Embed Size (px)
Citation preview

NOTES & TIPS
A DNA Assay Based on FluorescenceResonance Energy Transfer and DNATriplex Formation
Mengsu Yang,1 Li-Qiang Ren, Minghui Huang,Richard Y. C. Kong, and Wang Fun FongDepartment of Biology and Chemistry, City Universityof Hong Kong, 83 Tat Chee Avenue, Kowloon,Hong Kong, People’s Republic of China
Received August 11, 1997
Conventional methods for detection of mutations inDNA samples, such as Southern hybridization, directsequencing, and single-strand conformation polymor-phism analysis, are usually time consuming and expen-sive and usually involve DNA probes labeled with radio-active isotopes. Therefore, solution-based non-radioactiveDNA assays are desirable for the research and diagnosticlaboratories. In this report, we demonstrate the feasibil-ity of combining the sequence specificity of DNA triplexformation with the sensitivity of the fluorescence reso-nance energy transfer (FRET)2 method for the detectionof a specific mutation in the human p53 gene. DNA tri-plex formation refers to the sequence-specific recognitionof certain double-stranded DNA (dsDNA) sequences bysingle-stranded oligonucleotides. The most commonstructural motif is based on the binding of pyrimidine-rich oligonucleotides to the major groove of homopurine/homopyrimidine dsDNA through the formation ofHoogsteen hydrogen bonds (T*A2T and C1*G2C basetriplets) (1). Single triplet mismatches between an oli-gonucleotide and a DNA duplex have been shown todestabilize triple helix formation under appropriateconditions (2). FRET refers to the transfer of electronicexcitation energy from a fluorescence donor group to anunexcited acceptor group through dipole–dipole inter-actions, the efficiency of which depends on the orienta-tion of the donor and acceptor transition dipoles, thedistances between the donor and acceptor molecules,and the spectral properties of the donor and acceptor
(3). The energy transfer causes a decrease in the emis-sion intensity of the donor-labeled probe and an in-crease in the emission intensity of the acceptor-labeledprobe. Previous studies have shown that both DNAhybridization and DNA triplex formation may be mon-itored by FRET measurements (4, 5).
Inactivation of the p53 tumor suppressor protein is acommon event in the development of diverse types ofhuman cancers (6). A documented p53 mutation in hep-atocellular carcinoma cases consists of an 8-bp deletionstarting at the third position of codon 285 within exoneight of the 1760-bp human p53 gene (7). The deletiondisrupts a 12-bp homopurine/homopyrimidine sequencewhere a nearby 12-bp homopurine/homopyrimidine se-quence remains intact (Fig. 1). It has been shown byelectrophoresis and chemical protection methods that oli-gonucleotides bind to individual homopurine domainswithin this stretch of DNA sequence (8, 9). The proximityof the two homopurine sites in the p53 gene offers apossibility for FRET using donor-labeled and acceptor-labeled DNA probes. We have designed two oligonucleo-tide probes to specifically bind to the wild-type homopu-rine sequences I and II by means of the pyrimidine triple-helix motif (Fig. 1). Because sequence I is disrupted bythe 8-bp deletion, probe 1 is expected to form only 7 of 12possible base triplets with the mutant p53 duplex, whileit is fully matched with the wild-type duplex. Thus, bind-ing of probe 1 to sequence I might be used to differentiatebetween the wild-type and the mutant p53 sequence,with probe 2 providing a positive control.
Synthetic oligonucleotides were used to construct du-plex models (33-bp) that contain DNA sequences corre-sponding to the wild-type and the 8-bp deletion mutantforms of the p53 gene (Fig. 1). Fluorescently labeledprobes (8–12 nucleotides, nt) were prepared by auto-mated DNA synthesis (Pharmacia Gene Assembler Spe-cial) using b-cyanoethyl phosphoramidite derivatives andchemical labeling procedures. Briefly, 59-hexylamine-linked oligonucleotides were prepared with hexylamine-modified phosphoramidite reagents (Pharmacia Biotech)in the last coupling step of the synthesis. Modified con-trolled pore glass (CPG) reagents (Clontech) were used tomodify the 39-terminus of an oligonucleotide with desiredamino linkers. The NH2-modified oligonucleotides weretreated with a 200-fold excess of tetramethylrhodamine(TMR) or 5-carboxyfluorescein (FL) succinimidyl esters(Molecular Probes) to generate 59- or 39-TMR-labeledprobe 1 and 59-FL-labeled probe 2, respectively. The dye-
1 To whom correspondence should be addressed. Fax: (852)2788-7406.
2 Abbreviations used: FRET, fluorescence resonance energy transfer;dsDNA, double-stranded DNA; nt, nucleotide; TMR, tetramethyl-rhodamine; FL, 5-carboxyfluorescein; HCC, heptocellular carcinoma.
272 ANALYTICAL BIOCHEMISTRY 259, 272–274 (1998)ARTICLE NO. AB982646
0003-2697/98 $25.00Copyright © 1998 by Academic Press
All rights of reproduction in any form reserved.
labeled oligonucleotides were separated from the excessdye molecules by gel filtration using Sephadex-G25 col-umns (Bio-Rad) and further purified by reverse-phaseHPLC (Waters). Duplex DNA were prepared by anneal-ing the complementary strands (1:1) at 90°C for 10 min ina typical association buffer containing 40 mM Tris–ac-etate (pH 5.2), 100 mM MgCl2, and 1 mM spermine–4mM HCl. The dye-labeled probes (1.0 3 1026 M) wereallowed to equilibrate with the unlabeled duplex DNAsolutions (2.9 3 10210 to 6.0 3 1026 M) and the emissionintensities before and after the addition of the dsDNAwere measured using an SLM 4800C spectrofluorometer,with an excitation wavelength of 480 nm and emissionwavelengths of 490–650 nm. The relative changes in theemission of FL (520 nm) and TMR (584 nm) were re-corded accordingly.
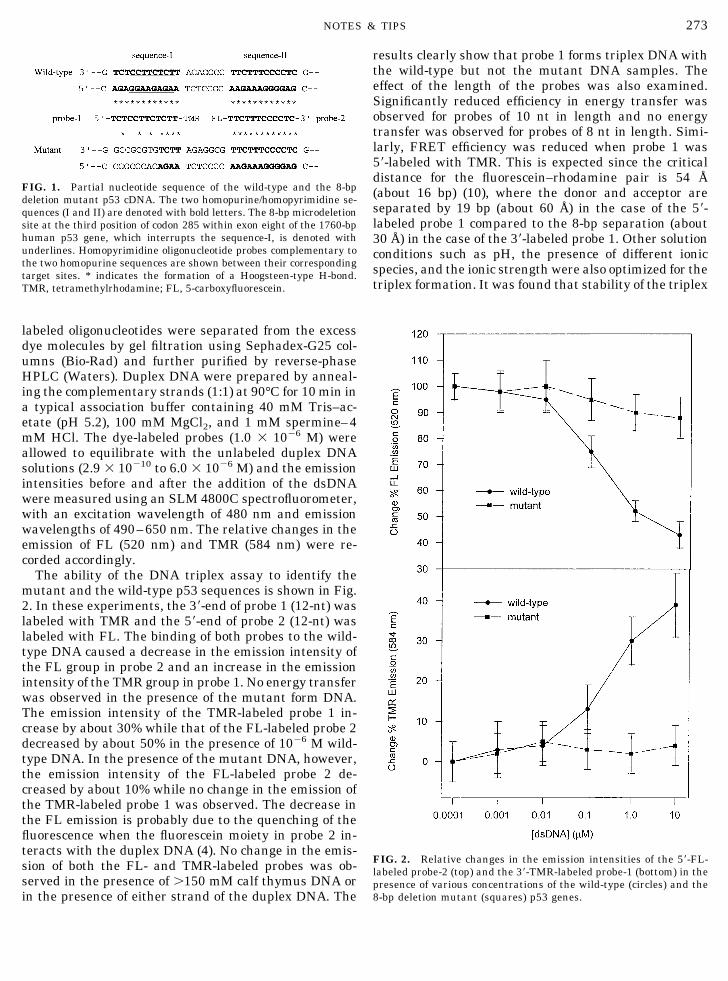
The ability of the DNA triplex assay to identify themutant and the wild-type p53 sequences is shown in Fig.2. In these experiments, the 39-end of probe 1 (12-nt) waslabeled with TMR and the 59-end of probe 2 (12-nt) waslabeled with FL. The binding of both probes to the wild-type DNA caused a decrease in the emission intensity ofthe FL group in probe 2 and an increase in the emissionintensity of the TMR group in probe 1. No energy transferwas observed in the presence of the mutant form DNA.The emission intensity of the TMR-labeled probe 1 in-crease by about 30% while that of the FL-labeled probe 2decreased by about 50% in the presence of 1026 M wild-type DNA. In the presence of the mutant DNA, however,the emission intensity of the FL-labeled probe 2 de-creased by about 10% while no change in the emission ofthe TMR-labeled probe 1 was observed. The decrease inthe FL emission is probably due to the quenching of thefluorescence when the fluorescein moiety in probe 2 in-teracts with the duplex DNA (4). No change in the emis-sion of both the FL- and TMR-labeled probes was ob-served in the presence of .150 mM calf thymus DNA orin the presence of either strand of the duplex DNA. The
results clearly show that probe 1 forms triplex DNA withthe wild-type but not the mutant DNA samples. Theeffect of the length of the probes was also examined.Significantly reduced efficiency in energy transfer wasobserved for probes of 10 nt in length and no energytransfer was observed for probes of 8 nt in length. Simi-larly, FRET efficiency was reduced when probe 1 was59-labeled with TMR. This is expected since the criticaldistance for the fluorescein–rhodamine pair is 54 Å(about 16 bp) (10), where the donor and acceptor areseparated by 19 bp (about 60 Å) in the case of the 59-labeled probe 1 compared to the 8-bp separation (about30 Å) in the case of the 39-labeled probe 1. Other solutionconditions such as pH, the presence of different ionicspecies, and the ionic strength were also optimized for thetriplex formation. It was found that stability of the triplex
FIG. 1. Partial nucleotide sequence of the wild-type and the 8-bpdeletion mutant p53 cDNA. The two homopurine/homopyrimidine se-quences (I and II) are denoted with bold letters. The 8-bp microdeletionsite at the third position of codon 285 within exon eight of the 1760-bphuman p53 gene, which interrupts the sequence-I, is denoted withunderlines. Homopyrimidine oligonucleotide probes complementary tothe two homopurine sequences are shown between their correspondingtarget sites. * indicates the formation of a Hoogsteen-type H-bond.TMR, tetramethylrhodamine; FL, 5-carboxyfluorescein.
FIG. 2. Relative changes in the emission intensities of the 59-FL-labeled probe-2 (top) and the 39-TMR-labeled probe-1 (bottom) in thepresence of various concentrations of the wild-type (circles) and the8-bp deletion mutant (squares) p53 genes.
273NOTES & TIPS

DNA depends on the pH and the concentrations of sperm-ine and Mg21, consistent with literature reports (11–14).
The FRET-based assay was applied to screen 21clinical DNA samples obtained from patients diag-nosed pathologically with hepatocellular carcinoma(HCC). HCC tissue samples (0.1–0.2 g) were powderedin liquid nitrogen and suspended in RSB buffer (10 mMTris–HCl, 10 mM NaCl, 25 mM EDTA, pH 7.4) with 1%SDS and proteinase K at 37°C for 5 h. Then, 5 M NaClwas added into the mixture to reach a final concentra-tion of 100 mM. DNA was extracted with phenol/chlo-roform, precipitated with 2.5 vol ethanol, and dissolvedin double-distilled water. Two primers correspondingto the related sequences in exon 8 of the p53 gene (15)were used for a subsequent PCR experiment: 59-GCCT-GTCCTGGGAGA-39 and 59-GGGCAGCTCGTGGTG-39. PCR amplification was performed in a total volumeof 100 ml containing approximately 2 mg DNA digestedwith restriction enzyme BamHI (Promega), 33 pmol ofeach primer, 200 mM each deoxynucleotide triphos-phate, 2.4 units of Taq polymerase, and 1 3 Taq poly-merase buffer (Promega) in 2 mM MgCl2 solution. ThePCR was carried out for 40 cycles with the first cycle at94°C (1 min), 50°C (1 min), and 72°C (1 min) and thesubsequent 39 cycles at 94°C (30 s), 55°C (45 s), and72°C (1 min). Following the final reaction cycle, thePCR samples were incubated for another 5 min at72°C. The above conditions generated the PCR productwith a concentration . 10 mM, as determined by UV-A260. The 75-bp PCR product was allowed to equili-brate with the dye-labeled probes according to themethod described above. Fluorescence energy transferwas observed in all samples, indicating that the 8-bpdeletion mutation does not appear in the DNA sam-ples. The results were confirmed by restriction enzymedigestion as the wild-type p53 gene contains a recog-nition site for MboII endonuclease (GAAGA) but not inthe deletion mutant form. The PCR products (5 ml)were digested with 75 units of restriction enzymeMboII (Promega) at 37°C for at least 6 h in a totalvolume of 50 ml. After digestion, the PCR productswere ethanol-precipitated, vacuum-dried, dissolved in20 ml water, and then separated on a 15% polyacryl-amide gel. Two bands of about 30 and 40 bp, corre-sponding to the restriction fragments, were visualizedin all samples by ethidium bromide staining.
In summary, the combination of DNA triplex forma-tion with FRET measurement provides an alternativeto solid-based hybridization assays for the detection ofmutations in homopurine DNA sequences. The solu-tion-based assay is simple to perform, uses few re-agents, and does not require DNA denaturation andhybridization steps. In addition, the background noiseusually associated with blot-hybridization techniquesdue to nonspecific adsorption of labeled reagents tosolid supports is minimized.
Acknowledgments. This work is supported by the Hong KongGovernment Industry Department and the City University of HongKong. We thank H. Zhou of the Cancer Research Center, XiamenUniversity, China, for the clinical HCC tissue samples.
REFERENCES
1. LeDoan, T., Perrouault, L., Praseuth, D., Habhoub, N., Decout,J.-L., Thuong, N. T., Lhomme, J., and Helene, C. (1987) NucleicAcids Res. 15, 7749–7760.
2. Moser, H. E., and Dervan, P. B. (1987) Science 238, 645–647.3. Styrer, L. (1978) Annu. Rev. Biochem. 47, 819–846.4. Morrison, L. E., and Stols, L. M. (1993) Biochemistry 32, 3095–
3104.5. Yang, M., Ghosh, S., and Millar, D. P. (1994) Biochemistry 33,
15329–15337.6. Oliner, J. D., Kinzer, K. W., Meltzer, D. L., and Vogelstein, B.
(1992) Nature 80, 358–361.7. Bressac, B., Kew, M., Wands, J., and Ozturk, M. (1991) Nature
350, 429–431.8. Olivas, W. M., and Maher, L. J. (1994) BioTechniques 16, 128–
130.9. Olivas, W. M., and Maher, L. J. (1994) Biochemistry 33, 983–988.
10. Lakowicz, J. R. (Ed.) (1991) Topics in Fluorescence Spectroscopy,Vol. 2, Plenum Press, New York.
11. Xodo, L., Manzini, G., and Quadrifoglio, F. (1990) Nucleic AcidsRes. 18, 3557–3564.
12. Pilch, D. S., Brousseau, R., and Shafer, R. H. (1990) NucleicAcids Res. 18, 5743–5749.
13. Roberts, R. W., and Crothers, D. M. (1991) Proc. Natl. Acad. Sci.USA 88, 9397–9401.
14. Singleton, S. F., and Dervan, P. B. (1992) J. Am. Chem. Soc. 114,6957–6965.
15. Harlow, E. D., Williamson, N. M., Ralston, R., Helfman, D. M.,and Adams, T. E. (1985) Mol. Cell. Biol. 5, 1601–1610.
A Direct Microfluorometric Method forMeasuring Subpicomole Amounts ofNicotinamide Adenine DinucleotidePhosphate, Glucose, and Glycogen
Long Thanh Nguyen,* D. George Stephenson,†and Gabriela M. M. Stephenson*,1
*Department of Chemical Sciences, Victoria University ofTechnology, P.O. Box 14428, MCMC, Melbourne, Victoria8001, Australia; and †School of Zoology, La TrobeUniversity, Melbourne, Australia
Received September 23, 1997
The major objective of this study was to develop anaccurate and reproducible microfluorometric pyridine nu-
1 To whom correspondence should be addressed. Fax: (03) 96884995. E-mail: [email protected].
274 NOTES & TIPS
ANALYTICAL BIOCHEMISTRY 259, 274–278 (1998)ARTICLE NO. AB982634
0003-2697/98 $25.00Copyright © 1998 by Academic Press
All rights of reproduction in any form reserved.





























