Embed Size (px)
DESCRIPTION
c
Citation preview

Seediscussions,stats,andauthorprofilesforthispublicationat:http://www.researchgate.net/publication/8145490
StructureandfunctionsoftheGNATsuperfamilyofacetyltransferases
ARTICLEinARCHIVESOFBIOCHEMISTRYANDBIOPHYSICS·FEBRUARY2005
ImpactFactor:3.04·DOI:10.1016/j.abb.2004.09.003·Source:PubMed
CITATIONS
251
DOWNLOADS
220
VIEWS
145
7AUTHORS,INCLUDING:
MatthewVetting
AlbertEinsteinCollegeofMedicine
57PUBLICATIONS1,667CITATIONS
SEEPROFILE
LuizPedroSoriodeCarvalho
TheFrancisCrickInstitute
40PUBLICATIONS1,184CITATIONS
SEEPROFILE
Availablefrom:MatthewVetting
Retrievedon:19August2015

www.elsevier.com/locate/yabbi
Archives of Biochemistry and Biophysics 433 (2005) 212–226
ABB
MinireviewStructure and functions of the GNAT superfamilyof acetyltransferasesq
Matthew W. Vetting, Luiz Pedro S. de Carvalho, Michael Yu, Subray S. Hegde,Sophie Magnet, Steven L. Roderick, John S. Blanchard*
Department of Biochemistry, Albert Einstein College of Medicine, 1300 Morris Park Avenue, Bronx, NY 10461, United States
Received 3 August 2004, and in revised form 2 September 2004Available online 7 October 2004
Abstract
The Gcn5-related N-acetyltransferases are an enormous superfamily of enzymes that are universally distributed in nature andthat use acyl-CoAs to acylate their cognate substrates. In this review, we will examine those members of this superfamily that havebeen both structurally and mechanistically characterized. These include aminoglycoside N-acetyltransferases, serotonin N-acetyl-transferase, glucosamine-6-phosphate N-acetyltransferase, the histone acetyltransferases, mycothiol synthase, protein N-myristoyl-transferase, and the Fem family of amino acyl transferases.� 2004 Elsevier Inc. All rights reserved.
Keywords: Acetyltransferase; Three-dimensional structure; Steady-state kinetics; GNAT; Histone acetyltransferase; Chemical mechanism; Structureand function of enzymes; Protein modification; Antibiotic resistance
The GNAT superfamily
The firstmembers ofwhat is now termed theGCN5-re-lated N-acetyltransferase (GNAT)1 superfamily wereidentified as aminoglycoside acetyltransferases in bacteriathat became resistant to the action of the antibiotics gen-tamicin and kanamycin [1]. Numerous genes encodingaminoglycosideN-acetyltransferases that regioselectivelyacetylated one of the 4–5 amino groups in aminoglyco-sides were cloned and sequenced in the 1980s and shown
0003-9861/$ - see front matter � 2004 Elsevier Inc. All rights reserved.
doi:10.1016/j.abb.2004.09.003
q This work was supported by Grants AI33696 and AI60899 (toJ.S.B.) from the National Institutes of Health. M. Yu was supportedby Training Grant T32 GM08572 from the National Institutes ofHealth.
* Corresponding author. Fax: +1 718 430 8565.E-mail address: [email protected] (J.S.
Blanchard).1 Abbreviations used: GNAT, GCN5-related N-acetyltransferase;
HAT, histone acetyltransferase; AgNATs, aminoglycoside N-acety-ltransferases; SNAT, serotonin N-acetyltransferase; AANAT, aryl-alkylamine N-acetyltransferase; YGCN5, yeast GCN5; UDP–MPP,UDP–UDP–N-acetylmuramyl pentapeptide.
to contain four amino acid ‘‘motifs’’ spanning approxi-mately 100–120 residues, in spite of their low overall se-quence homology [2]. Subsequently, they were shown toexhibit sequence homology to a class of eukaryotic tran-scription factors, the first being the yeast GCN5 in 1992[3]. The activity of a Tetrahymena homologue of the yeastGCN5 as an N-acetyltransferase that acetylated histoneswas reported in 1995 [4], providing a direct link betweenhistone acetylation and transcriptional regulation. Thesubsequent revelation that yGCN5 was, in fact, a histoneacetyltransferase (HAT) provided a model for HATrecruitment to specific promoters by DNA-bound acti-vating proteins [5]. The numerous genome sequencingefforts of the last decade, coupled with powerful bioinfor-matics approaches to protein superfamily identification,have revealed some 10,000 members of the GNAT familyin all kingdoms of life [see http://supfam.mrc-lmb.cam.ac.uk/SUPERFAMILY].
To date, over two dozen members of the family havebeen structurally characterized, revealing a structurallyconserved fold comprised of an N-terminal strand
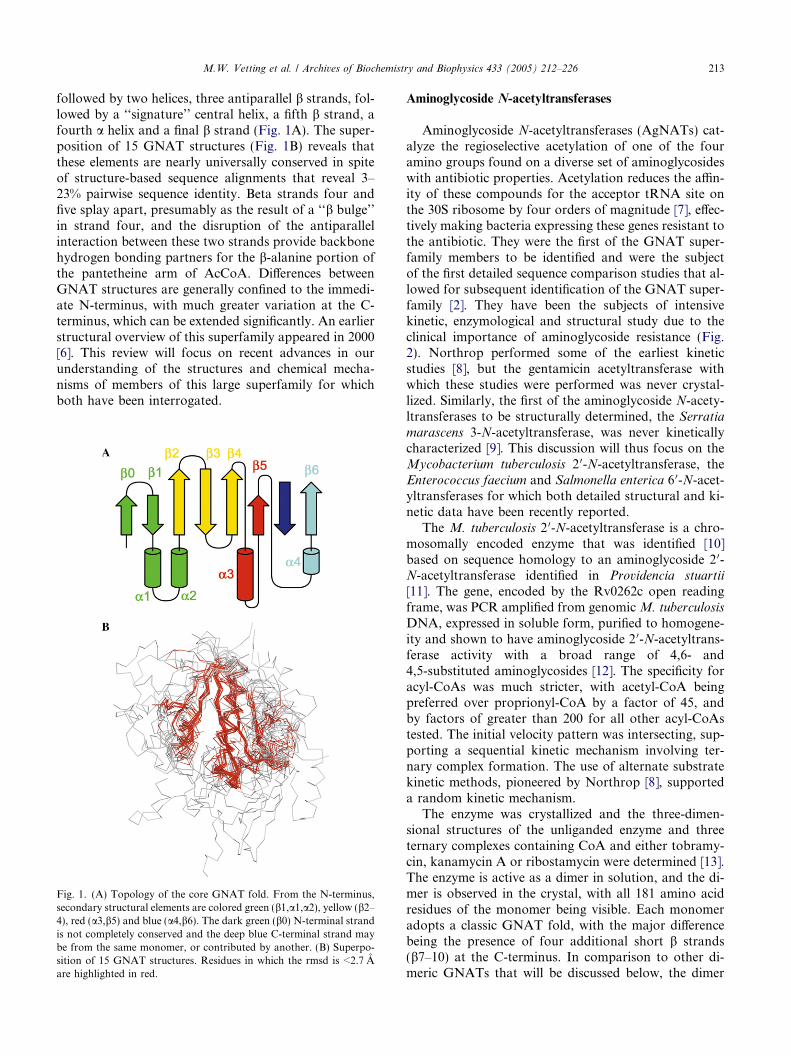
M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226 213
followed by two helices, three antiparallel b strands, fol-lowed by a ‘‘signature’’ central helix, a fifth b strand, afourth a helix and a final b strand (Fig. 1A). The super-position of 15 GNAT structures (Fig. 1B) reveals thatthese elements are nearly universally conserved in spiteof structure-based sequence alignments that reveal 3–23% pairwise sequence identity. Beta strands four andfive splay apart, presumably as the result of a ‘‘b bulge’’in strand four, and the disruption of the antiparallelinteraction between these two strands provide backbonehydrogen bonding partners for the b-alanine portion ofthe pantetheine arm of AcCoA. Differences betweenGNAT structures are generally confined to the immedi-ate N-terminus, with much greater variation at the C-terminus, which can be extended significantly. An earlierstructural overview of this superfamily appeared in 2000[6]. This review will focus on recent advances in ourunderstanding of the structures and chemical mecha-nisms of members of this large superfamily for whichboth have been interrogated.
Fig. 1. (A) Topology of the core GNAT fold. From the N-terminus,secondary structural elements are colored green (b1,a1,a2), yellow (b2–4), red (a3,b5) and blue (a4,b6). The dark green (b0) N-terminal strandis not completely conserved and the deep blue C-terminal strand maybe from the same monomer, or contributed by another. (B) Superpo-sition of 15 GNAT structures. Residues in which the rmsd is <2.7 Aare highlighted in red.
Aminoglycoside N-acetyltransferases
Aminoglycoside N-acetyltransferases (AgNATs) cat-alyze the regioselective acetylation of one of the fouramino groups found on a diverse set of aminoglycosideswith antibiotic properties. Acetylation reduces the affin-ity of these compounds for the acceptor tRNA site onthe 30S ribosome by four orders of magnitude [7], effec-tively making bacteria expressing these genes resistant tothe antibiotic. They were the first of the GNAT super-family members to be identified and were the subjectof the first detailed sequence comparison studies that al-lowed for subsequent identification of the GNAT super-family [2]. They have been the subjects of intensivekinetic, enzymological and structural study due to theclinical importance of aminoglycoside resistance (Fig.2). Northrop performed some of the earliest kineticstudies [8], but the gentamicin acetyltransferase withwhich these studies were performed was never crystal-lized. Similarly, the first of the aminoglycoside N-acety-ltransferases to be structurally determined, the Serratia
marascens 3-N-acetyltransferase, was never kineticallycharacterized [9]. This discussion will thus focus on theMycobacterium tuberculosis 2 0-N-acetyltransferase, theEnterococcus faecium and Salmonella enterica 6 0-N-acet-yltransferases for which both detailed structural and ki-netic data have been recently reported.
The M. tuberculosis 2 0-N-acetyltransferase is a chro-mosomally encoded enzyme that was identified [10]based on sequence homology to an aminoglycoside 2 0-N-acetyltransferase identified in Providencia stuartii
[11]. The gene, encoded by the Rv0262c open readingframe, was PCR amplified from genomicM. tuberculosis
DNA, expressed in soluble form, purified to homogene-ity and shown to have aminoglycoside 2 0-N-acetyltrans-ferase activity with a broad range of 4,6- and4,5-substituted aminoglycosides [12]. The specificity foracyl-CoAs was much stricter, with acetyl-CoA beingpreferred over proprionyl-CoA by a factor of 45, andby factors of greater than 200 for all other acyl-CoAstested. The initial velocity pattern was intersecting, sup-porting a sequential kinetic mechanism involving ter-nary complex formation. The use of alternate substratekinetic methods, pioneered by Northrop [8], supporteda random kinetic mechanism.
The enzyme was crystallized and the three-dimen-sional structures of the unliganded enzyme and threeternary complexes containing CoA and either tobramy-cin, kanamycin A or ribostamycin were determined [13].The enzyme is active as a dimer in solution, and the di-mer is observed in the crystal, with all 181 amino acidresidues of the monomer being visible. Each monomeradopts a classic GNAT fold, with the major differencebeing the presence of four additional short b strands(b7–10) at the C-terminus. In comparison to other di-meric GNATs that will be discussed below, the dimer

Fig. 2. (A) The reaction catalyzed by the M. tuberculosis aminoglycoside 2 0-N-acetyltransferase with kanamycin B, a 4,6-disubstituteddeoxystreptamine-containing aminoglycoside. The central deoxystreptamine ring is shown in red. (B) The reaction catalyzed by the E. faecium
and S. enterica aminoglycoside 2 0-N-acetyltransferases with kanamycin B. (C) The structure of ribostamycin, a 4,5-disubstituted deoxystreptamine-containing aminoglycoside.
214 M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226
interface is generated not only by elements of the C-ter-minal portion of the molecule but also the N-terminaland three central b strands (b2–4).
The structures of the ternary complexes, solved andrefined at resolutions ranging from 1.5 to 1.8 A, revealedfor the first time the nature of the interactions betweenthe bound aminoglycosides and an aminoglycoside ace-tyltransferase [13]. Aminoglycosides bind in a smallpocket and the vast majority of interactions are madebetween the central deoxystreptamine and primed 2,6-dideoxy-2,6-diamino-glucose ring (Fig. 3A). This pro-vides a structural rationale for the broad substratespecificity of the enzyme for both 4,6- and 4,5-disubsti-tuted aminoglycosides. There are few direct interactionsbetween the substituents of these two rings and enzymeside chains; rather a large number of bound water mol-ecules mediate the interactions between the anionic en-zyme side chains of D35, D40, E82, D152, and D179.The 2 0-amino group of bound aminoglycosides is ca.3.5–4.1 A from the thiol of bound CoA, a distance fullycompatible with direct nucleophilic attack on the thioes-ter carbonyl of AcCoA. The pantetheine arm of CoAinteracts with backbone amide and carbonyl groupsof b4, and is sandwiched between the splayed strandsof b4 and b5. The conformation of the loop connectingb4 and a3 provides five amide backbone hydrogenbonds to the pyrophosphate group of bound CoA,and this ‘‘P-loop’’ is a part of one of the conserved se-quence motifs and a conserved structural feature ofthe GNAT superfamily. A second structurally conserved
feature of the GNAT superfamily is the presence of a ‘‘bbulge’’ in b4, which in this case orients the backboneamides of G83 and V84 towards the carbonyl of the thi-oester of AcCoA. The backbone amide of V84 is suit-ably positioned to hydrogen bond to the carbonyl ofthe thioester, and additionally stabilizes the developingnegative charge on the oxygen atom in the tetrahedralintermediate. The thiol of bound CoA is within hydro-gen bonding distance of the phenolic hydroxyl groupof Y126, suggesting that this residue may serve to pro-tonate the thiolate anion formed in the reaction. Thecarboxyl group of the C-terminal W181 is hydrogenbonded to tightly bound water molecules that link thiscarboxyl group to the 2 0-amino group of bound amino-glycoside. This suggests that the C-terminal carboxylatemay function as the general base, and that the boundwater molecules serve as a ‘‘proton wire’’ [6] to allowfor proton transfer within the active site.
The E. faecium 6 0-N-acetyltransferase was discoveredin strains of the organism that exhibited low-level resis-tance to aminoglycosides, and the regioselectivity ofcatalysis was implicated by these strains sensitivity toaminoglycoside analogues containing a 6 0-hydroxyl sub-stituent (e.g., lividomycin) [14]. The chromosomally en-coded gene was heterologously expressed in Escherichia
coli, and the enzyme purified to homogeneity [15]. Theenzyme exhibited unusually broad specificity and mod-est selectivity for aminoglycosides, with all substratestested exhibiting kcat/Km values that differed by less thana factor of ten. The kinetic mechanism is ordered with

Fig. 3. Ribbon diagrams of aminoglycoside N-acetyltransferases. (A)M. tuberculosis 2 0-N-acetyltransferase in complex with CoA andribostamycin (1M4G.pdb). (B) E. faecium 6 0-N-acetyltransferase incomplex with CoA (1N71.pdb). (C) S. enterica 6 0-N-acetyltransferasein complex with CoA and ribostamycin (1S3Z.pdb). In Figs. 3, 7, 9 and11, the same coloring scheme is used for individual sections ofsecondary structure (see Fig. 1) and for bound ligands. Those segmentswhich are typical of the GNAT fold are colored green fi yel-lowfi redfi blue moving from N to C-terminus. Additional N-terminal and C-terminal residues are colored orange and salmon,respectively. AcCoA or CoA or derivatives are rendered as sticks withgray carbons whilst the substrates or inhibitors are shown as stickswith green carbons. Ribbon diagrams were created with the programPyMol (DeLano Scientific; http://pymol.sourceforge.net/).
M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226 215
AcCoA binding to the enzyme, followed by aminoglyco-side substrate [16]. Acetylated aminoglycoside is re-leased followed by CoA. Chemistry is not ratelimiting, as evidenced both by very small solvent kineticisotope effects (<1.3–1.4) and a large dependence of themaximum velocity on solvent microviscosity, arguingthat physical steps, likely product dissociation, are the
rate-limiting steps [16]. On the basis of both sequencealignments and the structure of the CoA binary complex(see below), a number of site-directed mutant forms ofthe enzyme were prepared to probe the involvement ofspecific residues in catalysis [17]. Of these, the replace-ment of E72 with an alanine residue and Y147 witheither an alanine or phenylalanine residue had the mostdramatic effects. In the case of the E72A mutant, therewas no significant effect on the kcat/Km value for Ac-CoA, but a decrease in kcat/Km for aminoglycoside sub-strate that was as large as 290-fold. Curiously, for someaminoglycoside substrates, this substitution had essen-tially no effect (e.g., neomycin). This suggested thatrather than functioning as a general base, E72 functionsin aminoglycoside recognition. On the other hand, theY147A exhibited both a 10-fold effect on the kcat/Km va-lue of AcCoA, and a larger effect on the relative kcat/Km
values for all aminoglycosides tested, ranging from a250- to 3300-fold decrease. Given the alignment ofY147 in the 6 0-N-acetyltransferse and Y126 of the 2 0-N-acetyltransferase, it is quite likely that Y147 servesas the general acid that protonates the thiolate anionof CoA.
The three-dimensional structure of the E. faecium 6 0-N-acetyltransferase was solved for the full-length 182residue, selenomethionine-substituted enzyme-AcCoAcomplex at 2.7 A resolution [18]. The structure revealeda compact GNAT domain with AcCoA bound betweenthe splayed strands of b4 and b5 (Fig. 3B). The ‘‘bbulge’’ located on strand 4 included residues H74 andP75, which is in the cis conformation. Although activeas the dimer in solution this structure provided no infor-mation about the catalytically relevant dimer. The en-zyme was also reported to catalyze acetyltransfer tohistones and poly-LL-lysine, the latter exhibiting a kcatof 0.002 s�1, compared to the rate of 0.4 s�1 with amino-glycosides. Crystallization of the enzyme with CoA wasaccomplished, and the space group in which the complexcrystallized allowed the dimer to be identified [19]. TheC-terminal b strands at the dimer interface are orderedb5, b7, b6, b6 0, b7 0, and b5 0, thus generating a continu-ous 14-stranded b sheet in the dimer.
Like the E. faecium 6 0-N-acetyltransferase, the geneencoding the S. enterica 6 0-N-acetyltransferase was iden-tified in a clinical strain that exhibited resistance to abroad range of 6 0-amino-containing aminoglycosides[20]. The chromosomally encoded gene, at the end of along operon, was transcriptionally silent in sensitivestrains. However, a massive 60 kbp chromosomal dele-tion that placed the constitutive nmpC promoterapproximately 2 kbp upstream of the gene, resulted inobservable mRNA transcripts for the gene in resistantstrains. The gene was subsequently PCR amplifiedand the solubly expressed protein was purified and char-acterized [21]. The enzyme was shown to catalyze regio-selective 6 0-N-acetylation of a broad range of 4,6- and

216 M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226
4,5-disubstituted aminoglycosides, but showed very highselectivity for acetyl-CoA over other acyl-CoAs. The en-zyme exhibited an intersecting initial velocity pattern,and product and dead-end inhibition studies revealed arandom binding of AcCoA and aminoglycoside. Sub-strate activation was observed for several aminoglyco-side substrates, suggesting that CoA release, normallythe rate-limiting step, could be enhanced at high amino-glycoside concentrations. Very small solvent isotopeeffects on the kinetic parameters supported the rate-lim-iting release of CoA. Direct binding of acyl-CoAs wasdemonstrated by both fluorescence titrations and iso-thermal titration calorimetry [22], which also allowedthe synergistic binding of aminoglycosides to the CoAbinary complex to be demonstrated.
The selenomethionine-substituted enzyme was crys-tallized, and the three-dimensional structure of theCoA binary complex and the ternary CoA-ribostamycincomplex were determined using MAD phasing methods[23]. The 145 amino acid monomer exhibited a classicGNAT fold, but the enzymatically active dimer was gen-erated via a b6 strand exchange observed previouslyonly for dimeric histone acetyltransferases (Fig. 3C).CoA, although not added in the crystallization drops,was observed in both subunits, with its pantetheinearm between the splayed b4 and b5 strands and interact-ing with main chain elements of b4. Electron density wasclearly observed for the aminoglycoside in the structur-ally determined ternary complex, with a ribostamycinmolecule bound at each of the two active sites, wherethe aminoglycoside interacts with side chains from bothmonomers due to the location of the active site withinthe dimer interface. As observed for the M. tuberculosis
2 0-N-acetyltransferase, the majority of interactions weremade between the ring substituents of the central deoxy-streptamine and 2,6-dideoxy-2,6-diamino-glucose rings.While several of these interactions were directly betweenthe carboxylate side chains of E79 and E136, additionalinteractions via intervening water molecules with en-zyme side chains were also observed. The 6 0-aminogroup was positioned 3.4 A from the thiol of boundCoA, a position consistent with the regiospecificity ofthe enzyme and the chemical reaction catalyzed by theenzyme. A model of bound AcCoA could be generatedand suggested that the carbonyl of the thioester wouldinteract with the main chain amide of I81. In contrastwith M. tuberculosis 2 0-N-acetyltransferase the ‘‘bbulge’’ in strand four (G80, E79) results in two consec-utive backbone carbonyls pointing towards the activesite. No residue that could serve the function of the gen-eral acid is present in the vicinity of the thiol of boundCoA, suggesting water-mediated protonation of theleaving thiolate anion is occurring. D115 is the mostlikely candidate for the general base, and it is hydro-gen-bonded to a water molecule, which in turn, is hydro-gen bonded to the 6 0-amino group via hydrogen bonds
that are <2.65 A in length. Finally, a comparison ofthe mode of interaction of ribostamycin with both theS. enterica 6 0- and M. tuberculosis 2 0-N-acetyltransferaseprovided a structural basis for the regiospecificity exhib-ited by the two enzymes [23].
As mentioned above, the formation of the activedimer by strand exchange had not been observed previ-ously in the structures of aminoglycoside acetyltransfe-rases, but had been observed in a tetrameric histoneacetyltransferase [24]. Expression of the S. enterica ami-noglycoside 6 0-N-acetyltransferase in pET28a generatesthe full-length enzyme with N-terminal His6 affinitytag and thrombin cleavage site. The structure of the en-zyme in one crystal form revealed that portions of thethrombin cleavage site interact with an adjacent dimerin the crystal lattice at the dimer interface where ribosta-mycin was observed in the ternary complex. The interac-tions between the N-terminal peptide and the adjacentenzyme molecule were similar to those observed betweenribostamycin and the enzyme, and included interactionsfrom both monomers of the dimer. Comparison of thedimer of the S. enterica 6 0-N-acetyltransferases withother GNAT structures revealed that the highest struc-tural homology was with the yeast Hpa2 histone acetyl-transferase [24]. This raised the question of whether thebacterial aminoglycoside N-acetyltransferase could cata-lyze protein acetylation. In fact, the enzyme catalyzesboth self-acetylation and the robust acetylation ofeukaryotic histone proteins. Finally, the enzyme wasshown to catalyze the peracetylation of the human his-tone H3 20-mer peptide, with some regioselectivity ofacetyl transfer. It remains to be determined what thetrue physiological roles, and substrates, for these chro-mosomally encoded ‘‘aminoglycoside’’ acetyltransfe-rases are.
Serotonin N-acetyltransferase
Serotonin N-acetyltransferase (SNAT, also referredto as arylalkylamine N-acetyltransferase, AANAT) cat-alyzes the penultimate step in the biosynthesis of melato-nin, a hormone proposed to play roles in circadianrhythms, and human mood and behavior. Serotonin,5-hydroxy-tryptamine is acetylated at the primaryamine, and subsequently methylated by hydroxyindole-O-methyltransferase to generate melatonin (Fig. 4).The N-acetyltransferase reaction has been suggested tolimit the rate of melatonin biosynthesis, and thus hasemerged as a target for inhibitor design [25,26].
The sheep gene encoding SNAT was identified in1998 [27] and the recombinant enzyme expressed in E.
coli. Using tryptamine as an alternate substrate, thereaction was shown to occur via a ternary complexmechanism. The kinetic mechanism was shown to beordered with acetyl-CoA binding to the free enzyme

Fig. 4. Biosynthesis of melatonin.
M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226 217
followed by the binding of tryptamine [27]. The productinhibition patterns observed using N-acetyl-tryptaminewere consistent with the ordered release of N-acetyl-tryptamine followed by CoA. The pH dependence ofthe kinetic parameters, kcat and kcat/Km, were subse-quently determined. The turnover rate was sensitive tothe protonation of a single group exhibiting a pK valueof 6.9, while kcat/Km exhibited a bell-shaped profile,indicative of two ionizable groups, one of which mustbe protonated and exhibited a pK value of 7.3, and asecond, which must be deprotonated and exhibited apK value of 8.5 [28]. The group observed in the kcatpH profile is very likely a catalytic base, required fordeprotonation of the positively charged amine. Theanalysis of the dependence of the reaction on the con-centration of added sucrose, a microviscosogen, sug-gested that the catalytic reaction was fast usingtryptamine as substrate, relative to physical steps, pre-sumably product release.
The finding that SNAT uses a ternary complex mech-anism suggested that bisubstrate analogues would bindtightly to the enzyme. Tryptamine was treated withbromoacetyl bromide to generate the N-bromoacetyl–tryptamine, and addition of CoA generated theexpected, stable thioether between CoA and N-acetyl-tryptamine (Fig. 5). The bisubstrate analogue, whenexamined using variable concentrations of acetyl-CoA,inhibited SNAT with a 90 nM Ki value [29]. The ques-tion of whether SNAT could catalyze the formation ofthe bisubstrate analogue was probed using N-bromoace-tyl-tryptamine and CoA as substrates for the ‘‘alkyl-transferase’’ reaction. Although the non-enzymaticreaction does occur and can be used synthetically, addi-tion of the enzyme accelerates the reaction by a factor of>10,000-fold [30]. This alkyltransferase reaction exhib-ited Michaelis–Menten kinetics for both substrates. Asurprising feature of this reaction was its robustness,since a priori, one would expect the reaction to rapidlyslow down due to the accumulation of the tight-binding
Fig. 5. Synthesis of bisu
bisubstrate analogue. Inhibition studies using a series ofsynthetic bisubstrate homologues revealed that while theacetyltransferase reaction was strongly inhibited at 20–60 nM concentrations of inhibitors, the alkyltransferasereaction was only inhibited at 10–20 lM concentrationsof these same inhibitors. Based on this data, the authorsargued that there was a separate alkyltransferase site,distinct from the acetyltransferase active site. In a de-tailed mechanistic investigation of the alkyltransferasereaction [31], the leaving group dependence wasexamined using a series of halo-acetyl-tryptamines,and revealed the expected order of reactivity for thenon-enzymatic reaction, with iodo-acetyl-tryptaminereacting slightly faster than bromo-acetyl-tryptamineand approximately 100 times faster than chloro-acetyl-tryptamine. The enzyme catalyzed reaction exhibiteddistinctly different leaving group behavior withBr > I > Cl, but with all rates being only 8-fold different.The mechanism of the alkyltransferase reaction followeda ternary complex mechanism, and the pH dependenceof kcat and kcat/Km were similar to those observed forthe acetyltransferase reaction. The presence of a cata-lytic base to promote thiol deprotonation for the alkyl-transferase reaction is expected to serve a similarfunction to amine deprotonation in the acetyltransferasereaction.
Finally, the contributions of various residues locatedat, or near to, the carbonyl oxygen and thioether sulfurof the bound bisubstrate analogue were investigatedusing site-directed mutagenesis. Two histidine residues,H120 and H122, which are conserved in most SNAT se-quences were evaluated for their potential role in catal-ysis[32]. The first studies revealed that their replacementwith glutamine (H122Q) had no effect on Vmax, and upto a 25-fold effect on Km. Tyrosine 168 is also conservedin most SNAT sequences, and removal of this functionalgroup in the Y168F mutant form of the enzyme reducedVmax 30-fold, and increased the Km for tryptamine by27-fold. This suggested that Y168 may function as a
bstrate analogues.

Fig. 7. Ribbon diagrams of serotonin and glucosamine-6-phosphateN-acetyltransferase. (A) Serotonin N-acetyltransferase (1L0C.pdb) incomplex with the bisubstrate analog CoA-S-acetyl tryptamine. (B)Dimeric glucosamine-6-phosphate N-acetyltransferase with boundAcCoA (1I12.pdb) and a modeled glucosamine-6-phosphate basedon the position of N-acetylglucosamine-6-phosphate in the ternarycomplex with CoA (1I1D.pdb).
218 M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226
general acid, serving to protonate the leaving thiolateanion of CoA following decomposition of the tetrahe-dral intermediate formed by attack of the primary amineof serotonin on the acetyl-CoA thioester (Fig. 6). The re-sults of Y168, H120 and H122 mutagenesis were inde-pendently confirmed [33] with essentially identicalresults. However, the simultaneous mutation of bothH120 and H122 to either glutamine or alanine residuesreduced kcat by factors of 8 and 270, respectively. Theauthors suggested that either of the two histidine resi-dues may function to assist in water-mediated aminedeprotonation, serving redundant functions. The analy-sis of the pH dependence of the kinetic parametersexhibited by the Y168F and H122Q fully supportedthe roles of Y168 as a general acid and H120/H122 asassisting in amine deprotonation.
The three-dimensional X-ray crystal structures of atruncated variant of sheep SNAT (residues 28–201) at2.5 (apoenzyme) and 1.8 A resolution (binary complexwith bisubstrate inhibitor shown) appeared in 1999 fromthe Dyda group [32]. These structures confirmed a se-quence analysis predicting SNAT to be a member ofthe GNAT superfamily, with all of the secondary struc-tural elements that define the acetyl-CoA binding site(see above). The structures of the apo and bisubstratecomplexes are similar with a rms deviation over138 Ca positions of 1.2 A. However, the binding ofthe CoA portion of the bisubstrate analogue induces asignificant rearrangement in the loop connecting a1and a2. A short three-residue segment of b sheet, thatoccupies in the apo form what becomes the CoA bindingsite, is used to extend a1 by one and one half turns. Asalt bridge formed between E54 and R131 stabilizes thisconformation, and residues of the former b sheet,including F56, now form part of the hydrophobic bind-ing pocket for tryptamine or other aryl- and alkylam-ines. This binding of acetyl-CoA induces majorstructural rearrangements at the active site that ‘‘com-plete’’ the serotonin binding site, providing a structuralbasis for the observed ordered binding of acetyl-CoAfollowed by tryptamine.
As with other GNAT superfamily members SNATexhibits the prototypical ‘‘V-shaped’’ groove where thepantetheine arm of CoA binds, a ‘‘b bulge’’ on strand
Fig. 6. The chemical mechanism of
4, as well as interactions between the carbonyl of eitheracetyl-CoA or, in this case, the carbonyl of the thioetherof the bisubstrate analogue with the backbone amide ofL124 on strand 4 (Fig. 7A). This interaction serves toboth position the thioester for attack, and polarize thecarbonyl making the carbon atom more susceptible tonucleophilic attack by the amine. Y168 is positionedappropriately to serve as the general acid and mutagen-esis studies referred to above, confirm this function. Therole of H120 and H122 as general bases, also discussedabove, is mediated by an extensive network of tightlybound water molecules that extend from both these res-idues to the tryptamine primary amine that is the locusof acetylation. Such a ‘‘proton wire’’ [6] has been ob-served in the structures of the aminoglycoside acety-ltransferases. Finally, in independently determined
serotonin N-acetyltransferase.

M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226 219
structures of SNAT with several bisubstrate analoguesbound [34], multiple conformations of the tryptamineportion of the analogue were observed.
Glucosamine-6-phosphate N-acetyltransferase
In Saccharomyces cerevisiae, UDP–N-acetylglucos-amine is generated from glucosamine-6-phosphate bythree enzymes that first acetylate the 2-amino groupand then uridylylate the N-acetyl-glucosamine1-phos-phate after intramolecular phosphate transfer (Fig. 8).In bacteria, a bifunctional enzyme, encoded by the glmU
gene, carries out the acetylation of glucosamine-1-phos-phate and the subsequent uridylylation [35]. The yeastgna1-encoded glucosamine-6-phosphate N-acetyltrans-ferase has been shown to be an essential gene for growth[36]. Sequence alignments with other acetyltransferasesshowed that activity was completely lost in the Y143Amutant. In 2001, the Bourne group solved the three-di-mensional structure of the enzyme and both the AcCoAbinary, and CoA-GlcNAc-6-P structures [37]. Thesethree structures were refined to 2.4, 1.3, and 1.8 A reso-lution, respectively. The apo enzyme crystal form re-vealed a dimeric enzyme composed of 159 residuemonomers that exhibited a classic GNAT fold, but theb6 strands were exchanged in the dimer, reminiscent ofthe Hpa2 and aminoglycoside 6 0-N-acetyltransfersestructures discussed above (Fig. 7B). The classic strandsplaying between b4 and b5 was observed as was the‘‘b bulge’’ involving backbone amides of both D99and I100 that generate the oxyanion hole. Y143 waswithin hydrogen bond distance from the sulfur atomof bound AcCoA, and its role as the general acid usedto protonate the leaving thiolate anion is the likely rea-son for the Y143A mutant showing no demonstrableactivity. In the ternary complex, a typical arrangementof ordered water molecules was observed linking the 2-amino group of bound NacGln-6-P to the side chainof E122, which is positioned similar to the general basesin numerous other GNATs discussed above. Curiously,the E122A mutant is fully active [36], and the authorsascribe the WT activity of the mutant to the low pK va-lue of GlcNAc-6-P (ca. 7.8) compared to other GNATsubstrates. The GlcNAc-6-P binding site is at the dimerinterface, and residues from both subunits make interac-tions with the bound sugar phosphate. In contrast tomany other GNATs, however, the side chains that inter-
Fig. 8. Biosynthesis of UDP–N-a
act with the phosphate group of GlcNAc-6-P effectivelytruncate the channel at the dimer interface in which bothAcCoA and GlcNAc-6-P bind. This, and the higher pKvalue of the e-amino group of lysine, is very likely to bethe reason that GNA1 is incapable of acetylatinghistones.
Histone acetyltransferases
The recognition that several transcriptional co-acti-vators, including yeast and Tetrahymena GCN5 [38],p300/CBP [39], P/CAF [40] and others were capable ofregioselective histone acetylation led to a flurry of ki-netic, mechanistic and structural studies of these en-zymes in the past six years. The members of theGNAT superfamily are one of four human HAT fami-lies, but have received extensive investigation. Althoughin both lower and higher eukaryotes the GNAT domainis often only part of a much larger protein, the ability todissect and study the isolated domains has made de-tailed mechanistic studies possible. Acetylation of theamino terminal histone ‘‘tails’’ removes positive charge,and it is likely that this results in weaker binding of thenucleosome core particles, composed of histones, toDNA. These loci are then free to interact with the com-plex transcriptional machinery, resulting in enhancedtranscription and translation of protein products [5].This may be an incomplete description of the functionof both histone acetylation and histone acetyltransfe-rases, since other proteins can be acetylated by GNATsuperfamily members, including non-histone proteins,general transcription factors and the tumor suppressorp53 [41].
The earliest kinetic studies were on the catalytic do-main (residues 99–262) of yeast GCN5 [42]. YeastGCN5 (yGCN5) exhibits regioselective acetyltransferto K14 of histone H3 and to K8 and 16 of histone H4[43]. The kinetic mechanism was shown to be sequential,and as we have noted throughout, this appears to be analmost universal kinetic mechanism for GNAT super-family members. The catalytic activity of yGCN5 wassensitive to EDAC, a group specific modifying reagentwith reasonable specificity for carboxyl groups in en-zymes. Alignment of members of the GNAT superfam-ily HATs revealed a relatively conserved glutamic acidresidue (E173 in yGCN5) that might function as a gen-eral base, assisting in the deprotonation of the e-amino
cetyl-glucosamine in yeast.

Fig. 9. Ribbon diagrams of histone N-acetyltransferases. (A) YeastHat1 histone acetyltransferase in complex with AcCoA (1BOB.pdb).(B) One dimer of tetrameric yeast Hpa2 histone acetyltransferase incomplex with AcCoA (1QSM.pdb). (C) Yeast GCN5 acetyltransferase(1YGH.pdb). (D) Tetrahymena GCN5 histone acetyltransferase incomplex with CoA and a histone H3 peptide (1QSN.pdb). (E) YeastEas1 histone acetyltransferase in complex with AcCoA (1MJB.pdb).
220 M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226
group of the acetylatable lysine residue. Both kcat andkcat/Km were pH-dependent, decreasing as the result ofthe protonation of a single enzyme residue. Site-directedmutagenesis to generate the isosteric, but non-ionizableglutamine residue (E173Q) reduced the activity by >300-fold relative to the non-enzymatic rate. This stronglysuggests a catalytic role for E173 as a general base [42].
In 2000, both the Denu and Cole [44,45] groups pub-lished studies that showed the kinetic mechanisms ofboth yGCN5 and P/CAF, respectively, to be OrderedBi–Bi. In both cases acetyl-CoA binds first, followedby histone peptide. Chemistry ensues and products arereleased in the order acetylated histone followed byCoASH. For P/CAF, the bisubstrate cross-linked ana-logue of acetyl-CoA and the N-terminal 20-mer wasshown to exhibit 30 nM binding affinity, however bisub-strate analogues that were shorter (7 and 12 residues)exhibited much weaker inhibitory activity [44]. Regiose-lective acetylation of K14 was demonstrated for the 20residue N-terminal peptide of histone H3 and confirmedusing the K14A peptide as a dead-end inhibitor. There isno effect of changing solvent microviscosity, suggestingthat kcat (2.8 s�1 for P/CAF and 1.7 s�1 for GCN5) islimited by the chemical step. Single turnover studies ofthe P/CAF catalytic domain demonstrated no burst ofproduct formed, consistent with rate-limiting chemistry[46].
The yeast Hat1 histone acetyltransferase was the firstof the HATs to be structurally characterized in 1998[47]. yHat1 regioselectively acetylates K12 of histoneH4, but in other species, Hat1 orthologues can acetylateK5 as well as K12 of histone H4. The full-length en-zyme, composed of residues 1–320, was expressed andcrystallized in the presence of AcCoA. The structurewas refined against 2.3 A data and revealed a bi-domainstructure composed of an N-terminal domain and the C-terminal GNAT domain from residues 122–320 (Fig.9A). The GNAT domain consisted of a six-stranded bsheet, surrounded on both sides by a helices. Bound Ac-CoA was bound in what is now recognized as its normal,‘‘kinked’’ conformation, and no clear electron densitywas observed for the 3 0-AMP portion of AcCoA. A sin-gle hydrogen bond was observed between the carbonylof the thioester and the backbone amide of F220. Ithad been suggested that regioselective acetylation wasachieved due to the presence of a sequence recognitionmotif (G · GK12 · G) on histone H4. A long channelwas observed leading into the active site, and modelingof a peptide revealed that this channel could accommo-date the six residues proposed to encompass the recogni-tion motif. In this model, the e-amino group of lysinewas positioned close enough to directly attack the thio-ester linkage, and residues of the H4 histone tail(GLGK12GG) were easily accommodated without sig-nificant steric clashes. No obvious candidates for eithera general acid or base were observed, with the side chain
of Q258 being closest to the sulfur atom of boundacetylCoA.
The following year, the structures of four HATs werepublished by the Ramakrishnan and Marmorsteingroups. The full-length yeast Hpa2 HAT was solved

M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226 221
using the selenomethionine-substituted recombinant en-zyme as both the apoenzyme (2.9 A) and as the AcCoAbinary complex (to 2.4 A). The enzyme is a dimer insolution, while the binary complex exists as a tetramerin solution and in the crystal, with unusual contactsmade between the adenine rings of the AcCoA mole-cules in adjacent dimers [24]. Residues 7/8-156 are ob-served in the monomers, which exhibit a typicalGNAT fold (Fig. 9B). The ‘‘P-loop’’ located betweena3 and b4 was disordered in the apoenzyme crystal,but showed well defined density in the binary complexas the result of hydrogen bonds to the oxygen atomsof the pyrophosphate moiety of AcCoA from the back-bone amide nitrogens of V101, G103, A104, G105, andG106. The carbonyl oxygen of the AcCoA thioester ishydrogen bonded to the backbone amide nitrogen ofL93, and residues N91 and D92 form a ‘‘b bulge’’ onstrand four adjacent to this interaction. The phenolichydroxyl group of Y139 is close to the sulfur atom ofbound AcCoA, suggesting that it may function as a gen-eral acid to assist in the appropriate partitioning of thetetrahedral intermediate. No obvious candidate residuethat could function as a general base was noted, how-ever the active site has a net positive electrostatic poten-tial, in contrast to most other GNATs. This featuremight serve to assist in the deprotonation of the e-aminogroup of the lysine residue as it entered the active site.
The human P/CAF histone acetyltransferase catalyticdomain comprising the 165 amino acid ‘‘core’’ (residues493–658 of the 832 amino acid full-length protein) wascrystallized as the CoA binary complex [48]. The struc-ture was solved using the Tetrahymena GCN5 structure(see below) as the search model using molecular replace-ment methods and refined to 2.3 A. E570 was observedto be positioned �11 A from the likely position of thethioester, and may serve the function of a general basevia intervening water molecules. A single interactionwas proposed between the backbone amide of C574and the carbonyl group of AcCoA. The non-conserved,‘‘non-core’’ N- and C-terminal regions were proposed tobe responsible for providing substrate specificity.
The namesake of the GNAT superfamily, the yeastGCN5, was determined at 1.9 A resolution [49]. In addi-tion to the similarity of the fold to other GNATs, E173was proposed to be the active site base (Fig. 9C). It issurrounded by non-polar residues, suggesting that itspK value would be perturbed upward compared to freein solution. The E173Q mutant was shown to havebackground histone acetyltransferase activity, and in-creased the doubling time of strains containing from2.5 h for wild-type strains to 3.6 h, similar to strains con-taining the gcn5 knockout (3.8 h).
The crystal structure of the catalytic domain (residues48–210) of the Tetrahymena GCN5, was solved at 1.7 Aresolution, as were the AcCoA binary complex at 2.0 Aresolution and the CoA-histone H3 peptide ternary
complex, KSTGGKAPRKQ, where the emboldened Kis the site of acetylation, to 2.2 A resolution [50]. Thepeptide was bound at the long channel previously pro-posed as the site of peptide binding, and although elec-tron density for the first three residues was not observed,the overall conformation is that of a random coil. Mostof the interactions between the peptide and enzyme arevia main chain atoms of the peptide, especially those be-tween K14 and the adjacent five C-terminal residues.E122, equivalent to E173 of the yGCN5, is again sur-rounded by non-polar residues, and a tightly boundwater molecule is hydrogen bonded to both E122 andthe amide backbone of Y160. The carbonyl of boundAcCoA is hydrogen bonded to the backbone amide ofL126, polarizing it for nucleophilic attack, and poten-tially stabilizing the anionic tetrahedral intermediate.The reaction mechanism proposed from this structureinvoked general base-assisted deprotonation of the e-amino group of K14 by E122 via the intervening watermolecule. No residues that could function as the generalacid were observed; supporting the kinetic observationsthat kcat was dependent only on the ionization state ofthe general base. Finally, a structure of this same en-zyme in complex with a bisubstrate analogue synthe-sized and kinetically evaluated by Cole and colleagues[44] was determined at 2.2 A resolution [51], as were acomplex containing CoA and a histone H3 peptide(Fig. 9D). Together with the structures of the variousbinary and ternary complexes discussed previously, thisallowed a complete description of the conformationalchanges occurring during AcCoA binding, peptide bind-ing, catalysis and product release. The implications forinhibitor design were discussed in this paper.
To conclude this discussion of histone acetyltransfe-rases, a final structure of a histone acetyltransferase,the yeast Esa1, will be discussed. Esa1 is a member ofa second, sequence-unrelated family of eukaryotic his-tone acetyltransferases; the MYST subfamily. Esa1 isthe catalytic subunit of the NuA4 complex that specifi-cally acetylates histone H4, is an essential gene in yeast[52], and is involved in G2/M cell cycle progression [53].The three dimensional structure of the recombinant pro-tein containing residues 160–435 in complex with CoAwas reported in 2000 by Marmorstein and colleagues,and revealed that it was a member of the GNAT super-family [54]. A predominantly b-strand N-terminal and apredominantly a-helical C-terminal extension were ob-served in addition to the central GNAT core (Fig. 9E).E338 was observed to occupy a position similar toE579 in P/CAF and E255 in yHat1, suggesting its roleas a general base. Indeed, the E338Q mutant exhibitedonly background histone acetyltransferase activity. Ina second study [55], these authors also noted the pres-ence of a cysteine residue at the active site, as has beenobserved in other GNAT superfamily members [21].Replacement of the C304 residue with either a serine

222 M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226
residue (C304S) or alanine residue (C304A) had equallydevastating effects on acetyltransferase activity. Crystal-lization of wild-type Esa1 in the presence of AcCoA re-vealed the presence of CoA at the active site and theformation of the S-acetyl-Cys304. Intriguingly, thestructure obtained with the E338Q mutant in the pres-ence of AcCoA yielded the bound intact AcCoA mole-cule, suggesting a role for E338 in promoting acetyltransfer from AcCoA to C304. These studies were con-firmed in solution studies using both [3H-methyl]-Ac-CoA and mass spectrometric analysis of mutants togenerate the mono-acetylated H3 histone 19-mer pep-tide. Finally, the initial velocity pattern was parallel,supporting a ping–pong kinetic mechanism [55]. Theauthors proposed that E338 functions as the generalbase, and via several intervening water molecules func-tions to deprotonate the thiol of C304, allowing for anisoenergetic acetyl transfer from AcCoA to C304. Theprotonated E338 would then protonate the leaving thiolof CoA. Restored to its anionic form, E338 could thenfunction again as a base, removing the e-amino groupof the lysine to be acetylated, thus permitting attackon the acetyl-enzyme and transfer to the histone. Thisremarkable chemical mechanism is the only documentedping–pong reaction amongst the GNAT superfamilymembers to date, but points to the catalytic versatilitythat nature will reveal even within a strictly conservedstructural framework.
Mycothiol synthase
Mycothiol is the major low molecular weight thiol inActinomycetes, including M. tuberculosis. The biosyn-thesis of mycothiol occurs in four steps: (1) an uncharac-terized coupling of myo-inositol and GlcNAc togenerate the pseudo-disaccharide, (2) the deacetylation
Fig. 10. Biosynthesis
of the N-acetyl group, (3) the formation of a peptidebond between the 2-amino group and LL-cysteine, and fi-nally (4) the acetylation of the a-amino group of LL-cys-teine [56] (Fig. 10). The enzyme that catalyzes the finalstep was identified and is encoded by the Rv0819 (mshD)gene in M. tuberculosis [57]. Interestingly, the 315 aminoacid sequence of mycothiol synthase is both twice aslong as a core GNAT sequence and also was predictedto contain two GNAT motifs. The recombinantly ex-pressed enzyme appears to function in solution as amonomer, and both gel filtration and dynamic lightscattering confirmed this [58]. The three-dimensionalstructure of the enzyme revealed two GNAT domains,an N-terminal domain (residues 1–140) and a C-termi-nal domain (residues 141–315) linked by a long loopconnecting the end of b6 of the N-terminal GNAT do-main to the beginning of b1 0 in the C-terminal GNATdomain (Fig. 11A). The b6 strands are exchanged acrossthe dimer interface between the two GNAT domains.Crystals of the AcCoA complex were also generatedand in this structure both the N- and C-terminal do-mains contain bound AcCoA [58]. While all the ele-ments for a functional GNAT enzyme are present inthe C-terminal domain, the AcCoA in the N-terminaldomain adopts a very different conformation, with theacetyl group rotated 90� from its normal orientation,and inaccessible to an incoming substrate. The typical‘‘b bulge’’ universally present in all GNAT structuressolved to date is also missing, suggesting that the N-ter-minal domain is catalytically non-functional. AcCoAbound to the C-terminal domain is observed in its typi-cal orientation, with the carbonyl group interacting withthe backbone amide of L238 and with Y294 positionednear the sulfur atom of AcCoA to potentially functionas the general acid. Thus, while containing two GNATdomains in an ‘‘internal dimer’’ that both can bind Ac-CoA, one of the domains appears to be non-functional.
of mycothiol.
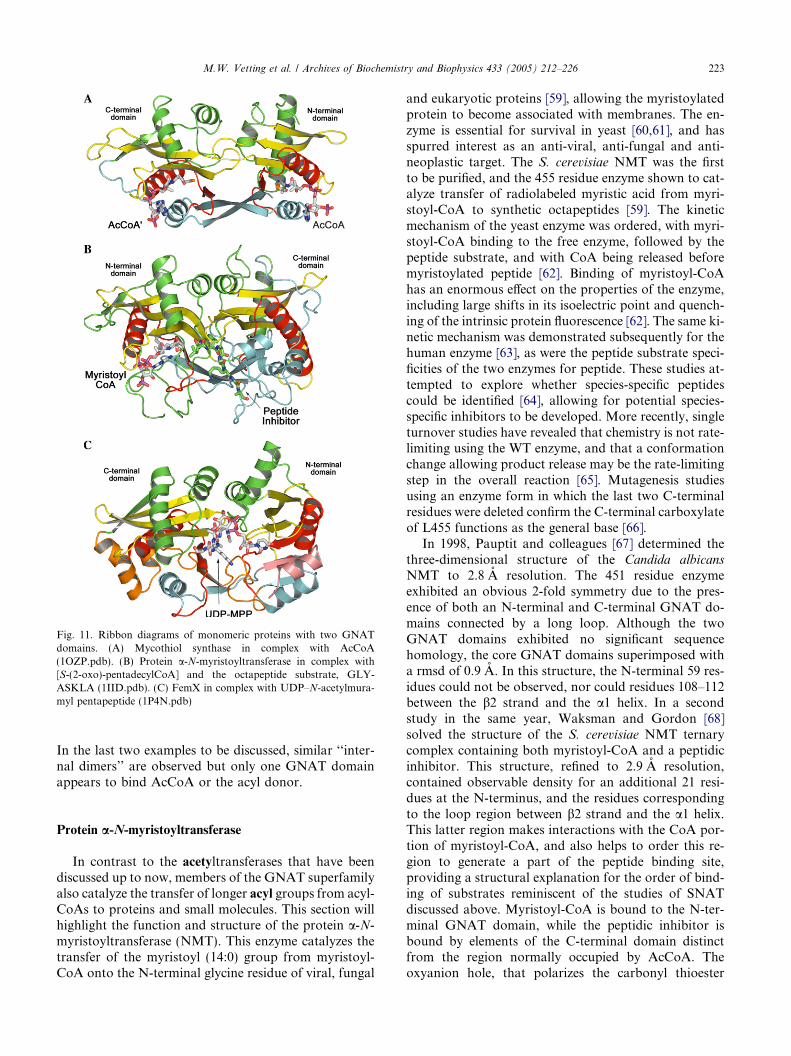
Fig. 11. Ribbon diagrams of monomeric proteins with two GNATdomains. (A) Mycothiol synthase in complex with AcCoA(1OZP.pdb). (B) Protein a-N-myristoyltransferase in complex with[S-(2-oxo)-pentadecylCoA] and the octapeptide substrate, GLY-ASKLA (1IID.pdb). (C) FemX in complex with UDP–N-acetylmura-myl pentapeptide (1P4N.pdb)
M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226 223
In the last two examples to be discussed, similar ‘‘inter-nal dimers’’ are observed but only one GNAT domainappears to bind AcCoA or the acyl donor.
Protein a-N-myristoyltransferase
In contrast to the acetyltransferases that have beendiscussed up to now, members of the GNAT superfamilyalso catalyze the transfer of longer acyl groups from acyl-CoAs to proteins and small molecules. This section willhighlight the function and structure of the protein a-N-myristoyltransferase (NMT). This enzyme catalyzes thetransfer of the myristoyl (14:0) group from myristoyl-CoA onto the N-terminal glycine residue of viral, fungal
and eukaryotic proteins [59], allowing the myristoylatedprotein to become associated with membranes. The en-zyme is essential for survival in yeast [60,61], and hasspurred interest as an anti-viral, anti-fungal and anti-neoplastic target. The S. cerevisiae NMT was the firstto be purified, and the 455 residue enzyme shown to cat-alyze transfer of radiolabeled myristic acid from myri-stoyl-CoA to synthetic octapeptides [59]. The kineticmechanism of the yeast enzyme was ordered, with myri-stoyl-CoA binding to the free enzyme, followed by thepeptide substrate, and with CoA being released beforemyristoylated peptide [62]. Binding of myristoyl-CoAhas an enormous effect on the properties of the enzyme,including large shifts in its isoelectric point and quench-ing of the intrinsic protein fluorescence [62]. The same ki-netic mechanism was demonstrated subsequently for thehuman enzyme [63], as were the peptide substrate speci-ficities of the two enzymes for peptide. These studies at-tempted to explore whether species-specific peptidescould be identified [64], allowing for potential species-specific inhibitors to be developed. More recently, singleturnover studies have revealed that chemistry is not rate-limiting using the WT enzyme, and that a conformationchange allowing product release may be the rate-limitingstep in the overall reaction [65]. Mutagenesis studiesusing an enzyme form in which the last two C-terminalresidues were deleted confirm the C-terminal carboxylateof L455 functions as the general base [66].
In 1998, Pauptit and colleagues [67] determined thethree-dimensional structure of the Candida albicansNMT to 2.8 A resolution. The 451 residue enzymeexhibited an obvious 2-fold symmetry due to the pres-ence of both an N-terminal and C-terminal GNAT do-mains connected by a long loop. Although the twoGNAT domains exhibited no significant sequencehomology, the core GNAT domains superimposed witha rmsd of 0.9 A. In this structure, the N-terminal 59 res-idues could not be observed, nor could residues 108–112between the b2 strand and the a1 helix. In a secondstudy in the same year, Waksman and Gordon [68]solved the structure of the S. cerevisiae NMT ternarycomplex containing both myristoyl-CoA and a peptidicinhibitor. This structure, refined to 2.9 A resolution,contained observable density for an additional 21 resi-dues at the N-terminus, and the residues correspondingto the loop region between b2 strand and the a1 helix.This latter region makes interactions with the CoA por-tion of myristoyl-CoA, and also helps to order this re-gion to generate a part of the peptide binding site,providing a structural explanation for the order of bind-ing of substrates reminiscent of the studies of SNATdiscussed above. Myristoyl-CoA is bound to the N-ter-minal GNAT domain, while the peptidic inhibitor isbound by elements of the C-terminal domain distinctfrom the region normally occupied by AcCoA. Theoxyanion hole, that polarizes the carbonyl thioester

224 M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226
and stabilizes the tetrahedral intermediate formed afterattack by the peptide a-N-glycyl residue is generatedby the main chain amide backbones of both F170 andL171. The amine of the peptidic inhibitor is within2.5 A of the C-terminal carboxylate of L455, suggestingthat it is the general base responsible for proton abstrac-tion from the N-terminal amino group of the substrate.The interactions of the peptidic inhibitor and enzyme ex-plained the strong preference for serine at position 6 ofthe peptide since this side chain interacts with the imid-azole function of H221, and the preference for a cationicresidue at position 7 of the peptide substrate, since thelysine side chain in the inhibitor is positioned near acluster of anionic residues that includes D106, D108and D417. Finally, the structure of a ternary complex(Fig. 11B) containing a bona fide octapeptide substrate,GLYASKLA, and the non-hydrolyzable myristoyl-CoAanalogue, [S-(2-oxo)-pentadecylCoA] was solved at2.5 A resolution [69].
The Fem family of acyltransferases
In Gram-positive species of bacteria, biosynthesis ofthe branched cell wall is an elaborate process that in-cludes the generation of the UDP–N-acetylmuramylpentapeptide and its further elaboration by the exten-sion of up to five additional amino acids in isopeptidelinkage with either the e-amino group of lysine, d-aminogroup of ornithine or amino acid of L,LL,L-diaminopime-late residue in position three of the pentapeptide. Thesepeptide extensions provide greater flexibility to thecross-linked cell wall than in the directly cross-linkedcell walls of Gram-negative species. The FEM familyof enzymes catalyze this unique reaction in which theacyl donor is an amino acylated tRNA. Little wasknown of their mechanism of action since the enzymeswere difficult to express and purify, were membrane-as-sociated and whose substrates were difficult to generateand assays difficult to perform. However, the purifica-tion of the soluble Lactobacillus viridescens FemX andits subsequent cloning in 2001 [70] allowed the first ki-netic and mechanistic investigations to be performed.This enzyme uses the soluble UDP–N-acetylmuramylpentapeptide (UDP–MPP) as substrate rather than thelipid-linked sugar pentapeptides used by other Fem fam-ily members. The kinetic mechanism was shown to besequential, with UDP–MPP binding before Ala-tRNAa-
la [71]. The pH dependence of the reaction was bell-shaped, with observation of both a group whose proton-ation abolished activity (pK 5.5) and a group whosedeprotonation abolished activity (pK 9.3). This sug-gested a mechanism in which after nucleophilic attackby the e-amino group of lysine on the alanyl-tRNA es-ter, general acid/base catalysis assisted in the transferof the amino acid to the pentapeptide.
In 2002, the three-dimensional structure of the Staph-ylococcus aureus FemA enzyme was solved at 2.1 A res-olution [72]. Although exhibiting no significant sequencehomology to other structurally described proteins, thestructure revealed two GNAT domains (ca. 1–144 andca. 145–395), with a third domain between b3 0 and b4 0
consisting of a pair of antiparallel a helices (residues246–307). The two GNAT domains can be superim-posed with rmsd of 2.4 A, and the overall structure bearssimilarity to both NMT and mycothiol synthase, dis-cussed above. The antiparallel a helices were proposedto function as a flexible platform to bind the amino-ac-ylated tRNA substrate. Since all previous structures forGNAT superfamily members implicated this fold in thebinding of acylCoA, this remarkable structure providesa unique structural anomaly. No evidence exists frommechanistic studies that implicate or suggest AcCoAor CoA as a catalytic component. A second structureof the Weissella viridescens FemX appeared earlier thisyear. Earlier predictions, based on sequence alignments,suggested that the coiled-coil region would not be pres-ent in the FemX orthologue [71], and this was confirmedin the structure of the 335 residue enzyme, determined inapo form, and in complex with UDP–MPP at 1.7 and1.9 A resolution, respectively [73]. The 60-residuecoiled-coil observed in FemA is replaced in FemX witha tight five-residue loop. The two GNAT domains arestructurally similar to the aminoglycoside, serotoninand histone acetyltransferases discussed above. Thebound UDP–MPP is observed at the cleft between thetwo domains, and makes interactions primarily withthe N-terminal GNAT domain (Fig. 10C). The positionof UDP–MPP binding is somewhat reminiscent of theposition where the peptide substrate is bound in theNMT ternary complex (Fig. 11B), and would also besomewhat similar to the position of histone peptidebinding at the dimer interface. The details of aminoacylated tRNA binding are unclear, although a channelthat can accommodate the amino-acylated CCA tail ofthe charged tRNA can be modeled into the structureto allow for a reasonable distance of approach to thee-amino group of lysine, whose side chain is not clearlyobserved in the final electron density map.
Conclusions
The GNAT superfamily is one of the largest enzymesuperfamilies recognized to date, with over 10,000 repre-sentatives from all kingdoms of life. In spite of verymodest degrees of overall primary sequence homology,the basic structure of the GNAT fold is extraordinarilyconserved, and serves two nearly universal functions: tobind the pantetheine arm of acyl-CoA and to hydrogenbond to and polarize the carbonyl of the thioester. Bind-ing of acyl-CoA induces sometimes-dramatic changes in

M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226 225
the conformation of regions of the fold, primarily in theconformation of a1/a2 and the adjoining loop, and occa-sionally in the ‘‘P loop’’ which coordinates the pyrophos-phate moiety, and this can generate additional structuralelements that are necessary for cognate substrate recog-nition and binding. The kinetic and chemical mecha-nisms of nearly all GNATs proceeds via the additionof both the acyl-CoA and acetylatable substrate to forma ternary complex, followed by nucleophilic attack bythe amine and elimination of the thiol of CoA. In mostof the cases for which data has been reported, the ther-modynamically favorable catalytic reaction is not rate-limiting. There are examples of general base catalysis,examples of general acid catalysis, examples where bothmay be involved, and examples where neither appear tobe necessary. The number and types of substrates isimpressive given that in any single organism, only a smallfraction of the substrates for the genome-encodedGNATs are known. Finally, as demonstrated by theESA1 histone acetyltransferase and the Fem amino acyl-transferases, the conserved structural framework extantin all GNAT superfamily members may be modified toallow for changes in chemical mechanism and the struc-ture of the acyl group donor.
There is clearly much more to be learned about thepotential manifold of chemistries and physiological sub-strates of this large enzyme superfamily. Thus, whilethere are a large number of known substrates for GNATsuperfamily members, including GNATs that acylateother antibiotics, small molecules and the homoserinelactones, in any single organism, very few GNATs havebeen functionally characterized. In E. coli, arguable oneof the most thoroughly studied organisms, only one ofthe 26 predicted GNATs has been biochemically charac-terized. Three additional GNATs have been ascribed thefunction of acetylating three ribosomal proteins, S5,S18, and L12. The other 22 have no known, or hypoth-esized function. Methods to identify the substrates forGNAT superfamily members in both eubacteria andeukaryotes are being pursued in our laboratory.
References
[1] J. Davies, G.D. Wright, Trends Microbiol. 5 (1997) 234–240.[2] K.J. Shaw, P.N. Rather, R.S. Hare, G.H. Miller, Microbiol. Rev.
57 (1993) 138–163.[3] S.L. Berger, B. Pina, N. Silverman, G.A. Marcus, J. Agapite, J.L.
Regier, S.J. Triezenberg, L. Guarente, Cell 70 (1992) 251–265.[4] J.E. Brownell, C.D. Allis, Proc. Natl. Acad. Sci. USA 92 (1995)
6364–6368.[5] J.E. Brownell, C.D. Allis, Curr. Opin. Genet. Dev. 6 (1996) 176–
184.[6] F. Dyda, D.C. Klein, A.B. Hickman, Annu. Rev. Biophys.
Biomol. Struct. 29 (2000) 81–103.[7] B. Llano-Sotelo, E.F. Azucena Jr., L.P. Kotra, S. Mobashery,
C.S. Chow, Chem. Biol. 9 (2002) 455–463.[8] K. Radika, D.B. Northrop, J. Biol. Chem. 259 (1984) 12543–
12546.
[9] E. Wolf, A. Vassilev, Y. Makino, A. Sali, Y. Nakatani, S.K.Burley, Cell 94 (1998) 439–449.
[10] J.A. Ainsa, E. Perez, V. Pelicic, F.X. Berthet, B. Gicquel, C.Martin, Mol. Microbiol. 24 (1997) 431–441.
[11] K. Franklin, A.J. Clarke, Antimicrob. Agents Chemother. 45(2001) 2238–2244.
[12] S.S. Hegde, F. Javid-Majd, J.S. Blanchard, J. Biol. Chem. 276(2001) 45876–45881.
[13] M.W. Vetting, S.S. Hegde, F. Javid-Majd, J.S. Blanchard, S.L.Roderick, Nat. Struct. Biol. 9 (2002) 653–658.
[14] Y. Costa, M. Galimand, R. Leclercq, J. Duval, P. Courvalin,Antimicrob. Agents Chemother. 37 (1993) 1896–1903.
[15] G.D. Wright, P. Ladak, Antimicrob. Agents Chemother. 41(1997) 956–960.
[16] K.A. Draker, D.B. Northrop, G.D. Wright, Biochemistry 42(2003) 6565–6574.
[17] K.A. Draker, G.D. Wright, Biochemistry 43 (2004) 446–454.[18] L.E. Wybenga-Groot, K. Draker, G.D. Wright, A.M. Berghuis,
Struct. Fold Des. 7 (1999) 497–507.[19] D.L. Burk, N. Ghuman, L.E. Wybenga-Groot, A.M. Berghuis,
Prot. Sci. 12 (2003) 426–437.[20] S. Magnet, P. Courvalin, T. Lambert, J. Bacteriol. 181 (1999)
6650–6655.[21] S. Magnet, T. Lambert, P. Courvalin, J.S. Blanchard, Biochem-
istry 40 (2001) 3700–3709.[22] S.S. Hegde, T.K. Dam, C.F. Brewer, J.S. Blanchard, Biochemistry
41 (2002) 7519–7527.[23] M.W. Vetting, S. Magnet, E. Nieves, S.L. Roderick, J.S. Blan-
chard, Chem. Biol. 11 (2004) 565–573.[24] M.L. Angus-Hill, R.N. Dutnall, S.T. Tafrov, R. Sternglanz, V.
Ramakrishnan, J. Mol. Biol. 294 (1999) 1311–1325.[25] W. Zheng, P.A. Cole, Curr. Med. Chem. 9 (2002) 1187–1199.[26] W. Zheng, P.A. Cole, Bioorg. Chem. 31 (2003) 398–411.[27] J. De Angelis, J. Gastel, D.C. Klein, P.A. Cole, J. Biol. Chem.
273 (1998) 3045–3050.[28] E.M. Khalil, J. De Angelis, P.A. Cole, J. Biol. Chem. 273 (1998)
30321–30327.[29] E.M. Khalil, P.A. Cole, J. Am. Chem. Soc. 120 (1998) 6195–6196.[30] E.M. Khalil, J. De Angelis, M. Ishii, P.A. Cole, Proc. Natl. Acad.
Sci. USA 96 (1999) 12418–12423.[31] W. Zheng, K.A. Scheibner, A.K. Ho, P.A. Cole, Chem. Biol. 8
(2001) 379–389.[32] A.B. Hickman, M.A. Namboodiri, D.C. Klein, F. Dyda, Cell 97
(1999) 361–369.[33] K.A. Scheibner, J. De Angelis, S.K. Burley, P.A. Cole, J. Biol.
Chem. 277 (2002) 18118–18126.[34] E. Wolf, J. De Angelis, E.M. Khalil, P.A. Cole, S.K. Burley, J.
Mol. Biol. 317 (2002) 215–224.[35] L.R. Olsen, S.L. Roderick, Biochemistry 40 (2001) 1913–1921.[36] T. Mio, T. Yamada-Okabe, M. Arisawa, H. Yamada-Okabe, J.
Biol. Chem. 274 (1999) 424–429.[37] C. Peneff, D. Mengin-Lecreulx, Y. Bourne, J. Biol. Chem. 276
(2001) 16328–16334.[38] J.E. Brownell, J. Zhou, T. Ranalli, R. Kobayashi, D.G. Edmond-
son, S.Y. Roth, C.D. Allis, Cell 84 (1996) 843–851.[39] A.J. Bannister, T. Kouzarides, Nature 384 (1996) 641–643.[40] X.J. Yang, V.V. Ogryzko, J. Nishikawa, B.H. Howard, Y.
Nakatani, Nature 382 (1996) 319–324.[41] D.E. Sterner, S.L. Berger, Microbiol. Mol. Biol. Rev. 64 (2000)
435–459.[42] K.G. Tanner, R.C. Trievel, M.H. Kuo, R.M. Howard, S.L.
Berger, C.D. Allis, R. Marmorstein, J.M. Denu, J. Biol. Chem.274 (1999) 18157–18160.
[43] A.F. Neuwald, D. Landsman, Trends Biochem. Sci. 22 (1997)154–155.
[44] O.D. Lau, A.D. Courtney, A. Vassilev, L.A. Marzilli, R.J. Cotter,Y. Nakatani, P.A. Cole, J. Biol. Chem. 275 (2000) 21953–21959.

226 M.W. Vetting et al. / Archives of Biochemistry and Biophysics 433 (2005) 212–226
[45] K.G. Tanner, M.R. Langer, Y. Kim, J.M. Denu, J. Biol. Chem.275 (2000) 22048–22055.
[46] K.G. Tanner, M.R. Langer, J.M. Denu, Biochemistry 39 (2000)11961–11969.
[47] R.N. Dutnall, S.T. Tafrov, R. Sternglanz, V. Ramakrishnan, Cell94 (1998) 427–438.
[48] A. Clements, J.R. Rojas, R.C. Trievel, L. Wang, S.L. Berger, R.Marmorstein, EMBO J. 18 (1999) 3521–3532.
[49] R.C. Trievel, J.R. Rojas, D.E. Sterner, R.N. Venkataramani, L.Wang, J. Zhou, C.D. Allis, S.L. Berger, R. Marmorstein, Proc.Natl. Acad. Sci. USA 96 (1999) 8931–8936.
[50] J.R. Rojas, R.C. Trievel, J. Zhou, Y. Mo, X. Li, S.L. Berger,C.D. Allis, R. Marmorstein, Nature 401 (1999) 93–98.
[51] A.N. Poux, M. Cebrat, C.M. Kim, P.A. Cole, R. Marmorstein,Proc. Natl. Acad. Sci. USA 99 (2002) 14065–14070.
[52] E.R. Smith, A. Eisen, W. Gu, M. Sattah, A. Pannuti, J. Zhou,R.G. Cook, J.C. Lucchesi, C.D. Allis, Proc. Natl. Acad. Sci.USA 95 (1998) 3561–3565.
[53] A.S. Clarke, J.E. Lowell, S.J. Jacobson, L. Pillus, Mol. Cell Biol.19 (1999) 2515–2526.
[54] Y. Yan, N.A. Barlev, R.H. Haley, S.L. Berger, R. Marmorstein,Mol. Cell 6 (2000) 1195–1205.
[55] Y. Yan, S. Harper, D.W. Speicher, R. Marmorstein, Nat. Struct.Biol. 9 (2002) 862–869.
[56] D. Sareen, G.L. Newton, R.C. Fahey, N.A. Buchmeier, J.Bacteriol. 185 (2003) 6736–6740.
[57] T. Koledin, G.L. Newton, R.C. Fahey, Arch. Microbiol. 178(2002) 331–337.
[58] M.W. Vetting, S.L. Roderick, M. Yu, J.S. Blanchard, Prot. Sci.12 (2003) 1954–1959.
[59] D.A. Towler, S.P. Adams, S.R. Eubanks, D.S. Towery, E.Jackson-Machelski, L. Glaser, J.I. Gordon, Proc. Natl. Acad.Sci. USA 84 (1987) 2708–2712.
[60] J.K. Lodge, E. Jackson-Machelski, D.L. Toffaletti, J.R. Perfect,J.I. Gordon, Proc. Natl. Acad. Sci. USA 91 (1994) 12008–12012.
[61] R.A. Weinberg, C.A. McWherter, S.K. Freeman, D.C. Wood,J.I. Gordon, S.C. Lee, Mol. Microbiol. 16 (1995) 241–250.
[62] D.A. Rudnick, C.A. McWherter, W.J. Rocque, P.J. Lennon,D.P. Getman, J.I. Gordon, J. Biol. Chem. 266 (1991) 9732–9739.
[63] W.J. Rocque, C.A. McWherter, D.C. Wood, J.I. Gordon, J. Biol.Chem. 268 (1993) 9964–9971.
[64] J.K. Lodge, E. Jackson-Machelski, M. Higgins, C.A. McWherter,J.A. Sikorski, B. Devadas, J.I. Gordon, J. Biol. Chem. 273 (1998)12482–12491.
[65] T.A. Farazi, J.K. Manchester, J.I. Gordon, Biochemistry 39(2000) 15807–15816.
[66] T.A. Farazi, J.K. Manchester, G. Waksman, J.I. Gordon,Biochemistry 40 (2001) 9177–9186.
[67] S.A. Weston, R. Camble, J. Colls, G. Rosenbrock, I. Taylor, M.Egerton, A.D. Tucker, A. Tunnicliffe, A. Mistry, F. Mancia, E.de la Fortelle, J. Irwin, G. Bricogne, R.A. Pauptit, Nat. Struct.Biol. 5 (1998) 213–221.
[68] R.S. Bhatnagar, K. Futterer, T.A. Farazi, S. Korolev, C.L.Murray, E. Jackson-Machelski, G.W. Gokel, J.I. Gordon, G.Waksman, Nat. Struct. Biol. 5 (1998) 1091–1097.
[69] T.A. Farazi, G. Waksman, J.I. Gordon, Biochemistry 40 (2001)6335–6343.
[70] S.S. Hegde, T.E. Shrader, J. Biol. Chem. 276 (2001) 6998–7003.
[71] S.S. Hegde, J.S. Blanchard, J. Biol. Chem. 278 (2003) 22861–22867.
[72] T.E. Benson, D.B. Prince, V.T. Mutchler, K.A. Curry, A.M. Ho,R.W. Sarver, J.C. Hagadorn, G.H. Choi, R.L. Garlick, Structure(Cambr.) 10 (2002) 1107–1115.
[73] S. Biarrotte-Sorin, A.P. Maillard, J. Delettre, W. Sougakoff, M.Arthur, C. Mayer, Structure (Cambr.) 12 (2004) 257–267.





























![[11] ABB Review 3_2008](https://img.pdfslide.us/doc/110x75/5527a726550346c3358b4854/11-abb-review-32008.jpg)