Embed Size (px)
Citation preview

4th Southern School on Computational Chemistry
13
AAbbssttrraaccttss
for
oral amp poster presentations
4th Southern School on Computational Chemistry
15
Understanding Intermolecular Perturbation Theory
William H Adams
Wright and Rieman Chemistry Laboratories Rutgers University
New Brunswick NJ 08903
The first calculation of an interatomic potential by perturbation methods was published almost 80 years ago [1] the first general formal theory just three years later [2] The fundamental problems confronting the perturbation theory approach however began to be recognized only 33 years ago [3-5] and it was not until the 1990s that intermolecular perturbation theories were examined to see if any overcame the fundamental problems [6] The ones that did were unsatisfactory for another reason It is only in the last three years that intermolecular perturbation theories have been published which are designed to deal with the fundamental problems in a satisfactory manner [78] We begin this lecture with a review of why perturbation theory is thought to be well suited to the calculation of intermolecular potentials Then we define the simplest intermolecular perturbation theory the Polarization Approximation (PA) explaining briefly how it was imagined to work then in detail how it fails It is the PA theory which defines the fundamental problems that must be solved We consider two of the old alternatives to the PA theory showing how one fails to solve the problem and why the other is unacceptable as a solution Finally if time permits we will tell you a little about the new perturbation theories
1 S C Wang Phys Z 28 663 (1927) 2 R Eisenschitz and F London Z Phys 60 491 (1930) 3 P Claverie Int J Quantum Chem 5 273 (1971) 4 J D Morgan III and B Simon Int J Quantum Chem 17 1143 (1980) 5 W Kutzelnigg J Chem Phys 73 343 (1980) 6 W H Adams Int J Quantum Chem S24 165 (1990) Chem Phys Lett 229 472 (1994)
Int J Quantum Chem 60 273 (1996) 7 K Patkowski B Jeziorski and K Szalewicz J Mol Struct (Theochem) 547 293 (2001) K
Patkowski B Jeziorski T Korona and K Szalewicz J Chem Phys 117 5124 (2002) K Patkowski B Jeziorski and K Szalewicz to be published
8 W H Adams Theor Chem Acc 108 225 (2002)
4th Southern School on Computational Chemistry
16
Interactions of Some Biomaterials with Caffeine in Aqueous Solutions at Different Temperatures
Anwar Ali Soghra Hyder and Saba Sabir
Department of Chemistry Jamia Millia Islamia (Central University) New Delhi-110025 India
Densityρ viscosity η and refractive index nD for glycine alanine serine and valine (010
020 030 040 and 050M) have been determined in 005M caffeine + water solvent mixtures at
25 30 35 400C The density data have been used to compute apparent molar volume φv
limiting apparent molar volume φ0v and the slope S
v using Massonrsquos equation The viscosity
data have been analysed by means of Jones-Dole equation The values of Falkenhagen
Coefficient A and Jones-Dole Coefficient B thus obtained are used to interprete the solute-
solute and solute-solvent interactions respectively The transition -state theory was applied to
obtain the activation parameters of viscous flow ie free energy of activation per mole of solvent
∆micro10and solute ∆micro2
0 The enthalpy ∆H and entropy ∆S of activation of viscous flow were
also computed for the system Refractive index was used to calculate molar refractivity RD of
the mixtures The results have been interpreted in the light of various interactions occurring
between the components of the mixtures under study
4th Southern School on Computational Chemistry
17
Physico-Chemical Studies of Glycine in Alkanediols + Water Mixtures at Different Temperatures
Anwar Ali Soghra Hyder and Shahla Khan
Department of Chemistry Jamia Millia Islamia (Central University) New Delhi-110025 India
Densitiesρ viscosities η and refractive indices nD of 01-05M glycine in aqueous (30
vv) ethan -12-diol propan -12-diol and butan-13-diol have been measured in the temperature
range from 298 to 313K The experimental values of ρ and η were used to calculate the values of
apparent molar volume φv partial molar volume at infinite dilution φ0v partial molar volume of
transferφ0v(tr) from aqueous diols to water A and B coefficients of the Jones-Dole equation free
energies of activation of viscous flow ∆micro10 and ∆micro2
0 per mole of solvent and solute
respectively hydration number Hn enthalpy ∆H and entropy ∆S of viscous flow nD data
were used to calculate molar refractivity RD The results have been interpreted in the light of
solute-solute and solute-solvent interactions The structure breaking property of the solute is also
taken into account
4th Southern School on Computational Chemistry
18
A Theoretical Investigation of the Structure and Properties of Ascorbic Acid (Vitamin C)
RN Allen MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 JR Lynch Street Jackson MS 39217
Pure ascorbic acid (AA) or vitamin C is a white crystalline solid which is water soluble AA is an extremely interesting molecule which has many important biological functions It is necessary for the production of collagen in connective tissue helps promote a healthy immune system in humans and helps prevent scurvy AA also functions as an antioxidant which it helps protect the body against damaging free radicals Ascorbic acid has been associated with prevention of many degenerative disease like cataracts certain cancers and cardiovascular diseases
Geometries of neutral tautomeric and anionic species of AA were optimized at the Density Functional Theory level using the B3LYP method The radical species were studied using the unrestricted B3LYP method Single point energy calculations were also performed using the MP2 and UMP2 method for all species All calculation utilized the 6-311++G(dp) basis set Nature of stationary points on the potential energy surfaces (PESs) was ascertained by harmonic vibrational frequency analysis all structures were found minima The Tomasirsquos polarized continuum model was used to examine the effects of aqueous solvation on the relative stability of interested species The electronic charge distribution molecular electrostatic potential ionization potential and electron affinity are also reported
O
O
O O
OO
CC
CC
C
C
4th Southern School on Computational Chemistry
19
Anchoring the Potential Energy Surface of the Water Trimer
Julie A Anderson and Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Six cyclic stationary points on the water trimer potential energy surface have been fully optimized at the MP2 level with the aug-cc-pVQZ basis set In agreement with previous work harmonic vibrational frequencies indicate two structures are minima Three are transition states connecting minima on the surface The remaining stationary point is a third-order saddle point The one- and n-particle limits of the electronic energies of each of these six structures were obtained by systematically varying basis sets and theoretical methods The former limit was estimated with the cc-pVXZ and aug-cc-pVXZ families of basis sets (X = 2-7) MP2 CCSD(T) and BD(TQ) calculations helped approach the latter Core correlation effects have also been assessed at the MP2 level with the cc-pCVXZ series of basis sets (X = 2-5) These data were combined in focal point analyses to provide highly accurate dissociation energies and relative energies for these stationary points
4th Southern School on Computational Chemistry
20
Two as Good as Four Relativistic Theory for Electrons Only
Maria Barysz and Andrzej J Sadlej
Department of Quantum Chemistry Institute of Chemistry Nicolaus Copernicus University Torun Poland
Relativistic quantum mechanics is routinely formulated in terms of the four-component one-electron wave functions These four-component vectors (spinors) form the basis for most advanced relativistic calculations in quantum chemistry The 4-component formalism is by no means trivial and imposes several inconvenient conditions on the form of the basis set functions into which each spinor component is to be expanded
Quite a gain in both the computational time and programming effort can be achieved by using the two-component formalism ie the formalism in which only the so--called large component of each spinor appears explicitly
However until recently the two-component methods have been merely approximations to the fully relativistic 4-component approaches A new approach which permits the reduction of the 4-component thoery to the exact two-component form will be surveyed and discussed The newly developed method permits a complete separation of the negative and positive energy spectra of the Dirac hamiltonian with arbitrarily high accuracy leading to what can be called a relativistic quantum theory for electrons only This means that all of relativistic quantum chemistry can be done in much easier two-component form The method can be also used to generate the spectrum of the negative energy states and opens new possibilities for the investigation of QED corrections References 1 M Barysz and A J Sadlej J Mol Struct (Theochem) 573 (2001) 181 2 M Barysz and A J Sadlej J Chem Phys 116 (2002) 2696 3 G Pestka and A J Sadlej J Mol Struct (Theochem) 592 (2002) 7 4 M Barysz in Theoretical Chemistry and Physics of Heavy and Superheavy Elements Eds U Kaldor and S Wilson Kluver Dordrecht 2003 pp 349 - 397
4th Southern School on Computational Chemistry
21
Computational Studies on Nitrogen-Substituted Steroidal Structures
Angela Bell
Auburn University Auburn AL
Using several steroidal structures as models nitrogen was introduced into their framework at
particular positions Variation in the location of the nitrogen could prove to be critical to the
compoundrsquos overall functionality A number of steroid structures containing nitrogen
azasteroids possess known pharmaceutical values such as anti-microbial andor anti-
inflammatory properties This project serves to support the experimental synthesis on one or
more of these compounds Theoretical calculations were pursued through the utilization of
PCModel semi-empirical as well as ab initio methods To bridge the gap between the
computing and bench environment computational studies will be undertaken to help determine a
reasonable synthetic strategy
4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
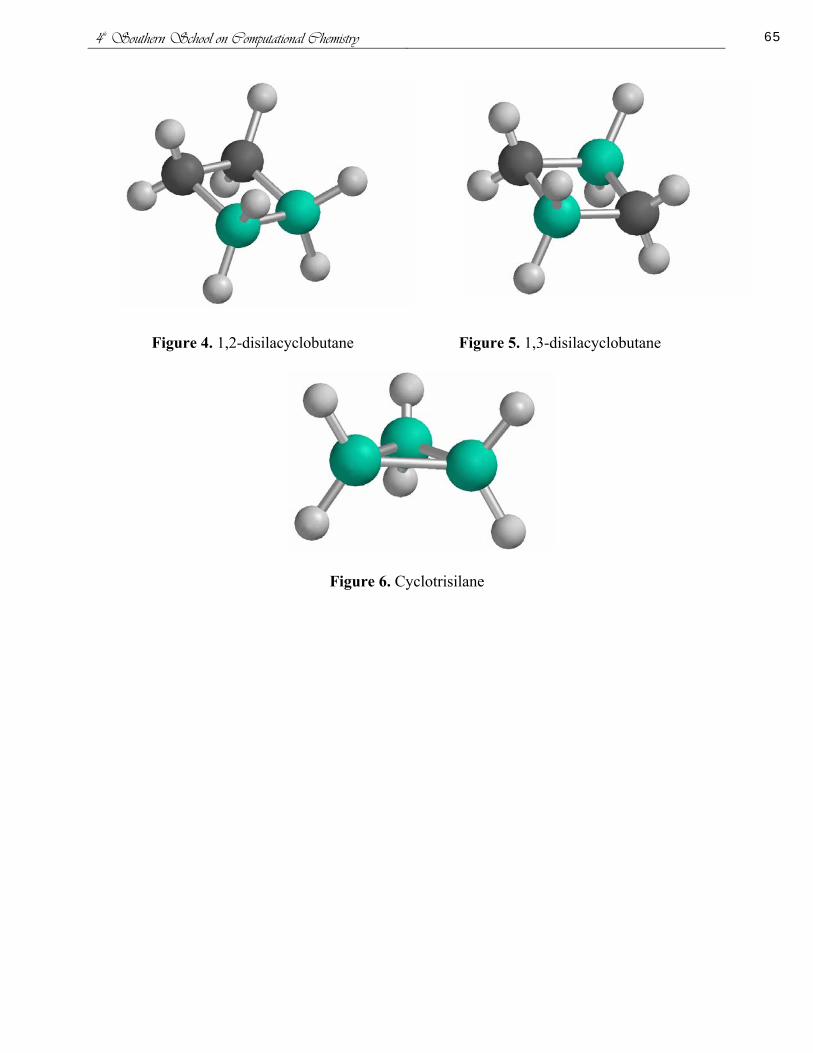
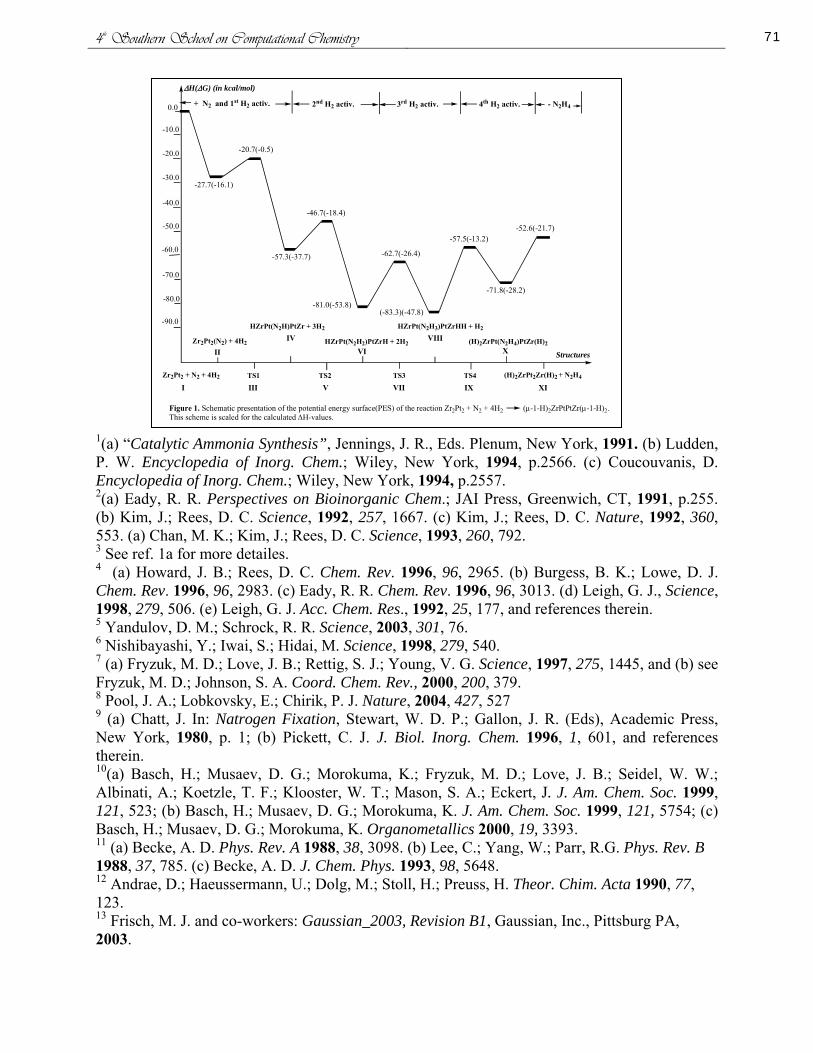
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
15
Understanding Intermolecular Perturbation Theory
William H Adams
Wright and Rieman Chemistry Laboratories Rutgers University
New Brunswick NJ 08903
The first calculation of an interatomic potential by perturbation methods was published almost 80 years ago [1] the first general formal theory just three years later [2] The fundamental problems confronting the perturbation theory approach however began to be recognized only 33 years ago [3-5] and it was not until the 1990s that intermolecular perturbation theories were examined to see if any overcame the fundamental problems [6] The ones that did were unsatisfactory for another reason It is only in the last three years that intermolecular perturbation theories have been published which are designed to deal with the fundamental problems in a satisfactory manner [78] We begin this lecture with a review of why perturbation theory is thought to be well suited to the calculation of intermolecular potentials Then we define the simplest intermolecular perturbation theory the Polarization Approximation (PA) explaining briefly how it was imagined to work then in detail how it fails It is the PA theory which defines the fundamental problems that must be solved We consider two of the old alternatives to the PA theory showing how one fails to solve the problem and why the other is unacceptable as a solution Finally if time permits we will tell you a little about the new perturbation theories
1 S C Wang Phys Z 28 663 (1927) 2 R Eisenschitz and F London Z Phys 60 491 (1930) 3 P Claverie Int J Quantum Chem 5 273 (1971) 4 J D Morgan III and B Simon Int J Quantum Chem 17 1143 (1980) 5 W Kutzelnigg J Chem Phys 73 343 (1980) 6 W H Adams Int J Quantum Chem S24 165 (1990) Chem Phys Lett 229 472 (1994)
Int J Quantum Chem 60 273 (1996) 7 K Patkowski B Jeziorski and K Szalewicz J Mol Struct (Theochem) 547 293 (2001) K
Patkowski B Jeziorski T Korona and K Szalewicz J Chem Phys 117 5124 (2002) K Patkowski B Jeziorski and K Szalewicz to be published
8 W H Adams Theor Chem Acc 108 225 (2002)
4th Southern School on Computational Chemistry
16
Interactions of Some Biomaterials with Caffeine in Aqueous Solutions at Different Temperatures
Anwar Ali Soghra Hyder and Saba Sabir
Department of Chemistry Jamia Millia Islamia (Central University) New Delhi-110025 India
Densityρ viscosity η and refractive index nD for glycine alanine serine and valine (010
020 030 040 and 050M) have been determined in 005M caffeine + water solvent mixtures at
25 30 35 400C The density data have been used to compute apparent molar volume φv
limiting apparent molar volume φ0v and the slope S
v using Massonrsquos equation The viscosity
data have been analysed by means of Jones-Dole equation The values of Falkenhagen
Coefficient A and Jones-Dole Coefficient B thus obtained are used to interprete the solute-
solute and solute-solvent interactions respectively The transition -state theory was applied to
obtain the activation parameters of viscous flow ie free energy of activation per mole of solvent
∆micro10and solute ∆micro2
0 The enthalpy ∆H and entropy ∆S of activation of viscous flow were
also computed for the system Refractive index was used to calculate molar refractivity RD of
the mixtures The results have been interpreted in the light of various interactions occurring
between the components of the mixtures under study
4th Southern School on Computational Chemistry
17
Physico-Chemical Studies of Glycine in Alkanediols + Water Mixtures at Different Temperatures
Anwar Ali Soghra Hyder and Shahla Khan
Department of Chemistry Jamia Millia Islamia (Central University) New Delhi-110025 India
Densitiesρ viscosities η and refractive indices nD of 01-05M glycine in aqueous (30
vv) ethan -12-diol propan -12-diol and butan-13-diol have been measured in the temperature
range from 298 to 313K The experimental values of ρ and η were used to calculate the values of
apparent molar volume φv partial molar volume at infinite dilution φ0v partial molar volume of
transferφ0v(tr) from aqueous diols to water A and B coefficients of the Jones-Dole equation free
energies of activation of viscous flow ∆micro10 and ∆micro2
0 per mole of solvent and solute
respectively hydration number Hn enthalpy ∆H and entropy ∆S of viscous flow nD data
were used to calculate molar refractivity RD The results have been interpreted in the light of
solute-solute and solute-solvent interactions The structure breaking property of the solute is also
taken into account
4th Southern School on Computational Chemistry
18
A Theoretical Investigation of the Structure and Properties of Ascorbic Acid (Vitamin C)
RN Allen MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 JR Lynch Street Jackson MS 39217
Pure ascorbic acid (AA) or vitamin C is a white crystalline solid which is water soluble AA is an extremely interesting molecule which has many important biological functions It is necessary for the production of collagen in connective tissue helps promote a healthy immune system in humans and helps prevent scurvy AA also functions as an antioxidant which it helps protect the body against damaging free radicals Ascorbic acid has been associated with prevention of many degenerative disease like cataracts certain cancers and cardiovascular diseases
Geometries of neutral tautomeric and anionic species of AA were optimized at the Density Functional Theory level using the B3LYP method The radical species were studied using the unrestricted B3LYP method Single point energy calculations were also performed using the MP2 and UMP2 method for all species All calculation utilized the 6-311++G(dp) basis set Nature of stationary points on the potential energy surfaces (PESs) was ascertained by harmonic vibrational frequency analysis all structures were found minima The Tomasirsquos polarized continuum model was used to examine the effects of aqueous solvation on the relative stability of interested species The electronic charge distribution molecular electrostatic potential ionization potential and electron affinity are also reported
O
O
O O
OO
CC
CC
C
C
4th Southern School on Computational Chemistry
19
Anchoring the Potential Energy Surface of the Water Trimer
Julie A Anderson and Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Six cyclic stationary points on the water trimer potential energy surface have been fully optimized at the MP2 level with the aug-cc-pVQZ basis set In agreement with previous work harmonic vibrational frequencies indicate two structures are minima Three are transition states connecting minima on the surface The remaining stationary point is a third-order saddle point The one- and n-particle limits of the electronic energies of each of these six structures were obtained by systematically varying basis sets and theoretical methods The former limit was estimated with the cc-pVXZ and aug-cc-pVXZ families of basis sets (X = 2-7) MP2 CCSD(T) and BD(TQ) calculations helped approach the latter Core correlation effects have also been assessed at the MP2 level with the cc-pCVXZ series of basis sets (X = 2-5) These data were combined in focal point analyses to provide highly accurate dissociation energies and relative energies for these stationary points
4th Southern School on Computational Chemistry
20
Two as Good as Four Relativistic Theory for Electrons Only
Maria Barysz and Andrzej J Sadlej
Department of Quantum Chemistry Institute of Chemistry Nicolaus Copernicus University Torun Poland
Relativistic quantum mechanics is routinely formulated in terms of the four-component one-electron wave functions These four-component vectors (spinors) form the basis for most advanced relativistic calculations in quantum chemistry The 4-component formalism is by no means trivial and imposes several inconvenient conditions on the form of the basis set functions into which each spinor component is to be expanded
Quite a gain in both the computational time and programming effort can be achieved by using the two-component formalism ie the formalism in which only the so--called large component of each spinor appears explicitly
However until recently the two-component methods have been merely approximations to the fully relativistic 4-component approaches A new approach which permits the reduction of the 4-component thoery to the exact two-component form will be surveyed and discussed The newly developed method permits a complete separation of the negative and positive energy spectra of the Dirac hamiltonian with arbitrarily high accuracy leading to what can be called a relativistic quantum theory for electrons only This means that all of relativistic quantum chemistry can be done in much easier two-component form The method can be also used to generate the spectrum of the negative energy states and opens new possibilities for the investigation of QED corrections References 1 M Barysz and A J Sadlej J Mol Struct (Theochem) 573 (2001) 181 2 M Barysz and A J Sadlej J Chem Phys 116 (2002) 2696 3 G Pestka and A J Sadlej J Mol Struct (Theochem) 592 (2002) 7 4 M Barysz in Theoretical Chemistry and Physics of Heavy and Superheavy Elements Eds U Kaldor and S Wilson Kluver Dordrecht 2003 pp 349 - 397
4th Southern School on Computational Chemistry
21
Computational Studies on Nitrogen-Substituted Steroidal Structures
Angela Bell
Auburn University Auburn AL
Using several steroidal structures as models nitrogen was introduced into their framework at
particular positions Variation in the location of the nitrogen could prove to be critical to the
compoundrsquos overall functionality A number of steroid structures containing nitrogen
azasteroids possess known pharmaceutical values such as anti-microbial andor anti-
inflammatory properties This project serves to support the experimental synthesis on one or
more of these compounds Theoretical calculations were pursued through the utilization of
PCModel semi-empirical as well as ab initio methods To bridge the gap between the
computing and bench environment computational studies will be undertaken to help determine a
reasonable synthetic strategy
4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
16
Interactions of Some Biomaterials with Caffeine in Aqueous Solutions at Different Temperatures
Anwar Ali Soghra Hyder and Saba Sabir
Department of Chemistry Jamia Millia Islamia (Central University) New Delhi-110025 India
Densityρ viscosity η and refractive index nD for glycine alanine serine and valine (010
020 030 040 and 050M) have been determined in 005M caffeine + water solvent mixtures at
25 30 35 400C The density data have been used to compute apparent molar volume φv
limiting apparent molar volume φ0v and the slope S
v using Massonrsquos equation The viscosity
data have been analysed by means of Jones-Dole equation The values of Falkenhagen
Coefficient A and Jones-Dole Coefficient B thus obtained are used to interprete the solute-
solute and solute-solvent interactions respectively The transition -state theory was applied to
obtain the activation parameters of viscous flow ie free energy of activation per mole of solvent
∆micro10and solute ∆micro2
0 The enthalpy ∆H and entropy ∆S of activation of viscous flow were
also computed for the system Refractive index was used to calculate molar refractivity RD of
the mixtures The results have been interpreted in the light of various interactions occurring
between the components of the mixtures under study
4th Southern School on Computational Chemistry
17
Physico-Chemical Studies of Glycine in Alkanediols + Water Mixtures at Different Temperatures
Anwar Ali Soghra Hyder and Shahla Khan
Department of Chemistry Jamia Millia Islamia (Central University) New Delhi-110025 India
Densitiesρ viscosities η and refractive indices nD of 01-05M glycine in aqueous (30
vv) ethan -12-diol propan -12-diol and butan-13-diol have been measured in the temperature
range from 298 to 313K The experimental values of ρ and η were used to calculate the values of
apparent molar volume φv partial molar volume at infinite dilution φ0v partial molar volume of
transferφ0v(tr) from aqueous diols to water A and B coefficients of the Jones-Dole equation free
energies of activation of viscous flow ∆micro10 and ∆micro2
0 per mole of solvent and solute
respectively hydration number Hn enthalpy ∆H and entropy ∆S of viscous flow nD data
were used to calculate molar refractivity RD The results have been interpreted in the light of
solute-solute and solute-solvent interactions The structure breaking property of the solute is also
taken into account
4th Southern School on Computational Chemistry
18
A Theoretical Investigation of the Structure and Properties of Ascorbic Acid (Vitamin C)
RN Allen MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 JR Lynch Street Jackson MS 39217
Pure ascorbic acid (AA) or vitamin C is a white crystalline solid which is water soluble AA is an extremely interesting molecule which has many important biological functions It is necessary for the production of collagen in connective tissue helps promote a healthy immune system in humans and helps prevent scurvy AA also functions as an antioxidant which it helps protect the body against damaging free radicals Ascorbic acid has been associated with prevention of many degenerative disease like cataracts certain cancers and cardiovascular diseases
Geometries of neutral tautomeric and anionic species of AA were optimized at the Density Functional Theory level using the B3LYP method The radical species were studied using the unrestricted B3LYP method Single point energy calculations were also performed using the MP2 and UMP2 method for all species All calculation utilized the 6-311++G(dp) basis set Nature of stationary points on the potential energy surfaces (PESs) was ascertained by harmonic vibrational frequency analysis all structures were found minima The Tomasirsquos polarized continuum model was used to examine the effects of aqueous solvation on the relative stability of interested species The electronic charge distribution molecular electrostatic potential ionization potential and electron affinity are also reported
O
O
O O
OO
CC
CC
C
C
4th Southern School on Computational Chemistry
19
Anchoring the Potential Energy Surface of the Water Trimer
Julie A Anderson and Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Six cyclic stationary points on the water trimer potential energy surface have been fully optimized at the MP2 level with the aug-cc-pVQZ basis set In agreement with previous work harmonic vibrational frequencies indicate two structures are minima Three are transition states connecting minima on the surface The remaining stationary point is a third-order saddle point The one- and n-particle limits of the electronic energies of each of these six structures were obtained by systematically varying basis sets and theoretical methods The former limit was estimated with the cc-pVXZ and aug-cc-pVXZ families of basis sets (X = 2-7) MP2 CCSD(T) and BD(TQ) calculations helped approach the latter Core correlation effects have also been assessed at the MP2 level with the cc-pCVXZ series of basis sets (X = 2-5) These data were combined in focal point analyses to provide highly accurate dissociation energies and relative energies for these stationary points
4th Southern School on Computational Chemistry
20
Two as Good as Four Relativistic Theory for Electrons Only
Maria Barysz and Andrzej J Sadlej
Department of Quantum Chemistry Institute of Chemistry Nicolaus Copernicus University Torun Poland
Relativistic quantum mechanics is routinely formulated in terms of the four-component one-electron wave functions These four-component vectors (spinors) form the basis for most advanced relativistic calculations in quantum chemistry The 4-component formalism is by no means trivial and imposes several inconvenient conditions on the form of the basis set functions into which each spinor component is to be expanded
Quite a gain in both the computational time and programming effort can be achieved by using the two-component formalism ie the formalism in which only the so--called large component of each spinor appears explicitly
However until recently the two-component methods have been merely approximations to the fully relativistic 4-component approaches A new approach which permits the reduction of the 4-component thoery to the exact two-component form will be surveyed and discussed The newly developed method permits a complete separation of the negative and positive energy spectra of the Dirac hamiltonian with arbitrarily high accuracy leading to what can be called a relativistic quantum theory for electrons only This means that all of relativistic quantum chemistry can be done in much easier two-component form The method can be also used to generate the spectrum of the negative energy states and opens new possibilities for the investigation of QED corrections References 1 M Barysz and A J Sadlej J Mol Struct (Theochem) 573 (2001) 181 2 M Barysz and A J Sadlej J Chem Phys 116 (2002) 2696 3 G Pestka and A J Sadlej J Mol Struct (Theochem) 592 (2002) 7 4 M Barysz in Theoretical Chemistry and Physics of Heavy and Superheavy Elements Eds U Kaldor and S Wilson Kluver Dordrecht 2003 pp 349 - 397
4th Southern School on Computational Chemistry
21
Computational Studies on Nitrogen-Substituted Steroidal Structures
Angela Bell
Auburn University Auburn AL
Using several steroidal structures as models nitrogen was introduced into their framework at
particular positions Variation in the location of the nitrogen could prove to be critical to the
compoundrsquos overall functionality A number of steroid structures containing nitrogen
azasteroids possess known pharmaceutical values such as anti-microbial andor anti-
inflammatory properties This project serves to support the experimental synthesis on one or
more of these compounds Theoretical calculations were pursued through the utilization of
PCModel semi-empirical as well as ab initio methods To bridge the gap between the
computing and bench environment computational studies will be undertaken to help determine a
reasonable synthetic strategy
4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
17
Physico-Chemical Studies of Glycine in Alkanediols + Water Mixtures at Different Temperatures
Anwar Ali Soghra Hyder and Shahla Khan
Department of Chemistry Jamia Millia Islamia (Central University) New Delhi-110025 India
Densitiesρ viscosities η and refractive indices nD of 01-05M glycine in aqueous (30
vv) ethan -12-diol propan -12-diol and butan-13-diol have been measured in the temperature
range from 298 to 313K The experimental values of ρ and η were used to calculate the values of
apparent molar volume φv partial molar volume at infinite dilution φ0v partial molar volume of
transferφ0v(tr) from aqueous diols to water A and B coefficients of the Jones-Dole equation free
energies of activation of viscous flow ∆micro10 and ∆micro2
0 per mole of solvent and solute
respectively hydration number Hn enthalpy ∆H and entropy ∆S of viscous flow nD data
were used to calculate molar refractivity RD The results have been interpreted in the light of
solute-solute and solute-solvent interactions The structure breaking property of the solute is also
taken into account
4th Southern School on Computational Chemistry
18
A Theoretical Investigation of the Structure and Properties of Ascorbic Acid (Vitamin C)
RN Allen MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 JR Lynch Street Jackson MS 39217
Pure ascorbic acid (AA) or vitamin C is a white crystalline solid which is water soluble AA is an extremely interesting molecule which has many important biological functions It is necessary for the production of collagen in connective tissue helps promote a healthy immune system in humans and helps prevent scurvy AA also functions as an antioxidant which it helps protect the body against damaging free radicals Ascorbic acid has been associated with prevention of many degenerative disease like cataracts certain cancers and cardiovascular diseases
Geometries of neutral tautomeric and anionic species of AA were optimized at the Density Functional Theory level using the B3LYP method The radical species were studied using the unrestricted B3LYP method Single point energy calculations were also performed using the MP2 and UMP2 method for all species All calculation utilized the 6-311++G(dp) basis set Nature of stationary points on the potential energy surfaces (PESs) was ascertained by harmonic vibrational frequency analysis all structures were found minima The Tomasirsquos polarized continuum model was used to examine the effects of aqueous solvation on the relative stability of interested species The electronic charge distribution molecular electrostatic potential ionization potential and electron affinity are also reported
O
O
O O
OO
CC
CC
C
C
4th Southern School on Computational Chemistry
19
Anchoring the Potential Energy Surface of the Water Trimer
Julie A Anderson and Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Six cyclic stationary points on the water trimer potential energy surface have been fully optimized at the MP2 level with the aug-cc-pVQZ basis set In agreement with previous work harmonic vibrational frequencies indicate two structures are minima Three are transition states connecting minima on the surface The remaining stationary point is a third-order saddle point The one- and n-particle limits of the electronic energies of each of these six structures were obtained by systematically varying basis sets and theoretical methods The former limit was estimated with the cc-pVXZ and aug-cc-pVXZ families of basis sets (X = 2-7) MP2 CCSD(T) and BD(TQ) calculations helped approach the latter Core correlation effects have also been assessed at the MP2 level with the cc-pCVXZ series of basis sets (X = 2-5) These data were combined in focal point analyses to provide highly accurate dissociation energies and relative energies for these stationary points
4th Southern School on Computational Chemistry
20
Two as Good as Four Relativistic Theory for Electrons Only
Maria Barysz and Andrzej J Sadlej
Department of Quantum Chemistry Institute of Chemistry Nicolaus Copernicus University Torun Poland
Relativistic quantum mechanics is routinely formulated in terms of the four-component one-electron wave functions These four-component vectors (spinors) form the basis for most advanced relativistic calculations in quantum chemistry The 4-component formalism is by no means trivial and imposes several inconvenient conditions on the form of the basis set functions into which each spinor component is to be expanded
Quite a gain in both the computational time and programming effort can be achieved by using the two-component formalism ie the formalism in which only the so--called large component of each spinor appears explicitly
However until recently the two-component methods have been merely approximations to the fully relativistic 4-component approaches A new approach which permits the reduction of the 4-component thoery to the exact two-component form will be surveyed and discussed The newly developed method permits a complete separation of the negative and positive energy spectra of the Dirac hamiltonian with arbitrarily high accuracy leading to what can be called a relativistic quantum theory for electrons only This means that all of relativistic quantum chemistry can be done in much easier two-component form The method can be also used to generate the spectrum of the negative energy states and opens new possibilities for the investigation of QED corrections References 1 M Barysz and A J Sadlej J Mol Struct (Theochem) 573 (2001) 181 2 M Barysz and A J Sadlej J Chem Phys 116 (2002) 2696 3 G Pestka and A J Sadlej J Mol Struct (Theochem) 592 (2002) 7 4 M Barysz in Theoretical Chemistry and Physics of Heavy and Superheavy Elements Eds U Kaldor and S Wilson Kluver Dordrecht 2003 pp 349 - 397
4th Southern School on Computational Chemistry
21
Computational Studies on Nitrogen-Substituted Steroidal Structures
Angela Bell
Auburn University Auburn AL
Using several steroidal structures as models nitrogen was introduced into their framework at
particular positions Variation in the location of the nitrogen could prove to be critical to the
compoundrsquos overall functionality A number of steroid structures containing nitrogen
azasteroids possess known pharmaceutical values such as anti-microbial andor anti-
inflammatory properties This project serves to support the experimental synthesis on one or
more of these compounds Theoretical calculations were pursued through the utilization of
PCModel semi-empirical as well as ab initio methods To bridge the gap between the
computing and bench environment computational studies will be undertaken to help determine a
reasonable synthetic strategy
4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
18
A Theoretical Investigation of the Structure and Properties of Ascorbic Acid (Vitamin C)
RN Allen MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 JR Lynch Street Jackson MS 39217
Pure ascorbic acid (AA) or vitamin C is a white crystalline solid which is water soluble AA is an extremely interesting molecule which has many important biological functions It is necessary for the production of collagen in connective tissue helps promote a healthy immune system in humans and helps prevent scurvy AA also functions as an antioxidant which it helps protect the body against damaging free radicals Ascorbic acid has been associated with prevention of many degenerative disease like cataracts certain cancers and cardiovascular diseases
Geometries of neutral tautomeric and anionic species of AA were optimized at the Density Functional Theory level using the B3LYP method The radical species were studied using the unrestricted B3LYP method Single point energy calculations were also performed using the MP2 and UMP2 method for all species All calculation utilized the 6-311++G(dp) basis set Nature of stationary points on the potential energy surfaces (PESs) was ascertained by harmonic vibrational frequency analysis all structures were found minima The Tomasirsquos polarized continuum model was used to examine the effects of aqueous solvation on the relative stability of interested species The electronic charge distribution molecular electrostatic potential ionization potential and electron affinity are also reported
O
O
O O
OO
CC
CC
C
C
4th Southern School on Computational Chemistry
19
Anchoring the Potential Energy Surface of the Water Trimer
Julie A Anderson and Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Six cyclic stationary points on the water trimer potential energy surface have been fully optimized at the MP2 level with the aug-cc-pVQZ basis set In agreement with previous work harmonic vibrational frequencies indicate two structures are minima Three are transition states connecting minima on the surface The remaining stationary point is a third-order saddle point The one- and n-particle limits of the electronic energies of each of these six structures were obtained by systematically varying basis sets and theoretical methods The former limit was estimated with the cc-pVXZ and aug-cc-pVXZ families of basis sets (X = 2-7) MP2 CCSD(T) and BD(TQ) calculations helped approach the latter Core correlation effects have also been assessed at the MP2 level with the cc-pCVXZ series of basis sets (X = 2-5) These data were combined in focal point analyses to provide highly accurate dissociation energies and relative energies for these stationary points
4th Southern School on Computational Chemistry
20
Two as Good as Four Relativistic Theory for Electrons Only
Maria Barysz and Andrzej J Sadlej
Department of Quantum Chemistry Institute of Chemistry Nicolaus Copernicus University Torun Poland
Relativistic quantum mechanics is routinely formulated in terms of the four-component one-electron wave functions These four-component vectors (spinors) form the basis for most advanced relativistic calculations in quantum chemistry The 4-component formalism is by no means trivial and imposes several inconvenient conditions on the form of the basis set functions into which each spinor component is to be expanded
Quite a gain in both the computational time and programming effort can be achieved by using the two-component formalism ie the formalism in which only the so--called large component of each spinor appears explicitly
However until recently the two-component methods have been merely approximations to the fully relativistic 4-component approaches A new approach which permits the reduction of the 4-component thoery to the exact two-component form will be surveyed and discussed The newly developed method permits a complete separation of the negative and positive energy spectra of the Dirac hamiltonian with arbitrarily high accuracy leading to what can be called a relativistic quantum theory for electrons only This means that all of relativistic quantum chemistry can be done in much easier two-component form The method can be also used to generate the spectrum of the negative energy states and opens new possibilities for the investigation of QED corrections References 1 M Barysz and A J Sadlej J Mol Struct (Theochem) 573 (2001) 181 2 M Barysz and A J Sadlej J Chem Phys 116 (2002) 2696 3 G Pestka and A J Sadlej J Mol Struct (Theochem) 592 (2002) 7 4 M Barysz in Theoretical Chemistry and Physics of Heavy and Superheavy Elements Eds U Kaldor and S Wilson Kluver Dordrecht 2003 pp 349 - 397
4th Southern School on Computational Chemistry
21
Computational Studies on Nitrogen-Substituted Steroidal Structures
Angela Bell
Auburn University Auburn AL
Using several steroidal structures as models nitrogen was introduced into their framework at
particular positions Variation in the location of the nitrogen could prove to be critical to the
compoundrsquos overall functionality A number of steroid structures containing nitrogen
azasteroids possess known pharmaceutical values such as anti-microbial andor anti-
inflammatory properties This project serves to support the experimental synthesis on one or
more of these compounds Theoretical calculations were pursued through the utilization of
PCModel semi-empirical as well as ab initio methods To bridge the gap between the
computing and bench environment computational studies will be undertaken to help determine a
reasonable synthetic strategy
4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
19
Anchoring the Potential Energy Surface of the Water Trimer
Julie A Anderson and Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Six cyclic stationary points on the water trimer potential energy surface have been fully optimized at the MP2 level with the aug-cc-pVQZ basis set In agreement with previous work harmonic vibrational frequencies indicate two structures are minima Three are transition states connecting minima on the surface The remaining stationary point is a third-order saddle point The one- and n-particle limits of the electronic energies of each of these six structures were obtained by systematically varying basis sets and theoretical methods The former limit was estimated with the cc-pVXZ and aug-cc-pVXZ families of basis sets (X = 2-7) MP2 CCSD(T) and BD(TQ) calculations helped approach the latter Core correlation effects have also been assessed at the MP2 level with the cc-pCVXZ series of basis sets (X = 2-5) These data were combined in focal point analyses to provide highly accurate dissociation energies and relative energies for these stationary points
4th Southern School on Computational Chemistry
20
Two as Good as Four Relativistic Theory for Electrons Only
Maria Barysz and Andrzej J Sadlej
Department of Quantum Chemistry Institute of Chemistry Nicolaus Copernicus University Torun Poland
Relativistic quantum mechanics is routinely formulated in terms of the four-component one-electron wave functions These four-component vectors (spinors) form the basis for most advanced relativistic calculations in quantum chemistry The 4-component formalism is by no means trivial and imposes several inconvenient conditions on the form of the basis set functions into which each spinor component is to be expanded
Quite a gain in both the computational time and programming effort can be achieved by using the two-component formalism ie the formalism in which only the so--called large component of each spinor appears explicitly
However until recently the two-component methods have been merely approximations to the fully relativistic 4-component approaches A new approach which permits the reduction of the 4-component thoery to the exact two-component form will be surveyed and discussed The newly developed method permits a complete separation of the negative and positive energy spectra of the Dirac hamiltonian with arbitrarily high accuracy leading to what can be called a relativistic quantum theory for electrons only This means that all of relativistic quantum chemistry can be done in much easier two-component form The method can be also used to generate the spectrum of the negative energy states and opens new possibilities for the investigation of QED corrections References 1 M Barysz and A J Sadlej J Mol Struct (Theochem) 573 (2001) 181 2 M Barysz and A J Sadlej J Chem Phys 116 (2002) 2696 3 G Pestka and A J Sadlej J Mol Struct (Theochem) 592 (2002) 7 4 M Barysz in Theoretical Chemistry and Physics of Heavy and Superheavy Elements Eds U Kaldor and S Wilson Kluver Dordrecht 2003 pp 349 - 397
4th Southern School on Computational Chemistry
21
Computational Studies on Nitrogen-Substituted Steroidal Structures
Angela Bell
Auburn University Auburn AL
Using several steroidal structures as models nitrogen was introduced into their framework at
particular positions Variation in the location of the nitrogen could prove to be critical to the
compoundrsquos overall functionality A number of steroid structures containing nitrogen
azasteroids possess known pharmaceutical values such as anti-microbial andor anti-
inflammatory properties This project serves to support the experimental synthesis on one or
more of these compounds Theoretical calculations were pursued through the utilization of
PCModel semi-empirical as well as ab initio methods To bridge the gap between the
computing and bench environment computational studies will be undertaken to help determine a
reasonable synthetic strategy
4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
20
Two as Good as Four Relativistic Theory for Electrons Only
Maria Barysz and Andrzej J Sadlej
Department of Quantum Chemistry Institute of Chemistry Nicolaus Copernicus University Torun Poland
Relativistic quantum mechanics is routinely formulated in terms of the four-component one-electron wave functions These four-component vectors (spinors) form the basis for most advanced relativistic calculations in quantum chemistry The 4-component formalism is by no means trivial and imposes several inconvenient conditions on the form of the basis set functions into which each spinor component is to be expanded
Quite a gain in both the computational time and programming effort can be achieved by using the two-component formalism ie the formalism in which only the so--called large component of each spinor appears explicitly
However until recently the two-component methods have been merely approximations to the fully relativistic 4-component approaches A new approach which permits the reduction of the 4-component thoery to the exact two-component form will be surveyed and discussed The newly developed method permits a complete separation of the negative and positive energy spectra of the Dirac hamiltonian with arbitrarily high accuracy leading to what can be called a relativistic quantum theory for electrons only This means that all of relativistic quantum chemistry can be done in much easier two-component form The method can be also used to generate the spectrum of the negative energy states and opens new possibilities for the investigation of QED corrections References 1 M Barysz and A J Sadlej J Mol Struct (Theochem) 573 (2001) 181 2 M Barysz and A J Sadlej J Chem Phys 116 (2002) 2696 3 G Pestka and A J Sadlej J Mol Struct (Theochem) 592 (2002) 7 4 M Barysz in Theoretical Chemistry and Physics of Heavy and Superheavy Elements Eds U Kaldor and S Wilson Kluver Dordrecht 2003 pp 349 - 397
4th Southern School on Computational Chemistry
21
Computational Studies on Nitrogen-Substituted Steroidal Structures
Angela Bell
Auburn University Auburn AL
Using several steroidal structures as models nitrogen was introduced into their framework at
particular positions Variation in the location of the nitrogen could prove to be critical to the
compoundrsquos overall functionality A number of steroid structures containing nitrogen
azasteroids possess known pharmaceutical values such as anti-microbial andor anti-
inflammatory properties This project serves to support the experimental synthesis on one or
more of these compounds Theoretical calculations were pursued through the utilization of
PCModel semi-empirical as well as ab initio methods To bridge the gap between the
computing and bench environment computational studies will be undertaken to help determine a
reasonable synthetic strategy
4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
21
Computational Studies on Nitrogen-Substituted Steroidal Structures
Angela Bell
Auburn University Auburn AL
Using several steroidal structures as models nitrogen was introduced into their framework at
particular positions Variation in the location of the nitrogen could prove to be critical to the
compoundrsquos overall functionality A number of steroid structures containing nitrogen
azasteroids possess known pharmaceutical values such as anti-microbial andor anti-
inflammatory properties This project serves to support the experimental synthesis on one or
more of these compounds Theoretical calculations were pursued through the utilization of
PCModel semi-empirical as well as ab initio methods To bridge the gap between the
computing and bench environment computational studies will be undertaken to help determine a
reasonable synthetic strategy
4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
22
In Silico Pharmacophore Development and Identification of Antimalarial Agents by Three Dimensional Multi-Conformer
Database Searches
Apurba K Bhattacharjee Mark G Hartell Daniel A Nichols Rickey P Hicks John E van Hamont and Wilbur K Milhous
Department of Medicinal Chemistry Division of Experimental Therapeutics Walter Reed Army Institute of Research Silver Spring MD 20910-7500 USA
The current global situation with respect to malaria indicates that about two billion people are exposed to the disease and more than 1 million people die from it every year The situation is rapidly worsening mainly due to non-availability of effective drugs and development of drug resistance of a large number of non-immune people in areas where malaria is frequently transmitted Therefore much effort and attention are needed for discovery and development of new and less toxic antimalarial drugs
We have developed a widely applicable three-dimensional QSAR pharmacophore model for antimalarial activity from a set of 17 substituted antimalarial indolo[21-b]quinazoline-612-diones (tryptanthrins) by using CATALYST These compounds exhibited remarkable in vitro activity (lt 100 ngmL) against sensitive and multidrug-resistant Plasmodium falciparum malaria The pharmacophore which contains two hydrogen bond acceptors (lipid) and two hydrophobic (aromatic) features was found to map well onto many well-known antimalarial drug classes including quinolines chalcones rhodamine dyes Pfmrk cyclin dependent kinase inhibitors malarial FabH inhibitors and plasmepsin inhibitors The phamacophore allowed searches for new antimalarial candidates from multiconformer 3D databases and enabled custom designed synthesis of new potent analogues
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for antimalarial activity of the tryptanthrins
Aromatic Hydrophobic
Aromatic Hydrophobic
H-Bond Acceptors
Figure 1 Pharmacophore for antimalarial activity
4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
23
In addition we also present a 3D pharmacophore model for chloroquine (CQ) resistance reversal ability This was developed from a training set of 17 diverse resistance reversal agents The training set includes imipramine desipramine and fifteen of their analogues all of them showed CQ-resistance reversal ability of varying degrees The CQ IC50 values covered activities ranging from 23 to 497 ngml determined against the W2 clone of Plasmodium falciparum in the presence and absence of 500 ngml of test compound The generated pharmacophore model indicates that two aromatic hydrophobic interaction sites on the tricyclic ring and a hydrogen bond acceptor (lipid) site at the side chain preferably on a nitrogen atom are necessary for potent activity Stereoelectronic properties calculated by using the AM1semi-empirical procedure are found to be consistent with the model particularly the electrostatic potential profiles characterized by a localized negative potential region by the side chain nitrogen atom and a large region covering the aromatic ring The calculated data further revealed that aminoalkyl substitution at the N5-position of the heterocycle and a secondary or tertiary aliphatic aminoalkyl nitrogen atom with two or three carbon bridge to the heteroaromatic nitrogen (N5) are required for potent ldquoresistance reversal activityrdquo The lowest energy conformer for the seventeen structures was determined and optimized to afford stereoelectronic properties such as molecular orbital energies electrostatic potentials atomic charges proton affinities octanol-water partition coefficients (log P) and structural parameters A fairly good correlation appears to exit between resistance reversal activity and intrinsic basicity of the nitrogen atom at the trycyclic ring system frontier orbital energies and lipophilicity of the molecules
PROTECT PROJECT SUSTAINPROTECT PROJECT SUSTAIN
Experimental Therapeutics
WRAIRWRAIR
Pharmacophore for CQ-Resistance Reversal
Hydrophobic Hydrophobic
H-bond acceptor
Figure 2 Pharmacophore for chloroquine resistance reversal
References 1 Bhattacharjee AK Hartell MG Nichols D Hicks RP Stanton B van Hamont J E Milhous W K
Structure-activity relationship study of antimalarial indolo [21-b]quinazoline-612-diones (tryptanthrins) Three dimensional pharmacophore modeling and identification of new antimalarial candidates Eur J Med Chem 39 (2004) 59-67
2 Bhattacharjee AK Kyle DE Vennerstrom JL Milhous WK A 3D QSAR Pharmacophore Model and Quantum Chemical Structure Activity Analysis of Chloroquine(CQ)-Resistance Reversal J Chem Info Comput Sci 2002 42 1212-1220
4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657

4th Southern School on Computational Chemistry
24
The Investigation of Meso-tetra(hydroxyphenyl)chlorin Vertical Excitation States in Water
James R Black
Auburn University Auburn AL 36849
Meso-tetra(hydroxyphenyl)chlorin (m-THCP) is a photosensitizer used in photodynamic
therapy in cancer treatment The accepted mechanism of tumor destruction involves the
formation of excited singlet oxygen via excited state energy transfer from m-THCP to oxygen
The excited states of m-THCP are investigated using two computational methods ZINDO and
TD in water The optimized geometry was calculated using HFAM1 and the results were
compared to the experimental values given by Howe
4th Southern School on Computational Chemistry
25
Ab Initio Study of Thermochemistry of Solid Solutions Substituting Solubility of Silicon in the Aluminum and Iron
VI Bolshakov1 VVRossikhin2 EOVoronkov2 SI Okovyty3
1Dnepropetrovsk State Academy of Building and Architecture49635 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
3Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine
The solid solutions formation should be studied theoretically because of the physical experiment complexity The process of solid solutions formation is considered by modeling of this one as chemical reaction between an unsubstituted cell and substituting atoms as reagents
Xk + Yl rarr Xk-l Yl + Xl
where X is the metal-solvent atom Y is the substituting atom k is the number of metal-solvent atoms l is the number of the substituting atoms
Proposed simulation is one of the ways that gives a possibility to perform ab initio quantum-chemical calculations of thermochemical quantities two of the more helpful of them are thermal enthalpy and Gibbs free energy The results are presented in Table 1 have been calculated on the base of periodic unrestricted Hartree-Fock method as implemented in the G98W program [1] and which could be used for predicting the thermostability of solid solutions are considered
Table 1 Enthalpy and Gibbs free energy of some substitution reactions
Reaction Si- place ∆Hr (kcalmol) ∆Gr (kcalmol) Al14 + Si rarr Al13Si + Al node -314 -879 Al14 + Si rarr Al13Si +Al side center +2573 +2196 Fe9 + Si rarr Fe8Si + Fe node -8722 -8848 Fe9 + Si rarr Fe8Si +Fe side center -5961 -5522
Obtained results allow evaluating a relative degree of thermostability for the solid solutions
and determining the more probable place of location for the substituting atom Using derived thermochemical date the equilibrium content of silicon in the aluminium and
iron has been estimated theoretically by the formula [2]
lnNSi = ∆SSiR - ∆HSiRT
where NSi is the number of atoms ∆SSi is the entropy change on substituted atom ∆HSi is the enthalpy change on substituted atom R is the gas constant T is the absolute temperature
4th Southern School on Computational Chemistry
26
Figure 1 Solubility of silicon in the aluminium and the iron
- is defined the iron - is defined the aluminium It is necessary to note that there is at least a qualitative agreement between the obtained
dependences and well-known experimental facts References 1Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 2Physical Metallurgy Edited by RWCahn North-Holland Publishing Company
Amsterdam1965
4th Southern School on Computational Chemistry
27
Active Site-Inhibitor Modeling Using a Customized HIV-Protease Polypeptide
1Deborah Bryan 2Jason Ford-Green 2Jesse Edwards 3John West 4Ben M Dunn
1 Department of ChemistryAHPCRC 2Department of BiologyAHPCRC 3Department of Chemistry Florida AampM University Tallahassee FL USA 4Department of Molecular Biology
and Biochemistry University of Florida Gainesville FL USA 32608
In an effort to develop unique HIV protease inhibitors Dunn et al have synthesized
customized polypeptides One of these inhibitors capable of adapting to mutations in the HIV
protease will be studied in this work Using molecular modeling we will explore whether these
inhibitors induce a stable conformation in the HIV protease In particular quantum mechanics
and molecular mechanics methods will be used to accomplish this task This work will discuss
those results Also molecular dynamics on the inhibitor and the HIV protease using molecular
mechanics will be conducted to begin simulations of the mechanism of inhibition
4th Southern School on Computational Chemistry
28
Selected Aspects of Cisplatin Hydration Quantum Chemical Approach to Thermodynamic and Kinetic Characteristics
Jaroslav V Burdaa Michal Zeizingera and Jerzy Leszczynskib
aDepartment of Chemical Physics and Optics Faculty of Mathematics and PhysicsCharles University Ke Karlovu 3 121 16 Prague 2 Czech Republic
bDepartment of Chemistry Jackson State University 1325 J R Lynch Street Jackson Mississippi 39217-0510 USA
Several computational schemes were chosen for examining hydration scheme of square-planar Pt(II) and Pd(II) complexes
First hydration of selected platinum complexes ([PtCl4]2- [Pt(NH3)4]2+ and cistransplatin ndash PtCl2(NH3)2) have been studied Up to two solvent molecules have been considered to replace the ligands In order to be able to draw conclusions about pH changes in the course of the hydration process both H2O and OH- species were considered in the solvating process The quasi-Gaussian 3 theory was adapted for the pseudopotential treatment of platinum complexes Since a heavy element was present in the complexes an additional stabilization due to the spin-orbit coupling and core-polarization potentials have been evaluated above the scheme of G3 treatment This spin-orbit coupling stabilization amounts to 2-5 kcalmol but does not qualitatively change the hydration preferences In accord with the experiment neutral Pt(NH3)2(OH)2 was found to be the most stable complex for hydration of both cis- and transplatin
As a second hydration surface of analogous palladium complexes has been examined by advanced quantum-chemical calculations Preliminary geometry optimizations were carried using the second-order Moslashller-Plesset level of theory with frozen core approximation with the 6-31G basis set for H N O and Cl atoms Pd was described with the Stuttgart relativistic pseudopotential with a basis set of corresponding quality for the explicitly treated electrons Final re-optimization of all the species considered in the hydration scheme was done at the MP2 (full) level Then the reaction surfaces of the structures localized by optimizations were constructed utilizing the MP4 single point evaluations with additional inclusion of diffuse functions The computed results were compared with corresponding data of analogous platinum complexes The Pd and Pt energy surfaces resemble each other to a surprisingly large extent Practically all qualitative trends such as cistrans energy ordering are identical and the solvation energies of Pd and Pt species differ only by a few (at most 10) kcalmol Concerning the markedly different biochemical and pharmacological roles of Pt- and Pd-based compounds our basic conclusion is that the difference between cisplatin and analogous palladium complexes cannot be rationalized considering the energetics (thermodynamic properties) of hydration because these properties do not differ significantly
In third step ab initio study on hydration (a metal-ligand replacement by water molecule or OH- group) of cis- and transplatin and their palladium analogues was performed within a neutral pseudomolecule approach (eg metal-complex + water as reactant complex) Subsequent replacement of the second ligand was considered Optimizations were performed at MP26-31+G(d) level with single-point energy evaluation using the CCSD(T)6-31++G(dp) approach For the obtained structures of reactants transition states (TS) and products both thermodynamic (reaction energies and Gibbs energies) and kinetic (rate constants) characteristics were estimated It was found that all the hydration processes are mildly endothermic reactions - in the first step they require 87 and 102 kcalmol for ammonium and chloride replacement in cisplatin and 138
4th Southern School on Computational Chemistry
29
and 178 kcalmol in the transplatin case respectively Corresponding energies for cispalladium amount to 52 and 98 kcalmol and 110 and 177 kcalmol for transpalladium Based on vibrational analyses at MP26-31+G(d) level Transition State Theory rate constants were computed for all the hydration reactions A qualitative agreement between the predicted and known experimental data was achieved It was also found that the close similarities in reaction thermodynamics of both Pd(II) and Pt(II) complexes (average difference for all the hydration reactions ca 18 kcalmol) do not correspond to the TS characteristics The TS energies for examined Pd(II) complexes are about 97 kcalmol lower in comparison with the Pt-analogues This leads to 106
times faster reaction course in the Pd cases This is by 1 or 2 orders of magnitude more than the results based on experimental measurements
In the last step COSMO model was used Palladium complexes involved in the 1st hydration step and all platinum complexes were reoptimized within DFT approach ndash B3LYP functional and the same 6-31+G(d) basis set Similarly the CCSD(T)6-31++G(dp) approach was combined with COSMO model and water environment for energy and MO analyses Substantial improvement of predicted characteristics for hydration reactions was obtained
4th Southern School on Computational Chemistry
30
Solvation Studies of Anti-HIV Prodrugs
1Michael Cato 1Jesse Edwards 1Ashley Moorer 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee FloridaUSA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee FloridaUSA 32307
Newly synthesized prodrugs aimed at delivering the well know nucleoside AZT to the active
site of the HIV virus has been developed by Dr Henry J Lee et al Following the prodrug
scheme the intact drug will deliver the lethal AZT portion after undergoing a metabolic reaction
In order to proceed with this mechanism the drug must first be made available to the active site
by passing through the membrane of the host cell Therefore structure of the intact drug
becomes critical This work will examine the lowest energy conformations at various dielectric
constants using molecular mechanics The change (from 1 to 10) in dielectric constant will
mimic the solvation process However due to the novelty of each compound high-level
computational studies have not been performed on these compounds In order to verify the
structure the accuracy of the lower level molecular mechanics calculations higher level quantum
mechanics calculations need to be performed In this study semi-empirical PM3 and AM1
calculations will be performed in order to compare the theories These results will also be
compared to Molecular Mechanics results
4th Southern School on Computational Chemistry
31
DFT Calculations of the Deamination of Cytosine
David M Close
Department of Physics East Tennessee State University Johnson City TN
Deamination of cytosine in DNA generates uracil Replication of these deaminated products produces a CG rarr TA transition mutation Spontaneous deamination of cytosine is rather slow In single stranded DNA deamination of cytosine occurs with an activation energy of 28 plusmn1 kcalmole at 37 oC [1] Cells use uracil-DNA glycosylase to prevent the build-up of uracil in DNA
Methylated cytosine residues undergo mutations at a significantly higher rate CpG duinucleotides constitute approximately 2 of the human genone However point mutations in the human germline and in human tumors reveal that approximately 13 of all point mutations occur specifically as a CpG rarr TpC or as a CpG rarr CpA transition It is estimated that 5-Methyl Cytosines undergo mutations at a rate 10-40 times higher than any other unmethylated base [2] The rate of mutation is attributable to the high rate of deamination of 5-Methyl Cytosine resulting in a thymine residue Since thymine is a normal constituent of DNA it is not always recognized as the aberrant base in the GT mismatch resulting from the 5-Methyl Cytosine deamination event
In the present study attempts are made to model the deamination of cytosine and 5-MethylCytosine Calculations were performed using DFT at the B3LYP6-31+G(dp) with the Gaussian 98 suite of programs [3] Frequency calculations were performed at the same level of theory to ensure that the systems represent true minima on the potential energy surfaces
It is known that for cytosine monomers in neutral and acidic solution deamination proceeds via an intermediate protonated at the N3 position of cytosine [4] The computations begin by positioning a water molecule in the vicinity of N3 and C4 (Fig 1) Here the N3bullbullbullH bond is 192Ǻ and the N4-HbullbullbullO bond is 199Ǻ Next the water protonates the N3 via a transition state shown in Fig 2 In the transition state shown here C4bullbullbullOH is 227 Ǻ and HObullbullbullN3 is 272 Ǻ
Figure 1 Cytosine + H2O Figure 2 Water has protonated N3
Next the OH- forms a tetrahedral intermediate at C4 (Fig 3)
4th Southern School on Computational Chemistry
32
Figure 3 Tetrahedral Intermediate at C4 This is followed by a second proton transfer to the gtC4-NH2 (Fig4) In the transition state shown here C4-ObullbullbullH is 132 Ǻ and HbullbullbullNH2 is 123 Ǻ
Figure 4 TS2
The energetics of these steps will be discussed and compared with the deamination of 5-Methyl Cytosine (resulting in a thymine residue)
Water can bring about the hydrolysis of all exo-amino groups of the DNA bases Cytosine and adenine are hydrolytically deaminated to uracil and hypoxanthine Yet neither C nor A are considered to be mutagenic hotspots since their deamination is efficiently repaired It is important to note however that 5-Methyl Cytosine is a mutagenic hotspot since its deamination cannot be distinguished from any other thymine
This work is supported by PHS Grant RO1 CA36810-17 awarded by the National Cancer
Institute DHHS Helpful discussions with Leonid Gorb are gratefully acknowledged
LA Frederico TA Kunkel and BR Shaw Biochemistry 29 2532 (1990) PW Laird Molecular Medicine Today 223 (1997) MJ Frisch et al Gaussian 98 Gaussian Pittsburgh PA 1998 R Shapiro and RS Klein Biochemistry 5 2358 (1966)
4th Southern School on Computational Chemistry
33
Ab Initio Ionization Energy Thresholds of DNA and RNA Bases in Gas Phase and in Aqueous Solution
Carlos E Crespo-Hernaacutendez12 Rafael Arce1 Yasuyuki Ishikawa1 Leonid Gorb3 Jerzy Leszczynski3 and David M Close4
1 Center for Molecular Modeling and Computational Chemistry Department of Chemistry University of Puerto Rico San Juan PR
2 Department of Chemistry The Ohio State University Columbus Ohio 3 Computational Center for Molecular Modeling Structure and Interactions Department of
Chemistry Jackson State University Jackson MS 4 Department of Physics East Tennessee State University Johnson City TN
We report the ionization energy thresholds for the DNA and RNA bases both in gas and in aqueous phase at HF and MP2 levels of theory using standard 6-31++G(dp) basis set The primary scope of the work was to find suitable methods for obtaining accurate ionization energies in an aqueous environment Our results show that the use of the spin projection procedure to correct the open shell systems for contamination by higher spin states significantly improves the calculated ionization energies We show further that long-range bulk polarization interactions have a significant role in the stabilization of the first ionization energy of the DNA and RNA bases The stabilization by water solvation of the vertical and adiabatic radical cations of the bases ranges from 215 to 258 eV and from 212 to 279 eV relative to the gas phase results respectively Taking into account the stabilization of the free electron by the water molecules the adiabatic ionization energies of the bases in aqueous solution were estimated to be 527 505 491 481 and 442 eV for uracil thymine cytosine adenine and guanine respectively Although the emphasis is on calculations of the ionization energy thresholds of the bases in aqueous solution the ionization energies on the gas phase are revisited taking into account the non-planarity of the neutral bases and correction for spin contamination This correction provides practically experimental accuracy into calculated ionization energies
4th Southern School on Computational Chemistry
34
Momentum Space Chemistry
Ernest R Davidson
University of Washington
Momentum and position are complementary variables in quantum mechanics The wave
function may alternatively be regarded as a function either of the position of the electrons or of
their momentum Chemists typically think only about position as the variable but solid state
physicists usually think about the momentum For calculations using Gaussian basis sets it is
easy to convert between the two ways of expressing the wave function
Several chemists have looked at wave functions in momentum space in an attempt to get
some insight Generally these attempts have failed to provide much information In this lecture
we will look at experiments on the Compton scattering of high-energy X-rays from single
crystals of ice and at so-called Electron Momentum Spectroscopy (EMS)of some simple
molecules These experiments provide some information about the momentum density in
molecules The Compton scattering experiment was widely claimed to prove that the hydrogen
bond is covalent We will see why theory does not support that interpretation EMS is claimed to
show pictures of individual orbitals in molecules and is one of the few experiments where
orbitals are claimed to be observed We will look at examples to see the extent to which this is
true
4th Southern School on Computational Chemistry
35
Stabilities Strain Energies and Isomerization Barriers of Some trans-Cycloalkenes
Steven Davis
Department of Chemistry and Biochemistry University of Mississippi
University MS 38677
The thermal isomerization of tricyclo[410027]heptane and bicyclo[320]hept-6-ene were
studied using ab initio methods at the multiconfiguration self-consistent field level The lowest
energy pathway for thermolysis of both structures proceedes through the (EZ)-13-
cycloheptadiene intermediate Ten transition states were located which connect these three
structures to the final product (ZZ)-13-cycloheptadiene Three reaction channels were
investigated which included the conrotatory and disrotatory ring opening of
tricyclo[410027]heptane and bicyclo[320]hept-6-ene and trans double bond rotation of (EZ)-
13-cycloheptadiene The activation barrier for the conrotatory ring opening of
tricyclo[410027]heptane to (EZ)-13-cycloheptadiene was found to be 40 kcalmol while the
disrotatory pathway to (ZZ)-13-cyclohetpadiene was calculated to be 55 kcalmol The
thermolysis of bicyclo[320]hept-6-ene via a conrotatory pathway to (EZ)-13-cycloheptadiene
had a 35 kcalmol barrier while the disrotatory pathway to (ZZ)-13-cyclohetpadiene had a
barrier of 48 kcalmol The barrier for the isomerization of (EZ)-13-cycloheptadiene to
bicyclo[320]hept-6-ene was found to be 12 kcalmol while that directly to (ZZ)-13-
cycloheptadiene was 20 kcalmol
4th Southern School on Computational Chemistry
36
Experimental and Theoretical Investigation of the Structure of 2rsquoBromoacetophenone
Aviane Flood Ming-Ju Huang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University
Jackson MS 39217
An ldquounknownrdquo compound was dissolved in CDCl3 referenced to tetramethylsilane (TMS) and analyzed using 1H NMR and 13C NMR spectra using the Bruker DPX-250 AVANCE spectrometer The 13C NMR spectra was used to run a DEPT (Distortionless Enhancement by Polarization Transfer) 135 experiment The 1H NMR spectra was used to run a two dimensional COSY (Correlation Spectroscopy) experiment Additional two dimensional experiments were conducted HETCOR (Heteronuclear Correlated Spectroscopy) and COLOC (Correlation via Long-range Coupling) using the 1H NMR and 13C NMR spectra The structure of the compound was then determined using the number and types of carbons and hydrogen collected from the experiment The bond connectivity was then derived using the two dimensional NMR experiments
Geometry optimizations were then carried out at the Hartree Fock (HF) and Becke Lee Yang and Parr hybrid functional (B3LYP) levels of theory employing the 6-31G and 6-311G basis sets The NMR shielding tensors were then collected without symmetry restrictions on the optimized structures using the GIAO (Gauge Independent Atomic Orbital) CSGT (Continuous Set of Gauge Transformations) and IGAIM methods The chemical shifts were then calculated by subtracting the absolute values of the isotropic shielding constants (ppm) from the calculated TMS shielding constants (ppm) The results obtained from the experiment were used to conclude that 2rsquobromoacetophenone was the compound identified in the experiment We report the experimental and theoretical chemical shifts bond distances and correlation coefficients
4th Southern School on Computational Chemistry
37
Theoretical Study of Interactions of Methyl-Cytosine with Na+ Cation and Water Molecules
A D Fortnera A Michalkovaab L Gorba J Leszczynskia
aComputational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
b Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
The interactions of methyl-cytosine (MC) and his imino tautomeric form (MCT) with the non-hydrated and hydrated Na+ by 1-6 water molecules cation (it represents the first hydration shell on this cation) have been studied Methyl-cytosine is one of prototinic molecules which could mimic the corresponding nucleotide in DNA or RNA molecules It is a pyrimidine derivative consisting of a single six member ring containing both nitrogen and carbon atom and an amino group The structure and dynamics of nucleic acid molecules are influenced by a variety of factors Among them interactions involving nucleic acids bases are particularly important This work is intended to study of the interactions interaction energies corrected by the basis set superposition error changes of geometry and electron density of MC and MCT caused by the interactions with the Na+ cation and water molecules The calculations have been performed at the B3LYP and MP2 levels of theory in conjunction with 6-31G(d) and cc-pVDZ basis sets The studied systems were fully optimized
The optimized structure of the systems contain MC or MCT the Na+ cation and water molecule is presented in Figure 1 (the MC-Na-W and MCT-Na-W models) In both systems the water molecule is oriented to MC so that creates one hydrogen bond with the nitrogen atom of the circle of MC and with the nitrogen atom of the NH group of MCT In both systems water molecule remains in the hydration shell of the Na+ cation The interactions of MC and MCT with cation and water molecules results in changes of geometry and electron density of MC We have found interaction energies of systems of MC and his tautomer with cation and water molecule We have found that the presence of this cation and water molecule can lead to the transfer of hydrogen of the N-H group of MCT to another nitrogen atom of the outside N-H group and so creates the MC molecule The reaction pathway of this transfer is presented in Figure 1 from reactant (the MCT-Na-W model) through transition state (the MCT-Na-W(ts) model) into the product titled as the MC-Na-W system obtained at the B3LYP6-31G(d) level The structure of the transition state is such that the hydrogen atom of the water molecule remains to be bonded with nitrogen of the outside N-C group of MCT The value of the activation energy obtained at the B3LYP6-31G(d) level is about 7 kcalmol It reveals that this reaction pathway is realistic
Biological significance of this study is in finding that the presence of cations and water molecules affects the properties of MC (as one can see from the comparison of the difference between total energies of isolated MC and MCT (about 2 kcalmol) and the MC-Na-W and MCT-Na-W systems (about 19 kcalmol)) so that MC is more mutagenic and has much larger activity
4th Southern School on Computational Chemistry
38
Figure 1 The scheme of transformation from the reactant (MCT-Na-W) through transition state (MCT-Na-W(ts)) into product (MC-Na-W) for the hydrogen transfer between imino tautomer of methyl-cytosine and methyl-cytosine obtained at the B3LYP6-31G(d) level
MCT-Na-W -672833973029 kcalmol
MC-Na-W -672864232031 kcalmol
7 kcalmol
19 kcalmol
MCT-Na-W(ts) -672822442763 kcalmol
4th Southern School on Computational Chemistry
39
Conformational Study of Thioformic Anhydride by Computational Methods
Gurvinder Gill and Eric A Noe
Department of Chemistry Jackson State University Jackson MS 39217
The conformations of thioformic anhydride have been studied at the HF and MP2 levels with
the Gaussian 98 program The optimized geometries relative free energies dipole moments and
the free-energy barriers were obtained for the EZ EE and ZZ conformations and their
corresponding transition states at various levels of theory At the MP26-311++G(2d2p) level
the EE conformation is higher in free energy relative to the most stable ( EZ ) conformation by
102 kcalmol Both the EZ and the EE conformations are nearly planar A third conformer ZZ
has optimized OCSC dihedral angles of 96deg and a relative free energy of 36 kcalmol The free-
energy barriers leading to topomerization of the EZ conformation were 67 and 86 kcalmol
depending on the pathway The dipole moments of the EE EZ and ZZ conformations were 216
290 and 414 D respectively
This work was supported by NSF - CREST Grant No HRD-9805465
4th Southern School on Computational Chemistry
40
Pharmacophore Development of Various Steroid-Based Pharmaceuticals
1Sharye Glynn 1Jesse Edwards 2Henry J Lee 2Dong-Hoon Ko
1Department of ChemistryAHPCRC 2College of Pharmacy and Pharmaceutical Sciences Florida AampM University Tallahassee FL USA 32307
The use of steroids (Glucocorticoids-GC) as an anti-inflammatory has been well established
Despite the effectiveness of steroids in this capacity there are serious toxic side effects The
mechanism of action of this unique class of compound has yet to be determined in full detail
However several researchers have proposed that an uncharacterized receptor forms a complex
(GC-R complex) with the glucocorticoid which initiates a mechanism preventing apoptosis of
granulocytes Due to the lack of information regarding the GC-R complex we have begun to
develop pharmacophores for a series of steroids In particular we will begin to examine
differences in the activity between these steroids upon the replacement of hydrogen in the 11β
position with a fluoro group The activity of the fluoro-replaced compounds possess an activity
about 30 times that of the hydrogen steroid compounds However the toxicity of these
compounds increases significantly Molecular mechanics and semi-empirical techniques are used
to determine the optimal structures between these two types (hydrogenfluoro) of steroids
Simple correlations between activity and structure will be generated using these computational
techniques
4th Southern School on Computational Chemistry
41
Combined ab Initio Molecular Dynamics and Quantum-Chemical Study of Selected Chemical Processes of Biological Importance
L Gorb1 O S Suhanov2 O Isaev1 A Furmanchuk1 I Tuntildeoacuten3 M F Ruiz-Lopez4 O V Shishkin2 and J Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University POBox 17910 1325 JRLynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
3Unidad Investigacioacuten Efectos del Medio Dept Quiacutemica Fiacutesica IcMol Universitat de Valegravencia 46100 Burjassot (Valegravencia) SPAIN
4Equipe de Chimie et Biochimie Theoriques UMR CNRS-UHP No7565 Faculte des Sciences et Techniques Boulevard des Aiguillettes BP 239 54506 Vandoeuvre-les-Nancy France
Combination of conventional and molecular dynamics (MD) ab inito calculations based on density-functional theory (DFT) emerges as a valuable mean for simulations in the different fields of chemistry and biology In this regards we discuss the strengths as well as the limitations of the DFT and DFTndashMD method basing on the recent applications that we have started in our lab
The following chemical processes have been considered 1 Water assisted formation of peptide bond The reaction path of the formic acid and amonia interaction that result in the formation of
peptide bond has been designed for the reaction complex that includes four water molecules Since it is believed that the formation of a zwitterionic intermediate plays a key role in the formation of peptide bond the molecular dynamic simulations have been carried out for those complex in a box of discrete water molecules The employed force field was based on the QMMM method The zwitterionic intermediate was described quantum mechanically at the DFT level and the water molecules were described by a molecular mechanics potential (the TIP3P43 potential was assumed) The role of static and dynamic solvent effects has been analyzed
At quantum-chemical level it was found that specific interactions with single water molecule decrease the value of the proton transfer barrier approximately twice At the MD simulation the profound dynamic solvent effect was found It significantly decreases the rate constant calculated from pure quantum-chemical data
2 Water molecule transitions in the monohydrates of Guanine The phenomenon of water molecule jumps in monohydrates of guanine using quantum-
chemical DFT technique and ab initio molecular dynamics (the CarndashParrinello) approach has been investigated The quantum-chemical calculations were applied in two ways They included and did not include the counterpoise correction at the step of optimizations
We have found that standard Berny algorithm of geometry optimization which does not include counterpoise correction yelds the values of water transition barrier (∆Gne) (B3LYPcc-pvdz harmonic approximation) in a range between 100 and 66 kcalmol that correspond to average lifetime in a range of 09 ps till 10x104 ps Geometry optimization that takes into account the counterpoise correction results in two ndash three fold decreasing of (∆Gne) After those correction the ∆Gne values are changed in the range 04 ndash 34 kcalmol with the corresponding
4th Southern School on Computational Chemistry
42
decrease of lifetime in the range of 03 ps till 47 ps There is also significant difference in hydration free energy enthalpies and parameters of hydration bonds
The ab-initio MD simulation reveals that resident time for monohydrates is in very good accordance with lifetime avaluated from B3LYPcc-pVDZ calculations that includes counterpoise correction In addition the MD data allow us to overcome the limits of harmonic approximation As the results we predict the dissociation into components for three among six comsidered monohydrates
We have also found that for some transitions the lowest vibrational mode along the inversion coordinate is slightly above the value of barrier In such a case the position of water protons might not be sufficiently localized and some monohydrates should be considered as structurally not rigid
3 Thermodynamics of stepwise water addition to methycytocine Based on B3LYP6-31G(d) calculations of stepwise hydration the model of the first
hydration shell of 1-methyl-cytosine at room temperature has been proposed The first solvation shell of 1-methyl-cytosine consists of three water molecules All of them are located in the area which provides hydrogen bonds with guanine during the formation of WatsonndashCrick base pair In other words three water molecules hydrate 1-methyl-cytosine specifically at room temperature
Car-Parrinello molecular dynamics simulations confirms the stability of 1-methylcytosine trihydrate However in contrast to quantum-chemical data we have found some other water binding places which could extend the first hydration shell of cytosine to more then three water molecules
4 Double proton transfer in AT DNA base pair The mechanism of double proton transfer in AT DNA base pair has been investigated at
DFTB3LYP level and at the DFTBLYP level using Car ndashParrinello molecular dynamics The most remarkable result which is obtained at canonical DFT level suggests that the local miminum corresponding to AT structure on the potential energy surface vanishes on the of the Gibbs free energy surface
According to Car-Parrinello molecular dynamics the rate of the proton transfer from rare tautomeric form AT to canonic AT structure simulated at 25 300 and 400K is very high The process lasts about 012 ps at 300K and 022 ps at 25K
Another important issue which is discussed is the lifetime of canonical AT structure According to quantum ndash chemical calculations the process of formation of AT structure from isolated A and T is characterized by the negative value of ∆G in the range of -2 kcalmol This result is also consistent with Car ndashParrinello molecular dynamic simulation since AT base pair remains stable and did not dissociate into components during 32 ps simulation
4th Southern School on Computational Chemistry
43
Structural and Theoretical Study of 2-Methoxy-2-Phenylacetophenone by NMR and Computational Chemistry
Jelani Griffin and Ming-Ju Huang
Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
The structure of 2-methoxy-2-phenylacetophenone was analyzed by NMR spectroscopy
Series of spectra of 1D and 2D NMR were taken for analysis Each carbon and hydrogen was
assigned based on the spectra taken and their chemical shifts were compared with other reports
The structure of 2-methoxy-2-phenylacetophenone has been fully optimized ab initio methods
The 1H and 13C NMR chemical shifts of 2-methoxy-2-phenylacetophenone were calculated by
means of GIAO CSGT and IGAIM The results from theoretical calculations were compared
with experimental data and the structure was compared with the theoretical molecular properties
2-methoxy-2-phenylacetophenone
4th Southern School on Computational Chemistry
44
A Quantum Monte Carlo Study of Compounds with Biological and Thermochemical Implications
Glake A Hill Jr ab Alexander C Kollias b William A Lester Jr b and Jerzy Leszczynski a
aPitzer Center for Theoretical Chemistry University of California Berkeley Berkeley CA 94720
bComputational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
Quantum Monte Carlo (QMC) is a stochastic method for the solution of the electronic Schroumldinger equation
ˆ H Φ = EΦ Here ˆ H is the Hamiltonian operator for the molecular system under consideration and E and
Φ have the usual meaning There are two main approaches in the QMC methodology ndash variational Monte Carlo
(VMC) and diffusion Monte Carlo (DMC) Based on the variational principle the VMC approach yields the ground state energy as the minimum of the functional
E Φ[ ]=Φint
HΦd
r x
ΦΦdr x int
where Φ has some particular symmetry The optimization procedure must preserve the
symmetry and the calculated VMC energy is greater than or equal to the lowest energy eigenstate of that symmetry
Integral evaluation is realized in the Monte Carlo method by summation of local energy
EL x( ) =ˆ H Φ x( )Φ x( )
values over a set of randomly distributed space points or ldquowalkersrdquo The usual representation of Φ in the approach is a product of a HF or a post-HF wave function with a function that depends explicitly on inter-particle ndash electron-electron electron-nucleus and even electron-other-nucleus ndash coordinates The latter ldquocorrelationrdquo function takes the form of a Jastrow factor or Schmidt-Moskowitz function The former factor has the form
ΨJ = exp minus u rij( )i lt jsum
⎡
⎣ ⎢ ⎢
⎤
⎦ ⎥ ⎥
where u r( ) is a function of r chosen variationally to minimize the energy The summation is over the system of electrons and nuclei
The DMC approach has its origin in the solution of the time-dependent Schroumldinger equation in imaginary time The ground state wave function in this approach is formed by consecutive application of the time propagator eminus H minusET( )τ where ET is a trial energy and imaginary time step τ has to be small on the energy scale The strict condition of a small time step and the goal of a chemical accuracy in calculated energies (whose error is sought to be ~1 kcalmol) make the DMC method more demanding in CPU time than the VMC method and to all but the most extensive basis set quantum chemical approaches A benefit of the DMC method is high accuracy that is not governed by the limitations of basis set quantum chemical methods such as
4th Southern School on Computational Chemistry
45
strong dependence on basis set quality or type and extent of many-particle representation of the wave function
The accuracy of the DMC method is governed by the nodal structure of the independent-particle part of the trial wave function
Optimization of correlation function parameters is accomplished with fixed sample optimization using the absolute deviation (AD) functional that minimizes the energy of ΨT and is given by
AD =1N
ET minus ELii
sum
Here N is the number of walkers ELi is the local energy of the ith configuration and ET is
reference energy chosen to minimize fluctuations Excited-state studies utilizing QMC methods are limited To our knowledge the only
excited-state study of a bio-molecule has been our work on porphyrin in which we computed the energy difference between the ground-state singlet and lowest triplet state and that between the ground-state and second excited singlet state using DMC
Reynolds et al provided some of the earliest results with the singlet-triplet splitting in methylene (CH2) With a single determinant and Jastrow function the DMC method yielded a singlet-triplet transition energy in better accord with experiment than available SCF and post-SCF procedures Later Grimes et al demonstrated the capability to compute an excited state of the same symmetry as a lower state QMC also rivaled multideterminant methods and often surpassed these approaches in the accuracy of computed atomization energies
Recently Needs and coworkers have utilized DMC to study the electronic excitation of hydrogenated silicon cluster It was concluded that given a reasonable trial wavefunction QMC gives impressively accurate excited transitions comparable to the best alternative computational approaches and detailed experimental data
In the course of this study we shall present fully the usefulness of the VMC method for the calculation of biological compounds and thermochemical data The DMC method will provide data that will serve as validation standard for this purpose 32 compounds from the G2 set are studied and compared to B3LYP and MP2 values These results will be discussed Finally ground state properties of Cytosine will be presented
4th Southern School on Computational Chemistry
46
Conventional Strain Energy in the Diphosphetanes Thiaphosphetanes and Thiadiphosphetanes
Patricia L Honea Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for the cis and trans isomers of 12-diphosphetane (Figure 1) and 13-diphosphetane (Figure 2) were determined within the isodesmic homodesmotic and hyperhomodesmotic models The project was extended to include 12-thiaphosphetane (Figure 3) and 13-thiaphosphetane (Figure 4) and the cis and trans isomers of 123-thiadiphosphetane (Figure 5) and 124-thiadiphosphetane (Figure 6) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies were computed for all pertinent molecular systems using SCF theory second-order perturbation theory and density functional theory The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons were employed 6-311G (dp) and 6-311+G(2df2pd) Additionally single point coupled-clustered calculations using the larger of the two basis sets were used to investigate the effects of higher-order electron correlation
Our results indicate that sulfur appears to increase the conventional strain energy as the diphosphetanes are less strained than the thiaphosphetanes and the thiadiphosphetanes In the thiadiphosphetanes stabilizing factors of the systems appear to influence the conventional strain energy as the most stable isomers are the least strained The opposite occurs in the diphosphetanes where the most stable isomers the 12-diphosphetanes are the most strained However if only the two 12 conformations are considered the least strained is the most stable The same trend is seen if just the 13 conformations are considered Overall the 12-diphosphetanes are more strained than the13-diphosphetanes because of increased Baeyer strain We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 cis-12-diphosphetane trans-12-diphosphetane
4th Southern School on Computational Chemistry
47
cis-13-diphosphetane
Figure 2 cis-13-diphosphetane trans-13-diphosphetane
Figure 3 12-thiaphosphetane Figure 4 13-thiaphosphetane
Figure 5 cis-123-thiadiphosphetane trans-123-thiadiphosphetane
4th Southern School on Computational Chemistry
48
Figure 6 cis-124-thiadiphosphetane trans-124-thiadiphosphetane
4th Southern School on Computational Chemistry
49
Conventional Ring Strain in Unsaturated Four-Membered Rings
Shelley S Huskey and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton MS 39058
In order to study the effect of unsaturation on the ring strain in small cyclic molecules the conventional strain energies for cyclobutene azetidine-1-ene (Figure 1) phosphetane-1-ene (Figure 2) azetidine-2-ene (Figure 3) and phosphetane-2-ene (Figure 4) are determined within the isodesmic homodesmotic and hyperhomodesmotic models Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular system using SCF theory second-order perturbation theory and density functional theory (DFT) The DFT functional employed is Beckersquos three parameter hybrid functional using the LYP correlation functional Two basis sets both of triple-zeta quality on valence electrons are employed 6-311G(dp) and 6-311+G(2df2pd) Finally the calculated strain energies are compared to those of cyclopropane cyclobutane azetidine and phosphetane
We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Figure 2
Figure 3 Figure 4
4th Southern School on Computational Chemistry
50
Insight into the Dispersion Energies of Hydrogen and Carbon Dimer Interactions
Cynthia Jeffries1 Glake Hill2 Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Jackson State University Jackson MS 39217
2Pitzer Center for Theoretical Chemistry Department of Chemistry University of California Berkeley CA 95401
Dispersion has been of great interest to those that are studying weak interactions in a number
of systems Scientists of nanostructures computational biology and fuel cell research are often
limited because of an inadequate description of this term In the present research the interaction
energies of hydrogen and carbon dimers at different angles and distances are elucidated and
studied to provide an empirical formula to better predict its magnitude The idea is to see it at
multiple angles and distances and compare the energies of each one to see how much change of
dispersion energy has occur between each molecule These calculations were run on Gaussian 98
using HF and CCSDT levels of theory using aug-cc-pVDZ aug-cc-pVTZ and aug-cc-pVQZ
Results will be discussed
4th Southern School on Computational Chemistry
51
Reduction of Dinitrotoluenes Theoretical DFT Investigation
Olexandr Isayev Leonid Gorb and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University 1400 JR Lynch St Jackson MS 39217
The development of cleanup technologies for a disposal of explosives is a challenge for environmental science Such development involves the coordination of experimental and theoretical investigations to integrate both technological and fundamental aspects of key process Although the major processes affecting the natural and engineering treatment of explosives have been investigated qualitatively many issues remain unsolved regarding a reaction mechanism
It is known that several single-electron-transfer are involved in the reactions of Dinitrotoluenes (DNTs) reduction These electron transfers can be either oxidative or reductive In this particular study we concentrate on reductive pathways Because the initial electron-transfer step is often the rate determining step and its rate constant is correlated with energetic of the reaction Therefore a key molecular descriptor in modeling electron transfer kinetics is the one-electron redox potential
Free Gibbs Energy for reduction reactions of 24- and 26-dinitrotoluenes have been calculated at B3LYP 6-311+G(d) level of theory All values of enthalpies and Gibbs free energies were adjusted by zero-point and thermal corrections The one-electron redox potentials were evaluated from free energy cycle scheme On the basis of these calculations the environmental fate of DNTs was estimated
4th Southern School on Computational Chemistry
52
Theoretical Study of Adsorption of Methyl tert-buthyl Ether on Broken Clay Minerals Surfaces
L D Johnsona A Michalkovaab L Gorbb J Leszczynskib
a Institute of Inorganic Chemistry Slovak Academy of Sciences Dubravska cesta 9 842 36 Bratislava Slovak Republic
b Computational Center of Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 J R Lynch Street P O Box 17910 Jackson MS 39217 USA
The adsorption of methyl tert-buthyl ether (MTBE) on the broken surfaces of clay minerals have been studied at the B3LYP6-31G(d) and MP26-31G(d) levels MTBE a widely used gasoline additive is recently being scrutinized for potential environmental damage in groundwater and in the atmosphere Clay minerals are layered aluminosilicates with the ability of the interlayer surfaces to accommodate organic molecules and modify the structure and reactivity Theoretical simulations of adsorption of MTBE on broken clay minerals surfaces can lead to understanding of interactions between this molecule and clay minerals and related issues as source characterization fate and transport process of MTBE on clay minerals The broken mineral surfaces have been simulated by representative cluster models of tetrahedra and octahedra of dickite (11 dioctahedral clay mineral of the kaolinite group) with Si4+ Al3+ and Mg2+ as the central cations These surfaces are of great interest because they are expected to have significantly higher chemical activity than regular ones The models of mineral were constructed so that contain terminal hydroxyl group water molecule or oxygen atom since these three terminations are the main situations which can occur on broken clay minerals surfaces For the reason to obtain electroneutral cluster with different termination than OH group this group was substituted by hydrogen atom The systems of MTBE with clay mineral fragment were fully optimized
The interactions of MTBE with the mineral fragments were found The molecule is stabilized mostly by the formation of multiple C-HhellipO hydrogen bonds between the C-H groups of MTBE (proton-donors) and oxygen atom of terminated hydroxyl groups of the mineral fragment (proton-acceptors) These hydrogen bonds are characterized by longer HhellipO distances than it is in regular O-HhellipO hydrogen bonds In Figure 1 is displayed an example of the studied interacting systems which presents the optimized structure of MTBE interacting with electroneutral tetrahedral Si(OH)4 fragment of mineral Different termination of cluster models results in different numbers of formed hydrogen bonds between MTBE and the mineral fragment In the case of the [Mg(H2O)6]2+ system also two O-HhellipO hydrogen bonds are formed between the oxygen atom of MTBE and terminating hydroxyl groups The adsorption results in changes of MTBE conformation and in the polarization and the electron density redistribution The interaction energies corrected by basis set superposition error have been found MTBE is relatively weakly stabilized on broken minerals surfaces as a consequence of the formation only weak intermolecular interactions The most stable is the system that contains the [Mg(H2O)6]2+ mineral fragment because of the formation of more strength O-HhellipO hydrogen bonds
4th Southern School on Computational Chemistry
53
Figure 1 The optimized structure of adsorbed MTBE on the electroneutral tetrahedral Si(OH)4 fragment of dickite
4th Southern School on Computational Chemistry
54
The Stability of Oxadispirocycle Isomers
Abby K Jones and Gregory S Tschumper
Chemistry and Biochemistry University of Mississippi 107 Coulter Hall University MS 38677 USA
A series of electronic structure computations have been carried out on various isomers and
conformers of oxadispirocycles (see figure) To help develop a much needed scheme that can
explain the relative stability of these species we have also examined substituted cyclohexane and
tetrahydropyran systems that mimic the environment of the central ring in the oxadispirocycles
The data for these simple monocyclic systems reveal some surprising stabilizing and
destabilizing influences that can be used to rationalize the observed stability of the
oxadispirocycles
Cis and Trans Oxadispirocycles
4th Southern School on Computational Chemistry
55
Theoretical Investigation on the Reactivity of a Stable Silylene with Halomethanes
Hyun Joo Michael L McKee
The reactions of a stable silylene 13-diaza-2-silacyclopent-4-en-2-ylidene 1 with
halomethanes (CH3X X = Cl Br I) are studied by using hybrid density functional B3LYP
method Both reaction pathways of silylene insertion into H3C-X bond to form 11 adducts 2
and disilane 3 formation are considered minimal reaction pathways are investigated to uncover
the reaction mechanisms for each halocarbon To explain the different reactivity population
analyses on the reaction intermediates and transition states are carried out by utilizing natural
population analyses (NPA)
N
Si
N
+ CH3X
H
H
N
Si
N
H
H
N
Si
N
H
H
N
Si
N
H
H
CH3
XX
CH3
or
X =Cl Br I
1 2 3
4th Southern School on Computational Chemistry
56
The Mechanism of Ketone-Catalyzed Epoxidation of Olefins with Carorsquos Acid A Computational DFT Study
Y Kholod1 S Okovytyy12 Yu Paukku1 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Caros acid (peroxomonosulphuric acid H2SO5) and its potassium salt are wide used agents for epoxidation of alkenes (1) Some organic compounds such as ketones and amines can catalyze this process It is generally assumed that ketone interacts with H2SO5 to form dioxirane which farther reacts with alkene to form epoxide molecule (2)
CH2CH2
CCH3 CH3
O
C CH3CH3
O O CH2CH2
CCH3 CH3
O
CH2
O
CH2
CH2
O
CH2 (2)
(1)H2SO5
H2SO4
H2SO5
H2SO4
However inner mechanisms of these processes are still insufficiently known Thus in present
work we have used quantum-chemical calculations at UBHandHLYP6-31G(d) level of theory to explore the potential energy surfaces (PES) of ethylene epoxidation as well as dimethylketone oxidation by peroxomonosulphuric acid to compare activation barriers of both non-catalytic and ketone-catalyzed epoxidation processes
Analyzing of PES has sown that the reaction (1) has revealed the diradical character of the transition state (TS1) which has an unsymmetrical open chain structure For the reaction (2) symmetrical (TS2) has been located
TS1
TS2
In order to estimate the efficiency of catalyze activation barriers of both of pathways (1 2) have been compared
4th Southern School on Computational Chemistry
57
Binding Energies of Monovalent and Divalent Cations with TNT
L Jami Lewis and David H Magers
Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Trinitrotoluene is considered a teratogen and mutagen and therefore a major environmental hazard when it seeps into ground water And yet TNT is prevalent at artillery ranges bomb sights and anywhere explosives are used for any purpose military or civil The only current EPA-approved method for remiaditing TNT from soil is incineration which is quite expensive Other treatments are currently being investigated involving base hydrolysis of TNT However little is know about the mechanism of this reaction Base hydrolysis always occurs in the presence of high concentrations of monovalent and divalent cations Studies have shown that the intermediates of the alkaline hydrolysis of TNT can occur through radicals that have been observed in tight associated with monovalent cations In the present study we began our investigation of this process by calculating the binding energy of TNT to such cations using SCF and density functional methods (DFT) The DFT functional employed was Beckersquos three parameter hybrid functional using the LYP correlation functional The ground-state geometry and the corresponding electronic energy of trinitrotoluene the energies of the Li Na K Mg and Ca cations and the optimum geometries and energies of a TNT dimer with each cation were calculated These initial computations yielded four different optimized structures (Figures 1-4) The magnesium and calcium complexes were the only two that were similar
Figure 1 Initial optimized structure of lithium cation with TNT dimer
Figure 2 Initial optimized structure of sodium cation with TNT dimer
4th Southern School on Computational Chemistry
58
Figure 3 Initial optimized structure of potassium cation with TNT dimer
Figure 4 Initial optimized structure of magnesium cation with TNT dimmer Each of these optimized structures was then used as a starting point for further geometry
optimizations with each of the other four cations with TNT dimmer In each case the most stable was used to determine the binding energy The most common conformation of these is a structure similar to that of the original lithium structure but with the two monomers twisted relative to each other (Figure 5) Every computation was performed at both the SCF and DFT levels of theory with three basis sets 3-21G(d) 6-31G(dp) and 6-311G(dp) Future work will include calculating the visible spectra of possible intermediates found in the base hydrolysis reaction of TNT using ZINDOS
Figure 5 New global minimum We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
59
Structural Studies of Novel Steroid-Nucleoside Conjugates Alkylated Derivatives
1Tia Lewis 1Jesse Edwards 1Desiree Paramore 2Henry Joung Lee 2Zhengqing You
1Department of ChemistryAHPCRC Florida AampM Tallahassee Florida USA 32307 2College of Pharmacy and Pharmaceutical Sciences Florida AampM Tallahassee
Florida USA 32307
In an attempt to develop anti-HIV agents devoid of serious toxic effects HJ Lee et al
synthesized these three novel compounds along the anti-drug scheme
AZT conjugated to Cholenic Acid (Conjugate1) P-16 acid (Conjugate 2) and P-21-oic acid
(Conjugate 3) where P is an abbreviation for Prednisolone After initial studies on these
compounds further modifications were made in order to examine the effect of subtle changes to
the structure of these compounds on the activity Three derivatives of the initial compounds were
created synthetically by adding an additional alkyl group between the ester bridge connecting
AZT to the steroid or acid Also in an effort to increase the activity according to that shown in
Conjugate 1 the steroid portion of the molecule was modified This provided the compound with
increased flexibility This work will examine the effect of these modifications on the structure
In previous computational studies on Conjugates 1 through 3 a dual binding site was
hypothesized A comparison between this work and previous work will be examined
4th Southern School on Computational Chemistry
60
Conformational Energetics of Naphthylquinolines
M Jeanann Lovell G Reid Bishop and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
A library of naphthylquinoline derivatives satisfying hypothesized structural criteria for triplex DNA selectivity have been designed and synthesized by Dr Lucjan Strekowski of Georgia State University Proposed structural characteristic criteria promoting intercalation between bases of triplex DNA include a large aromatic surface area an unfused flexible ring system cationic and crescent shape High-throughput competition dialysis experiments among fourteen of these test compounds demonstrated that the replacement of the secondary amine function found in LS8 (Figure 1) with an ether oxygen producing MHQ12 (Figure 2) greatly increased selectivity towards triplex DNA Preliminary semi-empirical studies show a correlation of enhanced triplex DNA selectivity with an increase in rotational flexibility of the side chain of the derivative compound
Figure 1 LS8 ndash amine linkage Figure 2 MHQ12 ndash ether linkage The binding study has been extended to include two additional compounds OZ121 (Figure
3) and G106 (Figure 4) OZ121 is identical to the highly selective MHQ12 except that a sulfur atom replaces the ether oxygen G106 contains an amide linkage between the naphthylquinoline and the side chain Here we present results from computational studies designed to examine the dynamic flexibility of the naphthylquinoline side-chain for the four compounds containing amine ether thiol or amide linkages Calculations are performed to determine the energy of each compound with varying dihedral angles between the side chain and the naphthylquinoline Beginning from optimized geometries the specific dihedral angle is frozen at 5-degree increments for values between 0 and 360 degrees and the rest of the structure is reoptimized to yield the energy barrier of the side-chain rotation and the approximate dihedral angle at which the top of the barrier lies Calculations are performed using semiempirical theory SCF theory and density functional theory
4th Southern School on Computational Chemistry
61
Figure 3 OZ121 ndash thiol linkage
Figure 4 G106 ndash amide linkage In the future results from these computational studies of all four derivatives will be coupled
with thermodynamic binding studies to determine Quantitative Structure Activity Relationships in an effort to design synthesize and test the best naphthylquinoline derivative that will bind tightly and selectively to triplex DNA
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
62
Conformational Flexibility in Naphthylquinoline Derivatives
David H Magers
Computational Chemistry Group Department of Chemistry and Biochemistry Mississippi College Clinton Mississippi
Certain naphthylquinoline derivatives have been shown to bind tightly and selectively to triplex DNA Original structural criteria for triplex DNA intercalation included a crescent shape to mimic the binding site dicationic character a large aromatic surface area and a flexible ring system Our studies have been designed to validate these hypothesized characteristics in several of the derivatives synthesized to date Specifically we have computed the energy barriers associated with the ring dihedral determining crescent versus linear conformations (Figure 1) Our initial findings predict the linear form is energetically more stable although the energy barrier between conformations is very small
Our conformational studies were then extended to the barrier of rotation of the side chain the
R group Results have led us to a new design criterion namely that the flexibility of the side chain may be key to triplex selectivity By freezing the chain dihedral angle at five-degree intervals we have computationally determined the energy barrier of rotation for four of the naphthylquinoline test compounds These are LS8 (Figure 2) MHQ12 OZ121 and G106 These systems differ only in the part of the side chain which links to the quinoline ring MHQ12 is formed by replacing the amine linkage in LS8 with an ether linkage OZ121 has a thiol linkage and G106 has an amide linkage Barriers of rotation of the side chain were computed using optimized linear conformations and crescent conformations of each system In order to incorporate indirectly the binding environment of the intercalator without specifically including the receptor future work will compute the rotational barrier of the side chains with the ring dihedral frozen at angles suggested by 4D-QSAR results based on molecular mechanics simulations All computations in the current study are performed using SCF theory and density functional theory with Beckersquos three parameter hybrid functional using the LYP correlation functional Three basis sets are employed 3-21G 6-31G(dp) and 6-311G(dp)
N
R
N
R
Crescent ConformationDihedral Angle between rings at or near 180o
Linear ConformationDihedral Angle between rings at or near 0o
Figure 1
4th Southern School on Computational Chemistry
63
Figure 2 LS8
We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
64
Conventional Strain Energy in Small Heterocycles of Carbon and Silicon
David H Magers and Crystal B Coghlan
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The conventional strain energies for three- and four-membered heterocycles of carbon and silicon are determined within the isodesmic homodesmotic and hyperhomodesmotic models These include silacyclopropane (Figure 1) disilacyclopropane (Figure 2) silacyclobutane (Figure 3) 12-disilacyclobutane (Figure 4) and 13-disilacyclobutane (Figure 5) Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory second-order perturbation theory (MP2) and density functional theory The DFT functional employed is Beckersquos three-parameter hybrid functional using the LYP correlation functional Two basis sets both of triple zeta quality on valence electrons are employed 6-311G (dp) and 6-311+G(2df2pd) Results indicate that silicon reduces the conventional strain energy of cyclobutane most likely because the Baeyer strain is reduced since the silicon can accommodate a small bond angle more easily than carbon However silicon substitution increases the conventional strain energy in cyclopropane perhaps by destroying the stabilizing factor of sigma delocalization Finaly the conventional strain energy for cyclotrisilane (Figure 6) is computed to determine if the stability returns when the ring is again a homocycle We gratefully acknowledge support from NSF EPSCoR (EPS-0132618)
Figure 1 Silacyclopropane Figure 2 Disilacyclopropane
Figure 3 Silacyclobutane
4th Southern School on Computational Chemistry
65
Figure 4 12-disilacyclobutane Figure 5 13-disilacyclobutane
Figure 6 Cyclotrisilane
4th Southern School on Computational Chemistry
66
Modeling Nitrogenase with the Fe8NS9+ Сluster Can DFT
Calculations Make a Contribution to Understanding the Mechanism
Michael L McKee
Department of Chemistry Auburn University Auburn AL 36849
The DFT method has been used to develop and test a mechanism for the action of nitrogenase The molybdenum atom is replaced by iron and the external ligands have been removed to yield a Fe8NS9
+ cluster The biological reaction (below) is modeled
Nitrogenase + N2 + 8H+ + 8e- rarr Nitrogenase + 2NH3 + H2 by assuming that the protonelectron additions can be simulated by a source of hydrogen
atoms The active catalysis is formed by the addition of three hydrogen atoms to the middle belt of bridging sulfur atoms (see below for cluster with N2 coordinated) Results will be presented for the production of molecular hydrogen (which is known to occur in the absence of N2) and the production of ammonia from N2
4th Southern School on Computational Chemistry
67
Computed Electrostatic Potentials on the Inner And Outer Surfaces of Carbon BoronNitrogen and CarbonBoronNitrogen Nanotube
Models
Jane S Murray Pat Lane Monica C Concha and Peter Politzer
Department of Chemistry University of New Orleans New Orleans LA 70148
Over the course of the past three years we have computed the electrostatic potential on
molecular surfaces of a variety of open and closed carbon boronnitrogen and
carbonboronnitrogen nanotube models at the HFSTO-5GHFSTO-3G level These models
have been of the (55) and (60) type both closed and open and (61) (71) (81) and (80) open
models the open tubes have hydrogens at the ends We have found that the carbon (55) (n0)
and (n1) nanotube models have very weak electrostatic potentials while the boronnitrogen and
carbonboronnitrogen models have a variety of patterns that exhibit strong positive and negative
potentials on the outer surfaces and strong positive potentials on the inner surfaces In this
poster we will show an interesting feature that is found for the (n0) open carbon nanotubes
which do not have full symmetry The surface electrostatic potentials of these models show a
distinctive polarized pattern This anomaly will be presented and discussed
4th Southern School on Computational Chemistry
68
Sputtering of an Amorphous Polyethylene Surface mdash A Molecular Dynamics Study
Michael T Mury and Steven J Stuart
Hunter Laboratories Clemson University Clemson SC 29634
Molecular dynamics simulations are performed to study the bombardment of an amorphous
polyethylene surface by an argon atom The adaptive intermolecular reactive empirical bond
order (AIREBO) potential is used to perform these simulations This model includes
intermolecular forces as well as covalent bonding forces in order to allow for the study of bond
breaking and formation in the condensed phase Several sputtering simulation studies have been
performed in the literature but few have used the AIREBO model and few studies have been on
polymer substrates The effect of the argon bombardment on the surface is examined as well as
the mass yield from the surface and the types of species which are sputtered from the surface In
order to determine if the results differ among boundary conditions periodic open and
ldquoabsorbing ldquo periodic boundary conditions were used in the simulations The three boundary
conditions yielded similar results for the simulations These results as well as initial studies on
substrate temperature incident atom angle and incident atom energy will be shown
4th Southern School on Computational Chemistry
69
Theoretical Prediction of a New Dinitrogen Reduction Process The Utilization of four Dihydrogen Molecules and Zr2Pt2 Cluster
Djamaladdin G Musaev
Cherry L Emerson Center for Scientific Computation Emory University 1515 Dickey Dr Atlanta GA 30322
Activation of the NequivN triple bond of dinitrogen and its chemical transformations under mild conditions is one of the most fascinating task of the modern chemical and biochemical sciences and challenges the scientists for over a century1 The reduced nitrogen finds applications in fuels fertilizers and dyes However still the major part of all the nitrogen required in human nutrition derives from the biological nitrogen fixation2 An industrial analog of the biological nitrogenation is the Haber-Bosch process3 which accounts for almost all the industrial dinitrogen fixation However this process operates at very high temperature and pressure and is economically less effective
The search for the more economical and alternative methods of the nitrogen fixation in the mild conditions continues Currently have accumulated much more accurate fundamental and practical knowledge on the bonding modes and reactivity of the dinitrogen molecule Many of these reactions have been extensively reviewed4 However the recent reactions of the transition-metal coordinated dinitrogen molecule with electrophiles5 coordinated6 and free78 dihydrogen molecule need to be briefly mentioned
Reaction of the coordinated N2 molecule with electrophiles is a core process of the many known nitrogen fixation reactions The mechanism of this process is reasonably well established especially with single (η1-manner end-on) bound dinitrogen complexes It is believed that this process occurs via stepwise mechanism (known as Chatt mechanism) by the protonation of the coordinated dinitrogen and reduction with the electrons flowing from the metal ion9 Recently Yandulov-Schrock reported5 the catalytic reduction of the N2 molecule coordinated to the triamidoamine-Mo(III) complex with the electrophiles It was demonstrated that N2 reduction occurs at a sterically protected single Mo-center that cycled from Mo(III) through Mo(VI) states
The fixation of the coordinated-N2 molecule with H2 molecule is even more challenging Recently it was shown that the combining two (and more) transition metal centers with could be indispensable for the dinitrogen reduction by dihydrogen
Indeed Fryzuk and co-workers7 have reported that the dihydrogen molecule added to the dinuclear metal-N2 complex [P2N2]Zr(micro-η2-N2)Zr[P2N2] FI where [P2N2] = PhP(CH2SiMe2NSiMe2CH2)2PPh reacts with the dinitrogen and leads to a new complex with bridging ZrHZr and N-H bonds [P2N2]Zr(micro-η2-N2H)Zr[P2N2](micro-H) FII Our extensive computational studies10 of the mechanism of this reaction on the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 F1 showed that the reaction of hydrogen molecule proceeds via a 21 kcalmol barrier at the ldquometathesis-likerdquo four-center transition state (involving the two H-atoms and one N- and one Zr centers) and produces the diazenido-micro-hydride
complex [p2n2]Zr(micro-η2-N2H)(micro-12-H)Zr[p2p2] F2 in agreement with the experiment7 However the calculations clearly demonstrated that the experimentally observed diazenido-micro-hydride complex F2 should not be the only product of the reaction (at least for the model complex) Indeed the calculations show that the H2 molecule (second one) could be added to the complex F2 with almost the same (in fact with slightly less 195 kcalmol) energy barrier
The product of the second H2 addition is the complex [p2n2](micro-H)Zr(micro-η2-N2H2)Zr micro-H)[p2p2]
4th Southern School on Computational Chemistry
70
F3 with two bridging NH units and two terminal Zr-H bonds Since the addition of the first H2 molecule to F1 is known to occur at laboratory conditions we predicted that the addition of the second hydrogen molecule to F1 (or the addition of the H2 molecule to F2) should occur as easily as the addition of the first H2 molecule to F1 Furthermore the calculations suggested that the addition of the third H2 molecule to F1 is kinetically less favorable than the first two but it is still a feasible at the appropriate dihydrogen pressure and temperature Based on these results we concluded that the model complex [p2n2]Zr(micro-η2-N2)Zr[p2n2] where [p2n2] = (PH3)2(NH2)2 can easily react with two (and perhaps three) hydrogen molecules under the appropriate laboratory conditions
Recently Chirik and co-workers8 have beautifully demonstrated that the dizirconium complex indeed can reduce dinitrogen to ammonia by utilizing dihydrogen molecules upon the proper regulation of the ligand environment of the Zr-centers The authors have demonstrated that the reduction of (η5-C5Me4H)2ZrCl2 with sodium amalgam under 1 atm of N2 produces [(η5-C5Me4H)2Zr]2(micro2η2η2-N2) complex C1 which easily adds two dihydrogen molecules and produces [(η5-C5Me4H)2ZrH]2(micro2η2η2-N2H2) C2 Heating solutions of the resulted complex C2 in boiling heptane for 5 min resulted in loss of one equivalent of H2 and cleavage of the N-N bond to form the complex [(η5-C5Me4H)2Zr]2(micro2-NH2)(micro2-N) C3 Strikingly warming the heptane solution of the complex C2 resulted in formation of ammonia Thus nitrogen fixation has been accomplished under mild conditions using H2 and variation of the ligand environment of the di-zirconium
Here I continue my efforts on the dinitrogen hydrogenation and report a new N2 reduction process utilizing four H2 molecules and Zr2Pt2 cluster I discuss the mechanism of the reaction Zr2Pt2 + N2 + 4H2 using B3LYP method11 and the large basis sets12 All calculations were performed using the quantum chemical package GAUSSIAN-200313
In brief it was shown that the reaction starts from the coordination of the N2 molecule to the Zr-centers of I The resulting complex Zr2Pt2(micro12-N2) II activates four dihydrogen molecules and produces the (micro-1-H)2ZrPt(micro-12-N2H4)PtZr(micro-1-H)2 X complex (see Figure 1) The activation barriers corresponding to the first second third and fourth H2 molecules are predicted to be ∆H=70 (∆G=156) 106 (193) 183 (274) and 258 (346) kcalmol respectively The dissociation energy of the hydrazine molecule from the product X is calculated to be 192(65) kcalmol The entire reaction Zr2Pt2 + N2 + 4H2 rarr (micro-1-H)2ZrPt2Zr(micro-1-H)2 + N2H4 is found to be exothermic by 526(217) kcalmol and proceed via 258 (346) kcalmol rate-determining barrier corresponding to activation of the fourth H2 The dinitrogen reduction to hydrazine by four dihydrogen molecules and Zr2Pt2 cluster is predicted to be feasible at the modern experimental conditions
We encourage experimentalists to check out our theoretical predictions
4th Southern School on Computational Chemistry
71
1(a) ldquoCatalytic Ammonia Synthesisrdquo Jennings J R Eds Plenum New York 1991 (b) Ludden P W Encyclopedia of Inorg Chem Wiley New York 1994 p2566 (c) Coucouvanis D Encyclopedia of Inorg Chem Wiley New York 1994 p2557 2(a) Eady R R Perspectives on Bioinorganic Chem JAI Press Greenwich CT 1991 p255 (b) Kim J Rees D C Science 1992 257 1667 (c) Kim J Rees D C Nature 1992 360 553 (a) Chan M K Kim J Rees D C Science 1993 260 792 3 See ref 1a for more detailes 4 (a) Howard J B Rees D C Chem Rev 1996 96 2965 (b) Burgess B K Lowe D J Chem Rev 1996 96 2983 (c) Eady R R Chem Rev 1996 96 3013 (d) Leigh G J Science 1998 279 506 (e) Leigh G J Acc Chem Res 1992 25 177 and references therein 5 Yandulov D M Schrock R R Science 2003 301 76 6 Nishibayashi Y Iwai S Hidai M Science 1998 279 540 7 (a) Fryzuk M D Love J B Rettig S J Young V G Science 1997 275 1445 and (b) see Fryzuk M D Johnson S A Coord Chem Rev 2000 200 379 8 Pool J A Lobkovsky E Chirik P J Nature 2004 427 527 9 (a) Chatt J In Natrogen Fixation Stewart W D P Gallon J R (Eds) Academic Press New York 1980 p 1 (b) Pickett C J J Biol Inorg Chem 1996 1 601 and references therein 10(a) Basch H Musaev D G Morokuma K Fryzuk M D Love J B Seidel W W Albinati A Koetzle T F Klooster W T Mason S A Eckert J J Am Chem Soc 1999 121 523 (b) Basch H Musaev D G Morokuma K J Am Chem Soc 1999 121 5754 (c) Basch H Musaev D G Morokuma K Organometallics 2000 19 3393 11 (a) Becke A D Phys Rev A 1988 38 3098 (b) Lee C Yang W Parr RG Phys Rev B 1988 37 785 (c) Becke A D J Chem Phys 1993 98 5648 12 Andrae D Haeussermann U Dolg M Stoll H Preuss H Theor Chim Acta 1990 77 123 13 Frisch M J and co-workers Gaussian_2003 Revision B1 Gaussian Inc Pittsburg PA 2003
00
-100
-200
-300
-400
-500
-600
-700
-800
+ N2 and 1st H2 activ 2nd H2 activ 3rd H2 activ
Zr2Pt2 + N2 + 4H2
Zr2Pt2(N2) + 4H2
TS1
HZrPt(N2H)PtZr + 3H2
TS2
HZrPt(N2H2)PtZrH + 2H2
TS3
HZrPt(N2H3)PtZrHH + H2
-277(-161)
-207(-05)
-573(-377)
-467(-184)
-810(-538)(-833)(-478)
-627(-264)
-575(-132)
-718(-282)
4th H2 activ
TS4
(H)2ZrPt(N2H4)PtZr(H)2
Structures
I
II
III
IV
V
VI
VII
VIII
IX
X
∆H(∆G) (in kcalmol)
-900
-526(-217)
- N2H4
XI(H)2ZrPt2Zr(H)2 + N2H4
Figure 1 Schematic presentation of the potential energy surface(PES) of the reaction Zr2Pt2 + N2 + 4H2 (micro-1-H)2ZrPtPtZr(micro-1-H)2 This scheme is scaled for the calculated ∆H-values
4th Southern School on Computational Chemistry
72
AIM Electron Density Analysis of the Structure and Bonding in Alicyclic Epoxides
SI Okovytyy12 LK Svjatenko2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Alicyclic epoxides are used as syntons for obtaining of the biologic activity compounds [1 2] The strain and polarity of oxiranes provide them the widely transformation spectra by interaction with electrophilic and nucleophilic reagents The investigation of the dependence of physical and physico-chemical properties of epoxides on their geometry is an actual task of organic chemistry Dispite the large numbers of topological studies on methyl-substituted oxiranes the electron density of alicyclic epoxides have not investigated
The atom in molecules (AIM) approach has been employed to characterize the structures and bonding of alicyclic epoxides (1-7) on PBE1PBE6-31+G electron distributions Tables 1 and 2 present the local properties at bond peripheral critical points and ring critical points of epoxides
O O O
OO О О
2 1
4
5
6
7
3
1 2 3 4 5 6 7 Table 1 Local properties (au) at bond peripheral critical points of epoxides (1-7)
C-O C-C Compound ρ(r)b nabla2ρ(r)b Н(r)b ε b ρ(r)b nabla2ρ(r)b Н(r)b ε b
1 02491 -03706 -03294 07040 02557 -05301 -02250 02697 2 02456 -03572 -03169 07443 02619 -05684 -02325 02246 3a 02487
(02451) -03600
(-03612) -03291
(-03161)06394
(06985)02569 -05460 -02243 02429
4a 02498 (02428)
-03770 (-03532)
-03318 (-03071)
06522 (06522)
02607 -05688 -02303 02256
5 02426 -03506 -03047 07678 02632 -05744 -02348 01957 6 02442 -03418 -03141 07585 02603 -05562 -02297 02063 7 02460 -03846 -03204 06378 02502 -05102 -02132 02180
a For compound C-O bonds is not equivalent The epoxidic rings in these compounds possess three (3-1) bond critical points (BCPs)
between the pairs of bonded nuclei of oxirane ring linked by the lines of maximum electron density and ring critical point (RCP) The position of BCP on bond line is closer to nucleus of less electronegative atom
The value of electron density ρ(r) at BCP correlates with the bond line length and bond order The negative value of Laplasian of the electron density nabla2ρ(r) is found in the internuclear region along C-O and C-C bond lines and in the region of the oxygen nucleus lone electron pairs
4th Southern School on Computational Chemistry
73
The covalent character of the C-O and C-C bonds confirmed by the negative value of the Laplasian local energy density H(r) and high value of the electron density at BCPs The strength of the C-C bond increases in the following row 7lt1lt3lt6lt4lt2lt5 The strength of the C-O bond increases in the following row 5lt4lt6lt3lt2lt7lt1 Thus decreasing of the reactivity of epoxidic compounds in reaction without activation of the epoxidic oxygen could be expected in the last row
Linear correlation between electron density at BCP and bond length has been found to be reasonable for C-O (r= 09774) and C-C (r= 09616) bonds in compounds (1-7) The electron density at BCP decreases as the length C-O and C-C bonds increases
Table 2 Local properties (au) at ring critical points of epoxides (1-7)
Compound ρ(r)r nabla2ρ(r)r Н(r)r εr η 1 02120 02702 -01312 12395 8436 2 02112 02931 -01287 14629 8413 3 02099 02902 -01284 13180 8389 4 02104 02976 -01276 14327 8377 5 02091 03024 -01258 15764 8383 6 02096 03008 -01267 15015 8399 7 02054 03044 -01216 12654 8302
The electron density at the BCP and RCP are very similar while the Laplasian at RCP is
small in magnitude and positive indicating that the electron density is not accumulated in this point but shifted towards the interacting atoms and concentrated in the atomic basins
The high value of electron delocalization index η suggest that electron density in oxiranes concentrate all over the ring plane The value point out the increasing of the surface delocalization σndashelectron density (σ-aromaticity) in the compounds 7lt4lt5lt3lt6lt2lt1
Weakening of the C-C bond will cause an increasing of its ellipticity ε with the effect that density is pushed into the area of two C-O bonds as observed in the following compounds row 5 2 4 3 1
The surface delocalization of σndashelectron density is directed parallel to the C-C bond enhancing the πndashcharacter of the C-O bonds and reducing that of the C-C bond The increasing of the ring and the C-O bond ellipticities and the decreasing of the C-C bond ellipticity leading to enhancing πndashcharacter of the epoxidic ring as observed in the following compounds row 3lt4lt2lt6lt5
The value of nabla2ρ(r) is parallel to ρ(r) in its behavior becoming slightly less negative as the electron density at the BCP decreases The C-O and C-C bonds of the ring have substantial ellipticities reflecting the electron density delocalization over the surface of the ring
Endo-epoxynorbornane (6) in contrast to exo-epoxynorbornane (5) possesses additional RCP linking with the O C5 nuclei and with the C5-C6 BCP by the gradient paths (Fig1)
4th Southern School on Computational Chemistry
74
a b Figure 1 Molecular graphs of exo-23-epoxynorbornane (a) endo-23-epoxynorbornane (b) The electron density in this addition RCP is small in magnitude (ρ(r)=00176 au) and
Laplasian is positive (nabla2ρ(r)=00864 au) indicating that the electron density in this RCP is depleted The high value of the ellipticity at RCP (εr= 90148) points out that delocalization of electron density is directed from ring center to the oxygen nucleus and to the C5-C6 bond region This yield the increasing of the oxygen chemical shift of the endo-epoxynorbornane (6) in contrast to one of the exo-epoxynorbornane (5) The difference of the oxygen chemical shifts of stereoisomeric epoxynorbornanes is 66 ppm
References Shotwell JB Hu S Medina E Tetrahedron Lett ndash 2000 ndash Vol41 N 49 ndash P9639ndash9643 Uchiyama M Kimura Y Ohta A Tetrahedron Lett ndash 2000 ndash Vol41 N 51 ndash P10013-
10017
4th Southern School on Computational Chemistry
75
Nature of the Transition Structure for Alkene Epoxidation by Peroxynitrous Acid and Dimethyldioxirane Comparison with
Peroxyformic Acid Epoxidation
S Okovytyy12 Y Kholod1 L Gorb2 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA
Epoxidation of alkenes by peroxy acids and substituted dioxiranes are familiar and synthetically useful methods for synthesis of epoxidic compounds Recently peroxynitrous acid also was proposed as promising epoxidative agent This is quite instable compound which undergoes facile hemolytic O-O bond fission to form OH and NO2 and therefore could be much more effective oxidant than peroxy acids and dioxiranes We present the results of comparative study of these three peroxides in reactions with ethylene
Exploring of the potential energy surface of ethylene epoxidation performed at CASSCF
UQCISD and UCCSD levels of theory has shown that reactions have revealed the diradical character of the transition states (TS1-TS3) which have an unsymmetrical open chain structure Calculations of epoxidation reactions using cost-effective DFT level with UBHandHLYP functional also lead to usimetrical diradical transition states The geometrical parameters of the transition states calculated at UBHandHLYP level of the theory are close to those found at the UQCISD and CCSD levels UBHandHLYP6-31G(d) spin-corrected values of activation barriers are in good agreement with ones calculated at MCQDPT2(1212)6-311++G(dp)-CASSCF(1212)6-311++G(dp) level
4th Southern School on Computational Chemistry
76
The Mechanism of Unimolecular Decomposition of CL-20 (24681012-hexanitro-24681012-hexaazaisowurtzitane)
A Computational DFT Study
S Okovytyy12 Y Kholod1 J Saloni2 MQasim3 HFredrickson3 J Leszczynski2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA 3US Army ERDC Vicksburg Mississippi 39180 USA
The recently developed explosive 24681012-hexanitro-24681012-hexaazaiso-wurtzitane (HNIW CL-20) (1) is used for military purposes The toxicity and potential carcinogenicity of this compound has led to concerns about its fate in the environment and the potential for human exposure One of the major transformation processes of CL-20 in the environment is unimolecular decomposition Because of the highly exothermic nature of explosive materials it is difficult to investigate many aspects of their reactivity with current experimental techniques Quantum-chemistry methods offer risk-free and accurate means by which one can study their behavior structures and properties Knowledge of intermediates understanding of complex chemical processes and estimation of the influence of different environmental factors on the reactivity of explosives are essential both to predict their behavior and enhance their removal from the environment
In the present work a quantum-chemical modeling of CL-20 unimolecular decomposition has
been carried out As it was shown by Wu and Fried the DFT methods give satisfactory values of potential energies of NndashN bond rupture which is the dominant channel in gas-phase decomposition of cyclic nitroamines1 Calculations have been performed at B3LYPcc-PVTZB3LYP6-31G(d) level of theory
During investigation all possible channels of structure (1) destruction have been modeled and energies of alternative reactions have been compared at each considered step Fig 1 shows the most favorable pathway of the abovementioned process including energies of corresponding reactions
4th Southern School on Computational Chemistry
77
Figure 1 Structures of local minima on the potential energy surface of the most favorable
pathway of CL-20 transformation to 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10) and corresponding energies of reactions calculated at B3LYPcc-PVTZB3LYP6-31G(d) level of theory in kcalmol
For CL-20 unimolecular degradation process we have found several types of reactions
homolytic cleavage of an NndashN bond accompanied by eliminating of NO2bull and a corresponding
radical HONO elimination to form a neutral unsaturated nitroamine and CndashC and CndashN bonds breaking leading to the ring opening
Since the molecular structure of CL-20 has NO2 groups of two different types four groups in two five-member rings and two in the six-member one it is likely that there will be preferential elimination of one type of NO2 group over the other There are also two possible types of HONO elimination reactions We have explored all these possibilities
At the first step of the study the energies of both of N2ndashN13 (analogue N6ndashN15 N8ndashN16 N12ndashN18) and N4ndashN14 (analogue N10ndashN17) bond rupture processes that are accompanied by elimination of NO2
bull or HONO species have been computed It has been concluded that the transformation 1rarr2 is the least endothermic one among all possible reactions
Then C1ndashC7 C5ndashC9 N4ndashC5 and C7ndashN8 bond cleavage of the structure (2) has been modeled The minimal energy corresponds to C1ndashC7 bond breaking reaction (2rarr3 in Fig 1) Simultaneously C1-N2 bond gains double character
The elimination of NO2bull group from N8-atom and C2ndashN8 double bond formation leads to
significant stabilization of the system due to transformation of a radical (3) to a neutral molecule (4)
Further two different types of decomposition of (4) have been investigated NO2bull-particle
elimination (rupture of N8ndashN16 or N10ndashN17 bonds) with formation of corresponding radicals and
4th Southern School on Computational Chemistry
78
HONO elimination (breaking of N8ndashN16 and C9ndashH N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds) with formation of a neutral molecule with three double bonds The HONO elimination reactions in this case are less endothermic then reactions of NO2
bull elimination Structure (5) is 475 kcalmol more favorable than the both products formed by HONO elimination due to N10ndashN17 and C9ndashH or N10ndashN17 and C11ndashH bonds ruptures Stability of structure (5) could be explained by conjugation of two double bonds C1=N12 and N10=C11
The break of N4-N14 and C5-H bonds with HONO and neutral molecule (6) formation is more advantageous if comped to NO2
bull elimination from (5) the first pathway is exothermic while the second one is endothermic
During the next steps two successive NO2bull-radical eliminations follow and yield molecules
(structure 7 and 9) which stabilize by the hydrogen migration from C3 and C9 atoms to N2 and N10 atoms accordingly (transformations 6rarr7 and 9rarr10)
CL-20 unimolecular decomposition results in formation the aromatic compound 15-dihydrodiimidazo[45-b4rsquo5rsquo-e]pyrazine (10)
The formation of the aromatic structure (10) has been confirmed by UVVIS and FTIR spectra received by Qasim and et al2
References Wu C J Fried LE J Phis Chem A 1997 101 8720 Qasim M Furey J Fredrickson HL McGrath C Bajpai R submitted to press
4th Southern School on Computational Chemistry
79
Ab Initio and NMR 19F Study of Comparative Stability of P and As Pentafluorohydroxocomplexes
SI Okovyty1 AVPlakhotnyk2 VVRossikhin2
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
In spite of existence of a close analogies range in physical-chemical behaviors of V-group p-elements fluorocomplexes in particular arsenic and phosphorus in a top oxidation level detail investigation of their behavior in solutions results in some significant differences Lange and Von Vezer [1 2] have sown that stepwise processes of complex forming in water fluorine-phosphorus systems proceed through stages of tetrahedral oxofluorine anions forming
H3PO4 + HF H2PO3F + H2O (1) H2PO3F + H2O HPO3F minus + H3O+ (2)
HPO3Fndash + HF PO2Fminus2 + H3O+ (3)
These ones transform to hexafluorphosphat ndash anion under increasing of hydrogen fluoride
concentration
PO2Fminus2 + 4 HF PF
minus6 + 2H2O (4)
Subsequently during study of processes proceeding during generation as well as destruction
(hydrolysis) of hexafluorphosphates analogical products have been fixed in some fluorocomplexes systems containing water molecules [3] Thus after the stage of the first fluoride-ion substitution and penthaphosphate formation according to equations (5 6)
PF6 + H2O fast
-PF5OH2 + F (5)
-
PF5OH2 + H2O
slow
PF5OH + H3O (6)- +
destruction of this particle takes place following by quick moving of three fluoride ions
away from an inner coordination sphere and decreasing of coordination number of P (V) to four
PF5OH minus + H2O PO2Fminus2 + 3 HF (7)
At the same time penthafluorohydroxoarsenates and tetrafluorodihydroxoarsenates as well
as their alkylderivation extracted by the number of authors [4 5] are stable compounds There
are intensive signals for AsF5OH minus and AsF4(OH)minus2 particles in 19F NMR spectra [6 7]
Undoubtedly in contrast to analogical phosphor compounds fluoroarsenate complexes demonstrate tendency to proceeding of a traditional process of stepwise ligand substitution
4th Southern School on Computational Chemistry
80
This significant difference in stability of arsenic and phosphorus penthafluorohydroxocomplexes as well as absence of literature data about this phenomenon has induced us to carry out calculations of geometric and electron structure of abovementioned complexes and additional experimental study using NMR spectroscopy
Optimization of structures of titled compounds has been carried out using DFT-B3LYP approximation in G98W program [8] Enthalpy ∆Hr and free energy of a process (8) ∆Gr as well as ionization potentials of complexes have been calculated (see Table 1)
EF5 + OH minus EF5OH minus (8)
where E = P or As for gas phase and water solution
Table 1 Calculated values of Enthalpy (∆Hr) and free energy (∆Gr) of a process (8) and ionization potentials of complexes
Complex -∆Hr kcalmol
-∆Gr kcalmol
I eV
PF5OH minus 15374 14301 971 Gas phase
AsF5OH minus 16353 15255 982
PF5OH minus 10046 8886 928 Water solution
AsF5OH minus 10599 9381 968 Despite some decreasing of complexes stability in water solution if compare to gas phase
they are still enough high thermodynamic stable Increasing of stability in the row PF5OH minus rarr AsF5OH minus is concerned with intrinsic
increasing of acceptor ability of corresponding Lewisrsquos acids in the row PF5 rarr AsF5 [9 10] and cannot explain above described difference in physical-chemical behaviors of arsenic and phosphorus fluorocomplexes
We suggested the possibility of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH minus complexes through transition state shown in Fig1
Figure 1 Transition state of intrasphere displacement of hydrogen from oxygen to fluorine in EF5OH
minuscomplexes (E = P or As)
A calculated gas phase activation barrier value ∆Gdegne for PF5OH minus is lower then for
AsF5OH minus It sets conditions for significant (more then on four orders) increasing of penthafluorohydroxophosphate lability in comparison with penthafluorohydroxoarsenate
Fig 2 and 3 show NMR spectra of fluoroarsenate systems as well as lithium hexafluorophosphate hydrolyzing in organic aprotonic medium containing 05 H2O
4th Southern School on Computational Chemistry
81
In the case of fluoroarsenate systems in addition to HF (singlet at 1600 ppm) signals of cis-
and trans- AsF4(OH)minus2 (at 446 ppm and 485 ppm correspondingly) as well as a group of
signals of AsF5OHminus
(568 ppm) are fixed In the case of fluorophosphates systems forming the
following products of hydrolysis have been observed PO2Fminus2 (doublet at ndash761 ppm JF-P
9118 Hz) PO3Fminus2
(doublet at ndash832 ppm JF-P 9765 Hz) as well as singlet of HF (-1531 ppm) Forming of complexes like PF5OH
minus solvated PF5 or POF3 practically has not been
observed
Figure 2 Figure 3
References 1 W Lange Ber B 62 1084 (1929) 2 Van Vezer Phosphorous and its compounds Moscow 1962 p 686 3 N N Shakhmatova V N Plachotnik Ye G Ilyin Coordination chemistry vol 15 11
p 1504 (1989) 4 HM Dess RW Parry J Amer Chem Soc vol 79 7 p 1589 (1957) 5 L Kolditz M Gitter Z Chem V 7 5 s 201 (1967) 6 B N Chernyshev N G Vasilyuk Ye G Ilyin Coordination chemistry vol 12 10 p
1394 (1988) 7 Tovmash N F Kokunov Yu V Plakhotnyk A V Coordination chemistry vol 21 10
(1995) 8 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA 9 KO Christe DA Dixon D Mc Lemove WW Wilson JA Sheehy JA Boatz J Fluor
Chem vol 101 p 151 (2000) 10 V N Plakhotnyk A A Yevtukh V V Rossikhin Yu V Kokunov A V Plakhotnyk
Ukr Khim Zhurn vol 69 11 ndash 12 p 78 (2003)
4th Southern School on Computational Chemistry
82
Electron Density Analysis of Tetracyclic Nitriles
SI Okovytyy12 LK Svjatenko2 LIKasyan2 J Leszczynski1
1Dnepropetrovsk National University Dnepropetrovsk 49050 Ukraine 2Computational Center for Molecular Structure and Interactions
Jackson State University Jackson Mississippi 39217 USA Tetracyclic compounds (1-3) are used for synthesis of the biologically active compounds [1]
Since endo-nitrile group has a small effect on chemical shift of Н12s and Н12a protons these protons must be close to equivalent However there found unusually large difference of chemical shift of bridge protons at carbon C12 which need to be explain In order to solve this problem the atoms in molecules theory (AIM) was employed to compute the atomic properties of tetracyclic compounds (1-3) on PBE1PBE6-31+G electron distributions
The topological analysis of the electron density reveals the existence of a bond paths linking the carbon atoms С9 С10 and hydrogen atom Н12s in the compounds (1 2) and bond paths linking hydrogen atoms H9 H10 and Н12s in the compound (3 Fig1)
Figure 1 Superposition of the contour lines (thin) of the charge density in the Н9nsdotsdotsdotH12ssdotsdotsdotH10n atomic interaction lines area with the molecular graphs (bold) in endo-4-cyanotetracyclo-[621136027]dodecane (3) Nuclei are represented by cross and the symbol triangle represents the locations of bond critical points
10
9
87
11
2
6
1
3
12
54H
H
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
O
HH
HH
HH
HaHs
H
HHs
Ha
Hn
HxH
CN
HH
1 2 3
Н10
C10 C9
Н9
C12
Н12s
4th Southern School on Computational Chemistry
83
Electron charge density at С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) bond critical points order of magnitude lower than those calculated for covalent bonds The corresponding Laplasians of the charge density are small in magnitude and positive indicating that the charge density is not accumulated in these points but concentrated towards the interacting nuclei The positive values of the local energy density are in line with this interpretation pointing to the association of these atomic interaction lines with closed-shell interactions
Closed-shell interactions С9sdotsdotsdotН12ssdotsdotsdotС10 (1 2) and Н9nsdotsdotsdotH12ssdotsdotsdotH10n (3) cause the Н12s proton deshielding which reflected in larger value of the proton Н12s chemical shift if compare to Н12a proton chemical shift (∆δН12sН12a sim15 ppm) Dependence of chemical shift of hydrogen atom H12s on its electron density for compounds (1-3) has been found
Reference
1 Kasyan LI Okovytyy SI Kasyan АО Golodaeva EA Uzlenko OV Bulletin of Dnipropetrovsk University Chemistry - 2000 - N 5 - P 3 - 8
4th Southern School on Computational Chemistry
84
Two-Dimensional Magnetoexcitons in the Presence of Rashba Spin-Orbit Interaction
O Olendski and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 Jackson MS 39217 USA
We study theoretically the effect of Rashba spin-orbit interaction on quantum well exciton in
a strong magnetic field We find that in the presence of an in-plane field component the
interplay between Zeeman and Rashba terms leads to a drastic change in the magnetoexciton
energy spectrum When separation between adjacent Landau levels with opposite spins becomes
of the order of the magnetoexciton binding energy the bright and dark exciton dispersions
exhibit anticrossing resulting in a pronounced minimum at finite momentum for the higher-
energy eigenstate With varying in-plane field the anticrossing moves to zero momentum
leading to a spin-orbit-induced splitting of the excitonic absorption spectrum Using algebra of
exciton creation and annihilation operators matrix elements of the Hamiltonian are calculated
exactly in the basis of one-exciton wavefunctions For strong field the full matrix reduces to the
4X4 form describing interaction between relevant spin-split Landau levels and the excitonic
spectrum is then calculated numerically
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by
ARO under grant DAAD19-01-2-0014
4th Southern School on Computational Chemistry
85
Roughness of the Film Growth on an Adsorbing Substrate with Kinetic Reactions in an Aqueous Solution of
Hydrophobic and Polar Groups
RB Pandey
Department of Physics and Astronomy University of Southern Mississippi Hattiesburg MS 39406-5046
Using computer simulations on a discrete lattice interface width and roughness of the film growth on an adsorbing substrate are studied Hydrophobic (H) polar (P) and water (W) components are considered with their appropriate molecular weights and interactions in an effective solvent medium
For example there is an attractive interaction between the polar groups and the water components and a repulsive interaction between the hydrophobic groups and water particles Constituent particles execute their stochastic motion with the Metropolis algorithm Periodic boundary conditions are used along the transverse directions while top boundary is open with an adsorbing impenetrable substrate at the bottom Evaporation of water component is allowed
The mixture is equilibrated with the non-covalent interactions before initiating a kinetic interaction The reaction initiates with covalent bonding of the constituents with the substrate and proceeds upward A covalently bonded particle becomes immobile The rate of reactions and the concentration of water are varied The density of the film grows as the reaction proceeds From the density profiles interface width of the film is estimated The interface width grows very fast initially but saturates eventually in the asymptotic time limit
The saturated interface width is a measure of the roughness of the film and is sensitive to rate of reaction and the water concentration Attempts are made to provide the empirical dependence of the roughness on the water concentration and the reaction rate
4th Southern School on Computational Chemistry
86
Polyhedral Oligomeric Silsesquioxane (POSS) Monomers with Atomic Alkali and Halogen Impurities
Sung Soo Park Chuanyun Xiao and Frank Hagelberg
Computational Center for Molecular Structures and Interactions Department of Physics Atmospheric Sciences and General Science
Jackson State University Jackson MS 39217
Polyhedral Oligomeric Silsesquioxane (POSS) molecules have found numerous technological applications In particular it has been shown that POSS molecules bond to organic polymers forming long chains that extend through the polymer and result in a nanostructured organic-inorganic hybrid In these units the POSS chains act like nanoscale reinforcing fibers producing extraordinary gains in heat resistance The use of POSS segments in plastics results in improved physical properties of these materials specifically in the enhancement of fire retardation and of usage temperatures as well as in increased hardness of the plastics Further various research projects have focused on metal-substituted POSS species which have been demonstrated to exhibit catalytic activity [1]
In this contribution we address the question to what extent the geometric energetic and electronic properties of POSS monomers can be influenced by addition of atomic alkali and halogen impurities More specifically we investigated POSS and POSS analogous systems of the type XA8H8O12 and XA8H8O12 ( X = Li Na K or F Cl A = C Si Ge) with exohedral as well as endohedral impurities respectively at the B3LYP6-31G and the B3LYPLANL2DZ level Endohedral structures are strongly preferred by the halogen containing systems Turning from halogen to alkali metal atom impurities one finds a reversal of this order of stabilities Here the exohedral structure results in general as more stable than the endohedral alternative A stability crossover however is found for a combination of a sufficiently large cage and small alkali metal atom impurity Thus for Ge8H8O12 doped with Li+ the exohedral geometry emerges as more stable than the endohedral variant [1] T Kudo MS Gordon JPhys Chem A 105 11276 (2001)
4th Southern School on Computational Chemistry
87
The Influence of Water on the DoublendashProton Transfer in Methylated GuaninendashCytosine Base Pair An ab Initio Study
Yevgeniy Podolyan Leonid Gorb Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Jackson State University Department of Chemistry 1325 Lynch St Jackson MS 39217
Double-proton transfer in DNA pairs is a process in which proton of one base is transferred to the other one and vice versa As an intramolecular proton transfer in DNA bases double-proton transfer can also lead to mutations Since the hydrogen atoms in the position 9 of purine and 1 of pyrimidine bases are replaced with a sugar moiety in DNA the bases methylated at corresponding positions should possess the properties more closely resembling those of the nucleotides
The current study of the double-proton transfer in methylated GuaninendashCytosine (mGmC) base pair has been performed at B3LYP6-31G(d) level of theory We have optimized local minima for isolated mGmC and the ones hydrated with 1 2 4 and 6 water molecules (see Fig 1) Complete potential energy surface (PES) scans have been performed for a gas-phase proton transfer in isolated base pair and the one hydrated with 2 water molecules at the same level of theory
The analysis of the relative energies and the PES scans shows only small changes in the pathway of the double-proton transfer in the mono- and dihydrated methylated base pairs as compared to isolated one The presence of 4 water molecules causes significant deformations of the rare form of base pair making it impossible to study the double-proton transfer in this species On the other hand 6 water molecules which form a complete hydration shell stabilize the zwitterionic form (the one in which only one proton is transferred) greater than the rare form The last finding is in line with the conclusion from the previous study of double-proton transfer in GuaninendashCytosine base pair using PCM model to simulate water surrounding
Figure 1 The most stable species of canonic mGmC base pair hydrated with 1 2 4 and 6 water molecules
4th Southern School on Computational Chemistry
88
Finite-Size Effects in Surface-Enhanced Raman Scattering from Molecules Adsorbed on Noble-Metal Nanoparticles
V N Pustovit K M Walker and T V Shahbazyan
Department of Physics Jackson State University PO Box 17660 JacksonMSi 39217 USA
Surface-enhanced Raman scattering (SERS) from molecules adsorbed on small metal particles has attracted increasing interest during past two decades The main SERS mechanism has electromagnetic (EM) origin and is due to the strong surface plasmon (SP) local field near the metal surface [1] (see also [2] for review of all SERS mechanisms) Recent observations of enormous (up to 1015) enhancement of single-molecule Raman scattering [3] as well as emerging possibilities of nanoparticle-based Raman sensors [4] have prompted a new interest in single particle SERS and in particular in finite-size effects in small nanoparticles Although classical EM enhancement is size-independent quantum corrections due to the discreteness of the electron spectrum result a weaker enhancement in small nanoparticles
Here we describe a novel finite-size quantum-mechanical mechanism that leads to a relative increase of SERS in small noble-metal particles This mechanism stems from different effect that the confining potential has on s-band and d-band electrons Namely the spillout of delocalized s-electrons beyond the classical nanoparticle boundary results in an incomplete embedding of sp-electron distribution in the background of localized d-electrons whose density profile follows more closely the classical shape (see Fig 1) In the absence of d-electron population in the nanoparticle surface layer the effective dielectric constant is reduced relative to the bulk giving rise to a blue shift of the SP resonance in Ag nanoparticles [5]
Figure 1 Schematic representation of electron density profiles in Ag nanoparticles The effective nanoparticle radius for s-electrons is larger than for d-electrons Local fields in the vicinity of metal surface are enhanced due to the reduction of screening in the surface layer
Here we demonstrate that SERS in noble-metal nanoparticles is strongly affected by the interplay of electron confinement and screening effects Indeed a reduction of d-electron screening in the surface layer leads to a stronger SP local field acting on a molecule located in a close proximity to the metal surface This results in an additional enhancement of the Raman signal Importantly such an enhancement becomes more pronounced for small nanoparticles due to the larger ratio of surface layer to overall nanoparticle size We have developed a theory for SERS that incorporates the surface layer effect within the framework of two-region model [5] Figure 2 shows the results of our numerical calculations of the Raman enhancement factor for Ag nanoparticles with radii in the range from 10 to 40 nm
4th Southern School on Computational Chemistry
89
Supported by NSF under grants DMR-0304036 DMR-0305557 and HRD-0318519 and by ARO under grant DAAD19-01-2-0014
Figure 2 Size-dependence of Raman enhancement factor calculated using two-region model for various surface layer thicknesses a (normalized with respect to a=0 nm R=10 nm value) For finite a SERS from small nanoparticles is stronger References [1] G C Schatz and R P Van Duyne in ldquoHandbook of Vibrational Spectroscopyrdquo J J
Chalmers and P R Griffiths eds (Wiley 2002) [2] K Kneipp H Kneipp I Itzkan R R Dasari and M S Feld ldquoUltrasensitive Chemical
Analysis by Raman Spectroscopyrdquo Chem Rev 99 2957 (1999) [3] S Nie and S R Emory ldquoProbing single molecules and single nanoparticles by surface-
enhanced Raman scatteringrdquo Science 275 1102 (1997) [4] Y C Cao R Jin and C A Mirkin ldquoNanoparticles with Raman Spectroscopic Fingerprints
for DNA and RNA Detectionrdquo Science 297 1536 (2002) [5] A Liebsch ldquoSurface-plasmon dispersion and size dependence of Mie resonance Silver
versus simple metalsrdquo Phys Rev 48 11317 (1993)
4th Southern School on Computational Chemistry
90
Using CL-20 as the Model UVVISFTIR Spectrophotometry Supports Theoretical Predictions as to InitialIntermediate Steps in
Chemical Transformation of Cyclic and Cage Cyclic Nitramines
Mohammad (Mo) Qasim1 Herbert L Fredrickson1 John Furey1 Ray Castellane1 Chris McGrath1 Jim Szecsody2
1USACE ERDC 3909 Halls Ferry Road Vicksburg MS 2Pacific Northwest National Laboratories Richland WA
Applying appropriate experimental methods to verify both hypothesis and theoretical study is useful in proving concepts and in ascertaining what is chemically possible Since molecular structures exhibit characteristic spectra the hypothesis that molecular structure under homologous conditions determines preferred degradation pathways that can be theoretically predicted was tested by first relating chemicalphysical properties to types and sites of reactivity then comparing obtained data with experimental results Changes in UVVIS spectra if consistent with predictions would constitute experimental verification
The hypothesis was first examined through semi-empirical predictive methods using 24681012-hexanitrohexaazoiso-wurtizane (CL-20) as the experimental model MOPAC quantum mechanical and classical force field mechanics were used to investigate most likely first tier intermediates through comparisons as to bond lengths and angles heat of formation steric energy dipole moments solvent accessibility and electrostatic potential surfaces partial charges and HOMOLUMO energies
Experimentally obtained spectral data UVVIS measured concentrations and followed the course of reactions while Stopped Flow spectrophotometry followed the rates of CL-20 degradation to fast-forming transition states and initialintermediate productsmdashto verify first tier transformation products of CL-20 due to two competing modes of degradation through reactions due to i) addition of (sodium) hydroxide ions (OH-) and to addition of ii) photo-induced free radicals
i) CL-20 breakdown due to OH- FTIR followed CL-20 degradation through alkali hydrolysis measuring changes in functional groupsmdashnitro and amino carbon-carbon (C-C) and carbon-nitrogen (C-N)mdashindicating breaking and formation of bonds Arbitrarily we call the three C-C bonds of CL-20 the two base bonds on opposite sides of the cyclohexane ring C1-C2 and C3-C4 respectively whereas the C5-C6 bond represents the ldquoatticrdquo of the two cyclopentane rings Different compounds result depending on which C-C breaks The preferred breakdown is through the C5-C6 bond joining the ldquoatticrdquo peaks of the two clyclopentane rings resulting in a compound having lower formation and force field energies 134578-hexanitrododedahydroiimidazo[45-b4rsquo5rsquo-e]pyrazine This breakdown then results in stretching the other two C-C bonds and also in stretching the nitro group ring N-N bonds Either OH- replace the nitro groups or (preferred pathway) they abstract protons and eliminate nitro groups via the E2 mechanism FTIR data reveal that at lower OH- concentrations (less than 21 molar ratio of OH- to CL-20) the resulting C-H bond stretching is most aliphatic (less than 3000cm-1) while at higher OH- concentrations (10 N1000 ppm of CL-20) the FTIR spectra shows that most of the C-H stretching is more than 3000cm-1 indicating the formation of an aromatic intermediate with a C-C bondmdashsimilar to that in TNT Accompanying FTIR data show a decrease in C-N intensity at 1049cm-1 Both UV and FTIR spectra of CL-20 when reacted with high concentrations of OH- strongly suggest the formation of an aromatic three-ring intermediate 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine Calculations show that this structure
4th Southern School on Computational Chemistry
91
exists in two forms cis and trans or maybe the two conformers boat and chair Formation of the aromatic structure is also strongly supported by UVVIS spectral characteristicsmdashthe intense yellow-green (375nm) color appearing when 11 by volume of 10 N NaOH was added to 1000 ppm of CL-20 in dichloromethane The FTIR spectra were obtained from the evaporated organic phase whereas the UVVIS spectra were obtained from the aqueous phase
Additional also included UV spectra reveal a concentration-dependent shift toward longer wavelengths upon addition of OH- suggesting stepwise removal of nitro groups and formation of double bonds until the aromatic compound 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine is formed However upon further addition of OH- an abrupt shift to shorter wavelengths occurs suggesting a breaking of the 15-dihydrodiimidazo[454rsquo5rsquo-e]pyrazine into smaller aromatic compounds
Different less important non-aromatic cyclic competing products of CL-20 can be formed by breaking the other two C-C bonds of the cyclohexane ring Simultaneous breaking of both base C-C bonds results in a bi-cycloheptagonal ring intermediate whereas breaking either of the base C-C bonds results in a compound consisting of two opposing cycloheptagonal rings a cyclononagonal and a cyclopentagonal ring
ii) Addition of photo-induced free radicals A monochromatic irradiation system developed for the study photo-induces free radical reactions through irradiation at wavelengths of maximum absorption Calculations indicate the free radical mechanism (symmetrical bond-breaking) is more apt to occur upon increase in number of symmetrical C-C (preferred) then N-N bonds contained within the molecule This bond-breaking may occur either sequentially or simultaneously Calculations also indicate that the less polar the bond the greater is the predisposition toward free radical bond-breaking The free radical degradation mechanism aspect of the experimental study is currently underway
In conclusion UVFTIR spectral data including FTIR measurements of changes in functional groups during appearancesdisappearances of intermediate species so far have verified both hypothesis and theoretical predictions Our theoretical predictions were also verified by electron spray MS (Szecsody et al)
4th Southern School on Computational Chemistry
92
When bottom cyclohexane ring breaks
Diimidazole aromatic upon further addition of OH detailed in following
First stage breaking differentC-C bonds CL20
Two forms cis trans of breaking attic (5-6) C-C bond
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
15-dihydrodiimidazo[45-b45-e]pyrazine
N
NN
NH
N
HN
By partial Charges
Detailed description of the Diimidazole compound
4th Southern School on Computational Chemistry
93
absorbance of 440 mgL CL20MeOH in several NaOH solutions
0
05
1
15
2
25
3
35
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
abso
rban
ce
CL20 in MeOH
CL20-001 N NaOH
CL20-001N + 30 minutes
CL20-01N NaOH
CL20-01N + 30 minutes
CL20-1N NaOH
CL20-1N NaOH
CL20-1N + 30 minutes
CL20-1N + 30 minutes
Comparison of UVVis Spectra Reaction of CL20 with NaOH
0
05
1
15
2
25
3
35
4
200 250 300 350 400 450 500 550 600 650 700
wavelength (nm)
Abs
orba
nce
OH-CL20
OH-MeOH
CL20-MeOH
MeOH
UVVIS Spectra of Cl 20 with different Concentrations of NaOH
FTIR Spectra of CL20 with Different Concentrations of NaOH
4th Southern School on Computational Chemistry
94
Theoretical Study of Diatomic Molecules XY (X=N P As and Y=Se Te) and its Cation (XY+) and Anion (XYndash)
Methods and Basis Effect
M Rekhis O Ouamerali
Laboratoire de Physicochimie Theacuteorique et Chimie Informatique Faculteacute de Chimie USTHB BP32 El Alia 16111 Bab Ezzouar Alger Algerie
In the present study ab-initio and density functional theory were applied to the diatomic
molecules XY formed by combining pairs of fifth and sixth groups atoms
(X=N P As Y=Se Te) and their cation (XY+ ) and anion (XY- ) Considering their position
in the periodic chart of elements the neutral entities are radicals and their ground electronic
states is ΠΧ2 whereas XY+ and XY- have respectively +ΣΧ1 and minusΣΧ3 ground electronic
states
The internuclear distances fundamental vibrational frequencies dissociation energies
ionization potential and electron affinity have been determined including computations using
restricted Hartree-Fock closed- and open- shell Moller-Plesset perturbation and DFT theory
using several bases sets with effective core potentials for tellurium
The optimized geometries and the calculated fundamental vibrations agree closely with
experimental information where available
The results are in agreement with the expectations of the periodic chart of elements Atomic
charges analysis show for tellurium atom the qualities of both metal and non metal element In
fact the absolute value of the atomic charge of Te is the greatest in the ionic species XTe plusmn
4th Southern School on Computational Chemistry
95
Continuing Studies of Dimethyl-substituted Cyclobutanes and the gem-Dimethyl Effect
Ashley L Ringer and David H Magers
Mississippi College Department of Chemistry and Biochemistry Clinton Mississippi
The gem-dimethyl effect is the acceleration of cyclization by substituents in the chain and is often used in organic synthesis as a ring-closing effect In the study calculations on methylcyclobutane (Figure 1) 11-dimethylcyclobutane (Figure 2) cis- and trans-12-dimethylcyclo-butane (Figure 3) and cis- and trans-13 dimethylcyclobutane (Figure 4) are performed to determine if this effect is a thermodynamic effect caused by lower strain energy or a kinetic effect that simply lowers the activation barrier for the cyclization reaction 11-dimethylcyclobutane is a four-membered carbon ring with gem-dimethyl substituents
Figure 1 Figure 2 methylcyclobutane 11-dimethylcyclobutane
Figure 3 cis-12-dimethylcyclobutane trans-12-dimethylcyclobutane
4th Southern School on Computational Chemistry
96
Figure 4 cis-13-dimethylcyclobutane trans-13-dimethylcyclobutane
One hypothesis suggests that the strain energy is lower because the methyl groups have more freedom to move in the ring than in the open chain Optimum equilibrium geometries harmonic vibrational frequencies and corresponding electronic energies are computed for all pertinent molecular systems using SCF theory density functional theory (DFT) and second-order perturbation theory (MP2) Comparisons are made between the conventional strain energy of cyclopropane and cyclobutane methylcyclobutane and the dimethyl-substituted cyclobutanes
SCF and DFT calculations indicate that 11-dimethylcyclobutane is approximately 3 to 35
kcalsmol less strained than methylcyclobutane which in turn is 3 to 35 kcalsmol less strained than cyclobutane that is there is at least some thermodynamic component to the gem-dimethyl effect The study continues by calculating conventional strain energies for the 12- and the 13-dimethylcyclobutanes Additionally the optimum geometries of all relevant systems are examined particularly noting the bonding angles in the rings to study the relevance of geometry to the gem-dimethyl effect We gratefully acknowledge the support of NSF EPSCoR (EPS-0132618)
4th Southern School on Computational Chemistry
97
Theoretical and Experimental Investigation of DonorndashAcceptor Li+ Cation Interaction with Bidentate Aprotonic Solvent
VN Plakhotnyk LD Tarasova VV Rossikhin
Dnepropetrovsk National University of Railway Transport Dnepropetrovsk 49010 Ukraine
Solutions of lithium salts in aprotonic solvents are wide used as electrolytes for lithium and lithium-ionic batteries [1 2] The problem of charge transfer in interelectrode space of batteries is closely interconnected with nature of interparticle interactions in electrolytes Ion-dipole interactions of a Li+ cation with solvent molecules play a special role among them [3 4]
Concentration of charge bearers to a first approximation is determinated by proceeding of competing processes of cation-anion association and solvation Presence of solvent molecules able to bidentate coordination in solution leads to forming of their complexes with Li+ cations containing chelate bonds [5]
We have considered a possibility of Li+ cations to form complexes with 12-dimethoxyethane (DME) as well as dimethylcarbonate (DMC) which are often using as components of electrolytes for lithium-ionic batteries Calculations of electronic and geometric structures of source molecules and complexes with ratio of lithiumsolvent equals to 12 as well as thermodynamic parameters of Li+ ndash DME and Li+ ndash DMC interaction reactions have been performed using the self-consistent field method of Hartree-Fock-Roothaan in G98W program [6]
From the optimized structures of Li+ complexes with DMC and DME (1 2) it is certainly that in both cases oxygen atoms bonded with methyl groups are electron donors and presence of carbonyl group in the case of DMC leads to electron density displacement along the С rarr О bond and decreasing of donor ability of oxygen atoms taking part in donor-acceptor bonding to Li+ cation
1 2
A following table demonstrates the thermodynamics of complexes forming reflecting the abovementioned circumstance
4th Southern School on Computational Chemistry
98
Table 1 Complex - ∆Hr kcalmol - ∆Gr kcalmol
Li(DME)2+ 1246 1044
Li(DMC)2+ 906 735
The observed difference in stability of Li+ ndash DME and Li+ ndash DMC complexes has been
confirmed by means of determination of temperature-concentration arias of LiBF42DMC and LiBF42DME crystalsolvates existence
Experiments have been carried out using a salt exemplar of 999 purity and thoroughly dehydrated solvents containing no more then 30 ppm H2O It has been sown that in the case of DMC crystalsolvate destruction occurs under ndash100С in area of 3044 - 3193 LiBF4 while DME crystalsolvate is stable in wide region of concentration (from 097 to 5107 LiBF4) and undergo congruent melting under + 300С
References 1 Skundin A M Electrochemical power engineering 2001 vol 1 1-2 pp 5 -15 2 Kedrinskiy I A Yakovlev V G Li ndash ionic accumulators Platina Press Krasnoyarsk
2002 266 p 3 Plakhotnyk VN Tovmash N F Kovtun Yu V Reports of Russian Academy of Science
1987 vol 292 6 p 1426 4 Plakhotnyk VN Tovmash N F Mishustin A N Goldshtejn I P Braverman O V
Damje V N Electrochemistry 1988 vol 24 7 р 964 5 Matsuda Y Nakashima H Morita M Takasu Y J Electrochem Soc 1981 vol 128
12 р 2552 6 Frisch M J at al (1998) Gaussian 98 Gaussian Inc Pittsburgh PA
4th Southern School on Computational Chemistry
99
Rare (Hydroxo) Tautomers of Nucleotides
Teri L Robinson1 Leonid Gorb1 Oleg Shishkin2 and Jerzy Leszczynski1
1Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217
2Institute for Scintillation Materials National Academy of Science of Ukraine 60 Lenina Ave Kharkiv 61001 UKRAINE
Two strands of phosphate and sugar coiled around each other in a helical manner and held together by hydrogen bonding between pairs of nitrogenous bases is defined as DNA Four bases are present and are classified as purines or pyrimidines Purines are adenine (A) and guanine (G) Pyrimidines are thymine (T) and cytosine (C) In guanine and thymine there are alternate molecular structures based on different locations of a particular hydrogen atom These alternate structures can be present in the keto and enol forms These two forms are known as tautomeric structure Tautomers in its rare form is a base in the nucleotide that can form different hydrogen bonds leading to mismatch pairing For example a rare form of A can pair with C and a rare form of T can pair with G ndash this believed to be the cause of transversion and transition mutations
Herein we have obtained and analyzed the molecular structures of rare (hydroxo) tautomers of 2-deoxyribonucleotides The molecular structure of different conformers of isolated lsquorarersquo 2rsquo-deoxyribonucleotides was optimized using B3LYP6-31G(d) method Results of calculations reveal that geometrical parameters and relative stability of conformers significantly depend on the nature of nucleobase its orientation conformation of the furanose ring and charge of the phosphate group Analysis of the electron density distribution in nucleotides reveals an existence of a number of intramolecular hydrogen bonds Relative stability of conformers is determined by nature of nucleobases its orientation and character of intramolecular hydrogen bonds Influence of negative charge of the phosphate group on geometrical parameters and relative stability of conformers has been analyzed Results of calculation reveal that geometry and relative energy of conformers of nucleotides may be easily tuned by charge of the phosphate group
4th Southern School on Computational Chemistry
100
Efficient AO-Formulation of MP2-Gradients
Svein Saeboa KrzysztofWolinskibd Peter Pulaybc and Jon Bakerbc
aDepartment of Chemistry Mississippi State University Mississippi State MS 39762 bParalell Quantum Solutions LLC 2013 Green Acres Suite A Fayetteville AR 72703
cDepartment of Chemistry and Biochemistry University of Arkansas Faytetteville AR 72703 dDepartment of Chemistry University of Lublin Lublin Poland
We have recently implemented an efficient AO-formulation of MP2-gradients The formulation can be used for any set of internal orbitals (eg localized orbitals) but currently implementation has only been completed for Canonical internal orbitals The orbital-invariant form of second-order Moslashller-Plesset perturbation theory can be derived from the Hylleraas functional form of the second-order energy The functional form is very convenient for the formulation of the gradient and the present derivation essentially follows our formulation from 19861
The formalism as well as its computer implementation will be described in detail The program is currently only running on a single CPU However we will provide examples of MP2-gradient calculations on systems with ~100 atoms and about ~1000 contracted basis functions carried out on a PC and timings and test-results will be provided
Parallelization of the program is in progress and when this is completed we expect that geometry optimization of systems with several hundred atoms can be routinely carried out on small PC-clusters at the MP2 level using suitable (~TZ+P or better) atomic basis sets
1Pulay P and Saebo S ldquoOrbital-invariant formulation and second-order gradient evaluation in Moller-Plesset perturbation theoryrdquo Theor Chim Acta 69 357 (1986)
4th Southern School on Computational Chemistry
101
Electron Impact Ionization at Intermediate and High Energies
Bidhan C Saha
Department of Physics Florida AampM University Tallahassee FL-32307
In recent years considerable attentions have been drawn to evaluate the electron impact ionization cross-sections for atomic and ionic systems for application to model the astrophysical and fusion plasmas [1 2] Inadequacy of experimental results and their scattered nature ndash both in energies and targets ndash have led to the development of numerous analytic models There are few rigorous quantal treatments for lighter targets [3 4] but their implementations to heavier targets are yet to be done So there is a demand for development of sufficiently accurate and simple procedures [5-8] to generate extensive sets of cross-sections for different species at a wider range of energies These cross sections are needed in many areas of applied sciences and technological problems [910] such as fusion plasmas modeling of radiation effects in material and medical physics plasmas etching of semiconductors mass spectrometry fluorescent lamps ion gauges atmospheric physics astrophysics etc
The binary-encounter-dipole (BED) model for electron-impact ionization of Kim and Rudd [11] combines a modified form of the Mott cross section and the Bethe-dipole cross section The relativistic effects become important for both medium and heavier atomic and ionic targets as the K-shell binding energies of the atoms increase The same effect becomes important at higher energies even for light targets So at high energies the effect of relativity remains very important Using relativistic corrections due to Moller [12] Kim et al [13] have applied it to various targets we name that model as RBED in this study
For a comparison we also use the relativistic modification if the Vriensrsquo binary-encounter approximation (BEA) model [14] and denote it RBEA in this study
In Fig 1 we have shown our results are compared with other theoretical findings So far we know there are no experimental single ionization cross sections for this ionic target
Figure 1
4th Southern School on Computational Chemistry
102
From Clusters to Crystals Theoretical Investigation on Molecular Structure of Sodium Halides
Julia Saloni12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Sodium halide clusters have been studied in different ways experimentally using Mass
Spectroscopy Raman Spectroscopy and also Ultra Fast Liquid Chromatography methods and
theoretically using ab initio or Molecular Dynamics methods
This work presents Density Functional Theory calculations on molecular structure of small
sodium bromide and iodide clusters Ground state geometries for all molecules were calculate
using B3LYP method in cc-pvtz (Na) and SDB-aug-cc-pvtz (BrI) basis sets
It is well known that bonds in crystal unit between sodium and halide (Na-X X=FClBrI)
have electrostatic nature bonds in clusters show the same property Although interactions
between more complexes structures of sodium halides manifest different nature It is observed in
Jahn-Teller Effect Many Body Interaction calculations are performed to characterize mentioned
above type of bonding
The analysis of collected data is going to answer the question where cluster ends and crystal
begins
4th Southern School on Computational Chemistry
103
Theoretical Study of the Reaction between CCl2 Carbene with NO
Hasan Sayin and Michael L McKee
Department of Chemistry Auburn University AL 36849
The reaction between CCl2 and nitric oxide is studied by optimizing minima and transition
states with DFT level and carrying out high-level calculations We tried to investigate rate
constant of this reaction which is known experimentally as 11x10-12 cm3molecule-1s-1
4th Southern School on Computational Chemistry
104
Molecular Modeling of Molecular Sponges
1Trineshia Sellars 1Jesse Edwards
1Department of ChemistryAHPCRC Florida AampM University Tallahassee FL USA 32307
The use of polymers as absorbates has been developed quite extensively for SiO2 with
environmental applications We will examine the use of other polymeric materials as absorbates
including several acrylates using molecular modeling techniques The monomer units of several
polymeric materials will be presented at the molecular mechanics level while varying the
dielectric constant in order to determine the most suitable starting structure for polymerization
The dielectric constant changes serve as solvation of the polymeric surface thus allowing the
determination of surface interactions between potential polymeric molecular sponges and various
absorbates These interactions will be presented
4th Southern School on Computational Chemistry
105
Novel Aromatic 78-Diazapentalenes
Yinghong Sheng1 Ray Butcher2 Haijun Jiao3 Bakhtiyor Rasulev1 Jiande Gu1 Jerome Karle2 and Jerzy Leszczynski1
1) The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University P O Box 17910 1400 J R Lynch Street Jackson Mississippi 39217 2) Laboratory for the Structure of Matter Code 6030 Naval Research
Laboratory Washington DC 20375-5341 3) LeibnizminusInstitut fuumlr Organische Katalyse an der Universitaumlt Rostock eV Buchbinderstrasse 5minus6 18055 Rostock Germany
Conjugated bicyclic hydrocarbons such as pentalene are of particular interests in the modern organic as well as physical and theoretical organic chemistry1 Parent pentalene is a thermally unstable compound belonging to the class of destabilized anti-aromatic systems with 8-π electrons2 Stabilization of the pentalene can be achieved by steric shielding (eg by tert-butyl groups)3 or by the introduction of donor groups in the 1 (or 346) positions and acceptor groups in the 2 (or 5) position4
1
2
34
5
6 7
8
Replacing of two carbon atoms on the pentalene rings leads to diazapentalenes such as 16- 34- 12- 36- 25- and 78-diazapentalenes Among them 16- 34- 12- 36- and 25-diazapentalenes have been extensively studied5 but no literature has been reported on 78-diazapentalene These kinds of bicyclic hetero conjugated systems are expected to exhibit the specific features and to have potential applications On the basis of the replacement pattern 78-replacement results in a 10-π electron system which is expected to be stabilized and aromatic while all other replacements which do not change the number of the π-electron are anti-aromatic as their parent pentalene system
Recently we have successfully isolated the nitro-substituted 78-diazapentalenes such as 1-nitro-78- 134-tris(nitro)- and 1346-tetrakis(nitro)-78-diazapentalenes respectively In addition to the enhanced aromatic stabilization it is expected that 78-diazapentalene should show the similar feature as the pentalene ie electron-donating groups such as amine group would stabilize the 78-diazapentalene further The electron-withdrawing groups destabilize the system However all attempts to isolate the electron-donating group substituted 78-diazapentalene failed Therefore it is of interest to assess how the nitro groups stabilize the 78-diazapentalene
Its analogue 25-diazapentalene also called pyrrolo[34-c]pyrrole is expected to be non-aromatic6 1346-tetradonor-25-diacceptor-substituted pentalenes has been reported to exhibit aromatic stabilization and a delocalized π-bonding system7
In this study the aromatic stabilities of pentalene 25-diazapentalene and 78-diazapentalene as well as their substituted derivatives are investigated Whether a compound is aromatic or anti-aromatic is not a simple binary answer and there is no unique and generally accepted definition of aromaticity89 It has been almost generally accepted that compounds of aromaticity are more stable than their chain analogues Mostly the molecular geometry of aromatic compounds is characterized by the equalized bond lengths In addition it has a π-electron ring current that is
4th Southern School on Computational Chemistry
106
induced when the molecule is exposed to an external magnetic filed which leads to an increased magnetic susceptibility and 1H NMR chemical shifts These lead to the quantitative criteria commonly used to assess the aromaticity of a molecule which can be derived from the energetic geometrical and magnetic properties2a10 Energetic criteria are usually based on homodesmotic and isodemic11 as well as isomerization reactions12 From geometrical point of view a greater bond length alternation is usually assumed to be associated with a decrease in aromatic characteristics13
Magnetic criteria are quite effective in characterizing aromaticity as the ability to sustain a diatropic ring current These are the defining characteristics of aromatic species14 It has been approved that the magnetic criterion is the most specific and unambiguous manifestation of aromaticity and anti-aromaticity Hence magnetic properties such as anisotropic of the magnetic susceptibility (∆χ) exaltation of isotropic magnetic susceptibility (Λ) and a recently proposed quantity NICS (nucleus-independent chemical shift) are frequently used as aromaticity criteria2a15-18
The structures aromatic stabilities nucleus-independent chemical shifts as well as the ultraviolet absorptions of pentalene 25-diazapentalene and their substituted derivatives were studied at the B3LYP6-31G(d) level A novel compound 78-diazapentalene has been synthesized and its properties have been investigated Based on the experimental X-ray structure and the theoretical results one can draw the following conclusions
(i) Pentalene and 25-diazapentalene exhibit the pronounced bond length alternation while 78-diazapentalene possesses delocalized bond length distribution Electron-donating group delocalizes the bond length distribution in pentalene and 25-diazapentalene while electron-withdrawing group results in localized bond distances
(ii) Unlikely unsubstituted 78-diazapentalene exhibits no salient bond length alternation The C-C C-N and N-N bond lengths are in between the typical single bond and double bond In addition electron-withdrawing substituted 78-diazapentalene shows a comparative notable bond length alternation
(iii) Under the circumstance which homodesmotic reaction is unavailable for 78-diazapentalene the isomerization of the modified model compounds provides as the effective way to study the aromaticity of the studied compounds In addition this kind of isomerization helps to understand the system stabilitiesinstabilities from the aromatic stabilizationanti-aromatic destabilization as well as from the substituents On the contrary nitro group significantly stabilizes the 78-diazapentalene The stabilization is attribute to the lone-pairs on the nitrogen atoms and the π-conjugation of nitro group with the bucyclic π-system
(iv) The GIAOB3LYP6-31G(d) computed nucleus-independent chemical shift NICS(0) and NICS(1) implies the greater aromaticity of 78-diazapentalene than pentalene and 25-diazapentalene
(v) 78-diazapentalene is 4n+2 π-system while pentalene and 25-diazapentalenes are 4n π-systems
References 1 (a) Minkin V I Minyaev R M Chem Rev 2001 101 1247 (b) Geoffrey F Cloke N Pure Appl
Chem 2001 73 233 2 (a) Schleyer P v R Jiao H Pure Appl Chem 1996 68 209 (b) Slayden S W Liebman J F
Chem Rev 2001 101 1541 3 (a) Hafner K Suss H U Angew Chem Int Ed Engl 1973 12 576 (b) Falchi A Gellini C Salvi
P R Hafner K J Phys Chem 1995 99 14659 4 Gimarc B M J Am Chem Soc 1983 105 1979 5 (a) Matsumoto K Iida H Hinomoto T Uchida T J Chem Res Synop 1995 8 338 (b) Richter R
Sieler J Hansen L K et al Acta Chem Scand 1991 45 1 (c) Trofimen S J Am Chem Soc 1965 87 4393
4th Southern School on Computational Chemistry
107
6 (a) Closs F Gompper R Angew Chem Int Ed Engl 1987 26 552 (b) Closs F Gompper R Noumlth H Wagner H-U Angew Chem Int Ed Engl 1988 27 842
7 Kataoka M Ohmae T Nakajima T J Org Chem 1986 51 358 8 Krygowski T M Cyrański M K Chem Rev 2001 101 1385 9 Minkin V I Glukhovtsev M N Simkin B Y Aromaticity and Antiaromaticity Electronic and
Structural Aspects John Wiley amp Sons Inc New York 1994 10 Schleyer P v R Freeman P Jiao H Goldfuss B Angew Chem Int Ed Engl 1995 34 337 11 (a) George P Trachtman M Brett A M Bock C W J Chem Soc Perkin Trans 1977 2 1036
(b) Hehre W J Ditchfield W J Radom L Pople J A J Am Chem Soc 1970 92 403 (d) Fink W H Richards J C J Am Chem Soc 1991 113 3393
12 (a) Wakita K Tokitoh N Okazaki R Nagase S Schleyer P v R Jiao H J Am Chem Soc 1999 121 11336 (b) Havenith R W A Jiao H Jeneskens L W van Lenthe J H Sarobe M Schleyer P v R Kataoka M Necula A Scott L T J Am Chem Soc 2002 124 2366 (b) Schleyer P v R Puumlhlhofer F Org Lett 2002 4 2873
13 Krygowski T M Cyrański M K Czarnocki Z Haumlfelinger G Katritzky A R Tetrahedron 2000 56 1783
14 Pauling L J Chem Phys 1936 4 673 15 (a) Schleyer P v R Maerker C Dransfeld A Jiao H Hommes N J R v J Am Chem Soc
1996 118 6317 (b) Schleyer P v R Jiao H Hommes N J R v Malkin V G Malkina O L J Am Chem Soc 1997 119 12669 (c) Schleyer P v R Manoharan M Wang Z-X Kiran B Jiao H Puchta R van E Hommes N J R Org Lett 2001 3 2465
16 Arduengo A J III Dixon D A Kumashiro K K Lee C Power W P Zilm K W J Am Chem Soc 1994 116 6361
17 (a) Jiao H Hommes N J R v E Schleyer P v R Meijere A d J Org Chem 1996 61 2826 (b) Moran D Stahl F Bettinger H F Schaefer III H F Schleyer P v R J Am Chem Soc 2003 125 6746
18 Gonzales J M Barden C J Brown S T Schleyer P v R Schaefer III H F Li Q-S J Am Chem Soc 2002 125 1064
4th Southern School on Computational Chemistry
108
Theoretical Study of Excited States of Nucleic Acid Bases Electronic Transitions Geometries and Interaction with Water
Molecules
MK Shukla and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street Jackson MS 39217 (USA)
Purine (guanine (G) and adenine (A)) and pyrimidine (cytosine (C) and thymine (T) (uracil (U))) bases are molecular bricks of nucleic acid polymers Understanding of structures and functions of genetic polymers demands the detailed and reliable information about the physical chemical and biological properties of bases itself Methods of computational chemistry offer reliable and efficient tools to study and understand such properties It is especially important where experimental data are missing and theoretical tools can serve as a reliable guideline for complex experimental results
Electronic spectroscopic techniques have long been used to study structure and properties of nucleic acid bases There are rich experimental data available for bases and therefore they provide an excellent opportunity to test theoretical methods Impressive progress in theoretical methods has been made to study ground state properties of molecules However such situation is still far behind to study excited state properties Theoretical methods are especially useful for the area where experimental data are scant and application of computational tools would provide complementary information about such system For example it is generally not possible to determine excited state geometrical parameters for complex molecular system like nucleic acid bases experimentally only very limited information can be obtained Therefore in this area of research computational methods can be especially useful and may also provide valuable information to experimentalists In this work we have computed singlet electronic transition energies excited state geometries and interaction with water molecules both in the ground and excited states for nucleic acid bases and base pairs The phenomena of phototautomerism were also investigated The ground state geometries were optimized at the HF6-311G(dp) level while excited states calculations were performed at the CIS6-311G(dp) level In some cases different bases sets were also used The three water molecules were considered in the first solvation shell to model aqueous environment Nature of potential energy surfaces was ascertained by harmonic vibrational frequency analysis all geometries were found minima at the respective potential energy surfaces All calculations were performed using Gaussian-94 and 98 programs
The ground state molecular geometries were found planar except for the amino group (for guanine adenine and cytosine) which was found pyramidal In the excited state geometries were generally found appreciably nonplanar as compared to that in the ground state Further geometrical deformations were found different in the singlet ππ and nπ states
4th Southern School on Computational Chemistry
109
Guanine-N9H (S1(π-π)) Guanine-N9H (S2(n-π))
Adenine-N7H (S3(nπ)) Thymine (S2(ππ))
Figure1 Geometries of guanine adenine and thymine in selected excited state at the CIS6-311G(dp) level
C6
N3
Cytosine-N1H (S1(ππ)) Cytosine-N1H (S2(nπ))
N1
N3
O2
H41
N42003
1906
2056
2715
2018
2060
N1C2
N3
C4
N4
2091
2166
1996
3036
19293451
2078
Cytosine-N1H +3W (S0) Cytosine-N1H +3W (S2(nπ))
Figure 2 Cytosine-N1H tautomer and hydrated form in excited states
C2
N1N3 O6
H1
N1 C6
H61
H62
N6
C6 N1C2
N3C4
C5C6
4th Southern School on Computational Chemistry
110
Figure 3 Geometry in the lowest singlet ππ excited state for (from the bottom to top) GC base pair guanine monomer GG1 base pair (N1HN7 NH2C6O) and GG2 base pair (N1HC6O) at the CIS6-311G(dp) level
The effect of hydration was generally found to decrease amino group pyramidalization in the
ground state In the excited state molecular geometries for hydrated forms were revealed comparatively more planar than those of the isolated cases Further the mode of hydration was found to be significantly different in the singlet nπ excited state as compared to those in the ground and singlet ππ excited states Computed scaled (scaling factor 072) electronic transition energies were generally found to be in good agreement with the corresponding experimental data The geometries of the GC and GG base pairs were also optimized in the lowest singlet ππ excited state (excitation being localized at the guanine moiety) The geometrical deformations were generally found similar to that of the isolated guanine It is interesting to note that GG1 base pair geometry in the excited state was found nonplanar while the corresponding geometry for the GG2 base pair was found almost planar
4th Southern School on Computational Chemistry
111
Computational Search for a Stable Pentavalent-Pentavalent Phosphorus-Phosphorus Bond
Tatiana Shvareva
Chemistry Department Auburn University 179 Chemistry Bldg Auburn University Auburn Alabama 36849
The structure of Naphtho[18 ndashcd]-12-diphosphole and its derivatives have been studied as a
possible source of stabilization of rare pentavalent - pentavalent P-P bond PM3 and MNDOd
levels of theory were used to calculate the potential energy surfaces for these structures
substituted with different halogens and to find the thermodynamically and kinetically most stable
structures Results were compared with experimental data reported in literature
4th Southern School on Computational Chemistry
112
Structure and Properties of Fullerene Made with Substituted Atoms
Tomekia Simeon Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University
1400 J R Lynch Street Jackson MS 39217
Since their discovery in 1985 C60 has had an impact in chemistry biology and physics and has embarked a new era in carbon science The unique properties of fullerene molecules allow for the assumption that in the future they will be widely used for the creation of new materials Previous studies show that the addition of defects to fullerene structures could help reduce the strain of covalent bonds [1] The isomers of the C60 and C58 clusters the C59M type clusters and other carbon clusters have been synthesized but in significant quantities [2-4] Also the strong influence on the electronic structure is rendered by endohedral inclusions in C60 [5-6] Since the radius of the fullerene molecule is about 35 angstrom many atoms and small molecules can be placed inside the C60 sphere Fullerene molecules with various substituted defects are an area of science underexplored The purpose of this study is to elucidate the physical and chemical properties of modified C60 The following molecules have been selected C59B C59N C59P and C59Si The influence of substitute atoms on the electronic spectrum bonding patterns delocalization and localization of molecular orbitals and electrostatic potential will be discussed [1] A Perzuz-Garrido Journal of Physics Condensed Matter 14 (2002) 5077-5082 [2] P W Fowler and F Zerbetto Chem Phys Lett 243 (1995) 36 [3] D E Clemmer J M Hunter KB Shelimov and M F Jarrold Nature 372 (1994) 248-250 [4] Y Miyamoto N Hamada A Oshiyama and S Saitio Phys Rev B 46 (1992) 1749 [5] M Sauders H A Jimenez-Vazquez RJ Cross and R J Poreda Science 271 (1996) 1693 [6] J Cioslowski and E D Fleischmann J Chem Phys 94 (1991) 3730
4th Southern School on Computational Chemistry
113
Interactions between Uracil and Amino Acids A DFT and Topology Study
Andrea Sterling Jing Wang Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University Jackson MS 39217
Hydrogen-bonding interactions often make substantial contributions to the specificity of protein-nucleic acid complexes It could be used to uniquely distinguish the backbone structures
In this study the interactions between amino acids and the nucleic base Uracil(U) are investigated Eight models concerning the amino acids arginine sparaginesglutamine aspartic acidglutamic acid and serinethreonine are studied The optimization structures were located at three different density functional theory levels B3LYP6-311G (dp) MPW1PW916-311G (dp) and KMLYP6-311G (dp) Frequency analysis results suggest that they are the local energy minima on the potential surface
The H-bonds in the interactive models of Uracil with the amino acids are characterized by the BCPs density and the Laplacian of density via atoms-in-molecules (AIM) approach The interaction energies suggest that amino acids of arginine and aspartic acidglutamic acid bind with Uracil tighter than asparaginesglutamine and serinethreonine
U_asngln_1 U_asngln_2 U_serthr_1 U_serthr_2
U_arg_1 U_arg_2 U_aspglu_1 U_aspglu_2
4th Southern School on Computational Chemistry
114
Li+(Ar)n Complexes mdash Individuum or Missing Link between H+(Ar)n and Na+(Ar)n
Jaroslaw J Szymczak12 Szczepan Roszak12 and Jerzy Leszczynski1
1The Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University 1400 JR Lynch Street PO Box 17910 Jackson Mississippi 39217
USA 2Institute of Physical and Theoretical Chemistry Wroclaw University of Technology Wyb Wyspianskiego 27 50-370 Wroclaw Poland
Ab initio studies of molecular structures and properties of the Li+Arn complexes were carried
out The investigation of the Li+Ar dimer with the MP2(FULL)6-311G+(3df) method provides
satisfactory agreement with the available experimental data This conclusion is extrapolated for
larger Li+Arn (ngt1) clusters which are studied within this level of theory The reported
complexes are stable and deserve the future experimental efforts The obtained results indicate
that the consecutive complexes represent the most symmetrical structures possible with the
closing shell for the Li+Ar6 cation The dissociation energies as well as interaction energy
components follow systematic paths of changes The reveled clusters are different from their
H+Arn predecessors characterized by two solvation shells of 7 ligands total and are also different
from Na+Arn clusters for which the single shell may accommodate eight argons The Ar-Ar
interactions do not influence geometries of small complexes but are noticeable in larger
structures due to repulsive forces caused by the lack of space around the central cation
4th Southern School on Computational Chemistry
115
Guiding Chemical Reaction Path Searches with Graph Theory
Gregory S Tschumper
Department of Chemistry and Biochemistry University of Mississippi
University Mississippi 38655 USA
Ab initio electronic structure theory has evolved into a powerful laboratory tool capable of providing reliable chemical predictions However a priori knowledge of the topology of a given potential energy hypersurface (PES) is generally required In this way the predictive ability of electronic structure theory is limited by the creativity of chemists Standard methods are for example incapable of finding unspecified or unknown reaction paths
Attempts to correct this shortcoming of electronic structure theory have been ongoing for some time Both direct dynamics and isopotential searching have enjoyed success in this field However both have major drawbacks Direct dynamics may predict unexpected chemistry but only if initial conditions are properly specified That is discovery of unexpected reactions depends to a certain degree on chance Isopotential searching has also proved capable of predicting new chemistry and in a systematic way This technique however is computationally demanding in the extreme In addition both of these methods expend large amounts of computational resources sampling chemically uninteresting regions of the PES
Here we present an approach that incorporates the advantages of both direct dynamics and isopotential searching while circumventing their most troublesome draw backs We do so by using graph theory to systematically guide reaction path searches In principle graph theory provides the means for the a priori determination of all possible atomic patterns on a PES as well as convenient matrix representations of molecules electronic structure and chemical reactions As such graph theory has been used extensively in chemistry These uses have included reaction path searches for very specific classes of compounds1 Our current work focuses on generalizing these graph theoretical techniques to all types of molecules and also on determining previously unknown chemical reactions The mathematics behind this approach and the associated programming challenges will be the subject of this talk The current state of the method will be detailed 1 A T Balaban J Chem Inf Comput Sci 1985 25 334-343
4th Southern School on Computational Chemistry
116
Interactions between Fas2 Loop I and AChE as Revealed by Theoretical Study
Jing Wang and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions Department of Chemistry Jackson State University Jackson MS 39217 USA
Acetylcholinesterase (AChE) is an especially efficient serine hydrolase that terminates synaptic transmission at cholinergic synapses through catalyzing the breakdown of the neurotransmitter acetylcholine (ACh) The great speed of the enzyme is essential for rapid modulation of synaptic activity The X-ray crystallography of AChE reveals a narrow and deep active site gorge which is lined with aromatic residues and is about 20 Aring deep The AChE gorge has two separate ligand binding sites the acylation site and the peripheral site (see Fig 1) Fasciculin2 (Fas2) a selective peptidic inhibitor of acetylcholinesterase is a member of the three-fingered peptide toxin super family which is extracted from green mamba venoms Fas2 is found to bind to AChE at the peripheral site
The x-ray data provides an opportunity to examine in detail the interaction of this toxin with
its target site Mutant experimental studies revealed that Fas2 residues Thr8 Thr9 and Arg11 form an active interactive subset located at the tip of Loop I The structural analysis of the experimental data shed light on the interactions of Fas2 and AChE However it does not reveal to what extent each contact residue between Fas2 and AChE contributes to the overall binding energy Therefore a detailed characterization of the binding site is essential for a better understanding of the consequences of the Fas2rsquos binding upon the active catalytic function of AChE
In the present study the interactions at the interface between Loop I of Fas2 and AChE are theoretically explored According to the inspection of the crystallographical data for the interface of Fas2 LoopI and AChE it is supposed that three Fas2 residues (Thr8 Arg11 and Ala12) have
4th Southern School on Computational Chemistry
117
tight contact with AChE residues at this location Therefore three different models were constructed to probe the protein-protein interactions Model_I Model_II and Model_III represent the interactions of Fas2 residue Ala12 (Ala12(F)) vs AChE residue Glu73 (Glu73(A)) Arg11(F) vs Glu82(A) and Asn85(A) Thr8(F) vs Try70(A) Val71(A) and Asp276(A) respectively (Fig 2)
The three models were fully optimized at B3LYP6-311G(dp) level of theory (Fig 3)
Atoms-in-molecule (AIM) theory was employed to characterize the corresponding noncovalent hydrogen bonds through the BCPs densities and Laplacian of electron densities The total interaction energy of Fas2 LoopI with AChE is predicted to be -7846 kcalmol after the zero point and BSSE corrections It is supposed that Thr8(F) which contributes -4892 kcalmol energy to the total interaction energy of LoopIrsquos binding plays the most important role among the three Fas2 interaction sites of Ala12(F) Arg12(F) and Thr8(F) The energy decomposition results through Kitaura-Morokuma scheme suggest that the electrostatic component is the main contribution to the overall interaction energy
The cooperative effects of Model_III have been elucidated through the geometry structure AIM and energy decomposition approaches With the tightened structure of Model_III accompanied by the enhancing of the interaction energy positive cooperative effect is expected which leads to about 83 kcalmol increase in binding energy compared with the combination of individual subset interactions
4th Southern School on Computational Chemistry
118
4th Southern School on Computational Chemistry
119
Molecular Modeling of Crystal Growth Control in Biological Systems
Andrzej Wierzbicki
Department of Chemistry University of South Alabama Mobile Alabama 36688
Members of five kingdoms of organisms are known to produce minerals to support a wide
spectrum of functions and more than sixty minerals are known to be formed by living
organisms Structural and functional properties of biologically formed minerals are quite
remarkable and they have been the subject of intense research over the last forty years One of
the most important aspects of the formation of minerals in living organisms involves the control
over crystal morphology to serve specific functions
In this presentation we will discuss control over crystal growth morphology by molecular
adsorbates for several biologically important classes of crystals such as calcite hydroxyapatite
struvite calcium oxalate monohydrate and calcium pyrophosphate dihydrate Using various
techniques of molecular modeling and dynamic simulations we investigate the molecular nature
of the interactions leading to the control over crystal growth for these crystals Biological
relevance of these studies will be also discussed
4th Southern School on Computational Chemistry
120
Electron Transport in Porphyrines
Ilya Yanov and Jerzy Leszczynski
Computational Center for Molecular Structure and Interactions (CCMSI) Department of Chemistry Jackson State University
Recent developments in nanotechnology open
the possibility for production of nanoscale
sensors which provide instant and inexpensive
way to monitor environmental conditions and
to diagnose chemical and biological hazards
One of the widely proposed molecules
for design of nanosensors is the porphyrin (eg
US Pat No 5981202 64020376 6488891
6495102) Porphyrins (Figure 1) are nitrogen-
containing compounds derived from the parent
molecule tetrapyrroleporphin Utilization of the porphyrin complexes as the sensor is based on
the fact that the absorbance spectrum of metallo-porphyrins are affected by neighboring
molecules Factors causing an increase in pi-electron orbitals at the periphery of the porphyrin
tend to cause red shifts of the absorbance bands Red shifts are found to arise as a function of the
electron affinity of side chain substituents at positions 1-8 As pi electrons are withdrawn from
the periphery the spectrum blue shifts to shorter wavelengths [1-3]
A perspective new way of design of nanosensors is to incorporate porphyrin molecules in
electric circuits and obtain information amperometrically We present here the results of ab initio
study of the conductance of porphyrin molecules Comparison with the available experimental
and theoretical data is provided
1 Igarashi S and YotsuyanagiT (1993) Analytica Chimica Acta 281347-351 2 Mauzerall D (1965) Biohem 4 1801-1810 3 Karasevich IE Anisomova B L Rubailo VL and Shilov AE (1993) Kinetics and
Catalysis 34 651-657