Embed Size (px)
Citation preview

Supporting Information © Wiley-VCH 2006
69451 Weinheim, Germany

1
Evolving thermostable reverse transcriptase activity in a DNA polymerase scaffold Katharina B. M. Sauter and Andreas Marx* Department of Chemistry, University of Konstanz, 78457 Konstanz, Germany. [email protected]
MATERIALS AND METHODS
Library construction The Klentaq wild type construct was obtained by cloning the gene into the BsaI site of pASK-IBA37plus (IBA). The insert was sequenced to confirm that it had no mutations. Introduction of random mutations was performed by error-prone PCR. PCR reactions were performed using 5 units of Taq DNA polymerase (Fermentas), 10 mM Tris⋅HCl (pH 8.8), 50 mM KCl, 0.08 % Nonidet P40, 1.5 mM MgCl2, 50 µM MnCl2, 200 µM each of dNTP, 20 pM template pASK-IBA37plus KTQ wt and 500 nM each of primer (5’-ATG GTA CGT CTC AGC GCG CCC TGG AGG AGG CCC CCT-3’ forward, 5’-ATG GTA CGT CTC ATA TCA CTC CTT GGC GGA GAG CCA GTC-3’ reverse) in a 100 µl mixture. It was extended by 14 cycles of PCR (95 °C for 1 min, 65 °C for 1 min and 72 °C for 2 min). The product (1655 bp) was purified (MinEluteTM Reaction Cleanup Kit, QIAGEN) followed by digestion with BsmBI. The restriction fragment was isolated by electrophoresis in a 0.8 % agarose gel and purified (MinEluteTM Gel Extraction Kit, QIAGEN). The fragment was ligated with the BsaI digested pASK-IBA37plus by incubating at 22 °C for 1 h with T4 DNA Ligase (Fermentas). The resulting plasmid library was transformed into E. coli BL21 (DE3) Gold. Clones were picked from agar plates and separately grown overnight in 96 well plates containing LB medium (100 µg/ml carbenicillin). Klentaq mutants were parallely expressed in 1 ml cultures in 96 well plates by inducing with 200 µg/l AHT for 1 h. Lysis was done in a 600 µl mixture containing 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2 and 1 mg/ml lysozyme. The lysate was incubated on ice for 30 min followed by heat denaturation of all non-thermostable proteins at 72 °C for 45 min. After centrifugation, the lysate was directly used for screening. Screening for PCR active mutants Reaction mixtures (20 µl) for library screening contained 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2, 200 µM of each dNTP, 20 pM template MS2 (100mer, 5’-ATC GCT CGA GAA CGC AAG TTC TTC AGC GAA AAG CAC GAC AGT GGT CGC TAC ATA GCG TGG TTC CAT ACT GGA GGT GAA ATC ACC GAC AGC ATG AAG TCC G-3’), 100 nM of each primer (5’-ATC GCT CGA GAA CGC AAG TT-3’ forward, 5’-CGG ACT TCA TGC TGT CGG TG-3’ reverse) and 0.6x SYBRgreen I (Molecular probes). Reaction mixtures were dispensed in 384 well plates using an automated liquid handling device (Hamilton Microlab Star) followed by addition of 5 µl lysate solution. For negative controls, analogously treated cultures of XL10 gold cells harboring the non-coding vector pASK-IBA37plus were employed. After 50 cycles of PCR (95 °C for 30 s, 55 °C for 35 s and 72 °C for 40 s), fluorescence intensities were quantified using a fluorescence plate reader (Polarstar Optima, BMG Labtechnologies GmbH) with excitation at 485 nm and emission at 520 nm. Screening for reverse transcription (RT) active mutants Reaction mixtures (20 µl) contained 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2, 200 µM of each dNTP, 50 pg/µl template RNA from bacteriophage MS2 (Roche), 100 nM of each primer (5’-ATC GCT CGA GAA CGC AAG TT-3’ forward, 5’-CGG ACT TCA TGC TGT CGG TG-3’ reverse) and 0.6x SYBRgreen I (Molecular probes). Reaction mixtures were dispensed in 96 well PCR plates using an automated liquid handling device (Hamilton Microlab Star) followed by addition of 5 µl lysate solution.
Reactions were performed in a real-time PCR cycler (iCycler, BIO-RAD). After an initial reverse transcription cycle (95 °C for 30 s, 55 °C for 35 s and 72 °C for 15 min) and 50 PCR cycles (95 °C for 30 s, 55 °C for 35 s and 72 °C for 40 s), active mutants were identified by measuring the fluorescence melting curve of the amplified product. 3 % of the PCR active mutants showed RT activities to a varied extend. The most promising mutant (M1) was purified and further analyzed. Second round of mutation A second library was constructed and analyzed as described before using mutant M1 as template. This resulted in mutant M2. Purification of Klentaq mutants Klentaq wt and selected mutants were expressed as described above and purified using Ni-NTA agarose (BIO-RAD) following the manufacturer’s protocol under omission of imidazole during the lysis and wash steps (Figure S1). After buffer exchange to 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2 and 1 mM DTT containing 50 % glycerol, concentrations were measured using the nanoorange assay (Molecular Probes).

2
wt M1 M2
kDa 70 60
50
Figure S1: SDS-PAGE gel of purified Klentaq DNA polymerases (coomassie blue staining). 7.25 pmol of each DNA
polymerase were loaded. Reverse transcription PCR using template RNA from bacteriophage MS2 (as shown in Figure 1a) Reaction mixtures (50 µl) contained 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2, 200 µM of each dNTP, 50 pg/µl template RNA from bacteriophage MS2 (Roche) or 40 pM DNA template MS2 (100 nt, 5’-d(ATC GCT CGA GAA CGC AAG TTC TTC AGC GAA AAG CAC GAC AGT GGT CGC TAC ATA GCG TGG TTC CAT ACT GGA GGT GAA ATC ACC GAC AGC ATG AAG TCC G)-3’), 100 nM of each primer (5’-d(ATC GCT CGA GAA CGC AAG TT)-3’ forward, 5’-d(CGG ACT TCA TGC TGT CGG TG)-3’ reverse) and 50 nM Klentaq DNA polymerase. After an initial reverse transcription cycle (95 °C for 30 s, 55 °C for 35 s and 72 °C for 15 min) the product was amplified by 30-40 PCR cycles (95 °C for 30 s, 55 °C for 35 s and 72 °C for 40 s). Reverse transcription PCR using template RNA from E. coli 16s mRNA Reaction mixtures (50 µl) contained 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2, 200 µM of each dNTP, 40 ng/µl template RNA S16 from E. coli (Roche) or 40 pM DNA template S16 DNA (100 nt, 5’-d(CTG GCG GCA GGC CTA ACA CAT GCA AGT CGA ACG GTA ACA GGA AGA AGC TTG CTT CTT TGC TGA CGA GTG GCG GAC GGG TGA GTA ATG TCT GGG AAA CTG C)-3’), 100 nM of each primer (5’-d(CTG GCG GCA GGC CTA ACA CA)-3’ forward, 5’-d(GCA GTT TCC CAG ACA TTA CT)-3’ reverse) and 50 nM Klentaq DNA polymerase. After an initial reverse transcription cycle (95 °C for 30 s, 66 °C for 35 s and 72 °C for 15 min) the product was amplified by 20-40 PCR cycles (95 °C for 30 s, 66 °C for 35 s and 72 °C for 40 s).
nt 1007550
Klentaq wt M1
PCR cycles 20 3040 40 4020 3040 40 40
DNARNA
RNARNA -template
DNARNA
RNARNA -
Figure S2: RT-PCR activity of Klentaq wt and M1 with S16 RNA template from E. coli.
Primer extension assays with an RNA template, determination of reverse transcription activity (as shown in Figure 1b) Reaction mixtures (10 µl) contained 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2, 100 µM of each dNTP, 100 nM template RNA (50mer, 5’-AUC GCU CGA GAA CGC AAG UUC UUC AGC GAA AAG CAC GAC AGU GGU CGC UA-3’), 100 nM 5’-[32P]-labeled primer (5’-d(TAG CGA CCA CTG TCG TGC TT)-3’) and 50 nM Klentaq DNA polymerase. After incubation at 95 °C for 30 s, 55 °C for 35 s and 72 °C for 2-90 min, reactions were quenched by addition of 3 volumes of gel loading buffer (80% formamide, 20 mM EDTA) and product mixtures were analysed by 12 % denaturing PAGE. Activity was measured by quantifying the intensity of each band produced by the respective DNA polymerase using a Phosphorimager. From this quantification the amount of incorporated nucleotides was calculated. The total amount of incorporated nucleotides for each reaction equals the sum of incorporated nucleotide of each band. The presented results (Table S1) are from measurements that are at least three times independently repeated. Table S1: Relative reverse transcription activities of Klentaq DNA polymerases.
wild-type M1 M2 specific activity [fmol dNTPs/min] 1.2 ± 0.1 48.9 ± 4.8 27.5 ± 3.2
relative activity 1 40 22
3
DNA polymerase activity determination on DNA templates Reaction mixtures (20 µl) contained 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2, 100 µM of each dNTP, 150 nM template DNA (50mer, 5’-ATC GCT CGA GAA CGC AAG TTC TTC AGC GAA AAG CAC GAC AGT GGT CGC TA-3’), 100 nM 5’-[32P] primer (5’-TAG CGA CCA CTG TCG TGC TT-3’) and 0.1-5 nM Klentaq DNA polymerase. After incubation at 95 °C for 30 s, 55 °C for 35 s and 72 °C for 10 min, reactions were quenched by addition of 3 volumes of gel loading buffer (80% formamide, 20 mM EDTA) and product mixtures were analysed by 12 % denaturating PAGE. Activity was measured by quantifying the intensity of each band produced by the respective DNA polymerase using a Phosphorimager. From this quantification the amount of incorporated nucleotides was calculated. The total amount of incorporated nucleotides for each reaction equals the sum of incorporated nucleotide of each band. The presented results (Table S2) are from measurements that are at least three times independently repeated.
Table S2: Specific activities of Klentaq DNA polymerases [fmol dNTPs/fmol pol]with 10 min.
wild-type M1 M2
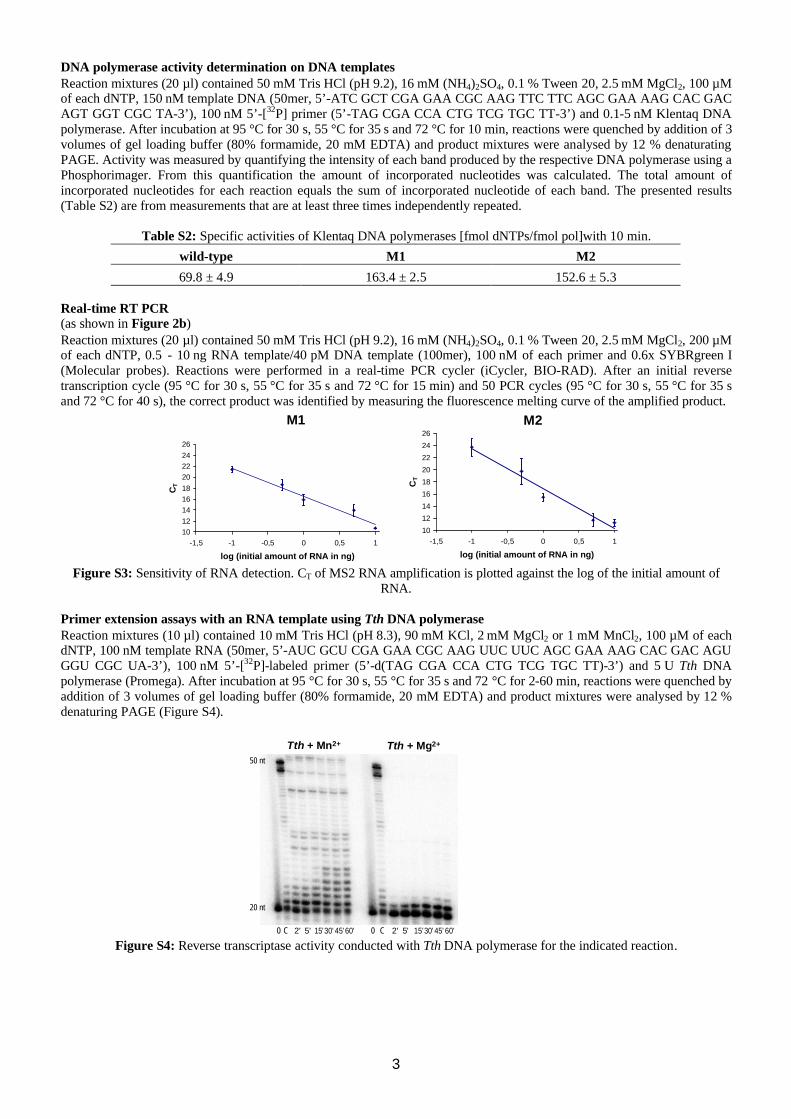
69.8 ± 4.9 163.4 ± 2.5 152.6 ± 5.3 Real-time RT PCR (as shown in Figure 2b) Reaction mixtures (20 µl) contained 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2, 200 µM of each dNTP, 0.5 - 10 ng RNA template/40 pM DNA template (100mer), 100 nM of each primer and 0.6x SYBRgreen I (Molecular probes). Reactions were performed in a real-time PCR cycler (iCycler, BIO-RAD). After an initial reverse transcription cycle (95 °C for 30 s, 55 °C for 35 s and 72 °C for 15 min) and 50 PCR cycles (95 °C for 30 s, 55 °C for 35 s and 72 °C for 40 s), the correct product was identified by measuring the fluorescence melting curve of the amplified product.
M1 M2
101214161820222426
-1,5 -1 -0,5 0 0,5 1
log (initial amount of RNA in ng)
CT
10
12
14
16
18
20
22
24
26
-1,5 -1 -0,5 0 0,5 1
log (initial amount of RNA in ng)
CT
Figure S3: Sensitivity of RNA detection. CT of MS2 RNA amplification is plotted against the log of the initial amount of
RNA. Primer extension assays with an RNA template using Tth DNA polymerase Reaction mixtures (10 µl) contained 10 mM Tris⋅HCl (pH 8.3), 90 mM KCl, 2 mM MgCl2 or 1 mM MnCl2, 100 µM of each dNTP, 100 nM template RNA (50mer, 5’-AUC GCU CGA GAA CGC AAG UUC UUC AGC GAA AAG CAC GAC AGU GGU CGC UA-3’), 100 nM 5’-[32P]-labeled primer (5’-d(TAG CGA CCA CTG TCG TGC TT)-3’) and 5 U Tth DNA polymerase (Promega). After incubation at 95 °C for 30 s, 55 °C for 35 s and 72 °C for 2-60 min, reactions were quenched by addition of 3 volumes of gel loading buffer (80% formamide, 20 mM EDTA) and product mixtures were analysed by 12 % denaturing PAGE (Figure S4).
2' 5' 15'30' 45'60'C0 2' 5' 15'30' 45' 60'C0
Tth + Mn2+ Tth + Mg2+
20 nt
50 nt
Figure S4: Reverse transcriptase activity conducted with Tth DNA polymerase for the indicated reaction.
4
Thermostability test Aliquots of Klentaq wild type and mutant enzymes in 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2 and 1 mM DTT containing 50 % glycerol were incubated at 95 °C for various time periods up to 90 min and then assayed for of DNA polymerase activity as described above. The results resulted from independently repeated experiments.
Table S3: Half-life of Klentaq DNA polymerases [min].
wild-type M1 M2
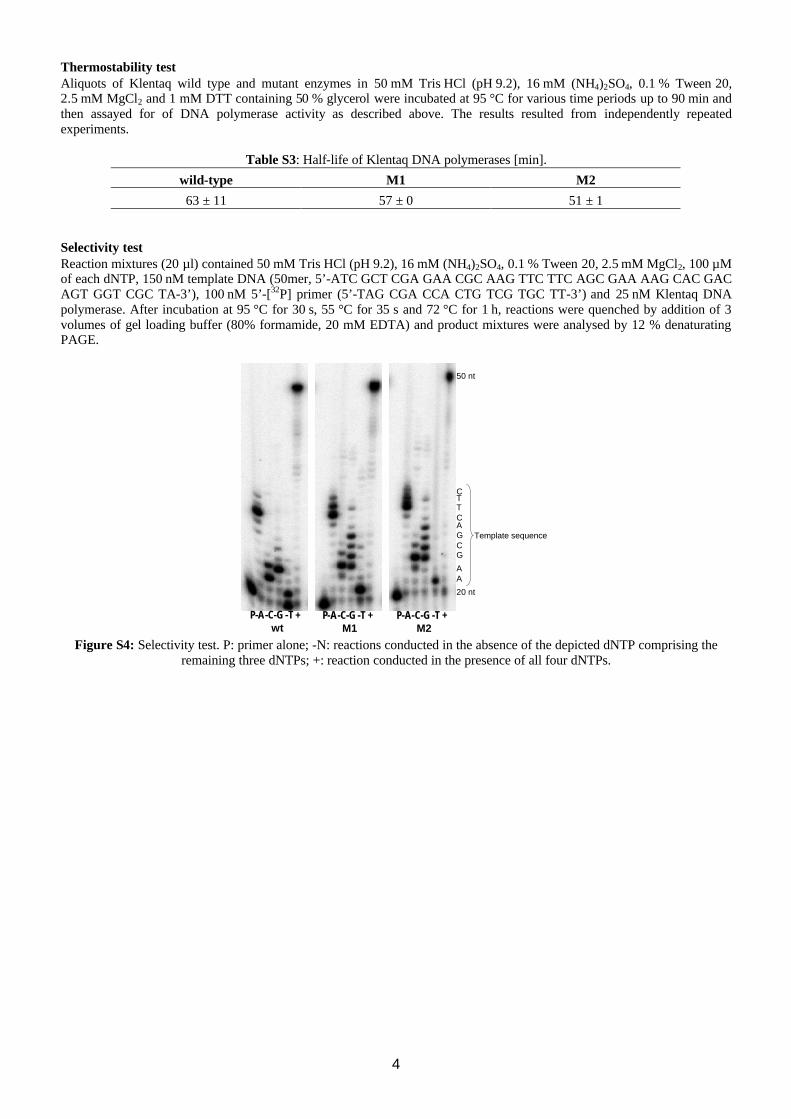
63 ± 11 57 ± 0 51 ± 1 Selectivity test Reaction mixtures (20 µl) contained 50 mM Tris⋅HCl (pH 9.2), 16 mM (NH4)2SO4, 0.1 % Tween 20, 2.5 mM MgCl2, 100 µM of each dNTP, 150 nM template DNA (50mer, 5’-ATC GCT CGA GAA CGC AAG TTC TTC AGC GAA AAG CAC GAC AGT GGT CGC TA-3’), 100 nM 5’-[32P] primer (5’-TAG CGA CCA CTG TCG TGC TT-3’) and 25 nM Klentaq DNA polymerase. After incubation at 95 °C for 30 s, 55 °C for 35 s and 72 °C for 1 h, reactions were quenched by addition of 3 volumes of gel loading buffer (80% formamide, 20 mM EDTA) and product mixtures were analysed by 12 % denaturating PAGE.
20 nt
A
GCGAC
CTT
50 nt
A
P-A-C-G -T +wt
P-A-C-G -T +M1
P-A-C-G -T +M2
Template sequence
Figure S4: Selectivity test. P: primer alone; -N: reactions conducted in the absence of the depicted dNTP comprising the
remaining three dNTPs; +: reaction conducted in the presence of all four dNTPs.










![69451 Weinheim, Germany - Wiley-VCHanomalous dispersion corrections were taken from the International Tables for X-ray Crystallography.[5] Structure solution, refinement and generation](https://img.pdfslide.us/doc/110x75/614409696cc38f259c25ead6/69451-weinheim-germany-wiley-anomalous-dispersion-corrections-were-taken-from.jpg)















![A NEW APPROAC TOWARDS MULTIPROPERTY · COMMUNICATIONS 920 WILEY-VCH Verlag GmbH, D-69451 Weinheim, 2001 1433-7851/01/4005-0920 $ 17.50+.50/0 Angew. Chem. Int. Ed. 2001, 40,No.5 ligation,[3]](https://img.pdfslide.us/doc/110x75/5fc72913c577425d7b108fcd/a-new-approac-towards-multiproperty-communications-920-wiley-vch-verlag-gmbh-d-69451.jpg)


