Embed Size (px)
Citation preview

Available online at www.sciencedirect.com
www.elsevier.com/locate/nmd
Neuromuscular Disorders 22 (2012) 361–367
Case report
A missense mutation in the skeletal muscle chloride channel 1(CLCN1) as candidate causal mutation for congenital
myotonia in a New Forest pony
Inge D. Wijnberg a,⇑, Marta Owczarek-Lipska b, Roberta Sacchetto c,Francesco Mascarello c, Francesco Pascoli c, Walter Grunberg d,
Johannes H. van der Kolk a, Cord Drogemuller b
a Department of Equine Sciences, Faculty of Veterinary Medicine, Utrecht University, Utrecht, The Netherlandsb Institute of Genetics, Vetsuisse Faculty, University of Berne, Switzerland
c Department of Experimental Veterinary Sciences, University of Padova, Padova, Italyd Department of Farm Animal Health, Faculty of Veterinary Medicine, Utrecht University, Utrecht, The Netherlands
Received 3 June 2011; received in revised form 26 September 2011; accepted 3 October 2011
Abstract
A 7-month-old New Forest foal presented for episodes of recumbency and stiffness with myotonic discharges on electromyography.The observed phenotype resembled congenital myotonia caused by CLCN1 mutations in goats and humans. Mutation of the CLCN1
gene was considered as possible cause and mutation analysis was performed. The affected foal was homozygous for a missense mutation(c.1775A>C, p.D592A) located in a well conserved domain of the CLCN1 gene. The mutation showed a recessive mode of inheritancewithin the reported pony family. Therefore, this CLCN1 polymorphism is considered to be a possible cause of congenital myotonia.� 2011 Elsevier B.V.
Keywords: Muscle spasm; Myotonic discharges; Electromyography; Becker; Thomsen; CLCN1; Horse
Open access under the Elsevier OA license.
1. Introduction
Myotonias are acquired or inherited neuromuscular dis-orders characterized by the slow relaxation of muscles aftervoluntary contraction or electrical stimulation. Geneticforms of congenital myotonia are characterized byimpaired function of chloride, sodium or potassium iontransport channels in the skeletal muscle membrane [1–3].It has been previously shown that sarcolemmal chlorideconduction is required to preserve membrane potentialand functional chloride channels are essential for restingpotential stabilization. Different mutations in genes that
0960-8966 � 2011 Elsevier B.V.
doi:10.1016/j.nmd.2011.10.001
⇑ Corresponding author. Address: Department Equine Sciences,Utrecht University, Yalelaan 114-116, 3584 CM Utrecht, The Nether-lands. Tel.: +31 30 253 1350; fax: +31 30 2537970.
E-mail address: [email protected] (I.D. Wijnberg).
Open access under the Elsevier OA license.
code for ion channels are involved in several hereditarymuscular diseases [1,2,4,5]. Human congenital myotoniais caused by recessive or dominant mutations of CLCN1
causing Becker’s (OMIM 255700) or Thomsen’s disease(OMIM 160800). The muscle chloride channel CLCN1 reg-ulates the electrical excitability of the skeletal muscle mem-brane [1,2,6,7].
The association between myotonia and chloride conduc-tion was first described in myotonic mice [8] and the firstmutation in the CLCN1 gene shown to cause myotoniawas found in domestic animals. The “fainting” goats whichdeveloped severe acute muscle stiffness and became immo-bile during vigorous movements or when startled showed amissense mutation of the CLCN1 gene (p.A885P) [4]. In1962 the first case of suspected equine myotonia wasreported [7]. As the phenotype of the New Forest ponyreported here strongly resembled the clinical signs and

362 I.D. Wijnberg et al. / Neuromuscular Disorders 22 (2012) 361–367
biochemical findings of both human and goat myotonia,we hypothesized that mutations in the CLCN1 gene mightalso be responsible for the clinical signs observed in thisanimal.
In this case study we report a missense mutation in theequine CLCN1 gene associated with a recessive inheritedmyotonia disease phenotype.
Fig. 1B. Posture of the patient at rest. Note the straight angle of thetarsus.
2. Case report
A 7-month-old New Forest pony stallion was presentedto the Equine Clinic of Utrecht University. During the first4–6 weeks of life the foal had developed normally. Mainabnormalities observed by the owner were recurrent epi-sodes of recumbency and difficulty rising to its feet as aresult of muscle stiffness. The frequency of these episodesof recumbency had increased in the months prior to presen-tation. At the time of clinical examination the foal wasfound to be well developed with above average musclemass when compared to other foals of the same breed.When stimulated (for example by opening of the stabledoor or when getting up) the foal seemed hyperreactiveand temporary protrusion of the third eyelid occurreddue to retraction of the eye uni- or bilaterally (Fig. 1A).During vocalization the whinnying ended in a high pitchedsqueaking, or a deep growling noise. The pelvic limbs wereextremely straight (minimal flexion of the tarsi) (Fig. 1B),and the foal walked reluctantly with a stiff gait. Whenwalking, the pelvic limbs did not always follow the trackof the thoracic limbs and the foal easily lost its balanceas a result of stiffness and occasionally lost its footing. Gen-eral examination revealed no other abnormalities thanretraction of the eye and protrusion of the third eyelidfor approximately 30 s following inspection of the conjunc-tiva. Neurological examination revealed normal mentation,no cranial nerve deficits and occasionally a wide-basedstance. Postural reactions were impossible to perform andpicking up the limbs was hampered by the muscle rigidity.
Fig. 1A. Retraction of the eyebulb due to the myotonia. Note theretracted eyebulb due to the myotonia.
Manual flexion of the proximal metacarpo/metatarso-pha-langeal joints was impossible. Passive movements of theneck and trunk were very limited and the foal was easilypulled off balance. Although the foal showed signs of weak-ness, ataxia was absent and the rigidity seemed to be themajor problem.
Blood biochemical analysis showed a plasma sodium[Na] concentration of 129 mmol/l [ref. 135–150 mmol/l],potassium [K] of 2.8 mmol/l [ref. 3.0–5.9 mmol/l], magne-sium of 0.77 mmol/l [ref. 0.80–1.20 mmol/l], chloride [Cl]of 99 mmol/l [ref. 96–107 mmol/l], total calcium [Ca] of3.97 mmol/l [ref. 2.4–3.3 mmol/l] and phosphate of1.48 mmol/l [[Pi]; ref. 0.8–1.8 mmol/l]. Blood obtainedimmediately after an episode of generalized stiffnessrevealed a [Na] of 138 mmol/l, a [K] of 4.1 mmol/l, a [Cl]of 95 mmol/l and a plasma glucose concentration of4.1 mmol/l (ref. 3.9–5.6 mmol/l). The fractional excretionof all electrolytes in urine was within normal limits andthe acid–base balance of the blood was normal. Muscleenzyme activities were not measured. DNA testing forthe mutation of the skeletal muscle sodium channel gene(SCN4A) causing hyperkalemic periodic paralysis in Quar-ter horses was negative. Electromyographic (EMG) needleexamination [9,10] using Viking Quest commercial EMGequipment showed prolonged insertional activity causedby spontaneous waxing and waning discharges after needleinsertion and as a result of muscle contraction in all mea-sured muscles including the subclavian and descending pec-toral muscle. The results of EMG examination wereconsistent with myotonia [2,5,11,12] (Fig. 2).
Routine morphological (Gomori trichrome, H&E) andhistochemical analysis (succinic dehydrogenase, cyto-chrome oxidase and m-ATPase) of biopsy samples fromthe M. semimembranosus, M. gluteus medius, M. vastuslateralis and M. triceps brachii caput longum were per-formed. There was no evidence of muscle damage, rhabdo-myolysis or central nuclei but extreme heterogeneity infiber diameters was noticed in all four muscle specimensexamined. In the caput longum of the M. triceps brachii
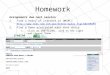
Fig. 2. (A) Myotonic discharges in the M. pectoralis descendens. (B) Myotonic discharges in the M. vastus lateralis. Dotted lines indicate the divisions,one division is 200 lV and 100 ms in 2A and 500 lV and 200 ms in 2B. The firing rate of the discharges is 72 Hz in 2A and 147 Hz in 2B.
Fig. 3. Histochemical staining of muscle sections from patient affected by myotonia. Panel a. M. semimembranosus; panel b: M. gluteus medius; panel c:M. vastus lateralis; panel d: M. triceps brachii Caput longum. Bar: 100 lm. Muscle samples were frozen in cold isopentane. Serial sections (10 lm) were cutin a cryostat and stained for myofibrillar adenosine triphosphatase (m-ATPase) after acid (pH 4.55) pre-incubation. Slow fibers (type 1) are dark whiletypes 2A and 2� are light. Images were acquired with a bright field microscope (Olympus Vanox AH-3, Japan), equipped with video camera and analyzedwith an image analysis software (Olympus DP-software-70).
I.D. Wijnberg et al. / Neuromuscular Disorders 22 (2012) 361–367 363

↑C-allele not present in 13 other horse breeds
A
B
Fig. 4. Pedigree and sequence analysis of genomic DNA of the missensemutation c.1775A>C in exon 15 of the equine CLCN1 gene of the case’sfamily presented in this study. (A) Pedigree of the case’s family presentedin this study. DNA samples were available from numbered horses only.The myotonic pony is shown in solid black box, the arrow indicated thepatient. The half black boxes are indicating the relatives heterozygous forthe mutation. (B) Sequence analysis of genomic DNA of the missensemutation c.1775A>C in exon 15 of the equine CLCN1 gene. Sequencechromatograms show the wildtype (1775AA; at the top), the carrier(1775A/C; in the middle) and the mutated (1775CC; at the bottom)individuals, respectively.
364 I.D. Wijnberg et al. / Neuromuscular Disorders 22 (2012) 361–367
two distinct type 1 fiber populations were observed. Thefirst consisted of fibers with a diameter of 50.36 ± 9.3 lm,whilst the second population (Fig. 3) consisted of fiberswith a very small diameter, 20.21 + 8.8 lm.
We hypothesized that a mutation in the CLCN1 genemay have been responsible for the clinical signs observedin the present case [7]. The analysed pedigree data sug-gested a monogenic autosomal recessive inheritance(Fig. 4A). Parents of the affected foal did not show anyclinical signs. EDTA blood samples of the affected foal,its dam, its sire, other related (n = 22) and unrelated(n = 21) New Forest ponies, and 56 horses of variousbreeds were examined (Supplementary Table 2). GenomicDNA was isolated using standard methods. PCR primerpairs were designed for the amplification of CLCN1 exonswith flanking introns (Table 1) as described previously [13].The equine CLCN1 gene has 23 exons (NCBI Gene ID:100050692), the total predicted transcript length is2970 bp (Genbank acc. XM_001915636) encoding a pro-tein of 989 aa (Genbank acc. XP_001915671). Initially,all exons and several introns of the equine CLCN1 genewere amplified using standard protocols applied to a totalof four DNA samples from the affected foal, its dam, itssire (Fig. 4A) and one unrelated control horse. The purifiedPCR products were directly sequenced from both direc-tions using the ABI BigDye v3.1 kit and an ABI 3730 cap-illary sequencer (LifeTechnologies, Rotkreuz, Switzerland).Sequence data were analysed using Sequencher 4.9 (Gene-Codes, Ann Arbor, MI, USA).
A total of three polymorphisms were identified, two sin-gle nucleotide substitutions in CLCN1 exon 15(c.1593A>G and c.1775A>C) and a three base pair inser-tion (c.2652_2653insCTT) in exon 23. No other mutationsin exonic or splice site regions of the CLCN1 gene wereidentified. The c.1593A>G transition did not result inamino acid exchange. The three family members (the foaland its parents) were homozygous for the mutant allele1593G and the unrelated control horse was homozygousfor the wildtype allele 1593A. This mutation was thereforenot associated with the disease. A similar genotype distri-bution was observed for the in-frame insertion(c.2652_2653insCTT) leading to an additional leucine resi-due after codon 884 which affects a less conserved proteinregion. In conclusion, these two sequence variants arehighly unlikely to be related to the proper function of theencoded protein.
Interestingly, the c.1775A>C transversion led to anamino acid replacement where aspartic acid is replacedby alanine (p.D592A). The affected pony was homozygousC/C for mutant allele1775C, the dam and the sire were het-erozygous A/C and the control horse was homozygous A/A for the wildtype allele 1775A (Fig. 4B).
To verify the association of the c.1775A>C mutationwith the clinical disease presentation an additional 42New Forest ponies and 56 horses of 13 different breedswere screened using the methods described above (Supple-mentary Table 2). None of the investigated animals was
homozygous for the mutant allele 1775C. Of the genotypedNew Forest ponies eight mares (including the mother) andone stallion (the sire of the affected foal) were heterozygous(A/C) for the mutant 1775C allele while all other horsesand ponies were homozygous A/A (Supplementary Table 2,Fig. 4A).
To test whether the observed missense mutation affectsCLCN1 splicing we amplified the entire CLCN1 cDNAof the affected foal. Total RNA extracted from four differ-ent skeletal muscles of the affected pony was subjected to areverse transcription-PCR (RT-PCR) to analyse theCLCN1 transcript. The entire coding sequence of 2970 bpof the CLCN1 gene was amplified using five overlapping
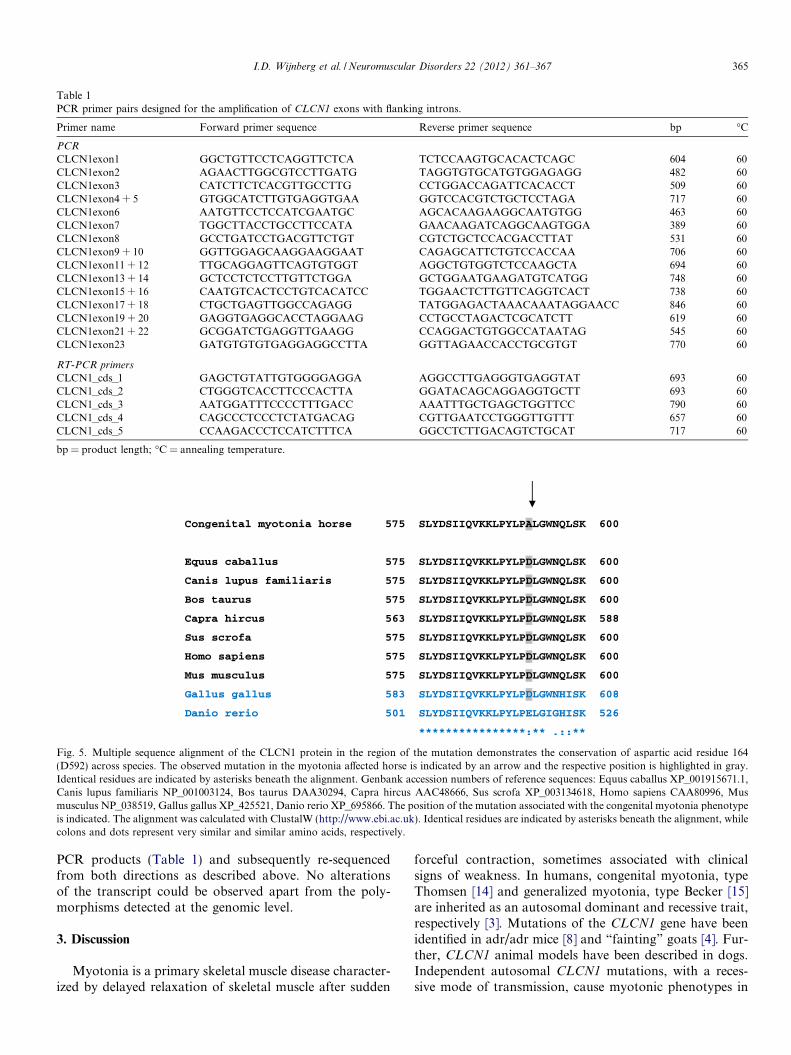
Table 1PCR primer pairs designed for the amplification of CLCN1 exons with flanking introns.
Primer name Forward primer sequence Reverse primer sequence bp �C
PCR
CLCN1exon1 GGCTGTTCCTCAGGTTCTCA TCTCCAAGTGCACACTCAGC 604 60CLCN1exon2 AGAACTTGGCGTCCTTGATG TAGGTGTGCATGTGGAGAGG 482 60CLCN1exon3 CATCTTCTCACGTTGCCTTG CCTGGACCAGATTCACACCT 509 60CLCN1exon4 + 5 GTGGCATCTTGTGAGGTGAA GGTCCACGTCTGCTCCTAGA 717 60CLCN1exon6 AATGTTCCTCCATCGAATGC AGCACAAGAAGGCAATGTGG 463 60CLCN1exon7 TGGCTTACCTGCCTTCCATA GAACAAGATCAGGCAAGTGGA 389 60CLCN1exon8 GCCTGATCCTGACGTTCTGT CGTCTGCTCCACGACCTTAT 531 60CLCN1exon9 + 10 GGTTGGAGCAAGGAAGGAAT CAGAGCATTCTGTCCACCAA 706 60CLCN1exon11 + 12 TTGCAGGAGTTCAGTGTGGT AGGCTGTGGTCTCCAAGCTA 694 60CLCN1exon13 + 14 GCTCCTCTCCTTGTTCTGGA GCTGGAATGAAGATGTCATGG 748 60CLCN1exon15 + 16 CAATGTCACTCCTGTCACATCC TGGAACTCTTGTTCAGGTCACT 738 60CLCN1exon17 + 18 CTGCTGAGTTGGCCAGAGG TATGGAGACTAAACAAATAGGAACC 846 60CLCN1exon19 + 20 GAGGTGAGGCACCTAGGAAG CCTGCCTAGACTCGCATCTT 619 60CLCN1exon21 + 22 GCGGATCTGAGGTTGAAGG CCAGGACTGTGGCCATAATAG 545 60CLCN1exon23 GATGTGTGTGAGGAGGCCTTA GGTTAGAACCACCTGCGTGT 770 60
RT-PCR primers
CLCN1_cds_1 GAGCTGTATTGTGGGGAGGA AGGCCTTGAGGGTGAGGTAT 693 60CLCN1_cds_2 CTGGGTCACCTTCCCACTTA GGATACAGCAGGAGGTGCTT 693 60CLCN1_cds_3 AATGGATTTCCCCTTTGACC AAATTTGCTGAGCTGGTTCC 790 60CLCN1_cds_4 CAGCCCTCCCTCTATGACAG CGTTGAATCCTGGGTTGTTT 657 60CLCN1_cds_5 CCAAGACCCTCCATCTTTCA GGCCTCTTGACAGTCTGCAT 717 60
bp = product length; �C = annealing temperature.
Fig. 5. Multiple sequence alignment of the CLCN1 protein in the region of the mutation demonstrates the conservation of aspartic acid residue 164(D592) across species. The observed mutation in the myotonia affected horse is indicated by an arrow and the respective position is highlighted in gray.Identical residues are indicated by asterisks beneath the alignment. Genbank accession numbers of reference sequences: Equus caballus XP_001915671.1,Canis lupus familiaris NP_001003124, Bos taurus DAA30294, Capra hircus AAC48666, Sus scrofa XP_003134618, Homo sapiens CAA80996, Musmusculus NP_038519, Gallus gallus XP_425521, Danio rerio XP_695866. The position of the mutation associated with the congenital myotonia phenotypeis indicated. The alignment was calculated with ClustalW (http://www.ebi.ac.uk). Identical residues are indicated by asterisks beneath the alignment, whilecolons and dots represent very similar and similar amino acids, respectively.
I.D. Wijnberg et al. / Neuromuscular Disorders 22 (2012) 361–367 365
PCR products (Table 1) and subsequently re-sequencedfrom both directions as described above. No alterationsof the transcript could be observed apart from the poly-morphisms detected at the genomic level.
3. Discussion
Myotonia is a primary skeletal muscle disease character-ized by delayed relaxation of skeletal muscle after sudden
forceful contraction, sometimes associated with clinicalsigns of weakness. In humans, congenital myotonia, typeThomsen [14] and generalized myotonia, type Becker [15]are inherited as an autosomal dominant and recessive trait,respectively [3]. Mutations of the CLCN1 gene have beenidentified in adr/adr mice [8] and “fainting” goats [4]. Fur-ther, CLCN1 animal models have been described in dogs.Independent autosomal CLCN1 mutations, with a reces-sive mode of transmission, cause myotonic phenotypes in

366 I.D. Wijnberg et al. / Neuromuscular Disorders 22 (2012) 361–367
Miniature Schnauzers [16] and in Australian Cattle Dogs[11]. The pony presented in this report demonstrated clini-cal signs and EMG changes strikingly similar to thoseobserved in myotonic goats, supporting our assumptionthat the disease phenotype in these two domestic speciesmight have been caused by independent CLCN1 genemutations [5,12]. Blood from the foal was examined forthe presence of mutations in the CLCN1 gene and foundto be homozygous for the missense mutation c.1755A>Cin exon 15. The associated CLCN1 mutation was probablyintroduced by the grandfather of the affected foal, as allcarriers identified so far could be traced back to this stal-lion (Fig. 4A). This putative founder was the paternalgrandfather and the maternal grand-grandfather of theaffected foal. Sequencing of unrelated controls of differentbreeds (Supplementary Table 2) showed that the CLCN1
c.1775A>C polymorphism was not present in other horses.The fact that none of the examined horses unrelated to theNew Forest pony breed was shown to be carrier of the dis-ease associated allele, indicates that this might be a quiterecent mutation, limited to a single breeding line whichhas not yet spread into the general population. It is possiblethat a spontaneous CLCN1 germline mutation occurred inthe assumed founder stallion.
The consequence of the c.1775A>C mutation was thesubstitution of aspartic acid for alanine in codon 592 ofthe C-terminal cytoplasmic domain of the equine CLCN1protein. There was no indication that this mutation affectssplicing of CLCN1. The affected amino acid position ishighly conserved across mammalian orthologues (Fig. 5).The human CLCN1 protein is enclosed by two related pro-tein domains, the voltage gated chloride channel and theCBS domain, respectively [17]. As CLCN1 functions as ahomooligomer, probably consisting of four subunits [17],it is possible that the codon 592 mutation affects the properformation of the functional ion channel. The potentialimpact of this myotonia associated amino acid substitutionwas evaluated by PolyPhen (http://genetics.bwh.har-vard.edu/pph/). The substitution affected a strongly con-served residue and was predicted to be “probablydamaging” with a PSIC score difference of 2.25, whichimplies that the substitution is very likely to affect proteinfunction or structure. The deficiency of functional chloridechannels is a very plausible explanation for the observedphenotype. The equine missense mutation seems to be per-fectly associated with the condition even though it islocated distant to the causative CLCN1 mutation describedin “fainting” goats [4,6,18].
The morphometric results resemble data reported forhuman patients affected by congenital myotonia: a selectiveatrophy of type I fibers has been observed in human skele-tal muscles [19] together with muscle fiber hypertrophy [1].However, breed and age differences do exist in muscle mor-phology [20] and muscle tissue of other New forest ponieswas not available to strengthen these conclusions. Humancongenital myotonia has also been reported to be resultof a heterogeneous trait: apart from the association of con-
genital myotonia with type I fiber atrophy, generalizedmuscle fiber hypertrophy was observed in three quartersof patients affected by recessive myotonia, whereas theremainder had normal muscle fiber size [1,21]. A moderatehypertrophy of myotonic fibers has previously beenreported in both myotonic goats, [22] and more recentlyin myotonic dogs [23]. Muscle enzyme activities were notavailable from this patient but are reported to vary fromnormal to moderately elevated [2,11] and therefore donot provide conclusive evidence.
In conclusion, this study reports the first case of congen-ital myotonia in horses for which the molecular genetic causewas analyzed and which was associated with a missensemutation of the equine CLCN1 gene. It illustrates the impor-tance of precise clinical recognition of putative inherited dis-eases and illustrates once again the value of the recentlydeciphered horse genome sequence in unraveling the molec-ular basis of such genetic anomalies. Our finding enablesdirect genetic testing and the eradication of this genetic dis-ease from the New Forest pony breeding population. Fur-thermore, this study provides an additional geneticallycharacterized large animal model for human myotonia.
Acknowledgements
The authors greatly appreciate the cooperation and sup-port of the breeders of the horses involved and the techni-cal assistance of E.R. Boom and the valuable help ofGiovanni Caporale and R. van den Boom.
Appendix A. Supplementary data
Supplementary data associated with this article can befound, in the online version, at doi:10.1016/j.nmd.2011.10.001.
References
[1] Lossin C, George Jr AL. Chapter 2 myotonia congenita. In: RouleauGuy, Gaspar Claudia, editors. Advances in genetics. Academic Press;2008. p. 25–55.
[2] F- Wu, Ryan A, Devaney J, Warnstedt M, Korade-Mirnics Z, PoserB, et al. Novel CLCN1 mutations with unique clinical and electro-physiological consequences. Brain 2002;125:2392–407.
[3] Kullmann DM. Neurological channelopathies. Annu Rev Neurosci2010;33:151–72.
[4] Beck CL, Fahlke C, George Jr AL. Molecular basis for decreasedmuscle chloride conductance in the myotonic goat. Proc Natl AcadSci USA 1996;93:11248–52.
[5] Cherian A, Baheti N, Kuruvilla A. Muscle channelopathies andelectrophysiological approach. Ann Indian Acad Neurol2008;11:20–7.
[6] Bryant SH, Conte-Camerino D. Chloride channel regulation in theskeletal muscle of normal and myotonic goats. Pflugers Archiv1991;417:605–10.
[7] Steinberg S, Botelho S. Myotonia in a horse. Science1962;137:979–80.
[8] Gronemeier M, Condie A, Prosser J, Steinmeyer K, Jentsch TJ,Jockusch H. Nonsense and missense mutations in the muscularchloride channel gene Clc- 1 of myotonic mice. J Biol Chem1994;269:5963–7.

I.D. Wijnberg et al. / Neuromuscular Disorders 22 (2012) 361–367 367
[9] Wijnberg ID, Van Der Kolk JH, Franssen H, Breukink HJ. Needleelectromyography in the horse compared with its principles in man: areview. Equine Vet J 2003;35:9–17.
[10] Wijnberg ID, van der Kolk JH, Franssen H, Breukink HJ. Electro-myographic changes of motor unit activity in horses with inducedhypocalcemia and hypomagnesemia. Am J Vet Res 2002;63:849–56.
[11] Finnigan DF, Hanna WJB, Poma R, Bendall AJ. A novel mutation ofthe CLCN1 gene associated with myotonia hereditaria in anAustralian cattle dog. J Vet Intern Med 2007;21:458–63.
[12] Arzel-Hezode M, Sternberg D, Tabti N, et al.. Homozygosity fordominant mutations increases severity of muscle channelopathies.Muscle Nerve 2010;41:470–7.
[13] Drogemuller C, Drogemuller M, Leeb T, et al.. Identification of amissense mutation in the bovine ATP2A1 gene in congenitalpseudomyotonia of Chianina cattle: an animal model of humanBrody disease. Genomics 2008;92:474–7.
[14] George Jr AL, Crackower MA, Abdalla JA, Hudson AJ, Ebers GC.Molecular basis of Thomsen’s disease (autosomal dominant myotoniacongenita). Nat Genet 1993;3:305–10.
[15] Koch MC, Ricker K, Otto M, et al.. Linkage data suggesting allelicheterogeneity for paramyotonia congenita and hyperkalemic periodicparalysis on chromosome 17. Hum Genet 1991;88:71–4.
[16] Rhodes TH, Vite CH, Giger U, Patterson DF, Fahlke C, George JrAL. A missense mutation in canine ClC-1 causes recessive myotoniacongenita in the dog. FEBS Lett 1999;456:54–8.
[17] Steinmeyer K, Lorenz C, Pusch M, Koch MC, Jentsch TJ. Multimericstructure of CIC-1 chloride channel revealed by mutations indominant myotonia congenita (Thomsen). EMBO J 1994;13:737–43.
[18] Camerino DC, Pierno S, De Luca A, Bryant SH. Antimyotonic effectsof tocainide enantiomers on skeletal muscle fibers of congenitallymyotonic goats. Neuromuscul Disord 2000;10:160–4.
[19] Brooke MH, Engel WK. The histographic analysis of human musclebiopsies with regard to fiber types. 3. Myotonias, myastheniagravis, and hypokalemic periodic paralysis. Neurology 1969;19:469–77.
[20] Rivero JLL, Galisteo AM, Aguera E, Miro F. Skeletal musclehistochemistry in male and female Andalusian and Arabian horses ofdifferent ages. Res Vet Sci 1993;54:160–9.
[21] Berchtold MW, Brinkmeier H, Muntener M. Calcium ion in skeletalmuscle: its crucial role for muscle function, plasticity, and disease.Physiol Rev 2000;80:1215–65.
[22] Atkinson JB, Swift LL, Lequire VS. Myotonia congenita. Ahistochemical and ultrastructural study in the goat: comparison withabnormalities found in human myotonia dystrophica. Am J Pathol1981;102:324–35.
[23] Vite CH, Cozzi F, Rich M, Klide AK, Volk SW, Lombardo R.Myotonic myopathy in a miniature Schnauzer: case report and datasuggesting abnormal chloride conductance across the muscle mem-brane. J Vet Intern Med 1998;12:394–7.