulating in the biological tissues of animals by the chemist Sören Jensen. In 1966, Jensen and his group at Stockholm University were tasked with identifying DDT and its metabolites in wildlife samples (Jensen, 1972). The observance of mysterious peaks in the gas chromatogram (initially coupled to an electron capture detector) led to further investigations utilizing MS. Based on the nature of the resulting highly specific mass spectra, it could be deduced that the unknown peaks
APCI
ESI
GC
APCI
GC
Polar
Ionic
Non-polar
101 102 103 104 105
Molecular Weight (Da)

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
25
contained multiple chlorines (Cl) due to the distinctive 35Cl:37Cl isotopic dis-tribution pattern and fragmentation from losses of Cl on the structure. From this data, structural elucidation could be performed on these unknown com-pounds, and they were identified as PCBs. Up to this point they had not been recognized to have been a contaminant in wildlife and the environment (Jensen, 1972). This early example shows the advantage of MS detection for the identification of contaminants in environmental samples.
MS is highly sensitive, specific, produces rapid results and is diverse in its applications (de Hoffmann and Stroobant, 2007). A MS system is com-posed, in a very basic sense, of an inlet, or means of generation and intro-duction of ions into the gas phase; a mass analyser e.g. quadrupole either single or multiple, which separates the ions according to their mass to charge ratio (m/z); and a detector, which records the ion events and their abundance as they arrive from the analyzer (de Hoffmann and Stroobant, 2007). Most currently available MS configurations can detect compounds at femto- to attomole levels (de Hoffmann and Stroobant, 2007). Many MS systems also contain a high-energy section before or in between the an-alyzer(s) where fragmentation of the parent molecule occurs (McLafferty and Tureček, 1993, de Hoffmann and Stroobant, 2007). Known as tandem mass spectrometry, or MS/MS, the fragment ions generated from this pro-cess can be separated in the second analyzer followed by detection. Speci-ficity is afforded by resolution between measured masses, which continually improves as designs advance, as well as the generation of fragments from a parent compound, providing structural information about the molecule (McLafferty and Tureček, 1993, de Hoffmann and Stroobant, 2007). MS data can be viewed practically immediately, with modern computing greatly improving the analytical tools required to deduce structural information from the spectrum. The wide range of applications that utilize MS can in part be attributed to the diversity of ion sources available (de Hoffmann and Stroobant, 2007). Also, multiple types of MS configurations and styles exist (Watson and Sparkman, 2007, de Hoffmann and Stroobant, 2007), and a more thorough description of some will be addressed later in this section.
Ionization sources have evolved over the years to include a wide array, each generating ions in a unique manner suited to different types of com-pounds. Choice of ionization type is in some cases dependent on the nature of sample introduction (i.e. GC, LC, direct insertion, etc.). Two fundamen-tal types of ionization, electron ionization (EI) and chemical ionization (CI) were described by McLafferty (1993) as being “hard” and “soft” ionization

26
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
techniques, respectively. They result in different forms of the ionized mol-ecule. EI uses a heated filament which produces a beam of energized elec-trons, at a potential of about 70 eV, with about 10 to 20 eV transferred to the molecules, to generate molecular ions of the analyte (M•+) (McLafferty and Tureček, 1993, de Hoffmann and Stroobant, 2007). This process occurs under vacuum and following ionization the ions then travel through ion focus electrodes, to the mass analyzer (McLafferty and Tureček, 1993, de Hoffmann and Stroobant, 2007). The term “hard” ionization refers to the fact that due to the excessive energetic potential applied in traditional EI to the molecules during ionization, noting that most molecules will ionize at 10 eV, fragmentation occurs, in some cases to the point where virtually no molecular parent ion exists (McLafferty and Tureček, 1993). So, although EI is suitable for most organic compounds, identification of molecules may be difficult if they experience this intense fragmentation (McLafferty and Tureček, 1993, de Hoffmann and Stroobant, 2007). Recent advances using “soft” EI (using 10-20 eV) by both Agilent and Markes International have focused on generating a stable electron beam at these lower eV levels to maximize signal from the more intact precursor molecule (Wang, 2016, Markes International, 2016) are promising but show little uptake in the field of POPs analysis to date. In the case of CI, precursor ions with much lower rates of fragmentation prior to reaching the source are observed (McLafferty and Tureček, 1993). In order to achieve CI, the analyte mole-cule traverses a free path and collides with in-source primary ions, produced by the introduction of reagent gases such as ammonia or methane (McLafferty and Tureček, 1993).
While EI and CI conditions are optimized for analytes introduced in the gas phase, they are often coupled to GC or a direct insertion probe, where high temperatures are used to introduce analytes into the ionization cham-ber. For compounds that are less volatile and utilize LC, such as PFAS, other ionization mechanisms are required. The most commonly used ionization modes for less volatile compounds are ESI and APCI, introduced in section 1.2.1. Unlike EI and CI, these ionization techniques occur under atmos-pheric pressure conditions and are collectively referred to as API (de Hoff-mann and Stroobant, 2007). For ESI the LC effluent feeds into a charged capillary tube, and, with the assistance of a nebulizer gas, droplets are sprayed from the capillary into an atmospheric pressure source region (Bru-ins, 1998). An electric field is produced in the region of the capillary tip by the placement of a counter-electrode near the charged capillary. This results in the application of a charge on the surface of the liquid droplets as they
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
27
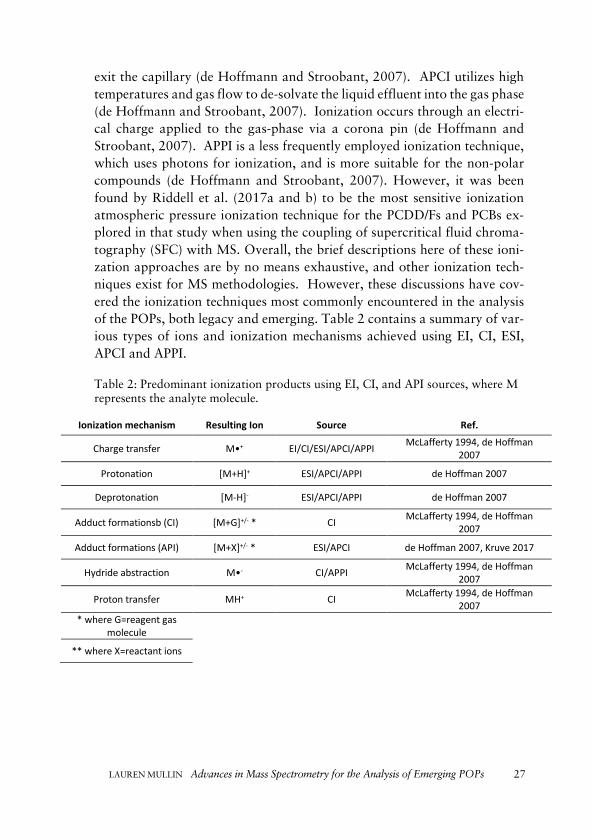
exit the capillary (de Hoffmann and Stroobant, 2007). APCI utilizes high temperatures and gas flow to de-solvate the liquid effluent into the gas phase (de Hoffmann and Stroobant, 2007). Ionization occurs through an electri-cal charge applied to the gas-phase via a corona pin (de Hoffmann and Stroobant, 2007). APPI is a less frequently employed ionization technique, which uses photons for ionization, and is more suitable for the non-polar compounds (de Hoffmann and Stroobant, 2007). However, it was been found by Riddell et al. (2017a and b) to be the most sensitive ionization atmospheric pressure ionization technique for the PCDD/Fs and PCBs ex-plored in that study when using the coupling of supercritical fluid chroma-tography (SFC) with MS. Overall, the brief descriptions here of these ioni-zation approaches are by no means exhaustive, and other ionization tech-niques exist for MS methodologies. However, these discussions have cov-ered the ionization techniques most commonly encountered in the analysis of the POPs, both legacy and emerging. Table 2 contains a summary of var-ious types of ions and ionization mechanisms achieved using EI, CI, ESI, APCI and APPI. Table 2: Predominant ionization products using EI, CI, and API sources, where M represents the analyte molecule.
Ionization mechanism Resulting Ion Source Ref.
Charge transfer M•+ EI/CI/ESI/APCI/APPI McLafferty 1994, de Hoffman 2007
Protonation [M+H]+ ESI/APCI/APPI de Hoffman 2007
Deprotonation [M-H]- ESI/APCI/APPI de Hoffman 2007
Adduct formationsb (CI) [M+G]+/- * CI McLafferty 1994, de Hoffman 2007
Adduct formations (API) [M+X]+/- * ESI/APCI de Hoffman 2007, Kruve 2017
Hydride abstraction M•- CI/APPI McLafferty 1994, de Hoffman 2007
Proton transfer MH+ CI McLafferty 1994, de Hoffman 2007
* where G=reagent gas molecule
** where X=reactant ions

28
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
1.3 Types of MS used for POPs analysis Numerous MS systems have been used in research settings over the years for POPs analyses. However, three should receive special attention due to their wide-spread and/or emerging use for the analysis of POPs. Magnetic sector, tandem-quadrupole (TQ) and time-of-flight (TOF) MS technologies will be discussed in the proceeding section.
1.3.1 Magnetic sector One of the earliest types of mass analysers, the magnetic sector is still widely employed in routine PCDD/F analysis, though has much evolved from its original form (Watson and Sparkman, 2007). The separation of masses in the magnetic sector is initiated by the acceleration of ions via an electrical potential difference (Vs) at the source site such that they have a given kinetic energy (Ek). The ions then pass through a magnetic field (of strength B), which separates ions based on their momentum (expressed by velocity (υ) multiplied by mass (m)) (McLafferty and Tureček, 1993, Watson and Sparkman, 2007 and de Hoffmann and Stroobant, 2007).
Due to double focussing of combined electrical sector and magnetic fields, modern magnetic sectors have been referred to as high resolution MS (HRMS) (Figure 2 is an example configuration). For the PCDD/Fs, mag-netic sectors have been in common use since the beginning of routine dioxin analysis in 1973 (Reiner et al., 2006). Indeed, the enhanced selectivity as a result of the instrumental resolution combined with notable sensitivity for these compounds (Reiner et al., 2006), makes magnetic sectors well suited to PCDD/F analysis in a range of complex matrices. As noted by Megson et al. (2016), single ion monitoring (SIM) is the widely used acquisition ap-proach which means analytes outside of the scheduled channels are lost—limiting the potential for non-targeted analyses that are of increasing inter-est for POPs and environmental contaminant analysis (Megson et al., 2016). Other general drawbacks include high cost, difficulty of use to non-expert users and commercial availability (Megson et al., 2016).

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
29
Figure 2: Schematic of double magnetic sector of the EB design, where M and M2 are two different masses. Reproduced from Watson and Sparkman (2007) as ref-erence.
1.3.2 Tandem quadrupole Another popular configuration for the analysis of the POPs utilizes some form of quadrupole MS, often in the form of TQ-MS. This system employs some means of fragmentation via collision-induced dissociation (CID) or alteration of the parent molecule from ionization to the arrival at the detec-tor (de Hoffmann and Stroobant, 2007). Figure 3 provides a basic sche-matic of this process. MS1 and MS2 are generally quadrupoles which func-tion as the mass selector and is comprised of four parallel rods with alter-nating positive and negative applied voltages (de Hoffmann and Stroobant, 2007). Multiple reaction monitoring (MRM) is a commonly employed ac-quisition approach for TQ-MS and following CID in the collision cell of the selected precursor mass, specified product ion masses are selected in MS2. Nominal mass resolution is attained on TQ-MS systems, and this technique can be considered low-resolution as a result.
E+
E-
Ion Source
B2
B1
MM2
Electric Sector

30
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Figure 3: Basic schematic of TQ-MS configuration. MS1 and MS2 represent the two quadrupole regions, or mass selectors, with fragmentation occurring in the collision cell in between (Watson and Sparkman, 2007).
For known masses of PCDD/Fs, TQ-MS analysis has been successful in
various matrices, in large part due to the recognizable and specific loss of CO37Cl from the parent molecule (Reiner et al., 2006, Watson and Spark-man, 2007). Also, a wide linear range spanning in most cases from 4 to 6 orders of magnitude for most TQ-MS systems makes them well suited for quantitative analysis (Watson and Sparkman, 2007, Megson et al., 2016). In fact, accurate quantitative analyses are the primary strength and use of TQ-MS systems. Various publications have employed TQ-MS successfully to quantify POPs in environmental and biological matrices (Dodder et al., 2006, Loos et al., 2009, Abb et al., 2011), and TQ-MS is one of the most widely used technique for new POPs and emerging contaminants (Megson et al., 2016, Lorenzo et al., 2018). More recently studies have employed the novel atmospheric pressure gas chromatography ionization source (de-scribed in Section 2.2) to enhance signal and specificity of the monitored analytes (Geng et al., 2014, van Bavel et al., 2015, Geng et al., 2016, Geng et al., 2017, Organtini et al., 2015, Organtini et al., 2015, Portoles et al., 2015 a and b, Portoles et al., 2016, Rivera-Austrui et al., 2017). Another practical consideration that results in the popularity of TQ-MS systems is the lower cost (Megson et al., 2016), as compared to that of a HRMS sys-tem.
1.3.3 Time-of-flight Gaining popularity in the field of contaminant analysis, TOF-MS systems have multiple configurations and utilize the time of an ion’s travel to deter-mine m/z (Watson and Sparkman, 2007, de Hoffmann and Stroobant, 2007). In a TOF-MS , the ions are accelerated and all have the same kinetic energy, but separate from one another during their travel under high vac-uum through a flight tube based on their mass (Watson and Sparkman 2007). Ions of the same mass then arrive in packets at the detector (Watson and Sparkman, 2007). Unlike the TQ-MS system described above, exact
MS1 MS2Collision Cell

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
31
mass can be determined using modern TOF instrumentation (Watson and Sparkman, 2007), typically to 4 decimal places. Exact mass theoretically allows for determination of the molecular formula of an unknown without as much ambiguity and without relying on fragmentation information as in a TQ-MS system. The measurement of a MS systems ability to separate and measure masses (resolution, or resolving power) is determined by the fol-lowing relationship:
R = m/∆m
(de Hoffmann and Stroobant, 2007)
Where ∆m is equal to the peak width of the mass measured at 50% (de Hoffmann and Stroobant, 2007). This measurement of resolution is re-ferred to as full width at half-maximum height (FWHM) (Watson and Sparkman, 2007). This differs from the measure of resolution on a mag-netic sector, where the valley between two peaks of a given mass is measured on a percentage basis (10% valley) of the peak height (de Hoffmann and Stroobant, 2007). The range of m/z values that can be monitored in a single experiment is broader as compared to the TQ-MS system (Watson and Sparkman, 2007). Full mass range is generally acquired in TOF-MS analy-sis, rather than specified nominal masses as in MRM experiments. Alt-hough the acquisition of a wide mass range (referred to as full scan or full spectrum) is not unique to TOF-MS systems, more information is gained due to the exact mass specificity. Figure 4 shows a basic TOF-MS schematic. Other features of TOF-MS systems include the introduction of a quadrupole prior to the flight tube, as well as collision cells, such that MS/MS experi-ments can be performed through CID (Watson and Sparkman, 2007, de Hoffmann and Stroobant, 2007). Additionally, full spectrum analysis is more sensitive on TOF-MS systems than on previously mentioned TQ-MS systems when scanning a mass range.

32
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Figure 4: Schematic of a Waters LCT premier TOF-MS. Image taken from appli-cation note 720000648EN on www.waters.com
TOF-MS for the analysis of POPs has been used in several older (Eljarrat et al., 2002, Focant et al., 2004, Hajšlová et al., 2007) and increasingly found in more recent studies focused on discovery of new POPs (Barzen-Hanson et al., 2015, Rotander et al., 2015, Newton et al., 2016, Fernando et al., 2016, Barzen-Hanson et al., 2017, Lorenzo et al., 2018). TOF-MS has great success in these studies with regards to identification of emerging POPs and other contaminants in both targeted and non-targeted studies, as a non-specific full mass range can be acquired, allowing for a potentially unlimited number of compound identifications. Isotopic distributions are also conserved, affording exceptional utility in the elucidation of Cl- and Br-containing molecules. For GCxGC methods which produce very narrow chromatographic peaks, use of TOF-MS operating at a rapid acquisition rate across the full experimental mass range is the ideal detector option (Fo-cant et al., 2004 and Megson et al., 2016). Lastly, recent hardware improve-ments such as increased ion focusing lens designs changes to TOF-MS in-strumentation increase sensitivity, while software advances accommodate data processing of highly rich non-targeted data independent acquisitions (DIA). With increasing interest in characterizing samples from the perspec-tive of a non-targeted approach, TOF-MS will likely see increasing use for POPs and environmental contaminant analyses.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
33
2.0 Alternative techniques for mass spectrometry-based analysis of POPs With the complexities of structures legacy and emerging POPs exhibit, the chromatographic separation and unequivocal MS-based detection has in-deed experienced its share of challenges. Chromatographic separation pro-vides the one challenge for isomers, particularly in the case of chiral com-pounds due to the nearly identical physico-chemical properties of enantio-mers (Eljarrat et al., 2008). As a result, there have been many innovative approaches to enhance separations, including chiral separations. Addition-ally, the analysis of emerging contaminants includes using less rigorous sam-ple clean-up techniques, resulting in ionization of various co-eluting com-pounds from the sample itself presenting a challenge in spectral deconvolu-tion and structural identification. Techniques which increase the specificity of identifications are thus becoming increasingly important in non-targeted data independent acquisitions. Overarching these considerations, any means to increase sensitivity are also desired. Alternative methods of chro-matography, ionization and gas-phase separations are finding utility in POPs analysis, namely supercritical fluid chromatography (SFC), atmos-pheric pressure gas chromatography, and ion mobility spectrometry (IMS). Upon introducing some of these advances, the following section highlights where these alternative techniques present opportunities for improvement of POPs and emerging contaminant analysis.
2.1 Supercritical Fluid Chromatography SFC is a separation technique based on the use, as its name would imply, of a supercritical fluid. This is a substance that has surpassed its critical point of temperature and pressure (Figure 5) (Lee and Markides, 1990). Not all substances are suited for practical conversion to a supercritical fluid, as the critical temperature or pressure required may exceed sustainable levels (Lee and Markides, 1990). At the critical point, the substance is equal parts liq-uid and gas, while beyond the critical point, the substance is neither liquid nor gas, instead exhibiting unique properties that are similar to both in dif-ferent respects (Lee and Markides, 1990). Those qualities that are most relevant to separations techniques are higher diffusivities and lower viscos-ities than liquids, while at the same time having higher viscosities than gases (Lee and Markides, 1990). Another factor influencing chromatographic ef-ficiency is density of the mobile phase. Density increases significantly when

34
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
comparing a supercritical fluid to a gas, on the order of 100 to 1,000 fold greater (Lee and Markides, 1990). This density increase results in improve-ments in molecular interactions as a result of shorter intermolecular dis-tances (Lee and Markides, 1990). The result of the properties of a super-critical fluid is a substance with enhanced solvating properties. The appli-cations of SFC utilize both packed and open tubular columns (Smith, 1999), but the following discussion will focus on packed columns.
Figure 5: Phase diagram adapted from Lee et al. (1990) indicating the critical point for an ambiguous substance, whereupon the formation of supercritical fluid is de-pendent on temperature and pressure.
The measurement of chromatographic efficiency for packed columns is described in detail by the van Deemter equation, displayed below:
H = A + B/u +Cu (Lee and Markides, 1990)
Where H is height equivalent of a theoretical plate (H.E.T.P.) (Van Deemter et al., 1956), A represents non-uniformity of flow (eddy diffusion), B is the fraction of h caused by longitudinal diffusion, C is the fraction of h caused by resistance to mass transfer in the mobile and stationary phases, and u is the velocity of the mobile phase (Lee and Markides, 1990). Faster flow rates are possible in SFC as compared to LC (Hühnerfuss and Shah, 2009), resulting in a higher u term and, thus, a lower h value.
Temperature
Pres
sure
Criti
cal
Pres
sure
Critical Temperature
Supercritical Fluid
Solid
Liquid
Gas

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
35
These terms used to describe chromatographic processes require some background on plate theory. Plate theory originates from distillation and was extrapolated to chromatography by Martin and Synge in 1941 (1941, Giddings, 1991). Further work to describe the particulars of chromato-graphic mass transfer was performed by van Deemter in 1956 (per the equa-tion in his name above) (Van Deemter et al., 1956). A theoretical plate (N) can be described as the occurrence of equilibrium between the mobile phase and solute on a column (Martin and Synge, 1941). In terms of chromato-graphic efficiency, a lower H value is desirable as this allows for a higher number of theoretical plates. This relationship is described in the below equation:
H=L/N (Andrés et al., 2015)
Where L is the column length in m. Further derivation of this equation
to solve for N=L/H, whereby a lower H can be seen to result in a higher N as described above.
These factors ultimately result in improved kinetic performance and unique chromatographic considerations for SFC as compared to LC. Per-renoud et al. (2012, 2013) explored this comparison using both 3.5 µm and 1.7 µm particle size columns for SFC and LC separations of small molecule pharmaceuticals. The SFC system using a smaller particle size column re-sults in the highest chromatographic efficiency, determined by the greatest range for the u term at the lowest H values. With regards to smaller particle diameters (dp) in both LC and SFC, a general decrease in mass transfer re-tardation is observed. This is because of a direct relationship between the resistance to mass transfer (C term, defined above) and dp (Perrenoud et al., 2012). For separation of stereoisomers, SFC is quite suitable not only be-cause of the enhanced solvating power, but also because of lower tempera-tures than GC analysis, theoretically affording a wider range of column en-antioselectivity (Hühnerfuss and Shah, 2009).
The use of SFC in environmental contaminant applications has had suc-cess for the analysis of 2,3,7,8-substituted PCDD/Fs, PCBs, polyaromatic hydrocarbons (PAHs), flame retardants, PFAS and pesticides in a range of complex matrices (Lee and Markides, 1990, Smith, 1999, Mullin et al., 2015, Riddell et al., 2015, Riddell et al., 2016, Riddell et al., 2017b, Yeung et al., 2017, Lorenzo et al., 2018). However, widespread adoption of SFC for the analysis of environmental contaminants has yet to occur, and in part could be due to regulatory methods indicating largely GC based methods

36
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
(Lee and Markides, 1990). With regards to chiral applications using SFC, most of these have been applied to pharmaceutical analysis (Smith, 1999), except for recent work by Riddell et al. (2016) separating HBCDD enanti-omers. Although most legacy POPs have yet to be explored for their ana-lytical possibilities using SFC, work by van Bavel et al. (1995 and 1996) investigated the use of supercritical fluid extraction (SFE) methods for the extraction of PCDD/Fs and PCBs. In this work, the contaminants’ extrac-tion from adipose tissue proved to be more efficient and less costly than the existing liquid extraction techniques.
Indeed, further investigation into SFC for POPs analysis is warranted. The interfacing of these modern SFC systems with MS affords for increased sensitivity, as has been seen on past SFC/MS systems (Lee and Markides, 1990, Smith, 1999). Figure 6 shows a schematic of one such newly intro-duced SFC/MS system.
Figure 6: Schematic of recently released for commercial use SFC system (the Ultra Performance Convergence Chromatography system from Waters Corporation), with MS coupling and supplemental make-up flow binary pump, illustration by Chris Hudalla formerly of Waters Corporation. PDA=photodiode array detector; CM=column manager; CCM=convergence man-ager, which houses the automated back pressure regulator (ABPR) and maintains the system pressure for the formation of supercritical CO2; SM=sample manager; BSM=binary solvent manager; PCM=pump control module.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
37
2.2 Atmospheric Pressure Gas Chromatography In gas chromatography-atmospheric pressure chemical ionization (GC-APCI), also referred to as APGC (atmospheric pressure gas chromatog-raphy), the GC effluent is swept into an atmospheric pressure MS source where ionization is induced by means of a corona discharge. A heated trans-fer line interfaces the GC analytical column to the MS source. Nitrogen is introduced into the ionization chamber from both the sample cone gas, aux-iliary gas and the heated transfer line from the GC interface. Ionization oc-curs through reactions between the charged nitrogen plasma (containing N2
+ and N4+ cations) generated from a corona pin discharge and analyte
molecules. The resulting ions are primarily the M+• ion for non-polar ana-lytes, as described below. Alternatively, introduction of a protic solvent (e.g. water) into the source will allow for protonation reactions generating the [M+H]+ ion for more polar molecules. Compared to the 70 eV energy typi-cally imparted during EI (McLafferty and Tureček, 1993), APGC is a “softer” technique resulting in less fragmentation (Li et al., 2015). Reduced fragmentation of the precursor ion within the source allows for controlled collision cell fragmentation. Sensitivity using APGC MS is increased over EI MS in part given the reduced fragmentation in the source. Moreover, the atmospheric pressure source design affords the switching between LC and GC inlets on a single MS system, increasing analyte coverage (Li et al., 2015). With regards to applications in POPs analysis, use of APCI interfaced with GC was first described by Mitchum et al. for the analysis of the 22 TCDD isomers (Mitchum et al., 1980). This and following studies (Korf-macher et al., 1983, 1984) highlighted the use of negative polarity APCI ionization to eliminate chromatographic interferences from other halogen-ated pollutants with the TCDD isomers in fish, pork and beef tallow, and snake eggs (Korfmacher et al., 1984, 1983; Mitchum et al., 1980). Recent work in biological and environmental matrices have assessed the use of APGC with positive polarity charge transfer ionization for the analysis of PCDD/Fs (van Bavel et al., 2015), PCBs and organochlorine pesticides (Geng et al., 2016) and PBDEs and their metabolites the methoxylated PBDEs (MeOH-PBDEs) (Geng et al., 2017). The work highlighted either equivalent or enhanced performance of using the APCI mechanism as com-pared to EI with regards to detection capability, as well as preservation of the intact analyte molecule as compared to EI. Similar trends, ultimately aiding in congener and isomer differentiation, were observed when analysed

38
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
mixed halogenated (bromo/chloro) PXDD/F species in both simulated do-mestic (Organtini et al., 2015 a, b) and industrial burn site samples (Fer-nando et al., 2016). Fernando et al. (2016) differentially implemented the use of negative ionization, as described in the final ionization equation above, generating the pseudo-molecular ion and enhanced specificity for isomer differentiation.
2.3 Ion Mobility Spectrometry IMS has seen renewed interest in recent years, especially with advances that enable it to be coupled with MS. IMS was originally termed plasma chro-matography (Collins and Lee, 2002), though the name has since changed to more accurately reflect its function. IMS separations are obtained by the application of an electrical potential to a gas-filled drift cell. Ions travel through the drift cell and arrive at a detector. The differing velocities of ions as they pass through the drift cell determine the drift time (measured in ms) and are a result of the number of collisions an ion has with the neutral gas molecules, such as N2, which populate the drift cell (Collins and Lee, 2002, Stach and Baumbach, 2002).
Different methods of ion mobility include drift time ion mobility spec-trometry (DT-IMS), field-assymetric waveform ion mobility spectrometry (FAIMS) which is also referred to as differential ion mobility (DMS) (Ah-med et al., 2019), and the more recently introduced travelling wave ion mo-bility spectrometry (TW-IMS) (Kanu et al., 2008). The use of DT-IMS is often not differentiated from IMS, as it was the initial form, and utilizes simply the movement of an ion through a gas filled drift cell at a single applied electrical field (Kanu et al., 2008). In FAIMS, an increased strength electrical field is applied between two electrodes (Collins and Lee, 2002, Kanu et al., 2008). The ions migration is different between the two elec-trodes dependent on its charge (and perpendicular to the drift gas direction), resulting in a divergent mobility depending on the electrode the ion is mov-ing towards (Kanu et al. 2008). In this way, mobility-based separation of ions occurs. TW-IMS utilizes a high electrical field that is moved through the IM cell segments, in this way causing ion mobility (Kanu et al., 2008). As a result, ions come in timed pulses through the IM cell through interac-tions with the waves of the electrical field (Kanu et al., 2008). TW-IMS utilizes lower pressures in the IM cell then the aforementioned techniques; this allows for a more efficient travel of ions to the detector (Kanu et al., 2008). Figure 7 illustrates both linear drift tube and travelling wave drift cell configurations.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
39
Figure 7: Schematic of linear drift tube and travelling wave ion mobility drift cells (adapted from D’Atri et al. (2018)).
An ions behavior in the IM cell is dependent on their shape, charge and size (Stach and Baumbach, 2002), and is defined by the term ΩD (collision-cross section, measured in units of Å2 and hereafter referred to as CCS). A simplified and non-absolute measurement of the various ions travelling through the IM cell can be determined as drift time (measured in ms), men-tioned previously. Drift gas composition is also an important factor dictat-ing the nature of an ion’s mobility, and various types have been employed, including N2, Ar, Ar/methane mixtures and CO2. The impact that the gas choice will have on the separation of ions is dependent on its density and the polarity of gas molecules (Collins and Lee, 2002, Kanu et al., 2008). The temperature of the gas in the IM region is a contributing factor for mobility values of an ion. Work by Bush et. al. (2010, 2012) using TW-IMS to standardize the determination of CCS for peptides has afforded for the use of an IM calibration procedure using DL-polyalanine, and more re-cently custom mixes designed for longer storage dates. Using such a calibra-tion procedure, relative recorded drift times of an ion can be adjusted to an absolute CCS value for that ion, irrespective of laboratory variations that will occur. This has been demonstrated in an inter-lab study conducted DT-IMS systems across four sites, analyzing metabolites, fatty acids, peptides and proteins and finding less than 1.5% error for CCS values as compared to those obtained on the reference system (Stow et al., 2017). These absolute
RF (-)
Anal
yser
Ion
sour
ce
Gas flow Gas flow
RF (+)Electric field
Linear Drift Tube Travelling Wave
Electric field

40
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
values can thus be utilized as an additional comparative value for compound identification (Bush et al., 2010, 2012). Use of CCS for this purpose has been demonstrated in various matrices, including for mycotoxins in cereals (Rhigetti et al., 2018), veterinary drugs in bovine urine (Tejada-Casado et al., 2018) and pesticides in fish (Regueiro et al., 2016). In these studies, certified reference materials are used to establish the experimental CCS val-ues prior to sample analysis. To further the use of CCS measurements, de-velopment of predictive models is an active area of current study. Diverging from computing-power intensive molecular modelling approaches utilized previously (Lapthorn et al., 2015), machine-learning techniques are on the rise. These programs rely on the input of previously established CCS values and identify key molecular descriptors which demonstrate an effect on the ions behavior in the drift cell (and thus its CCS value) (Mollerup et al. 2018, Bjilsma et al. 2017, Zhou et al. 2017 and 2018). Fidelity of these models with tested experimentally derived CCS values vary, and further develop-ment and refinement of this approach is certain to continue as they are of high potential utility to various MS studies performing structural elucida-tion and non-targeted analyses.
The potential of IM separations to differentiate between ions of the same mass also has much promise for the analysis of isomeric compounds and has been applied successfully to some POPs. Zheng et al. (2018) separated various isomeric PCBs, such as PCB-103 and PCB-126, PCB metabolites (OH-PCBs), and PAHs (Zheng et al., 2018) using DT-IMS. In the case of PCB-103 and -126, it was postulated that the separation occurred due to the more planar structure of PCB-126 versus PCB-103 which has only one -Cl at in the para- position and thus a more compact 3-D structure (Zheng et al., 2018). Using the DMS/FAIMS approach, branched isomers of PFOS and PFOA were separated in a much more rapid (three min.) time than tra-ditional chromatographic methods (Ahmed et al., 2019). In the case of ste-reoisomers, work performed on an IM quadrupole MS used the chiral mod-ifier (S)-(+)-2-butanol as an additive to N2 drift gas and resulted in the sep-aration of drift times for atenolol, serine, methionine, threonine, methyl-α-glucopyranoside, glucose, penicillamine, valinol, phenylalanine and trypto-phan enantiomers (Dwivedi et al., 2006). For these compounds, the ability to use a single chiral gas modifier showed promise as a rapid means for chiral separations without relying on chromatographic separations. The need for further investigation into using this system for compounds with multiple chiral centres and large molecules was, however, indicated (Dwivedi et al., 2006).

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
41
3.0 Aim of the Thesis The aim of this thesis was to develop methods addressing specific analytical gaps of emerging POPs. Documented analytical challenges for emerging POPs are vast and include the need for increased analyte coverage and spec-ificity, speed of analyses and increased sensitivity. These challenges span both targeted and non-targeted MS-based workflows and their implemen-tation across several matrices representing different environmental/biologi-cal compartments (human serum, whale blubber, water and dust). Work performed in this thesis incorporated two MS-coupled alternative tech-niques, SFC (Papers I and II) and IMS (Paper V), as well as revisiting basic LC-MS method development steps for analytically challenging new POPs (Papers III and IV) to address parts of these challenges. This required tar-geted optimization approaches for two cases, a novel brominated flame re-tardant (Papers I and II) and novel PFAS (Papers III and IV). In the final paper (V), a divergent focus used ion mobility derived gas-phase measure-ment for improving identification confidence in non-targeted acquisition for a wide array of emerging POPs and compounds of concern. Matrices repre-senting different environmental/biological compartments (human serum, whale blubber, water and dust) were addressed throughout the studies. Spe-cific objectives of each paper are described below: Paper I: Develop a rapid chromatographic separation for hexabromocy-clododecane (HBCDD) α−, β− and γ−diastereomers using SFC and optimize MS ionization conditions for best sensitivity. Demonstrate the method for quantification of HBCDD diastereomers in standards, whale blubber and human serum samples. Paper II: Develop a preparative scale SFC chiral separation method for HBCDD α−, β− and γ− (+) and (-) enantiomers and perform further charac-terization comparisons using analytical scale SFC and LC. Paper III: Provide a review of current analytical methods and global occur-rence for the emerging PFAS, hexafluoropropylene oxide-dimer acid (HFPO-DA). Discuss challenges, measurement in biotic and abiotic com-partments reported to date, and compare to legacy PFAS.

42
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Paper IV: Characterize various mobile phase additives to drive increased [M-H]- formation for HFPO-DA in order to improve LC-MS sensitivity and specificity. Paper V: Determine structural identification of various xenobiotic com-pounds present in dust using LC-QTOF-MS enabled with travelling-wave IMS. Discuss comparison of IMS-derived experimental gas-phase com-pound measurements to two predictive model values and the implications for supporting compound identification.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
43
4.0 Implementation of Alternative Techniques in Emerging POPs Analysis
4.1 Supercritical fluid chromatography for hexabromocyclodo-decane (HBCDD) The brominated flame retardant hexabromocyclododecane (HBCDD) was the third most widely used flame retardant in 2010, implemented largely in expanded and extruded polystyrene foam, as well as in furniture textiles (USEPA, HBCDD Action Plan, 2010). HBCDD is used as an additive to products, therefore leaching from these materials is likely (Alaee et al., 2003). Banned for general use (with the exception of a phase out period for extruded polystyrene foams and extruded polystyrene foams) by the Stock-holm Convention as of 2013 (Stockholm Convention, All POPs, 2019), HBCDD has been found to bioaccumulate and also to exist in numerous environmental compartments (Covaci et al., 2006). Investigations into the impact of HBCDD on mammalian health are ongoing, but initial studies have revealed reasons to be concerned. In the case of HBCDD, liver and thyroid hormone abnormalities have been observed in animal studies (Darnerud et al., 2003).
HBCDD is manufactured by the bromination of cyclododeca-1,5,9-triene (CDT) isomers (Covaci et al., 2006), and has a complex stereochemistry, with 6 stereogenic centres that result in 16 possible stereoisomers and 4 mesoforms (Heeb et al., 2005). The most predominant diastereomers in environmental and biological samples are α−,β− and γ− HBCDD (Figure 8), each containing an (+/-) enantiomeric pair (Figure 11). These stereoisomers are preferentially created from the bromination of trans,cis,cis-CDT (Heeb et al., 2005). By definition, diastereomers have different physical properties from one another (Solomons and Fryhle, 2008) and in the case of HBCDD, melting points range from 179-209°C in increasing order of the β−,α− and γ− diastereomers; water/octanol coefficients range from 5.07 to 5.47 in in-creasing order of α−,β− and γ−diastereomers; and water solubility ranges from 48.8 to 2.1 µg/L in decreasing order of α−,β− and γ−diastereomers (Eu-ropean Chemicals Agency, 2008). The proportion of these diastereomers varies depending on the sample type in which they are measured. In tech-nical blends, the γ−diastereomer predominates, followed by the α− and β−diastereomers (Eljarrat et al., 2008). Conversely, biological samples typ-ically exhibiting higher levels of the α− diastereomer (Covaci et al., 2006). One possible reason for this biological sample occurrence was postulated

44
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
by Zegers et al. (2005), who studied large marine mammals and proposed a stereoisomer specific biotransformation by the cytochrome P450 group of enzymes.
Figure 8: Stereoisomerism illustrated in the case of HBCDD (Heeb et al., 2008).
Aside from the previously described diastereomer proportional variances dependent on sample type, enantiomeric responses and behaviour of the predominant diastereomers have also been investigated in various biological matrices and is summarized by Xu et al. (2018). When equal amounts of both enantiomers exist in a formulation, it is considered a racemic mixture (Hühnerfuss and Shah, 2009, Solomons and Fryhle, 2008). The measure-ment of the interactions in a system of different enantiomers is achieved by calculating the ratio of one enantiomer with the total combined enantiomer pairs in a sample, called the enantiomer fraction (EF) (Dodder et al., 2006, Hühnerfuss and Shah, 2009). A racemic mixture will have an EF=0.5 of
Br
Br
Br
Br
Br
Br Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Br
Mirror
(-α) (+α)
(-β) (+β)
(+γ) (−γ)
RR
R
SS
R
S
S
S
S
R
R
S
RS
R
R
R
S
R
R
S
S
S
R
R
S
R
S
R
S
S
S
R
S
R

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
45
each enantiomer, while deviations in either direction suggest a process that favors one enantiomer over the other (Eljarrat et al., 2008). EFs deviating from 0.5 were observed namely for α− enantiomers (Xu et al., 2018). Due to near racemic mixtures of the same diastereomer observed in abiotic com-partments, it was postulated that the enantioselective signatures are at-tributable to metabolic processes (Xu et al. 2018). Although levels of β−HBCDD were not detected in appreciable amounts for the determination of EFs in these samples, Heeb et al. (2008) has found that racemization of this diastereomer occurs under thermal exposure conditions. The authors conclude that this should be considered in attempts to measure EFs for β−HBCDD.
HBCDD stereoisomers are typically analysed using liquid chromatog-raphy-mass spectrometry (LC-MS) with negative mode electrospray ioniza-tion. Ionization is characterised by a well conserved [M-H]- ion and under CID, the loss of -Br can be observed. Unlike the legacy and many other newly added POPs, gas chromatography (GC) analysis is unsuitable for HBCDD, as thermal interconversion under the high-temperature conditions has been observed for the diastereomers (Guerra et al., 2011). Upon reach-ing a temperature of 190°C, where α−,β− and γ−HBCDD attain a constant proportion of 78%, 14% and 8% respectively, independent of starting ma-terial proportions (Heeb et al., 2008). Due to the diastereomeric propor-tions, the accurate and reliable detection of the individual diastereomers is an important quality of analysis methods for HBCDD. Moreover, studies of enantiomeric behaviour of HBCDD have not been conclusive and further characterization across abiotic and biotic matrices are required. Though LC separations for both diastereomers and enantiomers have been successful, the investigation of SFC for a more rapid and efficient separation of stereo-isomers was embarked upon here.
4.1.1 Diastereomer separation using SFC The first assessment of SFC using supercritical CO2 for α−,β− and γ−HBCDD diastereomer separation is described in Paper I. Chromato-graphic method development was initiated under isocratic and gradient con-ditions across three reversed-phase column chemistries. It was that the HSS C18 column with a gradient achieved the optimum resolution between the three diastereomers. Resolution (Rs) between peaks was determined accord-ing to the United States Pharmacopeia (USP) definition of Rs=(2.0(Rt2-Rt1))/(W2+W1), where Rt=retention time, W2+W1=Sum of peak widths at

46
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
baseline between tangent lines drawn at 50% peak height (Waters Corpo-ration, Empower System Suitability Quick Reference Guide). Calculation was performed using Empower software (Waters Corporation). When com-pared to a RPLC method, both a reduction in run time (2.31 min. for SFC and 12 min. for RPLC) and increased resolution between diastereomers was observed. This is attributed to the increased diffusivity and reduced fluid viscosities of supercritical fluids over liquids which increase mass transfer of the analyte (Lee and Markides, 1990). Differences in diastereomer elu-tion order between SFC (α<γ<β) and RPLC (α<β<γ) methods were seen un-der the conditions used. The same elution order as observed for SFC have been reported in two other RPLC methods which had either lower column temperature or used a more retentive stationary phase (C30) (Dodder et al., 2006, Stapleton et al., 2006). These results suggest that this elution order is the result of increased interaction with the stationary phase. Figure 9 shows the elution order, resolution and retention times for both SFC and LC.
MS signal optimizations utilized post-column solvent input (referred to as the make-up flow), achieved through a T-union coupling with the LC effluent and controlled by a binary pump prior to flow into the MS source probe. Pressure at this union is controlled via an automated back-pressure regulator (ABPR) valve to mitigate pressure drop effects. The post column solution flow was used to supplement solvent such that an appropriate flow is available for ionization. In addition, the make-up flow solvent composi-tion was modified with additives which were tested for their enhancing or suppressing effects on ionization. It was found that use of 0.1% ammonium hydroxide increased HBCDD diastereomer signal with methanol and 2-pro-panol solvents, and 0.1% ammonium hydroxide with 2-propanol was opti-mal for aiding the ionization of HBCDD. Further ionization investigation was performed by comparing negative polarity ESI, APCI, and an interme-diate method electrospray/chemical ionization switching (ESCi™). As ob-served in previous RPLC studies of HBCDD ((Dodder et al., 2006, Guerra et al., 2011), ESI yielded the most intense [M-H]-
signal, APCI was the poor-est. Instrumental limits-of-detection and quantification (LOD/Q) as deter-mined with solvent standards were arrived at based on signal-to-noise (S/N) of 3 and 10, respectively, for the quantification MRM transition 650.6>80.9. LOD’s for all diastereomers were 100 fg on-column, and LOQs were 500 fg on-column for α− and γ−HBCDD and 250 fg on-column for β−HBCDD.
As a test of method efficacy, a small subset of whale blubber and human serum extracts prepared in previous studies (Rotander et al. 2012, Salihovic

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
47
et al., 2012) were analysed under the optimized conditions in MRM mode. One human serum and all six whale blubber samples were found to have levels of α−HBCDD above the LOQ. Presence of the α−HBCDD, as men-tioned earlier, dominates in biological matrices which this finding is in line with. The much lower quantity of HBCDD observed in the human serum (0.6 pg on-column) versus the whale blubber (8.3 to 85.9 pg on-column) samples is also reflective of findings by Johnson-Restrepo et al. (2008) and can be attributed in part to lipophilic nature of HBCDD (log KOW = 5.6 in technical product (USEPA, 2010)) making it more likely to accumulate in adipose tissue such as blubber. All identifications had a conservation of ion ratios between quantification and qualification MRM channels (640.6>80.9/640.6>87.9) within 20% of the value determined from the sol-vent standard calibration curve. This approach is a widely implemented step to further ensure correct analyte identification in matrix (Kaufmann et al., 2009).
Figure 9: Overlaid chromatograms of SFC and RPLC separations of α−,β− and γ−HBCDD illustrating the enhanced chromatographic resolution (Rs) for SFC at faster elution rates. In the case of the SFC separation, 1 µL of a 100 pg µL-1 tolu-ene solvent standard was injected, while the LC injection used a 10 µL injection of the same concentration methanol solvent standard.
4.1.2 Enantiomer separation using SFC Paper II describes the development for the chiral separation of (-/+) enanti-omers of α−,β− and γ−HBCDD as well as the two dominant meso-forms
Expanded CC Separation
CC LC
α−γ−
α−
β−
γ−
β−α−
β−
γ−
Rs=3.15
Rs=4.36
Rs=5.07
Rs=10.42
SFC
Expanded SFC Separation

48
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
(ε− and δ−) using SFC with supercritical CO2 for preparative and analytical scale methods. Chiral separations require a chiral stationary phase, and commercially available cellulose (CEL-1: cellulose tris-(3,5-dimethylphenyl-carbamate) and CEL-2: cellulose tris-(3-chloro-4-phenylmethylcarbamate)) and amylose (AMY-1: amylose tris-(3,5-dimethylphenylcarbamate) col-umns were screened using co-solvents amenable to SFC. It was found that the CEL columns were able to baseline separate the six HBCDD enantio-mers. Specifically, the analytical scale method used CEL-2 column with 2-propanol co-solvent, while the preparative scale method required CEL-1 column with acetonitrile co-solvent in order to achieve the necessary sepa-ration of β−HBCDD enantiomers. X-ray crystallography to confirm enan-tiomeric conformations was able to be performed for both α− and γ−HBCDD enantiomers following the growth of crystals derived from pre-parative scale effluents. β−HBCDD enantiomer crystals could not be grown, and the enantiomer identification was confirmed using isolated enantiomer fractions from the preparative separation on an established LC chiral method (Köppen et al. 2007).
It is postulated in Paper II that the bromine atom orientation is the major factor determining stereoisomer retention, driving the hydrogen bonding re-tention mechanism. Prediction of enantiomeric behavior in terms of confor-mational shape (meaning those enantiomers with similar conformations should have close elution profiles) is challenged for HBCDD due to the flex-ibility in cyclododecane ring structure. Lastly, it is also predicted that amyl-ose columns likely had poor resolution for HBCDD enantiomers as a result of less effective hydrogen bonding sites, versus uniformity of cellulose sta-tionary phases promoting more stable interactions.
The method for LC chiral HBCDD separation was employed using a Nu-celodex β−PM column, and the separation of the six stereoisomers plus two main mesoforms can be seen in Paper II, Figure 1. Elution order between the LC and SFC (Paper II, Figure 2) chiral methods, in a pattern similar to the diastereomer separation in Paper I, changes from (-)-α−<(-)-β−<(+)-α−<(+)-β-<-δ−<(+)-γ−<-ε−<(-)-γ for LC separation to (-)-α−<(+)-α<-δ−<(-)-γ−<(+)-γ-<-ε−<(-)-β−<-(+)-β−<(-)-γ for SFC separation. Baseline resolution was afforded for (+)-β−HBCDD from both (+)-γ−HBCDD and δ−HBCDD using the SFC method, while co-elution was observed under the LC condi-tions. This is of significance in terms of analytical specificity of HBCDD stereoisomer identification, especially in biological or environmental matri-ces which unequivocal separation would be required to make meaningful deductions regarding enantiomeric behaviour.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
49
4.2 Hexafluoropropylene oxide-dimer acid (HFPO-DA) PFAS have been widely used in surfactants for industrial processes and con-sumer commodities such as non-stick cookware, textiles and carpeting (Martin et al., 2004). They are comprised of a wide range of compounds which contain a partially or fully fluorinated alkyl chain as well as an ionic head-group moiety resulting in the unique property of being both hydro- and lipo-phobic (ampiphillic) (Buck et al., 2011, Martin et al., 2004), providing PFAS used in commodities their desired properties. PFAS are formed by either telomerization or electrochemical fluorination (Houde et al., 2006). The former process resulting in a mixture of branched, linear and short chain perfluoroalkyl compound forms (Houde et al., 2006); the latter process results in a mixture of straight, even-chained perfluoroalkyl iodides that are then used to manufacture fluorotelomer alcohols (FTOHs) and other products (Houde et al., 2006). Due to detection in the environ-ment and biota and observations of persistence (Buck et al., 2011, Houde et al., 2006) following release of these compounds from their processes and products of use, PFOS and PFOA along with various other long and short chain carboxylic and sulfonic acid PFAS have been identified as compounds of interest for monitoring and legislation. The primary manufacturer of PFOS, 3M, voluntarily phased out its production in 2002 (Martin et al. 2004) and the EPA 2010/2015 PFOA Stewardship Program is dedicated to phasing out its name-sakes’ production (Munoz et al., 2019).
However, demand for similar chemicals for numerous industrial pro-cesses has resulted in the increased use of replacement PFAS (Munoz et al., 2019). Described for the first time in the environment following a non-tar-geted analysis of the Cape Fear River, Strynar et al. (2015) elucidated sev-eral high-level per- and polyfluoroether carboxylic and sulfonic acid com-pounds (PFEC/SAs) not previously documented. One compound, hex-afluoropropylene oxide-dimer acid (HFPO-DA, also referred to by its am-monium salt trade name, GenX) was detected at particularly high levels. Since this study, the source of the elevated levels was linked to direct dis-charge into the CFR by its manufacturer, Chemours (Williamson, 2018), allowable according EPA stipulations on HFPO-DA waste removal if it was generated as a by-product (Hopkins et al., 2018). Resulting observations of drinking water contamination in that region caused the state government in North Carolina (the site of the Cape Fear River) to set a limit of 140 ng L-1 HFPO-DA in drinking water and actively seek removal of HFPO-DA con-tamination. As of 2018, the US EPA has updated its PFAS surface water analysis method 537.1 to include HFPO-DA (US EPA Method 537.1).

50
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Paper III describes the current concentrations reported for HFPO-DA across the eleven published studies. In addition to the above case, elevated concentrations ranging from 631-9,350 ng L-1 in surface water have been detected in China (Pan 2017, Song 2018, Heydebreck 2015), the Nether-lands (Heydebreck 2015, Gebbink 2017) and further testing in the Cape Fear River in the USA (Sun 2016, Hopkins 2018, McCord 2018). These concentrations are directly associated with fluorochemical production plant proximity, and samples taken after cessation of dumping in the Cape Fear River indicated a drop in detected HFPO-DA concentration (McCord 2018). Tap and surface water samples taken in areas where no known flu-orochemical production is occurring nearby have much lower or non-de-tected HFPO-DA concentrations. Figure 10 summarises the reported mean concentrations in water samples from available quantitative studies, as well as those for PFOA and another PFECA, hexafluoropropylene trimer acid (HFPO-TA). Biological samples tested include grass and leaves (Brandsma et al., 2019), human serum (Kato et al., 2018), urine (Kato et al., 2018, Pan et al., 2017) and carp muscle, liver tissue and blood (Pan et al., 2017). Like water samples, the grass/leave and carp samples were taken near a fluoro-chemical production plant and contained detectable levels of HFPO-DA as well as other PFAS (Brandsma et al., 2019, Pan et al., 2017). However, the human serum samples, for which no metadata was described, did not con-tain any detectable HFPO-DA and high detection rates of PFOA (Kato et al., 2018).

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
51
Figure 10: Mean concentrations (ng/L) of HFPO-DA (structure upper right), HFPO-TA and PFOA where reported from 2013-2018 global campaigns. Data is provided from: Sun et al., 2016, Brandsma et al., 2018, Heydebreck et al., 2015, Gebbink et al., 2017, Pan et al., 2017, Pan et al., 2018 and Song et al., 2018.
Discussed more fully in Paper III are the analytical approaches used for HFPO-DA and the unique challenges it presents as compared to other PFAS. Though the first publication describing the observation of HFPO-DA in the environment used high-resolution MS operating in full spectrum acquisition (Strynar et al., 2015), most studies have adopted HFPO-DA into existing PFAS targeted acquisitions using solid-phase extraction (SPE)-LC and elec-trospray negative polarity MRM acquisition on TQ-MS systems. SPE and LC methods utilize the same historically employed methods and reagents as legacy PFAS analysis, widely using weak-anionic exchange SPE and ammo-nium acetate or ammonium formate mobile phase additives. The monitored masses for MRM experiments are largely the nominal mass [M-H]- (329 m/z) and two predominant fragments, 285 m/z (loss of -CO2 following deprotonation) and 169 m/z (C3F7) generated from ether linkage breakage of the compound, and 119 m/z (C2F5). Sensitivity of these methods in terms of detection limits are described in Paper III Tables 1 and 2, and in brief, HFPO-DA detection limits are intermediate to less sensitive as compared to other monitored legacy and emerging PFAS. Two methods (Brandsma et al., 2019, USEPA Method 537.1) explicitly use the prominent 285 m/z fragment as precursor ions, for reasons discussed next.
Mean Concentrations (ng/L) of HFPO-DA, HFPO-TA and PFOA 2013-2018
Mea
n Le
vels
(ng/
L)

52
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
HFPO-DA ionization under the typical conditions described above re-sults in unique and potentially problematic ion species formation. As de-scribed by Strynar et al. (2015), the high-levels of HFPO-DA present in the Cape Fear River water sample resulted in a detected peak whose spectrum showed intense homodimer and homodimer sodium adduct ([2M-H]- and [2M-2H+Na]-) formation and minimal [M-H]- formation. Under the typical ESI- methods have been reports of these occurrences as well as unwanted in-source product ion formation, or fragmentation (ISF), found to be worse on some MS platforms than others (Sun et al., 2016). In total, this behavior contributes to poor detection of the [M-H]-. Alterations of source conditions such as cone voltage and gas flow yield little change in this behavior. This behavior has also been observed for other PFECAs (Strynar et al. 2015), presenting a class-wide challenge for identification. It can be postulated that numerous other PFECAs are used in industrial processes and their detection in environmental and biological samples with both TQ-MS and TOF-MS methods will likely continue. HFPO-DA is a representative of these com-pounds, which has a commercially available standard making directed method improvements possible. As a result, one aspect of basic LC-MS method development was revisited for HFPO-DA in Paper IV and is de-scribed in the next section.
4.2.2 Improvement to the ionization of HFPO-DA The main objective of Paper IV was to describe the results of exploring var-ious mobile phase additives to increase both signal and proportion of [M-H]- formation over current method results. Impacts of mobile phase com-position are not excluded to chromatographic performance and has also been shown to effect analyte ionization stability and efficiency (Mallet, 2003, Gao, 2005, Kamel, 1999, Kruve, 2017). The assessment was per-formed first through full scan acquisition on a TQ-MS system (Xevo TQ-D, Waters Corporation) using several commonly employed MS amenable mobile phase additives. Ammonium acetate, ammonium formate, ammo-nium bicarbonate (2 and 10 mM compositions), formic acid, acetic acid and ammonium hydroxide (0.1 and 0.5% compositions) were used as mobile phases for injections of 5.0 µg mL-1 HFPO-DA standard.
Paper IV describes the full results of this study, and in brief ammonium bicarbonate resulted in the repeatably highest signal of [M-H]-, lowest ho-modimer species formation and overall best monomer related species signal in total. An adduct identified using high-resolution MS was found to be the bicarbonate adduct [M-HCO3]- under ammonium bicarbonate conditions
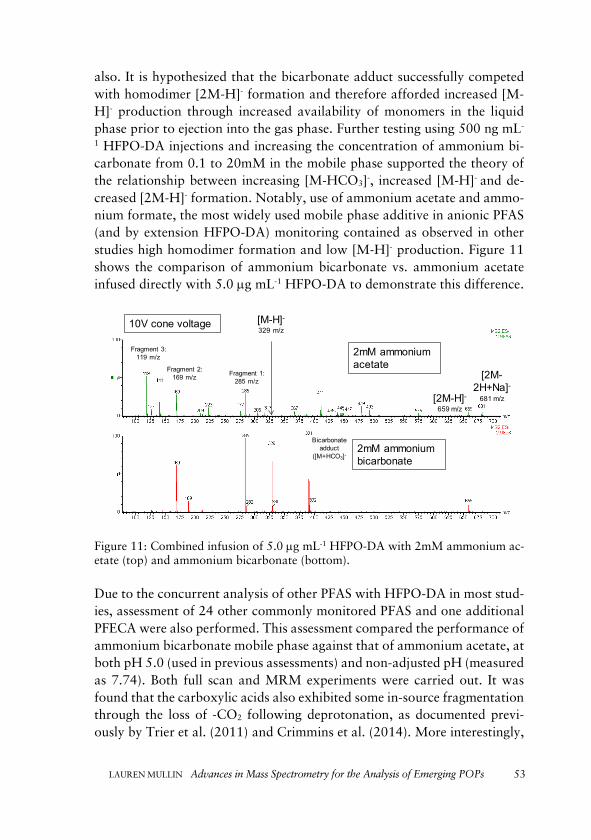
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
53
also. It is hypothesized that the bicarbonate adduct successfully competed with homodimer [2M-H]- formation and therefore afforded increased [M-H]- production through increased availability of monomers in the liquid phase prior to ejection into the gas phase. Further testing using 500 ng mL-
1 HFPO-DA injections and increasing the concentration of ammonium bi-carbonate from 0.1 to 20mM in the mobile phase supported the theory of the relationship between increasing [M-HCO3]-, increased [M-H]- and de-creased [2M-H]- formation. Notably, use of ammonium acetate and ammo-nium formate, the most widely used mobile phase additive in anionic PFAS (and by extension HFPO-DA) monitoring contained as observed in other studies high homodimer formation and low [M-H]- production. Figure 11 shows the comparison of ammonium bicarbonate vs. ammonium acetate infused directly with 5.0 µg mL-1 HFPO-DA to demonstrate this difference.
Figure 11: Combined infusion of 5.0 µg mL-1 HFPO-DA with 2mM ammonium ac-etate (top) and ammonium bicarbonate (bottom). Due to the concurrent analysis of other PFAS with HFPO-DA in most stud-ies, assessment of 24 other commonly monitored PFAS and one additional PFECA were also performed. This assessment compared the performance of ammonium bicarbonate mobile phase against that of ammonium acetate, at both pH 5.0 (used in previous assessments) and non-adjusted pH (measured as 7.74). Both full scan and MRM experiments were carried out. It was found that the carboxylic acids also exhibited some in-source fragmentation through the loss of -CO2 following deprotonation, as documented previ-ously by Trier et al. (2011) and Crimmins et al. (2014). More interestingly,
2mM ammonium acetate
2mM ammonium bicarbonate
[2M-H]-659 m/z
[M-H]-329 m/z
Fragment 1:285 m/z
Fragment 2:169 m/z
Fragment 3:119 m/z
10V cone voltage
Bicarbonate adduct
([M+HCO3]-
[2M-2H+Na]-
681 m/z

54
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
the carboxylic acids also formed [M-HCO3]- in addition to [M-H]- when using ammonium bicarbonate mobile phase additive. It is thought that this adduct formation occurs via a similar mechanism as known dimer for-mation propensity between carboxylic acid species (Faubel and Kisters, 1989). Overall, MRM total ion currents (TICs) were either most sensitive in ammonium bicarbonate (over half of the analytes) or ammonium acetate, non-adjusted pH except for perfluoro-1-octanesulfonamide (FOSA) which had the best response under ammonium acetate pH 5.0 adjusted conditions. Generally, TIC response was within 2x of minimum to maximum response conditions.
An initial test of ammonium bicarbonate method performance in the analysis of water extracts was also performed. Deionized, tap and surface (pond) water samples were extracted according to the method in McCord et al. (2018) and a solvent standard dilution series with three QC levels were prepared. Using a method monitoring 25 other PFAS in addition to native and isotopically labelled HFPO-DA, each with at least two transitions, Pa-per IV describes the results of quantification of QCs as well as water extract measurement repeatability. Overall, instrumental LOD/Qs determined us-ing solvent standards and based on signal-to-noise ratios of 3 and 10, re-spectively, were improved for HFPO-DA using ammonium bicarbonate mo-bile phase over the use of ammonium acetate (pH 5.0). Furthermore, in-creased in signal for both quantification (285>169) and qualifier (329>285 and 329>169) MRM channels was seen with ammonium bicarbonate mo-bile phase over both ammonium acetate pH levels. This is particularly im-portant considering the lower SPE recoveries reported in some cases for sur-face water samples (Heydebreck et al., 2015, Gebbink et al., 2017) thus requiring even more instrument method sensitivity. It is recommended that future method development efforts for PFAS and HFPO-DA consider as-sessment of mobile phase additives like ammonium bicarbonate over widely used ammonium acetate conditions.
4.3 Non-targeted acquisition of dust samples using TOF-MS As described in section 1.3.3, TOF-MS has been used in various environ-mental non-targeted acquisition studies of POPs and other compound clas-ses (Megson et al., 2016, Schymanski et al., 2015). Acting as one of the main HRMS instrument types employed in non-targeted studies (alongside Orbitrap MS), TOF and QTOF-MS affords accurate mass measurements and full spectrum acquisition, or DIA. This type of data generation is well suited to address studies of overall contaminant distribution and exposure

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
55
due to wide mass range (hence analyte) coverage and the ability to retro-spectively search mass lists (Andra et al., 2017, Acena et al. 2015). These studies are based on the premise that a vast number of xenobiotic chemicals are used in consumer and industrial processes and are likely to migrate from their point of use, as has been evidenced historically by POPs and other compounds of concern. The term used to describe lifetime environmental exposure to these chemicals is the exposome, first described by Dr. Chris Wild in 2005 (Sobus et al., 2017). Covering a wide range of environmental (Acena et al., 2015, Hernández et al., 2012, Krauss et al., 2010) and bio-logical (Andra et al., 2017) samples, several recent studies have demon-strated TOF-MS with DIA for assessment of the indoor environment con-taminant burden, specifically through the analysis of dust (Ouyang et al., 2017, Rager et al., 2016, Ubukata et al., 2015, Hilton et al., 2010, Moschet 2018, Rostkowski et al., 2019). Utilizing a mixture of GC and LC couplings with further variety in MS ionization sources (Rostkowski et al., 2019), these studies have uncovered a diverse range of contaminant classes includ-ing pesticides, legacy and emerging flame retardants and plasticizers and pharmaceuticals (Ouyang et al., 2017, Rager et al., 2016, Ubukata et al., 2015, Hilton et al., 2010, Moschet 2018, Rostkowski et al., 2019).
In order to make identifications, tactics to isolate masses of relevance from the large DIA m/z data table are required as a first step. One approach, targeted screening, uses a known list of compound masses with detection information like retention time, expected product ions, etc. to mine the data (Schymanski et al., 2015). Other approaches which are of great utility for studies looking at total contaminant profiling are referred to as suspect and non-targeted screening (Schymanski et al., 2015). It is worth noting that the term non-targeted acquisition used earlier specifically applies to the use of full spectral acquisition, rather than acquisition of a specific mass list. Sus-pect screening uses a mass list of likely compounds to be present in the sam-ple, where non-targeted screening isolates masses based on trending or other behavior followed by large chemical database searching (Schymanski et al., 2015). Methods for isolation include Kendrick mass defect (KMD) plotting, which is useful for isolating halogenated analogs based on their negative mass defect (Ubukata et al., 2015, Barzen-Hanson et al., 2017). Another method relies on the use of principal component analysis (PCA), a multi-variate analysis technique used to ordinate data (Gotelli and Ellison, 2013). When applied to GC- or LC-MS data, samples are grouped according to variance of exact mass/RT pairs in the sample set, each pair ideally repre-

56
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
senting a unique molecule. From the PCA, further comparison of group dif-ferences is performed which result in the isolation of exact mass/RT pairs that contribute to inter-sample variation. Regardless of the process used to isolate masses or if one is used at all (i.e., the entire data table is used), studies will conclude their non-targeted screening workflows with database searching such as NIST, EPA’s Distributed Structure-Searchable Toxicity (DSSTox) database, or vendor provided databases (Rager et al., 2016, Moschet et al., 2018, Ouyang et al., 2017). Criteria for compound identifi-cation are reliant on the inherent features of accurate mass data, namely a mass accuracy tolerance (often +/- 5 ppm) for averaged found m/z to theo-retical m/z and isotopic distribution fidelity (Acena et al., 2015).
Standardization of compound identification confidence in studies using non-targeted acquisition for environmental contaminant characterization have also been contributed to greatly to by the work of Schymanski et al. (2014, 2015). This work was necessitated by the inherent fact that proposed identifications were not always confirmed by solvent standards, a practi-cally required criteria for development of targeted acquisition methods. Al-ready described, targeted, suspect and non-targeted approaches derived identification results can be broken into levels of confidence ranging from 1(highly confident, confirmed with solvent standard) to 5 (low confidence, mass of interest with no structure proposed). According to this system and echoed throughout other non-targeted acquisition studies is the requirement of an authentic standard for full confirmation (Schymanski et al., 2014, 2015). This presents a challenge in that many emerging contaminants may not have such materials available. Paper V investigates one possible future avenue for increasing confidence in such identifications.
4.3.1 Application of Ion Mobility Spectrometry The use of TW-IMS coupled with LC-QTOF was investigated in Paper V for its ability to propose structural identifications of xenobiotic compounds in both household and e-waste processing facility dust samples. These sam-ple types represent different contaminant sources and levels, likely contain-ing a diverse set of characterized and uncharacterized xenobiotic chemicals. Following proposed structural identifications, comparison of the IMS ex-perimentally derived gas-phase measurement, CCS (described in Section 2.3) were made to predicted values. The goal of this work was to assess the fidelity of these predictions for diverse xenobiotic compounds as a future direction for assisting in compound identification support in non-targeted acquisition studies.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
57
The work was carried out using a DIA approach on a commercially avail-able IMS-enabled LC-QTOF. The LC system flow path was fitted with an in-line isolator column to hold-back system or mobile phase contamination prior to sample injection. Data was acquired from 50-1000m/z with a scan time of 0.25 sec in both negative and positive polarity ESI. Precursor and product ion spectra were collected over the course of the method run time, alternating between a collision energy of 4.0 eV and an elevated collision energy ramp of 20.0-55.0 eV. TW-IM separation for all ions occurred using nitrogen drift gas, and prior to analysis the drift cell was calibrated such that all ions’ drift times could be extrapolated to CCS values. QC samples to test both expected CCS value and mass accuracy conservation were ac-quired during both polarity data sets. Data was processed using a mixture of targeted and non-targeted screening techniques, described more fully in Paper V. Briefly, two targeted screening approaches used in-house libraries containing at the very least structural information, accurate mass product ions, CCS and in some cases RT. Non-targeted screening methods used ei-ther PCA to isolate exact mass/RT/CCS pairs that contributed significantly to dust sample type differences (i.e. household vs. e-waste processing facil-ity), or use of an isotope searching algorithm that isolated spectra based on the presence of -Br/-Cl distinctive isotope distribution pattern. Structural proposal for the non-targeted screening approach used ChemSpider data-base searching which included the EPA DSSTox database. CCS prediction was carried out in two programs which were derived from machine-learning approaches, created based on molecular features found to contribute to IMS separation behavior and trained with experimental CCS values from known compounds (Zhou et al., 2017, 2018). These programs require molecular input either by structural file (.mol or .sdf) or .csv of molecular descriptors that were obtained from ChemAxon.
In total, 29 xenobiotic compound identifications were proposed. The range of compound classes were, as expected, quite varied and included brominated phenols, pesticides, pharmaceuticals/personal care products (PPCPs), PFAS and organophosphorus compounds (OPs), some known OP-FRs. The OPs represented the largest class of compounds found. Paper V describes in detail the observation of OPs which have been widely docu-mented in environmental matrices and three which have not. Structural elu-cidation of OP precursor and product ions was facilitated by the observa-tion of common product ions. For the pesticides, the targeted identification approach utilized CCS values rather than retention times, due to differences
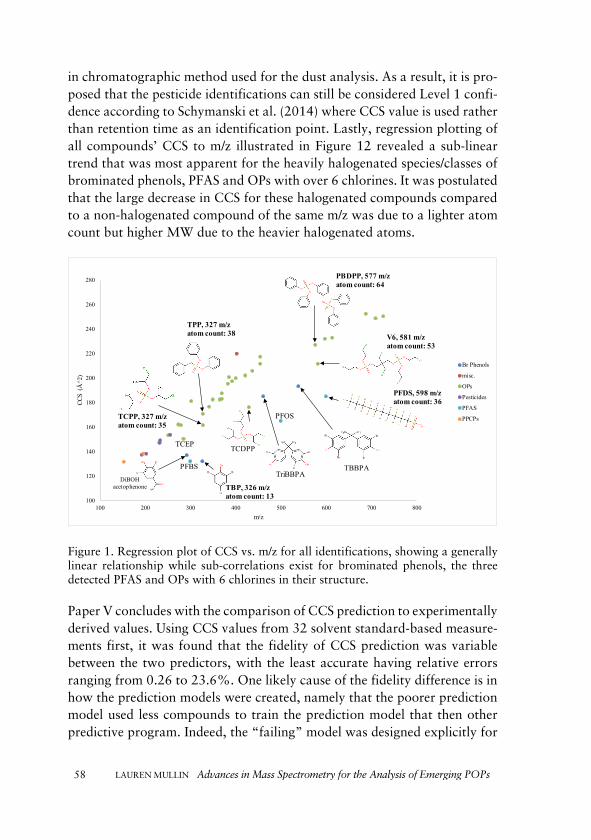
58
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
in chromatographic method used for the dust analysis. As a result, it is pro-posed that the pesticide identifications can still be considered Level 1 confi-dence according to Schymanski et al. (2014) where CCS value is used rather than retention time as an identification point. Lastly, regression plotting of all compounds’ CCS to m/z illustrated in Figure 12 revealed a sub-linear trend that was most apparent for the heavily halogenated species/classes of brominated phenols, PFAS and OPs with over 6 chlorines. It was postulated that the large decrease in CCS for these halogenated compounds compared to a non-halogenated compound of the same m/z was due to a lighter atom count but higher MW due to the heavier halogenated atoms.
Figure 1. Regression plot of CCS vs. m/z for all identifications, showing a generally linear relationship while sub-correlations exist for brominated phenols, the three detected PFAS and OPs with 6 chlorines in their structure. Paper V concludes with the comparison of CCS prediction to experimentally derived values. Using CCS values from 32 solvent standard-based measure-ments first, it was found that the fidelity of CCS prediction was variable between the two predictors, with the least accurate having relative errors ranging from 0.26 to 23.6%. One likely cause of the fidelity difference is in how the prediction models were created, namely that the poorer prediction model used less compounds to train the prediction model that then other predictive program. Indeed, the “failing” model was designed explicitly for
100
120
140
160
180
200
220
240
260
280
100 200 300 400 500 600 700 800
CCS
(Å^2
)
m/z
Br Phenols
misc.
OPs
Pesticides
PFAS
PPCPs
PFBS
PFOS
TriBBPA
TCDPP
TBP, 326 m/zatom count: 13
DiBOH acetophenone
TCEP
TPP, 327 m/zatom count: 38
PBDPP, 577 m/zatom count: 64
TBBPA
PFDS, 598 m/zatom count: 36
V6, 581 m/zatom count: 53
TCPP, 327 m/zatom count: 35

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
59
metabolites (Zhou et al., 2017, 2018), not necessarily heavily halogenated or aromatic compounds as observed here. Using the other prediction method, which demonstrated much closer fidelity (relative errors ranging from 0.26 to 5.15%), all proposed identification CCS values were com-pared. Figure 16 shows the relative error plot of the experimental CCS from samples vs. predicted CCS and experimental CCS from standards vs. pre-dicted CCS. Generally, the CCS values from samples relative error of CCS comparison to predicted CCS was within the range established when using standards. Though expectations for CCS relative error conservation when comparing solvent standard derived values to experimental values are 2%, no criteria for prediction models have been established. However, these promising results beget the recommendation for further development of pre-dictive models, specifically to include training sets with more diverse com-pound classes representing xenobiotic contaminants.
Figure 16. Relative error (%) for sample CCS vs. development model predicted CCS and solvent standard CCS vs. development model predicted CCS across compounds proposed in dust samples (b).
0
0.5
1
1.5
2
2.5
3
3.5
4
4.5
5
5.5Carbendazim
Diuron_ESI+TCPP
TBOEP
CDP
Imidacloprid
[Bis(2-hydroxyethyl)amino] methyl stearate
EDP, DPEHP
Caffeine
TPPO
PFDS
Acetominophen
Diuron_ESI-
Thiabendazole
Tris (2-propylphenyl)phosphate_CCS 217.3PFBS
2-tert-Butylphenyl diphenyl phosphateTCDPP, TDCP
TPP
TCEP
Benzyl hydrogen [2-(4-biphenylyl)-2-hydroxyethyl]phosphonate
PBDPP
2-ethylhexyl diphenyl phosphate (EDP,DPEHP)_ISF
2,6-Dibromo-4-[2-(3-bromo-4-hydroxyphenyl)propan-2-yl]phenol
TBBPA
PBDMPP
3',5'-Dibromo-4'-hydroxyacetophenone
V6
2,4,6-Tribromophenol
Tris (2-propylphenyl)phosphate_CCS 211.6
PFOS2-Isopropylphenyl diphenyl phosphate
% Error Distribution for sample CCS and standard CCS vs. development model predicted CCS
% error sample CCS vs. development model predicted CCS ABS % error standard CCS vs. development model predicted CCS ABS
b.


LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
61
5.0 Conclusions and Future Work Achieving the main objectives of this thesis involved divergent platforms and experiments, united in the common goal of addressing challenges for emerging POPs analysis. This was attained through using alternative tech-niques both in terms of hardware not previously applied to the specific chal-lenges here (SFC and IMS) as well as demonstrating that new problems can be approached with old method development solutions (such as assessment of different mobile phase additives). Though not exhaustive, demonstration for three out of the five papers in a relevant environmental or biological matrix was carried out to ensure proposed methodologies were applicable for the sample types they meant are to analyze further. These studies em-barked to improve sensitivity, specificity and efficiency of emerging POPs methodologies.
It was found that using SFC for the analysis of HBCDD stereoisomers provided a method with higher efficiency through faster run times. Practi-cally, this translates to generation of less organic solvent waste. Ideally, it also translates to higher sample through-put. In the case of HBCDD enan-tiomers, the specificity of analysis is greatly improved when using SFC over LC, and base-line resolution of all species was achieved in less than 15 minutes. Further implementation of these methods are recommended for large sample sets, from which the reduction in run time would be fully realized.
Assessing the unique ionization behavior of HFPO-DA as well as its global occurrence brought to light unique features of this highly publicized emerging PFAS. Preliminary method development using ammonium bicar-bonate mobile phase was able to replace homodimer formation with a sim-ilar mechanism using the bicarbonate adduct that appeared to increase the desired deprotonated pseudomolecular ion formation. Overall, this method showed improved sensitivity over existing method conditions. Though not performed in the scope of this thesis, method validation of this method and further method development comparisons with other PFAS are the next stage for this work. Furthermore, other ionization sources (either available now or in the future) should continuously be assessed for PFAS in general as need for increased sensitivity will surely continue.
The final study took a broader stance in the field of emerging POPs and contaminant analysis in general. Taking advantage of commercially availa-ble IMS-enabled QTOF-MS systems to gain gas-phase measurements of ions, characteristic gas-phase patterns for both well and lesser-characterized xenobiotic compounds were observed for the first time. This study is also

62
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
an early application of IMS-QTOF-MS for environmental analysis. In the context of other non-targeted studies, CCS values are a useful additional identification point, occurring in the gas phase and therefore unimpacted by changes in chromatographic method or sample type. Moreover, exciting de-velopments in predictive CCS programs include both increased accuracy over earlier models, and more facile access using structural files and molec-ular descriptors. Indeed, prediction accuracy for these types of programs, which are built based on training sets of experimentally derived CCS values, can only be improved with increased compound diversity such as highly halogenated molecules.
An exciting area for further study is the coupling of GC via atmospheric pressure chemical ionization (described in section 2.2) with IMS-enabled QTOF-MS instruments. Preliminary data not discussed in this thesis demonstrates conservation of CCS for [M+H]+ ions of pesticides analyzed under both LC and GC separation, whereas RT for both methods were as expected highly variable from one another. This same study could be ap-plied to other contaminants which can be chromatographically separated by either technique such as polyaromatic hydrocarbons (PAHs), thus providing complimentary measurement approaches. Most attractive about the coupling of GC in addition to LC to IMS QTOF-MS is the ability to cover wider analyte ranges, representing the full breadth of legacy and emerging POPs known to date. Other fields which involve the analysis of wide-ranging analyte classes such as metabolomics would similarly benefit from this approach. Along this same line of thinking, SFC coupling IMS is also currently feasible though has not been investigated for environmental contaminant analysis. Further characterization of these diverse chromato-graphic approaches in coupled to IMS would present findings around gas-phase characterization of both chiral and achiral separations.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
63
6.0 Acknowledgements First, I must thank my advisors, Leo Yeung and Ingrid Ericson Jogsten, for their thoughtful review of my manuscripts, talking me through a lot of thought processes and project ideas, and being endlessly kind and support-ive. Ingrid, you’ve been with me from the start of this long journey and I always have seen you as a guiding light. So many good memories over the years, across continents and countries. Bert van Bavel, you were the first to accept me as your student, and for that I am very grateful. Thank you.
Being employed as a scientist by Waters Corporation has opened so many doors to me, including the opportunity to do this industrial PhD. My men-tors Jennifer Burgess, Gareth Cleland and Rob Plumb have been amazing managers and guides, professionally and personally. Jen, you were the one who made it possible for me to embark on this journey in so many ways. You gave me confidence to fly when I didn’t know I could walk. I will never forget the ways you’ve nurtured me and listened. Gareth, you are like my work brother—made me laugh endlessly, taught me so so so much and someone I feel like I can trust with anything. Just thinking of all those good times in the lab, at conferences or just hanging out brings such a smile to my face. Rob, you have provided me with so many opportunities profes-sionally, given me the space to work on this thesis, and always provide ad-vice and support.
My dear family, you guys have endured long periods of not hearing from me as I worked through this. I love you all. Dad, Anne Marie, Mom, Isaac, Chris, Jess, Andrew, Nanny (Judy), and everyone else in the Mullin tribe. Myrtle, Minaz, Nadia and Adam you have accepted me as a member of your family, and I feel so lucky to be a part of it.
I have many friends that I want to thank, here are a few. Angela, you always give me the support to talk and then wise, honest advice I need. Baiba, you are a stand-up woman and a true friend—how lucky was I to meet you?! Kerri, you make everyday a good one just through being your fab self. Kasia, though time flies too fast between us, I always feel like we pick up where we left off. Ann and Maria, you always bring the sunshine. Kendon and Anunay, you just make me laugh. Don, top of the muffin to you. Marian and Uncle Doug Stevens you are both true role models to me. My many colleagues from Waters who have provided me great technical advice and discussion throughout: Dave Douce, Rhys Jones, Martin Palmer, Andy Jarrell, Mike Fogwill, Paul Rainville, Andy Aubin, Mike McCullagh, Jeff Goshawk and of course Hans Vissers. I’ve had the great fortune to work

64
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
with many collaborators who truly made this work and others possible: Karl Jobst, David Katz, Ruth Marfil-Vega, Dave Megson, Ronan Cariou, Bruno Le Bizec, Eric Reiner, Dawei Geng and Anna Kärrman. Nicole Rid-dell, a fellow industrial PhD candidate, you are a true super-star who could instantly provide me with a feeling of much-needed comradery.
More than anything, I acknowledge the deep love and support of my husband Adam. Everything we do together brings me joy, and we truly can talk about everything. Not only did you provide a lot of good discussion around this work, you also supported me in every way. Hey, you even were the one who trained me on APGC 😊😊. You are my best friend and my heart. I love you. Emi, my wonderful step-daughter, you have taught me more lessons than anyone else and watching you grow into the beautiful person you are is a gift.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
65
7.0 References Abb, M., Stahl, B., and Lorenz, W. 2011. Analysis of brominated flame
retardants in house dust. Chemosphere, 85(11), 1657-1663.
Aceña, J., Stampachiacchiere, S., Pérez, S., and Barceló, D. 2015. Advances in liquid chromatography–high-resolution mass spectrometry for quantitative and qualitative environmental analysis. Analytical and Bioanalytical Chemistry, 407(21), 6289-6299.
Ahmed, E., Kabir, K. M., Wang, H., Xiao, D., Fletcher, J., and Donald, W. A. 2019. Rapid separation of isomeric perfluoroalkyl substances by high-resolution differential ion mobility mass spectrometry. Analytica Chimica Acta, 1058, 127-135
Alaee, M., Arias, P., Sjödin, A., and Bergman, Å. 2003. An overview of commercially used brominated flame retardants, their applications, their use patterns in different countries/regions and possible modes of release. Environment International, 29(6), 683-689.
Andra, S. S., Austin, C., Patel, D., Dolios, G., Awawda, M., and Arora, M. 2017. Trends in the application of high-resolution mass spectrometry for human biomonitoring: an analytical primer to studying the environmental chemical space of the human exposome. Environment International, 100, 32-61.
Andres, A., Broeckhoven, K., and Desmet, G. 2015. Methods for the experimental characterization and analysis of the efficiency and speed of chromatographic columns: a step-by-step tutorial. Analytica Chimica Acta, 894, 20-34.
Barzen-Hanson, K. A., and Field, J. A. 2015. Discovery and implications of C2 and C3 perfluoroalkyl sulfonates in aqueous film-forming foams and groundwater. Environmental Science and Technology Letters, 2(4), 95-99.
Barzen-Hanson, K. A., Roberts, S. C., Choyke, S., Oetjen, K., McAlees, A., Riddell, N., McCrindle, R., Ferguson, P. L., Higgins, C. P., and Field, J. A. 2017. Discovery of 40 classes of per-and polyfluoroalkyl substances in historical aqueous film-forming foams (AFFFs) and AFFF-impacted groundwater. Environmental Science and Technology, 51(4), 2047-2057.

66
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Bijlsma, L., Bade R., Celma A., Mullin L., Cleland G., Stead S., Hernandez F., and Sancho J. V. 2017. Prediction of collision cross-section values for small molecules: Application to pesticide residue analysis. Analytical Chemistry, 89(12), 6583-6589.
Brack, W., Ait-Aissa, S., Burgess, R. M., Busch, W., Creusot, N., Di Paolo, C., Escher, B. I., Hewitt, M. L., Hilscherova, K., Hollender, J., Hollert, H., Jonker, W., Kool, J., Lamoree, M., Muschket, M., Neumann, S., Rostkowski, P., and Hollert, H. 2016. Effect-directed analysis supporting monitoring of aquatic environments—an in-depth overview. Science of the Total Environment, 544, 1073-1118.
Brack, W., Klamer H.J.C., Lopez de Alda, M., and Barceló, D. 2007. Effect-directed analysis of key toxicants in European river basins. A review. Environmental Science and Pollution Research-International, 14(1), 30-38.
Brandsma, S. H., Koekkoek, J. C., van Velzen, M. J. M., and de Boer, J. 2019. The PFOA substitute GenX detected in the environment near a fluoropolymer manufacturing plant in the Netherlands. Chemosphere, 220, 493-500.
Bruins, A. P. 1998. Mechanistic aspects of electrospray ionization. Journal of Chromatography A, 794(1-2), 345-357.
Buck, R. C., Franklin, J., Berger, U., Conder, J. M., Cousins, I. T., De Voogt, P., Jensen, A. A., Kannan, K., Mabury, S. A., and van Leeuwen, S. P. 2011. Perfluoroalkyl and polyfluoroalkyl substances in the environment: terminology, classification, and origins. Integrated environmental assessment and management, 7(4), 513-541.
Bush, M. F., Campuzano, I. D. and Robinson, C. V., 2012. Ion mobility mass spectrometry of peptide ions: effects of drift gas and calibration strategies. Analytical Chemistry, 84(16), 7124-7130.
Bush, M. F., Hall, Z., Giles, K., Hoyes, J., Robinson, C. V. and Ruotolo, B. T., 2010. Collision cross sections of proteins and their complexes: a calibration framework and database for gas-phase structural biology. Analytical Chemistry, 82(22), 9557-9565.
Collins, D., and Lee, M. 2002. Developments in ion mobility spectrometry–mass spectrometry. Analytical and Bioanalytical Chemistry, 372(1), 66-73.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
67
Covaci, A., Voorspoels, S., Ramos, L., Neels, H., and Blust, R. 2007. Recent developments in the analysis of brominated flame retardants and brominated natural compounds. Journal of Chromatography A, 1153(1-2), 145-171.
Darnerud, P. O. 2003. Toxic effects of brominated flame retardants in man and in wildlife. Environment International, 29(6), 841-853.
D'Atri, V., Causon, T., Hernandez‐Alba, O., Mutabazi, A., Veuthey, J. L., Cianferani, S., and Guillarme, D. 2018. Adding a new separation dimension to MS and LC–MS: What is the utility of ion mobility spectrometry? Journal of Separation Science, 41(1), 20-67.
de Hoffmann E. and Stroobant V., Mass spectrometry, Wiley Online Library, Third edn., 2007.
Dodder, N. G., Peck, A. M., Kucklick, J. R., and Sander, L. C. 2006. Analysis of hexabromocyclododecane diastereomers and enantiomers by liquid chromatography/tandem mass spectrometry: Chromatographic selectivity and ionization matrix effects. Journal of Chromatography A, 1135(1), 36-42.
Dorsey, J. G., and Dill, K. A. 1989. The molecular mechanism of retention in reversed-phase liquid chromatography. Chemical Reviews, 89(2), 331-346.
Dwivedi, P., Wu, C., Matz, L. M., Clowers, B. H., Siems, W. F., and Hill, H. H. 2006. Gas-phase chiral separations by ion mobility spectrometry. Analytical chemistry, 78(24), 8200-8206.
Eljarrat, E., and Barcelo, D. 2002. Congener‐specific determination of dioxins and related compounds by gas chromatography coupled to LRMS, HRMS, MS/MS and TOFMS. Journal of Mass Spectrometry, 37(11), 1105-1117.
Eljarrat, E., Guerra, P., and Barceló, D. 2008. Enantiomeric determination of chiral persistent organic pollutants and their metabolites. TrAC Trends in Analytical Chemistry, 27(10), 847-861.

68
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Ericson, I., Martí-Cid, R., Nadal, M., Van Bavel, B., Lindström, G., and Domingo, J. L. 2008. Human exposure to perfluorinated chemicals through the diet: intake of perfluorinated compounds in foods from the Catalan (Spain) market. Journal of Agricultural and Food Chemistry, 56(5), 1787-1794.
European Chemicals Agency (ECHA). 2008. Member state committee support document for identification of hexabromocyclododecane and all major diastereomers identified as a substance of very high concern. https://echa.europa.eu/documents/10162/1fc837ef-f922-476e-8e00-825ab60213c0.
Faubel, M., and Kisters, T. 1989. Non-equilibrium molecular evaporation of carboxylic acid dimers. Nature, 339(6225), 527-529.
Fernando, S., Green, M. K., Organtini, K., Dorman, F., Jones, R., Reiner, E. J., and Jobst, K. J. 2016. Differentiation of (mixed) halogenated dibenzo-p-dioxins by negative ion atmospheric pressure chemical ionization. Analytical chemistry, 88(10), 5205-5211.
Fiedler, H. 2003. The Hand Book of Environmental Chemistry, Persistent Organic Pollutants. Springer-Verlag, Berlin/Heidelberg.
Focant, J. F., Reiner, E. J., MacPherson, K., Kolic, T., Sjödin, A., Patterson Jr, D. G., Reese, S. L., Dorman, F. L., and Cochran, J. 2004. Measurement of PCDDs, PCDFs, and non-ortho-PCBs by comprehensive two-dimensional gas chromatography-isotope dilution time-of-flight mass spectrometry (GC× GC-IDTOFMS). Talanta, 63(5), 1231-1240.
Gao, S., Zhang, Z. P., and Karnes, H. T. 2005. Sensitivity enhancement in liquid chromatography/atmospheric pressure ionization mass spectrometry using derivatization and mobile phase additives. Journal of Chromatography B, 825(2), 98-110.
Gebbink, W. A., van Asseldonk, L., and van Leeuwen, S. P. 2017. Presence of emerging per-and polyfluoroalkyl substances (PFASs) in river and drinking water near a fluorochemical production plant in the Netherlands. Environmental science and technology, 51(19), 11057-11065.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
69
Geng, D., Ericson Jogsten, I., Kukucka, P., Hagberg, J., Roos, A., and van Bavel, B. 2014. Comparison on atmospheric pressure gas chromatography-tandem mass spectrometry (APGC-MS/MS) and high resolution mass spectrometry for the analysis of polybrominated diphenyl ethers (PBDEs). In Organohalogen Compounds (Vol. 76, pp. 1593-1596). Eco-Informa Press.
Geng, D., Jogsten, I. E., Dunstan, J., Hagberg, J., Wang, T., Ruzzin, J., Rabasa-Lhoret, R., and van Bavel, B. 2016. Gas chromatography/atmospheric pressure chemical ionization/mass spectrometry for the analysis of organochlorine pesticides and polychlorinated biphenyls in human serum. Journal of Chromatography A, 1453, 88-98.
Geng, D., Kukucka, P., and Jogsten, I. E. 2017. Analysis of brominated flame retardants and their derivatives by atmospheric pressure chemical ionization using gas chromatography coupled to tandem quadrupole mass spectrometry. Talanta, 162, 618-624.
Geyer, H. J., Rimkus, G. G., Scheunert, I., Kaune, A., Schramm, K. W., Kettrup, A., Zeeman, M., Muir, D. C., G, Hansen, L. G., and Mackay, D. 2000. Bioaccumulation and occurrence of endocrine-disrupting chemicals (EDCs), persistent organic pollutants (POPs), and other organic compounds in fish and other organisms including humans. In Bioaccumulation–New Aspects and Developments, 1-166. Springer, Berlin, Heidelberg.
Giddings, J. C. 1991. Unified separation science, 269-278. New York etc: Wiley.
Goscinny, S., Vandevijvere, S., Maleki, M., Van Overmeire, I., Windal, I., Hanot, V., Blaude, M. N., Vleminckx, C., and Van Loco, J. 2011. Dietary intake of hexabromocyclododecane diastereoisomers (α-, β-, and γ-HBCD) in the Belgian adult population. Chemosphere, 84(3), 279-288.
Gotelli, N. J., and Ellison, A. M. 2013. A Primer of Ecological Statistics, 2nd edition. Sinauer Associates, Inc. Publishers.
Griffiths, J. 2008. A brief history of mass spectrometry. Analytical Chemistry, 80(15), 5678-5683.

70
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Guerra, P., Eljarrat, E., and Barceló, D. 2011. Determination of halogenated flame retardants by liquid chromatography coupled to mass spectrometry. Trends in Analytical Chemistry, 30(6), 842-855.
Guilhaus, M., Selby, D., and Mlynski, V. 2000. Orthogonal acceleration time‐of‐flight mass spectrometry. Mass Spectrometry Reviews, 19(2), 65-107.
Guo, Y., and Kannan, K. 2015. Analytical methods for the measurement of legacy and emerging persistent organic pollutants in complex sample matrices. In Comprehensive Analytical Chemistry (Vol. 67, 1-56). Elsevier.
Hajšlová, J., Pulkrabová, J., Poustka, J., Čajka, T., and Randák, T. 2007. Brominated flame retardants and related chlorinated persistent organic pollutants in fish from river Elbe and its main tributary Vltava. Chemosphere, 69(8), 1195-1203.
Heeb, N. V., Schweizer, W. B., Kohler, M., and Gerecke, A. C. 2005. Structure elucidation of hexabromocyclododecanes—a class of compounds with a complex stereochemistry. Chemosphere, 61(1), 65-73.
Heeb, N. V., Schweizer, W. B., Kohler, M., and Gerecke, A. C. 2005. Structure elucidation of hexabromocyclododecanes—a class of compounds with a complex stereochemistry. Chemosphere, 61(1), 65-73.
Hernández, F., Sancho, J. V., Ibáñez, M., Abad, E., Portolés, T., and Mattioli, L. 2012. Current use of high-resolution mass spectrometry in the environmental sciences. Analytical and bioanalytical chemistry, 403(5), 1251-1264.
Heydebreck, F., Tang, J., Xie, Z., and Ebinghaus, R. 2015. Alternative and legacy perfluoroalkyl substances: differences between European and Chinese river/estuary systems. Environmental Science and Technology, 49(14), 8386-8395.
Hilton, D. C., Jones, R. S., and Sjödin, A. 2010. A method for rapid, non-targeted screening for environmental contaminants in household dust. Journal of Chromatography A, 1217(44), 6851-6856.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
71
Hopkins, Z. R., Sun, M., DeWitt, J. C., and Knappe, D. R. 2018. Recently detected drinking water contaminants: GenX and other per‐and polyfluoroalkyl ether acids. Journal‐American Water Works Association, 110(7), 13-28.
Houde, M., Martin, J. W., Letcher, R. J., Solomon, K. R., and Muir, D. C. 2006. Biological monitoring of polyfluoroalkyl substances: a review. Environmental Science and Technology, 40(11), 3463-3473.
Howard, P. H., and Muir, D. C. 2010. Identifying new persistent and bioaccumulative organics among chemicals in commerce. Environmental Science and Technology, 44 2277-2285.
Hühnerfuss, H., and Shah, M. R. 2009. Enantioselective chromatography—A powerful tool for the discrimination of biotic and abiotic transformation processes of chiral environmental pollutants. Journal of Chromatography A, 1216(3), 481-502.
Jensen, S. 1972. The PCB story. Ambio, 123-131.
Johnson-Restrepo, B., Adams, D. H., and Kannan, K. 2008. Tetrabromobisphenol A (TBBPA) and hexabromocyclododecanes (HBCDs) in tissues of humans, dolphins, and sharks from the United States. Chemosphere, 70(11), 1935-1944.
Jones, K. C., and De Voogt, P. 1999. Persistent organic pollutants (POPs): state of the science. Environmental Pollution, 100(1-3), 209-221.
Kamel, A. M., Brown, P. R., and Munson, B. 1999. Effects of mobile-phase additives, solution pH, ionization constant, and analyte concentration on the sensitivities and electrospray ionization mass spectra of nucleoside antiviral agents. Analytical chemistry, 71(24), 5481-5492.
Kanu, A.B., Dwivedi, P., Tam, M., Matz, L. and Hill, H.H., 2008. Ion mobility–mass spectrometry. Journal of Mass Spectrometry, 43(1), 1-22.
Kato, K., Kalathil, A. A., Patel, A. M., Ye, X., and Calafat, A. M. 2018. Per-and polyfluoroalkyl substances and fluorinated alternatives in urine and serum by on-line solid phase extraction–liquid chromatography–tandem mass spectrometry. Chemosphere, 209, 338-345.

72
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Kaufmann, A., Butcher, P., Maden, K., Widmer, M., Giles, K., and Uría, D. 2009. Are liquid chromatography/electrospray tandem quadrupole fragmentation ratios unequivocal confirmation criteria? Rapid Communications in Mass Spectrometry, 23(7), 985-998.
Köppen, R., Becker, R., Esslinger, S., and Nehls, I. 2010. Enantiomer-specific analysis of hexabromocyclododecane in fish from Etnefjorden (Norway). Chemosphere, 80(10), 1241-1245.
Korfmacher, W. A., Mitchum, R. K., Hileman, F. D. and Mazer, T., 1983. Analysis of 2, 3, 7, 8-tetrachlorodibenzofuran by fused silica GC combined with atmospheric pressure ionization MS. Chemosphere, 12(9), 1243-1249.
Korfmacher, W. A., Rowland, K. R., Mitchum, R. K., Daly, J. J., McDaniel, R. C. and Plummer, M. V., 1984. Analysis of snake tissue and snake eggs for 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin via fused silica GC combined with atmospheric pressure ionization MS. Chemosphere, 13(11), 1229-1233.
Krauss, M., Singer, H., and Hollender, J. 2010. LC–high resolution MS in environmental analysis: from target screening to the identification of unknowns. Analytical and bioanalytical chemistry, 397(3), 943-951.
Kruve, A., and Kaupmees, K. 2017. Adduct formation in ESI/MS by mobile phase additives. Journal of the American Society for Mass Spectrometry, 28(5), 887-894.
Langlois, I. 2007. Mass spectrometric isomer characterization of perfluorinated compounds in technical mixture, water and human blood (Doctoral dissertation, University of Basel).
Lapthorn, C., Pullen, F. S., Chowdhry, B. Z., Wright, P., Perkins, G. L., and Heredia, Y. 2015. How useful is molecular modelling in combination with ion mobility mass spectrometry for ‘small molecule’ ion mobility collision cross-sections? Analyst, 140(20), 6814-6823.
Lee, M. L., and Markides, K. E. 1990. Analytical supercritical fluid chromatography and extraction. Provo, Utah: Chromatography Conferences.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
73
Li, D. X., Gan, L., Bronja, A. and Schmitz, O. J., 2015. Gas chromatography coupled to atmospheric pressure ionization mass spectrometry (GC-API-MS): review. Analytica Chimica Acta, 891, 43-61.
Loos, R., Gawlik, B. M., Locoro, G., Rimaviciute, E., Contini, S., and Bidoglio, G. 2009. EU-wide survey of polar organic persistent pollutants in European river waters. Environmental pollution, 157(2), 561-568.
Lorenzo, M., Campo, J., and Picó, Y. 2018. Analytical challenges to determine emerging persistent organic pollutants in aquatic ecosystems. Trends in Analytical Chemistry, 103, 137-155.
Mallet, C. R., Lu, Z., and Mazzeo, J. R. 2004. A study of ion suppression effects in electrospray ionization from mobile phase additives and solid‐phase extracts. Rapid Communications in Mass Spectrometry, 18(1), 49-58.
Markes International, 2016. Select-eV: The next generation of ion source technology. https://www.markes.com/Products/Mass-spectrometry/Bench-TOF-Select/Select-eV.aspx. Accessed April 18 2019.
Martin, A. J. P., and Synge, R. M. 1941. A new form of chromatogram employing two liquid phases: A theory of chromatography. 2. Application to the micro-determination of the higher monoamino-acids in proteins. Biochemical Journal, 35(12), 1358.
Martin, J. W., Kannan, K., Berger, U. R. S., Voogt, P. D., Field, J., Franklin, J., Giesy, J. P., Harner, T., Muir, D. C. G., Scott, B., Kaiser, M., Jarnberg, U., Jones, K. C., Mabury, S., Schroeder, H., Simkic, M., Sottani, C., van Bavel, B., Karrman, A., Lindstrom, G., van Leeuwen, S., and Kaiser, M. 2004. Peer reviewed: analytical challenges hamper perfluoroalkyl research. Environmental Science and Technology, 248A-255A.
McCord, J., Newton, S., and Strynar, M. 2018. Validation of quantitative measurements and semi-quantitative estimates of emerging perfluoroethercarboxylic acids (PFECAs) and hexfluoroprolyene oxide acids (HFPOAs). Journal of Chromatography A, 1551, 52-58.
McEwen, C. N. and McKay, R. G., 2005. A Combination Atmospheric Pressure LC/MS: GC/MS Ion Source: Advantages of Dual AP-LC/MS: GC/MS Instrumentation. Journal of the American Society for Mass Spectrometry, 16(11), 1730-1738.

74
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
McLafferty, F. W., and Tureček, F., Interpretation of mass spectra, University Science Books, 1993.
McMaster, M.C. GC/MS A practical users guide, Wiley, Second edn., 2008.
Megson, D., Reiner, E. J., Jobst, K. J., Dorman, F. L., Robson, M., and Focant, J. F. 2016. A review of the determination of persistent organic pollutants for environmental forensics investigations. Analytica Chimica Acta, 941, 10-25.
Mitchum, R. K., Moler, G. F. and Korfmacher, W. A., 1980. Combined capillary gas chromatography/atmospheric pressure negative chemical ionization/mass spectrometry for the determination of 2, 3, 7, 8-tetrachlorodibenzo-p-dioxin in tissue. Analytical chemistry, 52(14), 2278-2282.
Mollerup, C. B., Mardal, M., Dalsgaard, P. W., Linnet, K., and Barron, L. P. 2018. Prediction of collision cross section and retention time for broad scope screening in gradient reversed-phase liquid chromatography-ion mobility-high resolution accurate mass spectrometry. Journal of Chromatography A, 1542, 82-88.
Morris, S., Bersuder, P., Allchin, C. R., Zegers, B., Boon, J. P., Leonards, P. E., and de Boer, J. 2006. Determination of the brominated flame retardant, hexabromocyclodocane, in sediments and biota by liquid chromatography-electrospray ionization mass spectrometry. Trends in Analytical Chemistry, 25(4), 343-349.
Moschet, C., Anumol, T., Lew, B. M., Bennett, D. H., and Young, T. M. 2018. Household dust as a repository of chemical accumulation: new insights from a comprehensive high-resolution mass spectrometric study. Environmental Science and Technology, 52(5), 2878-2887.
Muir, D. C., and Howard, P. H. 2006. Are there other persistent organic pollutants? A challenge for environmental chemists. Environmental Science and Technology, 40(23), 7157-7166.
Mullin, L., Burgess, J. A., Jogsten, I. E., Geng, D., Aubin, A., and van Bavel, B. 2015. Rapid separation of hexabromocyclododecane diastereomers using a novel method combining convergence chromatography and tandem mass spectrometry. Analytical Methods, 7(7), 2950-2958.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
75
Munoz, G., Liu, J., Duy, S. V., and Sauvé, S. 2019. Analysis of F-53B, Gen-X, ADONA, and emerging fluoroalkylether substances in environmental and biomonitoring samples: A review. Trends in Environmental Analytical Chemistry, e00066.
Newton, S., McMahen, R., Stoeckel, J. A., Chislock, M., Lindstrom, A., and Strynar, M. 2017. Novel polyfluorinated compounds identified using high resolution mass spectrometry downstream of manufacturing facilities near Decatur, Alabama. Environmental Science and Technology, 51(3), 1544-1552.
Organtini, K. L., Haimovici L., Jobst K. J., Reiner E. J., Ross, B., Ladak, A., Stevens, D., Cochran J. W., and Dorman, F. L. 2015. Comparison of atmospheric pressure ionization gas chromatography-triple quadrupole mass spectrometry to traditional high-resolution mass spectrometry for the identification and quantification of halogenated dioxins and furans. Analytical Chemistry, 87(15), 7902-7908.
Organtini, K. L., Myers, A. L., Jobst K. J., Reiner E. J., Ross, B., Ladak, A., Mullin, L., Stevens, D., and Dorman, F. L. 2015, Quantitative analysis of mixed halogen dioxins and furans in fire debris utilizing atmospheric pressure ionization gas chromatography-triple quadrupole mass spectrometry. Analytical Chemistry 87(20) 10368-10377.
Ouyang, X., Weiss, J. M., de Boer, J., Lamoree, M. H., and Leonards, P. E. 2017. Non-target analysis of household dust and laundry dryer lint using comprehensive two-dimensional liquid chromatography coupled with time-of-flight mass spectrometry. Chemosphere, 166, 431-437.
Pan, Y., Zhang, H., Cui, Q., Sheng, N., Yeung, L. W., Guo, Y., Sun, Y. and Dai, J. 2017. First report on the occurrence and bioaccumulation of hexafluoropropylene oxide trimer acid: An emerging concern. Environmental Science and Technology, 51(17), 9553-9560.
Pan, Y., Zhang, H., Cui, Q., Sheng, N., Yeung, L. W., Sun, Y., Guo, Y., and Dai, J. 2018. Worldwide distribution of novel perfluoroether carboxylic and sulfonic acids in surface water. Environmental Science and Technology, 52(14), 7621-7629.

76
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Perrenoud, A. G. G., Veuthey, J. L., and Guillarme, D. 2012. Comparison of ultra-high performance supercritical fluid chromatography and ultra-high performance liquid chromatography for the analysis of pharmaceutical compounds. Journal of Chromatography A, 1266, 158-167.
Perrenoud, A. G. G., Hamman, C., Goel, M., Veuthey, J. L., Guillarme, D., and Fekete, S. 2013. Maximizing kinetic performance in supercritical fluid chromatography using state-of-the-art instruments. Journal of Chroma-tography A, 1314, 288-297.
Portoles, T., Rosales, L. E., Sancho, J. V., Santos, F. J., and Moyano, E. 2015. Gas chromatography-tandem mass spectreomtry with atmospheric pressure chemical ionization for fluorotelomer alcohols and perfluorinated sulfonamides determination. Journal of Chromatography A, 1413, 107-116.
Portoles, T., Sales, C., Abalos, M., Saulo, J., and Abad, E. 2016. Evaluation of the capabilities of atmospheric pressure chemical ionization source coupled to tandem mass spectrometry for the determination of dioxin-like polychlorinatedbiphenyls in complex-matrix food samples. Analytical Chimica Acta, 937, 96-105.
Portoles, T., Sales, C., Gomara, B., Sancho, J. V., Beltran, J., Herrero, L., Gonzalez, M. J., and Hernandez, F. 2015. Novel analytical approach for brominated flame retardants based on the use of gas chromatography-atmospheric pressure chemical ionization-tandem mass spectrometry with emphasis in highly brominated congeners. Analytical Chemistry, 87, 9892-9899.
Rager, J. E., Strynar, M. J., Liang, S., McMahen, R. L., Richard, A. M., Grulke, C. M., Wambaugh, J. F., Isaacs, K. K., Judson, R., Williams, A. J., and Sobus, J. R. 2016. Linking high resolution mass spectrometry data with exposure and toxicity forecasts to advance high-throughput environmental monitoring. Environment International, 88, 269-280.
Regueiro, J., Negreira, N., and Berntssen, M. H. 2016. Ion-mobility-derived collision cross section as an additional identification point for multiresidue screening of pesticides in fish feed. Analytical Chemistry, 88(22), 11169-11177.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
77
Reiner, E. J., Clement, R. E., Okey, A. B., and Marvin, C. H. 2006. Advances in analytical techniques for polychlorinated dibenzo-p-dioxins, polychlorinated dibenzofurans and dioxin-like PCBs. Analytical and Bioanalytical chemistry, 386(4), 791-806.
Riddell, N., Mullin, L., van Bavel, B., Ericson Jogsten, I., McAlees, A., Brazeau, A., Synnott, S., Lough, A., McCrindle, R., and Chittim, B. 2016. Enantioselective analytical-and preparative-scale separation of hexabromocyclododecane stereoisomers using packed column supercritical fluid chromatography. Molecules, 21(11), 1509.
Riddell, N., van Bavel, B., Jogsten, I. E., McCrindle, R., McAlees, A., and Chittim, B. 2017. Coupling of supercritical fluid chromatography to mass spectrometry for the analysis of Dechlorane Plus: Examination of relevant negative ion atmospheric pressure chemical ionization mechanisms. Talanta, 171, 68-73. (a)
Riddell, N., van Bavel, B., Jogsten, I. E., McCrindle, R., McAlees, A., and Chittim, B. 2017. Coupling supercritical fluid chromatography to positive ion atmospheric pressure ionization mass spectrometry: Ionization optimization of halogenated environmental contaminants. International Journal of Mass Spectrometry, 421, 156-163. (b)
Riddell, N., van Bavel, B., Jogsten, I. E., McCrindle, R., McAlees, A., Potter, D., Tashiro, C., and Chittim, B. 2015. Comparative assessment of the chromatographic separation of 2, 3, 7, 8-substituted polychlorinated dibenzo-p-dioxins and polychlorinated dibenzofurans using supercritical fluid chromatography and high resolution gas chromatography. Analytical Methods, 7(21), 9245-9253.
Righetti, L., Bergmann, A., Galaverna, G., Rolfsson, O., Paglia, G., and Dall’Asta, C. 2018. Ion mobility-derived collision cross section database: Application to mycotoxin analysis. Analytica Chimica Acta, 1014, 50-57.
Ritter, L., Solomon, K. R., Forget, J., Stemeroff, M., and O’Lleary, C. 1995. A review of selected persistent organic pollutants. International Programme on Chemical Safety (IPCS). PCS/95.39. Geneva: World Health Organization, 65, 66.

78
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Rivera-Austrui, J., Martinez, K., Abalos, M., Sales, C., Portoles, T., Beltran, J., Saulo, J., Aristizabal, B.H., and Abad, E., 2017. Analysis of polychlorinated dibenzo-p-dioxins and dibenzofurans in stack gas emissions by gas chromatography-atmospheric pressure chemical ionization-triple-quadrupole mass spectrometry. Journal of Chromatography A, 1513, 245-249.
Rodan, B. D., Pennington, D. W., Eckley, N., and Boethling, R. S. 1999. Screening for persistent organic pollutants: techniques to provide a scientific basis for POPs criteria in international negotiations. Environmental Science and Technology, 33, 3482-3488.
Rostkowski, P., Haglund, P., Aalizadeh, R., Alygizakis, N., Thomaidis, N., Arandes, J. B., Nizzetto, P. B., Booij, P., Budzinski, H., Brunswick, P., Covaci, A., Gallampois, C., Grosse, S., Hindle, R., Ipolyi, I., Jobst, K., Kaserzon, S. L., Leonards, P., Lestremau, F., Letzel, T., Magner, J., Matsukami, H., Moschet, C., Oswald, P., Plassmann, M., Slobodnik, J., and Yang, C. 2019. The strength in numbers: comprehensive characterization of house dust using complementary mass spectrometric techniques. Analytical and Bioanalytical Chemistry, 411(10), 1957-1977.
Rotander, A., Ka rrman, A., Toms, L. M. L., Kay, M., Mueller, J. F., and Gómez Ramos, M. J. 2015. Novel fluorinated surfactants tentatively identified in firefighters using liquid chromatography quadrupole time-of-flight tandem mass spectrometry and a case-control approach. Environmental Science and Technology, 49(4), 2434-2442.
Rotander, A., van Bavel, B., Polder, A., Rigét, F., Auðunsson, G. A., Gabrielsen, G. W., Vikingsson, G., Bloch, D., and Dam, M. 2012. Polybrominated diphenyl ethers (PBDEs) in marine mammals from Arctic and North Atlantic regions, 1986–2009. Environment international, 40, 102-109. (a)
Rotander, A., van Bavel, B., Rigét, F., Auðunsson, G. A., Polder, A., Gabrielsen, G. W., Vikingsson, G., Mikkelsen, B., and Dam, M. 2012. Polychlorinated naphthalenes (PCNs) in sub-Arctic and Arctic marine mammals, 1986–2009. Environmental pollution, 164, 118-124. (b)

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
79
Rotander, A., van Bavel, B., Rigét, F., Auðunsson, G. A., Polder, A., Gabrielsen, G. W., Vikingsson, G., Mikkelsen, B. and Dam, M. 2012. Methoxylated polybrominated diphenyl ethers (MeO-PBDEs) are major contributors to the persistent organobromine load in sub-Arctic and Arctic marine mammals, 1986–2009. Science of the Total Environment, 416, 482-489. (c)
Salihovic, S., Lampa, E., Lindström, G., Lind, L., Lind, P. M., and van Bavel, B. 2012. Circulating levels of persistent organic pollutants (POPs) among elderly men and women from Sweden: results from the Prospective Investigation of the Vasculature in Uppsala Seniors (PIVUS). Environment international, 44, 59-67.
Schecter, A. 1994. Exposure assessment. In Dioxins and health (pp. 449-485). Springer, Boston, MA.
Schecter, A., Päpke, O., Harris, T. R., Tung, K. C., Musumba, A., Olson, J., and Birnbaum, L. 2006. Polybrominated diphenyl ether (PBDE) levels in an expanded market basket survey of US food and estimated PBDE dietary intake by age and sex. Environmental health perspectives, 114(10), 1515-1520.
Schymanski, E. L., Jeon, J., Gulde, R., Fenner, K., Ruff, M., Singer, H. P., and Hollender, J. 2014. Identifying small molecules via high resolution mass spectrometry: communicating confidence. Environmental Science and Technology, 48(4), 2097-2098.
Schymanski, E. L., Singer, H. P., Slobodnik, J., Ipolyi, I. M., Oswald, P., Krauss, M., Schulze, T., Haglund, P., Letzel, T., Grosse, S., Thomaidis, N. S., Bletsou, A., Zwiener, C., Ibanez, M., Portoles, T., de Boer, J., Reid, M. J., Onghena, M., Kunkel, W., Schulz, W., Guillon, A., Noyon, N., Leroy, G., Bados, P., Bogialli, S., Stipanicev, D., Rostkowski, P., and Hollender, J. 2015. Non-target screening with high-resolution mass spectrometry: critical review using a collaborative trial on water analysis. Analytical and Bioanalytical Chemistry, 407(21), 6237-6255.
Smith, R. M. 1999. Supercritical fluids in separation science–the dreams, the reality and the future. Journal of Chromatography A, 856(1-2), 83-115.

80
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
Sobus, J. R., Wambaugh, J. F., Isaacs, K. K., Williams, A. J., McEachran, A. D., Richard, A. M., Grulke, C. M., Ulrich, E. M., Rager, J. E., Strynar, M. J., and Newton, S. R. 2018. Integrating tools for non-targeted analysis research and chemical safety evaluations at the US EPA. Journal of exposure science and environmental epidemiology, 28(5), 411-426
Solomons, T. W. G., and Fryhle, C., 2008. Organic Chemistry, 9th ed. John Wiley and Sons.
Song, X., Vestergren, R., Shi, Y., Huang, J., and Cai, Y. 2018. Emissions, transport, and fate of emerging per-and polyfluoroalkyl substances from one of the major fluoropolymer manufacturing facilities in China. Environmental Science and Technology, 52(17), 9694-9703.
Stach, J. and Baumbach, J. I., 2002. Ion mobility spectrometry-basic elements and applications. Internation Journal of Ion Mobility Spectrometry, 5, 1-21.
Stapleton, H. M., Dodder, N. G., Kucklick, J. R., Reddy, C. M., Schantz, M. M., Becker, P. R., Gulland, B. J., and Wise, S. A. 2006. Determination of HBCD, PBDEs and MeO-BDEs in California sea lions (Zalophus californianus) stranded between 1993 and 2003. Marine Pollution Bulletin, 52(5), 522-531.
Stockholm Convention, All POPs. http://www.pops.int/TheConvention/ThePOPs/AllPOPs/tabid/2509/Default.aspx. Accessed April 19 2019.
Stockholm Convention, Chemicals proposed for listing under the Conven-tion. http://www.pops.int/TheConvention/ThePOPs/ChemicalsProposed-forListing/tabid/2510/Default.aspx. Accessed April 19 2019.
Stow, S. M., Causon, T. J., Zheng, X., Kurulugama, R. T., Mairinger, T., May, J. C., Rennie, E. E., Baker, E. S., Smith, R. D., McLean, J. A., Hann, S., and Fjelsted, J. C. 2017. An interlaboratory evaluation of drift tube ion mobility–mass spectrometry collision cross section measurements. Analytical Chemistry, 89(17), 9048-9055.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
81
Strynar, M., Dagnino, S., McMahen, R., Liang, S., Lindstrom, A., Andersen, E., McMillan L., Thurman, M., Ferrer, I., and Ball, C. 2015. Identification of novel perfluoroalkyl ether carboxylic acids (PFECAs) and sulfonic acids (PFESAs) in natural waters using accurate mass time-of-flight mass spectrometry (TOFMS). Environmental science and technology, 49(19), 11622-11630.
Stubbings, W. A., Riddell, N., Chittim, B., and Venier, M. 2017. Challenges in the analyses of organophosphate esters. Environmental Science and Technology Letters, 4(7), 292-297.
Sun, M., Arevalo, E., Strynar, M., Lindstrom, A., Richardson, M., Kearns, B., Pickett, A., Smith, C., and Knappe, D. R. 2016. Legacy and emerging perfluoroalkyl substances are important drinking water contaminants in the Cape Fear River Watershed of North Carolina. Environmental Science and Technology Letters, 3(12), 415-419.
Tejada-Casado, C., Hernández-Mesa M., Monteau F., Lara F. J., del Olmo-Iruela M., García-Campaña A. M., Le Bizec B., and Dervilly-Pinel G. 2018. Collision cross section (CCS) as a complementary parameter to characterize human and veterinary drugs. Analytica Chimica Acta, 1043, 52-63.
Ubukata, M., Jobst, K. J., Reiner, E. J., Reichenbach, S. E., Tao, Q., Hang, J., Wu, Z., Dane, J. A., and Cody, R. B. 2015. Non-targeted analysis of electronics waste by comprehensive two-dimensional gas chromatography combined with high-resolution mass spectrometry: Using accurate mass information and mass defect analysis to explore the data. Journal of Chromatography A, 1395, 152-159.
United States Environmental Protection Agency. 2010. Hexabromocyclododecane (HBCD) Action Plan. https://www.epa.gov/assessing-and-managing-chemicals-under-tsca/hexabromocyclododecane-hbcd-action-plan. Accessed April 28 2018.
United States Environmental Protection Agency. 2018. Method 537.1 Determination of selected per- and polyfluorinated alkyl substances in drinking water by solid phase extraction and liquid chromatography/tandem mass spectrometry (LC/MS/MS).

82
LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
van Bavel, B., Dahl, P., Karlsson, L., Harden, L., Rappe, C., and Lindstöm, G. 1995. Supercritical fluid extraction of PCBs from human adipose tissue for HRGC/LRMS analysis. Chemosphere, 30(7), 1229-1236.
van Bavel, B., Geng, D., Cherta, L., Nácher-Mestre, J., Portolés, T., Ábalos, M., Saulo, J., Abad, E., Dunstan, J., Jones, R., Kotz, A., Winterhalter, H., Malisch, R., Traag, W., Hagberg, J., Jogsten, I. E., and Beltran, J. 2015. Atmospheric-pressure chemical ionization tandem mass spectrometry (APGC/MS/MS) an alternative to high-resolution mass spectrometry (HRGC/HRMS) for the determination of dioxins. Analytical Chemistry, 87(17), 9047-9053.
van Bavel, B., Järemo, M., Karlsson, L., and Lindström, G. 1996. Development of a solid phase carbon trap for simultaneous determination of PCDDs, PCDFs, PCBs, and pesticides in environmental samples using SFE-LC. Analytical Chemistry, 68(7), 1279-1283.
Van Deemter, J. J., Zuiderweg, F. J., and Klinkenberg, A. V. 1956. Longitudinal diffusion and resistance to mass transfer as causes of nonideality in chromatography. Chemical Engineering Science, 5(6), 271-289.
Wang, M. 2016. Ion source for soft electron ionization and related systems and methods. Agilent Technologies, Inc. United States Patent Application Publishing, Pub No. US 2016/0172146 A1
Wania, F., and Mackay, D. 1999. The evolution of mass balance models of persistent organic pollutant fate in the environment. Environmental Pollution, 100(1-3), 223-240.
Waters Corporation. Empower System Suitability Quick Reference Guide. https://www.waters.com/webassets/cms/support/docs/71500031605ra.pdf. Accessed September 1 2019.
Watson, J. T., and Sparkman, O. D. 2007. Introduction to mass spectrometry: instrumentation, applications, and strategies for data interpretation. John Wiley and Sons.
Williamson, S. H. 2018. What’s in the Water? How Media Coverage of Corporate GenX Pollution Shapes Local Understanding of Risk. Critical Criminology, 26(2), 289-305.

LAUREN MULLIN Advances in Mass Spectrometry for the Analysis of Emerging POPs
83
Xu, C., Lin, X., Yin, S., Zhao, L., Liu, Y., Liu, K., Li, F., Yang, F., and Liu, W. 2018. Enantioselectivity in biotransformation and bioaccumulation processes of typical chiral contaminants. Environmental pollution. 243, 1274-1286.
Yeung, L. W., Stadey, C., and Mabury, S. A. 2017. Simultaneous analysis of perfluoroalkyl and polyfluoroalkyl substances including ultrashort-chain C2 and C3 compounds in rain and river water samples by ultra performance convergence chromatography. Journal of Chromatography A, 1522, 78-85.
Zegers, B. N., Mets, A., van Bommel, R., Minkenberg, C., Hamers, T., Kamstra, J. H., Pierce, G., and Boon, J. P. 2005. Levels of hexabromocyclododecane in harbor porpoises and common dolphins from western European seas, with evidence for stereoisomer-specific biotransformation by cytochrome P450. Environmental Science and Technology, 39(7), 2095-2100.
Zheng, X., Dupuis, K. T., Aly, N. A., Zhou, Y., Smith, F. B., Tang, K., Smith, R. D., and Baker, E. S. 2018. Utilizing ion mobility spectrometry and mass spectrometry for the analysis of polycyclic aromatic hydrocarbons, polychlorinated biphenyls, polybrominated diphenyl ethers and their metabolites. Analytica chimica acta, 1037, 265-273.
Zhou, Z., Tu, J., and Zhu, Z. J. 2018. Advancing the large-scale CCS database for metabolomics and lipidomics at the machine-learning era. Current Opinion in Chemical Biology, 42, 34-41.
Zhou, Z., Xiong X., and Zhu, Z. J. 2017. MetCCS predictor: a web server for predicting collision cross-section values of metabolites in ion mobility-mass spectrometry based metabolomics. Bioinformatics, 33(14), 2235-2237.


Publications in the series Örebro Studies in Chemistry
1. Bäckström, Mattias, On the Chemical State and Mobility of Lead and Other Trace Elements at the Biogeosphere/Technosphere Interface. 2002.
2. Hagberg, Jessika, Capillary zone electrophoresis for the analysis of low molecular weight organic acids in environmental systems. 2003.
3. Johansson, Inger, Characterisation of organic materials from incineration residues. 2003.
4. Dario, Mårten, Metal Distribution and Mobility under alkaline conditions. 2004.
5. Karlsson, Ulrika, Environmental levels of thallium – Influence of redox properties and anthropogenic sources. 2006.
6. Kärrman, Anna, Analysis and human levels of persistent perfluorinated chemicals. 2006.
7. Karlsson Marie, Levels of brominated flame retardants in humans and their environment: occupational and home exposure. 2006.
8. Löthgren, Carl-Johan, Mercury and Dioxins in a MercOx-scrubber. 2006.
9. Jönsson, Sofie, Microextraction for usage in environmental monitoring and modelling of nitroaromatic compounds and haloanisoles. 2008.
10. Ericson Jogsten, Ingrid, Assessment of human exposure to per- and polyfluorinated compounds (PFCs). Exposure through food, drinking water, house dust and indoor air. 2011.
11. Nilsson, Helena, Occupational exposure to fluorinated ski wax. 2012.
12. Salihovic, Samira, Development and Application of High-throughput Methods for Analysis of Persistent Organic Pollutants in Human Blood. 2013.
13. Larsson, Maria, Chemical and bioanalytical characterisation of PAH-contaminated soils. Identification, availability and mixture toxicity of AhR agonists. 2013.

14. Kalaitzaki, Argyro, Biocompatible microemulsions: formulation, encapsulation of bioactive compounds and their potential application. 2014
15. Saqib, Naeem, Distribution and chemical association of trace elements in incinerator residues and mining waste from a leaching perspective. 2016
16. Chatzidaki, Maria, Formulation and characterization of W/O nano-dispersions for bioactive delivery applications. 2016
17. Geng, Dawei, Gas Chromatography-Atmospheric Pressure Chemical Ionization-Tandem Mass Sepctrometry Methods for the Determination of Envrionmental Contaminants. 2016
18. Eriksson, Ulrika, Contribution of polyfluoroalkyl phosphate esters (PAPs) and other precursor compounds to perfluoroalkyl carboxylates (PFCAs) in humans and the environment. 2016
19. Riddell, Nicole, Packed Column Supercritical Fluid Chromatography: Applications in Environmental Chemistry. 2017
20. Bjurlid, Filip, Polybrominated dibenzo-p-dioxins and furans: from source of emission to human exposure. 2017
21. Stubleski, Jordan, Assessing the longitudinal trend of POP concentrations in humans using high-throughput sample preparation methods developed for low-volume samples. 2018
22. Persson, Josefin, Indoor air quality and chemical emissions of organic compounds in newly built low-energy preschools. 2018
23. Schönlau, Christine, Microplastics in the marine environment and the assessment of potential adverse effects of associated chemicals. 2019
24. Mullin, Lauren, Advances in Mass Spectrometry for the Analysis of Emerging Persistent Organic Pollutants. 2019





























