Embed Size (px)
Citation preview

ARTICLESPUBLISHED ONLINE: 13 JUNE 2010 | DOI: 10.1038/NMAT2778
A self-assembly pathway to alignedmonodomain gelsShuming Zhang1†, Megan A. Greenfield2†, Alvaro Mata3‡, Liam C. Palmer4, Ronit Bitton3,Jason R. Mantei1, Conrado Aparicio3, Monica Olvera de la Cruz1,2,3,4 and Samuel I. Stupp1,3,4,5*
Aggregates of charged amphiphilic molecules have been found to access a structure at elevated temperature that templatesalignment of supramolecular fibrils over macroscopic scales. The thermal pathway leads to a lamellar plaque structure withfibrous texture that breaks on cooling into large arrays of aligned nanoscale fibres and forms a strongly birefringent liquid. Bymanually dragging this liquid crystal from a pipette onto salty media, it is possible to extend this alignment over centimetresin noodle-shaped viscoelastic strings. Using this approach, the solution of supramolecular filaments can be mixed with cellsat physiological temperatures to form monodomain gels of aligned cells and filaments. The nature of the self-assemblyprocess and its biocompatibility would allow formation of cellular wires in situ that have any length and customized peptidecompositions for use in biological applications.
Inspired largely by biological systems, molecular self-assemblycontinues to be a theme of great interest in science. Thetargets differ broadly, from accessing ordered materials and
self-assembling devices1–3 to understanding howmisfolded proteinsself-assemble into stable fibres linked to human disease4,5. Long-range alignment of extracellular fibrils and cells in the heart6,brain and spinal cord7 must involve highly complex self-assemblymechanisms that remain largely unknown. Access to similar three-dimensional (3D) artificial systems of aligned fibrils and cells istherefore of scientific and biomedical interest8–10. Spontaneouslong-range alignment of molecules is known to occur in liquidcrystals11 but its fixation in the solid state normally requireschemical reactions12,13 that are not likely to be compatible withliving cells. Electrospinning of polymers faces similar challengesbecause it requires the use of high mechanical and electricalenergies14 that are not highly compatible with encapsulation ofliving cells. We report the discovery of a thermal pathway thatleads highly designable peptide-based small molecules in waterto form two-dimensional (2D) plaques with filamentous texturethat spontaneously template long-range alignment of bundlednanofibres on cooling. This liquid crystal of supramolecularfilaments can be mixed with cells at physiological temperaturesand drawn by hand from a pipette into salt solutions to formmonodomain gels of aligned filaments. Cells remain viable duringthe process and the monodomain gels can be drawn to arbitrarylengths and geometrical contours. We hypothesize that divalentions and slow relaxation times of the long nanofibre bundlesgenerated by this self-assembly pathway enable formation of themacroscopic monodomains.
We prepared 0.5–1.0 wt% aqueous solutions of peptideamphiphiles known to self-assemble into high-aspect-rationanofibres15,16. One peptide amphiphile molecule investigatedcontains the peptide sequence V3A3E3(CO2H) and a C16 alkyl tail
1Department of Materials Science and Engineering, Northwestern University, Evanston, Illinois 60208, USA, 2Department of Chemical and BiologicalEngineering, Northwestern University, Evanston, Illinois 60208, USA, 3Institute for BioNanotechnology in Medicine, Northwestern University, Chicago,Illinois 60611, USA, 4Department of Chemistry, Northwestern University, Evanston, Illinois 60208, USA, 5Department of Medicine, NorthwesternUniversity, Chicago, Illinois 60611, USA. †These authors contributed equally to this work. ‡Present address: Nanotechnology Platform, Parc Cientific,08028 Barcelona, Spain. *e-mail: [email protected].
at the peptide’s N-terminus, and its self-assembly into nanofibresis triggered by ions that screen the charged amino-acid residues,resulting in the formation of gels. The diameter of these nanofibres,which contain β-sheets near their hydrophobic core, is roughlyequivalent to the length of two peptide amphiphile moleculesand lengths in excess of micrometres. We heated the aqueoussolutions unscreened by added ions to 80 ◦C and kept them atthis temperature for 30min before cooling to 25 ◦C. After thisheat treatment, the solution viscosity increased threefold from 5to 15 cP. When calcium chloride was added to the heated andcooled peptide amphiphile solution, we observed the formationof a gel that was at least fourfold stiffer than one formed froman unheated solution (see Supplementary Information). Usingpolarized optical microscopy, we also found that gels or filmsformed from heated solutions contained large birefringent domains(tenths ofmillimetres; Fig. 1), whereas those formed fromunheatedsolutions appeared completely isotropicwith no birefringence.
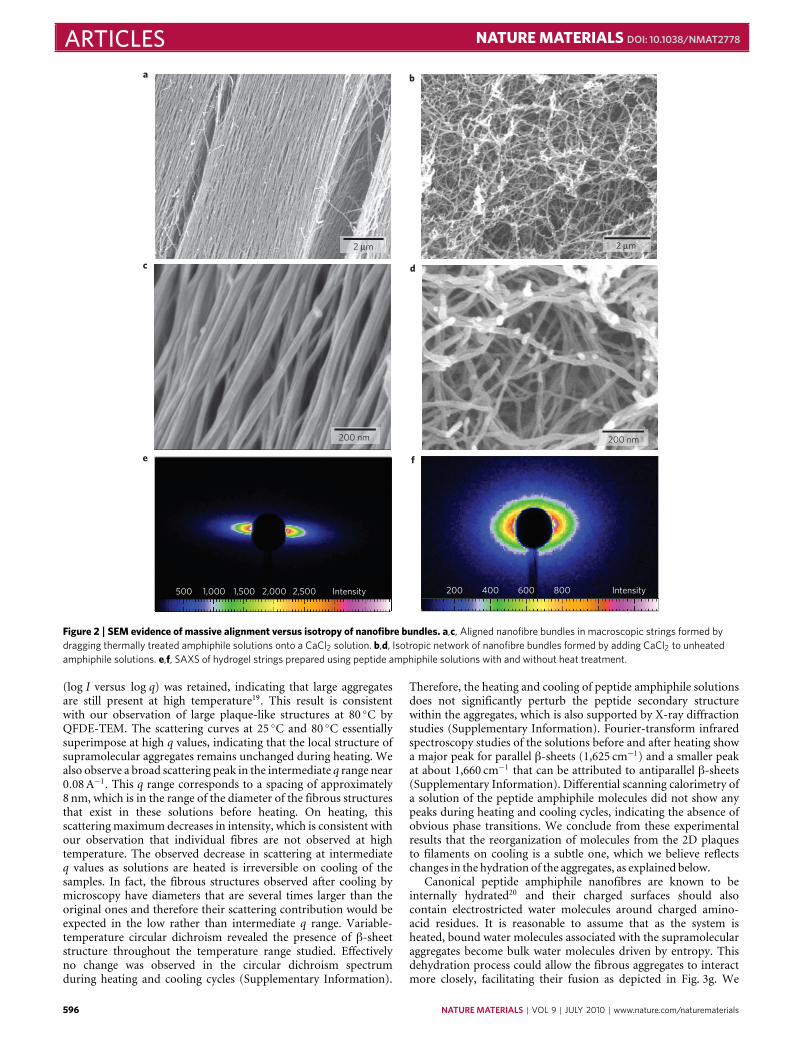
We observed that noodle-like strings of arbitrary length couldbe formed by manually drawing the aqueous peptide amphiphilesolution into a salty medium from a pipette (Fig. 1a,b). Whenthe solution was dragged on a surface covered by a thin layer ofthis medium (Fig. 1c), uniform birefringence was observed alongthe length of the string (Fig. 1h,i). This observation suggested thatmacroscopic alignment extending over centimetres was achieved.Using the same methods, the unheated solutions did not formmechanically stable string gels or show any birefringence. Scanningelectron microscopy (SEM) indicated that strings formed fromheated peptide amphiphile solutions contained extraordinarilylong arrays of aligned nanofibre bundles (Fig. 2a,b). In greatcontrast, unheated peptide amphiphile solutions formed matricesof randomly entangled nanofibres (Fig. 2d,e). To verify thisorientational order, we carried out small-angle X-ray scattering(SAXS) experiments and found that only strings generated from the
594 NATURE MATERIALS | VOL 9 | JULY 2010 | www.nature.com/naturematerials

NATURE MATERIALS DOI: 10.1038/NMAT2778 ARTICLESa
d e f
g h i
b c
5 mm 5 mm 5 mm
2 mm
4 mm
200 µm
200 µm 100 µm
200 µm
Figure 1 | Strings and gels with long-range internal alignment. a,b, A peptide amphiphile solution coloured with trypan blue injected intophosphate-buffered saline after heat treatment. c, The same solution dragged through a thin layer of aqueous CaCl2 to form a noodle-like string. d, A knotmade with peptide amphiphile string. e, Birefringence of a bubble gel observed between cross polars suggesting the presence of macroscopically aligneddomains. f, Similar domains in a gel film. g, Peptide amphiphile noodle spirals prepared on a spin coater. h, Birefringence of a single string suggestingalignment along the string axis. i, Light extinction between cross polars at the crosspoint of two noodles demonstrating uniform alignment in each.
heated solutions revealed alignment (Fig. 2c,f). In contrast, simplydragging a peptide amphiphile solution that had not been heateddoes not lead to significant alignment. Similar strings can be madewith other peptide amphiphile molecules, including those withbioactive epitopes, although their structural integrity depended onthe amino-acid sequence (Supplementary Information).
To gain a mechanistic understanding of the observed trans-formations, we examined the effects of heating on the peptideamphiphile solution structure by quick-freeze/deep-etch (QFDE)transmission electron microscopy17 (TEM). QFDE is a samplepreparation technique that allows high-resolution imaging of hy-drated structures while minimizing disruption of the sample result-ing from fixation or processing. The freshly dissolved peptide am-phiphile solution contained a variety of nanoscale, elongated objectsless than amicrometre in length (Supplementary Information).Mi-crographs of the peptide amphiphile solution equilibrated at 80 ◦Cfor 30min showed that the small aggregates largely disappeared; in-stead we observed thin ‘plaque-like’ structures up tomicrometres inboth length and width (Fig. 3a). Some portions of these 2D plaqueshad a periodic surface texture with a characteristic spacing of about7.5 nm, which corresponds to the expected diameter of a single
canonical nanofibre formed by the peptide amphiphile moleculesused15,18 (Fig. 3b). After cooling to room temperature, solutionswere clearly composed of aligned filaments (Fig. 3d). The filamentsdid not have the diameter of canonical nanofibres (7–8 nm) butwere instead tens of nanometres in diameter, and are thereforebundles of many cylindrical nanofibres. To further visualize the 3Dstructure of the plaque, we fixed the structure by adding calciumchloride at 80 ◦C and imaged the resulting structure by SEM.Although most of the sample cooled to ambient temperature wascomposed of large arrays of aligned nanofibres, some plaques werecaptured as well (Fig. 3e). These plaques measured about 40 nm inthickness and had lengths and widths comparable to those observedby QFDE-TEM. They often contained long parallel striations and,in some cases, appeared to be cracking into fibre bundles (Fig. 3f).These plaque structures were not observedwithout heating.
We explored the effect of heating on structure in peptideamphiphile solutions using SAXS, circular dichroism, differentialscanning calorimetry and Fourier-transform infrared spectroscopy.The X-ray scattering curve of peptide amphiphile solutions at 25 ◦C(Fig. 3c) shows a q−4 dependence within the low q range, indicativeof the presence of large aggregates. On heating to 80 ◦C, the−4 slope
NATURE MATERIALS | VOL 9 | JULY 2010 | www.nature.com/naturematerials 595
ARTICLES NATURE MATERIALS DOI: 10.1038/NMAT2778
a b
d
f
c
e
Intensity500 1,000 1,500 2,000 2,500 Intensity200 400 600 800
2 µm 2 µm
200 nm 200 nm
Figure 2 | SEM evidence of massive alignment versus isotropy of nanofibre bundles. a,c, Aligned nanofibre bundles in macroscopic strings formed bydragging thermally treated amphiphile solutions onto a CaCl2 solution. b,d, Isotropic network of nanofibre bundles formed by adding CaCl2 to unheatedamphiphile solutions. e,f, SAXS of hydrogel strings prepared using peptide amphiphile solutions with and without heat treatment.
(log I versus log q) was retained, indicating that large aggregatesare still present at high temperature19. This result is consistentwith our observation of large plaque-like structures at 80 ◦C byQFDE-TEM. The scattering curves at 25 ◦C and 80 ◦C essentiallysuperimpose at high q values, indicating that the local structure ofsupramolecular aggregates remains unchanged during heating. Wealso observe a broad scattering peak in the intermediate q range near0.08A−1. This q range corresponds to a spacing of approximately8 nm, which is in the range of the diameter of the fibrous structuresthat exist in these solutions before heating. On heating, thisscatteringmaximum decreases in intensity, which is consistent withour observation that individual fibres are not observed at hightemperature. The observed decrease in scattering at intermediateq values as solutions are heated is irreversible on cooling of thesamples. In fact, the fibrous structures observed after cooling bymicroscopy have diameters that are several times larger than theoriginal ones and therefore their scattering contribution would beexpected in the low rather than intermediate q range. Variable-temperature circular dichroism revealed the presence of β-sheetstructure throughout the temperature range studied. Effectivelyno change was observed in the circular dichroism spectrumduring heating and cooling cycles (Supplementary Information).
Therefore, the heating and cooling of peptide amphiphile solutionsdoes not significantly perturb the peptide secondary structurewithin the aggregates, which is also supported by X-ray diffractionstudies (Supplementary Information). Fourier-transform infraredspectroscopy studies of the solutions before and after heating showa major peak for parallel β-sheets (1,625 cm−1) and a smaller peakat about 1,660 cm−1 that can be attributed to antiparallel β-sheets(Supplementary Information). Differential scanning calorimetry ofa solution of the peptide amphiphile molecules did not show anypeaks during heating and cooling cycles, indicating the absence ofobvious phase transitions. We conclude from these experimentalresults that the reorganization of molecules from the 2D plaquesto filaments on cooling is a subtle one, which we believe reflectschanges in the hydration of the aggregates, as explained below.
Canonical peptide amphiphile nanofibres are known to beinternally hydrated20 and their charged surfaces should alsocontain electrostricted water molecules around charged amino-acid residues. It is reasonable to assume that as the system isheated, bound water molecules associated with the supramolecularaggregates become bulk water molecules driven by entropy. Thisdehydration process could allow the fibrous aggregates to interactmore closely, facilitating their fusion as depicted in Fig. 3g. We
596 NATURE MATERIALS | VOL 9 | JULY 2010 | www.nature.com/naturematerials

NATURE MATERIALS DOI: 10.1038/NMAT2778 ARTICLES
7.5 nm
0.01
0.10
a
c
e
g
i
h
j
f
d
b
I (cm
¬1 )
(a.
u.)
q (Ŭ1)0.100.01
25 °C before heating
80 °C for 2 min
25 °C after heating
40 nm
200 nm
100 nm500 nm
100 nm
200 nm
Figure 3 | Morphological changes resulting from thermal treatment. a, TEM obtained after a QFDE preparation of peptide amphiphile solution at 80 ◦Crevealing a micrometre-sized, sheet-like plaque structure. b, Higher-resolution QFDE-TEM of the sheet-like structures revealing a surface pattern with aperiodicity of about 7.5 nm. c, SAXS of peptide amphiphile solutions treated at different thermal conditions. d, QFDE-TEM of aligned nanofibre bundlestemplated by the sheet-like plaque after the peptide amphiphile solution was cooled to room temperature. e, SEM of plaques that were captured by addingCaCl2 at 80 ◦C. f, SEM of a plaque breaking into nanofibre bundles. g,h, Schematic representation of a plaque at high temperature (g) and its rupture intofused bundles on cooling (h). i,j, Schematic representation of the cross-section of a plaque formed by fused fibres (i) and of a fibre bundle (j).
believe the observed flat and textured plaques are the result ofthese fused, dehydrated fibres. During cooling, water moleculesshould simply rehydrate the supramolecular structures, but thisprocess actually generates filaments with diameters that are several
times greater than those of canonical nanofibres that exist beforethe thermal treatment (see the schematic in Fig. 3h). On thebasis of microscopy, the plaque appears to break into nanofibrebundles with diameters of approximately 40 nm (Fig. 2c) rather
NATURE MATERIALS | VOL 9 | JULY 2010 | www.nature.com/naturematerials 597

ARTICLES NATURE MATERIALS DOI: 10.1038/NMAT2778
than individual canonical fibres with diameters of about 8 nm.The diameter of the bundles corresponds to the plaque’s thicknessobserved by SEM (Fig. 3e).
The possibility of a transition from the plaque to a fused nanofi-bre bundle can be understood by computing the contributions tothe difference in free energy per thermal energy kBT per amphiphilefor a cylindrical fibre F f and a lamella F l,
F f−F l= (Fef−Fel)− (Fcf−Fcl)
where Fef and Fel are the electrostatic free energies of a fibre and alamella, respectively, and Fcf and Fcl are the cohesive free energiesof molecules in these two different morphologies. The fractionof ions condensed on the surface of peptide amphiphile fibresand plaques is estimated from the modified Poisson–Boltzmannequation21, which shows that the charges are neutralized bycounterions for both the plaque and the fibre (see SupplementaryInformation). Therefore, the transition should be dominated by thedifference in cohesive energies of lamellae and fibres,1Fc=Fcf−Fcl,which is given by,
1Fc=1HPA
kBT−[1SPA+1Swater]
where 1HPA is the enthalpy difference per peptide amphiphilemolecule between lamellar and fibre aggregates, and 1SPA and1Swater are the entropy differences of the peptide amphiphileand water molecules, respectively. As the β-sheet signature in thecircular dichroism spectrum does not change significantly duringcooling, we assumed that the internal energy of the β-sheet is similarin the fibre and the plaque. Therefore, the enthalpy differencebetween lamellae and fibres must originate from the coupling ofinteractions among peptide segments and hydrophobic tails. Thiscoupling is supported by our previous spectroscopic experimentsthat showed order can exist in the hydrophobic core of peptideamphiphile nanofibres and is enhanced by β-sheet orientationalong the fibre axis22. The fibre architecture could also optimizeinteractions among peptide segments and alkyl segments. Weestimate1HPA to be dominated by van der Waals forces, which areof the order of thermal energy. However, the increase in entropyof the peptide amphiphile and water molecules in the plaquestate can offset the enthalpy difference at elevated temperature.Specifically, the higher entropy in the plaque can originate ingreater translation of water molecules (1Swater), because there isless water interface per peptide amphiphile molecule than in thefibre structure, and therefore fewer restricted water molecules peramphiphile. This would reasonably predict a transition temperaturefrom fibrous to planar assemblies of the peptide amphiphiles (seeSupplementary Information).
The observed rupture of the plaque at lower temperatures intobundles of fibres that give rise to an aqueous lyotropic liquid crystalat an unusually low concentration suggests an unusual mechanismof membrane rupture. The plaque observed by QFDE-TEM at80 ◦C reveals a periodic surface texture with a characteristic spacingof about 7.5 nm, which corresponds to the expected diameter ofa single canonical nanofibre15. This strongly suggests the plaqueresults from the fusion of nanofibres as the dehydration occurs atelevated temperature. Themicroscopy also revealed the existence ofripples in the plaque of larger dimension than the fibres (Fig. 3a,b).We propose that fluctuations of the anisotropic plaque structurewith 1D fibrous texture are crucial for its metamorphosis intoarrays of highly aligned nanofibres. It is known that typicallyonly membranes in curved geometries such as cylinders breakby Rayleigh instabilities23; flat membranes generally rupture bycreating holes24. Therefore, the possible breaking mechanism of aplaque into bundled fibres that gives rise to a lyotropic liquid crystalis unique given its underlying anisotropic 1D substructure imparted
by the nanofibres composed of β-sheets. Long-range forces havebeen proposed to cause the rupture of surfactant membranes bymeans of concentration fluctuations25–27. The fibrous texture on thesurface, however, generates an anisotropic surface tension, whichmay lead to the formation of waves of fluctuating compositionon the surface similar to binary immiscible lipid membranes28.The waves resulting from the membrane tension appear as surfaceripples when the β-sheets align, and this may generate theconcentration of fluctuations required for rupture. However, thesize of the successful composition fluctuation has to be largeenough (larger than the membrane thickness D) to generate acritical size of a neck for rupture; otherwise the strain generatedby the composition fluctuations in the internal structure (theinterpenetrated bilayers) opposes the growth of the fluctuation29,and the lateral composition fluctuations are restored. One canassume that overall the breaking of the surface is due to a decrease inthe overall surface tension γ , which is the change of free energy (F)as the interface area (A) increases, ∂F/∂A. That is, the bundled-fibresurface tension γf is lower than that of the lamellar γl becauseof the increase of hydration and order of peptide amphiphilemolecules within the bundle. Unfortunately, the linear theory thatassumes γ is constant under a deformation of the interface is notappropriate to describe the breaking of a lamella. To a first-orderapproximation one can assume that the interfacial energy of thefluctuation that leads to the rupture of the plaque is of the order ofthe γf. Consider fluctuations perpendicular to the lamellar surfaceplane (x,y) described by a function h(x,y), which induce a decreasein γl–γf that may result in the rupture of the plaque. To inducerupture, the resulting free-energy change1F=Ff−F , is negative, or1F<0,whereFf is the free energy of the fluctuating plaque given by
Ff= (1/2)∫
dxdy γf(1+hx 2+hy 2)1/2 (1)
where hx = ∂h/∂x and hy = ∂h/∂y , and F the flat-plaque freeenergy is given by,
F = (γl/2)∫
dxdy
If we assume a 1D fluctuation along x of maximum amplitudeh0, which is half the thickness of the lamella D (h0 = D/2) andwavelength λ, then
h= h0exp(i2πx/λ) (2)
Therefore, by approximating (1+ hx 2+ hy 2)1/2 in equation (1) as(1+ hx 2/2) and assuming γf is a constant in the integrant of Ff,we find in equation (2) that after the integration of both Ff and F ,breaking occurs if1F/A6 0, or
1γ/2+γf(2πh0/λ)2/46 0 (3)
where1γ =γf−γl<0 andA is the total area of the plaque of volumeV = AD. Equation (3) gives a bound for the characteristic size λcabove which fluctuations lead to rupture, given by (2πh0/λc)2 =−21γ/γf or λc = h0π(2γf/(γl− γf))1/2; that is, all λ> λc lead torupture and all λ<λc would lead to stable plaques. As h0=D/2 atrupture and there is no long-range diffusion in the rupture process,λc is the most probable size for rupture, and, given that γf/(γl−γf)is of the order of one (see Supplementary Information), λc is of theorder of themembrane thicknessD, in agreement with the expectedsize of the breaking of a cylinder through aRayleigh instability.
It has been shown previously that liquid crystals can bealigned with elongational flow30. Theoretical models suggest thatduring uniaxial stretching of polymeric nematics, if the productof strain rate ε̇ and the conformational relaxation time λ isgreater than unity, one can expect high degrees of uniaxial
598 NATURE MATERIALS | VOL 9 | JULY 2010 | www.nature.com/naturematerials

NATURE MATERIALS DOI: 10.1038/NMAT2778 ARTICLES
∗ ∗ ∗
a b
c d
e f
0 ms
80 ms
160 ms
240 ms
320 ms
400 ms
500 μm
500 nm2 μm
150 μm 250 μm
1 cm
2,000 ms
1
2
3
Figure 4 | Cell alignment in strings of aligned filaments. a, Preferential alignment of encapsulated hMSCs along the string axis. b, Calcein-labelled alignedcells cultured in string. c, SEM images at different magnifications of a single cell in a string (inset is the zoom-out view; the arrow indicates the alignmentdirection). d, A conductive black string formed by dispersing carbon nanotubes in peptide amphiphile solutions before heating. The SEM micrograph on theright shows aligned nanofibre bundles in the black string. e, Top: calcium fluorescence image of HL-1 cardiomyocytes encapsulated in a noodle-like string.Below: successive spatial maps of calcium fluorescence intensity travelling at 80-ms intervals, showing the propagation of an electrical signal throughoutthe entire string and demonstrating a functional cardiac syncytium. f, Calcium fluorescence intensity signal in time at three points in the string marked bycolour in e, showing repeated propagating action potentials.
orientation31. In the context of this model, the product (knownas the Weissenberg number) would be greater than unity forthermally treated peptide amphiphile gel strings and less thanunity for gel strings formed with unheated samples. As we usedthe same longitudinal strain rate for both samples (Fig. 2c,d),the aggregates in thermally treated solutions must have longerrelaxation times. Thus, we hypothesize the formation of largerdiameter supramolecular bundles derived from the rupture of theplaque enables the fixing of macroscopic orientation over arbitrarylength in hand-drawn string gels. It is also interesting to note thatthe low strain rate used here (1–4 s−1) is ideal for supramolecularaggregates, because their non-covalent interactions may not bestable in some cases at the large strain rates commonly used in
electrospinning of polymers (104–105 s−1; ref. 32). The mechanismproposed here is fundamentally different from that observed ingel spinning of polymers33 and the polymerization of monomersin heated liquid-crystalline solution34. Although these strings wereproduced by hand drawing, this methodology could be extended toautomated printing technology.
We used the strings of aligned peptide amphiphile nanofibres todirect the orientation of cells in 3D environments. After dispersinghuman mesenchymal stem cells (hMSCs) in heated and cooledpeptide amphiphile solutions, we manually dragged these solutionsonto salty media (160mM NaCl and 5–10mM CaCl2) to formnoodle-shaped strings with encapsulated stem cells. These cellsremain viable during the process of string formation and start
NATURE MATERIALS | VOL 9 | JULY 2010 | www.nature.com/naturematerials 599

ARTICLES NATURE MATERIALS DOI: 10.1038/NMAT2778
to elongate along the string director axis within 12 h. Optical,fluorescence and electron microscopy (Fig. 4a–c) demonstratedthat both cell bodies and filopodia were aligned with peptideamphiphile nanofibre bundles in the extracellular space. Theseeffects on hMSCorientation are hypothesized to result from contactguidance along the preferentially orientedmatrix35. A ‘cellular wire’based on the noodle-like string could also serve as a bridge to directcells spatially for function or migration from one site to another;current methods to generate long-range 3D alignment of a syn-thetic, fibrous matrix over arbitrary lengths in the presence of livingcells are limited. To demonstrate further biological applications ofthe noodle construct, we formed a string in the presence of HL-1cardiomyocytes, a cell line with spontaneous electrical activity thatrequires extensive cell–cell contacts to propagate signals, and thenobserved the cellular behaviour over time36. The cells survived andproliferated extensively to fill the entire structure. By fluorescentlyvisualizing intracellular calcium concentration, we observed pock-ets of spontaneous electrical activity by day 6 and action potentialsthat were able to propagate throughout the entire macroscopicstructure by day 10 (Fig. 4e). Plots of the calcium fluorescence overtime show these propagating signals, demonstrating the presenceof a continuous functional cardiac syncytium (Fig. 4f). This findingsuggests potential for these ‘cellular wires’ in regenerative medicineapplications in which restoring electrical communication in tissuesis of critical importance, for example, in the treatment of cardiacarrhythmia or spinal cord injury. In addition to this ability topropagate electric signals, it is clear that the strong cell alignmentof the constructs, as found in muscle and nervous tissue, will alsobe helpful in developing regenerative strategies. In another examplewe created amonodomain peptide amphiphile string gel containingdispersed carbon nanotubes that retained the same degree of align-ment as in pure peptide amphiphile strings (Fig. 4d). These blackstrings had electrical conductivities of the order of 1–10 S cm−1 ina dry state, depending on the concentration of carbon nanotubes.The incorporation of carbon nanotubes into the structure withoutdisrupting the final construct suggests the use of the methodologyto align 1Dnanostructures within biocompatiblematrices.
We found in this work that supramolecular architectures ofself-assembling molecules can markedly change in a pathway-dependent manner. We specifically discovered a thermal pathwaythat converts isotropic solutions of peptide-containing moleculesto liquid crystals in which molecules group into long filamentsof bundled nanofibres. This transformation allows the formationof monodomain fibrous gels, which may contain viable cells aswell. These systems offer the potential to study phenomena inmacroscopically aligned arrays of cells and could also lead tothe development of therapies that require directed cell migration,directed cell growth or the spatial cell interconnections in tissuessuch as heart, brain and spinal cord.
Received 3 August 2009; accepted 29 April 2010;published online 13 June 2010
References1. Whitesides, G. M., Mathias, J. P. & Seto, C. T. Molecular self-assembly and
nanochemistry—a chemical strategy for the synthesis of nanostructures.Science 254, 1312–1319 (1991).
2. Reches, M. & Gazit, E. Casting metal nanowires within discrete self-assembledpeptide nanotubes. Science 300, 625–627 (2003).
3. Kim, S. O. et al. Epitaxial self-assembly of block copolymers on lithographicallydefined nanopatterned substrates. Nature 424, 411–414 (2003).
4. Nelson, R. et al. Structure of the cross-beta spine of amyloid-like fibrils.Nature435, 773–778 (2005).
5. Dobson, C. M. Protein folding and misfolding. Nature 426, 884–890 (2003).6. Chung, C. Y., Bien, H. & Entcheva, E. The role of cardiac tissue alignment in
modulating electrical function. J. Cardiovasc. Electr. 18, 1323–1329 (2007).7. Davies, S. J. A., Goucher, D. R., Doller, C. & Silver, J. Robust regeneration of
adult sensory axons in degenerating white matter of the adult rat spinal cord.J. Neurosci. 19, 5810–5822 (1999).
8. Feinberg, A. W. et al. Muscular thin films for building actuators and poweringdevices. Science 317, 1366–1370 (2007).
9. Merzlyak, A., Indrakanti, S. & Lee, S. W. Genetically engineered nanofiber-likeviruses for tissue regenerating materials. Nano Lett. 9, 846–852 (2009).
10. Bettinger, C. J., Langer, R. & Borenstein, J. T. Engineering substrate topographyat the micro- and nanoscale to control cell function. Angew. Chem. Int. Ed. 48,5406–5415 (2009).
11. Kato, T., Mizoshita, N. & Kishimoto, K. Functional liquid-crystallineassemblies: Self-organized soft materials. Angew. Chem. Int. Ed. 45,38–68 (2006).
12. Gin, D. L., Gu, W. Q., Pindzola, B. A. & Zhou, W. J. Polymerized lyotropicliquid crystal assemblies for materials applications. Acc. Chem. Res. 34,973–980 (2001).
13. Stupp, S. I. & Osenar, P. in Polymerization in Organized Media in Synthesis ofPolymers (ed. Schlüter, A. D.) (Wiley-VCH, 1999).
14. Greiner, A. & Wendorff, J. H. A fascinating method for the preparation ofultrathin fibres. Angew. Chem. Int. Ed. 46, 5670–5703 (2007).
15. Hartgerink, J. D., Beniash, E. & Stupp, S. I. Self-assembly and mineralization ofpeptide-amphiphile nanofibers. Science 294, 1684–1688 (2001).
16. Behanna, H. A., Donners, J. J. J. M., Gordon, A. C. & Stupp, S. I. Coassembly ofamphiphiles with opposite peptide polarities into nanofibers. J. Am. Chem. Soc.127, 1193–1200 (2005).
17. Ruberti, J. W. et al. Quick-freeze/deep-etch visualization of age-relatedlipid accumulation in Bruch’s membrane. Invest. Ophthalmol. Vis. Sci. 44,1753–1759 (2003).
18. Bull, S. R. et al. Magnetic resonance imaging of self-assembled biomaterialscaffolds. Bioconjugate Chem. 16, 1343–1348 (2005).
19. Glatter, O. & Kratky, O. (eds) Small Angle X-ray Scattering (Academic, 1982).20. Tovar, J. D., Claussen, R. C. & Stupp, S. I. Probing the interior of
peptide amphiphile supramolecular aggregates. J. Am. Chem. Soc. 127,7337–7345 (2005).
21. Cheng, H., Zhang, K., Libera, J. A., de la Cruz, M. O. & Bedzyk, M. J.Polynucleotide adsorption to negatively charged surfaces in divalent saltsolutions. Biophys. J. 90, 1164–1174 (2006).
22. Jiang, H. Z., Guler, M. O. & Stupp, S. I. The internal structure of self-assembledpeptide amphiphiles nanofibers. Soft. Matter. 3, 454–462 (2007).
23. Lenz, P. &Nelson, D. R. Hexatic undulations in curved geometries. Phys. Rev. E67, 031502 (2003).
24. Sandre, O., Moreaux, L. & Brochard-Wyart, F. Dynamics of transient pores instretched vesicles. Proc. Natl Acad. Sci. USA 96, 10591–10596 (1999).
25. De Wit, A., Gallez, D. & Christov, C. I. Nonlinear evolution equations for thinliquid films with insoluble surfactants. Phys. Fluids 6, 3256–3266 (1994).
26. Oron, A., Davis, S. H. & Bankoff, S. G. Long-scale evolution of thin liquid films.Rev. Mod. Phys. 69, 931–980 (1997).
27. Liu, Y. S., Li, M. H., Bansil, R. & Steinhart, M. Kinetics of phase transition fromlamellar to hexagonally packed cylinders for a triblock copolymer in a selectivesolvent.Macromolecules 40, 9482–9490 (2007).
28. Solis, F. J., Funkhouser, C. M. & Thornton, K. Conditions for overall planarityin membranes: Applications to multicomponent membranes with lamellarmorphology. Europhys. Lett. 82, 38001 (2008).
29. Mayes, A. M. & de la Cruz, M. O. Strain effects on the thermal stability of rodeutectics. Acta Metall. 37, 615–620 (1989).
30. Ide, Y. & Ophir, Z. Orientation development in thermotropic liquid crystalpolymers. Polym. Eng. Sci. 23, 261–265 (1983).
31. Larson, R. G. & Mead, D. W. The Ericksen Number and Deborah Numbercascades in sheared polymeric nematics. Liq. Cryst. 15, 151–169 (1993).
32. Reneker, D. H., Yarin, A. L., Fong, H. & Koombhongse, S. Bending instabilityof electrically charged liquid jets of polymer solutions in electrospinning.J. Appl. Phys. 87, 4531–4547 (2000).
33. Barham, P. J. & Keller, A. High-strength polyethylene fibres from solution andgel spinning. J. Mater. Sci. 20, 2281–2302 (1985).
34. Fujikake, H., Murashige, T., Sato, H., Kawakita, M. & Kikuchi, H. Molecularalignment enhancement phenomenon of polymer formed from a liquid crystalmonomer in a liquid crystal solvent. Appl. Phys. Lett. 82, 1622–1624 (2003).
35. Tranquillo, R. T. Self-organization of tissue-equivalents: the nature and role ofcontact guidance. Biochem. Soc. Symp. 65, 27–42 (1999).
36. Claycomb, W. C. et al. A cardiac muscle cell line that contracts and retainsphenotypic characteristics of the adult cardiomyocyte. Proc. Natl Acad. Sci. USA95, 2979–2984 (1998).
AcknowledgementsThis work was supported by the US Department of Energy-Basic Energy Sciences(DE-FG02-00ER45810, DE-FG02-08ER46539), National Institutes of Health(5-R01-EB003806, 5-R01-DE015920, 5-P50-NS054287), National Science Foundation(DMR-0605427), Department of Homeland Security Fellowship (M.A.G.),Non-Equilibrium Energy Research Center (NERC), an Energy Frontier Research Centerfunded by DOE-BES (award number DE-SC0000989 for L.C.P), NorthwesternUniversity’s NIH Biotechnology Training Program (pre-doctoral fellowship to J.R.M.),Ben Gurion University of Negev, Israel (post-doctoral fellowship for R.B.) and
600 NATURE MATERIALS | VOL 9 | JULY 2010 | www.nature.com/naturematerials

NATURE MATERIALS DOI: 10.1038/NMAT2778 ARTICLESGeneralitat de Catalunya (visiting scholar sponsorship for C.A.). Experiments made useof the following facilities at Northwestern University: J. B. Cohen X-ray DiffractionFacility, IMSERC, EPIC Facilities of the NUANCE Center, Keck Biophysics Facility,Biological Imaging Facility and the Institute for BioNanotechnology in Medicine and itsCleanroom Core Facility. The NUANCE Center is supported by the NSF-NSEC,NSF-MRSEC, Keck Foundation, the State of Illinois and Northwestern University. Weacknowledge facilities support by the Materials Research Center through NSF-MRSECgrant DMR-0520513. X-ray measurements were carried out at theDuPont–Northwestern–Dow Collaborative Access Team (DND-CAT) SynchrotronResearch Center located at Sector 5 of the Advanced Photon Source. DND-CAT issupported by the E. I. DuPont de Nemours, The Dow Chemical Company, the USNational Science Foundation through Grant DMR-9304725 and the State of Illinoisthrough the Department of Commerce and the Board of Higher Education Grant IBHEHECA NWU 96. Use of the Advanced Photon Source was supported by the USDepartment of Energy–Office of Basic Energy Sciences under ContractNo. W-31-109-Eng-38 and DE-AC02-06CH11357. Use of the BioCARS Sector 14 was
supported by the National Institutes of Health, National Center for Research Resources,under grant number RR007707. We also thank G. Darnell and S. Weigand for X-rayassistance and W. Claycomb for the generous gift of the HL-1 cells.
Author contributionsS.Z., M.A.G., A.M., L.C.P., R.B., C.A. and J.R.M. carried out experiments. M.A.G. andM.O.d.l.C. generated numerical data. M.A.G., M.O.d.l.C. and S.I.S developed atheoretical model. S.Z., M.A.G., L.C.P., R.B., J.R.M., M.O.d.l.C. and S.I.S. wrotethe paper.
Additional informationThe authors declare no competing financial interests. Supplementary informationaccompanies this paper on www.nature.com/naturematerials. Reprints and permissionsinformation is available online at http://npg.nature.com/reprintsandpermissions.Correspondence and requests for materials should be addressed to S.I.S.
NATURE MATERIALS | VOL 9 | JULY 2010 | www.nature.com/naturematerials 601





























