Embed Size (px)
Citation preview

A Simple Preparative Procedure for the Rapid Isolation of Phytosphingosine From the Yeast Hansenula ciferrii BENJAMIN WEISS and RICHARD L. STILLER, Division of Neuroscience, N.Y. State Psychiatric Institute and Department of Biochemistry, College of Physicians and Surgeons, Columbia University, New York, New York 10032
ABSTRACT
P h y t o s p h i n g o s i n e and dihydro- sphingosine were isolated as a mixture of their N-acetyl derivatives from the cells of the yeast Hansenula ciferrii. After alkaline hydrolysis of the N-acetyl com- pounds, the free base mixture was reacted with either acetone or p-nitrobenzalde- hyde. The reaction products consisting of the carbonyl derivative of phyto- sphingosine and free dihydrosphingosine were separated on an acetyl cellulose column. The acetone or p-nitrobenzalde- hyde derivative of phytosphingosine recovered from the benzene eluate con- tained no dibydrosphingosine as deter- mined by thin layer and gas liquid chro- matography. The operating time from preparation of the derivative through column purification was less than 3 hrs. Mass spectrometry of the acetone and p-nitrobenzaldehyde derivatives disclosed in each case that ring closure with the carbonyl reagent involved the amino and primary hydroxyl groups to give D-ribo- 2 - d i m e t h y 1- 4-( 2,3-dihy dro xyhexade cyl) oxazolidine and D-ribo-2-p-nitrophenyl- 4-(2,3-dihydroxyhexadecyl) oxazolidene, respectively.
INTRODUCTION AND DISCUSSION
The yeast Hansenula ciferrii produces partially (1) and fully acetylated derivatives of p h y t o s p h i n g o s i n e and dihydrosphingosine (2,3). The fully acetylated bases, after isolation from the culture medium, were separated by counter-current distribution (4). Recently, the free bases were resolved on a Silica Gel S c o l u m n by treatment with chloroform- methanol (9: 1) followed by two successive con- tinuous gradients containing varying pro- portions of chloroform-methanol-2M NH4OH (5). Since these methods involve considerable time, manipulations and analyses, we sought an easy chemical procedure for the resolution of these compounds. Accordingly, a procedure for the rapid separation of phytosphingosine, via its acetone or p-nitrobenzaldehyde derivative,
from dihydrosphingosine obtained from the cells of I1. ciferrii is now described. In addition, proof of the structure of the acetone and p- nitrobenzaldehyde derivatives of phytosphingo- sine is presented. In this study cells were employed as the source material because they contained about nine times more base than that present in the extracellular medium.
It was found that phytosphingosine forms a complex with acetone (6,7). Use of this reaction, it was reasoned, could be made in the separation of phytosphingosine from dihydro- sphingosine. We observed, however, that the complex was of limited stability under the initial conditions studied. A more stable deriva- tive of phytosphingosine was formed with p- nitrobenzaldehyde; dihydrosphingosine was unreactive under the same circumstances. The p-n i t robenzaldehyde derivative of phyto- sphingosine was separated quantitatively within 2 hr from dihydrosphingosine on an acetyl cellulose column by development with benzene; no dihydrosphingosine was present in the product obtained from the benzene eluate as determined by thin layer chromatography (TLC) and gas liquid chromatography (GLC). Similarly, the acetone derivative of phyto- sphingosine was separated from dihydro- sphingosine. The phytosphingosine obtained from the column could be stored until needed as the carbonyl derivative or as the sulfate salt which was formed by regeneration of the base from the derivative by treatment with dilute sulfuric acid.
Although analytical values were presented for the product formed from the reaction of acetone with phytosphingosine, no structural determinat ions were reported (6,7). We observed that the acetone derivative showed no UV absorption at 247 m/a for an azomethine linkage. The acetone and p-nitrobenzaldehyde derivatives gave negative ninhydrin reactions, had similar IR spectra with no absorption near 1600 cm-1 for a free amino group, and could be crystallized from various nonaqueous solvents. These observations, along with data from elementary and active hydrogen analyses, indi- cated that amino and hydroxyl groups were involved in ring formation with the carbonyl reagent. Attempts to ascertain which of the three hydroxyl groups participated in ring
782

PREPARATION OF PHYTOSPHINGOSINE 783
closure by permethylation of the p-nitrobenz- aldehyde derivative followed by ring cleavage and periodate oxidation were unsuccessful. The p-nitrobenzaldehyde derivative was recovered unchanged after treatment with sodium periodate in 90% aqueous methanol. Mass spectrometry of the acetone and p-nitrobenz- aldehyde derivatives disclosed in each case that ring closure with the carbonyl reagent involved the amino and primary hydroxyl groups to give D-ri b o -2-dimethyl-4-( 2,3-dihydro xyhe xadecyl ) oxazolidine and D-ribo-2-p-nitrophenyl-4-(2,3- dihydroxyhexadecyl) oxazolidine, respectively.
EXPERIMENTAL PROCEDURES
Materials and Methods. Hansenula ciferrii, mating type F-60-10, strain NRRL-Y-1031 was provided by L.J. Wickerham. Silicic acid, acetyl cellulose and Adsorbosil-1 were products of Mallinkrodt, Woelm and Applied Science Lab, respectively. The silicic acid was washed with chloroform:methanol (2:1) and activated by heating 24 hr at 120 C. All solvents were dis- tilled. TLC was conducted as previously described (8) on plates coated with Adsorbo- sil-1 and developed with chloroform-methanol (95:5) or chloroform-methanol-2M NH4OH (40:10:1) (9). Bands were detected by exposure to iodine vapor. The trimethylsilyl derivatives of the bases were analyzed by GLC (10) on 2.5% SE-30 on Gas Chrom Q, mesh 100/200. The column, injector and detector temperatures were maintained at 210 C, 270 C and 240 C, respectively. IR spectra were obtained on KBr discs with a Perkin Elmer IR Spectrophotometer. High resolution mass spectrometry was performed by Gollob Analytical Service.
Isolation of N-Acetyl Bases. Each of ten 1 liter Erlenmeyer flasks containing 500 ml of medium (2) was inoculated with the growth from a single slant and grown 72 hr at room temperature under vigorous aeration. The cells were collected by centrifugation and washed once with 300 ml of cold isotonic saline. To the cells suspended in methanol, 25 g]100 ml, was added 1 ml of 10 N KOH per 100 ml of sus- pension which was stirred magnetically 4 hr at room temperature. After standing overnight, the suspension was centrifuged and the precipi- tate was washed by centrifugation three times with 200 ml portions of methanol. The com- bined supernatant solutions, after addition of several drops of Dow antifoam A, were con- centrated below 55 C, on a water pump to approximately 150 ml. An equal volume of water was added and the solution was treated three times with 200 ml portions of ether. The
combined ether extracts were washed with water until neutral and the residue, after ether removal, was dried over P2Os and crystallized from 35 ml of acetonitrile; yield of crude N- acetyl bases from 52 g of dry cells, 780 mg (range 10 to 22 mg/g of dry cells). TLC showed N-acetylphytosphingosine and N-acetyldihydro- sphingosine with trace amounts of phyto- sphingosine and dihydrosphingosine; less polar impurities appeared at the front.
Purification of N-Acetyl Bases. A 30 g silicic acid column, 2.5 x 50 cm, packed from chloro- form, was loaded with 500 mg of crude N- acetyl bases in 30 ml of warm chloroform. The column was developed with 125 ml each of chloroform and methanol. The chloroform eluate was discarded and the residue obtained from concentration of the methanol eluate was crystallized from acetonitrile; yield 360 mg (range 67% to 74%).
Preparation of Free Bases. Purified N-acetyl bases, 720 mg, were refluxed 7 hr in 18 ml of ethanol and 2 ml of 2N aqueous KOH. An equal volume of water was added and the reaction mixture was treated three times with 30 ml portions of ether. After washing the com- bined ether extracts with water, the residue, obtained after removal of the solvent, was dried and washed by suspension in 35 ml of hot petroleum ether. Although further purification was unnecessary, they could be purified by application in 2% methanol in chloroform to a 20 g silicic acid column and developed with 125 ml each of 5% methanol in chloroform and methanol. The former eluate was discarded and the product was obtained by concentration of the methanol eluate; yield 544 mg (range 85% to 90%). The base mixture consisted of about 95% phytosphingosine and 5% dihydro- sphingosine as determined by GLC.
P-Nitrobenzaldehyde Derivative of Phyto- sphingosine. To 318 mg of free bases in 35 ml of warm petroleum ether, bp 68-74 C (Skelly- solve B), was added 164 mg of p-nitrobenz- aldehyde; the reaction mixture was heated until a clear solution resulted. After cooling to room temperature, the precipitate was collected by centrifugation and resuspended in hot petro- leum ether. The product obtained from the cool reaction mixture was dried, dissolved in 20 ml of benzene and applied to an 8.0 g acetyl cellulose column, 1.5 x 50 cm, which was packed in benzene and washed successively with 3 column volumes each of ethanol and benzene. After development with 125 ml of benzene the residue, obtained from concen- tration of the eluate, was dried and resuspended in hot petroleum ether. The cool suspension was centrifuged and the precipitate dried;yield
LIPIDS, VOL. 5, NO. 9

784 BENJAMIN WEISS AND RICHARD L. STILLER
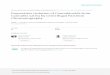
I 253
I 223
I I I - m 3 I I t i I 1 t
CH3 (CH2) 13 - C H - C H - C H - C H 2 CH3(CH2) 13 - I I I I OH OH HN 0
\ c / / \
02NC6H 4 H
160
I I 130
I I r - ' ~ 1 7 6 I I I I
C H - C H - C H - C H 2 I I I I
HN 0 OH OH \ / / \
CH 3 CH 3
A [3 FIG. 1. The mass spectra of the p-nitrobenzaldehyde (A) and acetone (B) derivatives of phyto-
sphingosine showed major peaks at m/e 193 and 100, respectively, which indicated participation of the amino and primary hydroxyl groups with the carbonyl reagent to give a substituted oxazolidine. See text for details.
356 mg (range 82% to 86%); mp 99-101 C. TLC revealed only one component, R F 0.9; GLC showed no dihydrosphingosine.
Analysis. Calculated for C25 H42 O5 N2 (450.3): C, 66.61; H, 9.40; O, 17.76; N, 6.22; Active H, 0.67. Found: C, 66.70; H, 9.50: O, 18.01 ; N, 6.26; Active H, 0.64.
Acetone Derivative of Phytosphingosine. Free bases, 318 mg, in 10 ml of acetone was heated to reflux, after cooling to room temper- ature, the acetone was removed under a stream of N 2 below 30 C. The dried residue was applied in 20 ml of hot benzene to an 8.0 g acetyl cellulose column. After washing the product into the column with several hot 15 ml portions of benzene, the column was developed with another 100 ml of benzene. The eluate was concentrated to dryness and the dried residue was crystallized from petroleum ether; yield 242 mg (range 70% to 74%); mp 102-104 C. TLC and GLC disclosed only one component.
Analysis . Calculated for C21H43NO 3 (357.3); C, 70.53; H, 12.12; O, 13.43; Active H, 0.84. Found: C, 70.08; H, 12.00; O, 13.66; Active H, 0.79. The acetone derivative of phytosphingosine obtained from a chilled acetone solution of the mixed bases melted at 105-107 C in agreement with that reported previously (7).
Regeneration of Phytosphingosine as Sulfate Salt. The p-nitrobenzaldehyde derivative, 180 rag, in 9.0 ml of 90% methanol and 1.0 ml of 0.5N aqueous sulfuric acid was heated to reflux. After cooling to room temperature, 10
ml of acetonitrile were added and the reaction mixture was centrifuged. The precipitate was washed successively by centrifugation with 3 ml portions of acetonitrile, two times, and once with ethanol. After drying, the precipitate was washed with 10 ml of hot petroleum ether; yield 127 mg (range 85% to 89%). GLC of the trimethylsilyl derivative formed directly from the sulfate salt showed only phytosphingosine, retention time 39 min.
Analysis. Calculated for C36Hs0N2Olo S (732.6); C, 58.96; H, 11.00; N, 3.82;O, 21.83; S, 4.37. Found: C, 58.52; H, 10.83; N, 4.02; O, 21.68; S, 4.34. The acetone derivative was treated in the same manner to yield the base sulfate.
Structure Proof of Derivatives of Phyto- sphingosine. Mass spectrometric analysis of the p-nitrobenzaldehyde derivative showed a major peak at m/e 193 which is the mass of a 5- membered ring (Fig. 1,A); this eliminated the hydroxyl on carbon atom 4 as a participant which would yield a 6-membered ring. If the hydroxyl on carbon atom 3 were involved, the ring mass would be 192 and the expected major peak would be at m/e 223 with smaller peaks at m/e 192 and 253. Since the fragment ion at m/e 193 was the largest with 223 and 253 progressively smaller, it was c ncluded that the terminal hydroxyl partici!,ated in ring formation. Similarly, the largest peak at m/e 100 corresponded to a 5-membered ring which involved the primary hydroxyl group of the acetone derivative (Fig. 1,B) with smaller peaks at m/e 130 and 160.
LIPIDS, VOL. 5, NO. 9

PREPARATION OF PHYTOSPHINGOSINE 785
ACKNOWLEDGMENT
This investigation was supported in part by Public Health Service Research Grant NB 06300-04 from the National Institutes of Neurological Diseases and Stroke.
REFERENCES
1. Greene, M.L., T. Kaneshiro and J.H. Law, Bio- chim. Biophys. Acta 98:582 (1965).
2. Wickerham, L.J., and F.H. Stodola, J. Bacteriol. 80:484 (1960).
3. Stodola, F.H., L.J. Wickerham, C.R. Scholfield and H.J. Button, Arch. Biochem.'Biophys. 98:176 (1962).
4. Stodola, F.H., and L.J. Wickerham, J. Biol. Chem. 235:2584 (1960).
5. Barenholz, Y., and S. Gatt, Biochim. Biophys. Acta 152:790 (1968).
6. Reindel, F., A. Weickmann, S. Picard, K. Luber and P. Turula, Ann. Chem. 544:116 (1940).
7. Carter, H.E., W.D. Celmer, W.E.M. Lands, K.L. Mueller and H.H. Tomizawa, J. Biol. Chem. 206:613 (1954).
8. Weiss, B., and R.L. Stiller, J. Lipid Res. 6:159 (1965).
9. Sambasivarao, K., and R.H. McCluer, Ibid. 4:437 (1963).
10. Carter, H.E., and R.C. Gaver, J. Lipid Res. 8:391 (1967).
~Received May 19, 19701
LIPIDS, VOL. 5, NO. 9