Embed Size (px)
Citation preview

Ab Initio Calculations of the Ground Electronic States of the C3Ar and C3Ne
Complexes
Yi-Ren Chen, Yi-Jen Wang, and Yen-Chu Hsu
Institute of Atomic and Molecular Sciences
Academia Sinica
Taiwan, R. O. C.
National Science Council, R. O. C.
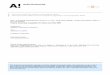
• The equilibrium geometry of the ground electronic state of the C3-Ar vdW complex has been determined by ab initio calculation at the level of CCSD(T)/cc-pVQZ.
3.916Å Ar
C
C
C
1.298Å
1.298Å
73.73o
179.3o
Zhang et.al., J. Chem. Phys.120, 3189(2004).
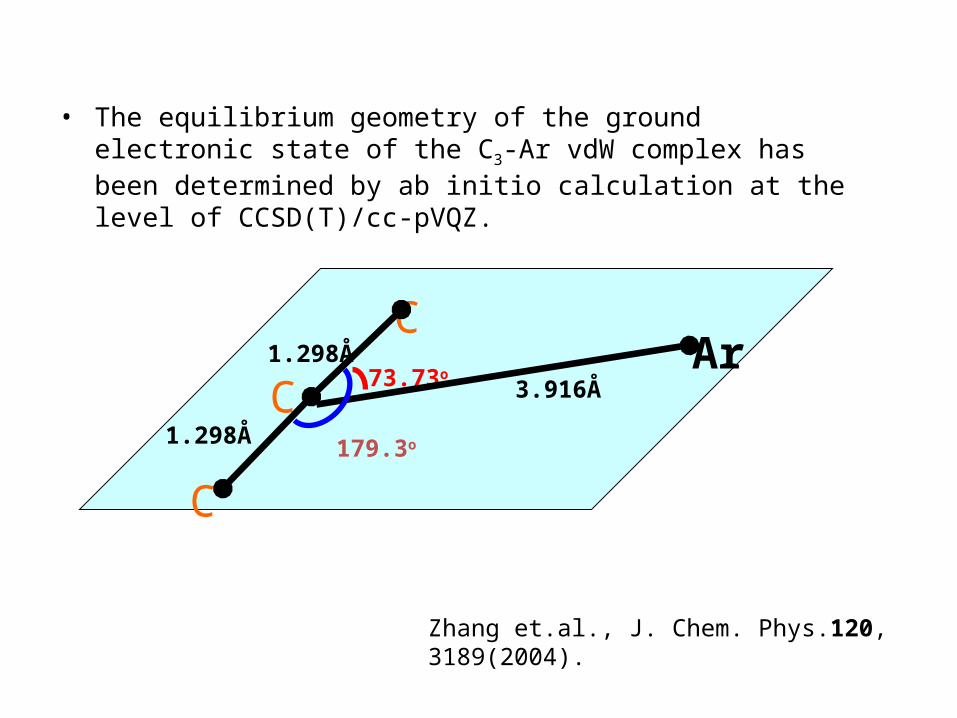
En
erg
y (c
m-1
)
0
100
200
300
400
500
600
700
800
(0,20,0)
(0,40,0)
(0,60,0)
(0,00,0)
C3
(0,11,0)
(0,31,0)
(0,80,0)
C3-KrC3-Ar
(2,4)=(0,1)
(1,1)
(1,2)
(1,3)
(1,4)
(1,5)
(1,6)
(1,7)
(1,8)
(1,9)
(0,1)
(1,3)
(1,4)
(1,5)
(1,6)
(1,7)
(1,8)
(1,9)
Zhang et.al., J. Chem. Phys.120, 3189(2004).Chao et.al., J. Chem. Phys.134, 074313(2011).

(i) Assume the C3-bending vibrational levels of the complex can be
described as a two-dimensional harmonic oscillator perturbed by the rare gas atom and the vibrationally averaged vdW stretch and vdW bend are independent of the C3-bending vibration.
(ii) Expand the interaction potential in terms of cosθ and cos2θ .
(iii) The observed C3 bending vibrational levels were used as the zero-order levels.
2rRVrRVrH 221
cos),(cos),(),( ,
C3Ar C3Kr V1=0.0 V1=0.0
V2=4.80.4 cm-1 V2=6.30.3 cm-1
PHO Model


En
erg
y (c
m-1
)
0
100
200
300
400
500
600
700
800
(0,20,0)
(0,40,0)
(0,60,0)
(0,00,0)
C3
(0,11,0)
(0,31,0)
(0,80,0)
C3-KrC3-Ar
(2,4)=(0,1)
(1,1)
(1,2)
(1,3)
(1,4)
(1,5)
(1,6)
(1,7)
(1,8)
(1,9)
(0,1)
(1,3)
(1,4)
(1,5)
(1,6)
(1,7)
(1,8)
(1,9)
C3-Ne
The levels observed in the C3-Ne complex cannot be assigned by this model!!
But,

Zhang et. al.,Mol. Phys. (2008)CCSD(T)/cc-pVTZ, 3S3P2dC-C-C=180°, ℓ(C-C)=1.29497Å2D-DVR

The A rotational constant of C3Ar should be the same as the B rotational constant of free C3, assuming the Ar atom only affects the bonding in the C3 part slightly.
A(C3Ar) is found to be larger than B(C3)
0.4549 cm-1 0.4305 cm-1
Therefore the top of the T is tilted.
With these values of A and B,simple geometry shows that the tilt angle is 12.4o
CC
C
Ar
12.4o
MI09

Computation Details1. Potential energy obtained from ab initio calculations C3Ar,
• CCSD(T)/cc-pVQZ using Molpro program• 46550 points in the internal coordinates: ρ (the bond angle of C3)=112-248°,
C r(C-C bond length of C3)= 1.2981Å , Ar R (the vdW bond length between Ar and the
r(fixed) center of mass of C3)=3.4-6.0 Å, R azimuth angle (the angle between the vector
R and the principal axis of C3)=0-180°,
Z colatitudes angle =0-90°.
r(fixed)
C • Equilibrium geometry of C3Ar: ρ=179.5°, R=3.9 Å, r= 1.2981Å, =74°, and =7°
C ρ
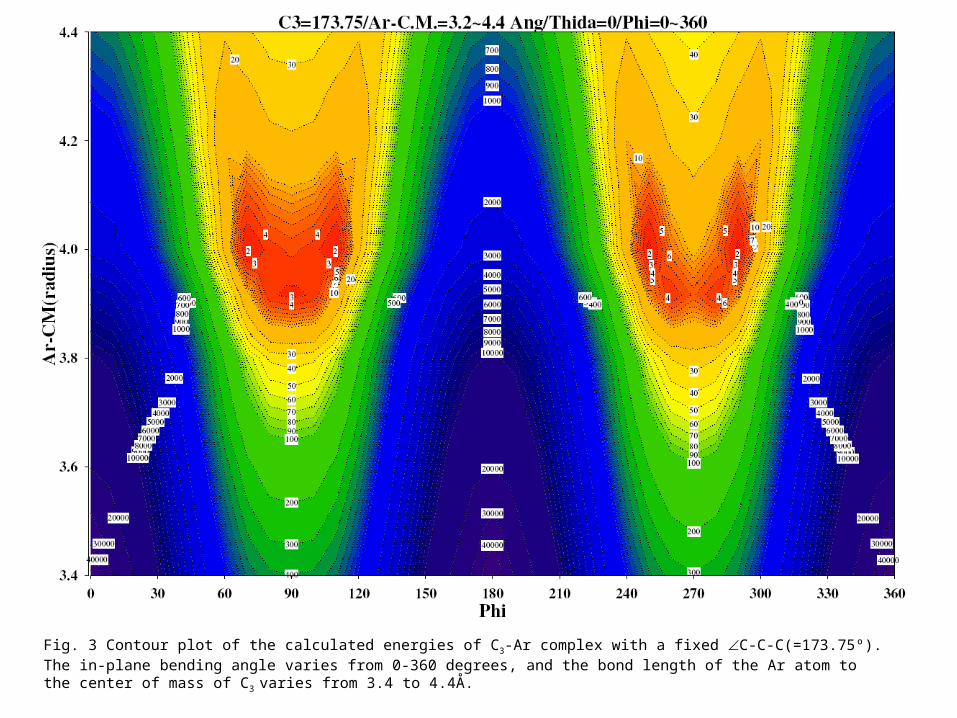
Fig. 3 Contour plot of the calculated energies of C3-Ar complex with a fixed C-C-C(=173.75º). The in-plane bending angle varies from 0-360 degrees, and the bond length of the Ar atom to the center of mass of C3 varies from 3.4 to 4.4Å.


Fig. 7. Contour plot of the calculated energies of C3-Ar complex with a fixed C-C-C(=160º) and a fixed bond length (3.6Å) between the Ar atom to the C. M. of C3. The in-plane bending angle varies from 0 to 360 degrees, and the out-of-plane bending angle varies from 0 to 90 degrees.

• The obtained potential energy points covering energies up to 19,736.8 cm-1 were fitted to an analytical function in terms of internal coordinates with one σ of 0.63 cm-1.
,
where , , R 0 =3.9 and
3.7Å for the Ar and Ne complexes, respectively.
• Assuming that the presence of the rare gas atom would not change the C-C stretch potential, the stretch potential of C3 monomer was added to the 4-D potential to obtain a full 6-D potential of C3-Rg.
V(r1,r2,R,,θ,)≈V(R,,θ,)+V(r1,r2,)-V()
Therein the stretch-bending potential of C3 was calculated at CCSD(T)/aug-cc-pVQZ in the range of (C-C) =1.1-1.5 Å and
C-C-C=70-179.5. This gives additional vibrational energy up to 7756cm-1. One standard deviation of the fitting error of C3 potential is 2.6cm-1.
(R, , , ) ( ) (sin ) cos sin ( ) ( ) cos sinm n p m n
lmn pmnV R l R
2( ) (1 exp( ( )) qo
qR f a R R gR
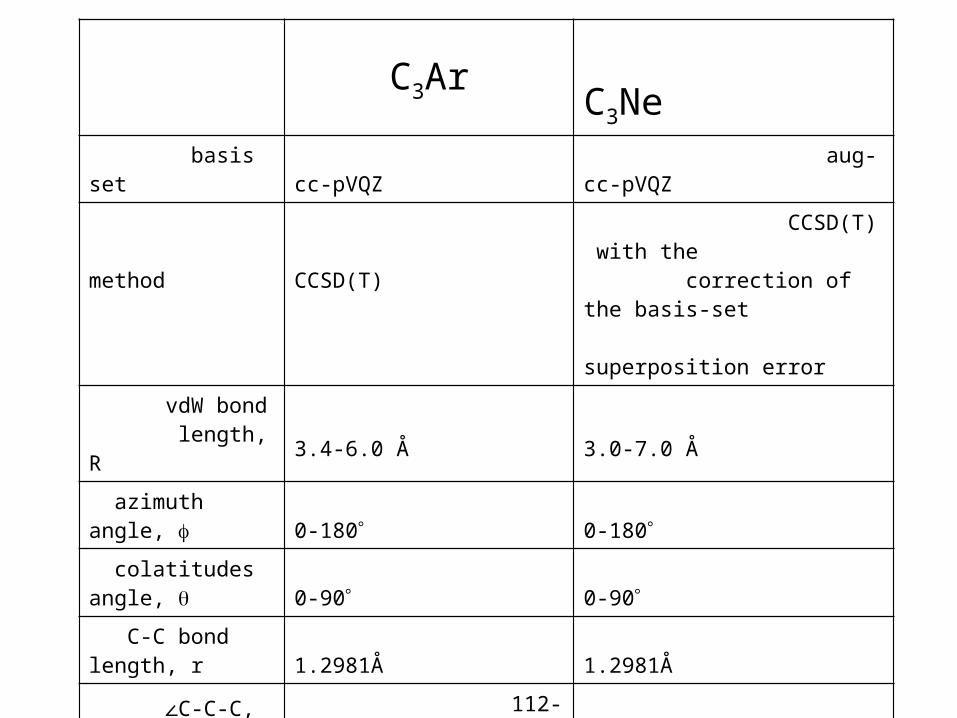
C3Ar C3Ne basis set cc-pVQZ aug-cc-pVQZ
method
CCSD(T)
CCSD(T) with the correction of the basis-set superposition error
vdW bond length, R 3.4-6.0 Å 3.0-7.0 Å
azimuth angle, 0-180 0-180
colatitudes angle, 0-90 0-90
C-C bond length, r 1.2981Å 1.2981Å
∠C-C-C, 112-248 106-254
# of ab initio points 46550 62178
Equilibrium geometry
ρ=179.5°, R=3.9 Å, r= 1.2981Å, =74°, and =7°
ρ=170°, R=3.6 Å, r= 1.2981Å, =80°, and =0°.
Energy range 0-19736.8 cm-1 0-26252 cm-1
PE fitting error 0.63 cm-1 0.86 cm-1

Conclusions
1. The 4D-potential energy functions of C3Ar and C3Ne were calculated at the level of CCSD(T)/cc-pVQZ or aug-cc-pVQZ. The C-C stretch potential energies were added to the 4D potential energies by assuming the vdW bend and stretch energies do not depend upon the C-C bond distances. This assumption was made because we are interested in the vibrational level structures of these two complexes for the first 800 cm-1 region.
2. An improved potential energy fit of C3Ne has been made by dividing the calculated PE into two regions such as energies below 200 cm-1 and energies above 200 cm-1. For instance, 67475 points were fit by 6384 parameters with one standard deviation of fit of 0.22 cm-1.
3. The equilibrium vdW bond length of C3Ar calculated by us is 3.9Å, longer than that (3.8Å) reported Zhang et. al.