Embed Size (px)
Citation preview

Abnormal Translocation of Tyrosinase and Tyrosinase-Related Protein 1 in Cutaneous Melanocytes of Hermansky±Pudlak Syndrome and in Melanoma Cells Transfected withAnti-Sense HPS1 cDNA
Rangaprasad Sarangarajan, Ashish Budev, Yang Zhao, William A. Gahl,* and Raymond E. BoissyDepartment of Dermatology, University of Cincinnati, Cincinnati, Ohio, and *Section on Human Biochemical Genetics, Heritable Disorders Branch,
NICHD, National Institutes of Health, Bethesda, Maryland, U.S.A.
Hermansky±Pudlak syndrome is an autosomal reces-sive disorder characterized by oculocutaneous albin-ism, a bleeding disorder, and, in some patients,ceroid storage and progressive lung disease.Although Hermansky±Pudlak syndrome exhibitslocus heterogeneity, most patients have mutations inthe HPS1 gene. Melanocytes in the basal epitheliallayer of skin from patients with different mutationsin the HPS1 gene exhibited occasional large com-plexes containing dihydroxyphenylalanine-positivecisterna and 50 nm vesicles. To characterize the roleof the HPS1 protein in cells, human HPS1 cDNAwas transfected into pigmented SK-MEL-188 mela-noma cells (M-188) in either the sense (S-188) or theantisense (A-188) orientation. Expression of the79 kDa HPS1 protein (in M-188 and S-188 cells) orlack of expression (in A-188 cells) was con®rmed byWestern blotting using two HPS1-protein-speci®cpolyclonal antibodies. Signi®cant reduction inexpression of HPS1 protein in A-188 cells resulted in
a signi®cant decrease in tyrosinase activity and mel-anin content compared with M-188 and S-188 cellsusing an intact cell assay for tyrosinase. In contrast,tyrosinase activities in cell lysates of M-188, S-188,and A-188 cells were not signi®cantly different.Knockout of HPS1 protein expression in A-188 cellscaused both tyrosinase and tyrosinase-related protein1 to be localized to large granular complexes in thecell cytosol and dendrites. Electron microscopeanalysis of the A-188 cells revealed that absence ofHPS1 protein resulted in the deposition of dihydrox-yphenylalanine reaction products (i.e., tyrosinase)con®ned to large membrane-bound structures withlimiting membranes. We conclude that lack of HPS1protein expression results in mistranslocation of tyro-sinase and tyrosinase-related protein 1 to largegranular complexes rather than melanosomes, com-promising melanin synthesis. Key words: albinism/hypopigmentation/traf®cking. J Invest Dermatol 117:641±646, 2001
Hermansky±Pudlak syndrome (HPS) is a rare auto-somal recessive disorder characterized by oculocu-taneous albinism, a bleeding disorder, and, in somepatients, ceroid storage disease and progressive lung®brosis (Shotelersuk and Gahl, 1998). The symp-
toms associated with HPS re¯ect structural and/or functionaldefects in a group of related organelles including melanosomes ofthe melanocyte (causing albinism), dense granules of the platelet(causing a bleeding disorder), and lysosomes of reticuloendothelialcells (causing ceroid storage). The hypopigmentation associatedwith HPS is extremely variable and involves the skin, hair, and eyes(Shotelersuk and Gahl, 1998). The characteristic clinical symptomsassociated with HPS can be due to mutations at different geneticloci including ADTB3A, which codes for a subunit of the coat
protein complex AP-3 (Oh et al, 1996; Huizing et al, 2000;Shotelersuk et al, 2000). The most common variant of HPS,however, is due to mutations in HPS1, a gene that encodes apredicted polypeptide of 700 amino acids with a molecular mass ofapproximately 80 kDa (Huizing et al, 2000; Oh et al, 2000). Thefunction of the HPS1 gene product is unknown, but the proteinappears to be nonglycosylated, cytosolic, and partly associated withmelanosomes (Oh et al, 2000). Melanocyte cultures lacking HPS1mRNA exhibit hypomelanization due to the translocation oftyrosinase and tyrosinase-related protein 1 (TRP-1) to membra-nous complexes rather than premelanosomes (Boissy et al, 1998b).Based upon these results, it has been suggested that HPS1p mayfacilitate traf®cking of melanocyte-speci®c gene products to thepremelanosome (Boissy et al, 1998b).
This study describes ultrastructural aberrations in humanmelanocytes bearing mutations in HPS1. In addition, the effectof inhibition of HPS1 protein (HPS1p) expression on traf®cking ofmelanocyte-speci®c gene products, tyrosinase activity, and melaninsynthesis was studied by transfecting pigmented melanoma cellswith HPS1 cDNA in an antisense orientation. This in vitro model ofHPS1p ablation recapitulates the in vivo melanocyte defects andpermits evaluation of the role of HPS1p in melanocytic cells.
Manuscript received February 23, 2001; revised March 27, 2001;accepted for publication April 4, 2001.
Reprint requests to: Dr. Raymond E. Boissy, Associate Professor ofDermatology, Department of Dermatology, University of CincinnatiCollege of Medicine, PO Box 670592, Cincinnati, OH 45267-0592.Email: [email protected]
Abbreviations: AP-3, adaptin complex 3; HPS, Hermansky±Pudlaksyndrome; TRP-1, tyrosinase-related protein 1.
0022-202X/01/$15.00 ´ Copyright # 2001 by The Society for Investigative Dermatology, Inc.
641

MATERIALS AND METHODS
Skin biopsies After informed consent, 4 mm skin biopsies wereobtained from normal volunteer donors and from two patients withHPS, one homozygous for the 16 bp duplication in HPS1 typical ofnorthwest Puerto Rican patients and one heterozygous for an A1195delmutation and an unknown mutation. Both patients have been describedpreviously (Boissy et al, 1998b).
Cell culture Pigmented melanoma SK-MEL-188 cells, a gift from Dr.Alan Houghton, Sloan Kettering Institute, New York, were maintainedin Dulbecco's minimum essential medium (DMEM) (Gibco BRL, GrandIsland, NY) supplemented with 10% fetal bovine serum (Gibco), 1%antibiotic/antimycotic solution (Gibco), and 1% MEM nonessentialamino acids (Gibco).
Vector construction and transfection A cDNA of approximately2.3 kb encoding the full length HPS1p was excised from pcDNA3.1/HisA, B, C and inserted into the EcoRI/EcoRI site of pcDNA3.1 (+)(Invitrogen, Carlsbad, San Diego) using standard molecular biologyprotocols. The orientations of inserts were con®rmed by restrictiondigests and sequencing. Cells were transfected with sense or antisenseHPS1 cDNA in pcDNA3.1 (+) using an Effectene reagent kit (Qiagen,Valencia, CA) according to the manufacturer's protocols. Selection of thetransfectants with 1.5 mg per ml G418 (Gibco) was started on day 3 afterthe initial transfection. Expression of HPS1p was con®rmed in thetransfected cells by Western blotting. Transfection ef®ciency ofapproximately 12%±15% was achieved by this method.
Dihydroxyphenylalanine (DOPA) histochemistry and electronmicroscopy Skin biopsies were cut into quarters and ®xed with half-strength Karnovsky's ®xative (Karnovsky, 1965) in 0.2 M sodiumcacodylate buffer at pH 7.2 for 24 h at 4°C. M-188, S-188, and A-188cells were seeded in Laboratory-Tek chamber slides (Nunc, Naperville,IL) coated with 1% pig gelatin and grown to approximately 80%±90%con¯uence (Boissy et al, 1998b). The cells were ®xed in the wells withhalf-strength Karnovsky's ®xative (Karnovsky, 1965) in 0.2 M sodiumcacodylate buffer at pH 7.2 for 30 min at room temperature. For DOPAhistochemistry, ®xed tissues and cells were incubated in a 0.1% solutionof L-DOPA twice for 2.5 h. The tissues and cells were washed threetimes in buffer and treated with 1.0% osmium tetroxide containing 1.5%potassium ferrocyanide (Karnovsky, 1971) for 30 min. The tissues andcells were washed, stained en bloc with 0.5% uranyl acetate for 30 min,dehydrated, and embedded in Eponate12. Areas of the Epon cast werecut out and mounted on Epon pegs and sectioned on an RMC MT6000-XL ultramicrotome. Ultrathin sections were then stained withaqueous solutions of uranyl acetate (2%) and lead citrate (0.3%) for15 min each and photographed using a JEOL JEM-100CX transmissionelectron microscope. All tissue processing supplies were purchased fromTed Pella (Tustin, CA).
Western blotting analysis Total cellular protein, from untransfectedSK-MEL-188 (M-188) cells and M-188 cells transfected with HPScDNA in either sense (S-188) or antisense (A-188) orientation, wasextracted using RIPA buffer. Equal amounts of protein were fractionatedon a 10% sodium dodecyl sulfate polyacrylamide gel. The proteins weretransferred to a nitrocellulose membrane and incubated with 10% nonfatdry milk in phosphate-buffered saline (PBS) and Tween-20 (PBST,pH 7.4 with 0.2% Tween-20) for 1 h at room temperature. Themembrane was probed with two polyclonal rabbit antibodies. One,called p-80, was generated against the peptide sequenceDDIQPSPRRARSSQN, corresponding to residues 253±267 of thehuman HPS1p (1:1000) (Dell'Angelica et al, 2000). The other, calledHPS-C, was generated against the carboxy terminus of the HPS1p(1:1000) in 2% nonfat milk/PBST solution at 4°C for 3 h. The bands ofinterest were visualized by indirect immuno-enzymatic staining using analkaline phosphatase labeled secondary antiserum followed by BCIP/NBT substrate (Kirkegaard and Perry, Gaithersburg, MD).
Tyrosine hydroxylase activities To quantitate tyrosinase activity, twoassay protocols were employed. In an in situ assay, intact cells wereincubated in medium containing 1.0 mCi per ml [3H]tyrosine (speci®cactivity, 54.2 Ci per mmol) for 24 h. In an in vitro assay, solubilized celllysates (in triplicate) were incubated in 1 ml of reaction mixture (intriplicate) containing 80±100 mg of protein, 1.0 mM L-DOPA, and 1.0mCi [3H]tyrosine at 37°C for 1 h. Each reaction was stopped by additionof an equal volume of 10% (wt/vol) solution of activated charcoal in0.2 N citric acid. The samples were centrifuged at 1500g in a Beckman5000 centrifuge for 5 min and the supernatants were passed over a
Dowex ion-exchange column followed by a wash of 0.1 N citric acid.After addition of 10 ml of scintillation ¯uid (Ultima Gold, PackardBiosciences, Groningen, The Netherlands), the radioactivity of the eluatewas counted in a Packard 1900 CA liquid scintillation analyzer.Tyrosinase activity was expressed as dpm per mg protein (in vitro assay)and dpm per cell (in situ assay).
Melanin content Equal numbers of M-188, S-188, and A-188 cellswere washed in ice-cold PBS and sonicated in 0.5% Nonidet P-40/PBSsolution. After determination of protein content using the bicinchoninicacid protein assay (Pierce Chemical, Rockford, IL), aliquots of the celllysates were solubilized in 0.5 ml of 0.1 N NaOH. The optical densityof the supernatant at 475 nm was compared with a standard curve byusing known concentrations of synthetic melanin, and results wereexpressed as micrograms of melanin per milligram of protein.
Immuno¯uorescence Cell cultures were plated on gelatin-coatedLaboratory-Tek (Nunc) chamber slides at 104 cells per 0.9 cm2 well andprocessed for indirect immuno¯uorescence the next day. The cells were®xed in 5% formalin for 10 min, permeabilized with 100% methanol for3 min, rinsed in PBS containing 1% bovine serum albumin (BSA)(Sigma, St. Louis, MO) three times, blocked with 10% normal goatserum in PBS/BSA, and incubated in primary antibody (as describedbelow) for 1 h. Specimens were rinsed with PBS/BSA and incubated ingoat antispecies-speci®c IgG (secondary antibody) conjugated to CyÔ2or CyÔ3 af®nity puri®ed antiserum (Jackson ImmunoResearchLaboratories, West Grove, PA) for 1 h. The slides were washed andmounted using Fluoromount (Southern Biotechnology Associates).Fluorescence images were acquired on a Zeiss LSM 510 confocalmicroscope (Carl Zeiss, Thornwood, NY) using identical parameters foruntransfected and transfected cells. Primary antibodies consisted ofhPEP7 (1:100) (a gift from Drs King and Oetting, Minnesota, MN),reactive against human tyrosinase, and MEL-5 (1:50) (Signet PathologicalLaboratories, Dedham, MA), reactive against human TRP-1 (Boissy et al,1998a).
RESULTS
Ultrastructural analysis of melanocytes in the basal layer ofHPS1 skin Skin biopsies obtained from a normal individual andfrom two patients with HPS1 were processed for DOPAhistochemistry and electron microscopy. Approximately 10% ofmelanocytes in the basal epithelial layer from both the HPS1patients exhibited an occasional large complex with DOPA-positive cisterna and 50 nm vesicles throughout the periphery ofthe HPS melanocytes (Figs 1a, b). These structures were absentfrom the control samples (Fig 1c).
Western blot analysis The absence of HPS1p from M-188 cellstransfected with the HPS1 antisense cDNA was con®rmed byWestern blot analysis using two polyclonal antibodies, identi®ed asp-80 and HPS-C. An expected band of approximately 79 kDa insize corresponding to HPS1p was detected in lanes containing M-188 and S-188 cell extracts using either p-80 or HPS-C antiserum(Fig 2). HPS1p expression was signi®cantly reduced in thepigmented melanoma SK-MEL-188 cells transfected withantisense HPS1 cDNA (A-188 lane in Fig 2).
Tyrosine hydroxylase activity and melanin content Aftertransfection, cells were pelleted prior to subculturing. The decreasein melanin in the A-188 cell cultures became visible at passage 6. Atthis passage, the in vitro tyrosinase activities of cell lysates from M-188, S-188, and A-188 cells were not signi®cantly different(Fig 3a). In contrast, tyrosinase activity assessed in intact cultures ofA-188 cells was 30% lower compared to activities determined inintact M-188 and S-188 cells (Fig 3b). The decrease in thetyrosinase activity in intact A-188 cells corresponded to a decreasein melanin synthesis as observed in the cell pellet (Fig 4a) and cellsuspension (Fig 4b) along with a 50% decrease in soluble melanincontent in these cells (Fig 4c).
Immuno¯uorescence At passage 6, intracellular localization oftyrosinase and TRP-1 in SK-MEL-188 (M-188) as well as in thosetransfected with sense (S-188) and antisense (A-188) HPS1 cDNAwas performed by immuno¯uorescence analysis using antibodies.There were no signi®cant differences in the patterns of expression
642 SARANGARAJAN ET AL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY

of either tyrosinase or TRP-1 in M-188 (Figs 4d, g, j) and S-188(Figs 4e, h, k) cells. In contrast, in A-188 cells, tyrosinase andTRP-1 were localized to distinct large granular structuresthroughout the cytoplasm and dendrites (Figs 4f, i, l).
DOPA histochemistry and electron microscopy Ultra-structural analysis of SK-MEL-188 (M-188) cells transfected withsense (S-188) or antisense (A-188) HPS1 cDNA was performedusing a combination of DOPA histochemistry and electronmicroscopy. DOPA reaction products were present in the trans-Golgi network and melanosome-like vesicles of M-188 and S-188cells (Figs 5a, b). In A-188 cells, DOPA reaction products were
observed within large membranous complexes of the cell body anddendrites as well as in the normal intracellular location (Figs 5c, d).These large membranous complexes were morphologically similarto those described in human HPS1 melanocytes in vivo (Fig 1) andin vitro (Boissy et al, 1998b).
DISCUSSION
The HPS represents a collection of autosomal recessive geneticdisorders, characterized clinically by oculocutaneous albinism and amild to severe bleeding diathesis due to absence of platelet densegranules. Some patients accumulate electron dense ceroid lipofuscinin various tissues, and some develop pulmonary ®brosis orgranulomatous colitis (Huizing et al, 2000). The genetic cause ofone type of HPS was ®rst identi®ed as a mutation in the HPS gene(currently classi®ed as HPS1), characterized by a 16 bp duplicationin a subpopulation of Puerto Rico inhabitants, resulting in absenceof HPS1p (Oh et al, 1996). Since then, numerous mutations invarious ethnic groups have been identi®ed (Huizing et al, 2000).Although mutations in the HPS1 gene affect skin pigmentation, therole of HPS1p in melanogenesis remains unclear. We havepreviously suggested that HPS1p is involved in mediating properintracellular traf®cking of melanocyte-speci®c gene products to thetarget organelle, the melanosome (Boissy et al, 1998b). This wasbased on the observation that melanocyte cultures established from
Figure 2. Expression of the HPS1 gene product in pigmentedmelanoma cells by Western blot analysis. SK-MEL-188 (M-188)cells were transfected with human HPS1 cDNA inserted into an EcoRI/EcoRI site of the pcDNA3.1 expression vector in the sense (S-188) andantisense (A-188) orientations. Following selection with G-418, the cellswere analyzed for expression of HPS1p by Western blot analysis usingHPS-C and p-80 antisera speci®c for HPS1p. As expected, both the M-188 and S-188 lanes contain a band of approximately 79 kDa usingeither the HPS-C or HPS-p80 antiserum. In contrast, this band wassigni®cantly reduced in the lane containing A-188 cell extract.
Figure 3. In vitro and in situ determination of tyrosinehydroxylase activities of tyrosinase in SK-MEL-188 (M-188) cellstransfected with human HPS1 cDNA in the sense (S-188) orantisense (A-188) orientations reveal altered activity. In vitro (celllysate) and in situ (intact cell) tyrosine hydroxylase activities of tyrosinasein SK-MEL-188 cells transfected with the human HPS1 cDNA in thesense (S-188) and antisense (A-188) orientations were determined asdescribed in Materials and Methods. The in vitro determinations in M-188,S-188, and A-188 were not signi®cantly different (A). Tyrosinehydroxylase activities in A-188 cells were signi®cantly lower (p < 0.05),however, compared to M-188 and S-188 as determined by the in situmethod (B). Tyrosine hydroxylase activities were expressed in dpm permg protein (in vitro method) and dpm per cell (in situ method).
Figure 1. HPS melanocytes exhibit DOPA-positive membranouscomplexes. Ultrastructure of melanocytes in the basal layer of theepidermis from an HPS patient homozygous for the 16 bp duplication inHPS1 (a), an HPS patient heterozygous for an A1195del mutation (b),and a control individual (c). Punch biopsies were processed for DOPAhistochemistry and electron microscopy. Melanocytes of the HPSpatients exhibited complexes with DOPA-positive cisterna (arrows) and50 nm vesicles (arrowheads) throughout the periphery of the HPSmelanocytes that were absent from the control. K, keratinocyte. Scale bar:(a, c) 2.0 mm; (b) 0.7 mm.
VOL. 117, NO. 3 SEPTEMBER 2001 MISTRANSLOCATION OF TYROSINASE AND TRP-1 IN HPS 643
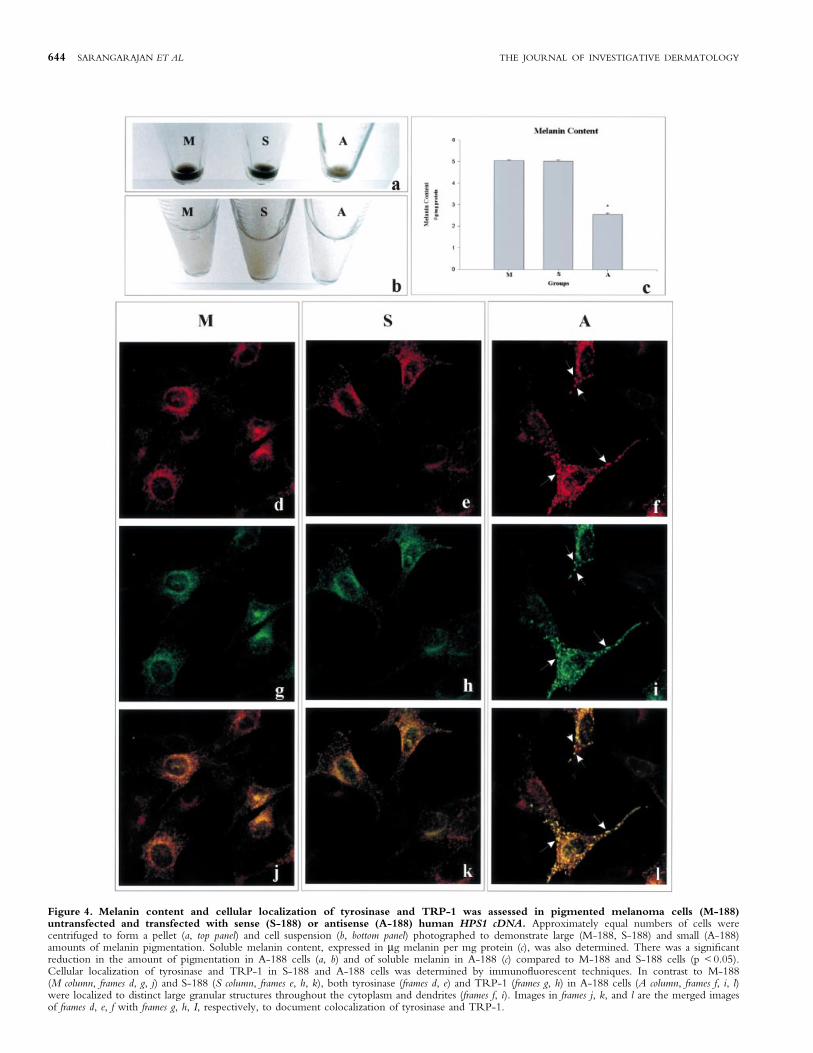
Figure 4. Melanin content and cellular localization of tyrosinase and TRP-1 was assessed in pigmented melanoma cells (M-188)untransfected and transfected with sense (S-188) or antisense (A-188) human HPS1 cDNA. Approximately equal numbers of cells werecentrifuged to form a pellet (a, top panel) and cell suspension (b, bottom panel) photographed to demonstrate large (M-188, S-188) and small (A-188)amounts of melanin pigmentation. Soluble melanin content, expressed in mg melanin per mg protein (c), was also determined. There was a signi®cantreduction in the amount of pigmentation in A-188 cells (a, b) and of soluble melanin in A-188 (c) compared to M-188 and S-188 cells (p < 0.05).Cellular localization of tyrosinase and TRP-1 in S-188 and A-188 cells was determined by immuno¯uorescent techniques. In contrast to M-188(M column, frames d, g, j) and S-188 (S column, frames e, h, k), both tyrosinase (frames d, e) and TRP-1 (frames g, h) in A-188 cells (A column, frames f, i, l)were localized to distinct large granular structures throughout the cytoplasm and dendrites (frames f, i). Images in frames j, k, and l are the merged imagesof frames d, e, f with frames g, h, I, respectively, to document colocalization of tyrosinase and TRP-1.
644 SARANGARAJAN ET AL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY

patients carrying mutations in HPS1 demonstrated altered intra-cellular traf®cking of tyrosinase and TRP-1 to large membranecomplexes. Recently, it was shown that in melanocytic cells HPS1pexists as a > 500 kDa complex consisting of a 200 kDa componentlocalized to the peri-nuclear area and associated with smallnoncoated vesicles as well as early stage melanosomes (but notwith mature melanosomes) (Oh et al, 2000). The authors suggestedthat HPS1p functions in the early stages of melanosome biogenesis.Concrete evidence regarding the exact function of HPS1p inmelanocytes and its role in melanogenesis is still elusive, however.This study was an attempt to characterize the role of HPS1p inmediating intracellular traf®cking of tyrosinase/TRP-1, as well asmelanocytic function, by knockout of HPS1p expression in apigmented melanoma cell line.
We initiated the study by characterizing the ultrastructuralcharacteristics of the melanocytes in intact skin of two HPSpatients, one homozygous for the 16 bp duplication and the otherheterozygous for an A1195del and an unidenti®ed mutation.Electron microscope analysis of the skin revealed large complexeswith DOPA-positive cisterna and 50 nm vesicles throughout theperiphery of the HPS melanocytes. This is consistent with ourprevious report demonstrating large membrane-bound complexescontaining DOPA reaction products and DOPA-positive ringsdelineated on both sides by limiting membranes in melanocytecultures from HPS patients (Boissy et al, 1998b). The ®ndingscon®rm that the ultrastructural abnormality previously observed inHPS1 melanocytes recapitulates the in vivo situation. The presenceof numerous giant melanosomes in a subset of Japanese patientscarrying mutations in HPS1 has been reported (Horikawa et al,2000).
Signi®cant reduction in HPS1p expression in pigmented mela-noma cells by transfection of HPS1 cDNA in the antisenseorientation provided an in vitro model of HPS1 to facilitatecharacterization of the role of HPS1p in intracellular traf®cking oftyrosinase/TRP-1. Signi®cant reduction in HPS1p expression inthe pigmented melanoma cells (M-188) transfected with HPS1antisense cDNA was con®rmed by Western blot analysis. Therewas no signi®cant difference in the tyrosine hydroxylase activities incell lysates of M-188, S-188, and A-188 cells, suggesting that lackof HPS1p does not in¯uence the level of tyrosinase proteinexpression in melanocytic cells. This is consistent with previousreports demonstrating normal tyrosine hydroxylase activities inlysates of HPS1 melanocytes (Boissy et al, 1998b). In contrast, thetyrosine hydroxylase activity of intact A-188 cells was signi®cantlyreduced compared to that of M-188 and S-188 cells. This isconsistent with previous reports demonstrating decreased tyrosinehydroxylase activities in intact HPS1 melanocyte cultures (Boissyet al, 1998b). As the melanosome is the primary site possessing thebiochemical conditions necessary for eliciting tyrosine hydroxylaseactivities associated with tyrosinase, the decrease in in situ tyrosinehydroxylase activity can be attributed to a decreased presence oftyrosinase in the melanosome. In addition, a signi®cant decrease inmelanin content in the A-188 cells (lacking expression of HPS1p)was observed. The differences in tyrosine hydroxylase activitiesassociated with in vitro and in situ determinations, along with thedecrease in melanin content in cells not expressing HPS1p, suggestaberrant targeting/localization of tyrosinase to the melanosome.
Immunohistochemical analysis of melanocyte cultures establishedfrom HPS1 patients demonstrated that both tyrosinase and TRP-1were localized in a coarse granular pattern with large aggregates
Figure 5. Electron microscopy of M-188, S-188, and A-188 cells processed by DOPAhistochemistry for the localization offunctional tyrosinase. SK-MEL-188 (M-188)cells transfected with sense (S-188) or antisense(A-188) human HPS1 cDNA were processed forelectron microscope analysis. DOPA reactionproducts were present in the trans-Golgi network(block arrows) and melanosomes (block arrowheads) ofM-188 (a), S-188 (b), and A-188 cells (c, d). TheA-188 cells (c, d), however, also exhibited thepresence of DOPA reaction products in largemembranous complexes (block arrows in c and d),in contrast to M-188 and S-188 cells. Scale bar: (a,b, c) 0.6 mm; (d) 0.45 mm.
VOL. 117, NO. 3 SEPTEMBER 2001 MISTRANSLOCATION OF TYROSINASE AND TRP-1 IN HPS 645

throughout the cell body that appear ultrastructurally as largemembranous complexes (Boissy et al, 1998b). Consistent with thisobservation, knockout of HPS1p (A-188 cells, Fig 4) resulted incon®nement of tyrosinase and TRP-1 expression to largemembranous complexes throughout the cell body and dendrites.Electron microscope analysis of the A-188 cells demonstrated thatabsence of HPS1p resulted in the deposition of DOPA reactionproducts (thereby localizing tyrosinase) in large membrane-boundstructures with limiting membranes. It has recently been speculatedthat melanocyte-speci®c proteins are traf®cked from the trans-Golgi network to the melanosome via a ``coated endosome'' (ofendosomal lineage) that served as an intermediate repository forthese proteins before their eventual sorting to the melanosome(Raposo et al, 2001). The large membranous complexes observedin A-188 cells could be viewed as a structure similar to the ``coatedendosome''. Absence of normal HPS1p would prevent thetraf®cking of tyrosinase to the melanosome and consequentlyaccumulates in these membrane complexes. Alternatively, thesemembranous complexes may represent residual bodies (of lysosomallineage) where degradation is occurring. The large membranouscomplexes reported herein (Figs 1, 5) as well as in cultured HPS1melanocytes (Boissy et al, 1998b) contain other inclusions inaddition to DOPA-positive elements that consist of 50 nm vesicles,compact aggregates of membranes, and mitochondria. Theseheterogeneous aggregates are characteristic of developing residualbodies (i.e., secondary lysosomes) that occur during autography.We speculate that the undirected tyrosinase in HPS1 melanocytesmay be subsequently traf®cked to the lysosome system fordegradation. Precise con®rmation of the large membranouscomplexes is warranted, however. Nevertheless, we can concludethat, with signi®cant reduction in the expression of HPS1p,tyrosinase is traf®cked to intracellular sites that are distinctlydifferent from melanosomes.
Biochemical and ultrastructural data obtained from this studyrecapitulate previous observations in skin biopsies as well as cultures
of melanocytes established from HPS1 patients. Thus, knockout ofthe HPS1p from the pigmented melanoma cell line represents aviable experimental model that can be utilized to evaluate the roleof HPS1p in intracellular traf®cking of melanocyte-speci®c geneproducts, tyrosinase and TRP-1 in particular, in the modulation ofpigmentation and melanocytic function.
REFERENCES
Boissy RE, Sakai C, Zhao H, Kobayashi T, Hearing VJ: Human tyrosinase relatedprotein-1 (TRP-1) does not function as a DHICA oxidase activity in contrastto murine TRP-1. Exp Dermatol 7:198±204, 1998a
Boissy RE, Zhao Y, Gahl WA: Altered protein localization in melanocytes fromHermansky±Pudlak syndrome: support for the role of the HPS gene product inintracellular traf®cking. Lab Invest 78:1037±1048, 1998b
Dell'Angelica EC, Aguilar RC, Wolins N, Hazelwood S, Gahl WA, Bonifacino JS:Molecular characterization of the protein encoded by the Hermansky±Pudlaksyndrome type 1 gene. J Biol Chem 275:1300±1306, 2000
Horikawa T, Araki K, Fukai K, Ueda M, Ueda T, Ito S, Ichihashi M: HeterozygousHPS1 mutations in a case of Hermansky±Pudlak syndrome with giantmelanosomes. Br J Dermatol 143:635±640, 2000
Huizing M, Anikster Y, Gahl WA: Hermansky±Pudlak syndrome and relateddisorders of organelle formation. Traf®c 1:823±835, 2000
Karnovsky MJ: A formaldehyde±glutaraldehyde ®xative of high osmolality for use inelectron microscopy. J Cell Biol 27:137(Abstract), 1965
Karnovsky MJ: Use of ferrocyanide-reduced osmium tetroxide in electronmicroscopy. J Cell Biol 51:146(Abstract), 1971
Oh J, Bailin T, Fukai K, et al: Positional cloning of a gene for Hermansky±Pudlaksyndrome, a disorder of cytoplasmic organelles. Nat Genet 14:300±306, 1996
Oh J, Liu Z-X, Feng GH, Raposo G, Spritz RA: The Hermansky±Pudlak syndrome(HPS) protein is part of a high molecular weight complex involved inbiogenesis of early melanosomes. Hum Mol Genet 9:375±385, 2000
Raposo G, Tenza D, Murphy DM, Berson JF, Marks MS: Distinct protein sortingand localization to premelanosomes, melanosomes and lysosomes in pigmentedmelanocytic cells. J Cell Biol in press, 2001
Shotelersuk, V, Gahl, WA: Hermansky±Pudlak syndrome: models for intracellularvesicle formation. Mol Genet Metab 65:85±96, 1998
Shotelersuk V, Dell'Angelica EC, Hartnell L, Bonifacino JS, Gahl WA: A newvariant of Hermansky±Pudlak syndrome due to mutations in a gene responsiblefor vesicle formation. Am J Med 108:423±427, 2000
646 SARANGARAJAN ET AL THE JOURNAL OF INVESTIGATIVE DERMATOLOGY





























