Embed Size (px)
Citation preview

1
A GENETIC INTERACTION BETWEEN THE CORE AND NS3 PROTEINS OF 1
HEPATITIS C VIRUS IS ESSENTIAL FOR PRODUCTION OF INFECTIOUS VIRUS 2
3
Daniel M. Jones1, Ali M. Atoom1, Xiaozhen Zhang2, Shyamasundaran Kottilil2 and 4
Rodney S. Russell1 5
6
1Immunology and Infectious Diseases, Faculty of Medicine, Memorial University of 7
Newfoundland, St. John’s, Newfoundland, Canada, A1B 3V6 and 2Immunopathogenesis 8
Section, Laboratory of Immunoregulation, National Institute of Allergy and Infectious 9
Diseases, National Institutes of Health, Bethesda, MD, USA 20892 10
11
Running title: Interaction between HCV core and NS3 proteins 12
Word count (abstract): 250 13
Word count (manuscript excluding references and figure legends): 6382 14
Corresponding author: [email protected] 15
16
17
18
Copyright © 2011, American Society for Microbiology and/or the Listed Authors/Institutions. All Rights Reserved.J. Virol. doi:10.1128/JVI.05313-11 JVI Accepts, published online ahead of print on 28 September 2011
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

2
ABSTRACT 19
By analogy to other members of the Flaviviridae family, the hepatitis C virus (HCV) core 20
protein is presumed to oligomerize to form the viral nucleocapsid, which encloses the 21
single-stranded RNA genome. Core protein is directed to lipid droplets (LDs) by Domain 22
2 (D2) of the protein and this process is critical for virus production. Domain 1 (D1) of 23
core is also important for infectious particle morphogenesis, although its precise 24
contribution to this process is poorly understood. In this study, we mutated amino acids 25
64-75 within D1 of core and examined the ability of these mutants to produce infectious 26
virus. We found that residues 64-66 are critical for generation of infectious progeny, 27
whereas 67-75 were dispensable for this process. Further investigation of the defective 28
64-66 mutant (termed JFH1T-64-66) revealed it to be incapable of producing infectious 29
intracellular virions, suggesting a fault during HCV assembly. Furthermore, isopycnic 30
gradient analyses revealed that JFH1T-64-66 assembled dense intracellular species of 31
core, presumably representing nucleocapsids. Thus, amino acids 64-66 are seemingly 32
not involved in core oligomerization/nucleocapsid assembly. Passaging of JFH1T-64-66 33
led to the emergence of a single compensatory mutation (K1302R) within the helicase 34
domain of NS3 that completely rescued its ability to produce infectious virus. 35
Importantly, the same NS3 mutation abrogated virus production in the context of wild-36
type core protein. Together, our results suggest that residues 64-66 of core D1 form a 37
highly specific interaction with the NS3 helicase that is essential for the generation of 38
infectious HCV particles at a stage downstream of nucleocapsid assembly. 39
40
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

3
INTRODUCTION 41
Hepatitis C virus (HCV) typically establishes chronic infections of the liver that frequently 42
lead to severe pathologies including cirrhosis and hepatocellular carcinoma. The current 43
combination therapy of pegylated-IFN-α and ribavirin is only partially effective and is 44
associated with numerous side effects. Treatment for HCV is currently transitioning to 45
the use of direct-acting antiviral (DAA) therapy, which has been specifically designed to 46
target viral proteins essential for HCV replication (30). While early results indicate that 47
DAA compounds show promise, a wider range of treatments targeting multiple aspects 48
of the viral life cycle would offer improved therapeutic options to infected individuals. In 49
this regard, a better understanding of HCV assembly could provide an alternate 50
exploitable target. 51
HCV is an enveloped virus possessing a single-stranded positive-sense RNA genome 52
that encodes a polyprotein of ~3000 amino acids (32). Cleavage of this polyprotein by 53
host- and virus-encoded proteases yields the structural (core, E1 and E2) and non-54
structural (NS2, NS3, NS4A, NS4B, NS5A and NS5B) proteins as well as p7, a small 55
peptide located between E2 and NS2 that is currently unassigned into either category. 56
The HCV core protein is presumed to oligomerize to form the viral nucleocapsid into 57
which the RNA genome is packaged during the early stages of virion assembly, 58
whereas E1 and E2 lie within the lipid envelope surrounding the nucleocapsid and 59
mediate cell attachment and entry (see (22) for review) . While no conclusive proof has 60
shown they comprise physical virion components, multiple studies have revealed the 61
importance of p7 and NS2 for the production of infectious virus particles (see (16) and 62
(4) for review). NS3-NS5B are essential for HCV RNA synthesis (25) and localize to 63
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

4
replication complexes (RCs) embedded within cellular endoplasmic reticulum (ER) 64
membranes (8, 13, 41, 48). However, many of the non-structural proteins are also 65
engaged in the generation of infectious virions, with reports of NS3 (14, 26, 53), NS4A, 66
(38) NS4B (17, 40) and NS5A (3, 27, 31, 50, 51) being essential for, or at least 67
contributing to, this process. It is therefore becoming apparent that the assembly of 68
infectious virus particles is a complex, multi-step mechanism involving the majority of 69
the HCV-encoded proteins. 70
Upon translation, HCV core (and the nascent polypeptide chain) is targeted to the ER 71
membrane by a signal peptide located between core and E1 where signal peptidase 72
cleavage liberates the immature, 21kDa form of the protein (43). Core is retained at the 73
ER membrane until further cleavage by signal peptide peptidase releases the signal 74
peptide to generate the mature, 19kDa core protein which then trafficks to the surface of 75
cellular lipid droplets (LDs) (15, 29). This interaction between core and LDs is essential 76
for virus production and is mediated by 2 amphipathic α-helices within the C-terminal 77
half of core (termed D2) (5). Meanwhile, the N-terminus of core (D1) harbours multiple 78
positively-charged residues and is speculated to (i) interact with viral RNA (10, 46, 47) 79
and (ii) harbour core-core interaction sites that permit assembly of the HCV 80
nucleocapsid (19, 28, 35, 36). Several studies have shown that mutations within both 81
domains of core can impair virion generation (2, 20, 34). However, while manipulation of 82
core D2 generally abrogates the critical association between core and LDs, the 83
mechanisms by which D1 residues affect virus production remain largely obscure. In an 84
attempt to gain a clearer insight into the role of core D1, we examined the contribution 85
of a select stretch of amino acids within this region to the generation of infectious HCV. 86
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

5
These mutants were extensively characterized and revealed a previously unreported 87
genetic interaction between core and NS3 that is essential for the production of 88
infectious virus particles. 89
90
MATERIALS AND METHODS 91
Plasmids and cloning. JFH1T harbours 3 amino acid changes (N417S [E2], N765D 92
[p7] and Q1012R [NS2]) that enhance infectious virus production and was derived from 93
the cell culture-adapted JFH1 strain JFH-AM1 (42). To create ΔE1/E2, JFH1T was used 94
as a template to amplify 2 fragments by PCR. Fragment 1 incorporated the AgeI 95
restriction site within the 5’UTR and introduced an AvrII site close to the N-terminus of 96
E1. Fragment 2 harboured a KpnI site within NS2 and introduced an AvrII site near the 97
C-terminus of E2. Following PCR amplification, fragments 1 and 2 were digested with 98
AgeI/AvrII and KpnI/AvrII, respectively. Fragments 1 and 2 were then ligated into a 99
JFH1T backbone digested with AgeI and KpnI in a 3-piece ligation reaction. The 100
resultant ΔE1/E2 construct contains the N-terminal 19aa of E1 and the C-terminal 82aa 101
of E2. Thus, the core/E1 and E2/p7 cleavage sites are maintained whilst infectious virus 102
production is abrogated. ΔGDD was created using the QuikChange II XL Site-Directed 103
Mutagenesis Kit (Stratagene). Complementary forward and reverse primers omitting the 104
9 nucleotides encoding the GDD sequence were used in a PCR reaction with JFH1T 105
providing the template. The resultant ΔGDD construct therefore harbours an in-frame 106
deletion of the GDD motif, effectively eliminating NS5B polymerase function. The 4 core 107
mutants (64-66, 67-69, 70-72 and 73-75) were also generated using the QuickChange II 108
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

6
XL Site-Directed Mutagenesis Kit using either JFH1T or JFH1 as a template. Here, 109
forward and reverse primers were designed to change blocks of 3 amino acids spanning 110
the 64-75 sequence to alanine (see Figure 1). All primer sequences are available upon 111
request. All plasmids were sequenced completely (both positive and negative strands) 112
to ensure authenticity of the constructs. 113
Cell culture. Huh-7.5 and S29 cells were propagated in Dulbecco’s modified Eagle’s 114
medium (DMEM, Invitrogen) supplemented with 10% foetal calf serum and 1% 115
penicillin/streptomycin to give DMEM complete (DMEMcomp). All cells were maintained 116
by incubation at 37ºC with 5% CO2. 117
Antibodies. The following antibodies were used in this study: mouse anti-HCV core 118
monoclonal antibody [B2] (Anogen); mouse anti-HCV NS3 monoclonal antibody 119
[C65371M] (Meridian Life Sciences); mouse anti-GAPDH monoclonal antibody [ab8245] 120
(Abcam); sheep anti-NS5A polyclonal antibody (a kind gift from Mark Harris, University 121
of Leeds, UK); goat anti-mouse and donkey anti-sheep IgG-HRP secondary antibodies 122
(Santa Cruz Biotechnology) and Alexa Fluor® 488 and 594 secondary antibodies 123
(Invitrogen). LDs were detected using LipidTOX (Invitrogen). For detection of core in 124
gradient analyses, mouse anti-HCV core monoclonal antibody [MA1-080] (Pierce 125
Research) was used. All antibodies were diluted 1/1000, except when B2 was used for 126
immunofluorescence, in which a 1/200 dilution was used. LipidTOX was used at a 1/66 127
dilution. 128
In vitro transcription and RNA transfection. 1x106 Huh-7.5 or S29 cells (per 129
transfection) were plated in 10cm cell culture dishes 24hrs prior to transfection. On the 130
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

7
day of transfection the media was removed and replaced with 2ml of serum-free DMEM. 131
Plasmid DNA encoding JFH1T, ΔE1/E2, ΔGDD and the 4 JFH1T (or JFH1) core mutants 132
(JFH1T-64-66, JFH1T-67-69, JFH1T-70-72 and JFH1T-73-75) was linearized by XbaI 133
digestion for 2 hrs at 37ºC. RNA was transcribed from 1µg of linearized constructs using 134
the T7 RiboMAX Express Large Scale RNA Production System (Promega). RNA 135
transcripts were transfected using Lipofectamine 2000 (Invitrogen) in accordance with 136
the manufacturer’s instructions (15µl lipofectamine per transfection). Lipofectamine/RNA 137
mixtures were added to cells and incubated at 37ºC with 5% CO2 for 4hrs. At this point, 138
the transfection mixture was removed and replaced with 6ml of DMEMcomp. Cells were 139
processed 72hrs post-transfection. For gradient analyses, 1.5x106 cells were seeded 140
per dish and harvested 48hrs post-transfection. 141
Infectious HCV titre determination. 24hrs prior to infection, 8-well chamber slides 142
(Lab-Tek) were seeded with Huh-7.5 cells (4x105 cells per well). On the day of infection 143
(72hrs post-transfection), cell supernatants were removed and passed through a Millex-144
HV 45µm filter (Millipore) before being serially diluted 10-fold in DMEMcomp. 100µl of 145
each dilution was then used to infect Huh-7.5 cells plated in chamber slides for 4hrs 146
before the infectious media was removed and replaced with fresh DMEMcomp. 72hrs 147
post-infection, cells were fixed and stained with anti-core antibody. Viral titres are 148
expressed as the number of focus-forming units (ffu) per ml of supernatant. 149
Preparation of intracellular lysates for infectious HCV titre determination and 150
gradient analysis. 72hrs post-transfection, cells were trypsinized, pelleted by 151
centrifugation at 400xg and resuspended in 1ml (for intracellular titre determination) or 152
500µl (for gradient analysis) DMEMcomp. These cells were then subjected to 4 freeze-153
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

8
thaw cycles (3mins freeze, 3mins thaw) before cell debris was pelleted by centrifugation 154
at 3000xg. Intracellular supernatants were then used to determine infectious titres (as 155
described above) or centrifuged through iodixanol gradients (see below). 156
Iodixanol gradient analyses. 500µl intracellular lysates prepared by freeze-thaw lysis 157
were layered over a 4.5ml continuous 10-50% iodixanol gradient (prepared using 158
OptiPrep Density Gradient Medium [Sigma] and Hanks’ Balanced Salt Solution 159
[Invitrogen]). Gradients were ultracentrifuged using a Beckman SW55 Ti rotor at 160
100,000xg for 16hrs at 4ºC. 10x500µl fractions were collected starting from the top of 161
each tube. Proteins were extracted from each fraction by methanol precipitation and 162
pelleted by centrifugation. Pellets were resuspended in 50µl SDS loading buffer, boiled 163
and then probed for core protein by Western blot analysis. RNA and virus 164
measurements were taken from a second gradient run in parallel to that used for core 165
detection. RNA levels were measured by Real-Time quantitative RT-PCR using JFH1-166
specific primers/probe in an ABI 7500 system as previously described (6) (primer 167
sequences and protocols available on request). 168
Indirect immunofluorescence. Cells grown on chamber slides or glass coverslips 169
were fixed in 100% acetone for 2mins, washed with phosphate buffered saline (PBS) 170
and incubated with primary antibody for 20mins. Following this, cells were washed with 171
PBS and incubated with secondary antibody conjugated to the appropriate fluorophore 172
for 20mins. Finally, cells were washed with PBS before being mounted using 173
Vectashield Hard Set Mounting Medium with DAPI (Vector Laboratories). When 174
examining core-LD association, cells were instead fixed using 4% paraformaldehyde for 175
20mins, followed by permeabilization with 0.1% Triton X-100 prepared in PBS for 176
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

9
15mins. Images were recorded using 10x magnification, except for the core-LD studies 177
where 100x magnification was used. 178
Preparation of samples for SDS-PAGE and Western blot analysis. For intracellular 179
protein detection, cells were trypsinized, pelleted by centrifugation at 8000xg and 180
resuspended in 300µl Passive Lysis Buffer (Promega) before incubation on ice for 181
30mins. Next, cell debris was pelleted by centrifugation and ~3% of resultant lysate was 182
mixed with SDS loading buffer at a 1:1 ratio and analyzed by SDS-PAGE. For detection 183
of core in supernatants, 6ml of supernatants taken from cells 72hrs post-transfection 184
were passed through a 0.45µm filter and layered over a 2ml 20% sucrose cushion. 185
Samples were ultracentrifuged using a Sorvall TH-641 rotor at 80,000xg for 4hrs at 4ºC. 186
Following this, supernatants and sucrose were discarded and the bottom of the tube 187
was washed with 30µl SDS loading buffer, of which 50% was loaded for SDS-PAGE. 188
RNA extraction, processing and sequencing. To identify compensatory mutations 189
that had arisen during passage of the core 64-66 mutant, supernatant from the P3 plate 190
was first used for 2 rounds of infection (I1 and I2 in Figure 7) to enrich the rescued virus 191
population. RNA was extracted from I2 supernatants using TRIzolLS (Invitrogen) and 192
long RT-PCR was performed as previously described (42, 49) Sequencing was 193
performed at the Centre for Applied Genomics at the Hospital for Sick Children 194
(Toronto). For the extraction of RNA from gradient fractions (Figure 6), the QIAamp Viral 195
RNA Mini Kit (Qiagen) was used in accordance with the manufacturer’s instructions. 196
197
198
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

10
RESULTS 199
Amino acids 64-66 of core are essential for infectious virus production. Several 200
groups have recently performed mutagenic analysis of the HCV core protein (2, 20, 34) 201
and these studies revealed a large number of amino acids to be important for infectious 202
virus production. We focussed our attention on a small stretch of residues within D1 of 203
core spanning amino acids 64-75. There were several reasons for this rationale. Firstly, 204
during long-term culture of wild-type JFH1, two independent studies identified a 205
potentially adaptive mutation (K74T) within this region ((55) and Emerson, SU, 206
unpublished data). Additionally, these residues have previously been shown to be 207
essential for virus production from a chimeric virus termed J6/JFH1, which produces 208
high viral titres in cell culture (34). Finally, forced evolution studies performed on mutant 209
viruses containing amino acid changes in this region of core identified compensatory 210
mutations in p7 and NS2, implying the existence of critical interactions between this 211
region of core and other viral proteins (34). 212
To test whether these amino acids were also essential in the background of a non-213
chimeric genome, we made 4 mutants (JFH1T-64-66, JFH1T-67-69, JFH1T-70-72 and 214
JFH1T-73-75) in which triplets of amino acids were mutated to alanine (Figure 1). In the 215
case of JFH1T-73-75, a valine was introduced at the final position since the original 216
amino acid was alanine. These mutants were generated in the background of JFH1T – a 217
JFH1 strain harbouring 3 adaptive mutations within E2 (N417S), p7 (N765D) and NS2 218
(Q1012R) that produces superior levels of infectious virus compared to wild-type JFH1 219
(42). To test whether these mutants were capable of producing infectious virus, Huh-7.5 220
cells were transfected with RNA from each mutant and visualized 72hrs later by indirect 221
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

11
immunofluorescence (Figure 2A). Detection of core indicated that >90% of cells were 222
HCV-positive when expressing RNA encoding JFH1T, JFH1T-67-69, JFH1T-70-72 or 223
JFH1T-73-75. By contrast, a low number of core-positive cells were observed when 224
transfected with JFH1T-64-66 RNA. This pattern of core expression was similar to that 225
visualized with ΔE1/E2, which is incapable of producing infectious particles. As 226
expected, cells transfected with replication-incompetent ΔGDD RNA exhibited no core 227
signal. To confirm these results quantitatively, supernatants from transfected cells were 228
used to determine virus titres for each viral RNA (Figure 2B). JFH1T-67-69, JFH1T-70-72 229
and JFH1T-73-75 all produced high titres of infectious virus, ranging from 3.4 – 7.3x105 230
ffu/ml, whereas JFH1T-64-66 generated titres of only 20 ffu/ml. As expected, cells 231
transfected with ΔE1/E2 and ΔGDD produced no detectable infectious virus. The effects 232
of core mutagenesis were not influenced by the adaptive mutations within JFH1T, since 233
an identical pattern of results were obtained when the mutations were introduced into 234
wild-type JFH1 RNA (Figure 2C). Finally, to ensure the reduced level of particle 235
production from JFH1T-64-66 was not a result of core being rendered unstable by the 236
mutagenesis, cell lysates were probed for the presence of core protein (Figure 2D). All 237
viruses with the exception of ΔGDD produced a detectable core band, indicating that 238
core was stably expressed in all cases. While the level of core was lower for JFH1T-64-239
66 compared to the other mutants and JFH1T, it was comparable to expression levels 240
exhibited by ΔE1/E2. Therefore, this lowered detection of core is not a consequence of 241
instability, but due to the lack of virus spread in cell culture as described above (Figure 242
2A). Taken together, these data indicate that core residues 64-66 are essential for the 243
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

12
generation of infectious virus whereas residues 67-75 contribute minimally to this 244
process. 245
246
JFH1T-64-66 cannot generate infectious intracellular particles. The reduced level of 247
infectious virion production by the JFH1T-64-66 mutant may result from several 248
possibilities. For instance, cells transfected with JFH1T-64-66 RNA might release HCV 249
particles into the supernatant as normal, but they may be non-infectious. Alternatively, 250
infectious HCV particles may be assembled but not released from cells. To address 251
these possibilities, supernatants from cells transfected with JFH1T, ΔE1/E2, ΔGDD and 252
each of the core mutants were subjected to ultracentrifugation through 20% sucrose 253
cushions to isolate any viral particles present. The resultant pellets were then probed for 254
the presence of core by Western blot (Figure 3A). Core was detectable in supernatants 255
from cells expressing JFH1T, JFH1T-67-69, JFH1T-70-72 or JFH1T-73-75. By 256
comparison, no protein was detectable in samples from ΔE1/E2, ΔGDD and JFH1T-64-257
66. This result argues against the release of non-infectious particles by JFH1T-64-66, 258
suggesting a fault with a step prior to virus release. To determine whether JFH1T-64-66 259
could form infectious intracellular particles, transfected cells were lysed by multiple 260
freeze-thaw cycles as previously described (12) and lysates were used for virus titre 261
measurement (Figure 3B). Intracellular titres for all RNAs were lower than the 262
extracellular values, and ranged from 4.3x104 – 1.6x105 ffu/ml for JFH1T, JFH1T-67-69, 263
JFH1T-70-72 and JFH1T-73-75. By comparison, JFH1T-64-66 produced intracellular 264
titres of <10 ffu/ml. Therefore, core residues 64-66 appear to be critical for the assembly 265
of infectious intracellular particles. 266
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

13
267
Analysis of core mutant viruses using a single-cycle virus production assay. Virus 268
titre data gathered using Huh-7.5 cells can amplify differences between infectious and 269
non-infectious genomes because these cells support multiple rounds of infection, and 270
as such can provide the mutant viruses with an opportunity to revert. To determine titres 271
from a single burst of virus production from each viral RNA, S29 cells were transfected 272
with JFH1T, ΔE1/E2, ΔGDD or the 4 core mutants. S29 cells are a subclone of Huh-7 273
cells that express almost no CD81, the essential HCV entry receptor, on their cell 274
surface (42) and are therefore 1000-fold less susceptible to HCV infection than Huh-7.5 275
cells. Consistent with this, observation of core staining in transfected cells indicated that 276
all constructs (with the exception of ΔGDD, where core was undetectable) were unable 277
to spread in cell culture, regardless of their capacity to generate infectious virus (Figure 278
4A). Both extracellular and intracellular virus titres obtained from transfected S29 cells 279
were lower than those from Huh-7.5 cells by ~1 log (compare Figures 4B and 3B), 280
which demonstrates the lack of amplification of virus titres that is normally observed in 281
Huh-7.5 cell cultures. Using this assay however, larger differences were observed 282
between the core mutants (Figure 4B). Although extracellular virus titres of JFH1T, 283
JFH1T-67-69 and JFH1T-70-72 were similar, levels of intracellular virus produced by the 284
core mutants were ~1 log lower compared to JFH1T. Thus, it is possible that residues 285
67-72 may partially contribute to infectious particle assembly, but are by no means 286
essential since extracellular virus titres remained comparable to those seen with JFH1T. 287
By contrast, residues 73-75 seemingly offer little or no role in infectious virus production, 288
as both intracellular and extracellular titres from 73-75 were similar to those of JFH1T. In 289
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

14
agreement with data obtained using Huh-7.5 cells, S29 cells transfected with RNA 290
encoding JFH1T-64-66 produced very little infectious extracellular virus (<10 ffu/ml) and 291
intracellular particles were undetectable. Finally, although core has no documented role 292
in viral RNA replication, it was still necessary to confirm that the effects on virus 293
production from JFH1T-64-66 were not a consequence of altered RNA synthesis. S29 294
cells were more appropriate than Huh-7.5 cells for this assay since they negate the 295
effects of virus spread, meaning viral proteins should only be expressed in cells that 296
were successfully transfected. Therefore, lysates from S29 cells were probed for the 297
presence of core protein 72hrs post-transfection (Figure 4C). With the exception of 298
ΔGDD, all viruses produced robust levels of core protein. Importantly, the level of core 299
produced by JFH1T-64-66 was comparable to that seen with JFH1T, indicating that the 300
low level of virus produced by JFH1T-64-66 did not result from aberrant RNA replication. 301
Overall, these results confirm that core residues 64-66 play an essential role in the 302
generation of infectious HCV particles. 303
304
Core expressed from JFH1T-64-66 localizes to the surface of lipid droplets. The 305
association of core with LDs is essential for the production of infectious virus and is 306
mediated by 2 amphipathic helices within D2 of the protein (5). Although residues 64-66 307
lay well outside this region, we wished to rule out the possibility that the block in virus 308
production from JFH1T-64-66 was due to an impact on the targeting of core to LDs. 309
Accordingly, cells transfected with RNA encoding JFH1T or JFH1T-64-66 were fixed and 310
probed for core and LDs before being examined by immunofluorescence confocal 311
microscopy (Figure 5). Core expressed from both JFH1T and JFH1T-64-66 was clearly 312
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

15
present on the surface of cellular LDs and examination of several cells revealed no 313
differences between them. Therefore, the decrease in virus production observed for 314
JFH1T-64-66 did not result from disrupting the association of core with LDs. 315
316
JFH1T-64-66 produces dense core-containing species similar to those observed 317
with JFH1T. The data outlined above suggest that core residues 64-66 are important for 318
the assembly of infectious particles at a stage following the recruitment of core to LDs. 319
Mutation of these residues could cause a defect (i) in core oligomerization that prevents 320
viral nucleocapsid construction or (ii) at a post-nucleocapsid assembly step. Therefore, 321
it was important to determine whether JFH1T-64-66 was able to form nucleocapsids, 322
especially considering this region has been previously implicated in core-core 323
interactions (28). To investigate this, intracellular lysates from cells harbouring JFH1T or 324
JFH1T-64-66 were ultracentrifuged through 10-50% iodixanol gradients and 10 fractions 325
were removed and analyzed for the presence of core, HCV RNA and intracellular 326
infectious virus (Figure 6). As a control, lysates from cells transfected with ΔE1/E2 were 327
also examined. For JFH1T, the highest concentration of core was present in fractions 1 328
(1.02g/cm3) and 7 (1.17g/cm3), with bands also detectable in fractions 6 and 8 329
(1.15g/cm3 and 1.20g/cm3 respectively, Figure 6A). We speculate that core species in 330
the lower fractions (6-8) represent various species of core oligomers/nucleocapsids, 331
since presumably these would be sufficiently dense to traverse farther into the gradient, 332
whereas protein in fraction 1 is likely to represent free core, LD-associated core and/or 333
lower-order core structures. Intracellular infectious particles were detected in all 334
fractions and peaked in fraction 6. This fraction also contained one of two peaks in viral 335
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

16
RNA, with the second being detected further up the gradient in fraction 2 (1.04g/cm3). 336
We speculate that the RNA peak in fraction 2 may represent a pool of HCV genomes 337
associated with structures such as replication complexes, since (i) these would be 338
expected to be rich in HCV RNA and (ii) low infectivity was associated with this fraction. 339
By contrast, the peak of RNA in fraction 6 correlates with both a high concentration of 340
infectious particles and dense core-containing species, strongly suggesting the 341
presence of assembled intracellular particles at this density. We consistently observed 342
that the peak of RNA and infectious titre (fraction 6) did not correlate with the peak of 343
core protein, implying that the core-containing species in fractions 7 and 8 may 344
represent nucleocapsids at earlier stages of maturation that have yet to increase in 345
buoyancy and associate with viral genomes. This theory is supported by the observation 346
that intracellular particles increase in buoyancy during viral egress as a result of the 347
continued addition of lipids to exiting HCV virions (12). 348
Analysis of JFH1T-64-66 iodixanol gradients revealed distributions of core and viral RNA 349
that were remarkably similar to that seen with JFH1T (Figure 6B). However, the reduced 350
ability of this mutant to produce virus meant that no infectious particles could be 351
detected in any fraction. Results obtained from JFH1T-64-66 were similar to those 352
observed with ΔE1/E2, which is believed to form intact nucleocapsids according to 353
gradient analysis (2) but not infectious particles due to the lack of envelope 354
glycoproteins. Overall, the data obtained from iodixanol gradient analyses show that 355
JFH1T-64-66 is still able to form dense intracellular species of core that are associated 356
with high levels of viral RNA. If these species indeed represent nucleocapsids, the data 357
suggest that this mutant is not compromised for their assembly. 358
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

17
359
Passaging JFH1T-64-66 leads to the emergence of a compensatory mutation 360
within NS3. In order to gain a clearer understanding as to how core residues 64-66 361
contribute to the production of infectious progeny, Huh-7.5 cells were transfected with 362
RNA encoding JFH1T-64-66 and serially passaged to encourage the emergence of 363
compensatory mutations that might correct the defect in virus production (Figure 7A). 364
Cells were passaged each time they reached confluency and virus production was 365
monitored by (i) detection of core protein in the transfected cells and (ii) measurement 366
of virus titre within the supernatants taken on the day of passage. Two independent 367
experiments were performed (referred to as 64-66 I and 64-66 II in Figure 7) since we 368
wished to determine whether different patterns of compensatory mutations would 369
emerge in independent cultures. For comparative purposes, cells were also transfected 370
with RNA from JFH1T or ΔGDD, although passaging was not performed on these 371
viruses. JFH1T exhibited core staining in >90% of cells and produced virus titres of 372
4.3x105 ffu/ml at passage 1 (P1, 3 days post-transfection) whereas core staining and 373
virus production were undetectable in cells harbouring ΔGDD (Figures 7B and C). As 374
observed previously, JFH1T-64-66 produced little virus at P1. However, virus production 375
from JFH1T-64-66 had increased by P2 (7 days post-transfection) and reached levels 376
comparable to JFH1T by P3 (12 days post-transfection) in both experiments. To enrich 377
any rescued virus present within the medium, supernatants from the P3 cells were used 378
to infect naive Huh-7.5 cells (I1) and 72 hrs later, supernatant from the I1 dish was used 379
to infect naive cells for a second round (I2, Figure 7A and D). Virus titres of I1 and I2 380
supernatants were comparable to those seen with JFH1T, indicating the likely 381
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

18
emergence of rescued virus that had corrected the defects in virus production (Figure 382
7D). At this point, RNA was extracted from the I2 supernatants and full HCV genome 383
sequencing was performed. Surprisingly, both experiments (I and II) revealed the 384
presence of a single amino acid change (K1302R, resulting from codon change AAA – 385
AGA at nucleotide position 4245) within the helicase domain of NS3. All original 386
mutations (64-66 in core and the adaptive mutations in E2, p7 and NS2) remained 387
present. 388
389
K1302R specifically rescues virus production from JFH1T-64-66. To determine 390
whether the K1302R mutation was responsible for restoring the generation of infectious 391
virus, this mutation was cloned into the original JFH1T-64-66 mutant to create JFH1T-64-392
66-KR (Figure 8A). Additionally, K1302R was introduced into JFH1T (JFH1T-KR) to 393
ensure the mutation was not acting in an adaptive manner and enhancing particle 394
generation independently of the 64-66 core mutation. RNAs encoding these constructs 395
were transfected into Huh-7.5 cells and visualized 72hrs later (Figure 8B). As observed 396
previously, only a minority of cells transfected with JFH1T-64-66 RNA were positive for 397
HCV core, consistent with the inability of this genome to generate infectious virus. By 398
comparison, JFH1T-64-66-KR RNA produced a high number of core-positive cells that 399
was reminiscent of the distribution observed with JFH1T, suggesting that K1302R was 400
able to rescue virus production in the background of the defective JFH1T-64-66 401
genome. Importantly, JFH1T-KR revealed a core-staining pattern similar to that seen in 402
cells transfected with RNA representing ΔE1/E2 or JFH1T-64-66, implying that K1302R 403
may be deleterious when expressed in a genome harbouring a wild-type core protein. 404
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

19
To confirm these data, intra- and extracellular infectious titres generated by each RNA 405
were determined (Figure 8C). While JFH1T-64-66 RNA produced 20 ffu/ml, virus titres 406
from JFH1T-64-66-KR were restored to 3.6x105 ffu/ml, comparable to levels obtained 407
with JFH1T (6.7x105 ffu/ml). Intriguingly, JFH1T-KR produced titres of only ~170 ffu/ml, 408
confirming that K1302R caused a strong defect in infectious virus production when 409
introduced into the JFH1T coding sequence. This defect occurred prior to the assembly 410
of intracellular infectious particles, since intracellular titres were reduced from 2.1x105 411
ffu/ml (JFH1T) to 15 ffu/ml (JFH1T-KR) in the presence of the K1302R mutation. To 412
determine whether K1302R altered NS3 stability or processing, cell lysates from S29 413
cells were analyzed by Western blot (Figure 8D). S29 cells were chosen for this assay 414
to bypass the effects of virus spread that would occur using Huh-7.5 cells. Intriguingly, 415
lower levels of NS3 were detected for mutants harbouring K1302R. This mutation 416
appeared to affect processing of the non-structural region in general since levels of 417
NS5A were also lower, whereas core (which is cleaved from the polyprotein by cellular 418
signal peptidase and not NS3) remained at levels comparable to genomes lacking 419
K1302R. To conclusively confirm that K1302R was able to rescue virus production from 420
JFH1T-64-66, supernatants from transfected cells were ultracentrifuged through 20% 421
sucrose cushions and the resultant pellets were probed for the presence of core (Figure 422
8E). Whereas core (i.e. virus particles) was undetectable in supernatants from JFH1T-423
64-66, a core band of equal intensity to that seen with JFH1T was observed for JFH1T-424
64-66-KR. By comparison, JFH1T-KR was unable to release detectable levels of core 425
into cell supernatants. Taken together, these results reveal that a single amino-acid 426
change within the helicase domain of NS3 is able to rescue virus production from a 427
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

20
genome containing a core mutation that abrogates generation of infectious virus. 428
Conversely, the same mutation appears to be incompatible with wild-type core protein. 429
430
DISCUSSION 431
Investigation into the processes that govern the production of infectious HCV particles 432
has only been possible since the development of a fully infectious cell culture system in 433
2005 (23, 52, 54), meaning this area of research is still in its infancy. Despite this, 434
significant steps have been taken towards understanding which viral and cellular factors 435
are important for infectious virion generation (see (4) and (16) for review). Here, we 436
show that a previously unreported genetic interaction between core and NS3 is 437
essential for the production of infectious progeny. This interaction was not required for 438
the recruitment of core to cellular LDs, nor was it important for the assembly of dense 439
core-containing species presumably representing viral nucleocapsids. Thus, it seems 440
that core and NS3 interact in a manner that contributes to virus production by an, as yet, 441
unknown mechanism. 442
Recently, several groups have attempted to determine the role of HCV core using 443
approaches that range from an alanine-scan of the majority of core, to mutating basic 444
amino acids within D1 of the protein (2, 20, 34). We also performed alanine-scanning 445
and targeted amino acids 64-75 of core D1 to examine their contribution to infectious 446
virus production. These residues have previously been shown to be important for 447
generation of virus (34) and lay within a region suggested to be involved in core 448
oligomerization (28). The results from this current study revealed that residues 64-66 449
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

21
were essential for the generation of infectious progeny, while amino acids 67-75 were 450
seemingly unimportant for this process. These results differ from those reported 451
previously, where amino acids 64-75 were all essential for, or substantially contributed 452
to, the generation of virus (34). In that study, the role of core in infectious virus 453
production was examined using a chimeric viral genome termed J6/JFH, which is 454
composed of core-NS2 sequences from the HCV genotype 2a strain J6, joined to the 455
NS3-NS5B region from JFH1 (also genotype 2a). By contrast, we mutated core in the 456
context of JFH1T – a genome that produces superior virus titres compared to JFH1, yet 457
differs by only 3 amino acids (42). Because all proteins within JFH1T are derived from 458
the same genome, it is likely that any results gained from mutational studies with this 459
construct offer a more authentic insight into the requirements for virus production since 460
presumably, no pre-existing protein incompatibilities exist. This may account for why 461
fewer amino acids were found to be essential for the generation of infectious virus from 462
JFH1T compared to previous studies utilising J6/JFH1. The effects of these mutations 463
were also confirmed in the background of wild-type JFH1, indicating that JFH1T 464
provides a suitable genetic background for the study of HCV particle assembly. 465
In this study, virus production could be completely rescued from the defective JFH1T-64-466
66 mutant upon the emergence of a single amino acid change in the helicase domain of 467
NS3 (K1302R [polyprotein numbering] or K272R [NS3 numbering]). A role for NS3 in 468
HCV assembly was first identified using an intergenotypic chimeric HCV genome 469
referred to as H-NS2/NS3-J (53), or later as HJ3 (26). This chimera harbours core-NS2 470
sequences from H77 (genotype 1a), whereas the NS3-NS5B coding region is derived 471
from JFH1 (genotype 2a). While initially unable to produce infectious virus, passaging of 472
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

22
HJ3 led to the emergence of a compensatory mutation within the helicase domain of 473
NS3 (Q1251L [polyprotein numbering], also referred to as Q221L [NS3 numbering]) that 474
rescued virus production from this chimeric genome (26, 53). Others have since found 475
that Q221L is able to enhance virus production from other chimeric genomes (44), or 476
suppress the effects of mutations that abrogate generation of virus (38). However, 477
Q221L also enhances virus production from unmodified JFH1 RNA (26) suggesting that 478
this particular NS3 mutation is a general enhancer of virus assembly, rather than a 479
mutation that merely corrects incompatibilities between intergenotypic chimeras. By 480
contrast, the K1302R mutation identified here functions by specifically correcting an 481
incompatibility between core and NS3, since (i) JFH1T-64-66 produced barely 482
detectable levels of virus in the absence of K1302R and (ii) introduction of K1302R into 483
a genome harbouring a wild-type core protein diminished virus production. Thus, a 484
genetic interaction between core residues 64-66 and NS3 residue 272 is apparently 485
essential for robust virus assembly. We propose that these 2 regions physically interact 486
in a manner that promotes the assembly of infectious virus particles. Indeed, one recent 487
study utilised purified proteins and several biochemical techniques to demonstrate that 488
core interacts with the helicase domain of NS3 and that this interaction was lost in the 489
presence of compounds that inhibited core oligomerization (33). It would therefore be 490
interesting to determine whether core protein expressed from JFH1T-64-66 or JFH1T 491
was able to interact with NS3 with and without the K1302R mutation. 492
Immunoprecipitation analyses with core and NS3 were attempted but proved 493
unsuccessful with the antibodies available to us. Therefore, attempts at addressing this 494
issue will be conducted in the future. Additionally, it was interesting to note that 495
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

23
genomes harbouring the K1302R compensatory mutation appeared to express lower 496
levels of both NS3 and NS5A compared to genomes lacking this mutation. Despite this 497
diminished level however, virus production from JFH1T-64-66 could be rescued to 498
almost wild-type levels in the presence of K1302R. It is currently unclear why a mutation 499
within the helicase domain (as opposed to the protease domain) of NS3 would affect 500
processing of the non-structural region. Similarly, it is odd that a mutation can diminish 501
the abundance of non-structural proteins within the cell, yet effectively rescue virus 502
production despite this. However, it has previously been reported that the non-structural 503
proteins exist in great excess compared to viral RNA in cells actively replicating HCV 504
subgenomic replicons (41). Therefore, it is possible that the diminished levels of NS3 505
and NS5A observed in the presence of K1302R have no overall consequence to the 506
HCV life cycle, and that the amounts of protein remaining are sufficient for their function. 507
The obvious question raised from this study is what precise role in virus assembly does 508
the interaction between core and NS3 play? Previous analysis of the HJ3 chimeric HCV 509
genome revealed it to be incapable of generating dense intracellular species of core, a 510
defect that could be corrected by the Q221L mutation in NS3 (26). This led the authors 511
to suggest that the NS3 helicase acts at a stage subsequent to the recruitment of core, 512
NS3 and NS5A to LDs, but preceding the assembly of nucleocapsids (26). However, 513
our data suggests that NS3 may serve additional functions during the assembly of 514
infectious HCV particles. JFH1T-64-66 was unable to effectively produce virions in the 515
absence of K1302R, yet, in agreement with the above study, this mutant still exhibited 516
targeting of core to LDs. In the absence of antibodies that work by immunofluorescence, 517
we were unable to determine whether NS3 and NS5A from JFH1T-64-66 were also 518
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

24
recruited to LDs. However, iodixanol gradient analysis revealed that JFH1T-64-66 519
formed dense core structures that were near identical to those seen with JFH1T. While 520
we cannot conclusively say that these structures represent bona fide nucleocapsids, the 521
fact that these fractions were also rich in viral RNA and infectious particles strongly 522
suggest this is the case. Therefore, it seems that while NS3 may contribute to 523
nucleocapsid assembly (26), the data obtained here suggest that this protein also 524
contributes to virus production in ways distinct from nucleocapsid formation. While the 525
helicase domain of NS3 has been extensively characterized (see (11) for a recent 526
review), the precise contribution of this helicase activity to the HCV life cycle is yet to be 527
defined. Similarly, the order of events that lead to the assembly of a fully infectious 528
virion is poorly understood. For example, while it is accepted that the targeting of core to 529
LDs is essential for virus production, it is still unclear whether these organelles provide 530
the scaffold upon which core oligomerizes to form the viral nucleocapsid. In fact, 531
available evidence suggests that core may begin to oligomerize at the ER membrane, 532
prior to it being targeted to the surface of LDs (1, 19). Since the complexes in which 533
viral RNA is replicated also reside at the ER membrane (8, 13, 41, 48), such an 534
approach would be logical since it would permit RNA packaging into assembling 535
nucleocapsids prior to their transport to LDs, where further assembly and/or maturation 536
processes may occur. If this were the case, the data presented here suggest that NS3 537
operates subsequent to nucleocapsid assembly and RNA packaging since JFH1T-64-66 538
core still formed dense species associated with a peak of viral RNA, yet no infectious 539
intracellular particles were present. Furthermore, while potential nucleocapsids were still 540
assembled upon the loss of the core-NS3 interaction, these structures must at some 541
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

25
point be degraded since no core was ever detected in the supernatants of cells 542
harbouring genomes where this interaction was abolished (as seen with JFH1T-64-66 543
and JFH1T-KR). This likely represents a mechanism by which HCV can avoid the 544
release of immature, non-infectious particles from infected cells, thereby preserving 545
valuable assembly/egress factors for correctly assembled nucleocapsids. 546
In summary, we have identified a genetic interaction between core and NS3 that is 547
essential for the production of infectious virus in cell culture. The notion that non-548
structural proteins may be involved in virus assembly has been demonstrated in related 549
viruses (21, 24, 37, 39), and therefore warrants further investigation in the case of HCV. 550
Inhibition of virus assembly is currently being considered as an alternative therapeutic 551
strategy for other viruses such as HIV (7, 9, 18, 45). Therefore, a finite protein-protein 552
interaction that is essential to infectious virion production, such as the one described 553
herein, may represent an attractive target for the development of future HCV therapies. 554
555
ACKNOWLEDGEMENTS 556
The authors thank Robert Purcell and Sue Emerson (NIH, USA) for providing the JFH-557
AM1 adapted strain of JFH1 and S29 cells, Takaji Wakita (National Institute of 558
Infectious Diseases and Toray Industries, Inc., Japan) for provision of the JFH1 559
infectious clone, and Charles Rice (Rockefeller University, USA and Apath, LLC, USA) 560
for provision of Huh-7.5 cells. The authors would also like to thank Jackie Vanderluit for 561
the use of her immunofluorescence microscope facility and Thomas Michalak for the 562
use of his gradient mixer (both from Memorial University, Canada). Finally, we thank 563
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

26
Mark Harris (University of Leeds, UK) for provision of the anti-NS5A antiserum and 564
Pablo Gastaminza (Centro nacional de biotecnologia, Spain) for crucial input and advice 565
regarding gradient preparation and interpretation. 566
567
REFERENCES 568
1. Ai, L. S., Y. W. Lee, and S. S. Chen. 2009. Characterization of hepatitis C virus core protein 569 multimerization and membrane envelopment: revelation of a cascade of core-membrane 570 interactions. J Virol 83:9923-39. 571
2. Alsaleh, K., P. Y. Delavalle, A. Pillez, G. Duverlie, V. Descamps, Y. Rouille, J. Dubuisson, and C. 572 Wychowski. 2010. Identification of basic amino acids at the N-terminal end of the core protein 573 that are crucial for hepatitis C virus infectivity. J Virol 84:12515-28. 574
3. Appel, N., M. Zayas, S. Miller, J. Krijnse-Locker, T. Schaller, P. Friebe, S. Kallis, U. Engel, and R. 575 Bartenschlager. 2008. Essential role of domain III of nonstructural protein 5A for hepatitis C 576 virus infectious particle assembly. PLoS Pathog 4:e1000035. 577
4. Bartenschlager, R., F. Penin, V. Lohmann, and P. Andre. 2011. Assembly of infectious hepatitis 578 C virus particles. Trends Microbiol 19:95-103. 579
5. Boulant, S., R. Montserret, R. G. Hope, M. Ratinier, P. Targett-Adams, J. P. Lavergne, F. Penin, 580 and J. McLauchlan. 2006. Structural determinants that target the hepatitis C virus core protein 581 to lipid droplets. J Biol Chem 281:22236-47. 582
6. Broering, R., X. Zhang, S. Kottilil, M. Trippler, M. Jiang, M. Lu, G. Gerken, and J. F. Schlaak. The 583 interferon stimulated gene 15 functions as a proviral factor for the hepatitis C virus and as a 584 regulator of the IFN response. Gut 59:1111-9. 585
7. Checkley, M. A., B. G. Luttge, F. Soheilian, K. Nagashima, and E. O. Freed. The capsid-spacer 586 peptide 1 Gag processing intermediate is a dominant-negative inhibitor of HIV-1 maturation. 587 Virology 400:137-44. 588
8. Egger, D., B. Wolk, R. Gosert, L. Bianchi, H. E. Blum, D. Moradpour, and K. Bienz. 2002. 589 Expression of hepatitis C virus proteins induces distinct membrane alterations including a 590 candidate viral replication complex. J Virol 76:5974-84. 591
9. Fader, L. D., R. Bethell, P. Bonneau, M. Bos, Y. Bousquet, M. G. Cordingley, R. Coulombe, P. 592 Deroy, A. M. Faucher, A. Gagnon, N. Goudreau, C. Grand-Maitre, I. Guse, O. Hucke, S. H. 593 Kawai, J. E. Lacoste, S. Landry, C. T. Lemke, E. Malenfant, S. Mason, S. Morin, J. O'Meara, B. 594 Simoneau, S. Titolo, and C. Yoakim. 2011. Discovery of a 1,5-dihydrobenzo[b][1,4]diazepine-595 2,4-dione series of inhibitors of HIV-1 capsid assembly. Bioorg Med Chem Lett 21:398-404. 596
10. Fan, Z., Q. R. Yang, J. S. Twu, and A. H. Sherker. 1999. Specific in vitro association between the 597 hepatitis C viral genome and core protein. J Med Virol 59:131-4. 598
11. Frick, D. N. 2007. The hepatitis C virus NS3 protein: a model RNA helicase and potential drug 599 target. Curr Issues Mol Biol 9:1-20. 600
12. Gastaminza, P., S. B. Kapadia, and F. V. Chisari. 2006. Differential biophysical properties of 601 infectious intracellular and secreted hepatitis C virus particles. J Virol 80:11074-81. 602
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

27
13. Gosert, R., D. Egger, V. Lohmann, R. Bartenschlager, H. E. Blum, K. Bienz, and D. Moradpour. 603 2003. Identification of the hepatitis C virus RNA replication complex in Huh-7 cells harboring 604 subgenomic replicons. J Virol 77:5487-92. 605
14. Han, Q., C. Xu, C. Wu, W. Zhu, R. Yang, and X. Chen. 2009. Compensatory mutations in NS3 606 and NS5A proteins enhance the virus production capability of hepatitis C reporter virus. Virus 607 Res 145:63-73. 608
15. Hussy, P., H. Langen, J. Mous, and H. Jacobsen. 1996. Hepatitis C virus core protein: carboxy-609 terminal boundaries of two processed species suggest cleavage by a signal peptide peptidase. 610 Virology 224:93-104. 611
16. Jones, D. M., and J. McLauchlan. 2010. Hepatitis C virus: assembly and release of virus 612 particles. J Biol Chem 285:22733-9. 613
17. Jones, D. M., A. H. Patel, P. Targett-Adams, and J. McLauchlan. 2009. The hepatitis C virus 614 NS4B protein can trans-complement viral RNA replication and modulates production of 615 infectious virus. J Virol 83:2163-77. 616
18. Keller, P. W., C. S. Adamson, J. B. Heymann, E. O. Freed, and A. C. Steven. 2011. HIV-1 617 maturation inhibitor bevirimat stabilizes the immature Gag lattice. J Virol 85:1420-8. 618
19. Klein, K. C., S. R. Dellos, and J. R. Lingappa. 2005. Identification of residues in the hepatitis C 619 virus core protein that are critical for capsid assembly in a cell-free system. J Virol 79:6814-26. 620
20. Kopp, M., C. L. Murray, C. T. Jones, and C. M. Rice. 2010. Genetic analysis of the carboxy-621 terminal region of the hepatitis C virus core protein. J Virol 84:1666-73. 622
21. Kummerer, B. M., and C. M. Rice. 2002. Mutations in the yellow fever virus nonstructural 623 protein NS2A selectively block production of infectious particles. J Virol 76:4773-84. 624
22. Lavie, M., A. Goffard, and J. Dubuisson. 2007. Assembly of a functional HCV glycoprotein 625 heterodimer. Curr Issues Mol Biol 9:71-86. 626
23. Lindenbach, B. D., M. J. Evans, A. J. Syder, B. Wolk, T. L. Tellinghuisen, C. C. Liu, T. Maruyama, 627 R. O. Hynes, D. R. Burton, J. A. McKeating, and C. M. Rice. 2005. Complete replication of 628 hepatitis C virus in cell culture. Science 309:623-6. 629
24. Liu, W. J., P. L. Sedlak, N. Kondratieva, and A. A. Khromykh. 2002. Complementation analysis 630 of the flavivirus Kunjin NS3 and NS5 proteins defines the minimal regions essential for 631 formation of a replication complex and shows a requirement of NS3 in cis for virus assembly. J 632 Virol 76:10766-75. 633
25. Lohmann, V., F. Korner, J. Koch, U. Herian, L. Theilmann, and R. Bartenschlager. 1999. 634 Replication of subgenomic hepatitis C virus RNAs in a hepatoma cell line. Science 285:110-3. 635
26. Ma, Y., J. Yates, Y. Liang, S. M. Lemon, and M. Yi. 2008. NS3 helicase domains involved in 636 infectious intracellular hepatitis C virus particle assembly. J Virol 82:7624-39. 637
27. Masaki, T., R. Suzuki, K. Murakami, H. Aizaki, K. Ishii, A. Murayama, T. Date, Y. Matsuura, T. 638 Miyamura, T. Wakita, and T. Suzuki. 2008. Interaction of hepatitis C virus nonstructural 639 protein 5A with core protein is critical for the production of infectious virus particles. J Virol 640 82:7964-76. 641
28. Matsumoto, M., S. B. Hwang, K. S. Jeng, N. Zhu, and M. M. Lai. 1996. Homotypic interaction 642 and multimerization of hepatitis C virus core protein. Virology 218:43-51. 643
29. McLauchlan, J., M. K. Lemberg, G. Hope, and B. Martoglio. 2002. Intramembrane proteolysis 644 promotes trafficking of hepatitis C virus core protein to lipid droplets. Embo J 21:3980-8. 645
30. Michaels, A. J., and D. R. Nelson. 2010. New therapies in the management of hepatitis C virus. 646 Curr Opin Gastroenterol 26:196-201. 647
31. Miyanari, Y., K. Atsuzawa, N. Usuda, K. Watashi, T. Hishiki, M. Zayas, R. Bartenschlager, T. 648 Wakita, M. Hijikata, and K. Shimotohno. 2007. The lipid droplet is an important organelle for 649 hepatitis C virus production. Nat Cell Biol 9:1089-97. 650
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

28
32. Moradpour, D., F. Penin, and C. M. Rice. 2007. Replication of hepatitis C virus. Nat Rev 651 Microbiol 5:453-63. 652
33. Mousseau, G., S. Kota, V. Takahashi, D. N. Frick, and A. D. Strosberg. 2011. Dimerization-653 driven interaction of hepatitis C virus core protein with NS3 helicase. J Gen Virol 92:101-11. 654
34. Murray, C. L., C. T. Jones, J. Tassello, and C. M. Rice. 2007. Alanine scanning of the hepatitis C 655 virus core protein reveals numerous residues essential for production of infectious virus. J 656 Virol 81:10220-31. 657
35. Nakai, K., T. Okamoto, T. Kimura-Someya, K. Ishii, C. K. Lim, H. Tani, E. Matsuo, T. Abe, Y. Mori, 658 T. Suzuki, T. Miyamura, J. H. Nunberg, K. Moriishi, and Y. Matsuura. 2006. Oligomerization of 659 hepatitis C virus core protein is crucial for interaction with the cytoplasmic domain of E1 660 envelope protein. J Virol 80:11265-73. 661
36. Nolandt, O., V. Kern, H. Muller, E. Pfaff, L. Theilmann, R. Welker, and H. G. Krausslich. 1997. 662 Analysis of hepatitis C virus core protein interaction domains. J Gen Virol 78 ( Pt 6):1331-40. 663
37. Patkar, C. G., and R. J. Kuhn. 2008. Yellow Fever virus NS3 plays an essential role in virus 664 assembly independent of its known enzymatic functions. J Virol 82:3342-52. 665
38. Phan, T., A. Kohlway, P. Dimberu, A. M. Pyle, and B. D. Lindenbach. 2011. The acidic domain of 666 hepatitis C virus NS4A contributes to RNA replication and virus particle assembly. J Virol 667 85:1193-204. 668
39. Pijlman, G. P., N. Kondratieva, and A. A. Khromykh. 2006. Translation of the flavivirus kunjin 669 NS3 gene in cis but not its RNA sequence or secondary structure is essential for efficient RNA 670 packaging. J Virol 80:11255-64. 671
40. Pokrovskii, M. V., C. O. Bush, R. K. Beran, M. F. Robinson, G. Cheng, N. Tirunagari, M. Fenaux, 672 A. E. Greenstein, W. Zhong, W. E. t. Delaney, and M. S. Paulson. 2011. Novel mutations in a 673 tissue culture-adapted hepatitis C virus strain improve infectious-virus stability and markedly 674 enhance infection kinetics. J Virol 85:3978-85. 675
41. Quinkert, D., R. Bartenschlager, and V. Lohmann. 2005. Quantitative analysis of the hepatitis C 676 virus replication complex. J Virol 79:13594-605. 677
42. Russell, R. S., J. C. Meunier, S. Takikawa, K. Faulk, R. E. Engle, J. Bukh, R. H. Purcell, and S. U. 678 Emerson. 2008. Advantages of a single-cycle production assay to study cell culture-adaptive 679 mutations of hepatitis C virus. Proc Natl Acad Sci U S A 105:4370-5. 680
43. Santolini, E., G. Migliaccio, and N. La Monica. 1994. Biosynthesis and biochemical properties of 681 the hepatitis C virus core protein. J Virol 68:3631-41. 682
44. Scheel, T. K., J. M. Gottwein, T. H. Carlsen, Y. P. Li, T. B. Jensen, U. Spengler, N. Weis, and J. 683 Bukh. 2011. Efficient culture adaptation of hepatitis C virus recombinants with genotype-684 specific core-NS2 by using previously identified mutations. J Virol 85:2891-906. 685
45. Shi, J., J. Zhou, V. B. Shah, C. Aiken, and K. Whitby. 2010. Small-molecule inhibition of human 686 immunodeficiency virus type 1 infection by virus capsid destabilization. J Virol 85:542-9. 687
46. Shimoike, T., S. Mimori, H. Tani, Y. Matsuura, and T. Miyamura. 1999. Interaction of hepatitis 688 C virus core protein with viral sense RNA and suppression of its translation. J Virol 73:9718-25. 689
47. Tanaka, Y., T. Shimoike, K. Ishii, R. Suzuki, T. Suzuki, H. Ushijima, Y. Matsuura, and T. 690 Miyamura. 2000. Selective binding of hepatitis C virus core protein to synthetic 691 oligonucleotides corresponding to the 5' untranslated region of the viral genome. Virology 692 270:229-36. 693
48. Targett-Adams, P., S. Boulant, and J. McLauchlan. 2008. Visualization of double-stranded RNA 694 in cells supporting hepatitis C virus RNA replication. J Virol 82:2182-95. 695
49. Tellier, R., J. Bukh, S. U. Emerson, R. H. Miller, and R. H. Purcell. 1996. Long PCR and its 696 application to hepatitis viruses: amplification of hepatitis A, hepatitis B, and hepatitis C virus 697 genomes. J Clin Microbiol 34:3085-91. 698
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

29
50. Tellinghuisen, T. L., K. L. Foss, and J. Treadaway. 2008. Regulation of hepatitis C virion 699 production via phosphorylation of the NS5A protein. PLoS Pathog 4:e1000032. 700
51. Tellinghuisen, T. L., K. L. Foss, J. C. Treadaway, and C. M. Rice. 2008. Identification of residues 701 required for RNA replication in domains II and III of the hepatitis C virus NS5A protein. J Virol 702 82:1073-83. 703
52. Wakita, T., T. Pietschmann, T. Kato, T. Date, M. Miyamoto, Z. Zhao, K. Murthy, A. Habermann, 704 H. G. Krausslich, M. Mizokami, R. Bartenschlager, and T. J. Liang. 2005. Production of 705 infectious hepatitis C virus in tissue culture from a cloned viral genome. Nat Med 11:791-6. 706
53. Yi, M., Y. Ma, J. Yates, and S. M. Lemon. 2007. Compensatory Mutations in E1, p7, NS2, and 707 NS3 Enhance Yields of Cell Culture-Infectious Intergenotypic Chimeric Hepatitis C Virus. J Virol 708 81:629-38. 709
54. Zhong, J., P. Gastaminza, G. Cheng, S. Kapadia, T. Kato, D. R. Burton, S. F. Wieland, S. L. 710 Uprichard, T. Wakita, and F. V. Chisari. 2005. Robust hepatitis C virus infection in vitro. Proc 711 Natl Acad Sci U S A 102:9294-9. 712
55. Zhong, J., P. Gastaminza, J. Chung, Z. Stamataki, M. Isogawa, G. Cheng, J. A. McKeating, and F. 713 V. Chisari. 2006. Persistent hepatitis C virus infection in vitro: coevolution of virus and host. J 714 Virol 80:11082-93. 715
716 717
FIGURE LEGENDS 718
Figure 1. Construction of core mutants. JFH1T contains 3 amino acid mutations (N417S 719
[E2], N765D [p7] and Q1012R [NS2]) that enhance infectious virus production 720
compared to JFH1. The amino acid sequence spanning core residues 63-76 are 721
depicted below the HCV polyprotein. Four mutants were constructed in the JFH1T 722
background (JFH1T-64-66, JFH1T-67-69, JFH1T-70-72 and JFH1T-73-75) with triplets of 723
residues converted to alanine in each. Mutated residues are shown in bold and 724
underlined. 725
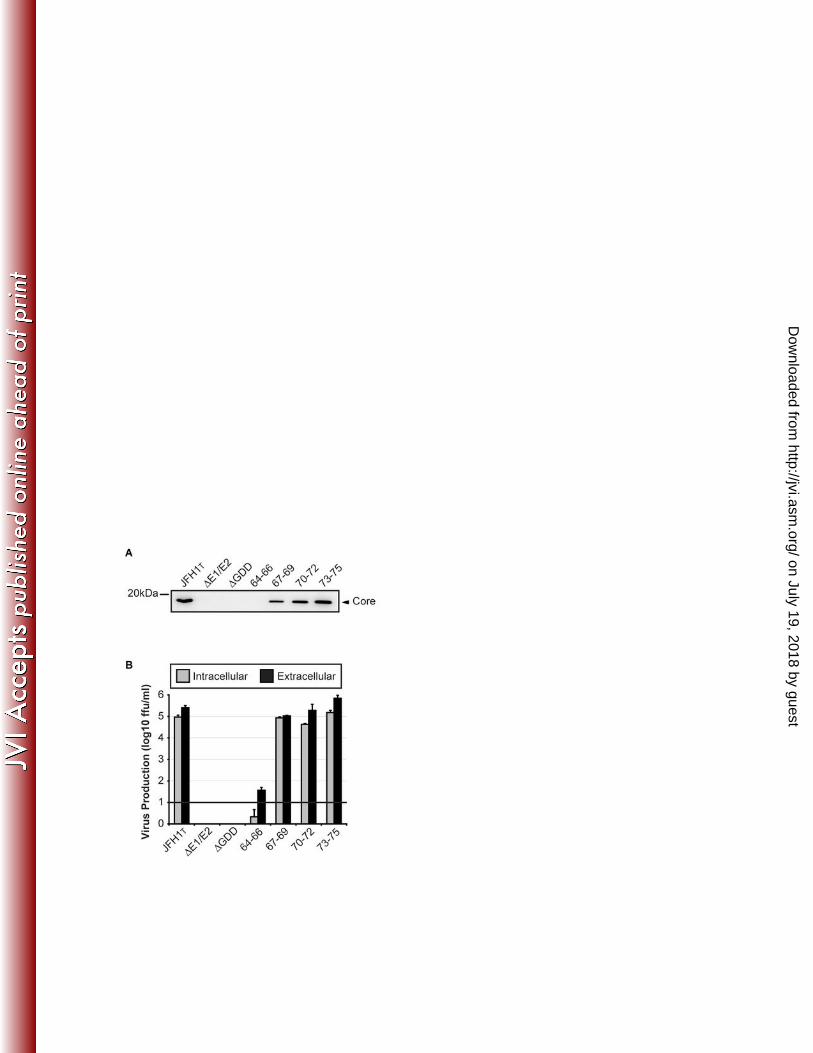
Figure 2. Core residues 64-66 are essential for robust production of infectious virus. [A] 726
Equal numbers of Huh-7.5 cells were transfected with RNA encoding JFH1T, ΔE1/E2, 727
ΔGDD or the 4 core mutants. 72hrs later, cells were fixed and probed with DAPI (blue) 728
and an antibody specific for HCV core (green). Scale bar, 100µm. [B] Supernatants 729
from cells transfected with core mutations in the background of JFH1T or [C] JFH1 were 730
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

30
harvested 72hrs post-transfection and serial dilutions of these supernatants were used 731
to infect naive Huh-7.5 cells. 72hrs later, infected cells were fixed and probed for the 732
presence of core protein to determine the number of focus-forming units (ffu) per ml of 733
supernatant. For all experiments of this kind, viral titres were measured in triplicate from 734
one experiment and error bars represent standard error of the mean. Experiments were 735
performed at least twice. The cut-off of the assay was 10ffu/ml and is indicated by the 736
solid line at log10=1. [D] Cell lysates were harvested 72hrs post-transfection and 737
subjected to Western blot analysis. For each RNA transfection, core and GAPDH were 738
detected. 739
740
Figure 3. Mutation of core residues 64-66 prevents release of virions into cell 741
supernatants and abrogates production of infectious intracellular particles. [A] 72hrs 742
post-transfection, viral particles released into the supernatants were isolated by 743
ultracentrifugation through a 20% sucrose cushion. The resultant pellet was probed for 744
the presence of core protein by Western blot. [B] Huh-7.5 cells transfected with each 745
RNA were harvested at 72hrs post-transfection and used to determine intracellular 746
levels of infectious virus. Extracellular titres were measured from supernatants taken at 747
the same time point. 748
Figure 4. Analysis of core mutants using a single-cycle virus production assay [A] 749
Equal numbers of S29 cells were transfected with RNA encoding JFH1T, ΔE1/E2, 750
ΔGDD or the 4 core mutants. 72hrs later, cells were fixed and probed for nuclei (blue) 751
and HCV core (green). Scale bar, 100µm. [B] Intra- and extracellular titres were 752
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

31
determined from S29 cell lysates or supernatants respectively at 72hrs post-753
transfection. [C] Cell lysates were harvested 72hrs post-transfection and subjected to 754
Western blot analysis. For each RNA transfection, core and GAPDH were detected. 755
Figure 5. Mutation of residues 64-66 does not affect the association between core and 756
LDs. Huh-7.5 cells were transfected with RNA from JFH1T or JFH1T-64-66 before being 757
fixed 72hrs later. Coverslips were stained using DAPI (blue), an antibody specific to 758
core (green) and LipidTOX for LD detection (red). Scale bar, 10µm. 759
Figure 6. Mutation of core residues 64-66 does not prevent formation of dense core-760
containing species. Intracellular lysates from cells transfected with [A] JFH1T, [B] 761
JFH1T-64-66 or [C] ΔE1/E2 were ultracentrifuged through 10-50% iodixanol gradients. 762
Each gradient was separated into 10 fractions, with fraction 1 representing the 763
uppermost fraction and 10 being the bottom. Intracellular titres (open triangles) and viral 764
RNA (open circles) were measured in each fraction while core was also detected by 765
Western blot. Values for titres are an average of 6 data sets while RNA values are an 766
average of 4. Error bars have been removed for purposes of clarity. The density (g/cm3) 767
of each fraction is indicated. 768
Figure 7. Passaging JFH1T-64-66 leads to the emergence of a compensatory mutation 769
within NS3. [A] A schematic representation of the method used to identify 770
compensatory mutations. Huh-7.5 cells were transfected with RNA encoding JFH1T-64-771
66. Cells were passaged each time they reached confluency and supernatants from the 772
day of split were used to determine infectious titres. For P1-P3, the cells themselves 773
were passaged, whereas supernatants were passed for I1 and I2 to enrich any rescued 774
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

32
virus. For I1, naive Huh-7.5 cells were infected using supernatants from the P3 dish, 775
since viral titres from JFH1T-64-66 had reached levels comparable to JFH1T by this 776
point. 72hrs later, supernatants were removed and used to infect naive cells a second 777
time (I2). RNA was harvested from I2 supernatants 72hrs post-infection and used for 778
sequencing. Two independent but identical experiments were performed, referred to as 779
64-66 I and II [B] Cells were fixed and core (green) and cell nuclei (blue) were 780
visualized on the day of cell passage. For comparison, cells transfected with JFH1T or 781
ΔGDD RNA at P1 are shown. Scale bar, 10µm. [C] Viral titres were determined from 782
supernatants taken at P1 (for JFH1T) or P1-P3 (64-66 I and II). [D] For 64-66 I and II, 783
P3 supernatants were used to infect naive Huh-7.5 cells (I1). 72hrs later, supernatants 784
were taken from the I1 dish and used to infect naive cells (I2) a second time. Titres from 785
each round of infection are shown. For comparison, titres from naive cells infected with 786
JFH1T P1 supernatants are also given. 787
Figure 8. A mutation within the helicase domain of NS3 rescues virus production from 788
JFH1T-64-66. [A] Passaging of JFH1T-64-66 led to the emergence of a single 789
compensatory mutation within NS3 (K1302R [polyprotein numbering] or K272R [NS3 790
numbering]). [B] Equal numbers of Huh-7.5 cells were transfected with RNA encoding 791
JFH1T, ΔE1/E2, ΔGDD, JFH1T-64-66, JFH1T-64-66-KR or JFH1T-KR. 72hrs later, cells 792
were fixed and cell nuclei (blue) and HCV core (green) were visualized. Scale bar, 793
100µm. [C] Supernatants and intracellular lysates were harvested from cells 72hrs post-794
transfection and intra- and extracellular titres were determined as previously described 795
[D] Cell lysates from transfected S29 sells were harvested 72hrs post-transfection and 796
subjected to Western blot analysis. For each RNA transfection, core, NS3, NS5A and 797
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from

33
GAPDH were detected. [E] 72hrs post-transfection, viral particles released into the 798
supernatants were isolated by ultracentrifugation through a 20% sucrose cushion. The 799
resultant pellet was probed for the presence of core protein by Western blot. 800
on July 19, 2018 by guesthttp://jvi.asm
.org/D
ownloaded from