Embed Size (px)
Citation preview

Proc. Natl. Acad. Sci. USAVol. 88, pp. 5979-5983, July 1991Genetics
Proteinase trapping: Screening for viral proteinase mutants bya complementation
(2A proteinase of human rhinovirus/amino acid substitutions/picornaviruses/lacZ operon/PCR mutagenesis)
HANS-DIETER LIEBIG*, TIM SKERN*, MARION LUDERER*, WOLFGANG SOMMERGRUBERt, DIETER BLAAS*,AND ERNST KUECHLER**Institute of Biochemistry, University of Vienna, Waehringerstrasse 17, A-1090 Vienna, Austria; and tErnst Boehringer Institut fuer Arzneimittelforschung,Dr. Boehringergasse 5 - 11, A-1120 Vienna, Austria
Communicated by Max L. Birnstiel, April 10, 1991
ABSTRACT Many virally encoded proteinases cleavethemselves out of a polyprotein, with cleavage occurring usu-ally at their own N terminus. This property was used to developan in vivo screening system using the lacZ gene fragment ofM13mpl8. When a fusion protein of the a fragment of fl-ga-lactosidase and an active 2A proteinase of human rhinovirus 2was expressed, a complementation was not affected, as the 2Aproteinase cleaved itself off the a fragment. However, fusion ofan inactive 2A prevented a complementation, as the 2Apolypeptide remained fused to the a fragment. After randommutation of the 2A gene by PCR amplification, mutants werescreened; M13 phage defective in a complementation wereobtained at an efficiency of 5% and were shown to containmutated 2A genes. Intermolecular cleavage was then examinedby expressing an a fragment-inactive proteinase fusion proteinas substrate for an active 2A proteinase expressed from an M13vector. a complementation indicated intermolecular process-ing of the 2A cleavage site on the a fragment-nactive protein-ase fusion protein. This versatile system thus allows the high-density screening of both active and inactive proteinase mu-tants, cleaving either intramolecularly or intermolecularly,and should be applicable to other proteinases of high specific-ity.
Viral strategies for condensing coding capacity are varied (1).Picornaviruses perform a limited proteolytic cleavage of apolyprotein that is derived from a single open reading framecoding for -2100 amino acids (2, 3). In rhinoviruses (the maincausative agents of the common cold) and polioviruses, thefirst proteolytic cleavage is carried out by the viral protein2A, which cleaves at its own N terminus; this cleavage occurson the growing peptide chain, so that the polyprotein isnormally not observed (4, 5). The remaining cleavages on thepolyprotein (including that at the C terminus of 2A) arecarried out by the viral protein 3C (6). In addition, the matureproteinase 2A is also involved in the shut-off of host-cellprotein synthesis through the inactivation of the p220 com-ponent of the eIF-4F cap binding complex (7-9). The mech-anism of the proteolysis of p220, which plays a role in theinitiation of cap-dependent translation, is unclear. The initi-ation of rhinovirus translation (which occurs internally),however, is not affected by p220 inactivation, as its RNA isnot capped; instead it is linked to a virally encoded protein,VPg, which is removed prior to translation, leaving pU as the5' terminus (2).
Mechanistically, the 2A and 3C proteinases have a cysteineresidue as the active site nucleophile; however, the sequencesurrounding it has a high similarity to that of serine protein-ases (5, 10). Furthermore, the amino acid sequence of picor-
naviral 2As can be modeled onto the three-dimensionalstructure of a-lytic proteinase (11). These viral proteins mayeven represent an evolutionary link between the serine andcysteine proteinases (10). In the absence of the three-dimensional molecular structure, mutational analysis repre-sents the best way ofgaining insight into their mechanism. Tothis end, we describe here a sensitive system for the detectionof proteinase mutants based on the lacZ screening system(12).
MATERIALS AND METHODSMaterials and General Methods. Restriction enzymes were
from Boehringer Mannheim orNew England Biolabs, and Taq(Thermus aquaticus) DNA polymerase was from Promega.Manipulation of DNA was by standard protocols (13). DNAwas sequenced by using T7 DNA polymerase (Pharmacia) and[a-32PjdATP (Amersham) as described (14). Site-directed mu-tagenesis was done with 2'-deoxycytidine 5'-[a-thio]triphos-phate (Pharmacia) according to the method of Taylor et al.(15). All manipulations were done in the Escherichia colistrain JM101; the concentrations of isopropyl ,B-D-thiogalactoside (IPTG) (Boehringer Mannheim) and5-bromo-4-chloro-3-indolyl ,B-D-galactoside (X-Gal) (Strata-gene) used for screening were 0.16 and 0.8 mM, respectively,unless stated otherwise.
Construction of Expression Vectors. The structures of theconstructions are shown in Fig. 1B. To generate M13/2AA2B, a DNA fragment containing the human rhinovirus 2(HRV2) nucleotides 3075-3704, the G-C tails from the orig-inal cDNA cloning, and 189 base pairs from the pEx34cvector was inserted into the Pvu I site of M13mpl8. Thisstrategy results in an in-frame fusion of the IacZ gene with theHRV2 cDNA; after induction with IPTG, a fusion protein isexpressed containing the first 45 amino acids of P-galacto-sidase (the a fragment), 19 amino acids from the multiplecloning site, the 28 C-terminal amino acids of VP1, all 142amino acids from 2A, the 39 N-terminal amino acids of 2B,and 19 amino acids encoded by the G-C tail and the pEx34cvector. An inactive variant M13/2A(Q134)A2B was madesimilarly by using a fragment containing the Arg-134 to Glnmutation, previously shown to inactivate 2A (5).M13/2A was constructed from M13/2AA2B as follows. The
multiple cloning site was partially deleted to remove the HindIllsite; the reading frame was maintained. The sequence coding foramino acids 42-44 of the a fragment was mutated from GCCCGC ACC to GCG TCG ACC to allow cleavage by Sal I, AccI, and HincII, changing amino acid 43 from Arg to Ser. An MluI site was introduced at the nucleotides corresponding to the 7thand 8th amino acids before the C terminus of VP1; the aminoacid sequence was not changed (5). Finally, the 2AA2B gene
Abbreviations: HRV, human rhinovirus; IPTG, isopropyl P-D-thiogalactoside; X-Gal, 5-bromo-4-chloro-3-indolyl fB-D-galactoside.
5979
The publication costs of this article were defrayed in part by page chargepayment. This article must therefore be hereby marked "advertisement"in accordance with 18 U.S.C. §1734 solely to indicate this fact.

Proc. Natl. Acad. Sci. USA 88 (1991)
INTRAMOLECULAR CLEAVAGE INTERMOLECULAR CLEAVAGE
FREED a(-FRAGMENT _
ICa-COMPLEMENTATION
TRAPPED a-FRAGMENT
__ + m\\\\\\\m
PLOcM 1 3/2AA2B
M1 3/2A(Q1 34)A2B
M 1 3/2A
Ml 3/2AA3GIy
LacZ45
AVP 1
2A28j 142
A2B +CX-COMPLEMENTATION
39 +
r* El 9_lllllK
R1 34:Q
CRI S
R43:S
He- -mR43:S A
+ +
FIG. 1. (A) Basis of proteinase trapping. The afragment and active proteinase parts of the fusionprotein are shown by the stippled and open portionsof the boxes, respectively; inactive proteinases arehatched. The proteinase cleavage site is shown bythe solid vertical line. (B) Structure of M13/2Aderivatives used in intramolecular cleavage. ThelacZ promoter/operator is marked with a solidarrow. The coding sequence of the lacZ/2A fusionsis boxed; the lacZ sequence is stippled, with theopen part representing the multiple cloning site.The rhinoviral sequences are as follows: verticallines, VP1; open, 2A; diamonds, 2B. The number ofamino acids in each component is given, and a plussign indicates that translation continues into thevector sequences. The vertical arrow between VP1and 2A indicates the position of the 2A cleavagesite; open arrows show the position of the oligonu-cleotide primers used for the PCR amplification. Arepresents the deletions of Gly-104, -107, and -108.The binding site for the antiserum PC20 is indicatedas a solid box. A jagged end indicates that thereading frame continues into vector sequences.Restriction enzyme sites used are shown on theconstruction M13/2A. A, Acc I; A, Apa I; B,BstEII; C, Hincll; H, HindIII; M, Mlu I; RI, EcoRI;S, Sal I. The degree of complementation observedwith each construction is given; this ranged be-tween a wild-type level (+ + +, dark blue plaques)and no complementation (-, white plaques).
was replaced with one coding for a 2A followed immediately bytwo stop codons (5). For the inactive variant M13/2AA3Gly, afragment was used in which residues Gly-104, -107, and -108 hadbeen deleted.M13/AlacZ2A and M13/AlacZ2AA&3Gly were constructed by
opening M13/2A and M13/2AA3Gly with EcoRI and Acc I; thisremoves the polylinker and amino acids 7-43 of the lacZfragment. After treatment with the Klenow enzyme, the largefragment was recircularized, regenerating the reading frame.pBR/2A- was made by introducing a DNA fragment
carrying the expression block of M13/2A into the Cla I/SalI 4282-base-pair fragment of pBR328. The region of theHRV2 protein VP2 recognized by the monoclonal antibody8F5 (ref. 16; amino acids 120-193 of VP2) was then clonedin-frame into the Apa I site of 2A.
Expression of Proteins and Detection on Western Blots. Pro-teins were expressed from M13 phage by stated techniques (13).After 6 hr of induction at 370C, the cells were harvested and theproteins were prepared for gel electrophoresis as described (5).Western blots were prepared with the mouse monoclonal an-tibody 8F5 (16) and the rabbit antiserum PC20 (5); the secondantibody was alkaline phosphatase coupled (Promega).
RESULTS
Effect of 2A Sequences on a Complementation. Langley etal. (17) showed that the deletion of amino acids 11-41 in the
3-galactosidase protein (referred to as the M15 B-galacto-sidase) could be complemented by a peptide containingamino acids 3-92. This peptide (a fragment) can be reducedfurther in size without affecting complementation. For in-stance, the pUC vectors developed by Vieira and Messing(12) express a peptide of 100 amino acids, only 59 of whichare derived from the a fragment. To establish a systemallowing the screening of large numbers of proteinase mu-tants, we reasoned that any modifications to the a fragmentthat hindered its diffusion or its association with the M15/3-galactosidase protein would reduce complementation. Asthe picornavirus proteinases are capable of cleaving them-selves off a growing peptide chain, the fusion of such aproteinase to the C terminus of the a fragment would notinfluence complementation. However, the fusion of an inac-tive proteinase would lead to a C-terminal extension of the afragment, which would be detrimental to complementation.The logic is illustrated in Fig. 1A.To test this hypothesis, the lacZ gene was fused to genes
coding for an active and inactive 2A proteinase from HRV2as shown in Fig. 1B. The fusion was made after the codon forthe 45th amino acid ofthe lacZ fragment; 28 amino acids fromthe C terminus of VP1 were included to ensure that 2Acleavage could take place. Competent E. coli JM101 cellswere transfected with DNA from the constructions in thepresence of the inducer IPTG and the substrate X-Gal; theresults are summarized in Fig. 1B. The truncation of the a
A
B
5980 Genetics: Liebig et al.

Proc. Natl. Acad. Sci. USA 88 (1991) 5981
fragment and the extension by 28 amino acids from VP1remaining after 2A had cleaved did not significantly affectcomplementation, as the plaques from the construction M13/2AA2B were dark blue. However, the plaques from theinactive proteinase construction M13/2A(Q134)A2B were
only faintly blue. The C-terminal extension of 200 aminoacids from the inactive 2A had apparently limited the abilityof the a fragment to complement. This restriction of the a
complementation by the proteinase is referred to as protein-ase trapping.
Reintroduction of an Arg residue at position 134 in theconstruction M13/2A(Q134)A2B was then undertaken bysite-directed mutagenesis. To eliminate the possibility ofreversion or contamination, the primer used contained an Argcodon (CGT instead of AGA) different from that of the wildtype. After in vitro mutagenesis, the DNA was transfectedinto JM101 cells in the presence of IPTG and X-Gal. Ap-proximately half of the plaques were blue, indicating that Argwas again present at position 134. This was confirmed byDNA sequencing; the 2A genes from phage obtained fromdark blue plaques contained the reintroduced Arg codon,whereas those from faintly blue plaques still possessed a Glncodon at this position. No other changes were observed in thesequence of the 2A.
Optimization and Characterization of the System. The ex-periment described above showed clearly that the presence ofthe 200-amino acid extension from the inactive 2A proteinasecould influence the ability of the a fragment to undergocomplementation. The system was then optimized as follows.First, Arg-43 of the a fragment was changed into Ser with thesimultaneous introduction of a Sal I site. Any proteinase cannow be fused to the lacZ gene, as the reading frame can bevaried by the appropriate choice of restriction enzyme;HincII and Acc I also recognize the Sal I site, but the cuts arestaggered by one nucleotide. In addition, the 2A/A2B genefragment was replaced with one coding for 2A followed bytwo stop codons. This allows the detection of the 2A proteinby the antiserum PC20, which only reacts when the Cterminus of 2A is free (5). As inactive 2A, a fragment wastaken in which glycine residues 104, 107, and 108 had beendeleted to eliminate the possibility of reversion. Finally, anMlu I site was introduced into the block 8 amino acids beforethe C terminus of VP1 to allow the 2A gene fragment and itscleavage site to be excised; this did not change the amino acidsequence.These modifications also affected the level of a comple-
mentation as blue plaques were produced with the wild-type2A and white ones were produced with the deletion mutant(Fig. 1B), making differentiation easier. Examination onWestern blots of the a fragment-2A fusion proteins inducedby IPTG with the antiserum PC20 revealed a band of 15 kDa,corresponding to mature 2A (Fig. 2). However, with theM13/2AA3GIy construction, the antiserum detected a bandof 24 kDa, corresponding to the sum of the molecular massesof the a fragment, the C-terminal amino acids of VP1 and 2A.
Screening for Inactive 2A Proteases After PCR Mutagenesis.PCR amplification of the a fragment/VP1/2A wild-type generegion from M13/2A was achieved by using the primerspositioned as in Fig. 1B (corresponding to M13mpl8 nucle-otides 6334-6350 and the complement of 6566-6583). Am-plification was for 40 cycles, upon which a further 3 units ofTaqDNA polymerase was added and amplification continuedfor another 10 cycles. The amplified DNA was then cleavedwith Mlu I and HindIII, giving a 450-bp fragment coding forthe last 8 amino acids ofVP1 and the entire 2A sequence. Thepurified fragment was ligated into the isolated 7.0-kilobaseMlu I/HindIII fragment ofM13/2A and the mixture was usedto transfect JM101 cells. Of the resulting plaques, =5% werewhite and 1% were only faintly blue; the remainder were notdistinguishable from wild-type blue plaques. Examination on
Ml 2 3
48_3626-
-24
_15
FIG. 2. Expression products of M13/2A and M13/2AA3GIy.Western blot of the proteins induced with 1 mM IPTG from JM101cells transfected with M13/2A (lane 1), M13/2AA3GIy (lane 2), andJM101 cells alone (lane 3) separated on a 15% polyacrylamide gel andprobed with the PC20 antiserum. The 24-kDa species is the un-cleaved product, and the 15-kDa product is the mature 2A protein.The positions and molecular masses (in kDa) of marker proteins (laneM) are shown on the left.
Western blots of the proteins produced from phage from 20white plaques on induction with IPTG revealed that theuncleaved form of 2A was produced (data not shown). Incontrast, a mature 2A in the induced proteins was expressedfrom each of six phage DNAs isolated from blue plaques,implying that 2A proteinase activity had not been affected.DNA sequencing of the VP1/2A genes from these 26
isolates was therefore carried out. Mutations in 2A werefound in all 20 phage DNAs with a white phenotype. Thenumbers of mutations found per 2A gene are summarized inTable 1 and the positions of the resulting missense mutationsare shown in Fig. 3. In the 6 phage DNAs obtained from blueplaques, 3 had wild-type 2A sequences and 1 had a silentmutation. Two possessed single amino acid changes (Ser-3 toCys and Ser-83 to Gly); presumably, these mutations do notaffect 2A function.Phage from four faintly blue plaques (after rescreening to
ensure that the color was reproducible) were also examined(Table 1 and Fig. 3). Amino acid changes were found in the2A genes of three of the phage DNAs. Of the three, oneexpressed 90% uncleaved 2A (indicating a reduced level offree a fragment) and two expressed only uncleaved 2A (datanot shown). This implied that the 2As were only marginallyactive, leading to the formation of insufficient mature 2A tobe detected on a Western blot but of sufficient a fragment togive a low level ofa complementation. The fourth mutant hada silent nucleotide change and expressed a mature 2A;changes in other parts of the a complementation system mayaccount for this faintly blue plaque.
Adaptation of the System for Intermolecular Cleavage. Theconstruction M13/2A is limited to the examination of intra-molecular cleavage. The system was therefore extended toenable intermolecular cleavage to be monitored, as depicted
Table 1. Nucleotide changes in VP1/2A genes of phage obtainedfrom white, faintly blue, and blue plaques
Number of mutations Plaque colorper 2A gene White Faintly blue Blue
0 - - 31 (silent) - 1 11 9* 2 21 + 1 (silent) 4 - -
2 6 1 -3 1 - -
Total 20 4 6
*Of the 9 white mutants bearing just 1 mutation, 2 resulted in stopcodons, 3 were deletions, and 4 contained an amino acid change.
Genetics: Liebig et al.
Proc. Natl. Acad. Sci. USA 88 (1991)
2A>A <-AVP'-' N
P CT TT S
T C R G
YC
B
R W S S S Lt T T tft
-28 -3 3 49 60/ 83 101 112 130 14261 1 8
78 84 8
2 13 4 5 6 673512-28 1 142
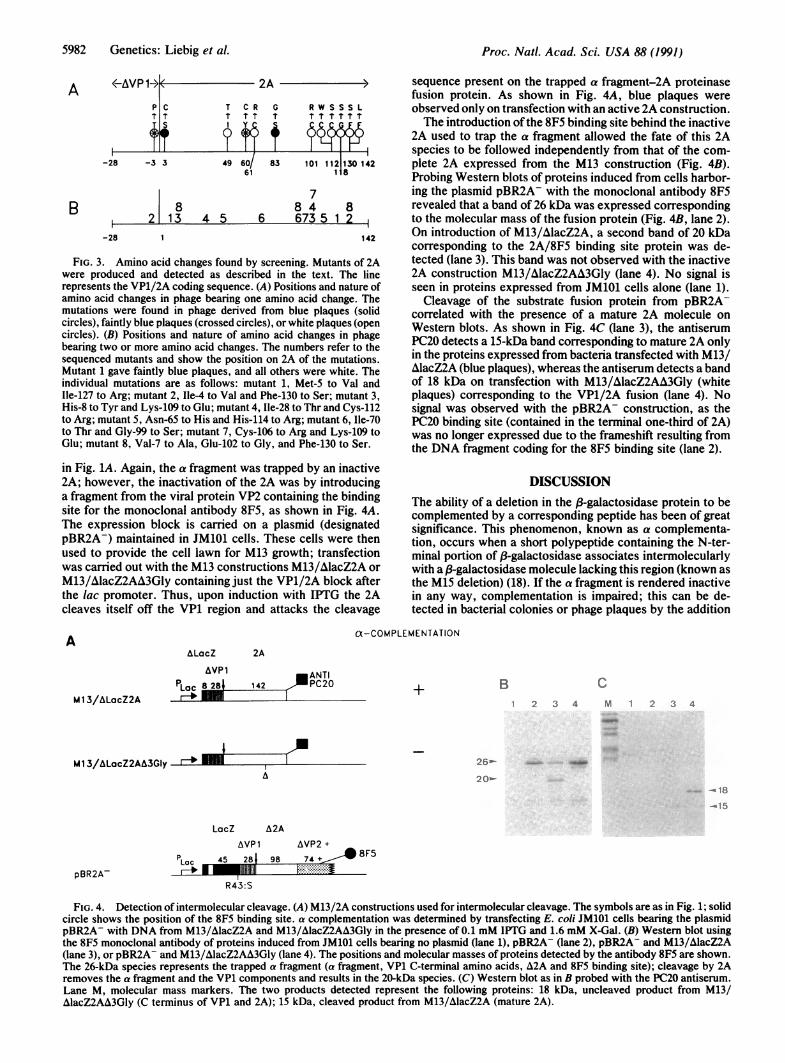
FIG. 3. Amino acid changes found by screening. Mutants of 2Awere produced and detected as described in the text. The linerepresents the VP1/2A coding sequence. (A) Positions and nature ofamino acid changes in phage bearing one amino acid change. Themutations were found in phage derived from blue plaques (solidcircles), faintly blue plaques (crossed circles), or white plaques (opencircles). (B) Positions and nature of amino acid changes in phagebearing two or more amino acid changes. The numbers refer to thesequenced mutants and show the position on 2A of the mutations.Mutant 1 gave faintly blue plaques, and all others were white. Theindividual mutations are as follows: mutant 1, Met-S to Val andIle-127 to Arg; mutant 2, Ile-4 to Val and Phe-130 to Ser; mutant 3,His-8 to Tyr and Lys-109 to Glu; mutant 4, Ile-28 to Thr and Cys-112to Arg; mutant 5, Asn-65 to His and His-114 to Arg; mutant 6, Ile-70to Thr and Gly-99 to Ser; mutant 7, Cys-106 to Arg and Lys-109 toGlu; mutant 8, Val-7 to Ala, Glu-102 to Gly, and Phe-130 to Ser.
in Fig. 1A. Again, the a fragment was trapped by an inactive2A; however, the inactivation of the 2A was by introducinga fragment from the viral protein VP2 containing the bindingsite for the monoclonal antibody 8F5, as shown in Fig. 4A.The expression block is carried on a plasmid (designatedpBR2A-) maintained in JM101 cells. These cells were thenused to provide the cell lawn for M13 growth; transfectionwas carried out with the M13 constructions M13/AlacZ2A orM13/AlacZ2AA3Gly containing just the VP1/2A block afterthe lac promoter. Thus, upon induction with IPTG the 2Acleaves itself off the VP1 region and attacks the cleavage
sequence present on the trapped a fragment-2A proteinasefusion protein. As shown in Fig. 4A, blue plaques wereobserved only on transfection with an active 2A construction.The introduction of the 8F5 binding site behind the inactive
2A used to trap the a fragment allowed the fate of this 2Aspecies to be followed independently from that of the com-plete 2A expressed from the M13 construction (Fig. 4B).Probing Western blots of proteins induced from cells harbor-ing the plasmid pBR2A- with the monoclonal antibody 8F5revealed that a band of 26 kDa was expressed correspondingto the molecular mass of the fusion protein (Fig. 4B, lane 2).On introduction of M13/AlacZ2A, a second band of 20 kDacorresponding to the 2A/8F5 binding site protein was de-tected (lane 3). This band was not observed with the inactive2A construction M13/AlacZ2AA3Gly (lane 4). No signal isseen in proteins expressed from JM101 cells alone (lane 1).Cleavage of the substrate fusion protein from pBR2A-
correlated with the presence of a mature 2A molecule onWestern blots. As shown in Fig. 4C (lane 3), the antiserumPC20 detects a 15-kDa band corresponding to mature 2A onlyin the proteins expressed from bacteria transfected with M13/AlacZ2A (blue plaques), whereas the antiserum detects a bandof 18 kDa on transfection with M13/AlacZ2AA3Gly (whiteplaques) corresponding to the VP1/2A fusion (lane 4). Nosignal was observed with the pBR2A- construction, as thePC20 binding site (contained in the terminal one-third of 2A)was no longer expressed due to the frameshift resulting fromthe DNA fragment coding for the 8F5 binding site (lane 2).
DISCUSSIONThe ability of a deletion in the P-galactosidase protein to becomplemented by a corresponding peptide has been of greatsignificance. This phenomenon, known as a complementa-tion, occurs when a short polypeptide containing the N-ter-minal portion of j-galactosidase associates intermolecularlywith a /3-galactosidase molecule lacking this region (known asthe M15 deletion) (18). If the a fragment is rendered inactivein any way, complementation is impaired; this can be de-tected in bacterial colonies or phage plaques by the addition
a-COMPLEMENTATION
ALacZ 2A
AVP 1 ANTI
PLac 8 28 142 PC20 + B1 2 3 4
Ml 3/ALacZ2AA3GIyA
26-
209-18
LacZ A2A
AVPl AVP2+p 45 28 987S + 8F5
R43:S
FIG. 4. Detection of intermolecular cleavage. (A) M13/2A constructions used for intermolecular cleavage. The symbols are as in Fig. 1; solidcircle shows the position of the 8F5 binding site. a complementation was determined by transfecting E. coli JM101 cells bearing the plasmidpBR2A- with DNA from M13/AlacZ2A and M13/AlacZ2AA3Gly in the presence of 0.1 mM IPTG and 1.6 mM X-Gal. (B) Western blot usingthe 8F5 monoclonal antibody of proteins induced from JM101 cells bearing no plasmid (lane 1), pBR2A- (lane 2), pBR2A- and M13/AlacZ2A(lane 3), or pBR2A- and M13/A&lacZ2AA&3Gly (lane 4). The positions and molecular masses of proteins detected by the antibody 8F5 are shown.The 26-kDa species represents the trapped a fragment (a fragment, VP1 C-terminal amino acids, A2A and 8F5 binding site); cleavage by 2Aremoves the a fragment and the VP1 components and results in the 20-kDa species. (C) Western blot as in B probed with the PC20 antiserum.Lane M, molecular mass markers. The two products detected represent the following proteins: 18 kDa, uncleaved product from M13/AlacZ2AA3Gly (C terminus of VP1 and 2A); 15 kDa, cleaved product from M13/AlacZ2A (mature 2A).
A
M 1 3/ALacZ2ACM 2 3 4
pBR2A-
5982 Genetics: Liebig et al.

Proc. Natl. Acad. Sci. USA 88 (1991) 5983
of a suitable substrate. Normally, insertions are made closeto the N terminus of the a fragment, causing the loss of thereading frame (12).We have modified a complementation to enable the activ-
ity of picornaviral proteinases to be monitored. Our ap-proach, referred to as proteinase trapping, was to introducethe HRV2 2A proteinase at the C terminus of the a fragment.The 2A proteinase presumably cleaves itself off while trans-lation of the fusion protein is still proceeding, leaving an afragment containing an extension of 28 amino acids derivedfrom the VP1 part of the cleavage site, which does not affectcomplementation. However, if the proteinase is inactive,there is a C-terminal extension in the optimized constructionsof 170 amino acids, which decreases or abolishes comple-mentation. The extent of the residual complementation wasfound to be dependent on the length of the a fragment and onpoint mutations within it.
Using PCR and proteinase trapping, inactive proteinasemutants were obtained at a frequency of5% and contained anaverage of 1.5 nucleotide changes per 2A gene. The muta-tions were spread throughout 2A and the VP1 part of thecleavage site. Interestingly, several mutations were found atthe C terminus of 2A, a region previously shown to beessential for activity but possessing no similarity to the serineproteinases. For example, amino acid Phe-130 (conserved inall picornaviral 2As) was found to be extremely sensitive tomutation, with the changes Phe to Ser and Phe to Leu bothrendering the enzyme inactive.The mechanism by which the 2A proteinases of rhinovi-
ruses and polioviruses induce cleavage of p220 and therebybring about the shut-off of host-cell synthesis is not under-stood. The ability to screen large numbers of mutants forintermolecular cleavage will allow the substrate specificity ofthe HRV2 2A to be compared with the intramolecular spec-ificity as recently defined (19). As all picornaviruses appearto have the same cellular target, it will be of interest tochallenge cleavage site variants of 2A with other 2As todefine closely a canonical sequence for intermolecular cleav-age. Recently, intermolecular cleavage by the human immu-nodeficiency virus proteinase has been demonstrated byinserting a decapeptide bearing the cleavage site into theentire ,3-galactosidase protein (20); the system described herehas the advantage that the natural environment of the cleav-age site is conserved. Furthermore, the presence of a pro-teinase cleavage site inside the 3-galactosidase protein doesnot allow the detection of intramolecular cleavage.
In short, we have developed a system for screening pro-teinase mutants that should be applicable to the determina-tion of structure-function relationships in any highly specificviral proteinase. Experiments to screen for second site re-vertants reactivating a disabled proteinase remain to be done.
It is also evident that the system can be used to monitor theerror frequency of heat-stable DNA polymerases.
We thank Drs. M. Zorn and P. Volkmann for supplying DNAfragments, L. Frasel and H. Auer for expert technical assistance, andZ. Rattler for fighting sloppy thinking. This work was supported bythe Osterreichischer Fonds zur Forderung der WissenschaftlichenForschung and Boehringer Ingelheim.
1. Watson, J., Hopkins, N. H., Roberts, J. W., Steitz, J. A. &Weiner, A. M. (1987) Molecular Biology of the Gene (Benja-min/Cummings Publishing, Menlo Park, CA), 4th Ed., Vol. 2.
2. Rueckert, R. R. (1990) in Virology, ed. Fields, B. N. (Raven,New York), 2nd Ed., Vols. 1 and 2, pp. 507-548.
3. Hellen, C. U. T., Kraeusslich, H.-G. & Wimmer, E. (1989)Biochemistry 28, 9881-9890.
4. Toyoda, H., Nicklin, M. J. H., Murray, M. G., Anderson,C. W., Dunn, J. J., Studier, F. W. & Wimmer, E. (1986) Cell45, 761-770.
5. Sommergruber, W., Zorn, M., Blaas, D., Fessl, F., Volkmann,P., Maurer-Fogy, I., Pallai, P., Merluzzi, V., Matteo, M.,Skern, T. & Kuechler, E. (1989) Virology 169, 68-77.
6. Hanecak, R., Semler, B., Anderson, C. & Wimmer, E. (1982)Proc. Natl. Acad. Sci. USA 79, 3973-3977.
7. Etchison, D., Hansen, E., Ehrenfeld, E., Edery, I., Sonenberg,N., Milburn, S. & Hershey, J. W. B. (1984) J. Virol. 51,832-837.
8. Kraeusslich, H.-G., Nicklin, M. J. H., Toyoda, H., Etchison,D. & Wimmer, E. (1987) J. Virol. 61, 2711-2718.
9. Sun, X.-H. & Baltimore, D. (1989) Proc. Natl. Acad. Sci. USA86, 2143-2146.
10. Gorbalenya, A. E., Blinov, V. M. & Donchenko, A. M. (1986)FEBS Lett. 194, 253-257.
11. Bazan, J. F. & Fletterick, R. J. (1988) Proc. Natl. Acad. Sci.USA 85, 7872-7876.
12. Vieira, J. & Messing, J. G. (1982) Gene 19, 259-268.13. Maniatis, T., Fritsch, E. F. & Sambrook, J. (1982) Molecular
Cloning:A Laboratory Manual (Cold Spring Harbor Lab., ColdSpring Harbor, NY).
14. Tabor, S. & Richardson, C. C. (1987) Proc. NatI. Acad. Sci.USA 84, 4767-4771.
15. Taylor, J. W., Ott, J. & Eckstein, F. (1985) Nucleic Acids Res.13, 8765-8785.
16. Skern, T., Neubauer, C., Frasel, L., Gruendler, P., Sommer-gruber, W., Zorn, M., Kuechler, E. & Blaas, D. (1987) J. Gen.Virol. 68, 315-323.
17. Langley, K. F., Villarecjo, M. R., Fowler, A. V., Zamenhof,P. J. & Zabin, I. (1975) Proc. Natl. Acad. Sci. USA 72,2154-1257.
18. Ullmann, A., Jacob, F. & Monod, J. (1967) J. Mol. Biol. 24,339-343.
19. Skern, T., Sommergruber, W., Auer, H., Volkmann, P., Zorn,M., Liebig, H.-D., Fessl, F., Blaas, D. & Kuechler, E. (1991)Virology 181, 46-54.
20. Baum, E. Z., Bebernitz, G. A. & Gluzman, Y. (1990) Proc.Natl. Acad. Sci. USA 87, 10023-10027.
Genetics: Liebig et al.





























