Embed Size (px)
Citation preview

ARTICLES
A Scalable Approach to Combinatorial Library Design for Drug Discovery
Puneet Sharma,*,†,§ Srinivasa Salapaka,‡,§ and Carolyn Beck†,§
Department of Industrial and Enterprise Systems Engineering, University of Illinois at Urbana Champaign,104 S. Mathews Avenue, Urbana, Illinois 61801, Department of Mechanical Science and Engineering,
1206 W. Green Street, Urbana, Illinois 61801, and Coordinated Science Laboratory, University of Illinois atUrbana Champaign, 1308 W. Main Street, Urbana, Illinois 61801
Received January 23, 2007
In this paper, we propose an algorithm for the design of lead generation libraries required in combinatorialdrug discovery. This algorithm addressessimultaneouslythe two key criteria of diversity and representativenessof compounds in the resulting library and is computationally efficient when applied to a large class of leadgeneration design problems. At the same time, additional constraints on experimental resources are alsoincorporated in the framework presented in this paper. A computationally efficient scalable algorithm isdeveloped, where the ability of the deterministic annealing algorithm to identify clusters is exploited totruncate computations over the entire data set to computations over individual clusters. An analysis of thisalgorithm quantifies the tradeoff between the error due to truncation and computational effort. Results appliedon test data sets corroborate the analysis and show improvement by factors as large as 10 or more, dependingon the data sets.
1. INTRODUCTION
In recent years, combinatorial chemistry techniques haveprovided some of the most important tools for assistingchemists in the exploration of huge chemical property spacesin search of new pharmaceutical agents. With the advent ofthese methods, a large number of compounds are accessedand synthesized using basic building blocks. Recent advancesin high-throughput screening approaches such as those usingmicro-/nanoarrays1 have given further impetus to large-scaleinvestigation of compounds for drug discovery. However,combinatorial libraries often consist of extremely largecollections of chemical compounds, typically several mil-lions. The time and cost associated with these experimentsmakes it practically impossible to synthesize each and everycombination from such a library of compounds. To overcomethis problem, chemists often work withVirtual combinatoriallibraries, which are essentially combinatorial databasescontaining an enumeration of all possible structures of agiven pharmacophore with all available reactants. They thenselect a subset of compounds from this virtual library ofcompounds and use it for physical synthesis and biologicaltarget testing. The selection of this subset is based on acomplex interplay between various objectives, which is castas a combinatorial optimization problem. To address thisproblem, computational optimization tools have been em-ployed to design libraries consisting of subsets of representa-tive compounds which can be synthesized and subsequently
tested for relevant properties, such as structural activity,bioaffinity, and aqueous solubility.
At the outset, the problem of designing such aleadgeneration library belongs to the class of combinatorialresource allocation problems, which have been widelystudied. These problems are typically cast as multiobjectiveoptimization problems with constraints. They arise in manydifferent areas such as minimum distortion problems in datacompression,2 facility location problems,3 optimal quadraturerules and discretization of partial differential equations,4
locational optimization problems in control theory,5,6 patternrecognition,7 neural networks,8 and clustering analysis.9 Theseproblems are nonconvex and computationally complex. It iswell-documented10 that most of them suffer from many localminima that riddle the cost surface. The feature thesecombinatorial resource allocation problems share is the goalof an optimal partition of the underlying domain togetherwith an optimal assignment of values from a finite set toeach cell of the partition. The distinguishing features of thisclass of problems come from having different conditions foroptimality and constraints, such as the distance metrics usedin the definition of coVerage, the number and types ofconstraints placed on the resources in the problem formula-tion, the necessity of computing global versus local optima,and the size and scale of the feasible domain. Thesedifferences add further problem-specific challenges to theunderlying combinatorial optimization problems.
The lead generation design problem is viewed as acombinatorial resource allocation problem since it requirespartitioning the underlying chemical property space spannedby compounds in the virtual library and ascribing a repre-sentative compound (i.e., a member of the lead generation
* Corresponding author phone: (217) 244 3364; e-mail: [email protected].
† Department of Industrial and Enterprise Systems Engineering.‡ Department of Mechanical Science and Engineering.§ Coordinated Science Laboratory.
27J. Chem. Inf. Model.2008,48, 27-41
10.1021/ci700023y CCC: $40.75 © 2008 American Chemical SocietyPublished on Web 12/06/2007

library) to each cell in the above partition. The main criterionfor constructing a lead generation library has traditionallybeen moleculardiVersity.11,12 As has been noted in theliterature,13,14 diversity as a sole criterion for selection canyield lead generation libraries with compounds that havelimited druglike properties, as this selection proceduredisproportionately favors outliers or atypical compounds.Though such a library contains maximally diverse com-pounds, it may be of little significance because of its limitedpharmaceutical applications. This drawback can be addressedby designing libraries that arerepresentatiVe of all potentialcompounds and their distribution in the property space.15-17
Thus, a good lead generation library design will have amanageable number of compounds with adequate diversityso that atypical compounds are duly represented to ensurethe synthesis of almost all potential drugs. At the same time,it will be representative of the distribution of compounds inthe virtual library. In addition to diversity and representative-ness, other requirements may also be considered that furtherconstrain the library design; for example, it may be requiredthat the library contains compounds that exhibit specificdruglike properties.14,18,19
The specific challenges, in addition to that of combinatorialoptimization, that lead generation design poses are (1)designing cost functions that reflect the notions of diversityand representation, (2) designing algorithms that are scalablein order to handle large data sets, and (3) incorporatingspecific constraints on compounds of lead generation (suchas constraints coming from limits on experimental resources).
Different multiobjective optimization methods have beenused previously for designing libraries. Stochastic methodssuch as simulated annealing20-23 and genetic algorithms24,25
have been proposed. Most of the stochastic methods attemptto avoid getting stuck in local minima by generating anensemble of states at every point and assigning a nonzeroprobability of transition to an “inferior” solution. Multiob-jective approaches to the problem are useful for includingseveral different design criteria in the algorithm. Taking intoconsideration the size of these combinatorial libraries, itbecomes essential to consider the scaling issues involved withany algorithm that solves the multiobjective optimizationproblems. Unfortunately, most of the methods mentionedabove do not address the key issues of diversity andrepresentativeness simultaneously, and these algorithms arenot scalable and often underperform when data sets are large.
This paper presents an algorithm for lead generation librarydesign (i.e., selecting a subset of compounds from a largerset of virtual compounds) which accounts simultaneously fordiversity as well as representativeness by formulating acombinatorial resource allocation problem with constraints.New procedures are presented that address the issues ofscalability and various constraints on the compounds in thelead generation library. We present an algorithm based onthe concept of deterministic annealing (DA)26,27 to cater tothe specific constraints and demands of combinatorialchemistry. The distinctive feature of the DA algorithm isthat it successfully avoids local minima. At the same time,it is typically faster than simulated annealing.28 We proposea scalable algorithm, which preserves the desirable featuresof DA and, at the same time, addresses the issue of quadraticcomplexity.29
This paper is organized as follows. In section 2, we providebackground information on some of the specifics of com-binatorial library design and the key issues that are to betackled while designing an algorithm. We then provide amathematical formulation of the library design problem. Theunderlying approach we employ for the solution of theselection problem, that is, the DA algorithm, is described insection 3.1. We then discuss our extensions and modificationsto the DA algorithm in section 3.2. The resulting scalablealgorithm, together with the tradeoff bounds, is discussed insection 3.3. Simulation results on a number of test data setsand comparisons with the nonscalable algorithm are pre-sented in section 4. Finally, we conclude the paper byrevisiting the key results obtained and identifying futuregoals.
2. BACKGROUND AND PROBLEM FORMULATION
Library design refers to the process of screening and thenselecting a subset of compounds from a given set of similaror distinct compounds (a virtual combinatorial library) fordrug discovery.30 The combinatorial nature of the selectionproblem makes it impractical to exhaustively enumerate eachand every possible subset to obtain the optimal solution. Forexample, in order to select 30 representative compounds froma set of 1000, we have1000C30 possibilities, that is, ap-proximately 3× 1025 different possible combinations fromwhich to choose. This makes the selection (based onenumeration) impractical and calls for efficient algorithmsthat make the optimal selection under given constraints. Themain aim of library design is to reduce the number ofcompounds for testing without compromising on the diver-sity, representativeness, and properties such as druglikebehavior in the subset chosen. It facilitates the process ofdrug discovery by replacing the synthesizing and subsequenttesting of all compounds by a much smaller set of repre-sentative compounds. On the basis of the current state ofdevelopment in the drug discovery process, library designis broadly classified into two main categories, namely, leadgeneration and lead optimization. The main purpose of theselibraries and their design criteria are discussed below.
2.1. Lead Generation Library Design.The developmentof a lead generation library usually involves the design andsynthesis of a large number of chemical compounds. It isrequired that these compounds be different from each other.The library containing these diverse compounds is then testedagainst a host of different biological agents. The mainobjective in designing such libraries is to obtain structurallydiverse compounds so as to be representative of the entirechemical space.
Lead optimization libraries are usually designed at a laterstage of the drug discovery process, when it is required toselect a subset of compounds that aresimilar to a given leadcompound(s). This results in an array of compounds whichare structurally and chemically similar to the lead. Thecriterion of similarity is generally used for designing targetedor focused libraries, with a single therapeutic target in mind.
2.1.1. Issues in Lead Generation Library Design.Thispaper deals with the problem of designing a library ofcompounds for the purpose of lead generation. The mostcommon method used to obtain such a library is to maximizethe diversity of the overall selection.11,12 It is based on the
28 J. Chem. Inf. Model., Vol. 48, No. 1, 2008 SHARMA ET AL .

strategy that the more diverse the set of compounds, the betterthe chance to obtain a lead compound with desired charac-teristics. As was noted earlier,13,14 such a design strategysuffers from an inherent problem which occurs due to thefact that using diversity as the only criterion may result in aset of compounds which are exceptions or singletons. Figure1 shows such a scenario. Here, the circles represent thedistribution of compounds in a descriptor space, while thecrosses denote compounds chosen according to the maximumdiversity principle. As can be seen from the figure, the clusterin the middle is not adequately represented in the finalselection. This selection focuses disproportionately on distantcompounds, which may be viewed as exceptions. From adrug discovery point of view, it is desirable for the leadgeneration library to contain more compounds from themiddle cluster (so as to adequately represent all the com-pounds) or to at least determine how proportionatelyrepresentative they are in order to make decisions on theamount of experimental resources to devote to these leadcompounds. A maximally diverse subset is of little practicalsignificance because of its limited pharmaceutical applica-tions. Hence, the criterion of representativeness should beconsidered along with diversity.15,16,31 To address thisproblem, we present an algorithm which selects a leadgeneration library that achieves high diversity and at the sametime specifies how representative (what percent of all thecompounds) each member in the library is. The processinvolves identifying different representative compound loca-tions in an iterative fashion and is discussed in followingsections.
The large size of combinatorial libraries calls for a scalableselection algorithm. We propose adiVide-and-conquerscheme for the implementation of the algorithm in whichthe underlying property space of the compounds is recur-sively and hierarchically partitioned into subdomains that areapproximately isolated from each other. The scheme quanti-fies the degree of isolation or equivalently the extent ofinteraction between the subdomains after each partitioningstep. This interaction term is used in specifying bounds onthe deviation from the results had the original DA algorithmbeen used on the entire property space. The implementationof the selection algorithm on each of these subsets that aresmaller in size than their parents in the hierarchy substantiallyreduces the computational time. The proposed algorithm iseasily adaptable to include additional criteria needed in thedesign of lead generation libraries. In addition to similarity
and diversity, other criteria includeconfinement, whichquantifies the degree to which the properties of a set ofcompounds lie between prescribed upper and lower ranges,32
and maximization of theactiVity of the set of compoundsagainst some predefined targets. Activity is usually measuredin terms of the quantitative structure of the given set. Thepresence of these multiple (and often conflicting) designobjectives makes the library design a multiobjective opti-mization problem with constraints.
2.2. Resource Allocation Problems.As was noted earlier,the lead generation library design problem belongs to theclass of combinatorial resource allocation problems. In orderto quantify the different criteria used for designing libraries(namely, diversity and similarity), it is required to defineappropriatemolecular descriptorswhich numerically char-acterize the various compounds in a chemical space.
2.2.1. Molecular Descriptors.Molecular descriptors areessential for quantifying the properties of different com-pounds in a chemical space. One-, two-, and three-dimensional descriptors are used to encode the chemicalcomposition, chemical topology, and three-dimensionalshape.33 A large number of descriptors can be used toeffectively characterize a chemical space. In practice, manyof these descriptors are correlated, and tools from dimen-sionality reduction (nonlinear mapping and principal com-ponent analysis methods34,35) are used to extract lowerdimensional representations that preserve theneighborhoodbehaVior. It has been previously shown that a two-dimensional molecular descriptor space exhibits a properneighborhood behavior which is essential for characterizingsimilarity and diversity properties between two compounds.23
In all of our simulations, we consider a 2D or 3D moleculardescriptor space. The Euclidean distance between two pointsin the space provides a good measure of the degree ofsimilarity between these two compounds. Thus, in thisscenario of a 2D descriptor space, the close neighbors of anactive compound will also be active. On the other hand, twocompounds which are far apart (in terms of the Euclideandistance) can be labeled as diverse. The 2D descriptor spaceprovides a means of quantifying the properties of differentcompounds (for more details see ref 23).
2.2.2. Problem Formulation.In its prototypical form, theproblem of selecting representative compounds for thepurpose of library design can be stated as follows.
GiVen a distribution of N compounds, xi, in a descriptorspaceΩ, find the set of M representatiVe compounds, rj, thatsolVes the following minimization problem:
Here d(xi,rj) represents an appropriatedistance metricbetween the representative compoundrj and the compoundxi, p(xi) is the relative weight that can be attached tocompoundxi (if all compounds are of equal importance, thenthe weightsp(xi) ) 1/N for eachi), andM is typically muchsmaller thanN. Alternatively, this problem can also beformulated as finding anoptimalpartition of the descriptorspaceΩ into M cells Rj and assigning to each cellRj arepresentative compoundrj such that the following costfunction is minimized:
Figure 1. A scenario depiciting the inherent problems with the“diversity” only criterion for lead generation library design.
minrj,1ejeM
∑i)1
N
p(xi) min1ejeM
d(xi,rj) (1)
COMBINATORIAL LIBRARY DESIGN J. Chem. Inf. Model., Vol. 48, No. 1, 200829

3. A SCALABLE ALGORITHM FOR MULTIOBJECTIVECOMBINATORIAL LIBRARY DESIGN
As was presented in section 2.2, the lead generation librarydesign problem can be viewed as a combinatorial resourceallocation problem with constraints. Due to the large sizeof such libraries, the solution calls for an algorithm thatscales up efficiently with the size of the underlying domain.The DA algorithm26,27 was introduced to solve the combi-natorial resource allocation problem (without addressing thescaling issues). A brief overview of the DA algorithm ispresented in section 3.1. Our scalable algorithm is based onthe original concepts of the DA algorithm, which we outlinehere.
3.1. Deterministic Annealing Algorithm. Realistic objec-tive functions in the context of lead library selection problemshave unpredictable surfaces with many local minima and,thus, require algorithms designed to avoid these local minima.The DA algorithm is suited for this purpose since it isspecifically designed to avoid local minima. This algorithmcan be viewed as a modification of another algorithm calledLloyd’s algorithm,2,36 which captures the essence of manyother algorithms typically employed for combinatorial re-source allocation problems. Lloyd’s algorithm is an iterativemethod which identifies two necessary conditions of theoptimal solution and then ensures that, at each iteration, thepartition of the domain and the representative compoundssatisfy these conditions, which are as follows:
1. Nearest-neighbor condition (Voronoi partitions). Thepartition of the descriptor space is such that each compoundin the descriptor space is associated with the nearestrepresentative compound.
2. Centroid condition. The location of each representativecompound,rj, is determined by the centroid of thejth cellRj.
In this algorithm, the initial step consists of randomlychoosing locations of representative compounds and thensuccessively iterating between the steps: (1) forming Voronoipartitions and (2) moving the representative compounds torespective centroids of cells until the sequence of locationsof representative compounds converge. It should be notedthat the solution depends substantially on the initial allocationas the locations in the successive iterations are influencedonly by near points of the domain and are virtuallyindependent offar points. As a result, the solutions fromthis algorithm typically get stuck in local minima.
The DA algorithm diminishes this local influence ofdomain members by allowing each memberxi ∈ Ω to beassociated with every representative compoundrj through aweighting parameterp(rj|xi). This weighting parameter canalso be interpreted as an association probability betweenrj
and xi. Thus, this algorithm does away with the hardpartitions of Lloyd’s algorithm. The DA formulation includesminimizing adistortion term:
which is similar to the cost function in eq 1. The termD is
referred to as distortion because of its parallels in rate-distortion theory.37 In addition to the distortion term, it alsoincludes an entropy term (H), given by
which measures the randomness of distribution of theassociated weights. The entropy is the highest when thedistribution of weights over each representative compoundis the same [p(rj|xi) ) 1/M] for eachxi, that is, when allxi
values have the same influence over every representativecompound. Maximizing the entropy is the key for selectinga maximally diverse subset of compounds. The notion ofdiversity in a given library of compounds has been previouslycharacterized using concepts from information theory.38 Inthese approaches, diversity is related to the informationcontent in a given subset (quantified by Shannon’s entropy39).
The DA algorithm solves the following multiobjectiveoptimization problem over iterations indexed byk, where
Tk is a parameter calledtemperaturewhich tends to zero ask tends to infinity. The cost functionF is calledfree energyas this formulation has an analog in statistical physics.40
Clearly, for large values ofTk, we mainly attempt tomaximize the entropy. AsTk is lowered, we trade entropyfor the reduction in distortion, and asTk approaches zero,we minimize D directly to obtain a hard (nonrandom)solution.Tk can be regarded as the Lagrange parameter inthis multiobjective optimization problem. Minimizing the freeenergy termF with respect to the weighting parameterp(rj|xi)is straightforward and gives theGibbsdistribution
where
Zi is called thepartition function. Note that the weightingparametersp(rj|xi) are simply radial basis functions, whichclearly decrease in value exponentially asrj and xi movefarther apart. The corresponding minimum ofF is obtainedby substituting forp(rj|xi) from eq 2
To minimizeF with respect to the representative compoundsrj, we set the corresponding gradients equal to zero, thatis, (∂F/∂rj ) 0); this yields the corresponding implicitequations for the locations of representative compounds
where
∑j)1
M
∑xi∈Rj
d(xi,rj) p(xi)
D ) ∑i)1
N
p(xi) ∑j)1
M
d(xi,rj) p(rj|xi)
H ) - ∑i)1
N
p(xi) ∑j)1
M
p(rj|xi) log p(rj|xi)
p(rj|xi) ) e-d(xi,rj)/Tk
Zi
Zi :) ∑j
e-d(xi,rj)/Tk (2)
F ) - Tk ∑i
p(xi) log Zi (3)
rj ) ∑i
p(xi|rj) xi, 1 e j e M
30 J. Chem. Inf. Model., Vol. 48, No. 1, 2008 SHARMA ET AL .

Note thatp(xi|rj) denotes the posterior probability calculatedusing Bayes’s rule and the above equations clearly conveythe centroidaspect of the solution.
The DA algorithm consists of minimizingF with respectto rj starting at high values ofTk, and then tracking theminimum of F while loweringTk. The steps at eachk are asfollows:
1. Fix rj and use eq 2 to compute the new weightsp(rj|xi)
2. Fix p(rj|xi) and use eq 4 to compute the representativecompound locationsrj
Representative compound locationsrj together with weight-ing parametersp(rj|xi) define the soft clustering solution tothe multiobjective clustering problem. As the temperatureTk is lowered further, these associations give way to hardpartitions, finally resulting in a solution where eachxi belongsto one of the clusters with unit weight.
Remark.The annealingmechanism in the DA algorithmenables it to successfully overcome the drawbacks of theLloyd’s type iterative process (K-means problems41), namely,its dependence on initial placement of cluster centers andits inability to avoid local minima. Thus, the DA algorithmresults in soft associations at each temperature level andreduces temperature till hard associations are formed (at zerotemperature), thereby solving the original problem.
At a fixed value of temperature, each iteration of the DAalgorithm is essentially equivalent to the Lloyd’s algorithm.In fact, the DA algorithm becomes equivalent to aK-meansalgorithm at asymptotic values of temperature (that is whenthe temperature variable goes to zero), with the representativecompound locations, obtained from the previous iteration,close to the centers of natural cluster. This clustering resultcould not have been achieved by a K-means algorithmbecause of an inability to navigate away from local minima.Thus each iteration in DA provides us with a better choicefor the representative compound locations, to be used in thenext iteration.
3.2. Algorithm Modifications for Constraints on Rep-resentative Compounds and Experimental Resources.Inthe original DA formulation, the individual representativecompounds are indistinguishable since each of them carryequal weight. However, capacity constraints often distinguishone representative compound from another in practicalsituations. In order to account for such capacity constraints,it is necessary to modify the DA algorithm. For the specificproblem of library design, one such scenario occurs whenwe want to address the issue of the representativeness ofindividual clusters in the final library design. In order toconstrain the size of each cluster, it becomes necessary todistinguish between the various locations of representativecompounds. Moreover, quantity constraints on the experi-mental resources also call for a modification of the DAalgorithm. In this section, we present two modifications ofthe original DA algorithm for tackling these issues.
3.2.1. Incorporating Representativeness.The necessityfor quantifying and incorporating representativeness isevident, as discussed in section 2.1. We incorporate repre-
sentativeness while identifying diverse compounds by speci-fying an additional parameterλj,1 e j e M, for eachcompound in the lead generation library. This parameterλj
quantifies the extent of representativeness of the compoundlocation rj; that is, it gives a measure of the number ofcompounds in the underlying data set that are representedby the compound locationrj. Thus, the resulting librarydesign will have compounds that will cover the entire dataset, where the compounds in the library that cover outlierswill have low representative weights and the compounds thatcover high-volume regular members in the data set will havehigh representative weights. In this way, the algorithm canbe used to identify diverse compounds through locationsrj
and at the same time determine how representative eachcompound in the library is.
This representativeness criterion is incorporated into thealgorithm by reinterpreting the DA algorithm. The DAalgorithm as presented above assumes that all the representa-tive compounds are identical. However, in the new inter-pretation, each locationrj can be weighted byλj, whichtranslates to the modified partition function in the algo-rithm:
The parametersλj, 1 e j e M, give relative weights (andtherefore relative representativeness) of locationsrj, 1 e je M,5,26 and without a loss of generality can be assumed tosum to 1. In this denotation,λj ) 0.2, for example, wouldmean that the representative compoundrj in the libraryrepresents 20% of the compounds in the data set. Themodified algorithm solves for locationsrj, which maximizethe diversity as before and, in addition, finds the weightsλj
that specify representativeness by solving the followingconstrained minimization problem:
such that
Note that, although the constraints do not seem to occur inthe cost function explicitly, they can be interpreted in termsof the DA algorithm where the partition function in eq 2 ismodified as in eq 5. These weighting constraints lead tomodified Gibbs distribution and free energy terms. Followinga procedure similar to that used in the original algorithm,we get
At each value of the temperature, the representative com-pound locationrj and the weightλj are given by
p(xi|rj) )p(xi) p(rj|xi)
∑k
p(xk) p(rj|xk)
(4)
Zi ) ∑j)1
M
λj e-d(xi,rj)/Tk (5)
minrj,1ejeM
∑i
p(xi) min1ejeM
d(xi,rj)
∑j)1
M
λj ) 1
p(rj|xi) )λj e-d(xi,rj)/Tk
∑j
λj e-d(xi,rj)/Tk
COMBINATORIAL LIBRARY DESIGN J. Chem. Inf. Model., Vol. 48, No. 1, 200831

Thus, we obtain locations for library designrj and theirrepresentative weightsλj. Experimental results in section 4demonstrate the efficacy of these algorithms, where theselection is depicted by locationrj in the descriptor spaceand the corresponding weightsλj are shown in pie charts.
3.2.2. Incorporating Constraints on Experimental Re-sources.The cost of chemical compounds and experimentalresources is significant and presents one of the mainimpediments in combinatorial diagnostics and drug synthesis.In fact, recent research in nanoinstrumentation has led tonanoarrays which bring together the many capabilities crucialfor rapid and high-volume research and productionsincluding design, prototyping, assembly, testing, and reliablereplication at the specificity of biomolecules.1,42 One of themain advantages of this approach is its economy, since verysmall amounts of biological and chemical agents are needed,thereby reducing the cost considerably. Still, differentcompounds require different experimental supplies which aretypically available in limited quantities. In this section, weinclude these constraints into our algorithm for lead genera-tion, as described below.
The library is classified intoq types corresponding toexperimental supplies required by the compounds for testing.We incorporate the supply constraints into the algorithm bytranslating them into direct constraints on each of therepresentative compounds. For example, thejth representativecompound can avail only an amount of thenth resource equalto Wjn. This type of a constraint is generally referred to as amulticapacity constraint.5
The modified optimization problem then is given by
such that
wherepn(xin) is the weight of the compound locationxi
n,which requires thenth type of experimental supply, andWjn
is the amount of thenth supply that thejth representativecompound can avail.
We proceed along the same lines as the DA algorithm bydefining the entropy term by
and minimizing the corresponding free energy, given byF) D - TkH. This procedure yields the Gibbs distribution ofthe form
Adding the constraints to this equation, we derive the newLagrangian given by
where qjn, 1 e n e q and 1 e j e M, are Lagrangemultipliers. Finally, the optimal locationsrj of representativecompounds for the lead generation library under experimentalconstraints are obtained by setting∂F′/∂rj ) 0. This givesthe following set of equations
which has the centroidal aspect in which averages are alsotaken about experimental supplies.
3.3. A Scalable Algorithm.As noted earlier, one of themajor problems with combinatorial optimization algorithmsis that of scalability, that is, the number of computationsscales up exponentially with an increase in the amount ofdata. In the DA algorithm, the computational complexity canbe addressed in two stepssfirst, by reducing the number ofiterations and, second, by reducing the number of computa-tions at every iteration. The DA algorithm, as describedearlier, exploits the phase transition feature26 in its processto decrease the number of iterations (in fact, in the DAalgorithm, typically the temperature variable is decreasedexponentially, which results in few iterations). The numberof computations per iteration in the DA algorithm isO(M2N),whereM is the number of representative compounds andNis the number of compounds in the underlying data set. Inthis section, we present an algorithm that requires fewercomputations per iteration. This amendment becomes neces-sary in the context of the selection problem in combinatorialchemistry, as the sizes of the initial data set are so large thatthe DA is typically too slow to be practical and often failsto handle the computational complexity.
We exploit the features inherent in the DA algorithm toreduce the number of computations in each iteration. Weuse the fact that, for a given temperature, the farther anindividual data point is from a cluster, the lower is itsinfluence on the cluster. This is evident from the form ofthe association probabilitiesp(rj|xi) in eq 2. That is, if twoclusters are far apart, then they have very little interactionbetween them. Thus, if we ignore the effect of a separatedcluster on the remaining data points, the resulting error willnot be significant. Here, note that ignoring the effects ofseparated regions (i.e., groups of clusters) on one anotherwill result in a considerable reduction in the number ofcomputations since the points that constitute a separatedregion will not contribute to the distortion and entropycomputations for the remaining points. Thus, the identifica-tion of separated regions in a data set enables us to processthe algorithm much more quickly for large data sets. Thissavings on the number of computations increases as thetemperature decreases since the number of separated regions,which are now smaller, increases as the temperature de-creases.
rj ) ∑i
p(xi|rj) xi
λj ) ∑i
p(rj|xi)p(xi), 1 e j e M
minrj
D ) ∑n
∑i
pn(xin) ∑
j)1
M
d(xin,rj) p(rj|xi
n) (6)
λjn ) Wjn, 1 e j e M, 1 e n e q (7)
H ) - ∑n
∑i
pn(xin) ∑
j)1
M
p(rj|xin) log p(rj|xi
n)
p(rj|xn) )λjn e-d(xn,rj)/Tk
∑k
λkn e-d(xn,rk)/Tk
F′ ) -1/T ∑n
∑i
log(∑j∑n
λjn e-d(xin,rj)/T) pn(xi
n) +
∑j
∑n
qjn(λjn - Wjn)
rj )
∑n
pn(xn) p(rj|xn) xndxn
∑n
pn(xn) p(rj|xn) dxn
32 J. Chem. Inf. Model., Vol. 48, No. 1, 2008 SHARMA ET AL .

The first step required to identify separated regions is todefine and quantify interaction that exists between the variousclusters. The next step is to group together sets of clustersamong which significant interaction exists, but which havesignificantly few interactions with other clusters. Once groupsof separateclusters are identified, the final step is to modifythe DA algorithm such that it ignores the effects of separategroups on one another. This modification significantlyreduces the number of computations to be performed by thealgorithm.
3.3.1. Cluster Interaction and Separation.In order tocharacterize the interaction between different clusters, it isnecessary to consider the mechanism of cluster identificationduring the process of the DA algorithm. As the temperature(Tk) is reduced after every iteration, the system undergoes aseries ofphase transitions(see ref 26 for details). The phasetransitions refer to the expansion of the set ofdistinctlocations of representative compounds at some critical valuesof temperature. We partition the underlying data set intoclusters by associating each distinct location to all the pointsin the data set that are closest; that is, we form theVoronoipartition using the nearest neighbor condition. In this way,we achieve finer partitions (smaller clusters), as the tem-perature decreases. More precisely, at high temperatures thatare above a precomputable critical value, all the representa-tive compounds are located at the centroid of the entireunderlying data set; therefore, there is only one distinctlocation for the representative compounds. As the temper-ature is decreased, successive phase transitions occur, whichlead to a greater number of distinct locations for representa-tive compounds and consequently finer clusters are formed.This provides us with a tool to control the number of clusterswe want in our final selection. It is shown26 that a clusterRi
splits at a critical temperatureTc when twice the maximumeigenvalue of the posterior covariance matrix, defined byCx|rj ) ∑i p(xi) p(xi|rj)(xi - rj)(xi - rj)T, becomes greaterthan the temperature value, that is, whenTc e 2λmax[Cx|ri].This is exploited in the DA algorithm to reduce the numberof iterations by jumping from one critical temperature to thenext without a significant loss in performance.
In the DA algorithm, the location of a representativecompound is primarily determined by the data points(compound locations) near to it since far-away points exertlittle influence, especially at low temperatures. The associa-tion probabilitiesp(rj|xi) determine the level of interactionbetween the clusterRj and the data pointxi. This interactionlevel decays exponentially with the increase in thedistancebetweenrj and xi. The total interaction exerted by all thedata points in a given space determines the relative weightof each cluster
wherep(rj) denotes the weight of clusterRj.We define the level of interaction that data points in cluster
Ri exert on clusterRj by
The higher this value is, the more interaction that exists
between clustersRi andRj. This gives us an effective wayto characterize the interaction between various clusters in adata set. In a probabilistic framework, this interaction valuecan also be interpreted as the probability of transition fromRi to Rj.
Consider them × m matrix (m e M)
In a probabilistic framework, this matrix can be considereda finite dimensional Markov operator, with the termAj,i
denoting the transition probability from regionRi to Rj. Figure2 shows the transition probabilities of the associated Markovchain. The higher the transition probability, the greater isthe amount of interaction between the two regions. Oncethe transition matrix is formed, the next step is to identifyregions, that is, groups of clusters, which are separate fromthe rest of the data. The separation is characterized by aquantity which we denote byε. We say a cluster (Rj) isε-separate if the level of its interaction with each of the otherclusters (Aj,i; i ) 1, 2, ...,n; i * j) is less thanε. Note thatthe value ε determines the number of separate regionsformed, if any, which in turn decides the error in distortiondue to the proposed algorithm with respect to that of theoriginal DA algorithm.
Alternative ways to identify invariant regions in theunderlying space, which we have explored but do not presenthere, can be motivated by concepts from graph theory. Onecan consider the Markov matrix as a undirected weightedgraph, with each entryAij representing the weight betweennode i and nodej. Tools from the spectral partitioning ofgraphs43-45 can be used to bipartition (or multipartition) thisresulting graph into invariant subgraphs. In order to getbalanced subgraphs, partitioning techniques such as normal-ized cuts46 can be used on this weighted graph. However,algorithms for the recursive bipartitioning of graphs resultin successive relaxations of the problem, thereby resultingin cumulative errors. Efficient algorithms for multiwaypartitions have not been exhaustively studied in the liter-ature.
3.3.2. Tradeoff between Error in Representative Com-pound Location and Computation Time.As was discussedin section 3.3, the greater the number of separate regions
p(rj) :) ∑i
N
p(xi,rj) ) ∑i
N
p(rj|xi) p(xi) (8)
εji ) ∑x∈Ri
p(rj|x) p(x) (9)
Figure 2. Markov chain.
COMBINATORIAL LIBRARY DESIGN J. Chem. Inf. Model., Vol. 48, No. 1, 200833

we use, the smaller the computation time for the modifiedalgorithm. At the same time, a greater number of separateregions results in a higher deviation in the distortion termof the proposed algorithm from the original DA algorithm.This tradeoff between a reduction in computation time andan increase in distortion error is systematically addressedbelow. We introduce the following notation on a subsetVof the domainΩ for a representative compoundrj:
Then, from the DA algorithm, the location of the representa-tive compound (rj) is determined by
Since the clusterΩj is separated from all the other clusters,the representative compound locationr′j will be determinedin the modified algorithm by
We obtain the component-wise difference betweenrj andr′j by subtracting terms. Note that the operations in eq 14,16, and 18 are component-wise operations. On simplifyingthe component-wise terms, we have
whereΩjc ) Ω[\]Ωj. Now note that
whereN is the cardinality ofΩ andMjc ) 1/N ∑xi∈Ωj
c xi. Wehave assumed thatx G 0 without any loss of generality sincethe resource allocation problem definition is independent oftranslation or scaling factors. Thus
Then, dividing through byN andM ) 1/N ∑xi ∈Ωxi gives
where
andεkj is the level of interaction between clusterΩj andΩk
as defined in eq 9.For a given data set, the quantitiesM, Mj, and Mj
c areknown a priori. For the error in representative compoundlocation |rj - r′j|/M to be less than a given valueδj (whereδj > 0), we must chooseηj such that
3.3.3. Algorithm. The modified algorithm is as follows:• Step 1: Initiate the DA algorithm and determine
representative compound locations together with the associa-tion probabilities.
• Step 2: When a split occurs (phase transition), identifyindividual clusters and use the association weights [p(rj|x)]to construct the transition matrix.
• Step 3: Use the transition matrix to identify separatedclusters, and group them together to form separated regions.Ωk will be separated fromΩj if the entriesAj,k andAk,j areless than a chosenεjk.
• Step 4: Apply the DA algorithm to each region,neglecting the effect of separate regions on one another.
• Step 5: Stop if the stopping criterion [such as numberof representative compounds (M), or computation time etc.]is met; otherwise, go to step 2.
Identification of the separate regions in the underlying dataprovides us with a tool to efficiently scale the DA algorithm.The number of computations at a given iteration is propor-tional to∑k)1
s Mk2Nk, whereNk is the number of compounds
andMk is the number of clusters in thekth group. For theoriginal DA algorithm, at any iteration, the number ofcomputations isM2N whereN ) ∑k)1
s Nk. Thus, the scalablealgorithm saves computations at each iteration. This differ-ence becomes bigger as temperature decreases since corre-sponding values ofNk decrease. That is, the scalablealgorithm effectively runssparallel DA algorithms, each witha relatively smaller number of data pointsNk. Moreover, asthe temperature decreases, the scalable algorithm runs anincreasing number of parallel DA algorithms on progressivelyshrinking subsets, which results in increasing savings oncomputations.
4. SIMULATION RESULTS
To demonstrate the advantages of the proposed algorithm,we have applied it on a number of data sets. We show thatthe algorithm we have proposed simultaneously addressesthe key issues of diversity and representativeness in clusteringthe descriptor space. At the same time, it also addressesscalability issues, which are imperative for huge data setsassociated with the drug discovery process.
4.1. Case 1: Design for Diversity and Representative-ness.As a first step, a fictitious data set was created topresent the “proof of concept” for the proposed optimization
Gj(V) :) ∑xi∈V
xip(xi) p(rj|xi) (10)
Hj(V) :) ∑xi∈V
p(xi) p(rj|xi) (11)
rj )Gj(Ω)
Hj(Ω)(12)
r′j )
∑xi∈Ωj
xip(xi) p(rj|xi)
∑xi∈Ωj
p(xi) p(rj|xi)
)Gj(Ωj)
Hj(Ωj)(13)
|rj - r′j| Emax[Gj(Ωj
c) Hj(Ωj), Gj(Ωj) Hj(Ωjc)]
Hj(Ω) Hj(Ωj)(14)
Gj(Ωjc) e ( ∑
xi∈Ωjc
xi) Hj(Ωjc) ) NMj
c Hj(Ωjc) (15)
|rj - r′j| Emax[NMj
c Hj(Ωj), Gj(Ωj)] Hj(Ωjc)
Hj(Ω) Hj(Ωj)(16)
) max[NMjc,
Gj(Ωj)
Hj(Ωj)][Hj(Ωjc)
Hj(Ω) ] (17)
|rj - r′j|MN
E max[Mjc
M,Mj
M]ηj (18)
ηj )
∑k*j
εkj
∑k
εkj
ηj eδj
N max[Mjc
M,Mj
M](19)
34 J. Chem. Inf. Model., Vol. 48, No. 1, 2008 SHARMA ET AL .
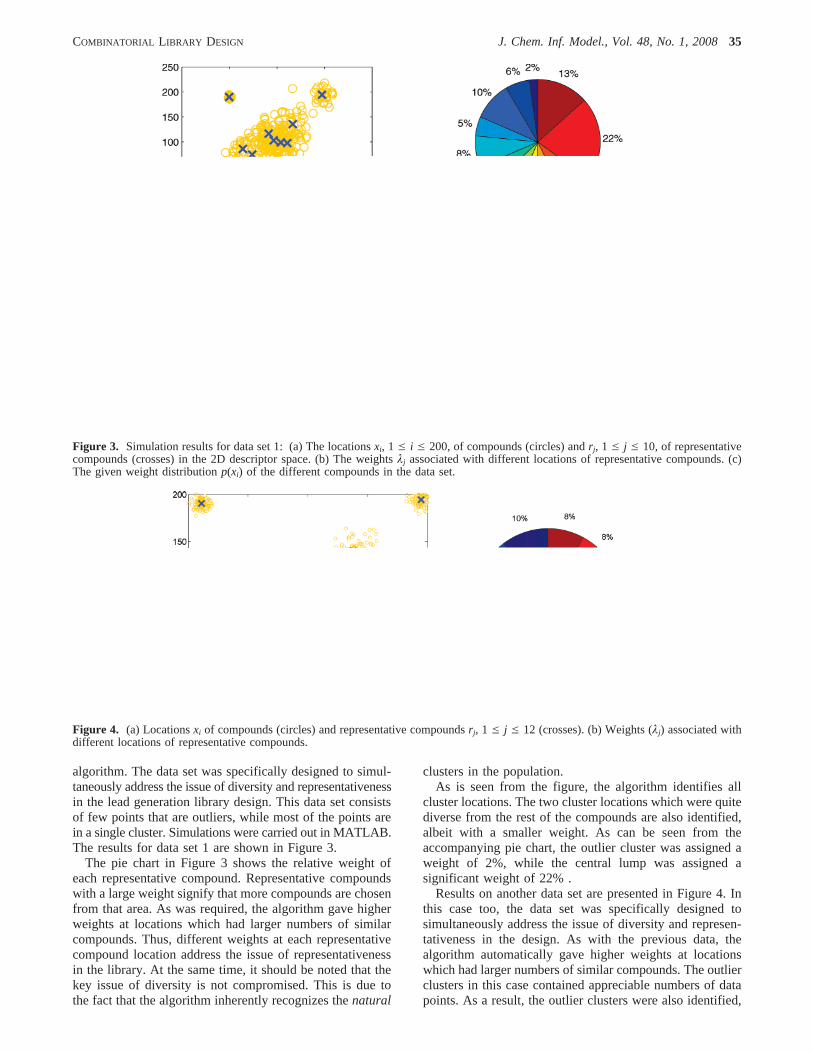
algorithm. The data set was specifically designed to simul-taneously address the issue of diversity and representativenessin the lead generation library design. This data set consistsof few points that are outliers, while most of the points arein a single cluster. Simulations were carried out in MATLAB.The results for data set 1 are shown in Figure 3.
The pie chart in Figure 3 shows the relative weight ofeach representative compound. Representative compoundswith a large weight signify that more compounds are chosenfrom that area. As was required, the algorithm gave higherweights at locations which had larger numbers of similarcompounds. Thus, different weights at each representativecompound location address the issue of representativenessin the library. At the same time, it should be noted that thekey issue of diversity is not compromised. This is due tothe fact that the algorithm inherently recognizes thenatural
clusters in the population.As is seen from the figure, the algorithm identifies all
cluster locations. The two cluster locations which were quitediverse from the rest of the compounds are also identified,albeit with a smaller weight. As can be seen from theaccompanying pie chart, the outlier cluster was assigned aweight of 2%, while the central lump was assigned asignificant weight of 22% .
Results on another data set are presented in Figure 4. Inthis case too, the data set was specifically designed tosimultaneously address the issue of diversity and represen-tativeness in the design. As with the previous data, thealgorithm automatically gave higher weights at locationswhich had larger numbers of similar compounds. The outlierclusters in this case contained appreciable numbers of datapoints. As a result, the outlier clusters were also identified,
Figure 3. Simulation results for data set 1: (a) The locationsxi, 1 e i e 200, of compounds (circles) andrj, 1 e j e 10, of representativecompounds (crosses) in the 2D descriptor space. (b) The weightsλj associated with different locations of representative compounds. (c)The given weight distributionp(xi) of the different compounds in the data set.
Figure 4. (a) Locationsxi of compounds (circles) and representative compoundsrj, 1 e j e 12 (crosses). (b) Weights (λj) associated withdifferent locations of representative compounds.
COMBINATORIAL LIBRARY DESIGN J. Chem. Inf. Model., Vol. 48, No. 1, 200835

but with a smaller weight than that of the central clusters(as indicated by the pie chart in Figure 4b). Each cluster isappropriately represented in the final selection.
Another feature of this algorithm is the flexibility itprovides in dealing withunique(i.e., far-away) clusters. Byproperly assigning the relative importance of clusters a priori,we can choose whether to include or reject these uniqueclusters in the library design. Though an immediate rejectionof unique clusters compromises the diversity of the library,there can be scenarios where the properties of these uniquecluster compounds are totally undesired in the lead generationlibrary. Thus, our approach gives us a means to deal withsuch scenarios effectively.
4.2. Case 2: Scalability and Computation Time.Takinginto consideration the huge size of combinatorial libraries,it becomes essential to consider the scaling issues involvedwith any clustering algorithm. As was pointed out earlier,the identification of separate regions in the lower dimensionaldescriptor space speeds up the algorithm (as it requires alesser amount of computation) and at the same time allowsfor larger data sets to be clustered. In order to demonstratethis fact, the algorithm was tested on a host of synthesizeddata sets. For the purpose of simulation, these data sets werecreated in the following manner.
The first set was obtained by identifying 10 randomlocations in a square region of size 400× 400. Theselocations were then chosen as the cluster centers. Next, the
size of each of these clusters was chosen, and all points inthe cluster were generated by a normal distribution ofrandomly chosen variance. A total of 5000 points comprisedthis data set. All the points were assigned equal weights [i.e.,p(xi) ) 1/N for all xi ∈ Ω]. Figure 5 shows the data set andthe representative compound locations obtained by theoriginal DA algorithm. The crosses denote the representativecompound locations (rj), and the pie chart gives the relativeweight of each representative compound (λj).
Our proposed algorithm starts with one representativecompound at the centroid of the data set. As the temperatureis reduced, the clusters are split, and separated regions aredetermined at each such split. Figure 6a shows the fourseparate regions identified by the algorithm (as described insection 3.3.1) at the instant when 12 representative compoundlocations have been identified. Figure 6b offers a comparisonbetween the two algorithms. Here, the crosses represent therepresentative compound locations (rj) determined by thenon-scalable DA algorithm.47 The circles represent thelocations (r′j) determined by the modified algorithm that wehave proposed. Note that the data set was partitioned into
Figure 5. (a) Locationsxi, 1 e i e 5000, of compounds (circles) andrj, 1 e j e 12, of representative compounds (crosses) in the 2Ddescriptor space determined from a nonscalable algorithm. (b) Relative weightsλj associated with different locations of representativecompounds.
Figure 6. (a) Separated regionsR1, R2, R3, andR4 (denoted by different colors) as determined by the proposed algorithm. (b) Comparisonof representative compound locationsrj and r′j obtained from two algorithms.
Table 1. Comparison between Nonscalable and ProposedAlgorithms
algorithm distortioncomputationtime (sec)
nonscalable DA 300.80 129.41proposed algorithm 316.51 21.53
36 J. Chem. Inf. Model., Vol. 48, No. 1, 2008 SHARMA ET AL .

four separated regions (represented by the four colors of thecircles). As can be seen from the figure, there is not muchdifference between the locations obtained by the twoalgorithms. The main advantage of the modified algorithmis in terms of the computation time of the algorithm and itsability to handle larger data sets. The results from the twoalgorithms have been presented in Table 1. As can be seenfrom Table 1, the proposed scalable algorithm uses just one-sixth of the time taken by the original (nonscalable) algorithmand results in only a 5.2% increase in distortion; this wasobtained forε ) 0.005. Both the algorithms were terminatedwhen the number of representative compounds reached 12.It should be noted here that, in case of the modifiedalgorithm, the computation time can be further reduced (bychangingε), but at the expense of error in distortion.
4.2.1. Further Examples.The scalable algorithm wasapplied on a number of different data sets. Results for threesuch cases have been presented in Figure 7. The data set incase 2 is comprised of six randomly chosen cluster centerswith 1000 points each. All the points were assigned equal
weights [i.e.,p(xi) ) 1/N for all xi ∈ Ω]. Figure 7a showsthe data set and the eight representative compound locationsobtained by the proposed scalable algorithm. The pie chartin Figure 7b gives the relative weight of each representativecompound location. Figure 7c offers a comparison betweenthe two algorithms. As in case 2, the data set in case 3 iscomprised of eight randomly chosen cluster locations with1000 points each. Both of the algorithms were executed tillthey identified eight representative compound locations inthe underlying data set. The clustering results are shown inFigure 7a-c. The data set in case 4 is comprised of two
Figure 7. (a, d, g) Simulated data set with locationsxi of compounds (circles) and representative compound locationsrj (crosses) determinedby a nonscalable algorithm. (b, e, h) Relative weightsλj of representative compound. (c, f, i) Comparison of representative locationsrj andr′j obtained from two algorithms.
Table 2. Distortion and Computation Times for Different Data Sets
case algorithm distortioncomputationtime (sec)
case 2 nonscalable DA 290.06 44.19proposed algorithm 302.98 11.98
case 3 nonscalable DA 672.31 60.43proposed algorithm 717.52 39.77
case 4 non-scalable DA 808.83 127.05proposed algorithm 848.79 41.85
COMBINATORIAL LIBRARY DESIGN J. Chem. Inf. Model., Vol. 48, No. 1, 200837

cluster centers with 2000 points each. Both of the algorithmswere executed till they identified 16 representative compoundlocations in the underlying data set. As was pointed outearlier, the main advantage of the modified algorithm is interms of the computation time and scalability. Results forthe three cases (from the two algorithms) have been presentedin Table 2. It should be noted that both of the algorithmswere terminated after a specific number of representativecompound locations had been identified. The proposedalgorithm took far less computation time when compared tothe nonscalable algorithm while maintaining less than 5%error in distortion.
The proposed algorithm has also been applied on higherdimensional data sets. Results for one such data set arepresented in Figure 8. The data set was created by iden-tifying eight random locations in a region of size 200× 200× 200. These locations were then chosen as the clustercenters. Next, the sizes of each of these clusters werechosen, and all points in the clusters were generated bynormal distributions of randomly chosen variance. A totalof 16 000 points comprised this data set. All of the pointswere assigned equal weights [i.e.,p(xi) ) 1/N for all xi ∈Ω]. Figure 8 shows the clustering results obtained by theproposed scalable algorithm. Here, the crosses represent therepresentative compound locations (rj) determined by ouralgorithm. The main advantage of the modified algorithmis in terms of the computation time of the algorithm and itsability to tackle larger data sets. The results from the twoalgorithms have been presented in Table 3. As can beseen from Table 3, our proposed scalable algorithm usesjust 54.2 s (as compared to 166.4 used by the nonscalablealgorithm) and results in only a 6.5% increase in distortion.It should be noted that both of the algorithms were ter-minated when the number of representative compounds
reached eight. In the case of the modified algorithm, thecomputation time can be further reduced at the expense oferror in distortion.
4.3. Case 3: Drug Discovery Data Set.This data set wasa modified version of the test library set.48 Each of the 50 000
Figure 8. Clustering of a 3D data set. Locationsxi of compounds (dots) and representative compoundsrj (crosses).
Table 3. Comparison between Nonscalable and ProposedAlgorithms
algorithm distortioncomputationtime (sec)
nonscalable DA 161.5 166.4proposed algorithm 172.1 54.2
Figure 9. Choosing 24 representative locations from the drugdiscovery data set.
Figure 10. Simulation results with constraints on experimentalresources.
38 J. Chem. Inf. Model., Vol. 48, No. 1, 2008 SHARMA ET AL .

members in this set were represented by 47 descriptors,which included topological, geometric, hybrid, constitutional,and electronic descriptors. These molecular descriptors werecomputed using the Chemistry Development Kit (CDK)Descriptor Calculator.49,50 As has been shown previously,these descriptors are well-suited for diversity- and similarity-based searching as they exhibit a neighborhood behavior.51
This 47-dimensional data was then normalized and projectedto a two-dimensional space. The projection was carried outusing principal component analysis on the higher dimensionaldata. Simulations were carried out on this two-dimensionaldata set. We applied our proposed scalable algorithm toidentify 25 representative compound locations from this dataset. The results are shown in Figure 9.
As was demonstrated in the previous section, the proposedalgorithm gave higher weights at locations which had largernumbers of similar compounds. Compounds which aremaximally diverse from all the others are identified with avery small weight. It should be noted that the nonscalableversion of the algorithm could not handle the number ofcomputations for this data set (we ran both the scalable andthe original DA algorithm using MATLAB on a 1.5 GHzIntel Centrino processor with 512 MB of RAM) due to thecombinatorial nature of the problem.
4.4. Case 4: Additional Constraints on RepresentativeCompounds.As was discussed in section 3, the multiob-jective framework of the proposed algorithm allows us toincorporate additional constraints in the selection problem.In this section, we have addressed two such constraints,namely, the experimental resources constraint and theexclusion/inclusion constraint. Results on simulated data setshave also been presented, wherein the proposed algorithmwas adapted to cater to these additional constraints in theselection criterion.
4.4.1. Constraints on Experimental Resources.A dataset was created to demonstrate the manner in which thealgorithm presented in section 3.2.2 accounts for theconstraints placed on experimental resources. Differentcompounds require different experimental supplies which aretypically available in limited quantities. As was discussedin section 3.2.2, in addition to diversity and representative-ness, we include these constraints on experimental suppliesinto our algorithm for selecting compounds. In this data set,the library has been divided into three classes on the basisof the experimental supplies required by the compounds fortesting, as shown in Figure 10 by different symbols. Itcontains a total of 280 compounds with 120 of the first class
(denoted by circles), 40 of the second class (denoted bysquares), and 120 of the third class (denoted by triangles).We incorporate experimental supply constraints into thealgorithm by translating them into direct constraints on eachof the representative compounds (eq 7). With these experi-mental supply constraints in place, the algorithm was usedto select 15 representative compound locations (rj) in thisdata set with capacities (Wjn) fixed for each class of resource.The crosses in Figure 10 represent the selection from thealgorithm in the wake of the capacity constraints for differenttypes of compounds. As can be seen from the selection, thealgorithm successfully addressed the key issues of diversityand representativeness together with the constraints that wereplaced due to experimental resources. To the authors’ bestknowledge, this is the first algorithm which specificallyaddresses this type of constraint on experimental resourcesin a multiobjective optimization framework.
4.4.2. Constraints on Exclusion and Inclusion of Cer-tain Properties. In the process of selecting a subset ofcompounds, there may arise scenarios where we would liketo inhibit selection of compounds exhibiting properties withincertain prespecified ranges. This constraint can be easilyincorporated in the cost function by modifying the distancemetric used in the problem formulation.
Consider a case in a 2D data set where each pointxi hasan associated radius (denoted byøij). The selection problem
Figure 11. (a) Modified “distance” metric for exclusion of specific regions. (b) Modified “distance” metric for exclusion and inclusion ofspecific regions.
Figure 12. Simulation results with exclusion constraint. Thelocationsxi, 1 e i e 90, of compounds (circles) andrj, 1 e j e 6,of representative compounds (crosses) in the 2D descriptor space.Blue circles represent undesirable properties.
COMBINATORIAL LIBRARY DESIGN J. Chem. Inf. Model., Vol. 48, No. 1, 200839

is fundamentally the same, but with the added constraint thatall of the selected points (rj) must be at least a distance oføij removed fromxi. The proposed algorithm can be modifiedto solve this problem by defining the distance function asfollows:
Such a distance function penalizes any selection (rj) whichis in close proximity to the data points. This is depicted inFigure 11a. For the purpose of simulation, a data set wascreated with points at 90 locations (xi, i ) 1, ..., 90). Theblue circle around the locationsxi denotes the region in theproperty space that is to be avoided by the selectionalgorithm. The objective was to select six representativecompounds from this data set such that the criterion ofdiversity and representativeness is optimally addressed in theselected subset. The selected locations are represented byblue crosses.
From Figure 12, note that the algorithm identifies the sixclusters under the constraint and that none of the clustercenters are located in the undesirable property space (denotedby blue circles). The same analysis can be extended to higherdimensional descriptor spaces, thereby making it possibleto exclude ranges of certain properties (e.g., molecular weightand solubility). Another distance metric is shown in Figure11b. As can be seen from the figure, it favors representativecompounds within a certain radius around a particularcompound. The flat region in the figure denotes this radius.Anything outside this region is penalized by this choice ofdistance metric. Depending on the scenarios encountered,the distance metric can be suitably modified to addressfurther such exclusion and inclusion constraints.
5. CONCLUSIONS
In this paper, we proposed an algorithm for the design oflead generation libraries. The problem was formulated in aconstrained multiobjective optimization setting and posed asa resource allocation problem with multiple constraints. Asa result, we successfully tackled the key issues of diversityand representativeness of compounds in the resulting library.Another distinguishing feature of the algorithm is its scal-ability, thus making it computationally efficient as comparedto other such optimization techniques. We characterized thelevel of interaction between various clusters and used it todivide the clustering problem with huge data size intomanageable subproblems with small sizes. This resulted insignificant improvements in the computation time andenabled the algorithm to be used on larger-sized data sets.The tradeoff between computation effort and error due totruncation is also characterized, thereby giving an option tothe end-user.
Currently, we are working on ways to formulate anoptimization problem where the computational expense formsa part of the total cost. This gives a way to prescribe howthe subsets need to be formed for a given tradeoff betweenthe computational expense and the deviation from thesolution that acts on the whole set (that is, which does notsubdivide the data and therefore does not save computationaltime). The main principle that the framework relies on isthe maximum entropy principle (MEP) developed extensively
by Jaynes.52,53The problem is viewed in a stochastic setting,and MEP proves to be a very useful tool for formulating theoptimization problem and finding an algorithm that solvesit. Formulating the lead generation library design problemin such a manner has enabled automation of thediVide-and-conquer rule, with predetermined bounds.
ACKNOWLEDGMENT
This work is partially supported by NSF ITR CollaborativeResearch Grant ECS-0426831 and a NSF ECS 0449310 CARgrant.
REFERENCES AND NOTES
(1) Demers, L.; Cioppa, G. D. Drug Discovery: Nanotechnology toAdvance Discovery R&D.Genet. Eng. News2003, 23.
(2) Gersho, A.; Gray, R.Vector Quantization and Signal Compression,1st ed.; Kluwer: Boston, Massachusetts, 1991.
(3) Drezner, Z.Facility Location: A SurVey of Applications and Methods;Springer-Verlag: New York, 1995; Springer Series in OperationsResearch.
(4) Du, Q.; Faber, V.; Gunzburger, M. Centroidal Voronoi Tessellations:Applications and Algorithms.SIAM ReV. 1999, 41, 637-676.
(5) Salapaka, S.; Khalak, A. Constraints on Locational OptimizationProblems. InProceedings of the IEEE Control and Decision Confer-ence; IEEE: Los Alamitos, CA, 2003.
(6) Cortes, J.; Martı´nez, S.; Karatas, T.; Bullo, F. Coverage Control forMobile Sensing Networks: Variations on a Theme.Proceedings ofthe Mediterranean Conference on Control and Automation; Mediter-ranean Control Association: Notre Dame, IN, 2002.
(7) Therrien, C.Decision, Estimation and Classification: An Introductionto Pattern Recognition and related topics, 1st ed.; Wiley: New York,1989; Vol. 14.
(8) Haykin, S.Neural Networks: A ComprehensiVe Foundation; PrenticeHall: Englewoods Cliffs, NJ, 1998.
(9) Hartigan, J.Clustering Algorithms; Wiley: New York, 1975.(10) Gray, R.; Karnin, E. Multiple local minima in vector quantizers.IEEE
Trans. Inf. Theory1982, IT-28, 256-361.(11) Willett, P. Computational tools for the analysis of molecular diversity.
Perspect. Drug DiscoVery Des.1997, 7/8, 1-11.(12) Blaney, J.; Martin, E. Computational approaches for combinatorial
library design and molecular diversity analysis.Curr. Opin. Chem.Biol. 1997, 1, 54-59.
(13) Rassokhin, D. N.; Agrafiotis, D. K. Kolmogorov-Smirnov statistic andits applications in library design.J. Mol. Graphics Modell.2000, 18,370-384.
(14) Lipinski, C. A.; Lomabardo, F.; Dominy, B. W.; Feeny, P. J.Experimental and Computational Approaches to estimate solubilityand permeability in drug discovery and development setting.AdV. DrugDeliVery ReV. 1997, 23, 2-25.
(15) Higgs, R. E.; Bemis, K. G.; Watson, I. A.; Wikel, J. H. Experimentaldesigns for selecting molecules from large chemical databases.J.Chem. Inf. Comput. Sci.1997, 37, 861-870.
(16) Snarey, M.; Terrett, N.; Willet, P.; Wilton, D. J. Comparison ofalgorithms for dissimilaritybased compound selection.J. Mol. GraphicsModell. 1997, 15, 372-385.
(17) Mount, J.; Ruppert, J.; Welch, W.; Jain, A. N. IcePick: A flexiblesurface-based system for molecular diversity.J. Med. Chem.1999,42, 60-66.
(18) Sheridan, R. P. The most common chemical replacements in drug-like compounds.J. Chem. Inf. Comput. Sci.2002, 2, 103-108.
(19) Wang, J.; Ramnarayan, K. Toward designing drug-like libraries: anovel computational approach for prediction of important structuralfeatures.J. Comb. Chem.1999, 1, 52-533.
(20) Waldman, M.; Li, H.; Hassan, M. Novel algorithms for the optimizationod molecular diversity of combinatorial libraries.J. Mol. GraphicsModell. 2000, 18, 412-426.
(21) Good, A. C.; Lewis, R. A. New methodology for profiling combina-torial libraries and screening sets: cleaning up the design process withHARPcik. J. Med. Chem.1997, 40, 3226-3236.
(22) Agrafiotis, D. K. Stochastic Algorithms for maximizing moleculardiversity.J. Chem. Inf. Comput. Sci.1997, 37, 841-851.
(23) Agrafiotis, D. Multiobjective Optimization of Combinatorial Libraries.IBM J. Res. DeV. 2001, 45, 545-566.
(24) Sheridan, R. P.; San Feliciano, S. G.; Kearsley, S. K. Designingtargeted libraries with Genetic Algorithms.J. Mol. Graphics Modell.2000, 18, 320-333.
d(xi,rj) ) (|xi - rj| - øij)2 (20)
40 J. Chem. Inf. Model., Vol. 48, No. 1, 2008 SHARMA ET AL .

(25) Brown, R. D.; Martin, Y. C. Designing combinatorial library mixturesusing Genetic Algorithms.J. Med. Chem.1997, 40, 2304-2313.
(26) Rose, K. Deterministic Annealing for Clustering, Compression,Classification, Regression and Related Optimization Problems.Proc.IEEE 1998, 86, 2210-39.
(27) Rose, K. Constrained Clustering as an Optimization Method.IEEETrans. Pattern Anal. Machine Intell.1993, 15, 785-794.
(28) Kirkpatrick, S.; Gelatt, C.; Vechhi, M. Optimization by SimulatedAnnealing.Science1983, 220, 671-680.
(29) Agrafiotis, D. K.; Lobanov, V. S.; Salemme, F. R. CombinatorialInformatics in the postgenomics era.Nat. ReV. 2002, 1, 337-346.
(30) Gordon, E.; Barrett, R.; Dower, W.; Fodor, S.; Gallop, M. Applicationsof Combinatorial Technologies to Drug Discovery. 2. CombinatorialOrganic Synthesis, Library Screening Strategies, and Future Directions.J. Med. Chem.1994, 37, 1385-1401.
(31) Clark, R. D. OptiSim: An Extended Dissimilarity Selection Methodfor Finding Diverse Representative Subsets.J. Chem. Inf. Comput.Sci.1997, 37, 1181-1188.
(32) Agrafiotis, D.; Lobanov, V. S. Ultrafast Algorithm for DesigningFocussed Combinatorial Arrays.J. Chem. Inf. Comput. Sci.2000, 40,1030-1038.
(33) Livingston, D. J. The characterization of molecular structures usingmolecular properties: A survey.J. Chem. Inf. Comput. Sci.2000, 40,195-209.
(34) Schlkopf, B.; Smola, A. J.; Mller, K.-R. Nonlinear component analysisas a kernel eigenvalue problem.Neural Comput.1998, 10, 1299-1319.
(35) Jolliffe, I. T. Principal Component Analysis; Springer-Verlag: NewYork, 2002; Springer Series in Statistics.
(36) Lloyd, S. Least squares quantization in PCM.IEEE Trans. Inf. Theory1982, 28, 129-137.
(37) Berger, T.Rate Distortion Theory: A Mathematical Basis for DataCompression; Prentice Hall: Englewood Cliffs, NJ, 1971.
(38) Lin, S. K. Molecular Diversity Assesment: logarithmic relations ofinformation and species diversity and logarithmic relations of entropyand indistinguishability after rejection of Gibbs paradox of entropymixing. Molecules1996, 1, 57-67.
(39) Shannon, C. E.; Weaver, W.The mathematical theory of communica-tion; University of Illinois Press: Urbana, Illinois, 1949.
(40) Rose, K. Statistical mechanics and phase transitions in clustering.Phys.ReV. Lett. 1990, 65, 945-948.
(41) MacQueen, J. B. Some Methods for Classification and Analysis ofMultivariate Observations. InProceedings of 5th Berkeley Symposiumon Mathematical Statistics and Probability; University of CaliforniaPress: Berkeley, CA, 1967.
(42) Lynch, M.; Mosher, C.; Huff, J.; Nettikadan, S.; Johnson, J.;Henderson, E. Functional protein nanoarrays for biomarker profiling.Proteomics2004, 4, 1695-702.
(43) Fiedler, M. Algebraic connectivity of graphs.Czech. Math. J.1973,23, 298-305.
(44) Fiedler, M. A property of eigenvectors of nonnegative symmetricmatrices and its application to graph theory.Czech. Math. J.1975,25, 619-633.
(45) Hendrickson, B.; Leland, M.Multidimensional Spectral Load Balanc-ing; Sandia National Laboratories: Albuquerque, NM, 1993.
(46) Shi, J.; Malik, J. Normalized Cuts and Image Segmentation.IEEETrans. Pattern Anal. Machine Intell.2000, 22, 888-905.
(47) Sharma, P.; Salapaka, S.; Beck, C. A Deterministic AnnealingApproach to Combinatorial Library Design for Drug Discovery. InProceedings of the American Control Conference; IEEE: Los Alami-tos, CA, 2005.
(48) McMaster HTS Lab Competition. HTS Data Mining and DockingCompetition. http://hts.mcmaster.ca/Downloads/82BFBEB4-F2A4-4934-B6A8-804CAD8E25A0.html (accessed June 2006).
(49) Guha, R. Chemistry Development Kit (CDK) Descriptor CalculatorGUI (v. 0.46). http://cheminfo.informatics.indiana.edu/rguha/code/java/cdkdesc.html (accessed October 2006).
(50) Steinbeck, C.; Hoppe, C.; Kuhn, S.; Floris, M.; Guha, R.; Willighagen,E. L. Recent Developments of the Chemistry Development Kit (CDK)- An Open-Source JAVA Library for Chemo and Bioinformatics.Curr.Pharm. Des.2006, 12, 2110-2120.
(51) Patterson, D. E.; Cramer, R. D.; Ferguson, A. M.; Clark, R. D.;Weinberger, L. E. Neighborhood Behavior: A Useful Concept forValidation of “Molecular Diversity” Descriptors.J. Med. Chem.1996,39, 3049-3059.
(52) Jaynes, E. T. Information Theory and Statistical Mechanics.Phys. ReV.1957, 106, 620-630.
(53) Jaynes, E. T. Information Theory and Statistical Mechanics II.Phys.ReV. 1957, 108, 171-190.
CI700023Y
COMBINATORIAL LIBRARY DESIGN J. Chem. Inf. Model., Vol. 48, No. 1, 200841





























